

Abstract
Diabetes remains one of the fastest growing chronic diseases and is a leading source of morbidity and accelerated mortality in the world. Loss of beta cell mass (BCM) and decreased sensitivity to insulin underlie diabetes pathogenesis. Yet, the ability to safely and directly assess BCM in individuals with diabetes does not exist. Measures such as blood glucose provide only a crude indirect picture of beta cell health. PET imaging could, in theory, allow for safe, direct, and precise characterization of BCM. However, identification of beta cell-specific radiolabeled tracers remains elusive. G-protein coupled receptor 44 (GPR44) is a transmembrane protein that was characterized in 2012 as highly beta cell-specific within the insulin-positive islets of Langerhans. Accordingly, radiolabeling of existing GPR44 antagonists could be a viable method to accelerate PET tracer development. The present study aims to evaluate and summarize published analogues of the GPR44 antagonist ramatroban to develop 18F-labeled PET tracers for BCM analysis. The 77 corresponding ramatroban analogues containing a fluorine nuclide were characterized for properties including binding affinity, selectivity, and pharmacokinetic and metabolic profile, and 32 compounds with favorable properties were identified. This review illustrates the potential of GPR44 analogues for the development of PET tracers.
Keywords: positron emission tomography (PET), G-protein coupled receptor 44 (GPR44), CRTH2, diabetes, beta cell mass (BCM), pancreatic islets, beta cell imaging, 18F-labeling, ramatroban analogues, PET probes
1. Introduction
1.1. Positron Emission Tomography (PET) Imaging for Beta Cell Mass (BCM)
Diabetes is a chronic disease characterized by the disruption of glucose homeostasis and a major cause of limb amputation, stroke, renal failure, and heart disease. Diabetes affected approximately 476 million people globally in 2017 and has become the seventh leading cause of death [1]. Type 1 diabetes (T1D) and type 2 diabetes (T2D) are two primary diagnoses, and pathology of both is caused by insulin dysregulation and subsequent hyperglycemia. In both T1D and T2D, the pathogenesis and severity of disease can be elucidated from beta cell mass (BCM). Access to BCM measurements in vivo is pivotal for determining the progression of BCM during disease pathogenesis and monitoring effects of diabetes treatments. However, post-mortem biopsies are currently the primary method to study BCM [2,3]. Routine biopsy sampling of living patients is restricted by the relatively inaccessible location of the pancreas in the body and the invasive nature of the procedure. In fact, a clinical study of T1D patients showed significant complications associated with the procedure, including bleeding and pancreatic leakage [4]. Another approach utilizing plasma markers, such as insulin, c-peptide, and glycated hemoglobin (HbA1c), fails to measure BCM accurately. The downstream metabolic processes which form these markers may hamper their correlation to BCM. These markers have also been shown to strongly fluctuate in association with metabolism [5]. As such, current methods do not allow for accurate and non-invasive in vivo longitudinal evaluation of BCM progression [3].
To overcome the limitations of current approaches to BCM analysis, positron emission tomography (PET) has been explored as a viable alternative. PET is a non-invasive, quantitative technique that assesses radioactive tracer concentrations in vivo. Since the tracer is presumably bound to a receptor, the receptor location and density can be mapped through computer analysis of the gamma rays. As such, PET imaging of BCM has the potential to follow the progression of metabolic diseases, support beta cell replacement technologies, and assist pharmaceutical drug development. The efficacy of PET imaging, however, depends in large part on the tracer selected, particularly because beta cells make up only 1% of the pancreas mass [6]. Accordingly, the application of PET to BCM analysis depends upon the development of a highly specific radiotracer targeting beta cells. While several cell-surface molecules, such as VMAT2 and GLP-1R, have been proposed, they have been found lacking in specificity [7,8]. Therefore, we are interested in identifying and assessing novel beta cell-specific molecular targets for PET tracer development.
1.2. G-Protein Coupled Receptor 44 (GPR44) and Related PET Tracers
In 1999, Marchese et al. conducted a PCR study to isolate novel G-protein coupled receptors (GPCR) based on oligonucleotide primers [9,10]. One of the GPCRs they identified was GPR44, a 7-helix transmembrane protein. In later studies, GPR44 was also referred to as prostaglandin D2 receptor 2 (DP2) and chemoattractant receptor-homologous molecule expressed on T helper type 2 cells (CRTH2). GPR44 was then applied for the investigation and treatment of metabolic and inflammatory diseases, and showed promising results. In 2012, the first study characterizing the expression of GPR44 in human islets used immunofluorescent staining to reveal that GPR44 is highly beta cell-specific, contained within the insulin-positive islets of Langerhans, and non-specific for glucagon [11]. Further studies continued to indicate that GPR44 could serve as a viable marker of BCM [12,13].
In 2001, binding assays revealed that the GPR44 agonist was pro-inflammatory mediator prostaglandin D2 (PGD2) [14,15]. PGD2 is a lipid molecule that has been implicated in asthma, allergic rhinitis, and other diseases involving type 2 inflammation. PGD2 binding to GPR44 causes an increase in cytoplasmic levels of Ca2+, which inhibits cAMP production and subsequently induces cell migration and activation [16]. As might be expected from their common binding to GPR44, PGD2 and other GPR44 ligands share many structural similarities. For instance, most GPR44 antagonists contain a carboxylic acid-derived region that is likely critical for binding [17]. An in silico study characterizing the binding mechanisms between GPR44 and two of its antagonists (Figure 1), fevipiprant (2-[2-methyl-1-[[4-methylsulfonyl-2-(trifluoromethyl)phenyl]methyl]pyrrolo[2,3-b]pyridin-3-yl]acetic acid) and TM-30089 ({3-[(4-fluoro-benzenesulfonyl)methyl-amino]-1,2,3,4-tetrahydro-carbazol-9-yl}acetic acid), (to be discussed in more detail in Section 2.2.2.), showed that the carboxylate group of the antagonist is possibly attracted to GPR44’s highly positively charged surface. The antagonist then enters the receptor through a gap between transmembrane helix 1 (TM1) and transmembrane helix 7 (TM7). Upon entering, the antagonist binds to a site that contains mostly aromatic residues, which matches well with the polar acetate group attached to a central aromatic group found in many GPR44 antagonists [18].
Figure 1.

Molecular structures of fevipiprant, TM-30089, ramatroban, [11C]AZ12204657 and [11C]MK-7246.
There exist a number of known and developed GPR44 ligands, but only two have been characterized as potential GPR44 PET tracers in Figure 1: [11C]AZ12204657 ([11C]- ((S)-2-(4-chloro-2-[2-chloro-4-(methylsulfonyl)phenoxy]phenoxy) propanoic acid) and [11C]MK-7246 ([11C]-{(7R)-7-[[(4-fluorophenyl)sulfonyl](methyl)amino]-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl}acetic acid). Interest in these ligands began when GPR44 antagonists AZ12204657 (IC50 = 2.5 nM and Kd = 2.9 µg), a structural analogue of the high-affinity GPR44 antagonist AZD3825, and MK-7246 ({(7R)-7-[[(4-fluorophenyl)sulfonyl](methyl)amino]-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl}acetic acid), a derivative of TM-30089, demonstrated specific binding to insulin-positive islets in human pancreatic sections [12,19,20]. These compounds were subsequently radiolabeled with carbon-11 and investigated as PET tracers in animal models. While MK-7246 will be discussed further in Section 2.2.3, it is worth noting that AZ12204657 has been developed into a PET tracer for BCM. In a mouse model with kidney capsule-engrafted human islets, [11C]AZ12204657 was shown to strongly bind to the capsule containing the transplants but expressed negligible binding to the non-transplanted kidney capsule [12,19]. This binding was confirmed to be GPR44-mediated, as it could be inhibited by pre-treatment with AZD3825. In addition, [11C]AZ12204657 demonstrated focal binding patterns consistent with insulin-positive areas in the pancreas of non-diabetic pigs.
However, the tracers studied insofar have been mainly labeled with carbon-11, which limits the application of these probes to a short half-life of 20 min and a viability of no longer than 2 h, after which radioactive imaging properties are lost. The application of 11C-labeling is further limited because PET tracers generally are suitable for clinical use within three half-lives, meaning that radiotracer quality testing should be set to be within 1 h [21]. Additionally, radiosynthesis of these probes requires artificial production of carbon-11 by an on-site cyclotron. These limitations do not allow tracers to be administered to multiple patients, delivered to remote sites, or implemented in preclinical validation assays.
Due to the limitations presented by 11C-labeled tracers, 18F-labeling is a more suitable and effective alternative for development of GPR44 PET tracers, as fluorine-18 has a half-life of 109.7 min that allows for up to 10 h of imaging. Additionally, fluorine-18 has a 97% positron emission (β+ decay), low maximum positron energy (0.635 MeV), favorable van der Waals radius (1.47 Å), and high electronegativity, with the corresponding high-energy bond with carbon (112 kcal/mol). The bonding with carbon is important, as it lends the radiolabeled tracer stability in high temperatures and oxidation resistance. As opposed to another prominent radioisotope, gallium-68, the low positron energy of fluorine-18 results in a short diffusion range (< 2.4 mm) for optimal imaging resolution [22]. Although the spatial resolution, sensitivity, contrast, and activity recovery coefficients do fluctuate depending on the PET tracer used, a comparison of fluorine-18 and gallium-68 imaging found that PET scanners performed better with fluorine-18, particularly high-resolution small animal PET scanners [23]. Currently, most of the PET agents used in clinical studies are 18F-labeled.
Development of 18F-labeled GPR44 PET tracers is of interest due to the positive results shown by these previous studies. Approaches to do so include modifying previously existing GPR44 antagonists and clinically evaluated drugs. In particular, the benefit of the drug approach is that the molecular structures and properties of such compounds have already been studied, and the appendage of the molecule may be simply replaced with a radioactive moiety to serve as a PET tracer.
The present paper aims to summarize current ramatroban-based GPR44 ligands containing a fluorine group and evaluate their potential as targets for the development of novel 18F-labeled PET tracers. An important consideration for 18F-labeling is that the potential molecule must already contain a fluorine group in the chemical structure to allow for 18F-labeling of the structures without significantly altering the pharmacological property of the molecule.
2. Ramatroban-Based Analogues for Potential GPR44 Binding
2.1. Ramatroban
In 1989, Rosentreter et al. at Bayer AG synthesized the novel indole sulfonamide, ramatroban ((3R)-3-(4-Fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-carbazolepropanoic acid). Ramatroban (Figure 1), also known as BAY u 3405, was first described as an antagonist of thromboxane receptor (TP) and later shown to also serve as an antagonist to GPR44. Initially, ramatroban was found to bind to TP after displacing the TP antagonist [3H]SQ29548 in a binding assay, resulting in an IC50 of 68 nM [24]. Additional binding assays with TP agonist U-46619 [25] showed stronger antagonist binding to TP, with a lower IC50 of 30 nM and Ki of 10 nM [26]. The Kd and Bmax of ramatroban were characterized as around 6 nM and 1177 binding sites/platelet, respectively [27].
Due to its binding to TP, ramatroban was developed for TP-mediated cardiovascular disorders. Specifically, ramatroban was found to inhibit platelet aggregation and deposition [28,29,30,31,32,33,34], contraction of smooth muscle [35,36,37], sudden death [38,39,40], myocardial infarctions [38,41,42,43], splenic artery occlusion shock [44], acute ischemia-reperfusion injury [42], and thrombosis [29,45,46,47,48,49]. In 2014, a study of constitutively active mutants (CAMs) in TP demonstrated ramatroban binding, in which ramatroban reduced basal activity [50]. In binding to TP, ramatroban targets TM1, and the carboxylic acid group of ramatroban forms polar attractions to TM2 and TM7 [51].
In vitro assays showed that ramatroban selectively inhibited PGD2-mediated responses. Displacement of TP agonists U-46619 and I-BOP demonstrated that ramatroban acts competitively on TP [52]. In this method, PGD2 concentration curves shifted to the right by a pA2 of 7–8.5 after ramatroban introduction, characteristic of a competitive inhibitors [53,54]. Ramatroban inhibition of PGD2, which binds to both TP and GPR44, led to the later discovery that ramatroban serves as a dual antagonist for TP and GPR44. Further confirmation was provided through investigations on PGD2-mediated mechanisms, including eosinophil shape change assays, and in vivo displacement [55].
However, ramatroban binding to GPR44 was about 16 times less selective than for TP. This binding affinity for GPR44 was elucidated in human embryonic kidney cells (HEK293 cells) transfected with GPR44, and the Ki value was found to be 290 nM [56]. The ramatroban IC50 value for GPR44 in various assays proved comparably weak at 100 nM [53,57]. Additionally, ramatroban exhibited a half-life in binding assays of 4.60 min [55]. The influence of ramatroban inhibition on GPR44 is particularly evident in studies of eosinophils, basophils, and T helper type 2 (TH2) cells migration or count [58,59,60,61]. In vitro flow cytometry found that ramatroban could inhibit eosinophil and basophil shape change [62,63], and inhibit mast cell-induced TH2 migration in vitro [64].
Overall, the poor selectivity for GPR44 (unacceptable dual binding to TP and GPR44) suggests ramatroban may not be optimal as a PET tracer for measuring BCM. However, to date, numerous compounds have been derived from ramatroban that suggests further directions for developing GPR44-specific PET tracers. Described below are fluorine nuclide-containing ramatroban analogues, which we defined as molecules either explicitly stated as ramatroban derivatives in the papers or molecules that contain the characteristic tetrahydrocarbazole group of ramatroban.
2.2. Ramatroban Analogues
2.2.1. Athersys, Inc.
In 2005, after ramatroban was identified as a GPR44 antagonist, Robarge et al. at Athersys, Inc. synthesized several ramatroban analogues to elucidate pharmacophores with promising affinity for GPR44. The analogues were created by changing the substituents and/or the location of the substitution [56], in which the scaffold of benzenesulfonamide was inserted into the phenyl group instead of the cyclohexane in ramatroban (Figure 2).
Figure 2.

