

Abstract
Mitochondrial diseases linked to mutations in mitochondrial (mt) tRNA sequences are common. However, the contributions of these tRNA mutations to the development of diseases is mostly unknown. Mutations may affect interactions with (mt)tRNA maturation enzymes or protein synthesis machinery leading to mitochondrial dysfunction. In human mitochondria, in most cases the first step of tRNA processing is the removal of the 5′ leader of precursor tRNAs (pre-tRNA) catalyzed by the three-component enzyme, mtRNase P. Additionally, one component of mtRNase P, mitochondrial RNase P protein 1 (MRPP1), catalyzes methylation of the R9 base in pre-tRNAs. Despite the central role of 5′ end processing in mitochondrial tRNA maturation, the link between mtRNase P and diseases is mostly unexplored. Here, we investigate how 11 different human disease-linked mutations in (mt)pre-tRNAIle, (mt)pre-tRNALeu(UUR), and (mt)pre-tRNAMet affect the activities of mtRNase P. We find that several mutations weaken the pre-tRNA binding affinity (KDs are approximately two- to sixfold higher than that of wild-type), while the majority of mutations decrease 5′ end processing and methylation activity catalyzed by mtRNase P (up to ∼55% and 90% reduction, respectively). Furthermore, all of the investigated mutations in (mt)pre-tRNALeu(UUR) alter the tRNA fold which contributes to the partial loss of function of mtRNase P. Overall, these results reveal an etiological link between early steps of (mt)tRNA-substrate processing and mitochondrial disease.
Keywords: RNase P, PRORP, pre-tRNA, tRNA processing, mitochondrial diseases, tRNA mutations
INTRODUCTION
Mitochondria serve as the powerhouse for the eukaryotic cell by synthesizing ATP. Due to its endosymbiotic origin, human mitochondria possess a circular genome (mtDNA) encoding two rRNAs, 22 tRNAs and 13 mRNAs that are essential for mitochondrial function. The mRNAs encode proteins involved in oxidative phosphorylation. Similar to the nuclear genome, mutation of mtDNA often leads to the development of diseases (Suzuki et al. 2011; Schapira 2012; Abbott et al. 2014; Finsterer and Kothari 2014; Chinnery 2015; Lightowlers et al. 2015). One in every 4000 children in the United States is born with a mitochondrial-related disease, and it is estimated that carriers of mitochondrial mutations are as prevalent as 1:200 in the healthy adult population (Elliott et al. 2008). Despite this frequency, the pathogenesis of these diseases is not clearly understood and treatment is limited (Schapira 2012; Chinnery 2015; Lightowlers et al. 2015). Therefore, further research is needed to understand the underlying cause of these diseases and to develop new therapeutic strategies. Importantly, the majority (>50%) of mitochondrial disease-associated mtDNA mutations are located in mitochondrial transfer RNA (tRNA) genes that occupy ∼10% of the mtDNA (Wittenhagen and Kelley 2003; Levinger et al. 2004a; Abbott et al. 2014). tRNAs are essential elements required for protein synthesis and they require several maturation steps before they become fully functional [9]. A link between mitochondrial diseases and impaired tRNA maturation in the mitochondria is beginning to emerge (Levinger et al. 2004a; Wang et al. 2011, 2013, 2018; Levinger and Serjanov 2012; Chatfield et al. 2015; Jiang et al. 2016; Saoji and Cox 2018).
The human mitochondrial genome encodes for three polycistronic units with tRNAs genes interspersed between rRNA and mRNA sequences (Ojala et al. 1981). These encoded polycistronic units contain no or only short noncoding sequences between tRNA genes and other coding regions, rendering 3′ and 5′ processing of tRNAs also important for the maturation of rRNAs and mRNAs. As a consequence, improper tRNA processing can influence maturation of other essential RNAs in the mitochondria (Wittenhagen and Kelley 2003; Levinger et al. 2004a). When RNA maturation is impaired, the amount of mature RNAs available for normal function such as protein translation is decreased, potentially leading to mitochondrial dysfunction. Mitochondrial impairment contributes to the failure of physiological processes that require high amounts of ATP and thereby to development of disease.
In human mitochondria, the first step of pre-tRNA maturation is the removal of the 5′ end of precursor tRNAs (pre-tRNAs) (Sanchez et al. 2011). This early step is catalyzed by the three-component enzyme, mitochondrial ribonuclease P (mtRNaseP) (Holzmann et al. 2008). In this complex, human MRPP3 (Mitochondrial RNase P Protein 3 or Protein Only RNase P, PRORP) harbors the catalytic center for 5′ end cleavage; however, it is only active in the presence of the subcomplex consisting of MRPP1 and MRPP2 (Mitochondrial RNase P Protein 1 and 2) (Holzmann et al. 2008). In addition to activating the 5′ end cleavage function of mtRNase P, the MRPP1/2 subcomplex independently catalyzes methyl transfer to the ninth base (m1A9/m1G9) of human mitochondrial tRNAs (Vilardo et al. 2012). In this subcomplex, MRPP2 (17β-hydroxysteroid dehydrogenase type 1, SDR5C1) is proposed to serve as a scaffold protein supporting the S-adenosyl-methionine (SAM)-dependent methyltransferase function of MRPP1 (TRM10C) (Vilardo et al. 2012).
mtRNase P is essential and plays a central role in mitochondrial function (Sanchez et al. 2011; Munch and Harper 2016; Rackham et al. 2016; Sen et al. 2016). Defects in 5′ pre-tRNA processing are implicated in several mitochondrial diseases, such as maternally inherited essential hypertension, mitochondrial myopathy, MELAS, and HSD10 disease (Bindoff et al. 1993; Rossmanith and Karwan 1998; Li and Guan 2010; Wang et al. 2011; Deutschmann et al. 2014a; Chatfield et al. 2015; Vilardo and Rossmanith 2015; Falk et al. 2016; Jiang et al. 2016). Mitochondrial diseases can be inherited in two ways, either by mutations in the nuclear genome or in the mitochondrial genome. In particular, mutations in the nuclear encoded MRPP2 are linked to HSD10 disease (Deutschmann et al. 2014b; Chatfield et al. 2015; Vilardo and Rossmanith 2015; Amberger et al. 2016). Strikingly, most clinically observed mutations in MRPP2 do not affect the dehydrogenase activity of MRPP2 but rather alter the methyltransferase and 5′ end cleavage activity of mtRNase P (Deutschmann et al. 2014b; Chatfield et al. 2015; Falk et al. 2016). In addition, a single-nucleotide polymorphism in the MRPP3 gene influences mitochondrial tRNA methylation patterns and potentially leads to differences in metabolism among individuals (Hodgkinson et al. 2014). Furthermore, disease linked mutations in human (mt)pre-tRNAs can also impair 5′ end pre-tRNA processing, although studies of such mutations are limited.
So far, it has been shown that the A4263G (mt)tRNAIle and A5655G (mt)tRNAAla mutations linked to hypertension can affect 5′ end cleavage and methyltransferase activity of the human mtRNase P (Wang et al. 2011; Jiang et al. 2016). These mutations decrease mature mitochondrial tRNA levels in patient cell lines, suggesting that the perturbed processing of tRNA is likely an underlying cause of this disease.
