

Summary
To identify disease-relevant T cell receptors (TCRs) with shared antigen specificity, we analyzed 778,938 TCRβ chain sequences from 178 non-small cell lung cancer patients using the GLIPH2 (grouping of lymphocyte interactions with paratope hotspots 2) algorithm. We identified over 66,000 shared specificity groups, of which 435 were clonally expanded and enriched in tumors compared to adjacent lung. The antigenic epitopes of one such tumor-enriched specificity group were identified using a yeast peptide-HLA A∗02:01 display library. These included a peptide from the epithelial protein TMEM161A, which is overexpressed in tumors and cross-reactive epitopes from Epstein-Barr virus and E. coli. Our findings suggest that this cross-reactivity may underlie the presence of virus-specific T cells in tumor infiltrates and that pathogen cross-reactivity may be a feature of multiple cancers. The approach and analytical pipelines generated in this work, as well as the specificity groups defined here, present a resource for understanding the T cell response in cancer.
Keywords: T cell receptor repertoire, TCR, cross-reactivity, NSCLC, TMEM161A, EBV, LMP2A, EntS, cancer, GLIPH2, T cell specificity, tumor-infiltrating lymphocyte
Graphical abstract
Highlights
-
•
The algorithm GLIPH2 enables analysis of shared TCR specificity and HLA prediction
-
•
Tumor-infiltrating T cells cross-react to EBV antigens and shared tumor antigens
-
•
EBV-specific T cells expanded in patients responding to immune checkpoint blockade
-
•
Cross-reactive CD8 T cells express GZMK
Chiou, Tseng, et al. analyze TCRβ chain sequences from 178 non-small cell lung cancer patients and identify shared specificity groups, which in turn enable antigen identification. One such antigenic epitope—a peptide from an epithelial protein—is cross-reactive to epitopes from Epstein-Barr virus and E. coli, suggesting that cross-reactivity may underlie the presence of pathogen-specific T cells in tumor infiltrates.
Introduction
Despite the widespread use of immunotherapies for treating cancer, our understanding of T cell specificities in this disease is very limited (Sharma and Allison, 2020). Antigen specificity is the key determinant of T cell function, but challenges posed by T cell receptor (TCR) diversity and human leukocyte antigens (HLAs) allele polymorphism have been major obstacles to understanding the full scope of antigens recognized by tumor-infiltrating T cells (Arstila et al., 1999; Robins et al., 2010). Tumor-infiltrating T cells that recognize mutated proteins (i.e., neoantigens), non-mutated tumor-associated antigens (TAAs), and viral antigens have been described (Coulie et al., 1994; 1995; Kawakami et al., 1994; Koziel et al., 1995; Murray et al., 1992; Rehermann et al., 1995; Savage et al., 2008; van der Bruggen et al., 1991; Wölfel et al., 1995). In tumors with no known viral etiology, prior reports have identified virus-specific T cells infiltrating tumors, including those that recognize influenza (flu), Epstein-Barr virus (EBV), or cytomegalovirus (CMV) (Andersen et al., 2012; Rosato et al., 2019; Scheper et al., 2019; Simoni et al., 2018). In these tumors, virus-specific tumor-infiltrating T cells are presumed to not recognize tumor antigens and are often referred to as “bystander cells” (Scheper et al., 2019; Simoni et al., 2018).
With respect to the search for TAAs, next-generation sequencing has enabled rapid sequencing of large numbers of TCR variable regions in tumor-infiltrating T cells, but challenges remain in making use of the data generated. This is in part due to hundreds or thousands of distinct TCR sequences that can recognize the same peptide-major histocompatibility complex (MHC) ligand (Song et al., 2017). To reduce this immense sequence diversity to a much smaller number of specificities, we developed an algorithm, GLIPH (grouping of lymphocyte interactions by paratope hotspots; Glanville et al., 2017), and an improved version (GLIPH2; Huang et al., 2020), that parses large numbers of TCR sequences into shared specificity groups that are highly likely to recognize the same peptide-MHC ligands. These shared specificity groups are established based on identical amino acid sequence motifs or strong homologies within the complementarity-determining region 3 (CDR3) of the TCRβ chain.
Here, we used GLIPH2 to identify over 66,000 high-quality, shared specificity groups from 778,938 CDR3β sequences found in 178 non-small cell lung cancer (NSCLC) patients with surgically resectable tumors (Reuben et al., 2020). Four hundred thirty five shared specificity groups were clonally expanded in the tumor compared to the adjacent lung tissue. Among those, CDR3β sequences containing a “S%DGMNTE” sequence motif were prioritized for antigen discovery using HLA-A∗02 yeast display library, where “%” denotes the amino acid that varied (Gee et al., 2018). T cells with the “S%DGMNTE CDR3β” motif responded to the non-mutated tumor antigen TMEM161A, as well as antigens from EBV and E. coli, demonstrating T cell cross-reactivity to TAAs and common pathogens. Furthermore, we uncovered a second example of cross-reactivity between an endogenous antigen and an EBV epitope and two other cases where EBV-specific CDR3β sequences were clonally expanded in patients who had clinically significant responses to anti-PD-1 treatment. This suggests that pathogen cross-reactivity may be an important feature in the interaction between neoplasia and T cell immunity. Overall, the approach presented here enables the comprehensive analyses of shared T cell specificities in human cancer and the identification of specific antigens using a yeast display library, with broader application to other cancer types.
Results
Defining shared specificity groups for tumor-infiltrating T cells in human lung cancer
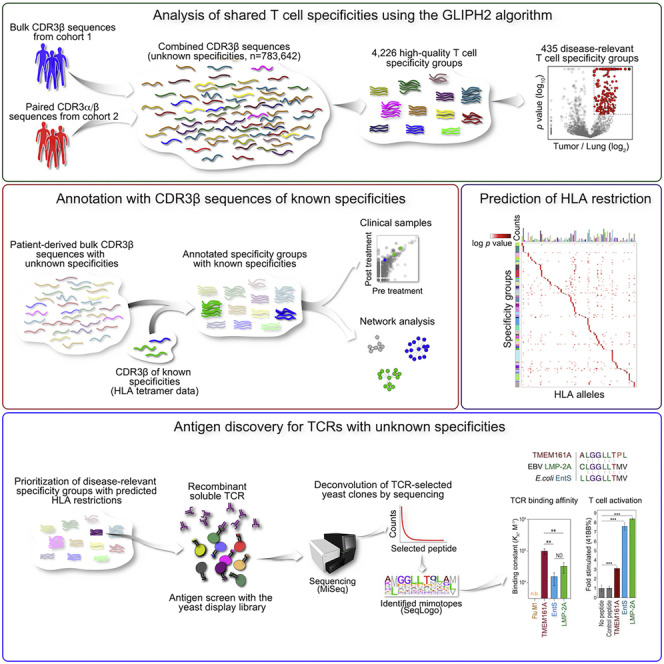
As described previously, GLIPH2 identifies CDR3β sequences that are highly likely to have shared peptide-MHC specificities based on local motifs and/or global homology (Glanville et al., 2017; Huang et al., 2020). To identify T cells recognizing shared tumor antigens in lung cancer, we applied GLIPH2 to a recently published MD Anderson Cancer Center (MDACC) dataset of 778,938 distinct CDR3β sequences from NSCLC tumors and from adjacent lungs. This clinical cohort represents 178 patients with surgically resectable disease and with available HLA data (Table S1) (Reuben et al., 2020). We first defined shared specificity groups with a set of specific filtering criteria and identified 66,094 shared specificity groups (Figure 1A; Table S2). To focus on the most disease-relevant TCRs, we further identified 4,226 specificity groups with evidence of clonal expansion, and of these, 435 were enriched in tumor compared to adjacent lung (Figures 1A and S1A; Table S3). Thus, the CDR3β members of these 435 tumor-enriched specificity groups are inferred to recognize yet undiscovered TAAs.
Figure 1.

Establishing specificity groups with CDR3β sequences from lung cancer patients
(A) Analysis of shared T cell specificities with the GLIPH2 algorithm. Step 1: 778,938 CDR3β sequences from the MDACC cohort as input for GLIPH2 analysis. Step 2: establish 66,094 specificity groups with multiple criteria (Figure S1A). Step 3: establish 4,226 clonally expanded specificity groups. Step 4: establish 435 clonally expanded, tumor-enriched specificity groups.
(B) Clinical relevance of tumor-enriched specificity groups in lung cancer. The most clonally expanded CDR3β sequences from tumors belonged to the 435 tumor-enriched specificity groups, whereas those from lung tissues of healthy donors and COPD patients did not. The trend was validated with tumors from a second NSCLC cohort (the TRACERx consortium, n = 202, validation). ∗∗∗p < 0.001; ∗p < 0.05 by paired t test. NS, not significantly different.
(C) Network analysis of 396 specificity groups annotated with CDR3β sequences from HLA tetramers with flu (red), EBV (green), and CMV (blue) antigens. Each dot is a specificity group, edges indicate the presence of identical CDR3β sequence(s) shared across two specificity groups.
(D) Percentage (%) of HLA-A∗02 or HLA-B∗08 tetramer-annotated specificity groups with significantly enriched the A∗02 (purple, left plot) or B∗08 (blue, right plot) supertype alleles, respectively. Specificity groups annotated with tetramers of other HLA alleles (other tetramer) were included for comparisons.
(E) Percentage of shared specificity between any two given MDACC NSCLC patients (% shared between any 2 patients, total n = 178) based on CDR3β membership in total specificity groups regardless of clonal expansion (n = 66,094), membership in clonally expanded specificity groups (n = 4,226), or comparison of identical CDR3β sequences. Boxes represent medians with the first (25th) and third (75th) quartiles.
(F and G) Bootstrapping of specificity group numbers (y axis, specificity group #) with varying sampling sizes (individuals sampled) for either HLA-A∗02+ or HLA-A∗02− NSCLC patients (F) or healthy donors (G, Emerson study). Data represent means with 3× standard errors from repeated sampling.
Next, we reasoned that T cells recognizing shared tumor antigens would undergo clonal expansion in NSCLC patients but not in individuals without cancer. We observed a significantly higher percentage of the expanded CDR3β clones in the MDACC NSCLC cohort (Figure 1B) belonging to the 435 tumor-enriched specificity groups compared to the remainder of less expanded TCRs. We made a similar observation in a validation cohort of 1,173,806 CDR3β sequences from 202 tumor samples representing 68 NSCLC patients (TRACERx; Joshi et al., 2019; Figure 1B). In contrast, adjacent lungs of cancer patients (not involved by tumor) (Figure S1B), lungs from healthy donors, or lungs from chronic obstructive pulmonary disease (COPD) patients (without cancer diagnoses) (Reuben et al., 2020) had fewer CDR3β clones that belonged to tumor-enriched specificity groups. (Figure 1B). Together, these data demonstrate that GLIPH2 successfully parsed a large dataset of CDR3β sequences into a few hundred tumor-enriched specificity groups with disease relevance to NSCLC.
Viral specificity group inferences from HLA tetramer datasets
To validate the shared specificity groups established by GLIPH2, we included CDR3β sequences from publicly available HLA tetramer databases in combination with the MDACC CDR3β sequences for a joint GLIPH2 analysis (Glanville et al., 2017; Shugay et al., 2018; Song et al., 2017). The publicly available tetramer CDR3β sequences primarily cover viral specificities and were experimentally shown to bind epitopes in the context of their respective HLAs. This allowed us to annotate some specificity groups with CDR3β sequences linked to unique epitopes in the context of their HLA and therefore infer the shared specificity of the remaining CDR3β members. The joint analysis annotated 394 of the 66,094 shared specificity groups (Figures 1A and 1C). Of these specificity groups, 71 were clonally expanded and annotated with 10 distinct tetramers (Figure S1C). We found that CDR3β sequences with inferred specificities to flu-, EBV-, or CMV-derived antigens collectively did not show biases in the tumor compared to the adjacent lung (data not shown). Furthermore, the estimated frequencies of these viral-specific CDR3β clones were well above the naive level (one in every 105–106) and on par with the previously reported ranges measured by HLA tetramer staining (data not shown) (Andersen et al., 2012; Rosato et al., 2019; Simoni et al., 2018). Thirteen of the 27 expanded flu M1-annotated specificity groups carry either the “RS” or “GxY” motifs known to be critical for the engagement with the flu-M158–66 peptide/HLA-A∗02 (Figure S1D) (Song et al., 2017). Network analysis organized these tetramer-annotated specificity groups with identical CDR3β sequence members into communities (Figures 1C and S1C). Specificity groups belonging to a given community were consistently annotated with identical HLA tetramers (Figures 1C, S1C, and S1D), indicating that some antigen specificity groups, albeit sharing distinct sequence motifs, are exhibiting the same specificity and HLA restriction. Among the 394 shared specificity groups annotated with tetramers, 588 out of 634 identical CDR3β sequence members (93%) connected specificity groups annotated with the same tetramer (Figures S1E and S1F). Among the 71 clonally expanded specificity groups annotated with tetramers, 92 out of 92 identical CDR3β sequence members (100%) connected groups annotated with the same tetramer (Figures S1C and S1G). This result indicates that while CDR3β sequences are not the sole determinant of specificity, GLIPH2 analysis of CDR3β sequences leads to correct specificity inferences in the vast majority of cases.