Molecular structures of compounds 1–6.
Compound 1 showed higher selectivity for GPR44 than ramatroban, with a 6 times greater affinity for GPR44 than TP. However, compound 1 still displayed a weak affinity to GPR44. In an hCRTH2 binding assay with HEK293 cells, the Ki value was high at 250 nM. The compound was further found to undesirably bind to TP with a Ki value of 1500 nM and express a high 92% human TP inhibition at a concentration of 50,000 nM. Similar to compound 1, a series of related compounds differing in the number and/or position of the fluorine group attached to the phenyl group was characterized (Figure 2). These compounds, however, all showed weak affinities for GPR44 with Ki values of 970 nM or greater, several times higher than the Ki value of ramatroban (see compounds 2, 3, 4, 5, and 6, with Ki of 1300, 970, 1700, 3600, and 4200 nM, respectively). Compound 3, which contained a 2-F phenyl group, had the lowest Ki value but still displayed an undesirable binding affinity (Figure 2). Overall, these compounds did not exhibit optimal properties desirable for a selective GPR44 PET tracer.
Another series of compounds 7–11 (Figure 3) similar to compound 1 replaced the carboxylic acid group of the propanoic acid with a sulfonic acid group (compound 7) or phosphonic acid group (compound 8). In addition, the propanoic acid was replaced with an acrylic acid analogue (compound 9) or with a methyl group substitution (compounds 10 and 11). All these compounds showed a less favorable GPR44 Ki of 2400 nM or greater, indicating weak or no affinity to GPR44. As such, these compounds are not recommended as potential GPR44 PET probes.
Figure 3.

Molecular structures of compounds 7–19.
The next series of compounds explored modifications of the chain length of the propanoic acid group (Figure 3). Decreasing the chain length by one carbon to replace the propanoic acid group with an acetic acid group (compound 12) increased the affinity for GPR44. This modification reduced the Ki for GPR44 to 30 nM, a significant improvement relative to ramatroban. Meanwhile, the affinity weakened when the chain length was increased by one carbon (compound 13) or two carbons (compound 14), which both resulted in poor binding to GPR44 with Ki values of 8300 nM and 4600 nM, respectively. Due to the better Ki value, compound 12 was further tested for TP selectivity and was found to demonstrate negligible off-target binding with a Ki > 20,000 nM and a 21% hTP inhibition at 50,000 nM compound concentration, indicating good selectivity for GPR44 over TP. Further modifications of compound 12 were made by adding either a methyl group (compound 15) or an ethyl group (compound 16), both of which resulted in unfavorable characteristics with Ki values of 4400 nM and 18,000 nM, respectively. Additional reduction in the number of carbons replaced the propanoic acid group directly with a carboxylic acid group, producing three benzoic acid isomers substituted at the ortho (compound 17), meta (compound 18) and para (compound 19) position of the benzene ring (Figure 3). The compounds did not exhibit promising affinity for GPR44, with high Ki values of 33,000, 27,000, and 6400 nM, respectively.
The affinity and selectivity for GPR44 increased when replacing the propanoic acid group with an acetic acid group to decrease the chain length by one carbon (compound 12, GPR44 Ki = 30 nM, TP Ki > 20,000 nM). Based on compound 12 (using the acetic acid group instead of the propanoic acid group), another group of compounds, 20–24 (Figure 4), had further modifications on the cyclohexane group by reducing one carbon (cyclopentane ring, compounds 23) or adding one carbon (cycloheptane ring, compound 24) to create a 5-membered or 7-membered ring. Compounds were also created by adding a substituent to the 6-membered ring (cyclohexane ring, compounds 20–22). These compounds all showed better Ki values and IC50 values compared to ramatroban, in which compound 20 (addition of a methyl group to the cyclohexane ring) had the most favorable Ki value of 13 nM. Compound 20’s affinity for GPR44 was further characterized in a Ca2+ assay measuring PGD2-mediated receptor activation, showing an IC50 of 9.7 nM. The above binding properties suggest that compound 20 binds well to GPR44. Compounds 24 (a 7-membered ring) and 21 (a phenyl group addition to the 6-membered ring) also showed favorable Ki values of 20 nM and 50 nM, respectively, and IC50 values of 22 nM and 22 nM, respectively. Compound 22 and 23, however, are less than optimal with higher Ki values of 150 nM and 200 nM, respectively, and IC50 values of 120 nM and 130 nM, respectively. The three compounds (20, 21, and 24) with the best results were further assessed for selectivity through TP binding assays, and all showed a Ki > 20,000 nM. Furthermore, for compounds 20 and 24, hTP inhibition at 50,000 nM were 30% and 28%, respectively, indicating that these compounds are not likely to bind off-target to TP. However, compound 21 demonstrated an unfavorable hTP inhibition of 66%.
Figure 4.

Molecular structures of compounds 20–24.
Recommendations
Based on the 2005 Robarge et al. study, compounds 12, 20, 21 and 24 are the most promising antagonists for GPR44, with strong binding affinity for GPR44 and optimal selectivity against TP. The modifications of ramatroban included decreasing the chain length by one carbon to replace the propanoic acid group with an acetic acid group (compound 12), adding a methyl group to the cyclohexane group (compound 20), adding a phenyl group to the cyclohexane group (compound 21), and adding one carbon to the cyclohexane group to create a 7-membered ring (compound 24). These compounds can be considered for further development as GPR44 PET tracers.
2.2.2. TM-30642, TM-30643, and TM-30089
Additionally, in 2005, Ulven et al. at 7TM Pharma developed a series of promising ramatroban analogues (Figure 5): TM-30642, TM-30643, and TM-30089 [65].
Figure 5.

Molecular structures of TM-30642, TM-30643 and TM-30089.
TM-30642 (3-{3-[(4-fluoro-benzenesulfonyl)-methyl-amino]-1,2,3,4-tetrahydro-carbazol-9-yl}-propionic acid) was synthesized by the addition of a methyl group to the nitrogen atom of the sulfonamide group in ramatroban (Figure 5). This structural modification was found to be advantageous, as it displayed a stronger affinity for GPR44 and weaker affinity for TP. In a [3H]PGD2 assay, TM-30642 exhibited an optimally low Ki of 1.9 nM. The IC50 value of 26 nM, elucidated in an assay that assessed inhibition second messenger inositol phosphate or cAMP production, was adequately low. In a bioluminescence resonance energy transfer (BRET), the IC50 value was also low at 15 nM. TM-30642 was determined to be a surmountable antagonist of GPR44 in [3H]PGD2 saturation isotherms, effects of which were modeled using Scatchard plots. In PGD2-mediated stimulation of [35S]GTPγS binding in transfected GPR44 cells, TM-30642 produced rightward shifts and no change in Bmax (maximum specific binding) in PGD2-stimulated inositol phosphate production, β-arrestin translocation, and ex vivo human eosinophil shape change, characteristic of competitive antagonists [55]. TM-30642 was further tested as a racemic mixture and demonstrated negligible off-target binding for both TP and DP1. The weak binding to TP was found through a [3H]SQ29548 assay, where TM-30642 demonstrated a Ki of 3000 nM. The results were confirmed in an assessment of TP agonist U-46619-induced inositol phosphate accumulation, in which the IC50 was desirably high at 4600 nM, indicating that TM-30642 had a weaker selectivity for TP than ramatroban. TM-30642 was also non-selective for DP1, with a Ki of 6100 nM and IC50 > 10,000 nM. Of note, this study indicates that all of the enantiomers of TM-30642 displayed negligible off-target binding to TP or DP1 [65]. The pharmacokinetic profile of TM-30642 has not been made available, but a half-life of 7.72 min was reported from a [3H]PGD2 displacement binding assay.
TM-30643 ([3-(4-fluoro-benzenesulfonylamino)-1,2,3,4-tetrahydro-carbazol-9-yl]-acetic acid) was synthesized by replacing the propanoic acid in ramatroban with acetic acid (Figure 5), rather than the additional methyl group in TM-30642. The shortening of the carboxylic acid chain in TM-30643 led to a 1000-fold increase in GPR44 activity relative to ramatroban [65]. The affinity of TM-30643 to GPR44 was high, with a Ki of 0.51 nM elucidated in a [3H]PGD2 assay. Along with the low Ki value, the IC50 was shown to be 3.8 nM in second messenger production and BRET assay. TM-30643 was also characterized as an insurmountable antagonist of GPR44, in which PGD2 affinity does not change but Bmax decreases upon administration. In an indirect nonequilibrium model where [3H]PGD2 association curves modeled antagonist dissociation, TM-30643 notably reduced rate constant Kapp2 of the second slower phase of association by about 1.5 × 106 times. TM-30643 additionally did not change dissociation of [3H]PGD2 in HEK293 cells, indicating that it is unlikely to function through allosteric interactions [55]. TM-30643 also shows a desirably weak affinity for TP, with a high Ki of 540 nM from a [3H]SQ29548 assay. Furthermore, the IC50 for TP similarly showed little off-target binding at 1700 nM in an assay for U-46619-induced inositol phosphate accumulation. TM-30643 also did not display off-target binding to DP1 at a Ki of 5300 nM and IC50 > 10,000 nM. Similar to TM-30642, TM-30643 was studied as a racemic mixture, indicating that all enantiomers displayed little specificity to TP or DP1 [65]. The promising GPR44 affinity and selectivity of TM-30643 led to further characterization of the compound. TM-30643 exhibited concentration-dependent displacement of [3H]PGD2 in HEK293 cells, with a long half-life of 6.75 × 106 min [55].
Encouraged by the favorable properties of TM-30642 and TM-30643, Ulven and Kostenis combined the features of both analogues: addition of a methyl group to the nitrogen atom and replacement of the propanoic acid with the shorter acetic acid group. This resulted in the development of the insurmountable GPR44 antagonist TM-30089, also known as CAY10471 (Figure 5). Compared with ramatroban, TM-30089 demonstrated stronger sub-nanomolar potency towards GPR44 and negligible off-target binding to TP and DP1. The affinity of TM-30089 to GPR44 was evaluated in equilibrium competition bindings assays that showed a highly favorable Ki value of 0.60 nM. An IC50 value of 1.2 nM was also obtained through the inhibition of PGD2-induced cAMP production. In a GPR44 [3H]PGD2 binding assay, the log pKi value of ramatroban was 0.58 less than the TM-30089 value of 8.96. For TP, the [3H]SQ29548 binding assay showed TM-30089 had a log pKi over 1.5 times less than that for ramatroban, demonstrating reduced off-target binding [66]. Similar to TM-30642 and TM-30643, TM-30089 is a racemic mixture with enantiomers that displayed significant affinity and selectivity for GPR44 over TP. TM-30089 was found to retain its pharmacological profile in mouse and rat orthologs of GPR44. Both in vivo studies in mice and in situ studies on guinea pigs revealed that TM-30089 inhibited characteristic responses associated with GPR44 activation. In a mouse model of allergic asthma, a 5 mg/kg oral dose of TM-30089 applied four times over two consecutive days inhibited tissue eosinophilia and mucous cell hyperplasia, indicating strong affinity for GPR44 [66]. Furthermore, a study of circadian entrainment found that mice exhibited similarly impaired light-induced phase advance for both GPR44 knockouts and mice administered with TM-30089 [67]. Again in a mouse model, response following oral administration of TM-30089 mirrored GPR44 knockout mice, this time by reducing renal fibrosis [68].
Recommendations
Due to favorable selectivity for GPR44 over TP and DP1, TM-30642, TM-30643, and TM-30089 can all be considered promising candidates for developing GPR44 PET tracers.
2.2.3. Merck Analogues
MK-7246
The R enantiomer of the reverse indole of TM-30089 was isolated from the racemic mixture and identified as MK-7246 by Gallant et al. from Merck in 2007 (Figure 6), which displayed a higher affinity for and selectivity toward GPR44 over TP than the S enantiomer [69]. Subsequent studies investigated the compound’s pharmacokinetic and metabolic profile. MK-7246 displayed a Ki value of 2.5 nM from GPR44 competition binding assays and strong binding affinity to GPR44 in recombinant mouse, rat, dog, cynomolgus monkey, and human with IC50 values of 9.2 nM, 4.6 nM, 6.3 nM, 6.9 nM, and 3.5 nM, respectively [70]. In addition to a favorable affinity for GPR44, MK-7246 displayed little off-target binding, with a 149-fold weaker affinity for the DP receptor and more than 1500-fold weaker affinity for other prostanoid receptors. MK-7246 also demonstrated antagonist potency in blocking DK-PGD2-induced inhibition of cAMP in recombinant cells overexpressing human GPR44 (IC50 = 3.0 nM), DK-PGD2-induced eosinophil shape change (IC50 = 2.2 nM), and DK-PGD2-induced CD11b expression on eosinophils and basophils (IC50 = 6.2 nM and 5.4 nM, respectively).
Figure 6.