Despite the potential link between mtRNase P and some other mitochondrial diseases, there are very few studies addressing the direct role of mtRNase P in disease (Wang et al. 2011; Jiang et al. 2016). Hence, here we have in vitro transcribed 11 human mitochondrial pre-tRNAs containing disease-linked mutations and investigated their effects on binding, methylation, and nuclease activity of human mtRNase P. We found that most of the investigated mitochondrial pre-tRNA mutations negatively affected the methylation and nuclease activity of the mtRNase P complex. Additionally, we have shown that a subset of these pre-tRNA mutations influenced tRNA folding and tertiary structure and this effect may account for the decrease in mtRNase P activity. Taken together, our study reveals that disease-linked mutations in (mt)tRNAs result in reduced mtRNase P activity in vitro and suggests that the contributions of these mutations to disease development may be related to modulated and disrupted mtRNase P function in vivo.
RESULTS
Disease-related mutations in human mitochondrial pre-tRNAs can influence substrate binding of mtRNase P
It has been long proposed that human mitochondrial tRNA mutations contribute to diseases by altering the structure of immature tRNAs and impairing recognition by processing and modification enzymes (Rossmanith and Karwan 1998; Kelley et al. 2000, 2001; Yasukawa et al. 2000; Wittenhagen and Kelley 2003; Levinger et al. 2004a; Yan et al. 2006; Wang et al. 2013, 2018; Abbott et al. 2014; Mustoe et al. 2015). However, how mutations affect binding to mtRNase P has not been evaluated thus far. To address this question we measured binding affinities (KD,app) of mtRNase P for pre-tRNAs containing a variety of disease-linked mutations through in vitro fluorescent binding assays.
We previously demonstrated that (mt)pre-tRNAIle, (mt)pre-tRNALeu(UUR), and (mt)pre-tRNAMet are substrates for mtRNase P when in vitro transcribed and unmodified (Karasik et al. 2019). Since these pre-tRNAs are also hotspots for mitochondrial disease mutations (Levinger et al. 2004b; Li et al. 2009; Li and Guan 2010; Wang et al. 2011, 2013), we investigated in vitro binding affinities of their mutant versions to mtRNase P. A representative set of mitochondrial disease tRNA mutations located in functional regions of (mt)tRNAs were chosen for this study (Fig. 1). The majority of the selected mutations are located in D- and T-loop and stem (tRNA elbow region; Fig. 1D), while one maps to the acceptor stem and another one is found in the first position in the 5′ end of the tRNA leader (N−1 position). These tRNA positions were already shown to be important for substrate recognition of the stand alone PRORPs (Klemm et al. 2017). Due to the conserved structure of human and plant PRORPs, we propose that mutations in these regions could also disrupt interactions of pre-tRNA and mtRNase P (Howard et al. 2013; Li et al. 2015; Reinhard et al. 2015; Karasik et al. 2016; Teramoto et al. 2020). In addition, these mutations also represent a wide spectrum of diseases with varying severity of outcomes, and therefore these studies could give an insight into the potential correlation between observed in vitro defects on mtRNase P and disease.
FIGURE 1.

Locations of the investigated mitochondrial tRNA mutations in the predicted secondary structure of (A) (mt)pre-tRNAIle, (B) (mt)pre-tRNALeu(UUR), and (C) (mt)pre-tRNAMet. Circles indicate methylation sites for the MRPP1/2 subcomplex. (D) Location of the mutations in the L-shaped tertiary structure of tRNAs. High-resolution structure of the investigated tRNAs are not available, therefore we used a conserved eukaryotic tRNAPhe with known structure (PDB ID: 1ehz) for generating the figure. Mutations are color coded according to the colors shown in A, B, and C. Purple represents mutation at the same position in (mt)pre-tRNALeu(UUR) (U3290C) and (mt)pre-tRNAIle (A4317G). We also note that the D-loop for (mt)pre-tRNALeu(UUR) and (mt)pre-tRNAMet is different in length from most canonical tRNAs and therefore the elbow region may fold differently than modeled here.
We have in vitro transcribed and 5′ fluorescently labeled wild-type and mutant (mt)pre-tRNAs and measured their binding affinity (KD,app) to mtRNase P based on previously established fluorescence polarization binding assays (Karasik et al. 2019; Liu et al. 2019). Four of the (mt)pre-tRNA mutations moderately increase the KD,app values by approximately two- to sixfold relative to the wild-type (mt)pre-tRNA sequences (mutations A3243G G3249A, A4317G, and C4320U) indicating reduced affinity to mtRNase P (Table 1; Fig. 2A; Supplemental Fig. 1). However, several of the mutations in (mt)pre-tRNAs (G3242A, G3250C, A3288G, U3290C, A4269G, A4401G) did not effect on the measured human mtRNase P binding affinity.
TABLE 1.
Disease-linked tRNA mutations affect binding and cleavage rates of mtRNase P

FIGURE 2.
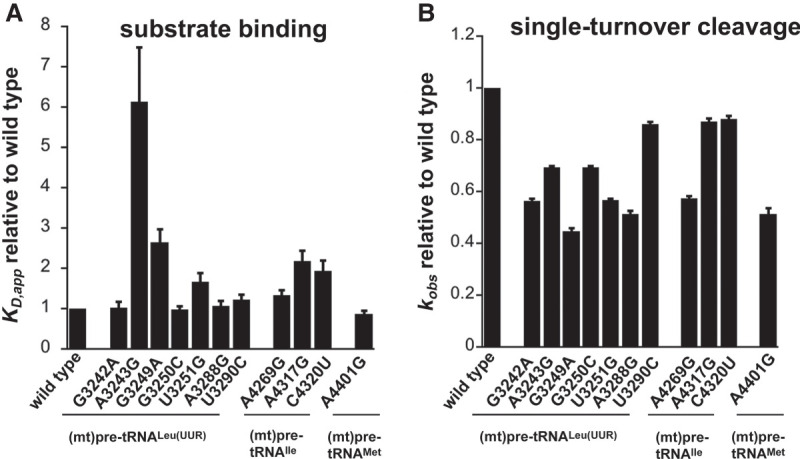
Disease-related tRNA mutations affect binding affinity and 5′ end pre-tRNA processing activity by the mtRNase P. (A) Binding affinities of mutant mitochondrial pre-tRNAs for mtRNase P using standard binding conditions. 20 nM fluorescently labeled substrates were preincubated with 150 nM MRPP1/2 for 5 min and then titrated with 0–5 µM MRPP3 as described in Karasik et al. (2019). Data were collected when binding reached equilibria from changes in fluorescence anisotropy. The KD values are calculated from the fit of the binding isotherm to the data and reported values are divided by the wild-type KD values. (B) Single-turnover cleavage rates of mitochondrial pre-tRNAs containing mutations catalyzed by mtRNase P using standard cleavage conditions. Reactions contained 20 nM fluorescently labeled substrate, 1 µM MRPP3, and 0.4 µM MRPP1/2. The kobs,cleavage values are calculated from the fit of the single exponential to the data and reported values are divided by the wild-type kobs,cleavage values.