HLA allele enrichment within TCR specificity groups makes robust inferences of HLA restriction
We next examined whether HLA allele enrichment within a specificity group accurately reflected the HLA context annotated by the tetramer. We quantified the enrichment of HLA supertypes across all clonally expanded specificity groups annotated with tetramer CDR3β sequences (Harjanto et al., 2014; Sidney et al., 2008). We focused on the HLA-A∗02 and HLA-B∗08 supertypes since these tetramer-defined HLA contexts were the most abundant in the MDACC dataset (Figure S1C). We reasoned that if a given specificity group was annotated by an HLA/peptide tetramer, there should be a higher probability of observing enrichment of HLA allele(s) belonging to the same supertype by GLIPH2. Indeed, 36.7% of all HLA-A∗02 tetramer-annotated specificity groups were enriched with HLA-A∗02 supertype alleles, whereas none of the groups annotated with non-A∗02 tetramers were enriched (Figure 1D). While 62.5% of HLA-B∗08 tetramer-annotated specificity groups were enriched with HLA-B∗08 supertype alleles, only 3.13% of the non-B∗08 tetramer-annotated groups were enriched (Figure 1D). Therefore, the enrichment of a given HLA allele within a specificity group accurately reflected the HLA context of the cognate antigen. Previous work has also validated the inferred HLA restricting element by expressing TCR heterodimers in reporter T cells and identifying their peptide-MHC specificities (Glanville et al., 2017).
Inferred T cell specificities enable robust comparisons of T cell repertoires across patients
One of the major advantages of establishing TCR specificity groups with GLIPH2 is that it greatly facilitates TCR repertoire analysis across individuals. In the MDACC lung cancer dataset, an average ∼0.4% of the repertoire was shared between any two patients (Figure 1E). The likelihood of measuring such shared specificities increased to 1.9% when considering the 4,226 shared specificity groups (enriched in clonally expanded TCR sequences) and to 5.3% when considering all 66,094 shared specificity groups (Figure 1E). This demonstrated that GLIPH2 captured shared specificities in the T cell repertoire to an extent that was not possible by only comparing CDR3β sequences across individuals.
Next, we reasoned that if a finite number of shared TCR specificities exist in a particular disease context, the number of specificity groups should reach saturation given enough patients. By bootstrapping from patients who carry at least one copy of the most prevalent HLA-A∗02:01 allele, we found that the number of HLA-A∗02:01-enriched specificity groups reaches saturation at ∼70 patients (Figure 1F). Repertoires from at least nine patients were needed to establish half of all the specificity groups (n = 77) (Figure 1F). In contrast, concurrent bootstrapping from A∗02:01-negative patients accounted for far fewer A∗02:01-enriched specificity groups (Figure 1F). In addition, bootstrapping from an independent, healthy cohort with comparable CDR3β sequencing depth did not reach saturation over similar sampling sizes, consistent with a higher prevalence of TCRs belonging to these specificity groups in NSCLC patients carrying the A∗02:01 allele (Figure 1G). Of note, the number of patients needed to establish half of specificity groups was dependent on the level of clonal expansion, the numbers of specificity groups, and the sequencing depth (Figures S1H–S1J). Thus, a complete set of TCR specificity groups could be established with finite patient numbers. Furthermore, these results showed that T cell specificity inference is strengthened by HLA allele enrichment.
Experimental validation of GLIPH2-inferred specificities
Given that experimental validation of T cell specificities requires TCRα/β pairs, we therefore performed single-cell TCR sequencing (scTCR-seq) from 15 early-stage NSCLC patients treated at Stanford (Table S4). Tumor-infiltrating T cells were prepared from surgically resected specimens and index sorted by fluorescence-activated cell sorting (FACS) before sequencing (Figure S2A). scTCR-seq yielded 4,704 paired CDR3α and CDR3β sequences. We combined these CDR3β sequences with the MDACC NSCLC sequences for a joint GLIPH2 analysis. We chose to validate four T cell clones belonging to three flu M1-annotated specificity groups (SV%SNQP, SIRS%YE, and S%RSTDT) and one EBV BMLF1-annotated specificity group (RTG%GNT). We used Jurkat 76 cells, deficient for both TCR⍺ and TCRβ, to express the four TCR candidates and co-cultured them with HLA-A∗02+ T2 cells pulsed with their respective peptides (Figures S2B and S2C). Three of them responded to their predicted antigens in the context of HLA-A∗02, showing the robustness of GLIPH2 for inferring T cell specificities (Figures S2B and S2C). Similar analyses of specificity group members in M. tuberculosis studies found that ∼80%–90% of the TCRs recognized the predicted peptide-MHC ligands (Glanville et al., 2017).
Characterization of tumor-enriched specificity groups
To identify disease-relevant specificity groups, we focused on the 435 tumor-enriched specificity groups that revealed a strong clonal bias in the tumor compared to the adjacent lung (Figures 2A and S1A). Using the transcriptome data available from 84 patients (total n = 178), we found that the percentage of T cells belonging to these tumor-enriched specificity groups correlated with gene set enrichment analysis (GSEA) hallmark signatures of cancer progression, including MYC and the cell cycle programs (Figures 2B). In contrast, using the specificity groups expanded in the adjacent lungs (n = 114), we failed to observe any significant correlation with the GSEA hallmark gene sets (data not shown). Thus, this result showed a correlation between the 435 tumor-enriched specificity groups and an aggressive, highly proliferative cancer phenotype. Next, we systematically examined the enrichments of all HLA alleles in the MDACC cohort for the 435 tumor-enriched specificity groups and found only one predominant allele in most cases (n = 202/435; Figures 2C and 2D). Of note, we found that in cases when motifs were enriched with multiple predominant alleles, e.g., those co-enriched with both HLA-B∗07:02 and HLA-C∗07:02 (Figure 2C), strong linkage disequilibrium in the associated HLA alleles could be observed.
Figure 2.

The TCR members of the tumor-enriched specificity group with the motif “S%DGMNTE” are inferred to recognize tumor antigen in the context of HLA-A∗02
(A) Left: volcano plot showing the comparison of the 4,226 clonally expanded specificity groups between tumor (T) and the adjacent lung (N) by Poisson test. The y axis represents the negative log10 converted p values of the Poisson test, and the x axis represents the log2 converted fold difference between tumor and adjacent lung (T/N). Dot size represents levels of clonal expansion. Tumor-enriched specificity groups (n = 435) are highlighted in red. Right: volcano plot of T/N comparison for CDR3β clonotypes. CDR3β clones of the 435 tumor-enriched specificity groups (left) are highlighted in red.
(B) Pearson correlations and the corresponding p values between the signature scores for the hallmark GSEA gene sets (n = 50) and the percentages of CDR3β clones belonging to the 435 tumor-enriched specificity groups. Significant comparisons are highlighted in red (p < 0.05).
(C) Heatmap showing the −log10 p values of top-enriched HLA allele(s) of the 435 tumor-enriched specificity groups. Top, number of MDACC patients carrying each indicated HLA alleles.
(D) Number of top-enriched HLA allele(s) found in each of the 435 tumor-enriched specificity groups.
(E) Volcano plot for the 4,226 NSCLC specificity groups as in (A, left). The tumor-enriched specificity groups significantly enriched with HLA-A∗02 alleles (p < 0.05 by Fisher’s exact test) are colored in green. The specificity group “S%DGMNTE” is highlighted.
(F) The distinct CDR3β sequence members of the “S%DGMNTE” specificity group. For each CDR3β sequence, the Vβ gene usage (Vβ), number of patients with each sequence (patient counts), number of HLA-A∗02+ patients (counts of HLA-A∗02+ cases/total), and the average clonal frequencies (% by patient) found in the adjacent lung, tumor, and peripheral blood are shown. ND, not detected. Bottom: p values for the enrichment of Vβ gene usage, HLA-A∗02 alleles, and the level of clonal expansion are shown.
Identification of a shared specificity group cross-reactive to tumor and pathogen-derived antigens in human lung cancer
Of the 435 tumor-enriched specificity groups, we prioritized those that fulfilled the criteria of (1) having a paired TCRα/β clonotype from the Stanford cohort and (2) significantly enriched with HLA-A∗02 alleles by Fisher’s exact test. This led us to focus on the specificity group with the “S%DGMNTE” CDR3β motif (Figures 2E and 2F). Hence, the candidate TCRα/β clonotype (referred to as TCR2) bearing the CDR3α sequence CAVLMDSNYQLIW and CDR3β sequence CASSGDGMNTEAFF was chosen for antigen identification (Figure 3A).
Figure 3.

Identification of tumor and pathogen-derived antigens recognized by a tumor-enriched TCR in human lung cancer
(A) Top: top-20 mimotopes from the 4th round of selection on an 11-mer yeast library are used to stimulate Jurkat-TCR2 cells. CD69 fold change is shown compared to unstimulated control. Bottom: ranked raw counts (log10) of the enriched mimotopes from the selection.
(B) Alignment of the top-two mimotopes with peptides from the human TMEM161A locus, EBV LMP-2A, and E. coli EntS. All peptides were 9-mers and predicted to bind HLA-A∗02 with high affinities.
(C) Left: representative FACS plots showing the stimulation of the Jurkat-TCR2 cells with 9-mers from the human TMEM161A locus (TMEM9-mer), LMP-2A of EBV (LMP9-mer), and EntS from E. coli (EntS9-mer); right: results of Jurkat-TCR2 cell stimulation in triplicate. Control PP, control peptide (GILGFVFTL); No PP, no peptide.
(D) Stimulation of primary T cells ectopically expressing TCR2 TCRα/β chains with either 9-mers (left) or full-length proteins (right). Stimulation of primary T cells expressing TCR14 by 293T-A∗02 cells expressing full-length FluM1 protein was shown as control. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by t test. Control PP, control peptide (GILGFVFTL).
(E) The binding of TCR2 to the indicated A∗02/9-mers was determined by biolayer interferometry. An overlay of binding traces over a concentration series of the indicated A∗02/9-mers from one representative experiment is shown. The data points are represented as open circles and the fits from a simple 1:1 Langmuir interaction model are indicated by solid lines. Each binding experiment was repeated three times.
(F) The equilibrium association constants (KA) of the surface plasma resonance as in (E). The flu M1 peptide showed no detectable binding (n.b.) to TCR2. Significance was determined by t test after one-way ANOVA. The reported p values were corrected for multiple comparisons. ∗∗p < 0.01. ND, not different. All error bars represent standard deviation of the mean.
To identify the cognate epitopes of the candidate clone TCR2, we screened a yeast library displaying peptides of four different lengths (8–11 amino acids) in the context of wild-type HLA-A∗02:01 (Gee et al., 2018). Four rounds of selection with a multimer of TCR2 led to the enrichment of peptide sequences (mimotopes) in the 11-mer library (Table S5). We performed an in vitro stimulation assay with the top-20 enriched mimotopes and showed that the top-two sequences “AMGGLLTQLAM” and “KLGGLLTMVGV” stimulated Jurkat cells expressing TCR2 (Jurkat-TCR2) (Figures 3A and S3A). A protein database search (UniParc) (UniProt Consortium, 2019) led to the identification of multiple endogenous 9-mers that shared close sequence similarities with the top-two mimotopes and were predicted to bind HLA-A∗02:01 with anchors separated by six instead of eight amino acids (Figures 3B and S3B). Indeed, 9-mer variants of the top mimotope stimulated Jurkat-TCR2 cells to comparable levels as the 11-mer counterpart (Figure S3C). This result indicated that the identified HLA-A∗02 antigens were de facto 9-mers.
We functionally validated all candidate endogenous peptide 9-mers resembling the top-two mimetopes (11-mer) (Figure S3B). We found that 9-mers from the mammalian protein TMEM161A (TMEM9-mer, ALGGLLTPL), the latent membrane protein 2a (LMP9-mer, CLGGLLTMV) from EBV, and the enterobactin exporter (EntS9-mer, LLGGLLTMV) from E. coli could all stimulate the Jurkat-TCR2 cells (Figures 3C, S3D, and S3E). These results demonstrated that TCR2 was cross-reactive to antigens from humans and pathogens. The accurate GLIPH2 inference of HLA restriction facilitated antigen discovery with the HLA-A∗02:01 yeast library.
To show that the full-length proteins TMEM161A, LMP2, and EntS could be processed, presented on HLA-A∗02:01, and activate specific T cells, we overexpressed these proteins in HLA-A∗02+ 293T cells and measured the responses of co-cultured primary T cells expressing TCR2. Similar to the pulsed peptides, 293T cells expressing full-length TMEM161A, LMP2, and EntS all stimulated the co-cultured TCR2-T cells, with TMEM161A appearing to be the weakest stimulator (Figure 3D). We further performed a biolayer interferometry to quantify the binding affinity of each cross-reactive epitopes to TCR2 and showed that the weakest stimulator TMEM9-mer revealed the most stable binding to TCR2 (Figures 3E and 3F). Thus, this result suggested a partial uncoupling of binding affinity and signaling strength, similar to the previous report (Sibener et al., 2018). In summary, we identified a tumor-enriched TCR specificity group with cross-reactivity to both a TAA and pathogen-derived antigens.
TMEM161A is overexpressed on human lung cancer
We found significantly higher levels of TMEM161A protein expression in human lung cancer compared to adjacent lung tissue (Figures 4A, 4B, and S4A). We also noted some heterogeneity in TMEM161A expression on some tumor sections (Figure S4B). We also examined TMEM161A gene expression in the Cancer Genome Atlas (TCGA) NSCLC dataset. Consistent with protein expression, we found higher levels of TMEM161A transcript in tumors compared to the adjacent lung. The level of TMEM161A expression was higher in squamous cell carcinomas (SCCs) of the lung compared to adenocarcinomas (Figure 4C). Whole-exome sequencing of specimens from the Stanford cohort did not identify any mutation within the coding region of the TMEM161A locus, supporting its role as a non-mutated TAA (Table S6). Similarly, less than 1% of deleterious mutations in the TMEM161A locus were found in the pan-lung cancer TCGA dataset (n = 6/1053; Figure S4C). In addition, TMEM161A expression in lung cancer associated with GSEA signatures related to cell proliferation programs and the proto-oncogene MYC targets, consistent with the general trend revealed by the 435 tumor-enriched specificity groups (Figures 2B, 4D, and 4E). In contrast, TMEM161A expression appeared to inversely correlate with gene sets related to inflammatory responses (Figures 4D and 4E). These data showed that TMEM161A is a TAA overexpressed in human NSCLC and associated with gene expression signatures such as MYC and cell cycle.