Molecular structure and synthesis of MK-7246 from ramatroban.
Administration of MK-7246 to mouse, rat, dog, sheep, and rhesus and cynomolgus monkey orally and intravenously demonstrated a good overall 24 h pharmacokinetic profile [70]. Favorable oral bioavailability above 50% was found in all species (F = 109%, 114%, 67%, and 57%, respectively), except in cynomolgus monkey for unknown reasons (F = 10%). While low plasma clearance following intravenous administration was observed in mouse (t1/2 = 2.8 h), rat (t1/2 = 5.6 h), rhesus monkey (t1/2 = 8.1 h), and sheep (t1/2 = 6.5 h), moderate plasma clearance was found in dog (t1/2 = 8.4 h). Ex vivo oral dosage of MK-7246 revealed a relatively good relationship between MK-7246 plasma levels and ex vivo inhibition of DK-PGD2-induced CD11b expression in blood eosinophils. In cynomolgus monkey, blood samples collected after oral administration of MK-7246 showed an IC50 value of 15 nM, and direct addition of MK-7246 to blood in vitro showed an IC50 value of 3.5 nM. In sheep, the IC50 values obtained were 107 nM following intravenous administration and 22.5 nM after direct in vitro administration. Overall, these results demonstrate that MK-7246 exhibits strong affinity and selectivity for human GPR44, antagonist activity on recombinant and endogenous GPR44, and good oral bioavailability and metabolic stability in various species.
Additional studies on the association kinetics of [3H]MK-7246 from recombinant human GPR44 measured an on rate of association (Kon) value of 0.0016 to 0.0017 min−1 × nM−1 and a t1/2[on] value of 19.8 to 20.9 min [70]. Comparatively, dissociation of [3H]MK-7246 had an off rate of dissociation (Koff) value of 0.0212 to 0.0216 min−1 × nM−1 and the t1/2[off] value of 32.2 to 33.9 min. Given the slower dissociation than association, these studies derived a low Kd (Koff/Kon) of 13.6 to 18.6 nM for [3H]MK-7246 at GPR44. In contrast, saturation analysis revealed a Kd value of 2.3 nM for [3H]MK-7246, which is similar to the MK-7246 Ki value of 2.5 nM from competition binding assays.
In vitro and in vivo PET studies performed for [11C]MK-7246 also demonstrated favorable results [71]. Autoradiography assays on human pancreatic sections with and without T2D showed receptor-mediated focal binding in areas with insulin positive islet of Langerhans, indicating the potential of [11C]MK-7246 to accurately quantify BCM. In rat, [11C]MK-7246 demonstrated rapid distribution in the blood, rapid excretion from the liver, and more gradual excretion through the kidney following intravenous administration. Most other tissues demonstrated low uptake and rapid washout. Biodistribution assays in rat by PET-MRI imaging predicted through dosimetry calculations a low radiation dosage in human, especially in radio-sensitive tissues such as red marrow. The largest predicted absorbed radiation doses were in the myocardium, liver, and small intestine of humans. Overall, the absorbed dose for the whole body was approximated as 0.0036 mSv of [11C]MK-7246. This could potentially allow for up to 7 PET examinations per year before reaching the clinical research annual limit of 10 mSv. In pig studies, [11C]MK-7246 showed similar excretion behavior from the liver and kidney and strong uptake from the small intestine. PET-CT investigations in pig demonstrated GPR44-mediated binding to the pancreas and off-target binding to the spleen. However, weaker binding was observed in the human spleen indicating that off-target spleen binding is not expected for human PET imaging. Abolishment of baseline [11C]MK-7246 uptake in pig using MK-7246 pretreatment showed a 66% reduction in the pancreas and 88% reduction in the spleen. While strong [11C]MK-7246 concentrations in the liver, duodenum, and jejunum may spillover into the PET imaging of small animals, the issue was not observed in pig PET imaging, suggesting that it may not pose a problem in human imaging.
Excretion of [11C]MK-7246 was found to occur mainly through the liver, bile, and intestines in both rat and pig studies [71]. The major metabolite excreted in a rat study was acyl glucuronide (90%), which displayed weak affinity for GPR44 (Ki > 7200 nM). Additionally, excreted were minor amounts of the parent MK-7246 (2.7%) and a decarboxylated metabolite (2.3%) [69]. Acyl glucuronide demonstrated low to moderate plasma clearance of 2.2–15 mL/min/kg, normal volume distribution of 0.5–4.8 L/kg, and normal plasma half-life from 5.6 to 11 h. The plasma exposure of acyl glucuronide compared to MK-7246 was found to vary among species and be highest in human primate (0.5–1.5) in comparison to dog (0.06) and Sprague Dawley rat (0.02). In various animals, the order of metabolic stability has been characterized as decreasing from the Sprague Dawley rat, beagle dog, cynomolgus monkey, to rhesus monkey and human.
Recommendations
In addition to having been well characterized, MK-7246 advantageously demonstrates potency and selectivity for GPR44 over other prostanoid receptors.
Merck Analogues in 2008
In 2008, a series of DP1 antagonists, described by Beaulieu et al. at Merck, were developed based off a 6-5-5 or 6-5-6 tricyclic scaffold. Among those, two enantiomers with a tetrahydrocarbazole group were synthesized (compounds 25 and 26, Figure 7), but the compounds exhibited poor affinity for GPR44. This may be inferred by the design of the study to develop DP1 antagonists. The binding for one could not be elucidated (compound 25), whereas the other demonstrated binding at 19,200 nM (compound 26). On the other hand, both exhibited strong affinity to DP1 [72]. Due to weak binding to GPR44 and off-target binding to DP1, these compounds should not be considered for development of GPR44 PET tracers. Other compounds in this series do not contain a tetrahydrocarbazole scaffold and, therefore, are not discussed in this study.
Figure 7.

Molecular structures of compounds 25 and 26.
Merck Analogues in 2011a
Encouraged by the promising results from MK-7246, Zaghdane et al. at Merck synthesized a series of novel GPR44 antagonists in 2011. These compounds were based on the structure of MK-7246, except with an amide group replacing the sulfonamide group [73].
The replacement of the sulfonyl group of MK-7246 with a carbonyl group yielded compound 27. Further addition of a CH2 group between the carbonyl and phenyl group produced compound 28, which showed promising properties (Table 1). The binding affinity of compound 28 for GPR44 was desirably strong with a Ki of 7.4 nM and IC50 of 12 nM, characterized through a cAMP assay in HEK293 cells. Additional studies for GPR44-related eosinophil shape change confirmed strong affinity with an IC50 at 4.8 nM. The selectivity of the compound was found to be optimal with IC50 values for DP1 and TP above 1000 nM. The pharmacokinetic profile of compound 16 showed issues with bioavailability, however, possibly due to limited access into the liver. In rat, the bioavailability was only 14%, but the clearance, distribution, and half-life were comparable to MK-7246 (0.38 mL/min/kg, 0.13 L/kg, and 4.6 h, respectively). The compound was also predicted to have no issues with drug-drug interaction, as there was little selectivity for CYP (IC50 > 50,000 nM). The metabolism of compound 28 was low, with 91% concentration remaining after 1 h and 60% glucuronidated. The study identified no hydroxylated metabolites produced from the compound or amide hydrolysis. However, compound 27 showed a weak affinity for GPR44 (Figure 8). The Ki and IC50 elucidated through a cAMP assay both were unsuitably high at 340 nM and 2500 nM, respectively. As such, the poor affinity of this compound led to discontinuation of its testing.
Table 1.
Binding affinity and functional antagonism on hCRTH2 for preliminary aryl amides and structure–activity relationship (SAR) on the amide linker (values were obtained from [73]).
| Compd. | hCRTH2 a (Ki) | cAMP b (IC50) | EOS c,d (IC50) |
|---|---|---|---|
| 27 | 340 nM | 2500 nM | |
| 28 | 7.4 nM | 12 nM | 4.8 nM |
| 29 | 3.0 nM | 6.2 nM | 6.0 nM |
| 30 | 2.0 nM | 2.0 nM | 4.4 nM |
| 31 | 1.8 nM | 4.0 nM | |
| 32 | 1.7 nM | 4.0 nM | 1.4 nM |
| 33 | 4.3 nM | 8.0 nM | 2.4 nM |
| 34 | 141 nM | ||
| 35 | 70 nM | ||
| 36 | 22 nM | ||
| 37 | 126 nM | ||
| 38 | 19 nM | ||
| 39 | 408 nM | ||
| 40 | 4.0 nM | 3.0 nM | 0.96 nM |
| 41 | 39 nM | 30 nM |
a Radioligand competition binding assay using membrane proteins from HEK293 (EBNA) cells stably expressing the receptor CRTH2 in a 10 mM solution of HEPES/KOH (all values are mean of two or more experiments). b Functional assay: the intracellular concentration of cAMP was determined using the 125I-cAMP scintillation proximity assay. The assay was performed in Hank’s balanced salt solution 25 mM HEPES containing 5000 nM Forskolin (Ki is an average of at least two independent titrations). c IC50s are an average of at least two independent titrations. d Whole blood eosinophil shape change assay.
Figure 8.

Molecular structures of compounds 27 and 28.
Methylation and replacement of the sulfone group by two methyl groups (29), a C3 (30), C4 (31) or C5 (32) cyclic group, or an oxygen containing 6-membered ring (33) produced compounds with optimal GPR44 affinity (Figure 9). The Ki values were favorably low at 3.0 nM, 2.0 nM, 1.8 nM, 1.7 nM, and 4.3 nM, respectively, with a comparable eosinophil shape change IC50 values of 6.0 nM, 4.4 nM, 1.0 nM, 1.4 nM and 2.4 nM. Furthermore, cAMP IC50 values of 6.2 nM, 2.0 nM, 4.0 nM, 4.0 nM and 8.0 nM for compounds 29, 30, 31, 32 and 33, respectively, confirmed that these compounds have a strong affinity for GPR44. Additional characterization reported that compounds 31, 32, and 40 (discussed below) have superior pharmacokinetic properties, especially oral bioavailability. For instance, compound 31 showed a high bioavailability (F = 93%). Further properties (Cl = 12 mL/min/kg, Vd = 2.6 L/kg, t1/2 = 2.6 h, and CYP3A4 and CYP C9 IC50 > 50,000 nM) were also in the optimal range.
Figure 9.

Molecular structures of compounds 29–39.
Additional compounds were synthesized by substitution of the phenyl ring outside the tetrahydrocarbazole group but were not further characterized because they exhibited weak affinity for GPR44 (Figure 9). The Ki values were 141 nM (compound 34), 70 nM (compound 35), 22 nM (compound 36), 126 nM (compound 37), 19 nM (compound 38) and 408 nM (compound 39).
Addition of a methyl group resulted in two diastereomers, but only the R-isomer (compound 40) exhibited favorable affinity values (Figure 10). The Ki value was low at 4.0 nM, and the IC50 values, elucidated through eosinophil shape change and cAMP production, were 0.96 nM and 3.0 nM, respectively. The selectivity of compound 40 for GPR44 was also promising, in which the compound exhibited at least 500-fold selectivity for GPR44 over IP, EP1, EP2, EP3, EP4 and FP. In vivo studies on compound 40 found optimal pharmacokinetic properties. The bioavailability (F) of 73%, drug clearance (Cl) of 1.1 mL/min/kg, volume of distribution (Vd) of 0.36 L/kg, and half-life of 4.7 h were all optimal or acceptable. Compound 40 should also have little drug-drug interaction, as its affinity for CYP 3A4 and CYP 2C9 showed an IC50 greater than 50,000 nM. By comparison, the other isomer (compound 41) showed a higher Ki value of 39 nM and IC50 value of 30 nM from a cAMP assay.
Figure 10.