Mitochondrial pre-tRNA mutations decrease 5′ end cleavage activity of mtRNase P
Mutations associated with hypertension in (mt)pre-tRNAIle and (mt)pre-tRNAAla affected 5′ tRNA processing by the mtRNase P (Wang et al. 2011; Jiang et al. 2016). Notably, even as little as ∼30% decrease in mtRNase P activity can manifest in mitochondrial disease. Despite the observed connection between 5′ end tRNA processing and diseases in human mitochondria, studies of how mutant forms of mitochondrial tRNAs are processed by mtRNase P are limited. Thus, we investigated the effect of selected disease-associated mutations (Fig. 1) in human mitochondrial pre-tRNAs on the 5′ end cleavage activity of mtRNase P. We used a previously optimized fluorescence polarization based single-turnover assay for mtRNase P that measures the reactivity of bound complex (mtRNase P•pre-tRNA) (Karasik et al. 2019). We found that the single-turnover activity, represented by the observed kinetic rate (kobs,cleavage), was reduced for all investigated tRNA mutants by 10% to 55% as compared to wild-type (Table 1; Fig. 2B; Supplemental Fig. 1). The greatest impact on nuclease activity catalyzed by mtRNase P was caused by (mt)pre-tRNA mutations, G3249A and A3288G, in D- and T-loops in (mt)pre-tRNALeu(UUR), respectively, and A4401G in the leader of (mt) pre-tRNAMet, resulting in ≥50% decrease in activity. Additionally, we also observed a modest decrease (30%–50%) in mtRNase P activity for mutations A3243A, G3242A, G3250C, and U3251C in (mt)pre-tRNALeu(UUR) and A4269G in (mt)pre-tRNAIle. However, some tRNA mutations, such as U3290C in (mt)pre-tRNALeu(UUR) and A3269G and A4317G in (mt)pre-tRNAIle, only slightly affected (∼10%) the 5′ pre-tRNA processing activity of mtRNase P. Taken together, a majority (8 out of 11) of investigated mutations modestly affected single-turnover 5′ tRNA cleavage efficiency of human mtRNase P.
Disease-linked (mt)pre-tRNALeu(UUR) mutations alter m1G9 methylation activity in the mitochondria
The MRPP1/2 subcomplex is predicted to methylate the m1A9/m1G9 position of most mitochondrial tRNAs; however, it is unknown if disease-linked mutations in pre-tRNAs can affect the activity of MRPP1/2. We have previously shown that both (mt)pre-tRNALeu(UUR) and (mt)pre-tRNAIle are methylated at their N9 position by mtRNase P based on single-turnover primer extension methylation assays (Karasik et al. 2019). We therefore tested whether disease-linked mutations alter the methylation efficiency of MRPP1/2 for the various mutant versions of these two (mt)pre-tRNAs. Since the presence of human MRPP3 in methylation assays does not change the single-turnover rate for (mt)pre-tRNAIle (Vilardo et al. 2012; Karasik et al. 2019), we assessed the effects of mitochondrial pre-tRNA mutations using the MRPP1/MRPP2 subcomplex without the presence of MRPP3 in the assays. A differential effect of mutations in (mt)pre-tRNAIle on MRPP1/2 methylation activity was observed. Specifically, MRPP1/2 single-turnover methylation rates (kobs,meth) of both C4320U and A4317G (mt)pre-tRNAIle mutants displayed negligible differences as compared to the wild-type (mt)pre-tRNAIle (Fig. 3A). In contrast, the presence of the A4269G mutation in (mt)pre-tRNAIle increased the MRPP1/2 single-turnover methylation rate by ∼2.5 fold (Fig. 3A). Next, we investigated single-turnover methylation rates for (mt)pre-tRNALeu(UUR) variants and mtRNase P. We previously found that the presence of MRPP3 in methylation assays enhances single-turnover methylation rates of the MRPP1/2 subcomplex for the wild-type (mt)pre-tRNALeu(UUR) and therefore we used equimolar concentrations of MRPP3 and the MRPP1/2 subcomplex in our methylation assays (Karasik et al. 2019). Most mutations in (mt)pre-tRNALeu(UUR) significantly decreased single-turnover kinetics to such an extent that determining single-turnover methylation rates was challenging. Therefore, we measured the fraction of single-turnover methylation at 1 h compared to methylation of wild-type (mt)pre-tRNALeu(UUR) (Fig. 3B,C). All disease-linked (mt)pre-tRNALeu(UUR) mutants reduced methyltransferase activity significantly for mtRNase P (∼40%–95%). However, we note that this may still be an underestimation of the effect of (mt)pre-tRNALeu(UUR) mutations on methyltranferase activity of mtRNase P since we compare their m1A9 methylation levels to fully methylated wild-type (mt)pre- tRNALeu(UUR). Taken together, all of the investigated disease-linked tRNA mutations in (mt)pre-tRNALeu(UUR) decrease single-turnover methyltransferase and/or cleavage activity of human mitochondrial mtRNase P.
FIGURE 3.

Disease-linked mutations in human (mt)pre-tRNALeu(UUR) decrease methylation efficiency catalyzed by the human mtRNase P complex. (A) Single-turnover methylation rates of mutant (mt)pre-tRNAIle variants relative to that of the wild-type (mt)pre-tRNAIle catalyzed by the MRPP1/2 subcomplex using standard reaction conditions. Reaction contained 800 nM unlabeled pre-tRNA and 5 µM MRPP1/2. (B) Quantification of methylation efficiency of mutant (mt)pre-tRNALeu(UUR) variants relative to wild-type after 60 min incubation with mtRNase P. Reaction contained 800 nM unlabeled pre-tRNA and 5 µM MRPP1/2 and MRPP3. (C) Representative gel showing methylation efficiencies of wild-type and mutant (mt)pre-tRNALeu(UUR) variants by human mtRNase P at 30 and 60 min using standard reaction conditions. Black arrows indicate two products of primer extension reactions; the read through (unmethylated pre-tRNA) and the “run-off” at the methylation sites (methylated pre-tRNA).
Mutations in mitochondrial pre-tRNAs can alter their folding and structural ensemble
Mutations in some mitochondrial tRNAs were shown to alter their structure which in turn may lead to decreased recognition by mitochondrial tRNA-binding enzymes (such as the 3′ end-cleavage enzyme RNase Z) (Levinger et al. 2004a; Levinger and Serjanov 2012; Wang et al. 2013, 2018). Therefore, we also assessed the potential contribution of selected disease-related mutations in inducing changes to the structural ensemble and stability via UV melting experiments. In general, tRNAs exhibit several unfolding transitions upon denaturation, manifesting as hyperchromicity that can be monitored at 260 nm (Stein and Crothers 1976; Mustoe et al. 2015). The first major transition is generally assigned to tertiary structure unfolding (Mustoe et al. 2015). Given that the tRNA structure, which is essential for efficient processing and functionality in protein synthesis, is stabilized by these tertiary contacts, we focused on dissecting this first transition of precursor (mt)tRNAs (Fig. 4).
FIGURE 4.

Disease-associated tRNA mutations alter the structure of pre-tRNAs. (A) Representative UV melting profile curves for the wild-type and mutant (mt)pre-tRNAIle variants. (B,C) Representative UV melting profile curves of wild-type and mutant (mt)pre-tRNALeu(UUR)s. (D) Representative UV melting profile curves of wild-type and mutant (mt)pre-tRNAMets.
Previous UV melting analysis of the wild-type (mt)pre-tRNAIle revealed that two major transitions, with a melting temperature of 47.7°C for the first transition, were observed (Fig. 4A; Karasik et al. 2019). Disease-associated tRNA mutations in (mt)pre-tRNAIle caused minor changes in the first observed transition as reflected by a lower maximum of the first-order derivative plots compared to that of the wild-type (Fig. 4A; Supplemental Figs. 2, 3). In addition, while we observed one pronounced first transition for wild-type (mt)pre-tRNAIle in the UV melting curve, the mutants appeared to have at least two separate transitions at lower temperatures (Supplemental Table 1). Although these transitions were overlapping and not well resolved, it is consistent with previous UV melting experiments carried out on wild-type and disease-causing mutant (mt)tRNASer variants, where introducing mutations into tRNASer caused the separation of the pronounced first melting transition into two transitions with a lower maximum of the first-order derivative plots and unfolding enthalpy (Mustoe et al. 2015). These slight changes in the UV melting curves of (mt)pre-tRNAIle mutants demonstrate an altered structural ensemble and folding pathway perhaps through disruption of interactions with Mg2+ or loss of H-bonding.