Figure 4.

TMEM161A protein is highly expressed in human lung cancer
(A) Representative images of TMEM161A immunohistochemistry on tumor (top) and the adjacent lung (bottom) sections from four patients. Scale bar, 100 μm. Rightmost panels: zoomed in images of patient A16 tumor with TMEM161A immunohistochemistry (top) and H&E staining on a serial section (bottom). Scale bar, 40 μm.
(B) Quantification of TMEM161A immunohistochemistry on sections from the Stanford NSCLC cohort (n = 11). Boxplots show medians with the first (25th) and third (75th) quartiles with individual data points. ∗∗∗p < 0.001.
(C) TMEM161A expression quantified by bulk RNA-seq of the indicated samples from TCGA (n = 958) is shown in boxplots. Adj-Ctrl, the adjacent lung control. TMEM161A expression normalized against Adj-Ctrl is shown. p values were calculated with the Wilcoxon Rank Sum test. ND, not significantly different. Boxplots represent medians with the first (25th) and third (75th) quartiles.
(D) Gene set enrichment analysis of the ranked gene list based on Pearson correlation with TMEM161A abundance in the pan-lung cancer TCGA dataset (n = 958). Left: hallmark gene sets with highest (blue) and lowest (red) normalized enrichment scores are indicated, and their enrichment curves are shown (right).
(E) Single-sample GSEA signature scores (Sig score) of two most and two least enriched hallmark signatures are plotted against TMEM161A expression. Pearson correlation coefficients are shown in plots (cor coef).
T cells recognizing TMEM161A antigen have the “S%DGMNTE” sequence motif
We further interrogated the TCR sequence identity of TMEM161A-specific CD8+ T cells in vivo and examined their clinical relevance. TMEM161A-specific T cells could be detected in 31/78 (40%) of HLA-A∗02+ patients in the MDACC NSCLC cohort. We used TMEM9-mer/HLA-A∗02 tetramers to sort T cells from the tumor of patient A6, where the TCR2 clone was first identified. scTCR-seq of TMEM9-mer/A02 tetramer+ T cells from tumor and the adjacent lung confirmed that they carried the “S%DGMNTE” motif, consistent with their recognition of TMEM161A in vivo (Figures S4D and S4E). We next examined how tumor characteristics impact the recruitment of T cells with the “S%DGMNTE” motif among patients who were HLA-A02+. We observed that T cells with the “S%DGMNTE” motif were observed more frequently in SCCs compared to adenocarcinomas, similar to the expression pattern of TMEM161A (Figures 4C and S4F). We also noted that the percentage of T cells with the “S%DGMNTE” motif in tumors with a mutation count of less than 500 was higher than in tumors with mutation count of greater than 500 (total n = 34), although this observation may be impacted by the association between total infiltrating T cell numbers and mutation burden (Figure S5A). Finally, although the presence of detected T cells with the “S%DGMNTE” motif in tumors alone did not predict patient outcome, we observed that T cells with the “S%DGMNTE” CDR3β motif were among 146 shared specificity groups enriched in patients without recurrence (Figures S5B–S5D).
CD8+ T cells with the “S%DGMNTE” motif were also detected in healthy donors
To characterize the cross-reactive TMEM161A-specific and pathogen-specific clonotypes, we used TMEM9-mer/HLA-A∗02 tetramers or EntS9-mer/HLA-A∗02 tetramers to sort CD8+ T cells from the peripheral blood of HLA-A∗02+ healthy donors and NSCLC patients by FACS (Figure 5A). We saw no difference in the frequency of HLA-A∗02/TMEM9-mer+ CD8 T cells in healthy donors and lung cancer patients (Figures 5B and 5C), suggesting that these T cells were likely maintained due to cross-reactivity to pathogen-derived antigens. Consistent with this, the frequencies of these specific T cells, as quantified by tetramers or GLIPH2, were approximately one in every 103–105 T cells (tetramer-measured: 0.0032%–0.0980%; GLIPH2-inferred: 0%–0.2643%), higher than naive level for human CD8+ T cells (Yu et al., 2015).
Figure 5.

Isolation and characterization of cross-reactive TMEM161A-specific T cells from peripheral blood of healthy donors and lung cancer patients
(A) Schematic showing the procedure used to capture antigen-specific T cell clones from HLA-A∗02+ healthy donors and NSCLC patients. Cells were sorted by FACS directly into 96-well plates for scRNA-seq and scTCR-seq.
(B) Representative FACS plots of T cells sorted with indicated tetramers from the PBMC of HLA-A∗02+ healthy donors (He65 and He66) or HLA-A∗02+ NSCLC patients (A6 and A17).
(C) Percentage of tetramer+ T cells from healthy donors (n = 11) and NSCLC patients (n = 7). Boxes represent medians with the first (25th) and third (75th) quartiles. NS, not significantly different.
(D) Percentage of distinct CDR3β sequences in tetramer-sorted T cells from healthy donors and NSCLC patient. Numbers in plots represent the cell counts.
(E) Indicated TCR clonotypes identified with tetramers were expressed in Jurkat cells and co-cultured with T2 cells pulsed with indicated 9-mers. y axis (fold stimulated) shows activation by CD69 fold change compared to unstimulated control. ∗∗∗p < 0.001. Ctrl peptide, control peptide (GILGFVFTL).
(F and G) Cell-mediated cytotoxicity of H1395 lung cancer cells. Primary T cells ectopically expressing TCR2α/β chains were co-cultured with the A∗02+ H1395 cancer cells and pulsed with either no peptide, TMEM9-mer, or LMP9-mer. Representative images (F) and results using cells from two different donors (G) are shown. ∗∗p < 0.01; ∗∗∗p < 0.001 by t test. Error bars represent standard deviation of the mean.
Regardless of which tetramer was used to sort peripheral blood T cells, the CDR3β sequences of the sorted cells consistently carried the “S%DGMNTE” motif (Figure 5D). In fact, we found a variety of CDR3β sequences sharing the “S%DGMNTE” motif where % could be a glycine, glutamate, or serine, confirming the diversity seen in the GLIPH2 analysis using the MDACC data (Figures 2F and 5D). Furthermore, single-cell RNA sequencing (scRNA-seq) data suggested that HLA-A∗02/TMEM9-mer+ cells mostly manifested effector T cell states, indicating that they had previously encountered their cognate antigens, even in healthy individuals (Figures S5E and S5F).
To functionally validate CDR3α/β sequences from the tetramer-sorted clones, we generated stable Jurkat cells expressing the TCRα/β chains identified with the tetramers. We then quantified their reactivities to both TMEM9-mer and pathogen-derived 9-mers in the context of HLA-A∗02:01. We found that the Jurkat cell clones with the “S%DGMNTE” CDR3β motif could respond to all cross-reactive peptides only when paired with the permissive TCR2α chain (CDR3α: CAVLMDSNYQLIW; Figure 5E). For example, we identified a CDR3α/β pair that did not carry the “S%DGMNTE” motif and recognized TMEM9-mer but not the microbial antigens (TCR16; Figure 5E). Finally, we quantified the cell-mediated cytotoxicity induced by the cross-reactive epitopes by co-culturing an HLA-A∗02+ lung cancer cell line H1395 with primary T cells expressing TCR2. Compared to the no peptide control, both LMP9-mer and TMEM9-mer induced more than 50% of target cell lysis (Figures 5F and 5G). Cancer cells pulsed with TMEM9-mer were weaker targets for cell-mediated cytotoxicity compared to those with LMP9-mer, consistent with the results of the T cell activation studies (Figures 3C and 3D). In summary, CD8+ T cells with the “S%DGMNTE” motif cross-reacted with the TMEM161A tumor antigen and the pathogen-derived antigens EntS and LMP2 when paired with the permissive α chain. Recognition of these cross-reactive antigens on HLA-A∗02 led to target cell lysis by CD8+ T cells with the “S%DGMNTE” motif.
Phenotypic characterization of TMEM161A-specific CD8+ T cells in lung cancer
We sequenced the full single-cell transcriptomes of 2,950 sorted, tumor-infiltrating T cells from 10 NSCLC patients using the SMART-seq method and acquired their paired CDR3α/β repertoires (Figure S2A) (Han et al., 2014; Stubbington et al., 2016). We identified 14 major cell states of which 13 could be mapped to those reported in a separate cohort (Figures 6A and S6A–S6C; Table S7) (Guo et al., 2018). Clusters c5, c6, c12 (CD8+ T cells with effector phenotypes), c7, and c10 (CD8+ T cells with resident memory phenotype) were among the most expanded (Figures 6B and 6C). To uncover the cell states of clones specific for shared antigens, we examined the scRNA-seq profiles of the TCR specificity groups members. We found that 2.9% of the T cells (n = 86/2950) belonged to the clonally expanded specificity groups (top, Figures 6D). Twelve of these T cells were members of the 435 tumor-enriched specificity groups, whereas 13 of these T cells were inferred to be specific to viral epitopes (Figures 1C and 6D). Interestingly, T cells belonging to the tumor-enriched specificity groups were biased toward the effector phenotype (c5) and differentially expressed EOMES, KLRG1, GZMK, and other genes expressed in activated natural killer cells (Figures 6D–6F; Table S7). Consistently, HLA-A∗02/TMEM9-mer tetramer-sorted CD8+ T cells from tumor also preferentially exhibited the effector T cell phenotype c5 (Figures 6D–6F). Pseudotime trajectories and activation/exhaustion signature scores indicate that these T cells adopt distinct cell states (Figures 6G and 6H). In comparison, T cells inferred to be virus-specific exhibited cell states that included effector (c5, c6, c12) and tissue resident-memory phenotypes (c7). In conclusion, TMEM161A-specific CD8+ T cells showed a range of effector T cell states in NSCLC, consistent with recognition of their cognate antigen in situ.
Figure 6.

Phenotypic characterization of the TMEM161A-specific CD8+ T cells
(A and B) Dimension reduction by Uniform Manifold Approximation and Projection (UMAP) of the scRNA-seq data from 2,950 sorted tumor-infiltrating T cells from 10 NSCLC patients (Stanford cohort). The identified cell clusters (n = 14) are labeled with distinct colors (A) and shown with varying dot sizes representing the level of clonal expansion (B).
(C) Clonality of the 2,950 sorted T cells as in (B) quantified as 1 - Pielou’s evenness.
(D) Breakdown of cell states for T cell clones of the 4,226 specificity groups defined in Figure S1A (top), viral-related specificity groups (second from top), the 435 tumor-enriched specificity groups (third from top), and TMEM9-mer/A∗02 tetramer-sorted CD8 T cells from tumor (bottom, patient A6).
(E) Heatmap showing differentially expressed genes for each cell cluster defined in (A). Select differential genes for cluster c5, c6, and c7 are highlighted.
(F) Stacked violin plot showing the expression of highlighted differential genes in (E) in all cell clusters.
(G) Pseudotime trajectory of CD8+ single cells by Monocle (v2.10.1).
(H) Exhaustion score versus activation score for CD8+ T cells sorted by the HLA-A∗02/TMEM9-mer tetramer (top right) and those that belong to tumor-enriched specificity groups (bottom right), colored by the cluster identity. Exhausted CD8+ T cells (c11) and activated CTL (c12) are shown for comparison.
Expansion of EBV-specific T cell clones in patients responding to immune checkpoint blockade
To see if pathogen-specific T cells might impact clinical responses to anti-PD1 checkpoint immunotherapy, we analyzed the TCR repertoire of two NSCLC patients who experienced a clinical response to treatment (Figure S7A). We sequenced paired CDR3α/β repertoires on both pre- and post-treatment blood samples and identified 102 CDR3β clonotypes that expanded in post-treatment samples (Figure 7A). Of these expanded clones, 41 belonged to 99 specificity groups identified in tumor-infiltrating T cell CDR3β repertoires (total n = 66,094; Figure S1A). We used tetramer-defined T cell CDR3β sequences to annotate these specificity groups and found 11 (total n = 99) containing 3 expanded CDR3β clones inferred to recognize EBV and flu antigens (Figure 7B). To validate the specificity inferences, we created two Jurkat cell clones expressing the TCRα/β chains inferred to recognize the EBV antigens and a T2 cell line expressing wild-type B∗35 (Figures 7B and S7B). Indeed, upon co-culture with the T2-B∗35 cells, both Jurkat-TCR27 and -TCR28 cells responded to the predicted EBV peptides (Figure 7C). Of note, these EBV-specific specificity groups were not only expanded post-treatment but also showed a bias in tumor compared to the adjacent lung, suggesting the potential cross-reactivities to unknown TAAs (Figure S7C). Furthermore, we found that the EBV-specific clone TCR15 (CDR3β: CSARTGVGNTIYF) identified from patient A11 (Figure S2C) was inferred to have the same antigen specificity as two previously reported clones detected in patients receiving immune checkpoint blockade at the time of clinical response (CDR3β: CSARVGVGNTIYF and CSARSGVGNTIYF) (Anagnostou et al., 2019). Our analysis suggested that these clones belonged to the “R%GVGNT” specificity group predicted to recognize EBV-BMLF1 (GLCTLVAML) in the HLA-A∗02 context. We further tested three similar epitopes from the human ORFeome that were predicted to bind HLA-A∗02 and found the endogenous “LLGTLVAML” from the human CLDN2 locus also stimulated the Jurkat-TCR15 clone (Figure S7D), indicating that TCR15 was indeed cross-reactive to both EBV and a TAA. In summary, these results indicated that pathogen-specific T cells in patients might play a role in the anti-tumor immune responses upon treatment with immune checkpoint inhibitors.
Figure 7.