Molecular structures of compounds 40 and 41.
Recommendations
The strong affinity and pharmacokinetic properties mark compounds 29–33 and 40 as promising candidates for PET tracers, with compounds 31 and 32 showing the most favorable Ki values. Therefore, all these compounds may be developed into potentially selective GPR44 PET tracers.
Merck Analogues in 2011b
In 2011, Simard et al. at Merck synthesized a series of compounds based on MK-7246 [74].
Compounds 42–45, which are regioisomers of one another with different placements of the N atom in the pyridine ring, were found to have weak affinities for GPR44 (Table 2). The affinity was most promising for compound 45, which has a 7-azaindole group, with a Ki value of 3.3 nM in binding assays. However, the Ki values were unfavorable for the other three compounds at 7189 nM (compound 42), 6725 nM (compound 43), and 139 nM (compound 44). The cAMP IC50 values correlated to the Ki values and were undefined (compounds 42 and 43), 351 nM (compound 44), and 3.4 nM (compound 45). Further characterization of compound 45 through the eosinophil shape change assay reported an IC50 value of 7.0 nM. There was also little to no off-target binding to TP and DP1. Specifically, the Ki of compounds 42–45 to TP were > 7000 nM, > 6800 nM, > 22,000 nM, and > 22,000 nM, respectively; the Ki for binding to DP1 were > 1300 nM, > 3400 nM, > 12,000 nM, and > 47,000 nM, respectively. Due to its strong affinity and selectivity for GPR44, compound 45 was further characterized for covalent binding, and the resulting value (52 pmol equiv/mg at 6 h and 50 equiv/mg at 24 h) indicated potential hepatotoxicity. Meanwhile, the CYP value was optimal at an IC50 > 50,000 nM, indicating little potential for drug-drug interaction.
Table 2.
Activities of azaindole sulfonamides and amides (values were obtained from [74]).
| Compd. | hCRTH2 a (Ki) | cAMP b (IC50) | EOS c (IC50) |
DP a (Ki) |
TP a (Ki) |
|---|---|---|---|---|---|
| 42 | 7189 nM | > 1300 nM | > 7000 nM | ||
| 43 | 6725 nM | > 3400 nM | > 6800 nM | ||
| 44 | 139 nM | 351 nM | > 12,000 nM | > 22,000 nM | |
| 45 | 3.3 nM | 3.4 nM | 7.0 nM | > 47,000 nM | > 22,000 nM |
| 46 | 1.8 nM | 3.2 nM | 3.3 nM | > 18,000 nM | > 27,000 nM |
| 47 | 1.9 nM | 3.6 nM | 15.8 nM | > 3800 nM | > 1000 nM |
| 48 | 3.4 nM | 5.7 nM | 1.2 nM | > 38,000 nM | > 71,000 nM |
| 49 | 21.1 nM | 73.8 nM | > 12,000 nM | > 22,000 nM | |
| 50 | 4.7 nM | 3.5 nM | 2.3 nM | > 10,000 nM | > 21,000 nM |
| 51 | 3.6 nM | 7.5 nM | 3.1 nM | > 3700 nM | > 6800 nM |
| 52 d | 5.1 nM | 4.4 nM | 2.4 nM | > 4000 nM | > 7200 nM |
| 53 | 3.4 nM | 4.7 nM | 1.2 nM | > 4000 nM | > 7200 nM |
| 54 | 3.9 nM | 4.5 nM | 3.4 nM | > 4000 nM | > 7200 nM |
| 55 | 11.5 nM | 7.0 nM | > 4000 nM | > 7200 nM |
a Radioligand competition binding assay using membrane proteins from HEK293 (EBNA) cells stably expressing the receptor hCRTH2 in a 10 mM solution of HEPES/KOH (all values are mean of two or more experiments). b Functional assay: the intracellular concentration of cAMP was determined using the 125I-cAMP scintillation proximity assay. The assay was performed in Hank’s balanced salt solution 25 mM HEPES containing 5000 nM Forskolin (all values are mean of two or more experiments). c Human whole blood eosinophil shape change assay (all values are mean of two or more experiments). d Diastereomeric mixture.
Compound 46 is a ramatroban analogue with a chlorine group that creates an electrophilic site on the azaindole. It exhibited strong affinity for GPR44 with a Ki value of 1.8 nM. Another compound with a fluorine substituted phenyl group in a corresponding position to the chlorine of compound 46 (compound 47) showed similarly strong affinity of Ki = 1.9 nM. The IC50 values, determined in a cAMP assay, were similar at 3.2 nM (compound 46) and 3.6 nM (compound 47). However, in eosinophil shape change assays, compound 46 exhibited a more favorable IC50 of 3.3 nM than the IC50 of 15.8 nM of compound 47 (Figure 11). All Ki values for TP and DP1 were greater than 1000 nM (compound 46: TP > 27,000 nM and DP > 18,000 nM; compound 47: TP > 1000 nM and DP > 3800 nM). Both compounds demonstrated favorable affinity and specificity for GPR44, with negligible binding to TP and DP1.
Figure 11.

Molecular structures of compounds 42–47.
Compounds 48–55 (Figure 12) were produced with an amide group and displayed optimal binding affinity. All Ki values were 11.5 nM or lower, except for compound 49, with the lowest value being 3.4 nM for compounds 48 and 53 (Table 2). The strong affinity indicated by the low Ki values was corroborated further in cAMP and eosinophil shape change assays, with IC50 values under 10 nM. All compounds showed little potential for off-target binding, as Ki values for DP1 and TP were all greater than 4000 nM. Unlike compound 48, however, the stereoisomer compound 49 had cAMP assay Ki and IC50 values of 21.1 nM and 73.8 nM, respectively, indicating weak affinity for GPR44. The difference between compounds 48 and 49 evidenced the stereo-sensitivity of GPR44 binding.
Figure 12.

Molecular structures of compounds 48–55.
The pharmacokinetic properties of the compounds were assessed in rat and demonstrated greater than 50% bioavailability in only three compounds. Compounds 48, 50 and 51 exhibited bioavailability of 84%, 156% and 63%, respectively. The pharmacokinetic profile of compound 48 was the foremost with a rapid clearance of 3.6 mL/min/kg, a low volume of distribution of 0.67 L/kg, and a half-life at 4 h. Based on its favorable pharmacokinetic profile, compound 48 was further tested for covalent binding (8 pmol equiv/mg at 6 h and 6 pmol equiv/mg at 24 h), CYP binding (IC50 > 50,000 nM), and Kobs (activation constant, representing CYP3A4 time-dependent inhibition, of < 0.004 min−1 at 50 mM). The lowered Kobs implies that compound 48 may have less risk for irreversible binding to the cytochrome active site and subsequent drug-drug interactions.
Recommendations
Compounds 45–48 and 50–55, particularly compounds 48 and 53, demonstrated desirable affinity and selectivity for GPR44. These compounds may be considered as candidates for developing GPR44 PET tracers to assess BCM.
2.2.4. AstraZeneca
In 2011, Luker et al. at AstraZeneca conducted a virtual screening to identify compounds with similar pharmacophore features as ramatroban [75]. The identified lead compound 56 (Figure 13) was then optimized through side chain modifications and changes around the acid to develop a series of zwitterions which included potent and selective GPR44 antagonists. Although compound 56 shows a moderate IC50 for GPR44 of 16 nM, compound 56 is not discussed in more detail as it does not contain any fluorine atoms for potential 18F-labeling.
Figure 13.

Molecular structures of compounds 56–61.
Compound 57, which replaced the -Cl of compound 56 with a para-CF3 subunit on the phenoxyacetic acid ring, had a favorable IC50 for GPR44 of 18 nM. It was further confirmed as an antagonist, rather than an agonist, of GPR44 through an eosinophil shape change assay, where it lowered the response rate of DK-PGD2 to less than 10%. The lipophilicity, measured through log D7.4 of 0.4, and clearance (both in rat hepatocytes and human liver microsomes) of < 4 µL/min/1 × 106 cells and 6 µL/min/mg, respectively, were both moderate. However, in a Ca2+ assay, the IC50 for compound 57 was found to be unacceptably high at 517 nM.
Compound 58 was produced through replacement of the sulfonyl group in compound 56 with a carbonyl group, and replacement of the chlorine group with the CF3 group [75]. Importantly, this resulted in minimal changes to potency, despite the expectation that the highly charged acids would require control of polar surface area in order to maintain optimal oral pharmacokinetic profiles [76]. Compound 58 demonstrated an IC50 value of 316 nM in radiometric binding assays [75]. As such, compound 58 showed an undesirably weak affinity for GPR44 (Table 3).
Table 3.
Preliminary SAR of compounds 57–61 (values were obtained from [75]).
| Compd. | CRTH2 Binding (IC50 a) | Log D7.4 | Rat Hep Clint b | Hum Mic Clint c | Agonism EOS Shape Change d | CRTH2 Ca2+ (IC50e) |
|---|---|---|---|---|---|---|
| 57 | 18 nM | 0.4 | <4 | 6 | IA | 517 nM |
| 58 | 316 nM | <3 | 5 | |||
| 59 | 10 nM | 1.0 | <3 | 11 | IA | |
| 60 | 1.0 nM | <3 | <3 | IA | 9 nM | |
| 61 | 0.3 nM | 0.8 | <3 | <3 | yes |
a Radiometric binding assay, n > 2 measurements. b Rat hepatocyte intrinsic clearance (µL/min/1 × 106 cells). c Human liver microsomes intrinsic clearance (µL/min/mg). d Agonism in eosinophil shape change, IA refers to <10% response of DK-PGD2. e Antagonism of PGD2-mediated Ca2+ flux in human CHO cells, n > 2 measurements.
The halogen substitution on the distal aryl ring using 4-fluorophenyl resulted in compound 59 (Figure 13), which demonstrated a low IC50 value of 10 nM. An additional substitution with a methyl group on the piperazine ring (compound 60) further decreased the IC50 to a desirably low value of 1.0 nM (Figure 13). The para-position was found to be more potent and metabolically stable. Replacement of the chlorine in compound 60 with a CF3 (compound 61) maintained similar activity, with an IC50 value of 0.3 nM (Figure 13). While other compounds did not display agonist activity and behaved as antagonists, compound 61 showed partial agonist activity in the eosinophil shape change assay, with a DK-PGD2 efficacy of around 40%.
Recommendations
The strong affinities of compounds 59–61 make them promising candidates for the development of GPR44 PET tracers.
2.2.5. Actelion Pharmaceuticals Ltd.
Setipiprant
In 2013, Fretz et al. at Actelion Pharmaceuticals Ltd. used a lead optimization program to screen 80,000 compounds from a GPCR library, looking for potential GPR44 antagonists. In screening for structure–activity relationship (SAR), particularly for oral bioavailability, the study discovered the precursor to a lipophilic naphthyl-containing compound, later known as setipiprant. Setipiprant (2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid or ACT-129968) is a derivative of indole-1-acetic acid, which is a selective inhibitor of GPR44 [77].
The GPR44 binding affinity of setipiprant was found to be promising. In HEK-293 cells expressing hCRTH2, Ca2+ and cAMP assays showed IC50 values of 30 nM and 80 nM, respectively. Additional binding studies in buffer (6 nM) and HSA (340 nM) for IC50 were also characterized, confirming desirable affinity. The selectivity of setipiprant to GPR44 was also encouraging, as IC50 values for other prostanoid receptors were consistently above 1200 nM. Specifically, a screen for DP1 (IC50 = 1290 nM, fSel = IC50 (hDP1)/IC50 (hCRTH2) = 215), EP1 (IC50 > 25,000 nM), EP2 (IC50 = 2600 nM), EP3 (IC50 > 25,000 nM), EP4 (IC50 > 10,000 nM), and TP (IC50 > 25,000 nM) binding showed weak affinity for prostaglandin receptors beside GPR44. Setipiprant is therefore a selective and potent inhibitor of GPR44.
The physicochemical properties of setipiprant were also identified, revealing a molecular weight of 402.42 Da. The elimination half-life of setipiprant was shown to be greater than 4 h following incubation in rat and human plasma and simulated intestinal fluid (SIF), but only greater than 1 h in simulated gastric fluid (SGF). Setipiprant’s intrinsic clearance was favorable at Cl values below 10 uL/min/mg/protein. Additional properties of low lipophilicity and high solubility were also regarded as optimal, at a ligand efficiency (LE) of 0.37, cLogP of 3.3, log D7.4 of 0.1, solubility in water of 50 ug/mL at pH 4.3, solubility in buffer of 90 ug/mL at pH 4, and solubility in buffer of 880 ug/mL at pH 7. An in vivo study using rats found intravenously administered setipiprant AUC(0−last) of 58,500 ng/h/m, plasma clearance of 1.3 mL/min/kg, and oral bioavailability of 44%. Later, in male beagle dog administered setipiprant orally and intravenously, the compound showed an AUC(0−last) of 91,100 ng/h/m, clearance of 1.3 mL/min/kg, and bioavailability of 55%. The higher exposure and slightly greater bioavailability but same clearance in dog and rat shows that setipiprant exhibits species-dependent pharmacokinetic properties. Of note is that a FLIPR assay against mouse, rat, guinea pig, and dog GPR44 revealed that setipiprant acts as a partial agonist on mouse and guinea pig receptors. This indicates that studies in vivo may be limited in choice of organisms.
The possibility of drug-drug interaction was shown to be low, with an in vitro CYP2C9 IC50 > 50,000 nM, HLM of 5 μL/min/mg protein, RLM of 5 μL/min/mg protein, and intrinsic clearance (Clint) in rat hepatocytes of 3.1 μL/min/106 cells. Setipiprant was also found to not accumulate in the body, as assessed by clinical trial [78]. The profile of setipiprant was further characterized in an open-label, 2-period, 2-way crossover, randomized study in which patients were administered 250 or 500 mg setipiprant, and elucidated the following properties in humans (Table 4) [79].
Table 4.
Plasma pharmacokinetic variables of setipiprant in healthy subjects after administration of a single 500 mg dose of setipiprant as a capsule (n = 20) or tablet (n = 20) (values were obtained from [79]).
| Variable | Capsule a | Tablet a | Tablet: Capsule b |
|---|---|---|---|
| Cmax, µg/mL | 6.44 (5.46–7.58) | 6.04 (4.72–7.74) | 0.94 (0.79–1.12) |
| tmax, h | 3.00 (1.50–5.00) | 3.50 (1.00–5.00) | 0.00 (−0.50–1.00) |
| t1/2, h | 11.12 (9.76–12.67) | 11.40 (10.54–12.34) | 1.03 (0.93–1.13) |
| AUC0−∞, µg∙h/mL | 31.50 (26.52–37.40) | 31.88 (26.54–38.31) | 1.01 (0.92–1.12) |
a Values indicate geometric means (95% CIs), except for tmax, for which median and range are presented. b Values indicate ratio of geometric means (90% CIs), except for tmax, for which difference of medians (90% CIs) is presented.
As a result of its promising properties, setipiprant continued to be assessed in clinical studies. A 3-centre, double-blinded, placebo-controlled, cross-over study of 18 allergic asthmatic males administered with 1000 mg setipiprant or matching placebo showed that in allergen-induced airway responses, the compound reduced the late asthmatic response, inhibiting the AUC on average by 25.6%, while no difference in the early asthmatic response or allergen-induced changes in exhaled nitric oxide was observed. Setipiprant also showed a protective effect against the allergen-induced airway hyperresponsiveness to methacholine [80]. A prospective, randomized, double-blind, placebo- and active-referenced (cetirizine) phase 2 trial and phase 3 trial of 100–1000 mg setipiprant investigated effects on allergic rhinitis, and showed significant, dose-related improvement in mean change from baseline day-time nasal symptom scores over 2 weeks for the phase 2 trial but no significant effect in the phase 3 trial. Additionally, total and individual night-time nasal symptom scores and day-time eye symptom scores symptom scores were significantly improved with setipiprant 1000 mg compared to placebo in the phase 2, but not the phase 3, trial. The study did show a favorable safety profile for setipiprant [81]. Another randomized, double-blind, placebo-controlled study of 2000 mg setipiprant administered orally also exhibited favorable safety in both single- and multiple-dose administration [78].
The inadequate efficacy exhibited by setipiprant as a pharmaceutical in the described phase III study, in addition to three phase II studies (including a 12-week study in 438 asthmatics), led to the termination of further studies in April 2012 [82].
Later studies characterized the metabolic breakdown of setipiprant, which discovered four main metabolites (M1-4, Figure 14). The metabolites were found following incubation of [14C] setipiprant with human hepatocytes, microsomes, and bactosomes expressing the human enzymes CYP2C8, CYP2C9, and CYP3A4. The CYP enzymes initially epoxide the naphthoyl ring of setipiprant, and the regioisomeric epoxide products are hydrated by human epoxide hydrolase 62 to produce M1 and changed by epoxide 63 to produce M2. The pre-hydration epoxide may also be subject to a hydride shift (NIH shift) that produces 4-hydroxy-naphthoyl metabolite M3. Other processes produce the 5-hydroxy-naphthoyl metabolite M4 [83].
Figure 14.