UV melting curves of wild-type (mt)pre-tRNALeu(UUR) and (mt)pre-tRNAMet exhibited at least three but at most four folding transitions (Fig. 4B,C; Supplemental Figs. 2, 3; Karasik et al. 2019). However, we found that most (mt)pre-tRNALeu(UUR) mutants almost entirely lack the first transition observed for wild-type (Fig. 2B). This suggests that introducing these mutations into (mt)pre-tRNALeu(UUR) can significantly affect folding and disrupt tertiary structure. For two of the (mt)pre-tRNALeu(UUR) mutants, A3243G and G3250C, we observed apparent first transitions at higher temperatures (∼50°C). It is possible these particular mutations altered folding that allowed stronger interactions in the tertiary structure.
We have also examined wild-type and A4401G (mt)pre-tRNAMet in UV melting experiments. We found that the mutant (mt)pre-tRNAMet exhibited a similar first transition melting temperature as wild-type (Tm A401G = 33.8 ± 1.1°C, Tm wild type = 32.6 ± 1.1°C) albeit with a higher maximum of the first-order derivative plots and unfolding enthalpies (ΔHA401G = 27.2 ± 1.6, ΔHwild type = 24.0 ± 7.0 J) (Fig. 4C). Mutation of this position also altered the second transition (assigned to unfolding of the secondary structure); we observed two distinct peaks at higher temperatures when A4401G is introduced. The observed differences between wild-type and A4401G (mt)pre-tRNAMet could be the consequence of base-pairing between the N−1 and the 3′ discriminator base that influences overall folding and structure in mutant (mt)pre-tRNAMet.
We further investigated the structural differences of selected disease-causing (mt)pre-tRNA mutations by native polyacrylamide gel electrophoresis (native PAGE) (Supplemental Fig. 4). We found that (mt)pre-tRNAs carrying the mutations A3243G or A4317G are represented as higher bands as compared to their wild-type counterparts suggesting that they have a more “open” conformation. This further suggests that these mutations may disrupt folding of the tertiary structure of these tRNAs. On the other hand, G3249A mutation of (mt)pre-tRNALeu(UUR) did not exhibit major changes in migration as compared to the wild-type. Since results of UV melting assays on this mutant (mt)pre-tRNA indicated altered folding, it is plausible that this mutant tRNA may adopt an alternative tertial structure that leads to the observed inefficiencies of mtRNase P activities, but similar native PAGE characteristics to wild-type (mt)pre-tRNALeu(UUR).
DISCUSSION
While mitochondrial tRNA gene mutations leading to diseases are numerous, there are only limited studies investigating the connection between those and mtRNase P (Wang et al. 2011; Jiang et al. 2016). Thus, we speculated that many more tRNA mutations in mtDNA linked to different diseases could affect mtRNase P function in the mitochondria. Here, we show that mutations in (mt)pre-tRNALeu(UUR) and (mt)pre-tRNAMet associated with distinct mitochondrial diseases in (mt)pre-tRNA genes significantly reduce binding, methylation and/or 5′ end processing activity of mtRNase P as well as influencing pre-tRNA folding and structure that may account for some of the decreased enzymatic activities of mtRNase P in vitro.
Selected mutations in (mt)pre-tRNALeu(UUR) are linked to several diseases with a wide range of disease outcomes (Table 1; Goto et al. 1990, 1992; Sweeney et al. 1993; Seneca et al. 2001; Gattermann et al. 2004; Gerber et al. 2010; Li and Guan 2010; Wortmann et al. 2012; Wang et al. 2013). Seven of the eleven selected mitochondrial tRNA mutations are located near or in the elbow region of human mitochondrial (mt)pre-tRNALeu(UUR) (Fig. 1) that was shown to be an important region for substrate recognition in plant PRORP homologs (Gobert et al. 2013; Imai et al. 2014; Klemm et al. 2017; Pinker et al. 2017). Therefore, these mutations have the potential to directly impair specific tRNA-mtRNase P interactions or influence the structural integrity of the elbow region of the pre-tRNA affecting proper tRNA-binding position or orientation by mtRNase P. For instance, A3243G and G3249A mutations could potentially disrupt predicted stacking and H-bonding interactions, respectively, between the T- and D-loop (Suzuki et al. 2011). Additionally, replacement of A to G could disrupt the U8–A14 interaction in the case of the A3243G mutation (Sterner et al. 1995). We also note that the D-loop for (mt)tRNALeu(UUR) is longer than that of the canonical tRNAs and thus the elbow region may adopt an alternative structure. However, future structural studies are needed to map the precise interactions in this region. In our biochemical assays, we found that all investigated (mt)pre-tRNALeu(UUR) mutations impacted single-turnover rates for m1G9 methylation (∼40%–95% as compared to wild-type activity) and decreased single-turnover 5′ end cleavage rates of mtRNase P by ∼10%–55%. However, these mutations in general had a smaller effect on the binding affinity, indicating that the structural changes affected the reactivity of the bound mtRNase P•mtpre-tRNA complex. This suggests that structural changes in mt-pre-tRNA caused by mutations are retained in the enzyme-bound complex. Consistent with this, all mutant (mt)pre-tRNALeu(UUR) variants show significantly altered UV melting profiles. This observation, as compared to the wild-type, reflect an altered ensemble of these mutant pre-tRNAs. Previously, we found that the wild-type (mt)pre-tRNALeu(UUR) melting temperature (Tm = ∼28°C) is significantly lower than human mitochondrial physiological temperature suggesting that this pre-tRNA is structurally unstable in the absence of other stabilizing factors, such as other protein partners or RNA modifications (Karasik et al. 2019). Our data suggest that the investigated mutations cause disturbances in folding and/or tertiary structure of pre-(mt)tRNALeu(UUR), as indicated by changes in the first transition observed in UV melting experiments. The severely affected folding and structure of investigated (mt)pre-tRNALeu(UUR) variants could explain the observed reduced 5′ end cleavage and methylation single-turnover rates for mtRNase P. However, we found that binding of these mutant (mt)pre-tRNALeu(UUR) variants to mtRNase P was not altered significantly, except for A3243G (mt)pre-tRNALeu(UUR). This suggests that the altered structural properties of the tRNALeu(UUR) variants only minimally affect substrate recognition. We propose that although most of these mutants still bind to mtRNase P, they are not oriented properly in the active sites of MRPP1 and PRORP for optimal methyltransferase and cleavage activity.
Most of the investigated pathogenic mutations in (mt)pre-tRNALeu(UUR) cause severe diseases with a wide range of outcomes, such as mitochondrial myopathies (G3250C, U3251G, and A3288G), Kearns-Sayre syndrome (G3249A), cardiomyopathy and ineffective hematopoiesis (A3242G), MELAS (mitochondrial encephalomyopathy, lactic-acidose, and stroke-like episodes) and diabetes with deafness (A3243G) (Goto et al. 1990, 1992; Sweeney et al. 1993; Seneca et al. 2001; Gattermann et al. 2004; Gerber et al. 2010; Li and Guan 2010; Wortmann et al. 2012; Wang et al. 2013). It is plausible that observed effects on mtRNase P activity in vitro contribute to the development of these diseases in vivo. In agreement, previous reports showed a link between mitochondrial disease and decreased amounts of (mt)tRNAs in vivo or accumulation of unprocessed intermediates, indicating that 5′ end processing by mtRNase P can become rate-limiting in the pre-tRNA maturation pathway in the mitochondria (Wang et al. 2013; Jiang et al. 2016; Sen et al. 2016).