Virus-specific CD8 T cell clones expanded in patients responding to anti-PD1 treatment
(A) Comparisons of pre- and post-treatment CDR3β clonal frequencies (in log10 percent) in the peripheral blood of patient M1 (left) and M2 (right). CDR3β clones inferred to recognize viral antigens are highlighted.
(B) Specificity groups containing expanded CDR3β clones post-treatment (column 5, CDR3β sequence) from patients M1 or M2 (column 6, Patient ID) that are annotated with viral tetramer CDR3β sequences (column 2–4, antigen and HLA alleles of the tetramers). Enrichment of the A∗02:01 or B∗35:01 allele is shown (last two columns, p values from the hypergeometric tests are shown). CDR3α/β sequences of the two EBV-related expanded clones from patient M2 are shown at the bottom.
(C) TCR27- (CDR3β: CASSTGDSNQPQHF, top panels) and TCR28- (CDR3β: CASSARTGELFF, bottom panels) Jurkat cell lines were created and tested for their reactivities to the predicted EBV antigens in the context of B∗35 as shown in (B). TCR27- and TCR28-Jurkat cells were co-cultured with T2-B∗35 cells pulsed with indicated peptides (above each plot). Level of activation was quantified with CD69 expression. Control peptide: LPFDFTPGY.
Discussion
While recent work on T cell specificities in cancer has focused on neoantigens that are typically unique to individuals, prior work also describes shared tumor antigens that are inappropriately expressed or overexpressed in tumors. Here, we developed an approach to systematically survey the TCR repertoire of a substantial number of NSCLC patients to uncover shared T cell specificities. Using the GLIPH2 algorithm, we first distilled this raw TCR sequence data into a much smaller and more useful collection of shared specificity groups with inferred HLA restrictions. We then prioritized disease-relevant TCR candidates for antigen discovery. The enormous diversity of the yeast library greatly facilitated antigen identification and the discovery of cross-reactive antigens. Unlike other MHC/peptide libraries built in mammalian cells, the yeast libraries incorporate close to 109 randomly permutated peptide sequences (Gee et al., 2018; Joglekar et al., 2019; Kula et al., 2019; Li et al., 2019). While previously the uncertainty of HLA restriction limited the success of antigen identification using the yeast library (Gee et al., 2018), we overcame this limitation by using GLIPH2 to infer the correct HLA context of the candidate TCR.
Using this approach in lung cancer, we discovered examples of TCRs cross-reactive to both tumor and microbial antigens. Thus, this seems to be a likely explanation for the reports of pathogen-specific T cells infiltrating tumors (Andersen et al., 2012; Rosato et al., 2019; Scheper et al., 2019; Simoni et al., 2018). We previously proposed that maintaining a broad T cell repertoire to defend against pathogens may rely heavily on TCR cross-reactivity (Su et al., 2013). T cells specific to self-antigens have been detected in the peripheral blood of healthy individuals, pruned but not clonally deleted in the thymus, potentially to avoid immunologic “blind spots” to pathogens (Sewell, 2012; Yu et al., 2015). Because cancer cells overexpress self-antigens, T cell specificity for self-antigens may partly explain why previous studies observed low reactivities of tumor-infiltrating T cells to autologous tumor (Scheper et al., 2019). In this study, we observed that TMEM161A-specific T cells were relatively weak responders to the self-antigen TMEM161A compared to antigens from EBV and E. coli. Despite this weak reactivity, the data presented here show that the binding affinity of TCR2 to the TMEM9-mer/A∗02:01 ligand is higher than LMP2 and EntS. This suggests that in tumors, the uncoupling of TCR binding from T cell activation may be yet another mechanism by which the natural course of specific responses against TAAs are dampened during tumor progression. This provides a possible explanation for why these T cells are localized to tumors where TMEM161A is overexpressed but where EBV and E. coli are likely absent. In this regard, previous reports show that EBV is rarely detected in lung cancer (Kheir et al., 2019) and E. coli is rarely detected in the lung sputum (Cameron et al., 2017; Dickson et al., 2016).
Previously, common pathogen-specific T cells found in tumors have been presumed to be “bystanders” and not specific for TAAs. Our data showed that T cell specificities for TAAs and pathogen-derived antigens were not mutually exclusive. Furthermore, these pathogen-specific T cells in tumors exhibited an effector phenotype rather than an exhausted or stressed state and lacked CD39 expression (Simoni et al., 2018). In this study, we described examples of cross-reactive T cells with weaker reactivities to shared, non-mutated tumor antigens compared to the cross-reactive microbial antigens. Despite this weaker reactivity, our data suggested that cross-reactive T cells might play a role in controlling cancer progression in the setting of anti-PD1 checkpoint blockade. Although it is still unclear what roles these cross-reactive T cells play in the anti-tumor immune response unleashed by immune checkpoint blockade, it is tempting to speculate that exposure to cross-reactive microbial antigens might overcome tolerance for non-mutated tumor or self-antigens (Ohashi et al., 1991; Röcken et al., 1992). The idea that pathogens could be the basis of immunotherapy was suggested originally from the work of William Coley who, in the late nineteenth century, pioneered a mixed bacterial vaccine termed Coley’s toxin for the treatment of cancer patients with some success (McCarthy, 2006). Recently the gut microbiome has been shown to be a key determinant of immunotherapy responses in cancer (Gopalakrishnan et al., 2018; Matson et al., 2018; Routy et al., 2018; Sivan et al., 2015; Vétizou et al., 2015). In pancreatic cancer, a unique microbiome composition has been observed in patients with longest survival after surgery (Riquelme et al., 2019). Cross-reactive T cells recognizing both tumor antigens and microbial antigens have also been shown to control tumor growth in mouse models (Bessell et al., 2020; Fluckiger et al., 2020). In addition, EBV and flu have recently been shown to induce anti-tumor immunity against shared TAAs (Choi et al., 2020; Newman et al., 2020). Additional studies are needed to understand whether or not there is a causal relationship between the microbe/TAA-cross-reactive T cells and clinical benefit from immune checkpoint blockade.
In summary, we present a resource for comprehensively characterizing TCRs from a large NSCLC patient cohort using methodologies that could be applied to any tumor type. Thus, we reduced almost 800,000 TCR sequences from 178 patients to over 66,000 specificities shared by three or more individuals. Of the 66,000 specificity groups we identified, we then subsetted these into 435 specificities that were enriched in the tumors versus adjacent lung. This number may represent a much smaller number of antigens since a given peptide-MHC ligand can elicit five or more different specificity groups (Glanville et al., 2017). We found an intriguing cross-reactivity between non-mutated tumor antigens and pathogens, which could explain recent puzzling results describing nominally virus-specific T cells infiltrating tumors (Simoni et al., 2018), implying, as does other data presented here, that this cross-reactivity may be a common phenomenon. This raises the prospect that memory T cells to these pathogenic epitopes could trigger a cross-reactive response against cancer. Perhaps during the early phase of neoplasia, pre-cancerous cells that happen to overexpress self-antigens (mutated or not) that are cross-reactive to similar antigens from EBV interact with these T cells to create a chronic, low-grade inflammatory tumor microenvironment. In support of this, we observe that cross-reactive T cells express high levels of granzyme K, which has been reported in the context of inflammatory diseases and aging (Corridoni et al., 2020; Mogilenko et al., 2020). Since inflammation is known to promote neoplasia, this could then facilitate the process by which some cells become malignant.
Limitations of the study
The GLIPH2 algorithm infers T cell specificities based on TCRβ sequences only. Thus, it captures only a portion of all input sequences. Although the current study focuses on T cells from lung cancer, many of the shared specificity groups generated, including the “S%DGMNTE” motif, are anticipated to overlap with other cancer types and can serve as a template for analysis. However, further studies are needed in order to establish a complete shared specificity landscape for other cancer types. Finally, we identified only a few examples of cross-reactive specificities and thus we cannot rule out the possibility that at least some pathogen-specific T cells in lung cancer infiltrates were not cross-reactive to TAA and therefore true “bystanders.”
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD4 antibody | Biolegend | Clone: OKT4 |
| Anti-CD4 antibody | Biolegend | Clone: RPA-T4 |
| Anti-CD8 antibody | Biolegend | Clone: SK1 |
| Anti-CD8 antibody | Biolegend | Clone: HIT8a |
| Anti-CD3 antibody | Biolegend | Clone: OKT3 |
| Anti-CD3 antibody | Biolegend | Clone: UCHT1 |
| Anti-CD45 antibody | Biolegend | Clone: H130 |
| Anti-CD25 antibody | Biolegend | Clone: BC96 |
| Anti-PD-1 antibody | Biolegend | Clone: EH12.2H7 |
| Anti-CD137 antibody | Biolegend | Clone: 4B4-1 |
| Anti-HLA-DR antibody | Biolegend | Clone: L243 |
| Anti-HLA-BC antibody | Thermo Fisher Scientific | Clone: B1.23.2 |
| Anti-TCRγδ antibody | Biolegend | Clone: B1 |
| Anti-TCR⍺/β | Biolegend | Clone: IP26 |
| Anti-CD19 antibody | Biolegend | Clone: H1B19 |
| Anti-CD14 antibody | Biolegend | Clone: M5E2 |
| Anti-CD38 antibody | Biolegend | Clone: HIT2 |
| Anti-CD69 antibody | Biolegend | Clone: FN50 |
| Anti-APC microbeads | Miltenyi Biotech | Cat#: 130-090-855 |
| Anti-CD3/CD28 microbeads | Thermo Fisher Scientific | Cat#: 11141D |
| Anti-TMEM161A antibody | abcam | Clone: EPR14369 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase III | Worthington Biochemical | Cat#: LS004182 |
| DNase I | Worthington Biochemical | Cat#: LS002007 |
| Zombie Aqua | Biolegend | Cat#: 423102 |
| Human TruStain FcX | Biolegend | Cat#: 422302 |
| Live/dead near-IR dye | Thermo Fisher Scientific | Cat#: L34975 |
| AMPure XP beads | Beckman Coulter | Cat#: A63881 |
| Recombinant hIL-2 | Peprotech | Cat#: 200-02 |
| Recombinant RNase Inhibitor | Takara Bio | Cat#: 2313A |
| ERCC RNA Spike-In Mix | Ambion/Life Technologies | Cat#: 4456740 |
| LNA-TSO | Exiqon | Cat#: 500100 |
| Ni-NTA resin | QIAGEN | Cat#: 30210 |
| BirA Biotin-protein ligase | Avidity | Cat#: BirA500 |
| Streptavidin MicroBeads | Miltenyi Biotech | Cat#: 130-048-101 |
| LS Columns | Miltenyi Biotech | Cat#: 130-042-401 |
| Polyethylenimine (PEI) | Millipore-Sigma | Cat#: 408727 |
| Opti-MEM | Thermo Fisher Scientific | Cat#: 31985062 |
| RetroNectin® Recombinant Human Fibronectin Fragment | Takara Bio | Cat#: T100A |
| FuGENE® 6 | Promega | Cat#: E2691 |
| Amicon® Ultra-15 Centrifugal Filter Unit (30 kDa filter) | Millipore-Sigma | Cat#: UFC903024 |
| Streptavidin | Thermo Fisher Scientific | Cat#: 434302 |
| Custom synthetic peptides | Alan Scientific | N/A |
| Flex-T HLA-A∗02:02 Monomer UVX | Biolegend | Cat#: 280003 |
| Betaine | Millipore-Sigma | Cat#: W422312 |
| Critical Commercial Assays | ||
| SMARTScribe Reverse Transcriptase kit | Takara Bio | Cat#: 639538 |
| KAPA Library Quantification kit | Roche | Cat#: KK2602 |
| AATI Fragment Analyzer | Agilent | Cat#: DNF-474-1000 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#: FC-131-1096 |
| MiSeq Reagent Kit v2 (300-cycles; yeast screen) | Illumina | Cat#: MS-102-2002 |
| MiSeq Reagent Kit v2 (500-cycles; scTCR-seq) | Illumina | Cat#: MS-102-2003 |
| Chromium Single-Cell V(D)J kit (for TCR) | 10x Genomics | Cat#: 1000005; 1000009; 120262; 1000084; 1000080; 1000014; 1000020 |
| Immunoseq assay (Deep) | Adaptive Biotechnologies | Cat#: hsTCRB |
| RosetteSep human T cell enrichment cocktail | Stem Cell Technologies | Cat#: 15061 |
| KAPA HyperPrep Kit | Roche | Cat#: KK8502 |
| SeqCap EZ MedExome Enrichment Kit | Roche | Cat#: 07676581001 |
| Gibson Assembly Cloning Kit | NEB | Cat#: E5510S |
| Zymoprep II kit | Zymo Research | Cat#: D2004 |
| In-Fusion Cloning | Takara Bio | Cat#: 638947 |
| Deposited Data | ||
| scRNA-seq data (tumor-infiltrating T cells from 10 Stanford lung cancer patients) | This paper | Database: GSE151537 (SuperSeries #GSE151538) |
| scRNA-seq data (tetramer-sorted T cells from peripheral blood) | This paper | Database: GSE151531 (SuperSeries #GSE151538) |
| Human reference genome NCBI build 37, GRCh38 | Genome Reference Consortium | https://genome.ucsc.edu/cgi-bin/hgTables |
| Bulk CDR3β sequences (n = 178 HLA-typed NSCLC patients, MDACC) | (Reuben et al., 2020); IMMUNOSEQ ANALYZER (Adaptive Biotechnologies) | https://clients.adaptivebiotech.com |
| HLA tetramer-derived CDR3β sequences | (Shugay et al., 2018) | https://vdjdb.cdr3.net |
| Reference CDR3β sequences for GLIPH2 | (Huang et al., 2020) | http://50.255.35.37:8080/tools |
| Bulk RNA-seq data from tumors (pan-lung cancer) | The Cancer Genome Atlas | https://portal.gdc.cancer.gov |
| GSEA hallmark gene sets | Broad Institute | http://www.gsea-msigdb.org/gsea/downloads.jsp |
| The UniProt Archive (UniParc) | The UniProt consortium | https://www.uniprot.org/downloads |
| Emerson CDR3β dataset | (Emerson et al., 2017) | https://clients.adaptivebiotech.com |
| TRACERx CDR3β sequences | (Joshi et al., 2019) | https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA544699 |
| Experimental Models: Cell Lines | ||
| Jurkat 76 cells | S.-A. Xue, University of College London | N/A |
| T2 cells | ATCC | Cat#: CRL-1992; RRID: CVCL_2211 |
| HLA-A∗02+ 293T cells | S. Feldman, Stanford University | N/A |
| 293T HEK cells | ATCC | Cat#: CRL-11268 |
| H1395 cells | ATCC | Cat#: CRL-5868; RRID: CVCL_1467 |
| Sf9 cells | ATCC | Cat#: CRL-1711 |
| Hi5 cells | Thermo Fisher Scientific | Cat#: BTI-TN-5B1-4 |
| Recombinant DNA | ||
| Custom gBlocks dsDNA fragments | IDT | N/A |
| pAcGP67a vector | BD Biosciences | N/A |
| Bestbac 2.0 | Expression systems | Cat#: 91-002 |
| Yeast A∗02 display library constructs | K.C. Garcia, Stanford University | N/A |
| Soluble TCR baculoviral constructs | K.C. Garcia, Stanford University | N/A |
| EF1a-MCS-GFP-PGK-puro lentiviral vector | (Witwicka et al., 2015) | Addgene#: 73582 |
| MSGV1 retroviral vector | S. Rosenberg, NIH | N/A |
| gag-pol plasmid (Δ8.9) | M.M. Winslow, Stanford University | N/A |
| pMD.G plasmid (VSV-G) | M.M. Winslow, Stanford University | N/A |
| Lenti-TMEM161A plasmid | GeneCopoeia | Cat#: EX-A1961-Lv241 |
| Software and Algorithms | ||
| GLIPH2 algorithm | (Huang et al., 2020) | http://50.255.35.37:8080 |
| R version 4.0.2 | CRAN | https://www.r-project.org/; RRID: SCR_001905 |
| star/2.7.1a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR; RRID: SCR_015899 |
| samtools/1.4 | (Li et al., 2009) | http://www.htslib.org; RRID: SCR_002105 |
| python/2.7.3 | Python Software Foundation | https://www.python.org; RRID: SCR_008394 |
| htseq-count (HTSeq 0.5.4p5) | (Anders et al., 2015) | https://htseq.readthedocs.io/; RRID: SCR_011867 |
| GSEA v2.2.2 | Broad Institute; (Subramanian et al., 2005) | RRID: SCR_003199 |
| GSVA/1.34.0 | (Hänzelmann et al., 2013) | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| FIJI/2.0.0-rc-69/1.52p | (Schindelin et al., 2012) | https://imagej.net/Fiji; RRID: SCR_002285 |
| TraCeR algorithm | (Stubbington et al., 2016) | https://github.com/Teichlab/tracer |
| HighV-QUEST | international ImMunoGeneTics information system (IMGT) | http://www.imgt.org/; RRID: SCR_018196 |
| FlowJo software | FlowJo, LLC | https://www.flowjo.com; RRID: SCR_008520 |
| varscan2/2.4.3 | (Koboldt et al., 2012) | http://varscan.sourceforge.net; RRID: SCR_006849 |
| gatk-3.7/MuTect2 | Broad Institute; (Cibulskis et al., 2013) | RRID: SCR_000559 |
| Strelka/2.9.10 | Illumina; (Saunders et al., 2012) | RRID: SCR_005109 |
| Seurat/3.1.4 | Bioconductor | RRID: SCR_016341 |
| Monocle/2.10.1 | Bioconductor | RRID: SCR_018685 |
| netMHCpan/4.0 | (Jurtz et al., 2017; Reynisson et al., 2020) | RRID: SCR_018182 |
| BLASTP | National Center for Biotechnology Information (NCBI); (Johnson et al., 2008) | RRID: SCR_001010 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Mark Davis (mmdavis@stanford.edu).