Metabolites of setipiprant [83]. The asterisk (*) denotes the position of the 14C atom in [14C]setipiprant.
A clinical study of 6 healthy male subjects orally dosed with a total of 1000 mg 14C-labeled setipiprant also showed that M1 and M2 were the main metabolites. M1 and M2 accounted for 20.0% and 15.3% of the administered radioactive dose. Of note is that M2 and other metabolites besides M1 were not determined in plasma, and even M1 exhibited concentrations consistently below 10% of those of unchanged setipiprant. Feces proved to be the primary recovery route for setipiprant, although the recovered amount of unchanged setipiprant in urine did account for 3.7% [84]. A second clinical study generally assessed that setipiprant was eliminated through a biphasic pattern with an elimination half-life between 10 h and 18 h, where steady-state conditions were reached after 2–3 days [78]. Due to the long elimination half-life, the metabolism of setipiprant is not expected to be a concern for PET imaging.
In the same study as setipiprant, Fretz et al. characterized a number of other compounds (Figure 15) [77]. 2-(3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid analogues were characterized for their affinity for GPR44. Fluorine-containing compounds 64 and 65 exhibited adequate IC50 values at 7 and 9 nM, respectively, measured in the presence of assay buffer solution. Additional characterization of compound 65 elucidated an IC50 value of 60 nM in binding in the presence of human serum albumin (HSA) and, an fHSA value of 6. Additionally, IC50 values of 50 nM, 150 nM, and 130 nM were found through a cell based Ca2+ flux assay, cAMP homogeneous time resolved fluorescence (HTRF) assay, and human eosinophil shape change assay, respectively. Regarding the selectivity for prostanoid receptors hDP1, hEP2, and hEP4, IC50 values were greater than 10,000 for each of hDP1, hEP2 β-arrestin, and hEP4 β-arrestin, indicating negligible off-target binding. Additionally, the fSel value was greater than 1100. Another fluorine-containing analogue, compound 66, showed a less favorable IC50 of 35 nM (Figure 15).
Figure 15.