On the other hand, we found that U3290C (mt)pre-tRNALeu(UUR) mutation, which is a possible hypertension factor and contributes to a milder disease, has less impact on mtRNase P binding and processing activity. However, the same mutation also influenced mtRNase P's methylation activity and altered the UV melting profile. This suggests that mutations severely impacting m1A9 methylation and folding could still manifest as mild diseases. It is plausible that combinations of further modifications of pre-tRNAs can stabilize their structure and would result in enhanced 5′ end processing in vivo. Therefore, the effect of these mutations on mitochondrial tRNA maturation pathways in vivo needs further testing (Fig. 5).
FIGURE 5.

Summary of affected mtRNase P activities of each (mt)pre-tRNA mutation in this study.
The mitochondrial tRNAIle gene is also a hotspot for mutations; A4269G and A4317G mutations are associated with fatal infantile cardiomyopathy (FICP) (Taniike et al. 1992; Yasukawa et al. 2000; Hino et al. 2004), and C4320U (mt)pre-tRNAIle mutation is associated with early-onset severe encephalomyopathy (Table 1; Santorelli et al. 1995). Thus, we also investigated these three (mt)pre-tRNAIle variants containing disease-associated mutations located near or in the elbow region of the pre-tRNA. It has been previously shown that the A4269G mutation in mature (mt)tRNAIle, associated with the mitochondrial disease FICP, reduces (mt)tRNAIle aminoacetylation, in vivo half-life and in vitro melting temperature (Yasukawa et al. 2000). In agreement with these findings, we have observed that this mutation affected the folding and structure of (mt)pre-tRNAIle based on UV melting experiments and modestly reduced 5′ end pre-tRNA cleavage; however, it did not influence the substrate affinity of mtRNase P based on our binding assays. The A4269G mutation is expected to disrupt local secondary tRNA structure by eliminating base-pairing at the base of the acceptor stem and consequently cause changes in the structure of this region. Therefore, integrity in this area may play a role in proper orientation of the substrate for 5′ end cleavage in the RNase P•substrate complex. On the other hand, single-turnover methylation rates for this mutant pre-tRNA were enhanced (∼2.5-fold). This can be potentially attributed to better access to the methylation site by MRPP1 due to a more open structural arrangement of the acceptor stem. We also found that A4317G and C4320U mutant (mt)pre-tRNAIle variants were bound less tightly to mtRNase P and had slightly altered UV melting profiles compared to the wild-type pre-tRNAIle. However, these mutations did not influence single-turnover methylation rates, and decreased 5′ leader cleavage activity of mtRNAs P by only ∼10%. This suggests that the structural changes in these mutations are not recapitulated in the enzyme-bound complex. Furthermore, these data indicate that these mutations may affect different functions in the mitochondria than 5′ end processing or R9 methylation of pre-tRNAs. Nevertheless, we note that previous studies found that a ∼20% decrease in single-turnover activity of mtRNase P in vitro for A4263G (mt)pre-tRNAIle mutant led to significant accumulation of 5′ uncleaved mitochondrial pre-tRNAs in vivo (Wang et al. 2011). Therefore, the possible involvement of mtRNase P in diseases linked to A4317G and C4320U tRNA mutations cannot be ruled out.
Plant homologs of MRPP3 have been show to interact with the N−1 base in the 5′ end leader of pre-tRNAs (Howard et al. 2016; Klemm et al. 2017). Therefore, we hypothesized that disease-linked mutations in this position could influence 5′ end tRNA cleavage in the mitochondria that could contribute to the development of diseases. Hence we also investigated the effects of mutation in mitochondrial (mt)pre-tRNAMet associated with hypertension (A4401G, found at the N−1 position) on mtRNase P activities (Table 1; Zhu et al. 2009b). We found that this mutation affected 5′ end cleavage, but not binding of mtRNase P. We posit that this mutation may influence the interaction between the 5′ end leader and MRPP3 by disrupting the proper orientation of the 5′ end leader bound to the active site of MRPP3, subsequently causing decreased 5′ end processing rates by mtRNase P. Since the folding and structure of this mutant pre-tRNA is also affected based on our UV melting data, subsequent steps in the mitochondrial tRNA maturation pathway are expected to also be influenced. Therefore, reduced 5′ end pre-tRNA processing could be one of the factors leading to hypertension in patients carrying A4401G mutation; however, this needs further testing.
Freshly transcribed mitochondrial polycistronic units punctuated by tRNAs are expected to be subject to a hierarchical cascade of RNA processing and modifications. The 5′ end pre-tRNA processing and methylation by mtRNase P are first in line after transcription and required for downstream processes in vivo (Sanchez et al. 2011; Reinhard et al. 2017). Therefore, many pre-tRNA mutations could affect and decrease mtRNase P function first before they can also impact other downstream processes. Additional negative effects of these mutations in downstream processes and enzymes are possible and the impact on the latter may compound the initial effect on mtRNase P activity multiplying the adverse consequences of these mutations. However, as observed here and elsewhere, tRNA maturation enzymes can still act on 5′ unprocessed pre-tRNAs in vitro. Due to this in vitro activity, it could be observed that downstream events from 5′ end processing, such as 3′ tRNA processing and addition of CCA at the 3′ end of the processed tRNA, in the mitochondrial tRNA maturation pathway also have reduced activities with pre-tRNAs containing disease-linked mutations (Levinger et al. 2003, 2004a,b; Sissler et al. 2004; Yan et al. 2006; Levinger and Serjanov 2012; Wang et al. 2013). In particular, some of the investigated mutations (such as A4317G in (mt)pre-tRNAIle [Levinger et al. 2003]) have been previously shown to have an inhibitory effect on 3′ tRNA maturation by human mitochondrial ELAC2 (Levinger et al. 2003, 2004b). We also observed differential effects on enzyme activities (5′ end processing and methylation) for the same pre-tRNA mutation. Therefore, it is likely that tRNA mutations in the mtDNA affect several steps of pre-tRNA maturation and development of diseases could be the consequence of these additive sequential effects on enzyme activity. There is a wide range of manifestation of mitochondrial diseases associated with tRNA mutations that may arise from differences in how severely these mutations affect the different steps in the tRNA maturation pathway. However, in vivo studies are needed to investigate the exact role of mtRNase P in human mitochondrial diseases and the potential of the additive effects on the mitochondrial tRNA maturation pathway.
Taken together, we have found that the in vitro activities of mtRNase P are impacted when mtRNase P encounters pre-tRNAs carrying human disease-linked mutations (Fig. 5). The observed impairment in mtRNase P activities could arise from changes in pre-tRNA structure when mutations are introduced. Our work potentially implicates the mtRNase P complex as a significant factor in the development of several mitochondrial diseases. Further understanding of the exact role of mtRNase P in the context of mitochondrial diseases is needed that may enable the development of new therapeutics in the future.