Materials availability
Further information and material requests should be addressed to Mark Davis (mmdavis@stanford.edu).
Data and code availability
The scRNA-Seq data from tumor-infiltrating T cells (n = 2950, GEO: GSE151537) and HLA tetramer-sorted peripheral blood T cells (n = 623, GEO: GSE151531) were deposited in the GEO database (SuperSeries accession number: GSE151538). The algorithm GLIPH2, reference CDR3β sequences, and tutorial are available from the following link: http://50.255.35.37:8080 (Huang et al., 2020).
Experimental model and subject details
Protocols for collection of human tissue and blood were approved by the Stanford Institutional Review Board (IRB 15166). Inclusion criteria included adult patients (age > = 18 years), known or suspected diagnosis of NSCLC, primary tumor > 2 cm, and consent for research. Patients receiving neoadjuvant therapy or patients with underlying lung infection, inflammatory, or fibrotic disease were excluded. Overall, 21 patients with surgically-resectable NSCLC treated at Stanford were included in this study. A table of patient characteristics is provided (Table S4). DNA was extracted from peripheral blood PBMC (QIAGEN) for HLA tying. In addition, we analyzed samples from 2 patients with advanced/metastatic disease treated with anti-PD1 antibody on IRB 21319. Patients experienced clinical benefit at 6 months after initiation of treatment.
Method details
Tissue processing
Tissue was processed within 2 h from surgery. Tissue was divided and one section for cell suspensions and another section for histology. Cell suspensions were generated by mincing of tissue followed by digestion with collagenase III (200 IU/mL) and DNase I (100 U/mL) (Worthington Biochemical) for 40 min in RPMI and passing through a 70-um filter. Sections for histology were fixed in 4% paraformaldehyde and transferred to 70% ethanol solution the following day.
FACS analyses
T cells were isolated from tumor single cell suspensions by antibody staining followed by cell sorting on a 5-laser FACSAria Fusion sorter (Stanford FACS Facility) purchased using funds from the Parker Institute for Cancer Immunotherapy. Tumor cell suspensions were stained in PBS with Zombie Aqua dye (Biolegend) for viability assessment. This was followed by staining in PBS with 2% FBS in Fc Blocking solution (Biolegend) plus the following antibodies: anti-CD4 (OKT4, Biolegend), anti-CD8 (SK1, Biolegend), anti-CD3 (OKT3, Biolegend), anti-CD45 (H130, Biolegend), anti-CD25 (BC96, Biolegend), anti-PD-1(EH12.2H7, Biolegend), anti-CD137 (4B4-1, BD Biosciences), anti-HLA-DR (L243, Biolegend). CD3+CD45+AquaZombie- cells were index sorted directly into 96-well plates preloaded with 4 uL of capture buffer, snap frozen on dry ice, and stored at −80°C. Ectopic HLA-B∗35 was detected with anti-HLA-BC monoclonal antibody (clone B1.23.2, Thermo Fisher Scientific). Transduced Jurkat 76 cells expressing exogenous TCRα/β chains were sorted on a FACSAria Fusion sorter at Stanford or a BD Biosciences Influx High Speed Cell Sorter at the Flow Cytometry Core Facility of the Cancer Institute of New Jersey.
Establishment of T cell specificity groups
The GLIPH2 algorithm was implemented for the establishment of T cell specificity groups using 778,938 distinct CDR3β sequences from the MD Anderson NSCLC dataset (Reuben et al., 2020). Briefly, by comparing with the reference dataset of 273,920 distinct CDR3β sequences (both CD4 and CD8) from 12 healthy individuals, GLIPH2 first discovered clusters of CDR3β sequences sharing either global or local motifs as previously described (Huang et al., 2020). The output of CDR3β clusters with shared sequence motifs is accompanied by multiple statistical measurements to facilitate the calling of high-confidence specificity groups, including biases in Vβ gene usage, CDR3β length distribution (relevant only for local motifs), cluster size, HLA allele usage, and clonal expansion. To establish high-confidence specificity groups with the NSCLC dataset, we prioritized TCR specificity groups with at least 3 distinct CDR3β members from a minimum of 3 different patients with significant biases in Vβ gene usage, and CDR3β clonal expansion in comparison with the reference dataset. This led to the discovery of 4,226 specificity groups that formed the basis for further analyses throughout the study.
Classification of TCRs and specificity groups
For CDR3β clonotypes, we included only distinct sequences from each MDACC patient with frequencies above 0.1% in tumors or adjacent lung samples in order to focus on the most expanded TCRs. In Figures 2A and 2B, we compared the abundance (rounded, normalized count) of each distinct TCR in the tumor versus the paired adjacent lung from the same patient. The p value for the comparison in abundance between tumor and the adjacent lung were calculated with the poisson.test function in R (alternative = “two.sided”). For specificity groups with clonal expansion (n = 4,226), a list of summed frequencies (up to 100%, rounded to integers) of all CDR3β members that belong to each specificity group was first created for both tumor and the adjacent lung from each MDACC patient. Poisson test was then used to calculate the p value for the comparison of these summed frequencies in the lists using the poisson.test function (Figures 2A and 2B).
Annotation of specificity groups
To annotate inferred specificity groups from lung cancer patients, we ran a combined GLIPH analysis using both the MD Anderson lung cancer patient CDR3β sequences and publicly available, tetramer-derived CDR3β sequences (Glanville et al., 2017; Shugay et al., 2018; Song et al., 2017). To do so, we first identified tetramer-derived CDR3β sequences that could form TCR specificity groups by running an independent GLIPH analysis with a total 10,051 CDR3β sequences from the tetramer datasets. This led to the formation of 395 specificity groups containing 1,561 CDR3β sequences. We then combined these 1,561 CDR3β sequences with the 778,938 CDR3β sequences from the MD Anderson lung cancer dataset for the aforementioned GLIPH2 analysis. Any specificity group that includes at least one CDR3β sequence from the tetramer data is considered “annotated” and would be assigned a specificity and HLA restriction according to the associated tetramer sequence(s). Of note, in all cases where multiple tetramer-derived CDR3β sequences were found in a given specificity group, there was only one dominant tetramer-defined specificity/HLA involved.
Validation of HLA restriction inference
For the tetramer-annotated specificity groups mentioned above (n = 71), we validated the inferences of HLA restriction made by the GLIPH2 algorithm against the HLA restriction informed by tetramers. Specificity groups annotated with HLA-A∗02 (n = 49 out of 71) or HLA-B∗08 (n = 8 out of 71) tetramers were chosen for the validation because they were the most prevalent. To validate a specificity group for enrichment with HLA-A∗02 alleles, we first constructed a contingency table with the number of patients in the specificity group carrying HLA-A∗02 supertype allele(s) and the number of patients without these alleles, number of all NSCLC patients carrying HLA-A∗02 supertype allele(s) (n = 79) and those who do not (n = 98). We then calculated p values using the hypergeometric test (phyper in R, lower.tail = FALSE). We reported the numbers of specificity groups significantly enriched with HLA-A∗02 supertype alleles (p < 0.05 by the hypergeometric test) as a fraction over the number of specific groups annotated with HLA-A∗02 tetramers (n = 18 out of 49). We also reported the numbers of specificity groups significantly enriched with HLA-A∗02 supertype alleles as a fraction over the number of specificity groups annotated with non-HLA-A∗02 tetramers (n = 0 out of 22). We repeated this process for the validation of specificity groups enriched with HLA-B∗08 supertype alleles. To identify top-enriched HLA allele(s) for a specificity group (Figures 2C and 2D), hypergeometric test was used to first uncover HLA allele(s) that are significantly enriched (phyper, lower.tail = FALSE). The highest value of fraction (# of patients carrying the allele within a specificity group / all patients within a specificity group) was determined and used to find top-enriched allele(s) with both p value < 0.05 and the highest fraction value.
HLA-A∗02:01 specificity group bootstrapping
To estimate the number of HLA-A∗02:01+ NSCLC patients needed to cover 50% of all HLA-A∗02:01-enriched specificity groups (n = 77), we carried out a bootstrapping process through random sampling of patients with incremental sampling sizes. First, we established 77 specificity groups (from the 4,226 NSCLC-enriched specificity groups) that were significantly enriched with the HLA-A∗02:01 allele (p < 0.05). Bootstrapping was conducted with random sampling (with replacement) of 1 through 160 patients for 100 times. For each sampling event, we tallied the sum of HLA-A∗02:01-enriched specificity groups found using the CDR3β sequences from the sampled patients (specificity count, Figures 1F and 1G). We then calculated the mean and the standard error of the specificity counts from the bootstrapping process. As an internal control, we repeated the bootstrapping process on the rest of HLA-A∗02:01- NSCLC patients. To compare with specificity groups from a healthy cohort, we used 989,816 distinct CDR3β sequences from 304 HLA-A∗02:01+ and 1,153,600 CDR3β sequences from 362 HLA-A∗02- healthy donors’ PBMC from a publicly available dataset (Emerson dataset, (Emerson et al., 2017)). To adjust for the differences in sequencing depth (below), 5000 distinct CDR3β sequences (with the highest frequencies) from each healthy donor were included for the GLIPH analysis. To address the influence of clonal expansion on specificity group quantification, we compared the bootstrapping results with the aforementioned HLA-A∗02:01-enriched specificity groups to an equal number of HLA-A∗02:01-enriched specificity groups without clonal expansion (n = 77). We used a similar strategy to address how the total number of specificity groups impacted this result. We performed bootstrapping using various enrichment cutoffs for HLA-A∗02 enrichment (p < 0.05, n = 1,267; p < 0.025, n = 319; p < 0.01, n = 71 specificity groups). Finally, to address the impact of sequencing depth on specificity group quantification, we down-sampled the total input CDR3β sequences randomly in the bootstrapping process by the indicated proportions (50%, 25%, 12.5%, or 0% down-sampled).