Molecular structures of setipiprant and compounds 64–74.
Extensive changes to the precursor compound were made to produce the synthesized compounds (Figure 15). Out of compounds 67–74, only compound 73 consistently produced IC50 values below 50 nM at 15 nM and 40 nM in two assays for Ca2+ flux and cAMP, respectively. The IC50 values for the other compounds 67–72 and 74 were determined using measurements of Ca2+ flux (40, 40, 1280, 20, 660, 70, and 65 nM, respectively) and cAMP (70, 95, not determined (nd), 380, nd, 160, and 160 nM, respectively). Additional binding studies in buffer (4, 9, 190, 11, 70, 4, 5, and 10 nM, respectively) and HSA (12, 60, 180, 70, 530, 180, 35, and 30 nM, respectively) for IC50 were also characterized (Table 5). The lower affinity of compounds 69 and 71 may be attributed to the substitution of chlorine and fluorine at the C6 and C8 position of the core, respectively. Additionally, setipiprant exhibited a substantially higher albumin shift up to a factor fHSA of 57; this appears to be associated with the presence of a naphthyl or a naphthylmethyl moiety. In addition, all antagonists inhibited the PGD2 induced shape change of human eosinophils in the hESC assay, and compounds 67, 68 and 73 were discovered to antagonize hESC most effectively with IC50 < 100 nM. All compounds reduced COX-1 (prostaglandin-endoperoxide synthase) enzyme activity by less than 30% at 10,000 nM compound concentration (all IC50 values were determined well above 10,000 nM).
Table 5.
SAR study exploring compounds 65 and 67–74, with IC50 binding data for hCRTH2 of selected compounds, measured in the presence and absence of human serum albumin (HSA) in the assay buffera (values were obtained from [77]).
| Compd. | hCRTH2 Receptor Interaction | Prostanoid Receptor Interaction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Binding in | Ca2+ Flux a | cAMP | hESC | hDP1 | fSel c | hEP2 | hEP4 | |||
| Buffer IC50 d | HSA IC50 d | fHSA b | IC50 d | IC50 d | IC50 d | Binding IC50 d | β-Arrestin IC50 d | β-Arrestin IC50 d | ||
| Setipiprant | 6 | 340 | 57 | 30 | 80 | 235 | 1290 | 215 | 2600 | >10,000 |
| 65 | 9 | 60 | 6 | 50 | 160 | 130 | >10,000 | >1100 | >10,000 | >10,000 |
| 67 | 4 | 12 | 3 | 40 | 70 | 34 | >10,000 | >2500 | >10,000 | >10,000 |
| 68 | 9 | 60 | 7 | 40 | 95 | 60 | >10,000 | >1100 | >10,000 | >10,000 |
| 69 | 190 | 180 | 1 | 1280 | nd | nd | 4100 | 22 | >10,000 | >10,000 |
| 70 | 11 | 70 | 6 | 20 | 380 | 180 | 2200 | 200 | >10,000 | >10,000 |
| 71 | 70 | 530 | 7 | 660 | nd | nd | 310 | 4.1 | 2700 | >10,000 |
| 72 | 4 | 180 | 45 | 70 | 160 | 275 | >10,000 | >2500 | >10,000 | >10,000 |
| 73 | 5 | 35 | 7 | 15 | 40 | 50 | >10,000 | >2000 | >10,000 | >10,000 |
| 74 | 10 | 30 | 3 | 65 | 160 | 210 | >10,000 | >1000 | 9500 | >10,000 |
nd = not determined.a IC50 values are given for the effects in the cell based Ca2+ flux assay, the cAMP homogeneous time resolved fluorescence (HTRF) assay, and the human eosinophil shape change assay. Data are given to demonstrate selectivity against prostanoid receptors hDP1, hEP1-4, and hTP2. The IC50 values represent the mean from at least three independent experiments if not stated otherwise. b Human serum albumin shift factor fHSA = IC50 (HSA)/IC50 (buffer). c Selectivity factor fSel = IC50 (hDP1)/IC50 (hCRTH2). d Units are nM.
Selectivity of compounds 67–74 was further determined in a screen against DP1 (IC50 > 10,000, > 10,000, 4100, 2200, 310, > 10,000, > 10,000, > 10,000 nM, fSel > 2500, > 1100, 22, 200, 4.1, > 2500, > 2000, and > 10,000, respectively), EP1 (IC50 > 25,000 nM for all in Ca2+ assays), EP2 (IC50 > 10,000, > 10,000, > 10,000, > 10,000, 2700, > 10,000, > 10,000, and 9500 nM, respectively in the β-arrestin and binding assays), EP3 (IC50 > 25,000 nM for all in Ca2+ assays), EP4 (IC50 > 10,000 nM for all in the β-arrestin and binding assays), and TP2 (IC50 > 25,000 nM for all). Of note is that FLIPR assays against mouse, rat, guinea pig, and dog GPR44 revealed that all compounds exhibited antagonistic effects when binding to GPR44 with comparable potencies, except for compound 68, which were identified as partial agonists on the mouse and the guinea pig receptor. This indicates that the choice of species used to develop PET tracers from compound 68 will have to be carefully considered. The physicochemical properties of the compounds with fSel > 200 in favor of hCRTH2 were further studied. The Clint values of compounds 67 and 68 in rat liver microsomes were low at 8 and less than 4 μL/min/mg protein, respectively. However, the Clint values of compounds 72 and 73 were high, indicating they may be prone to oxidative metabolism. The values of compounds 72 and 73 were between 23 and 63 μL/min/mg protein. However, none of the Clint values in rat hepatocytes were above 5.3 μL/min/106 cells, indicating low clearance in the liver. The plasma clearance (measured as Cl) for compound 67 was found to be detrimentally high (36 mL/min/kg). For compound 68, the low plasma clearance (5.9 mL/min/kg) and moderate exposure did not exempt it from a low oral bioavailability of 23%. Meanwhile, the exposure for compound 73 was dramatically low at 62 ng/h/mL and led to a bioavailability of 2%. Additional pharmacokinetic properties were elucidated from male beagle dogs orally administered 10 mg/kg and intravenously administered 1 mg/kg of compound 68 and setipiprant. The results show that the exposure was high at 9620 and 91,100 ng h−1 mL−1, respectively, and bioavailability was adequate at 64% and 55%, respectively. However, compound 68 demonstrated a greater clearance of 11 mL min−1 kg−1.
Recommendations
Setipiprant is an encouraging candidate for development of PET tracers to characterize BCM, particularly because it has been highly characterized as a potent compound both in vitro and in vivo. Among the analogues of setipiprant, compounds 64 and 65 presented favorable IC50 values, although additional characterizations will have to be made. It is worth noting that radiolabeling a trifluoromethyl group, such as those contained in compound 65, has been traditionally difficult due to isotopic dilution with fluorine-19. However, methods including using a novel radiofluorination reagent and reduction of base-cryptand concentration have been developed, through which one fluorine-18 is introduced into the molecule [85]. Other compounds may be used as a reference in developing new PET tracers, such as compounds 72 and 74, which exhibited excellent to moderate IC50 values, specificity for GPR44, preferable plasma clearance, and favorable bioavailability.
2.2.6. CT-133 (Compound 45)
CT-133 (namely, compound 45; {8-[(4-fluoro-benzenesulfonyl)-methyl-amino]-6,7,8,9-tetrahydro-pyrido[3,2-b]indolizin-5-yl}-sodium acetic acid) was further evaluated by Guo et al. at the CSPC Pharmaceutical Group in 2015 (Figure 16) [86]. The pharmacokinetics of CT-133 was also promising in vivo, with satisfactory bioavailability. In mouse, dog, and rat, oral administration of CT-133 showed bioavailability of 89.6%, 85.4%, and 71.6%, respectively, with little difference in repeated and single administration in rat. The safety profile was determined to be favorable, and there was no induction of metabolic enzymes. In another example of CT-133’s safety, rat dosed with up to 2000 mg/kg/day and dog dosed with up to 1000 mg/kg/day or 100 mg/kg/day for seven days showed no concerning reaction. Later, CT-133 was shown to be a potent antagonist for treatment of disease. In lipopolysaccharide-induced acute lung injury, 10 or 30 mg/kg CT-133 administered intragastrically suppressed neutrophil and macrophage cell count dose-dependently and migration to PGD2 locations in vitro and in mouse lungs [87,88].
Figure 16.
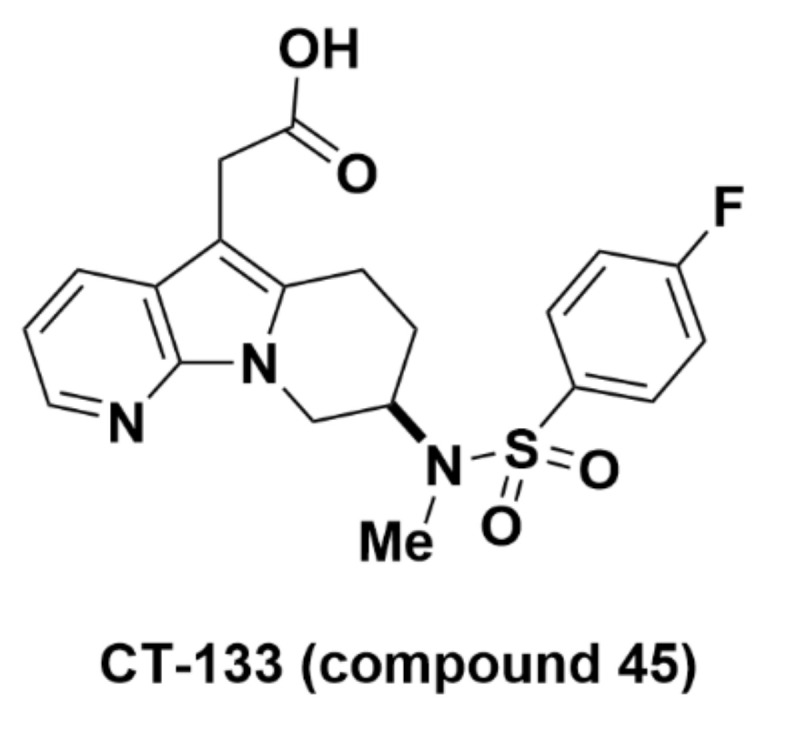
Molecular structure of CT-133 (compound 45).
In silico analysis provided further details into the mechanisms behind optimized binding of CT-133 to GPR44. CT-133 binds to a partially occluded region on extracellular GPR44 enclosed by the ligand entrance. Comparison of PGD2 and CT-133 binding on the region by estimates of the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and surface-exposure using the solvent accessible surface area (SASA) showed values of 1.80 Å (PGD2), 1.44 Å (CT-133), and 1.75 Å (unbound); 2.32 Å (PGD2), 2.17 Å (CT-133), and 3.10 Å (unbound); and 245805 Å2 (PGD2) and 187765 Å2 (CT-133), respectively. CT-133’s low RMSD value suggests that the compound significantly stabilizes the binding region, perhaps favoring CT-133 binding over PGD2. The stabilization is corroborated by the low RMSF and SASA values, representing the residues’ motion restriction and reduced orientations, respectively. In the active site, PGD2 and CT-133 comparison yielded RMSD values of 1.52 Å and 0.68 Å, respectively, again showing the higher stability and binding affinity of CT-133. Inhibitor qualities of CT-133 were confirmed by RMSD values (2.74 Å) of binding to helix 8, a region believed to assist inhibition of GPR44 function, which were lower than that for PGD2 (3.55 Å) or the unbound receptor (3.426 Å). Higher RMSF values of 4.53 Å for CT-133 relative to relative to 3.54 Å for PGD2 represented greater CT-133-mediated helix 8 flexibility. The RMSD and RMSF of CT-133 binding to helix 8 both represent more optimal inhibitor qualities.
The binding affinity for GPR44, estimated through a MM/PBSA-based approach, showed total free energy values of -67.50 kcal/mol for CT-133 and -50.12 kcal/mol for PGD2. The lower binding energy has been attributed to the 7-azaindole group that causes CT-133 to bind more favorably to GPR44 than PGD2 [89].
Recommendations
Due to its strong binding affinity and selectivity for GPR44 (see Section 2.2.3. Merck Analogues), in addition to confirmed pharmacokinetics in vivo, CT-133 is a promising compound for use in development of GPR44-specific PET tracers.
2.3. Radiosynthesis Strategies for 18F-Labeled Ramatroban-Based Analogues
The 77 selected ramatroban analogues containing a fluorine nuclide were characterized for properties including binding affinity, off-target binding, and pharmacokinetic and metabolic profile. Among these analogues, 32 compounds with favorable properties were recommended as potential 18F-labeled GPR44 PET tracers. Late-stage radiofluorination and the building block approach are two major strategies for radiosynthesis of 18F-labeled tracers. In recent years, novel and promising methodologies to implement the two strategies of aromatic radiofluorination have been developed and discussed in the comprehensive reviews by Preshlock et al., Brooks et al., and van der Born et al. [90,91,92]. Additionally, strategies are being actively developed and reported, which enable 18F-labeling in places that are traditionally challenging. For example, Chen et al. described a method for direct 18F-labeling [93]. Given these strategies, it would be beneficial to successfully generate the recommended potential 18F-labeled GPR44 analogues.
Our lab recently followed the strategy outlined above in selecting promising compounds to develop GPR44-specific PET radiotracers and utilized nucleophilic substitution to radiolabel with fluorine-18 a precursor (Precursor 1, 4-(N-(9-(carboxymethyl)-2,3,4,9-tetrahydro-1H-carbazol-3-yl)-N-methylsulfamoyl)-N,N,N-trimethylbenzenaminium-trifluoromethanesulfonate) of the selective GPR44 antagonist, TM-30089 (Ki = 0.6 nM) (Scheme 1). The resulting novel GPR44 tracer, [18F]-TM-30089 ([18F]-({3-[(4-fluoro-benzenesulfonyl)methyl-amino]-1,2,3,4-tetrahydro-carbazol-9-yl}acetic acid), demonstrated high radiochemical yield (30%) and radiochemical purity (> 98%). [18F]-TM-30089 biodistribution analysis additionally showed low tracer uptake in NOD/SCID mouse pancreas, matching the characteristic GPR44 expression present there. The manuscript is currently being prepared for publication.
Scheme 1.