MATERIALS AND METHODS
In vitro transcription and 5′ end labeling of pre-tRNAs
5′ end fluorescein labeled wild-type and mutant pre-tRNAs were prepared as previously described (Karasik et al. 2019) by in vitro transcription (Howard et al. 2012; Karasik et al. 2016). Pre-tRNA DNAs were commercially synthesized by Integrated DNA Technologies and used as a template for in vitro transcription reaction. The reaction contained 50 mM Tris-HCl pH 8.0, 4 mM MgCl2, 1 mM spermidine, 5 mM dithiothreitol (DTT), 4 mM ATP, 4 mM CTP, 4 mM UTP, 1 mM GTP, 4 mM guanosine-5′-O-monophosphorothioate (GMPS), 350 µg/mL purified T7 RNA polymerase, 0.4 nmole DNA template containing T7 promoter (in 1 mL reaction volume). After 4 h incubation at 37°C, the reaction was stopped, the pre-tRNAs were 5′ end labeled and then gel-purified as described before (Howard et al. 2012; Karasik et al. 2016). Typical labeling efficiencies were ∼1%–5%. Unlabeled pre-tRNAs were made similarly to labeled ones, except the transcription reaction contained 4 mM GTP and excluded GMPS in addition to omitting the 5′ end labeling step with fluorescein. The concentrations of total and labeled pre-tRNA were measured with absorbance using a Nanodrop (Thermo Scientific) spectrophotometer and calculated by using the following extinction coefficients: 909,100 cm−1 M−1 for (mt)pre-tRNAIle7:0, 927,300 cm−1 M−1 for (mt)pre-tRNALeu(UUR)6:0, 838,200 cm−1 M−1 for (mt)pre-tRNAMet6:0.
Protein expression and purification
Full length MRPP2 and His6-Δ39 MRPP1 lacking the predicted mitochondrial targeting sequences were cloned into pCDFDuet-1 (Novagen) vector (Liu et al. 2019). MRPP1 and 2 were coexpressed and purified together from E. coli (BL21 or Rosetta). First, expression was induced by addition of 200 µM isopropyl β-d-1-thiogalactopyranoside (IPTG) to transfected bacteria, followed by incubation overnight at 18°C. Bacterial cultures were collected and lysed as described before (Karasik et al. 2016, 2019; Liu et al. 2019). The MRPP1/MRPP2 complex was purified using a nickel affinity column (GE Healthcare) and the His-tag was removed from MRPP1 by incubation with TEV protease. MRPP1 and 2 formed a complex of ∼150 kDa that was further purified by gel-filtration (Superdex 200, GE Healthcare). His6-Δ95 MRPP3 cDNA lacking the amino-terminal disordered region was cloned into pMCSG7 vector and expressed and purified, as previously described (Karasik et al. 2019). In brief, MRPP3 expression was induced by addition of 200 µM IPTG to transfected E. coli BL21. Cells were harvested and lysed after overnight incubation similarly to the MRPP1/2 subcomplex. MRPP3 was purified by a nickel affinity column and the His-tag was removed by TEV-protease. Purity of the coexpressed MRPP1/2 subcomplex and MRPP3 was evaluated by SDS-PAGE (Sodium-dodecyl-sulfate polyacrylamide gel electrophoresis) and coomassie staining. Protein concentrations were measured by Nanodrop spectrophotometer (Thermo Scientific) using 56.4 kDa as molecular weight and ε280 = 82,500 cm−1 M−1 for MRPP3. The ratio of MRPP1/2 subunits as prepared is 1:4 based on analytical ultracentrifugation experiments (Liu et al. 2019). Therefore, the MRPP1/2 protein concentration was measured using 149 kDa as the molecular weight and extinction coefficient of 79,520 cm−1 mol−1.
5′ end cleavage single-turnover cleavage assays
Pre-tRNA substrates were refolded before each 5′ pre-tRNA cleavage reaction. First, pre-tRNAs were heated up to 95°C for 3 min, cooled down to ∼22°C (∼10–15 min), then folded by incubation in “cleavage reaction buffer” (30 mM MOPS pH 7.8, 150 mM NaCl, 1 mM MgCl2, 1 mM DTT) containing 1 mM Mg2+ for an additional 5 min at ∼22°C. All single-turnover reactions were conducted as previously reported at 28°C and saturating enzyme concentrations (Karasik et al. 2019). The assay temperature was chosen to be equal or be above the previously observed UV melting transitions (carried out under similar circumstances) to ensure proper folding of pre-tRNAs (Karasik et al. 2019). To initiate the reaction, a mixture of 500 nM MRPP1/MRPP2 complex and 100 nM to 2 µM Δ95 MRPP3 in cleavage reaction buffer was added to 20 nM fluorescently labeled pre-tRNA substrate. Changes in fluorescence polarization was measured at 28°C using the ClarioStar plate reader (BMG LabTech). Data from at least three independent experiments were analyzed using Kaleidagraph 4.1.3. Equation 1 was fit to the data to calculate the observed rate constant (kobs, cleavage) and the curve-fitting error.
| (1) |
Fluorescence polarization binding assays
Pre-tRNAs used in these experiments were folded as described above, except buffers used for folding contained 1 mM Ca2+ instead of Mg2+. All binding assays were performed in 30 mM MOPS pH 7.8, 150 mM NaCl, 6 mM CaCl2, 1 mM DTT (“binding buffer”) at 28°C (Karasik et al. 2019). Prior to each experiment 20 nM folded and fluorescently labeled pre-tRNAs were preincubated with recombinant 150 nM MRPP1/2 to reach equilibrium. Then we added increasing concentrations of MRPP3 (0–5 µM) and measured the fluorescence anisotropy values after an additional 5 min incubation. Data were corrected with the anisotropy measured in the absence of the protein but presence of 20 nM Fl-pre-tRNA. The binding isotherm (Eq. 2) was fit to the fluorescence polarization data from at least three independent experiments. Kaleidagraph 4.1.3 software was used to analyze data to calculate the dissociation constant (KD) and the standard error. In Equation 2, A is the observed anisotropy, Ao is the initial anisotropy, ΔA is the total change in anisotropy, P is the concentration of MRPP3, and KD,app is the apparent dissociation constant.
| (2) |
Primer extension methylation assays
Unlabeled pre-tRNAs were prepared by in vitro transcription and gel-based purification. Before each experiment pre-tRNAs were folded in 1 mM Ca2+ containing “binding buffer.” Primer extension methylation assays were carried out as described before (Karasik et al. 2019). In short, 800 nM folded pre-tRNAs were incubated together with near-saturating (5 µM) MRPP1/2 or the reconstituted mtRNase P complex in “binding buffer” and 25 µM S-adenosyl methionine as described before (Karasik et al. 2019). The reaction was terminated at different time points by heating the sample for 30 sec at 95°C then placing it on ice. Terminated methyltransfer reactions were subjected to primer extension followed by separation of products by urea-gel electrophoresis (Karasik et al. 2019). Sequences for primers that bind to the anticodon loop were the following for wild-type and mutant tRNAIle, tRNALeu(UUR) and tRNAMet, respectively: 6-FAM-CTATTATTTACTCTATC; 6-FAM-CCTCTGACTGTAAAG; 6-FAM-CGGGGTATGGGCCCG. Data from at least three independent experiments were analyzed using Kaleidagraph 4.1.3. Eq 1 was fit to the data to calculate the single-turnover rate constants (kobs, meth) and the curve-fitting error.
UV melting experiments
Pre-tRNAs were folded as described above before each experiment using a “UV melting buffer” of final concentration of 10 mM PIPES pH 7, 150 mM NaCl, 1 mM MgCl2 and 1 µM pre-tRNA. Melting curves were obtained by heating the samples at 1°C/min from 15°C to 95°C while absorbance at 260 nm was monitored at every 0.5 or 1°C using a Cary 300 spectrophotometer. Cooling from 95°C to 25°C (hysteresis) resulted in a curve comparable to that obtained by heating. Data were corrected with data from blank measurements and analyzed with fitUVData.py and Global Melt Fit (Draper et al. 2000; Mustoe et al. 2015). Most pre-tRNAs showed three or four transitions in agreement with previous findings (Mustoe et al. 2015). Average Tms and standard errors were calculated from at least three independent experiments and bootstrapping analysis was performed as described before using fitUVData.py and Global Melt Fit (Mustoe et al. 2015) to ensure the robustness of the data where it was applicable.