GSEA analysis of the TCGA data
Normalized gene expression data from bulk RNA-Seq analyses of human NSCLC resected tumors and adjacent lungs from the Cancer Genome Atlas (TCGA) were downloaded from the NCI GDC Legacy Archive (n = 1,017 for tumors and n = 110 for adjacent lungs). To conduct gene set enrichment analysis (GSEA) with the TCGA dataset, we first calculated the correlation coefficients between any gene and TMEM161A using the Pearson correlation. The sorted gene list based on the correlation coefficient with TMEM161A gene expression was then used for GSEA with the Preranked tool (v2.2.2, Broad Institute) and all hallmark gene sets (Subramanian et al., 2005). The signature scores were derived using the gene lists of indicated hallmark signatures with the single-sample GSEA (ssGSEA) method as described previously (Hänzelmann et al., 2013).
FACS sorting of antigen-specific CD8 T cells
Recombinant HLA-A∗02 monomer with UV exchangeable peptide were either synthesized as previously described (Altman and Davis, 2003) or purchased commercially (Biolegend). UV peptide exchange was performed over 20 min with 1 mM of peptide in PBS using Strategene UV Stratalinker 2400. Streptavidin conjugated fluorophore was added incrementally the following day for a final 4:1 molar ratio of MHC:streptavidin. Tetramer staining was performed in PBS plus 2% FBS in Fc Blocking solution (Biolegend) at room temperature for 1 h. For peripheral blood samples, cells were subsequently stained with anti-TCRγδ (B1, Biolegend), anti-CD19 (H1B19, Biolegend), anti-CD14 (M5E2, Biolegend), anti-CD3 (OKT3, Biolegend), anti-CD4 (RPA-T4, Biolegend), anti-CD8 (HIT8a, Biolegend), and live/dead near-IR dye (Invitrogen). For tumor samples, cells were stained with anti-CD4 (OKT4, Biolegend), anti-CD8 (HIT8a, Biolegend), anti-CD3 (UCHT1, Biolegend), anti-CD45 (H130, Biolegend).
Single-cell RNA-seq (scRNA-Seq)
Full transcriptomes from FACS sorted T cells at the single-cell level were generated according to the previously reported procedures with some modifications (Picelli et al., 2014). First strand cDNA was then generated with Takara’s SMARTScribe Reverse Transcriptase kit according to manufacturer’s protocol (Takara Bio). Notable changes from the previously reported Smart-Seq2 RT step includes: 2 mM of dNTP and 2 μM of oligo dT were included in the capture buffer; 1M of Betaine and additional 6 mM MgCl2 were included in the RT reaction buffer. The cDNA samples were then amplified with the KAPA Library Quantification kit for 22 – 25 cycles (Roche). We used 1 (of total 25/well) μL of amplified cDNA for single-cell TCR-sequencing and thus bypassing the RT step as reported previously (Han et al., 2014). To proceed with scRNA-Seq, full-length cDNA samples were first cleaned up with 0.6 – 0.8x volume of precalibrated AMPure XP beads (Beckman Coulter) to exclude DNA fragments smaller than 500 base pairs. We used the automatic liquid handler Biomek FXP Automated Workstation (Beckman Coulter) in order to eliminate cell-to-cell variabilities. The quality of purified full-length cDNA was validated with the AATI Fragment Analyzer (Agilent). Subsequently, we used the measurements from the Fragment Analyzer in order to normalize the cDNA input with a Mantis liquid handler (Formulatrix). We then consolidated the cDNA samples into a 384-well plate (LVSD) with a Mosquito X1 liquid handler (TTP labtech). After transfer, Illumina sequencing libraries were prepared using a Mosquito HTS liquid handler (TTP labtech). We used only 0.4 uL (of total 23 uL) of cDNA per well to make the full transcriptome libraries with the Nextera XT DNA Library Preparation Kit (Illumina, FC-131-1096). We used custom-made i5 and i7 unique 8-bp indexing primers (IDT) to multiplex 384 wells in a single sequencing run. The libraries were amplified on a C1000 Touch Thermal Cycler with 384-Well Reaction Module (Bio-rad). We checked the pooled libraries with the Agilent 2100 Bioanalyzer (Stanford PAN facility) and acquired paired-end sequences (150bp x 2) on a Hiseq 4000 Sequencing System (Illumina) purchased with funds from NIH (S10OD018220) for the Stanford Functional Genomics Facility (SFGF).
Single-cell TCR sequencing (scTCR-seq)
Single T cells were sorted and captured as described above in the method for scRNA-Seq sample preparation. Following first strand cDNA synthesis (Takara) and amplification (Roche), we used 1 uL (of total 25 uL/well) of amplified cDNA for single-cell TCR-sequencing and thus bypassing the RT step as reported previously (Han et al., 2014). Nested PCR was performed with TCRα/β primers carrying multiplexing barcodes that enabled pooled CDR3α/β sequencing in a single Miseq run. Paired sequencing reads were joined, demultiplexed, and mapped to the human TCR references from the international ImMunoGeneTics information system® (IMGT) with custom scripts as reported previously (Han et al., 2014). Paired CDR3αβ sequences from the resected tumor of patient A6 were derived using the Chromium Single-Cell V(D)J kit from the 10x Genomics according to the protocol from the manufacturer. For advanced/metastatic lung cancer patients treated with anti-PD1 therapy, bulk TCR sequencing was performed on pre- and post- treatment PBMCs with Immunoseq assay (Adaptive Biotechnologies, Seattle, WA). Single-cell TCR sequencing was performed on post-treatment samples sorted for CD38+HLA-DR+ cells, as described above.
Data analyses of scRNA-Seq results
Sequencing reads were first de-multiplexed and binned into separate FASTQ files that correspond with the full transcriptomes of individual T cells. STAR aligner (2.6.1d) (Dobin et al., 2013) was used to map the reads with default parameters against human genome reference GRCh38 (v21) from the UCSC genome browser. Mapped reads were sorted and indexed with samtools (1.4) (Li et al., 2009). Gene expression was first quantified by counting reads mapped to genes with htseq-count (HTSeq 0.5.4p5) using the following settings:–stranded = no–type = exon–idattr = gene_name–mode = intersection-nonempty (Anders et al., 2015). Unless otherwise stated, all single-cell T cell states were analyzed with Seurat (3.1.4) packages in R using raw read counts. To derive TCR repertoires from the scRNA-Seq results, reads mapped to both the TCRα and TCRβ genes were first reconstructed with the TraCeR algorithm as described previously (Stubbington et al., 2016). The reconstructed DNA sequences were then submitted to the IMGT to call gene segment usage and the CDR3 amino acid sequences through HighV-QUEST.
GLIPH2 analysis on TRACERx data
Raw FASTQ files (tumor, n = 202; adjacent lung, n = 63) with demultiplexed, joined reads of the bulk CDR3β nucleotide sequences from the TRACERx cohort of NSCLC were downloaded from the Short Read Archive as reported (Joshi et al., 2019). The amino acid sequences of CDR3β, V gene usage, and the error-corrected clonal counts were subsequently derived by using the Decombinator scripts established previously (Oakes et al., 2017). To quantify the percentages of tumor-enriched specificity groups shown in Figure 1C, we first conducted joint GLIPH2 analyses with combined CDR3β sequences from the MDACC cohort (n = 778,938) and the bulk CDR3β sequences from the TRACERx cohort (tumor, n = 1,173,806 CDR3β sequences; adjacent lung, n = 247,578 CDR3β sequences). The total percentages (%) of top-20 clonally expanded as well as the rest CDR3β clonotypes that belonged to the 435 tumor-enriched specificity groups were then derived for each tumor (n = 202) and the adjacent lung tissue (n = 63).
Soluble biotinylated TCRα/β synthesis
Soluble TCRα/β chains used for yeast selections were made as described previously (Gee et al., 2018). Briefly, synthetic gene blocks (gBlocks®) of N-terminal truncated TCRα or TCRβ chain V and modified C gene fragments were assembled into the baculoviral pAcGP67a construct (BD Biosciences) with Gibson assembly (New England BioLabs). The final baculoviral plasmid was co-transfected into Sf9 cells (ATCC) with Bestbac 2.0 (Expression systems) with FuGENE® 6 (Promega) to make the crude viral supernatant (P0). Subsequently, viruses were passaged at a dilution of 1:500 in 30-50 mL cultures at a density of 1 × 106 cells/mL to generate higher titer viruses (P1). To generate the soluble TCRα/β chains, up to 4 L of High Five (Hi5, ThermoFisher Scientific) cells were infected with P1 baculovirus at a dilution of 1:500-1:1000 at a density of 2 × 106 cells/mL for a week before protein purification. Recombinant TCRα/β chains were bound with Ni-NTA resin (QIAGEN) in the Hi5 cell media for 3 h at room temperature, washed with 20 mM imidazole in 1X HBS at pH 7.2, and eluded eluted in 200 mM imidazole in 1X HBS at pH 7.2. After buffer exchange to 1X HBS at pH 7.2 with a 30 kDa filter (Millipore-Sigma), purified proteins were biotinylated overnight with birA ligase in the presence of 100 μM biotin, 40 mM Bicine at pH 8.3, 10 mM ATP, and 10mM Magnesium Acetate at 4°C. Biotinylated proteins were purified by size-exclusion chromatography using an AKTAPurifier Superdex 200 column (GE Healthcare) and validated on a SDS-PAGE gel to confirm the stoichiometry and biotinylation with excess streptavidin.
Antigen discovery with the yeast library
To uncover the cognate antigens of the candidate TCRα/β, we used the yeast HLA-A∗02 libraries displaying highly diverse peptides of 4 different length (Gee et al., 2018). Briefly, we first expanded 4 separate naive HLA-A∗02 libraries carrying distinct lengths of peptides to beyond 10x diversities in SDCAA pH 6.0 before induction of the peptide-HLA-Aga2p composite proteins with SGCAA. Induced libraries were used for affinity-based selection with biotinylated soluble TCRα/β chains coupled to streptavidin-coated magnetic MACS beads (Miltenyi) in the presence of 0.5% bovine serum albumin and 1 mM EDTA to reduce the background. We cultured the selected yeast clones in SDCAA until confluency, then induced confluent cells in SGCAA for 2-3 days before the next round of selection. The selection was repeated four times and then enrichment of cognate antigens was confirmed with Sanger sequencing of 20 colonies. Once confirmed, we prepared the plasmid DNA from 5-10 × 107 yeast cells per round of selection by miniprep (Zymoprep II kit, Zymo Research). The peptide coding regions were PCR-amplified with composite oligos with Illumina P5/P7-Truseq indexed adapters and gel purified for pooled sequencing on a Miseq sequencer (2x150 V2 kit).
Lentiviral TCR transduction
TCR⍺ chain, P2A linker, and TCRβ chain fusion gene fragments were purchased from IDT and cloned into MCS of the EF1a-MCS-GFP-PGK-puro lentiviral vector (Glanville et al., 2017; Witwicka et al., 2015). HEK293T cells were plated on a 10-cm dish at a density of 7.5 × 106 cells in 10 mL of DMEM the day prior to transfection. 293Ts were co-transfected with 3.3 μg of the lentiviral plasmid, 2.5 μg of the gag-pol plasmid, and 0.83 μg of the VSV-G envelope plasmid pre-mixed with 33 μL of PEI in 120 μL of Opti-MEM (ThermoFisher Scientific). After 24 h, the medium was replenished and viral supernatant was collected 24 and 48 h later. TCR-deficient Jurkat cells (below) were transduced with viral supernatant, TCR expression was assessed by flow cytometry, and TCR-expressing cells were sorted based on the expression of GFP, CD3, and the transduced TCRα/β chains. For lentivirus expressing full-length EntS, LMP2, and FluM1, gene fragments were also purchased from IDT and cloned into MCS of EF1a-MCS-GFP-PGK-puro lentiviral vector. Lentivirus for expressing human TMEM161A (NM_017814) was purchased from GeneCopoeia. Lentivirus was produced as described above, and 293T cells stably expressing HLA-A∗02 (293A2) were transduced with viral supernatant. Transduced 293A2 cells were sorted based on GFP expression and used for in vitro T cell stimulation.
Retroviral TCR transduction
For retroviral-mediated expression of TCR2 in primary T cells, TCR⍺ chain, P2A linker, and TCRβ chain were PCR amplified from the lentiviral vector (described above) and cloned into the MCS of an MSGV1-based retroviral vector (gift from Steve Rosenberg laboratory) using In-Fusion Cloning (Takara). For retroviral-mediated expression of TCR14 in primary T cells, TCR⍺ chain, P2A linker, and TCRβ chain fusion gene fragments were purchased from IDT and cloned into MCS of an MSGV1-based retroviral vector.
Cell culture
The Jurkat 76 T cell line deficient for both TCR⍺ and TCRβ were provided by Dr. Shao-An Xue (Department of Immunology, University of College London).
Jurkat cells and primary T cells were grown in complete RPMI (ThermoFisher) containing 10% FBS, 25 mM HEPES, 290 μg/mL L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin, 1mM sodium pyruvate, and 1x non-essential amino acids. T2 cells were grown in IMDM (Fisher Scientific) with 20% FBS, 290 μg/mL L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin. 293T cells stably expressing HLA-A∗02 were provided by Dr. Steve Feldman (Stanford School of Medicine) and grown in DMEM (ThermoFisher) with 10% FBS, 290 μg/mL L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin.