Radiosynthesis of [18F]-TM-30089.
3. Conclusions
Taken together, the existing pool of ramatroban analogues containing fluorine groups could serve as a source of promising GPR44 18F-labeled PET tracers to investigate metabolic diseases longitudinally, monitor beta cell therapies, and evaluate pharmaceutical drug efficacy. To develop these compounds into PET tracers, direct and indirect radiolabeling methods may be utilized to incorporate fluorine-18. Our group followed the outlined approach and effectively developed [18F]-TM-30089 in an unpublished study. Therefore, there is great potential for future development of 18F-labeled PET tracers targeting GPR44 to accurately monitor and quantify BCM.
Author Contributions
Writing—original draft preparation, L.A.H., K.X.H., J.T. and J.L; writing—review and editing, L.A.H., K.X.H., J.T., F.K. and J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Wanek Family Project for Type 1 Diabetes (50286-2007672).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lin X., Xu Y., Pan X., Xu J., Ding Y., Sun X., Song X., Ren Y., Shan P.-F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020;10:1–11. doi: 10.1038/s41598-020-71908-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eriksson O., Laughlin M., Brom M., Nuutila P., Roden M., Hwa A., Bonadonna R., Gotthardt M. In vivo imaging of beta cells with radiotracers: State of the art, prospects and recommendations for development and use. Diabetologia. 2016;59:1340–1349. doi: 10.1007/s00125-016-3959-7. [DOI] [PubMed] [Google Scholar]
- 3.Rahier J., Guiot Y., Goebbels R.M., Sempoux C., Henquin J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008;10:32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 4.Krogvold L., Edwin B., Buanes T., Ludvigsson J., Korsgren O., Hyöty H., Frisk G., Hanssen K.F., Dahl-Jørgensen K. Pancreatic biopsy by minimal tail resection in live adult patients at the onset of type 1 diabetes: Experiences from the DiViD study. Diabetologia. 2014;57:841–843. doi: 10.1007/s00125-013-3155-y. [DOI] [PubMed] [Google Scholar]
- 5.Henquin J.-C., Dufrane D., Nenquin M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes. 2006;55:3470–3477. doi: 10.2337/db06-0868. [DOI] [PubMed] [Google Scholar]
- 6.Sweet I.R., Cook D.L., Lernmark Å., Greenbaum C.J., Krohn K.A. Non—Invasive imaging of beta cell mass: A quantitative analysis. Diabetes Technol. Ther. 2004;6:652–659. doi: 10.1089/dia.2004.6.652. [DOI] [PubMed] [Google Scholar]
- 7.Wei W., Ehlerding E.B., Lan X., Luo Q.Y., Cai W. Molecular imaging of beta-cells: Diabetes and beyond. Adv. Drug. Deliv. Rev. 2019;139:16–31. doi: 10.1016/j.addr.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eriksson O. GPR44 as a target for imaging pancreatic beta-cell mass. Curr. Diabetes Rep. 2019;19:1–8. doi: 10.1007/s11892-019-1164-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marchese A., George S.R., Kolakowski L.F., Lynch K.R., O’Dowd B.F. Novel GPCRs and their endogenous ligands: Expanding the boundaries of physiology and pharmacology. Trends Pharmacol. Sci. 1999;20:370–375. doi: 10.1016/S0165-6147(99)01366-8. [DOI] [PubMed] [Google Scholar]
- 10.Marchese A., Sawzdargo M., Nguyen T., Cheng R., Heng H.H., Nowak T., Im D.S., Lynch K.R., George S.R., O’Dowd B.F. Discovery of three novel orphan G-protein-coupled receptors. Genomics. 1999;56:12–21. doi: 10.1006/geno.1998.5655. [DOI] [PubMed] [Google Scholar]
- 11.Lindskog C., Korsgren O., Pontén F., Eriksson J.W., Johansson L., Danielsson A. Novel pancreatic beta cell-specific proteins: Antibody—Based proteomics for identification of new biomarker candidates. J. Proteom. 2012;75:2611–2620. doi: 10.1016/j.jprot.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Jahan M., Johnström P., Selvaraju R.K., Svedberg M., Winzell M.S., Bernström J., Kingston L., Schou M., Jia Z., Skrtic S., et al. The development of a GPR44 targeting radioligand [(11)C]AZ12204657 for in vivo assessment of beta cell mass. Eur. J. Nucl. Med. Mol. Imaging Res. 2018;8:113. doi: 10.1186/s13550-018-0465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellström-Lindahl E., Danielsson A., Pontén F., Czernichow P., Korsgren O., Johansson L., Eriksson O. GPR44 is a pancreatic protein restricted to the human beta cell. Acta Diabetol. 2015;53:413–421. doi: 10.1007/s00592-015-0811-3. [DOI] [PubMed] [Google Scholar]
- 14.Hirai H., Tanaka K., Yoshie O., Ogawa K., Kenmotsu K., Takamori Y., Ichimasa M., Sugamura K., Nakamura M., Takano S., et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor Crth2. J. Exp. Med. 2001;193:255–262. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monneret G., Gravel S., Diamond M., Rokach J., Powell W.S. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–1948. doi: 10.1182/blood.V98.6.1942. [DOI] [PubMed] [Google Scholar]
- 16.Kupczyk M., Kuna P. Targeting the PGD2/CRTH2/DP1 signaling pathway in asthma and allergic disease: Current status and future perspectives. Drugs. 2017;77:1281–1294. doi: 10.1007/s40265-017-0777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pettipher R., Whittaker M. Update on the development of antagonists of chemoattractant receptor—Homologous molecule expressed on Th2 Cells (CRTH2). From lead optimization to clinical proof-of-concept in asthma and allergic rhinitis. J. Med. Chem. 2012;55:2915–2931. doi: 10.1021/jm2013997. [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Yao D., Deepak R.K., Liu H., Xiao Q., Fan H., Gong W., Wei Z., Zhang C. Structures of the human PGD2 receptor CRTH2 reveal novel mechanisms for ligand recognition. Mol. Cell. 2018;72:48–59. doi: 10.1016/j.molcel.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eriksson O., Johnstrom P., Cselenyi Z., Jahan M., Selvaraju R.K., Jensen-Waern M., Takano A., Sorhede Winzell M., Halldin C., Skrtic S., et al. In vivo visualization of beta-cells by targeting of GPR44. Diabetes. 2018;67:182–192. doi: 10.2337/db17-0764. [DOI] [PubMed] [Google Scholar]
- 20.Jahan M. Ph.D. Thesis. Karolinska Institutet; Stockholm, Sweden: 2016. Development of Novel PET Radioligands for Visualizing Beta Cell Mass and Amyloid Plaques. [Google Scholar]
- 21.Matthews P.M., Rabiner E.A., Passchier J., Gunn R.N. Positron emission tomography molecular imaging for drug development. Br. J. Clin. Pharmacol. 2012;73:175–186. doi: 10.1111/j.1365-2125.2011.04085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson O., Kiesewetter D.O., Chen X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconj. Chem. 2015;26:1–18. doi: 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Crespo A. Comparison of gallium-68 and fluorine-18 imaging characteristics in positron emission tomography. Appl. Radiat. Isotopes. 2013;76:55–62. doi: 10.1016/j.apradiso.2012.06.034. [DOI] [PubMed] [Google Scholar]
- 24.Darius H., Michael-Hepp J., Meyer J. Receptor binding properties of the new and specific thromboxane receptor antagonist Bay U 3405. Agents Actions Suppl. 1992;37:157–161. doi: 10.1007/978-3-0348-7262-1_21. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Martin I., Kern O., Brooks S., Smith L.B., Valenzuela-Leon P.C., Bonilla B., Ackerman H., Calvo E. Biochemical characterization of AeD7L2 and its physiological relevance in blood feeding in the dengue mosquito vector, Aedes aegypti. Fed. Eur. Biochem. Soc. J. 2020 doi: 10.1111/febs.15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishizuka T., Matsui T., Okamoto Y., Ohta A., Shichijo M. Ramatroban (BAY u 3405): A novel dual antagonist of TXA2 receptor and CRTh2, a newly identified prostaglandin D2 receptor. Cardiovasc. Drug Rev. 2006;22:71–90. doi: 10.1111/j.1527-3466.2004.tb00132.x. [DOI] [PubMed] [Google Scholar]
- 27.Theis J.G., Dellweg H., Perzborn E., Gross R. Binding characteristics of the new thromboxane A2/prostaglandin H2 receptor antagonist [3H]BAY U 3405 to washed human platelets and platelet membranes. Biochem. Pharmacol. 1992;44:495–503. doi: 10.1016/0006-2952(92)90441-K. [DOI] [PubMed] [Google Scholar]
- 28.Seuter F., Perzborn E., Rosentreter U., Böshagen H., Fiedler V.B. Inhibition of platelet aggregation in vitro and ex vivo by the new thromboxane antagonist (3R)-3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-Carbazolepropanoic acid. Arzneimittelforschung. 1989;39:1525–1527. [PubMed] [Google Scholar]
- 29.Fiedler V.B., Perzborn E., Seuter F., Rosentreter U., Böshagen H. Reduction of in vivo coronary artery thrombosis by the novel thromboxane antagonist (3R)-3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-Carbazolepropanoic acid. Arzneimittelforschung. 1989;39:1527–1530. [PubMed] [Google Scholar]
- 30.Perzborn E., Fiedler V.B., Seuter F., Stasch J.P., Weber H., Sander E., Böshagen H., Rosentreter U. Characterization of Bay U 3405, a novel thromboxane A2/endoperoxide receptor antagonist. Stroke. 1990;21:IV143–IV145. [PubMed] [Google Scholar]
- 31.Pieters H., Roodt J.P., Badenhorst P.N., van Wyk V., Schall R., Lötter M.G., Hundt H.K., Nel C.J. Antithrombotic activity of Bay u3405, a thromboxane A2-antagonist, in patients with Dacron aortic grafts: A random controlled clinical trial. Thromb. Haemost. 1993;70:903–908. doi: 10.1055/s-0038-1649697. [DOI] [PubMed] [Google Scholar]
- 32.Escolar G., Albors M., Garrido M., Bioque G., Díaz Ricart M., Carretero M., Ordinas A. Inhibition of platelet-vessel wall interactions by thromboxane receptor antagonism in a human in vitro system: Potentiation of antiplatelet effects of aspirin. Eur. J. Clin. Investig. 1998;28:562–568. doi: 10.1046/j.1365-2362.1998.00327.x. [DOI] [PubMed] [Google Scholar]
- 33.Mitsuhashi M., Tanaka A., Fujisawa C., Kawamoto K., Itakura A., Takaku M., Hironaka T., Sawada S., Matsuda H. Necessity of thromboxane A2for initiation of platelet—Mediated contact sensitivity: Dual activation of platelets and vascular endothelial cells. J. Immunol. 2001;166:617–623. doi: 10.4049/jimmunol.166.1.617. [DOI] [PubMed] [Google Scholar]
- 34.Ulrych T., Böhm A., Polzin A., Daum G., Nüsing R.M., Geisslinger G., Hohlfeld T., Schrör K., Rauch B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011;9:790–798. doi: 10.1111/j.1538-7836.2011.04194.x. [DOI] [PubMed] [Google Scholar]
- 35.Aizawa H., Takata S., Shigyo M., Matsumoto K., Koto H., Inoue H., Hara N. Effect of BAY u3405, a thromboxane A2 receptor antagonist, on neuro-effector transmission in canine tracheal tissue. Prostaglandins Leukot. Essent. Fat. Acids. 1995;53:213–217. doi: 10.1016/0952-3278(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 36.Braun M., Schrör K. Bay U 3405 inhibits cerebral vasospasm induced by authentic thromboxane A2. Stroke. 1990;21:IV152–IV154. [PubMed] [Google Scholar]
- 37.Aizawa H., Inoue H., Matsumoto K., Koto H., Nakano H., Hara N. Thromboxane A2 antagonist inhibits leukotriene D4-induced smooth muscle contraction in guinea-pig lung parenchyma, but not in trachea. Prostaglandins Leukot. Essent. Fat. Acids. 1996;55:437–440. doi: 10.1016/S0952-3278(96)90128-3. [DOI] [PubMed] [Google Scholar]
- 38.Seuter F., Perzborn E., Fiedler V.B. Effect of Bay U 3405, a new thromboxane antagonist, on collagen–induced thromboembolism in rabbits. Stroke. 1990;21:IV146–IV148. [PubMed] [Google Scholar]
- 39.Seuter F., Perzborn E., Fiedler V.B., Rosentreter U., Böshagen H. Effect of BAY U 3405, a new thromboxane antagonist, on sudden death in rabbits. J. Lipid Mediat. 1991;3:283–288. [PubMed] [Google Scholar]
- 40.Ma X.L., Karasawa A., Lefer A.M. Mechanism of the protective action of Bay U 3405, a new specific thromboxane receptor antagonist, in arachidonate–induced sudden death. Methods Find. Exp. Clin. Pharmacol. 1991;13:105–110. [PubMed] [Google Scholar]
- 41.Fiedler V.B., Seuter F., Perzborn E. Effects of the novel thromboxane antagonist Bay U 3405 on experimental coronary artery disease. Stroke. 1990;21:IV149–IV151. [PubMed] [Google Scholar]
- 42.Squadrito F., Ioculano M., Altavilla D., Zingarelli B., Canale P., Campo G.M., Saitta A., Oriti S., Faggiotto A., Caputi A.P. Reduction of myocardial leukocyte accumulation and myocardial infarct size following administration of BAY U 3405, a thromboxane A2 receptor antagonist, in myocardial ischaemia–reperfusion injury. Inflamm. Res. 1993;39:143–149. doi: 10.1007/bf01998967. [DOI] [PubMed] [Google Scholar]
- 43.Fiedler V.B., Perzborn E., Seuter F. Protective effect of a novel thromboxane antagonist, BAY-U3405, on canine myocardial damage after coronary artery occlusion and reperfusion. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1991;11:77–84. [PubMed] [Google Scholar]
- 44.Canale P., Squadrito F., Altavilla D., Ioculano M., Campo G.M., Squadrito G., Urna G., Sardella A., Caputi A.P. Beneficial effects of BAY U 3405, a novel thromboxane A2 receptor antagonist, in splanchnic artery occlusion shock. Pharmacology. 1994;49:376–385. doi: 10.1159/000139256. [DOI] [PubMed] [Google Scholar]
- 45.Kotzé H.F., Lamprecht S., Badenhorst P.N., van Wyk V., Roodt J.P., Alexander K. In vivo inhibition of acute platelet-dependent thrombosis in a baboon model by Bay U 3405, a thromboxane A2-receptor antagonist. Thromb. Haemost. 1993;70:672–675. [PubMed] [Google Scholar]
- 46.Rote W.E., Mu D.X., Lucchesi B.R. Thromboxane antagonism in experimental canine carotid artery thrombosis. Stroke. 1993;24:820–827. doi: 10.1161/01.STR.24.6.820. [DOI] [PubMed] [Google Scholar]
- 47.Cissé-Thiam M., Drouet L. Comparative study of the antithrombotic effect of aspirin and Bay U3405, antagonist of a thromboxane A2 receptor. Dakar Med. 1999;44:25–27. [PubMed] [Google Scholar]