Native acrylamide gel electrophoresis
Pre-tRNAs were diluted in “cleavage reaction buffer” to contain 1 pmol of fluorescently labeled pre-tRNA in 10 μL of solution. The pre-tRNAs were refolded by heating at 95°C for 3 min, cooling on ice for 10–15 min, and incubating at ∼22°C for 30 min. Samples were prepared for gel electrophoresis by the addition of 2 μL of 50% glycerol and xylene cyanol loading dye. A 10% native gel was prepared using 29:1 acrylamide/bis-acrylamide and TBE (Tris-Borate-EDTA) buffer. Gel electrophoresis was performed at 15 W and 4°C for 4 h, followed by imaging with an Amersham Typhoon Biomolecular Imager (GE Healthcare).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Aranganathan Shamuganathan for supplying some of the MRPP1/2 enzyme preparation. We also thank Dr. Adrian Ferre-D'Amare, Dr. Robert Trachman, and Dr. Xin Liu for providing help for UV melting experiments. In addition, we are grateful to Dr. Robert Trachman for reading the manuscript and providing valuable feedback. Special thanks to Meredith Purchal for her help in performing densitometry. This work was supported by the National Institutes of Health (R01 GM117141 to M.K.), the National Institutes of Health (GM55387 to C.A.F.), the Robert A. Welch Foundation (A-1987 to C.A.F.), and the American Heart Association predoctoral fellowship (16PRE29890011 to A.K.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.077198.120.
REFERENCES
- Abbott JA, Francklyn CS, Robey-Bond SM. 2014. Transfer RNA and human disease. Front Genet 5: 158. 10.3389/fgene.2014.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger A, Deutschmann AJ, Traunfellner P, Moser P, Feichtinger RG, Kofler B, Zschocke J. 2016. 17β-Hydroxysteroid dehydrogenase type 10 predicts survival of patients with colorectal cancer and affects mitochondrial DNA content. Cancer Lett 374: 149–155. 10.1016/j.canlet.2016.02.011 [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Howell N, Poulton J, McCullough DA, Morten KJ, Lightowlers RN, Turnbull DM, Weber K. 1993. Abnormal RNA processing associated with a novel tRNA mutation in mitochondrial DNA. A potential disease mechanism. J Biol Chem 268: 19559–19564. 10.1016/S0021-9258(19)36552-4 [DOI] [PubMed] [Google Scholar]
- Chatfield KC, Coughlin CR, Friederich MW, Gallagher RC, Hesselberth JR, Lovell MA, Ofman R, Swanson MA, Thomas JA, Wanders RJ, et al. 2015. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 21: 1–10. 10.1016/j.mito.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF. 2015. Mitochondrial disease in adults: what's old and what's new? EMBO Mol Med 7: 1503–1512. 10.15252/emmm.201505079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschmann AJ, Amberger A, Zavadil C, Steinbeisser H, Mayr JA, Feichtinger RG, Oerum S, Yue WW, Zschocke J. 2014a. Mutation or knock-down of 17β-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum Mol Genet 23: 3618–3628. 10.1093/hmg/ddu072 [DOI] [PubMed] [Google Scholar]
- Deutschmann AJ, Amberger A, Zavadil C, Steinbeisser H, Mayr JA, Feichtinger RG, Oerum S, Yue WW, Zschocke J. 2014b. Mutation or knock-down of 17β-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum Mol Genet 23: 3618–3628. 10.1093/hmg/ddu072 [DOI] [PubMed] [Google Scholar]
- Draper DE, Bukhman YV, Gluick TC. 2000. Thermal methods for the analysis of RNA folding pathways. Curr Protoc Nucleic Acid Chem Chapter 11: Unit 11.13. 10.1002/0471142700.nc1103s02 [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. 2008. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet 83: 254–260. 10.1016/j.ajhg.2008.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Gai X, Shigematsu M, Vilardo E, Takase R, McCormick E, Christian T, Place E, Pierce EA, Consugar M, et al. 2016. A novel HSD17B10 mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol 13: 477–485. 10.1080/15476286.2016.1159381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Kothari S. 2014. Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol 177: 754–763. 10.1016/j.ijcard.2014.11.014 [DOI] [PubMed] [Google Scholar]
- Gattermann N, Wulfert M, Junge B, Germing U, Haas R, Hofhaus G. 2004. Ineffective hematopoiesis linked with a mitochondrial tRNA mutation (G3242A) in a patient with myelodysplastic syndrome. Blood 103: 1499–1502. 10.1182/blood-2003-07-2446 [DOI] [PubMed] [Google Scholar]
- Gerber B, Manser C, Wiesli P, Meier CA. 2010. A family with diabetes and heart failure. BMJ Case Rep 2010: bcr0120102613. 10.1136/bcr.01.2010.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert A, Pinker F, Fuchsbauer O, Gutmann B, Boutin R, Roblin P, Sauter C, Giege P. 2013. Structural insights into protein-only RNase P complexed with tRNA. Nat Commun 4: 1353. 10.1038/ncomms2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. 1990. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348: 651–653. 10.1038/348651a0 [DOI] [PubMed] [Google Scholar]
- Goto Y, Tojo M, Tohyama J, Horai S, Nonaka I. 1992. A novel point mutation in the mitochondrial tRNALeu(UUR) gene in a family with mitochondrial myopathy. Ann Neurol 31: 672–675. 10.1002/ana.410310617 [DOI] [PubMed] [Google Scholar]
- Hino N, Suzuki T, Yasukawa T, Seio K, Watanabe K, Ueda T. 2004. The pathogenic A4269G mutation in human mitochondrial tRNA(Ile) alters the T-stem structure and decreases the binding affinity for elongation factor Tu. Genes Cells 9: 243–252. 10.1111/j.1356-9597.2004.00718.x [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Idaghdour Y, Gbeha E, Grenier JC, Hip-Ki E, Bruat V, Goulet JP, de Malliard T, Awadalla P. 2014. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 344: 413–415. 10.1126/science.1251110 [DOI] [PubMed] [Google Scholar]
- Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. 2008. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474. 10.1016/j.cell.2008.09.013 [DOI] [PubMed] [Google Scholar]
- Howard MJ, Lim WH, Fierke CA, Koutmos M. 2012. Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci 109: 16149–16154. 10.1073/pnas.1209062109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ, Liu X, Lim WH, Klemm BP, Fierke CA, Koutmos M, Engelke DR. 2013. RNase P enzymes: divergent scaffolds for a conserved biological reaction. RNA Biol 10: 909–914. 10.4161/rna.24513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ, Karasik A, Klemm BP, Mei C, Shanmuganathan A, Fierke CA, Koutmos M. 2016. Differential substrate recognition by isozymes of plant protein-only Ribonuclease P. RNA (New York, NY) 22: 782–792. 10.1261/rna.055541.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Nakamura T, Maeda T, Nakayama K, Gao X, Nakashima T, Kakuta Y, Kimura M. 2014. Pentatricopeptide repeat motifs in the processing enzyme PRORP1 in Arabidopsis thaliana play a crucial role in recognition of nucleotide bases at TψC loop in precursor tRNAs. Biochem Biophys Res Commun 450: 1541–1546. 10.1016/j.bbrc.2014.07.030 [DOI] [PubMed] [Google Scholar]