In vitro stimulation of the Jurkat T cells
Jurkat 76 cells expressing the exogenous TCR of interest were sorted on CD3/GFP double-positive populations (Figure S3A) and co-cultured with T2 cells in complete RPMI as detailed above. To find homologous sequences of the identified epitopes, netMHCpan was used to predict the binding affinity of the homologous peptide to a given HLA allele (Jurtz et al., 2017; Reynisson et al., 2020). We used the BLASTP algorithm to perform the search for matching peptides in the UniParc protein database (Johnson et al., 2008). Peptides were dissolved in DMSO at 20 mM stock concentration and diluted to a final concentration of 2 μM. After 18 h of stimulation, cells were washed and stained with anti-CD3 (OKT3, Biolegend), anti-CD69 (FN50, Biolegend), and anti-TCR⍺/β (IP26, Biolegend) antibodies. Cells were acquired using FACS Fortessa (BD Biosciences) automated high throughput sampler or the Accuri C6 Plus flow cytometer (BD), and data analyzed using FlowJo software (Treestar).
Expression of TCRα/β on primary T cells
T cells were isolated from a leukoreduction system chamber from an HLA-A∗02 positive healthy donor from the Stanford institutional blood bank using the RosetteSep human T cell enrichment cocktail (Stem Cell Technologies) and viably stored in liquid nitrogen. For T cell activation, T cells were thawed and stimulated with anti-CD3/CD28 beads (Life Technologies) in the presence of IL-2 (100 IU/mL). On days 1 and 2, activated T cells were retrovirally transduced using Retronectin (Takara) coated plates in media containing 100 IU/mL IL-2. Anti-CD3/CD28 beads were removed on day 3 and media containing IL-2 were replenished once every 2 days. Following 8 days of in vitro expansion, T cells were co-cultured with 293A2 cells expressing full-length TMEM161A, EntS, LMP2, FluM1, or GFP alone at a 1:1 ratio. Following 18 h incubation, cells were stained with anti-CD3 (OKT3, Biolegend), anti-CD69 (FN50, Biolegend), anti-TCR⍺/β (IP26, Biolegend), anti-CD137 (4B4-1, BD Biosciences), and live/dead near-IR dye (Invitrogen). Data were acquired using FACS Fortessa (BD Biosciences) automated high throughput sampler and analyzed using FlowJo software (Treestar).
Binding affinity measurements using BLI
Binding affinity of TCR2 to the indicated pMHC monomers was determined by BLI using an Octet QK instrument (ForteBio). The purified, soluble TCR2 was captured onto amine reactive second-generation (AR2G) biosensors using the amine reactive second-generation reagent kit. The ligand-bound biosensors were dipped into a concentration series (20 μM followed by 4-fold dilutions) of the indicated analytes in PBST (PBS with 0.05% Tween-20) to determine the binding kinetics. A series of unliganded biosensors dipped into the analytes served as controls for referencing. In addition, signals from analyte binding to an irrelevant TCR was used for non-specific binding correction. The traces were processed using ForteBio Data Analysis Software.
In vitro cytotoxicity assay
Primary T cells were isolated from HLA-A∗02+ healthy donors. Cells were retrovirally transduced with TCR2 as described above. Following 9 days of in vitro expansion, cells were co-stained with TMEM9-mer/HLA-A∗02 tetramers (APC) and FluM1/A∗02 tetramers (PE) and enriched with anti-APC microbeads (Miltenyi Biotech), and enrichment confirmed with analysis on FACS Fortessa. The following day, TMEM9-mer/HLA-A∗02 tetramers-enriched T cells were co-cultured with H1395 lung adenocarcinoma cells at a 20:1 ratio in 96-well flat bottom plates for over 120 h. A minimum of triplicate wells were plated for each condition. Plates were imaged every 3 h using the InuCyte ZOOM Live-Cell analysis system (Essen Bioscience). Four images per well at 10x zoom were acquired at each time point. Total integrated GFP intensity per well was recorded and normalized to the starting measurement and plotted over time.
Immunohistochemistry of TMEM161A
TMEM161A staining of paraffin-embedded tissue was performed according to standard procedures by the Stanford Human Pathology/Histology Service Center.
Anti-TMEM161A antibody was stained at 1:50 (abcam ab180954), followed by HRP-conjugated secondary antibody. Tissue was counterstained with hematoxylin. Automated imaging analysis was performed using Fiji imaging processing package (Schindelin et al., 2012).
Whole-exome sequencing
Whole-exome sequencing of tumor DNA and matched germline leukocyte DNA was performed by inputting 75ng of sheared genomic DNA for library preparation with the KAPA HyperPrep Kit (Roche) with modifications to the manufacturer’s instruction, as described previously (Hellmann et al., 2020). Library-prepared samples were captured with the SeqCap EZ MedExome Kit (NimbleGen) according to the manufacturer’s instructions. Sequencing data were demultiplexed and mapped to hg19 using a custom bioinformatics pipeline, as described previously (Newman et al., 2014). VarScan 2 (Koboldt et al., 2012), Mutect (Cibulskis et al., 2013), and Strelka (Saunders et al., 2012) were used to call variants use default parameters. Variants called by at least two of the approaches were then filtered by requiring: 1) variant allele frequency of at least 2.5%, 2) at least 30X depth in both tumor and germline samples, 3) zero germline reads, and 4) a population allele frequency of less than 0.1% in the Genome Aggregation database (Lek et al., 2016).
Quantification and statistical analysis
Unless stated otherwise, all statistical analyses performed in finding high-confidence specificity groups with GLIPH2 were Fisher’s exact tests using the contingency tables with the CDR3β query set (specificity group) and the reference set (Huang et al., 2020). Poisson test was used to determine the representation bias in comparisons of distinct CDR3β sequences or specificity groups between tumors and adjacent lungs. Hypergeometric test (phyper in R, lower.tail = FALSE) was used to quantify the enrichment of HLA supertype alleles (Figure 1D) and to find top-enriched HLA allele(s) for each specificity group (Figures 2C and 2D). Student’s t test was used to assess the results from all in vitro assays. Statistical significance was defined as p value < 0.05.
Acknowledgments
This work was supported by Parker Institute for Cancer Immunotherapy, Virginia and D.K. Ludwig Fund for Cancer Research, the Howard Hughes Medical Institute, NIH U54 CA232568-01, NIH 5P30CA124435 (C.L.M.), NIH 5RO1-AI03867, NIH U19 AI057229, and a St. Baldrick’s-Stand Up to Cancer Dream Team Translational Research grant (SU2C-AACR-DT-27-17). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. C.L.M. and M.M.D. are members of the Parker Institute for Cancer Immunotherapy, which provided partial funding for this project. D.T. was funded by a Lung Cancer Research Foundation scientific grant, Ellie Guardino Cancer Foundation Award from the Stanford Cancer Institute, a Stanford School of Medicine Honorary Dean’s Fellowship, and a Conquer Cancer Foundation of ASCO Young Investigator Award. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the American Society of Clinical Oncology or the Conquer Cancer Foundation. This work was also funded in part by the National Cancer Institute of the National Institutes of Health Research Project grant (R01CA234629-01), the AACR-Johnson & Johnson Lung Cancer Innovation Science grant (18-90-52-ZHAN), the Cancer Prevention and Research Institute of Texas Multi-Investigator Research Award grant (RP160668), and the University of Texas Lung Specialized Programs of Research Excellence grant (grant number P50CA70907). Flow cytometry and cell-sorting services were provided by (1) the Stanford Shared FACS Facility supported by the NIH S10 Shared Instrument Grants (S10RR025518-01 and S10RR027431-01) and (2) the Flow Cytometry & Cell Sorting Core Facility, a shared resource of Rutgers - Robert Wood Johnson Medical School and the Rutgers Cancer Institute of New Jersey (P30CA072720-5921), and also supported by NIH Shared Instrumentation grant (1 S10 RR025468-01).
Author contributions
S.-H.C. and D.T. contributed equally. C.L.M. and M.M.D. are jointly supervising authors. M.M.D. and C.L.M. are both corresponding authors. S.-H.C., D.T., C.L.M., and M.M.D. conceived the project and wrote the manuscript. D.T., S.-H.C., A.R., V.M., I.S.M., P.T., and X.Y. performed experiments. S.-H.C. performed the bioinformatics. S.C. and R.S. helped with robotic-assisted SMART-seq2 data collection and analyses. X.Y. and D.N. assisted with yeast screens. C.W. wrote the GLIPH2 script. S.-H.C., D.T., K.C.G., E.S., I.L.W., C.L.M., and M.M.D. interpreted data. J.B.S., M.F.B., L.B., N.S.L., H.A.W., J.W.N., S.K.P., and J.A.B. contributed clinical samples. G.J.B. reviewed pathology. A.D. and P.H.S. assisted with immunohistochemistry data. S.-H.C., J.W., A.M.M., and D.M.L. assisted with scTCR-seq. R.R., L.L., D.D.K, and S.A.F. assisted with experimental design. B.Y.N. and M.D. contributed clinical samples and performed and interpreted exome-seq data. J.Z., A.R., I.I.W., J.V.H., and P.A.F. generated the TCR sequences, clinical data, and the exome-seq data of the MDACC NSCLC cohort. All authors reviewed and approved the final version of manuscript.
Declaration of interests
C.L.M. is a founder of, holds equity in, and receives consulting fees from Lyell Immunopharma and receives consulting fees from NeoImmuneTech, Nektar, Apricity, and Roche. J.W.N. reports research support from Genentech/Roche, Merck, Novartis, Boehringer Ingelheim, Exelixis, Takeda Pharmaceuticals, Nektar Therapeutics, Adaptimmune, and GSK and has served in a consulting or advisory role for AstraZeneca, Genentech/Roche, Exelixis Inc., Jounce Therapeutics, Takeda Pharmaceuticals, Eli Lilly and Company, Calithera Biosciences, Amgen, Regeneron Pharmaceuticals, Natera, and Iovance Biotherapeutics. H.A.W. has received research support from Celgene, Clovis Oncology, Genentech/Roche, Arrys Therapeutics, Novartis, Merck, BMS, Exelixis, Lilly, Pfizer, and has participated on the advisory boards of Helsinn, Mirati, Cellworks, Genentech/Roche, Merck, and ITMIG. N.S.L. has received research funding from Intuitive Foundation and Auspex Diagnostics. E.S. is a consultant for Lyell Immunopharma. L.L. is a consultant for Lyell Immunopharma. S.A.F. is consulting for Lonza PerMed and Samsara BioCapital. M.D. reports research funding from Varian Medical Systems and Illumina; ownership interest in CiberMed and Foresight Diagnostics; patent filings related to cancer biomarkers; paid consultancy from Roche, AstraZeneca, BioNTech, Genentech, Novartis, and Gritstone Oncology; and travel/honoraria from Reflexion. K.C.G. is founder of 3T therapeutics. I.I.W. has received honoraria from Genentech/Roche, Bayer, Bristol-Myers Squibb, AstraZeneca/Medimmune, Pfizer, HTG Molecular, Asuragen, Merck, GlaxoSmithKline, Guardant Health, Oncocyte, and MSD. I.I.W. is also supported by Genentech, Oncoplex, HTG Molecular, DepArray, Merck, Bristol-Myers Squibb, Medimmune, Adaptive, Adaptimmune, EMD Serono, Pfizer, Takeda, Amgen, Karus, Johnson & Johnson, Bayer, Iovance, 4D, Novartis, and Akoya. J.Z. reports grants from Merck and Johnson & Johnson, as well as adversary/consulting/Hornoraria fees from Bristol Myers Squibb, AstraZeneca, GenePlus, Innovent, OrigMed, and Roche outside the submitted work. This study was supported in part by a Cancer Prevention Research Institute of Texas Multi-Investigator Research Award (grant number RP160668) and the University of Texas Lung Specialized Programs of Research Excellence grant (grant number P50CA70907). S.-H.C., D.T., C.L.M., and M.M.D have a patent related to this work.
Published: March 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.02.014.
Supplemental information
Headers:Total patient number, number of distinct patients from whom all the CDR3bclones of the indicated specificity group are derived; Distinct CDR3b number, number of distinct CDR3bclones in the indicated specificity group; All CDR3 number, number of all CDR3bclones in the indicated specificity group; All distinct TCR list, list of all distinct CDR3bclones in the indicated specificity group; Vb p value, pvalue for the Vbgene enrichment in the indicated specificity group; Most freq Vb, the most prevalent Vbgene enriched in the indicated specificity group; Most freq Vb (#), number of CDR3bclones with the most prevalent Vbgene in the indicated specificity group; CDR3b length p value, p value for the bias in length distribution in the indicated specificity group; Expansion p value, pvalue for the bias in clonal expansion in the indicated specificity group.
Headers (A-L): same as Table S2; Enriched HLAs (by hypergeometric test), HLA allele(s) enriched in the indicated specificity group; # of enriched HLAs, number of distinct HLA allele(s) enriched in the indicated specificity group; Enriched HLA p value (by hypergeometric test), pvalue(s) for the enrichment(s) of the HLA allele(s); Enriched HLA fraction (0-1), fraction(s) of HLA allele(s) enriched in the indicated specificity group; log2FD (Tumor/Adj lung), log2converted fold difference between the summed CDR3bclonal frequencies in tumor and adjacent lung; p value by Poisson test (Tumor/Adj lung), pvalues of the CDR3bclonal difference probability (tumor/adjacent lung) calculated with Poisson test.