- 48.Seuter F., Perzborn E., Fiedler V.B. Effect of BAY U 3405, a new thromboxane antagonist, on arachidonic acid induced thromboembolism. Adv. Prostaglandin Thromboxane Leukot. Res. 1991;21:355–358. [PubMed] [Google Scholar]
- 49.Rounding H., Fiedler V.B. Improved coronary thrombolysis by tissue-type plasminogen activator in the presence of BAY U 3405. Eur. J. Pharmacol. 1991;198:207–210. doi: 10.1016/0014-2999(91)90623-X. [DOI] [PubMed] [Google Scholar]
- 50.Chakraborty R., Bhullar R.P., Dakshinamurti S., Hwa J., Chelikani P. Inverse agonism of SQ 29,548 and ramatroban on thromboxane A2 receptor. PLoS ONE. 2014;9:e85937. doi: 10.1371/journal.pone.0085937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan H., Chen S., Yuan X., Han S., Zhang H., Xia W., Xu Y., Zhao Q., Wu B. Structural basis for ligand recognition of the human thromboxane A2 receptor. Nat. Chem. Biol. 2018;15:27–33. doi: 10.1038/s41589-018-0170-9. [DOI] [PubMed] [Google Scholar]
- 52.Mckenniff M.G., Norman P., Cuthbert N.J., Gardiner P.J. BAY U3405, a potent and selective thromboxane A2 receptor antagonist on airway smooth muscle in vitro. Br. J. Pharmacol. 1991;104:585–590. doi: 10.1111/j.1476-5381.1991.tb12473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugimoto H., Shichijo M., Okano M., Bacon K.B. CRTH2-specific binding characteristics of [3H] ramatroban and its effects on PGD2, 15-deoxy-Δ12, 14-PGJ2 and indomethacin-induced agonist responses. Eur. J. Pharmacol. 2005;524:30–37. doi: 10.1016/j.ejphar.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 54.Mathiesen J.M., Ulven T., Martini L., Gerlach L.O., Heinemann A., Kostenis E. Identification of indole derivatives exclusively interfering with a G protein-independent signaling pathway of the prostaglandin D2 receptor CRTH2. Mol. Pharmacol. 2005;68:393–402. doi: 10.1124/mol.104.010520. [DOI] [PubMed] [Google Scholar]
- 55.Mathiesen J.M., Christopoulos A., Ulven T., Royer J.F., Campillo M., Heinemann A., Pardo L., Kostenis E. On the mechanism of interaction of potent surmountable and insurmountable antagonists with the prostaglandin D2 receptor CRTH2. Mol. Pharmacol. 2006;69:1441–1453. doi: 10.1124/mol.105.017681. [DOI] [PubMed] [Google Scholar]
- 56.Robarge M.J., Bom D.C., Tumey L.N., Varga N., Gleason E., Silver D., Song J., Murphy S.M., Ekema G., Doucette C., et al. Isosteric ramatroban analogs: Selective and potent CRTH-2 antagonists. Bioorg. Med. Chem. Lett. 2005;15:1749–1753. doi: 10.1016/j.bmcl.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 57.Sugimoto H., Shichijo M., Iino T., Manabe Y., Watanabe A., Shimazaki M., Gantner F., Bacon K.B. An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY U3405), inhibits prostaglandin D2-Induced eosinophil migration in vitro. J. Pharmacol. Exp. Ther. 2003;305:347–352. doi: 10.1124/jpet.102.046748. [DOI] [PubMed] [Google Scholar]
- 58.Vinall S.L., Townsend E.R., Pettipher R. A paracrine role for chemoattractant receptor-homologous molecule expressed on T helper type 2 cells (CRTH2) in mediating chemotactic activation of CRTH2+ CD4+T helper type 2 lymphocytes. J. Immunol. 2007;121:577–584. doi: 10.1111/j.1365-2567.2007.02606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schratl P., Royer J.F., Kostenis E., Ulven T., Sturm E.M., Waldhoer M., Hoefler G., Schuligoi R., Lippe I.T., Peskar B.A., et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J. Immunol. 2007;179:4792–4799. doi: 10.4049/jimmunol.179.7.4792. [DOI] [PubMed] [Google Scholar]
- 60.Whelan C.J. Evidence that 13–14 di-hydro, 15-keto prostaglandin D2-induced airway eosinophilia in guinea-pigs is independent of interleukin-5. Inflamm. Res. 2009;58:103–108. doi: 10.1007/s00011-009-8112-3. [DOI] [PubMed] [Google Scholar]
- 61.Lou H.-Q., Ying Y.-F., Hu Y. CRTH2 antagonist ameliorates airway inflammation in rats with asthma. J. Zhejiang Univ. Med. Sci. 2010;39:64–70. doi: 10.3785/j.issn.1008-9292.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 62.Boehm E., Sturm G.J., Weiglhofer I., Sandig H., Shichijo M., McNamee A., Pease J.E., Kollroser M., Peskar B.A., Heinemann A. 11-Dehydro-thromboxane B2, a stable thromboxane metabolite, is a full agonist of chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) in human eosinophils and basophils. J. Biol. Chem. 2004;279:7663–7670. doi: 10.1074/jbc.M310270200. [DOI] [PubMed] [Google Scholar]
- 63.Shiraishi Y., Asano K., Nakajima T., Oguma T., Suzuki Y., Shiomi T., Sayama K., Niimi K., Wakaki M., Kagyo J., et al. Prostaglandin D2-induced eosinophilic airway inflammation is mediated by CRTH2 receptor. J. Pharmacol. Exp. Ther. 2005;312:954–960. doi: 10.1124/jpet.104.078212. [DOI] [PubMed] [Google Scholar]
- 64.Gyles S.L., Xue L., Townsend E.R., Wettey F., Pettipher R. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+Th2 lymphocytes in response to mast cell supernatants. Immunology. 2006;119:362–368. doi: 10.1111/j.1365-2567.2006.02440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ulven T., Kostenis E. Minor structural modifications convert the dual TP/CRTH2 antagonist ramatroban into a highly selective and potent CRTH2 antagonist. J. Med. Chem. 2005;48:897–900. doi: 10.1021/jm049036i. [DOI] [PubMed] [Google Scholar]
- 66.Uller L., Mathiesen J.M., Alenmyr L., Korsgren M., Ulven T., Högberg T., Andersson G., A Persson C.G., Kostenis E. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir. Res. 2007;8:16. doi: 10.1186/1465-9921-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawaguchi C., Shintani N., Hayata-Takano A., Hatanaka M., Kuromi A., Nakamura R., Yamano Y., Shintani Y., Nagai K., Tsuchiya S., et al. Lipocalin-type prostaglandin D synthase regulates light-induced phase advance of the central circadian rhythm in mice. Commun. Biol. 2020;3:1–13. doi: 10.1038/s42003-020-01281-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ito H., Yan X., Nagata N., Aritake K., Katsumata Y., Matsuhashi T., Nakamura M., Hirai H., Urade Y., Asano K., et al. PGD2-CRTH2 Pathway Promotes Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2012;23:1797–1809. doi: 10.1681/ASN.2012020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gallant M., Beaulieu C., Berthelette C., Colucci J., Crackower M.A., Dalton C., Denis D., Ducharme Y., Friesen R.W., Guay D., et al. Discovery of MK-7246, a selective CRTH2 antagonist for the treatment of respiratory diseases. Bioorg. Med. Chem. Lett. 2011;21:288–293. doi: 10.1016/j.bmcl.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 70.Gervais F.G., Sawyer N., Stocco R., Hamel M., Krawczyk C., Sillaots S., Denis D., Wong E., Wang Z., Gallant M., et al. Pharmacological characterization of MK-7246, a potent and selective CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper type 2 Cells) Antagonist. Mol. Pharmacol. 2010;79:69–76. doi: 10.1124/mol.110.068585. [DOI] [PubMed] [Google Scholar]
- 71.Eriksson J., Roy T., Sawadjoon S., Bachmann K., Sköld C., Larhed M., Weis J., Selvaraju R.K., Korsgren O., Eriksson O., et al. Synthesis and preclinical evaluation of the CRTH2 antagonist [11C]MK-7246 as a novel PET tracer and potential surrogate marker for pancreatic beta-cell mass. Nucl. Med. Biol. 2019;71:1–10. doi: 10.1016/j.nucmedbio.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 72.Beaulieu C., Guay D., Wang Z., Leblanc Y., Roy P., Dufresne C., Zamboni R., Berthelette C., Day S., Tsou N., et al. Identification of prostaglandin D2 receptor antagonists based on a tetrahydropyridoindole scaffold. Bioorg. Med. Chem. Lett. 2008;18:2696–2700. doi: 10.1016/j.bmcl.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 73.Zaghdane H., Boyd M., Colucci J., Simard D., Berthelette C., Leblanc Y., Wang Z., Houle R., Lévesque J.F., Molinaro C., et al. New indole amide derivatives as potent CRTH2 receptor antagonists. Bioorg. Med. Chem. Lett. 2011;21:3471–3474. doi: 10.1016/j.bmcl.2011.03.085. [DOI] [PubMed] [Google Scholar]
- 74.Simard D., Leblanc Y., Berthelette C., Zaghdane M.H., Molinaro C., Wang Z., Gallant M., Lau S., Thao T., Hamel M., et al. Azaindoles as potent CRTH2 receptor antagonists. Bioorg. Med. Chem. Lett. 2011;21:841–845. doi: 10.1016/j.bmcl.2010.11.084. [DOI] [PubMed] [Google Scholar]
- 75.Luker T., Bonnert R., Paine S.W., Schmidt J., Sargent C., Cook A.R., Cook A., Gardiner P., Hill S., Weyman-Jones C., et al. Zwitterionic CRTh2 Antagonists. J. Med. Chem. 2011;54:1779–1788. doi: 10.1021/jm1014549. [DOI] [PubMed] [Google Scholar]
- 76.Kelder J., Grootenhuis P.D.J., Bayada D.M., Delbressine L.P.C., Ploemen J. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999;16:1514–1519. doi: 10.1023/A:1015040217741. [DOI] [PubMed] [Google Scholar]
- 77.Fretz H., Valdenaire A., Pothier J., Hilpert K., Gnerre C., Peter O., Leroy X., Riederer M.A. Identification of 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (Setipiprant/ACT-129968), a potent, selective, and orally bioavailable chemoattractant receptor-homologous molecule expressed on Th2 Cells (CRTH2) Antagonist. J. Med. Chem. 2013;56:4899–4911. doi: 10.1021/jm400122f. [DOI] [PubMed] [Google Scholar]
- 78.Sidharta P.N., Diamant Z., Dingemanse J. Single and multiple dose tolerability and pharmacokinetics of the CRTH2 antagonist setipiprant in healthy male subjects. Fundam. Clin. Pharmacol. 2014;28:690–699. doi: 10.1111/fcp.12079. [DOI] [PubMed] [Google Scholar]
- 79.Baldoni D., Mackie A., Gutierrez M., Theodor R., Dingemanse J. Setipiprant, a Selective oral antagonist of human CRTH2: Relative bioavailability of a capsule and a tablet formulation in healthy female and male subjects. Clin. Ther. 2013;35:1842–1848. doi: 10.1016/j.clinthera.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 80.Diamant Z., Sidharta P.N., Singh D., O’Connor B.J., Zuiker R., Leaker B.R., Silkey M., Dingemanse J. Setipiprant, a selective CRTH2 antagonist, reduces allergen–induced airway responses in allergic asthmatics. Clin. Exp. Allergy. 2014;44:1044–1052. doi: 10.1111/cea.12357. [DOI] [PubMed] [Google Scholar]
- 81.Ratner P., Andrews C.P., Hampel F.C., Martin B., Mohar D.E., Bourrelly D., Danaietash P., Mangialaio S., Dingemanse J., Hmissi A., et al. Efficacy and safety of setipiprant in seasonal allergic rhinitis: Results from phase 2 and phase 3 randomized, double-blind, placebo- and active-referenced studies. Allergy Asthma Clin. Immunol. 2017;13:1–15. doi: 10.1186/s13223-017-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Actelion Provides Update on CRTH2 Program. [(accessed on 17 April 2020)]; Available online: https://pipelinereview.com/index.php/2012040247547/Small-Molecules/Actelion-provides-update-on-CRTH2-program.html.
- 83.Risch P., Pfeifer T., Segrestaa J., Fretz H., Pothier J. Verification of the major metabolic oxidation path for the Naphthoyl group in chemoattractant receptor-homologous molecule expressed on Th2 Cells (CRTh2) antagonist 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (Setipiprant/ACT-129968) J. Med. Chem. 2015;58:8011–8035. doi: 10.1021/acs.jmedchem.5b00824. [DOI] [PubMed] [Google Scholar]
- 84.Hoch M., Wank J., Kluge I., Wagner-Redeker W., Dingemanse J. Disposition and metabolism of setipiprant, a selective oral CRTH2 antagonist, in humans. Drugs Res. Dev. 2013;13:253–269. doi: 10.1007/s40268-013-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pees A., Windhorst A.D., Vosjan M.J.W.D., Tadino V., Vugts D.J. Synthesis of [18 F]Fluoroform with high molar activity. Eur. J. Org. Chem. 2020;2020:1177–1185. doi: 10.1002/ejoc.202000056. [DOI] [Google Scholar]
- 86.Guo D., Liu L.Y. The in vivo profile of CT133, a potent, well tolerated, and selective CRTH2 antagonist for the treatment of allergic asthma and rhinitis. J. Allergy Clin. Immunol. 2015;135:Ab3. doi: 10.1016/j.jaci.2014.12.946. [DOI] [Google Scholar]
- 87.Hussain M., Xu C., Wu X., Lu M., Tang L., Wu F., Wu X., Wu J. A CRTH2 antagonist, CT-133, suppresses NF-kappaB signalling to relieve lipopolysaccharide-induced acute lung injury. Eur. J. Pharmacol. 2019;854:79–91. doi: 10.1016/j.ejphar.2019.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hussain M., Xu C., Yao M., Zhang Q., Wu J., Wu X., Lu M., Tang L., Wu F., Wu X. CRTH2 antagonist, CT-133, effectively alleviates cigarette smoke-induced acute lung injury. Life Sci. 2019;216:156–167. doi: 10.1016/j.lfs.2018.11.039. [DOI] [PubMed] [Google Scholar]
- 89.Issahaku A.R., Agoni C., Kumi R.O., Olotu F.A., Soliman M.E.S., A Fisayo O. Lipid-Embedded molecular dynamics simulation model for exploring the reverse prostaglandin D2 agonism of CT-133 towards CRTH2 in the treatment of Type-2 inflammation dependent diseases. Chem. Biodivers. 2020;17:1900548. doi: 10.1002/cbdv.201900548. [DOI] [Google Scholar]
- 90.Preshlock S.M., Tredwell M., Gouverneur V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev. 2016;116:719–766. doi: 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- 91.Van Der Born D., Pees A., Poot A.J., Orru R.V.A., Windhorst A.D., Vugts D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017;46:4709–4773. doi: 10.1039/C6CS00492J. [DOI] [PubMed] [Google Scholar]
- 92.Brooks A.F., Topczewski J.J., Ichiishi N., Sanford M.S., Scott P.J.H. Late-stage [18F] Fluorination: New solutions to old problems. Chem. Sci. 2014;5:4545–4553. doi: 10.1039/C4SC02099E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen W., Huang Z., Tay N.E.S., Giglio B., Wang M., Wang H., Wu Z., Nicewicz D.A., Li Z. Direct arene C-H fluorination with (18)F(-) via organic photoredox catalysis. Science. 2019;364:1170–1174. doi: 10.1126/science.aav7019. [DOI] [PMC free article] [PubMed] [Google Scholar]