References
- Altman J.D., Davis M.M. MHC-peptide tetramers to visualize antigen-specific T cells. Curr Protoc Immunol. 2003;Chapter 17:Unit 17.13. doi: 10.1002/0471142735.im1703s53. [DOI] [PubMed] [Google Scholar]
- Anagnostou V., Forde P.M., White J.R., Niknafs N., Hruban C., Naidoo J., Marrone K., Sivakumar I.K.A., Bruhm D.C., Rosner S. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Res. 2019;79:1214–1225. doi: 10.1158/0008-5472.CAN-18-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen R.S., Thrue C.A., Junker N., Lyngaa R., Donia M., Ellebæk E., Svane I.M., Schumacher T.N., Thor Straten P., Hadrup S.R. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 2012;72:1642–1650. doi: 10.1158/0008-5472.CAN-11-2614. [DOI] [PubMed] [Google Scholar]
- Arstila T.P., Casrouge A., Baron V., Even J., Kanellopoulos J., Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- Bessell C.A., Isser A., Havel J.J., Lee S., Bell D.R., Hickey J.W., Chaisawangwong W., Glick Bieler J., Srivastava R., Kuo F. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight. 2020;5:e135597. doi: 10.1172/jci.insight.135597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S.J.S., Lewis K.E., Huws S.A., Hegarty M.J., Lewis P.D., Pachebat J.A., Mur L.A.J. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS ONE. 2017;12:e0177062. doi: 10.1371/journal.pone.0177062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I.-K., Wang Z., Ke Q., Hong M., Paul D.W., Jr., Fernandes S.M., Hu Z., Stevens J., Guleria I., Kim H.J. Mechanism of EBV inducing anti-tumour immunity and its therapeutic use. Nature. 2020;590:157–162. doi: 10.1038/s41586-020-03075-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corridoni D., Antanaviciute A., Gupta T., Fawkner-Corbett D., Aulicino A., Jagielowicz M., Parikh K., Repapi E., Taylor S., Ishikawa D. Single-cell atlas of colonic CD8+ T cells in ulcerative colitis. Nat. Med. 2020;26:1480–1490. doi: 10.1038/s41591-020-1003-4. [DOI] [PubMed] [Google Scholar]
- Coulie P.G., Brichard V., Van Pel A., Wölfel T., Schneider J., Traversari C., Mattei S., De Plaen E., Lurquin C., Szikora J.P. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med. 1994;180:35–42. doi: 10.1084/jem.180.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie P.G., Lehmann F., Lethé B., Herman J., Lurquin C., Andrawiss M., Boon T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA. 1995;92:7976–7980. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson R.P., Erb-Downward J.R., Martinez F.J., Huffnagle G.B. The Microbiome and the Respiratory Tract. Annu. Rev. Physiol. 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson R.O., DeWitt W.S., Vignali M., Gravley J., Hu J.K., Osborne E.J., Desmarais C., Klinger M., Carlson C.S., Hansen J.A. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 2017;49:659–665. doi: 10.1038/ng.3822. [DOI] [PubMed] [Google Scholar]
- Fluckiger A., Daillère R., Sassi M., Sixt B.S., Liu P., Loos F., Richard C., Rabu C., Alou M.T., Goubet A.G. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science. 2020;369:936–942. doi: 10.1126/science.aax0701. [DOI] [PubMed] [Google Scholar]
- Gee M.H., Han A., Lofgren S.M., Beausang J.F., Mendoza J.L., Birnbaum M.E., Bethune M.T., Fischer S., Yang X., Gomez-Eerland R. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell. 2018;172:549–563.e16. doi: 10.1016/j.cell.2017.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville J., Huang H., Nau A., Hatton O., Wagar L.E., Rubelt F., Ji X., Han A., Krams S.M., Pettus C. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–98. doi: 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V., Spencer C.N., Nezi L., Reuben A., Andrews M.C., Karpinets T.V., Prieto P.A., Vicente D., Hoffman K., Wei S.C. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- Han A., Glanville J., Hansmann L., Davis M.M. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 2014;32:684–692. doi: 10.1038/nbt.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjanto S., Ng L.F., Tong J.C. Clustering HLA class I superfamilies using structural interaction patterns. PLoS ONE. 2014;9:e86655. doi: 10.1371/journal.pone.0086655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann M.D., Nabet B.Y., Rizvi H., Chaudhuri A.A., Wells D.K., Dunphy M.P.S., Chabon J.J., Liu C.L., Hui A.B., Arbour K.C. Circulating tumor DNA analysis to assess risk of progression after long-term response to PD-(L)1 blockade in NSCLC. Clin. Cancer Res. 2020;26:2849–2858. doi: 10.1158/1078-0432.CCR-19-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Wang C., Rubelt F., Scriba T.J., Davis M.M. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol. 2020;38:1194–1202. doi: 10.1038/s41587-020-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar A.V., Leonard M.T., Jeppson J.D., Swift M., Li G., Wong S., Peng S., Zaretsky J.M., Heath J.R., Ribas A. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods. 2019;16:191–198. doi: 10.1038/s41592-018-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn201. W5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K., de Massy M.R., Ismail M., Reading J.L., Uddin I., Woolston A., Hatipoglu E., Oakes T., Rosenthal R., Peacock T., TRACERx consortium Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019;25:1549–1559. doi: 10.1038/s41591-019-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y., Eliyahu S., Delgado C.H., Robbins P.F., Rivoltini L., Topalian S.L., Miki T., Rosenberg S.A. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc. Natl. Acad. Sci. USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheir F., Zhao M., Strong M.J., Yu Y., Nanbo A., Flemington E.K., Morris G.F., Reiss K., Li L., Lin Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers (Basel) 2019;11:759. doi: 10.3390/cancers11060759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel M.J., Dudley D., Afdhal N., Grakoui A., Rice C.M., Choo Q.L., Houghton M., Walker B.D. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J. Clin. Invest. 1995;96:2311–2321. doi: 10.1172/JCI118287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kula T., Dezfulian M.H., Wang C.I., Abdelfattah N.S., Hartman Z.C., Wucherpfennig K.W., Lyerly H.K., Elledge S.J. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell. 2019;178:1016–1028.e13. doi: 10.1016/j.cell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Bethune M.T., Wong S., Joglekar A.V., Leonard M.T., Wang J.K., Kim J.T., Cheng D., Peng S., Zaretsky J.M. T cell antigen discovery via trogocytosis. Nat. Methods. 2019;16:183–190. doi: 10.1038/s41592-018-0305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson V., Fessler J., Bao R., Chongsuwat T., Zha Y., Alegre M.L., Luke J.J., Gajewski T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006;26:154–158. [PMC free article] [PubMed] [Google Scholar]
- Mogilenko D.A., Shpynov O., Andhey P.S., Arthur L., Swain A., Esaulova E., Brioschi S., Shchukina I., Kerndl M., Bambouskova M. T Cells as Conserved Hallmark of Inflammaging. Immunity; 2020. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK(+) CD8. [DOI] [PubMed] [Google Scholar]
- Murray R.J., Kurilla M.G., Brooks J.M., Thomas W.A., Rowe M., Kieff E., Rickinson A.B. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J. Exp. Med. 1992;176:157–168. doi: 10.1084/jem.176.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Bratman S.V., To J., Wynne J.F., Eclov N.C., Modlin L.A., Liu C.L., Neal J.W., Wakelee H.A., Merritt R.E. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.H., Chesson C.B., Herzog N.L., Bommareddy P.K., Aspromonte S.M., Pepe R., Estupinian R., Aboelatta M.M., Buddhadev S., Tarabichi S. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA. 2020;117:1119–1128. doi: 10.1073/pnas.1904022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes T., Heather J.M., Best K., Byng-Maddick R., Husovsky C., Ismail M., Joshi K., Maxwell G., Noursadeghi M., Riddell N. Quantitative Characterization of the T Cell Receptor Repertoire of Naïve and Memory Subsets Using an Integrated Experimental and Computational Pipeline Which Is Robust, Economical, and Versatile. Front. Immunol. 2017;8:1267. doi: 10.3389/fimmu.2017.01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi P.S., Oehen S., Buerki K., Pircher H., Ohashi C.T., Odermatt B., Malissen B., Zinkernagel R.M., Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Rehermann B., Fowler P., Sidney J., Person J., Redeker A., Brown M., Moss B., Sette A., Chisari F.V. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J. Exp. Med. 1995;181:1047–1058. doi: 10.1084/jem.181.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A., Zhang J., Chiou S.H., Gittelman R.M., Li J., Lee W.C., Fujimoto J., Behrens C., Liu X., Wang F. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020;11:603. doi: 10.1038/s41467-019-14273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme E., Zhang Y., Zhang L., Montiel M., Zoltan M., Dong W., Quesada P., Sahin I., Chandra V., San Lucas A. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806.e12. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins H.S., Srivastava S.K., Campregher P.V., Turtle C.J., Andriesen J., Riddell S.R., Carlson C.S., Warren E.H. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci. Transl. Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röcken M., Urban J.F., Shevach E.M. Infection breaks T-cell tolerance. Nature. 1992;359:79–82. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- Rosato P.C., Wijeyesinghe S., Stolley J.M., Nelson C.E., Davis R.L., Manlove L.S., Pennell C.A., Blazar B.R., Chen C.C., Geller M.A. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat. Commun. 2019;10:567. doi: 10.1038/s41467-019-08534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B., Le Chatelier E., Derosa L., Duong C.P.M., Alou M.T., Daillère R., Fluckiger A., Messaoudene M., Rauber C., Roberti M.P. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Savage P.A., Vosseller K., Kang C., Larimore K., Riedel E., Wojnoonski K., Jungbluth A.A., Allison J.P. Recognition of a ubiquitous self antigen by prostate cancer-infiltrating CD8+ T lymphocytes. Science. 2008;319:215–220. doi: 10.1126/science.1148886. [DOI] [PubMed] [Google Scholar]
- Scheper W., Kelderman S., Fanchi L.F., Linnemann C., Bendle G., de Rooij M.A.J., Hirt C., Mezzadra R., Slagter M., Dijkstra K. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019;25:89–94. doi: 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell A.K. Why must T cells be cross-reactive? Nat. Rev. Immunol. 2012;12:669–677. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020;20:75–76. doi: 10.1038/s41577-020-0275-8. [DOI] [PubMed] [Google Scholar]
- Shugay M., Bagaev D.V., Zvyagin I.V., Vroomans R.M., Crawford J.C., Dolton G., Komech E.A., Sycheva A.L., Koneva A.E., Egorov E.S. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46(D1):D419–D427. doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibener L.V., Fernandes R.A., Kolawole E.M., Carbone C.B., Liu F., McAffee D., Birnbaum M.E., Yang X., Su L.F., Yu W. Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell. 2018;174:672–687.e27. doi: 10.1016/j.cell.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J., Peters B., Frahm N., Brander C., Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- Sivan A., Corrales L., Hubert N., Williams J.B., Aquino-Michaels K., Earley Z.M., Benyamin F.W., Lei Y.M., Jabri B., Alegre M.L. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I., Gil A., Mishra R., Ghersi D., Selin L.K., Stern L.J. Broad TCR repertoire and diverse structural solutions for recognition of an immunodominant CD8+ T cell epitope. Nat. Struct. Mol. Biol. 2017;24:395–406. doi: 10.1038/nsmb.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington M.J.T., Lönnberg T., Proserpio V., Clare S., Speak A.O., Dougan G., Teichmann S.A. T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods. 2016;13:329–332. doi: 10.1038/nmeth.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.F., Kidd B.A., Han A., Kotzin J.J., Davis M.M. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity. 2013;38:373–383. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B., Knuth A., Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- Vétizou M., Pitt J.M., Daillère R., Lepage P., Waldschmitt N., Flament C., Rusakiewicz S., Routy B., Roberti M.P., Duong C.P. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwicka H., Hwang S.Y., Reyes-Gutierrez P., Jia H., Odgren P.E., Donahue L.R., Birnbaum M.J., Odgren P.R. Studies of OC-STAMP in Osteoclast Fusion: A New Knockout Mouse Model, Rescue of Cell Fusion, and Transmembrane Topology. PLoS ONE. 2015;10:e0128275. doi: 10.1371/journal.pone.0128275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfel T., Hauer M., Schneider J., Serrano M., Wölfel C., Klehmann-Hieb E., De Plaen E., Hankeln T., Meyer zum Büschenfelde K.H., Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- Yu W., Jiang N., Ebert P.J., Kidd B.A., Müller S., Lund P.J., Juang J., Adachi K., Tse T., Birnbaum M.E. Clonal Deletion Prunes but Does Not Eliminate Self-Specific αβ CD8(+) T Lymphocytes. Immunity. 2015;42:929–941. doi: 10.1016/j.immuni.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Headers:Total patient number, number of distinct patients from whom all the CDR3bclones of the indicated specificity group are derived; Distinct CDR3b number, number of distinct CDR3bclones in the indicated specificity group; All CDR3 number, number of all CDR3bclones in the indicated specificity group; All distinct TCR list, list of all distinct CDR3bclones in the indicated specificity group; Vb p value, pvalue for the Vbgene enrichment in the indicated specificity group; Most freq Vb, the most prevalent Vbgene enriched in the indicated specificity group; Most freq Vb (#), number of CDR3bclones with the most prevalent Vbgene in the indicated specificity group; CDR3b length p value, p value for the bias in length distribution in the indicated specificity group; Expansion p value, pvalue for the bias in clonal expansion in the indicated specificity group.
Headers (A-L): same as Table S2; Enriched HLAs (by hypergeometric test), HLA allele(s) enriched in the indicated specificity group; # of enriched HLAs, number of distinct HLA allele(s) enriched in the indicated specificity group; Enriched HLA p value (by hypergeometric test), pvalue(s) for the enrichment(s) of the HLA allele(s); Enriched HLA fraction (0-1), fraction(s) of HLA allele(s) enriched in the indicated specificity group; log2FD (Tumor/Adj lung), log2converted fold difference between the summed CDR3bclonal frequencies in tumor and adjacent lung; p value by Poisson test (Tumor/Adj lung), pvalues of the CDR3bclonal difference probability (tumor/adjacent lung) calculated with Poisson test.
Data Availability Statement
The scRNA-Seq data from tumor-infiltrating T cells (n = 2950, GEO: GSE151537) and HLA tetramer-sorted peripheral blood T cells (n = 623, GEO: GSE151531) were deposited in the GEO database (SuperSeries accession number: GSE151538). The algorithm GLIPH2, reference CDR3β sequences, and tutorial are available from the following link: http://50.255.35.37:8080 (Huang et al., 2020).