- Jiang P, Wang M, Xue L, Xiao Y, Yu J, Wang H, Yao J, Liu H, Peng Y, Liu H, et al. 2016. A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol Cell Biol 36: 1920–1930. 10.1128/MCB.00199-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasik A, Shanmuganathan A, Howard MJ, Fierke CA, Koutmos M. 2016. Nuclear protein-only ribonuclease P2 structure and biochemical characterization provide insight into the conserved properties of tRNA 5′ end processing enzymes. J Mol Biol 428: 26–40. 10.1016/j.jmb.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasik A, Fierke CA, Koutmos M. 2019. Interplay between substrate recognition, 5′ end tRNA processing and methylation activity of human mitochondrial RNase P. RNA 25: 1646–1660. 10.1261/rna.069310.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley SO, Steinberg SV, Schimmel P. 2000. Functional defects of pathogenic human mitochondrial tRNAs related to structural fragility. Nat Struct Biol 7: 862–865. 10.1038/79612 [DOI] [PubMed] [Google Scholar]
- Kelley SO, Steinberg SV, Schimmel P. 2001. Fragile T-stem in disease-associated human mitochondrial tRNA sensitizes structure to local and distant mutations. J Biol Chem 276: 10607–10611. 10.1074/jbc.M008320200 [DOI] [PubMed] [Google Scholar]
- Klemm BP, Karasik A, Kaitany KJ, Shanmuganathan A, Henley MJ, Thelen AZ, Dewar AJ, Jackson ND, Koutmos M, Fierke CA. 2017. Molecular recognition of pre-tRNA by Arabidopsis protein-only Ribonuclease P. RNA 23: 1860–1873. 10.1261/rna.061457.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinger L, Serjanov D. 2012. Pathogenesis-related mutations in the T-loops of human mitochondrial tRNAs affect 3′ end processing and tRNA structure. RNA Biol 9: 283–291. 10.4161/rna.19025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinger L, Giege R, Florentz C. 2003. Pathology-related substitutions in human mitochondrial tRNAIle reduce precursor 3′ end processing efficiency in vitro. Nucleic Acids Res 31: 1904–1912. 10.1093/nar/gkg282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinger L, Morl M, Florentz C. 2004a. Mitochondrial tRNA 3′ end metabolism and human disease. Nucleic Acids Res 32: 5430–5441. 10.1093/nar/gkh884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinger L, Oestreich I, Florentz C, Morl M. 2004b. A pathogenesis-associated mutation in human mitochondrial tRNALeu(UUR) leads to reduced 3′-end processing and CCA addition. J Mol Biol 337: 535–544. 10.1016/j.jmb.2004.02.008 [DOI] [PubMed] [Google Scholar]
- Li R, Guan MX. 2010. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol Cell Biol 30: 2147–2154. 10.1128/MCB.01614-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liu Y, Li Z, Yang L, Wang S, Guan MX. 2009. Failures in mitochondrial tRNAMet and tRNAGln metabolism caused by the novel 4401A>G mutation are involved in essential hypertension in a Han Chinese Family. Hypertension 54: 329–337. 10.1161/HYPERTENSIONAHA.109.129270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Liu X, Zhou W, Yang X, Shen Y. 2015. Auto-inhibitory mechanism of the human mitochondrial RNase P protein complex. Sci Rep 5: 9878. 10.1038/srep09878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightowlers RN, Taylor RW, Turnbull DM. 2015. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 349: 1494–1499. 10.1126/science.aac7516 [DOI] [PubMed] [Google Scholar]
- Liu X, Wu N, Shanmuganathan A, Klemm BP, Howard MJ, Lim WH, Koutmos M, Fierke CA. 2019. Kinetic mechanism of human mitochondrial RNase P. bioRxiv 10.1101/666792 [DOI] [Google Scholar]
- Munch C, Harper JW. 2016. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534: 710–713. 10.1038/nature18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustoe AM, Liu X, Lin PJ, Al-Hashimi HM, Fierke CA, Brooks CL III. 2015. Noncanonical secondary structure stabilizes mitochondrial tRNASer(UCN) by reducing the entropic cost of tertiary folding. J Am Chem Soc 137: 3592–3599. 10.1021/ja5130308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470–474. 10.1038/290470a0 [DOI] [PubMed] [Google Scholar]
- Pinker F, Schelcher C, Fernandez-Millan P, Gobert A, Birck C, Thureau A, Roblin P, Giege P, Sauter C. 2017. Biophysical analysis of Arabidopsis protein-only RNase P alone and in complex with tRNA provides a refined model of tRNA binding. J Biol Chem 292: 13904–13913. 10.1074/jbc.M117.782078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood AM, Richman TR, Stewart JB, et al. 2016. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16: 1874–1890. 10.1016/j.celrep.2016.07.031 [DOI] [PubMed] [Google Scholar]
- Reinhard L, Sridhara S, Hallberg BM. 2015. Structure of the nuclease subunit of human mitochondrial RNase P. Nucleic Acids Res 43: 5664–5672. 10.1093/nar/gkv481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard L, Sridhara S, Hallberg BM. 2017. The MRPP1/MRPP2 complex is a tRNA-maturation platform in human mitochondria. Nucleic Acids Res 45: 12469–12480. 10.1093/nar/gkx902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmanith W, Karwan RM. 1998. Impairment of tRNA processing by point mutations in mitochondrial tRNALeu(UUR) associated with mitochondrial diseases. FEBS Lett 433: 269–274. 10.1016/S0014-5793(98)00928-4 [DOI] [PubMed] [Google Scholar]
- Sanchez MI, Mercer TR, Davies SM, Shearwood AM, Nygard KK, Richman TR, Mattick JS, Rackham O, Filipovska A. 2011. RNA processing in human mitochondria. Cell Cycle 10: 2904–2916. 10.4161/cc.10.17.17060 [DOI] [PubMed] [Google Scholar]
- Santorelli FM, Mak SC, Vazquez-Acevedo M, Gonzalez-Astiazaran A, Ridaura-Sanz C, Gonzalez-Halphen D, DiMauro S. 1995. A novel mitochondrial DNA point mutation associated with mitochondrial encephalocardiomyopathy. Biochem Biophys Res Commun 216: 835–840. 10.1006/bbrc.1995.2697 [DOI] [PubMed] [Google Scholar]
- Saoji M, Cox RT. 2018. Mitochondrial RNase P complex in animals: mitochondrial tRNA processing and links to disease. In RNA metabolism in mitochondria. Nucleic acids and molecular biology (ed. Cruz-Reyes J, Gray M), Vol. 34, pp. 47–71. Springer, Cham, Switzerland. [Google Scholar]
- Schapira AH. 2012. Mitochondrial diseases. Lancet 379: 1825–1834. 10.1016/S0140-6736(11)61305-6 [DOI] [PubMed] [Google Scholar]
- Sen A, Karasik A, Shanmuganathan A, Mirkovic E, Koutmos M, Cox RT. 2016. Loss of the mitochondrial protein-only ribonuclease P complex causes aberrant tRNA processing and lethality in Drosophila. Nucleic Acids Res 44: 6409–6422. 10.1093/nar/gkw338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneca S, Verhelst H, De Meirleir L, Meire F, Ceuterick-De Groote C, Lissens W, Van Coster R. 2001. A new mitochondrial point mutation in the transfer RNALeu gene in a patient with a clinical phenotype resembling Kearns-Sayre syndrome. Arch Neurol 58: 1113–1118. 10.1001/archneur.58.7.1113 [DOI] [PubMed] [Google Scholar]
- Sissler M, Helm M, Frugier M, Giege R, Florentz C. 2004. Aminoacylation properties of pathology-related human mitochondrial tRNALys variants. RNA 10: 841–853. 10.1261/rna.5267604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Crothers DM. 1976. Conformational changes of transfer RNA. The role of magnesium(II). Biochemistry 15: 160–168. 10.1021/bi00646a025 [DOI] [PubMed] [Google Scholar]
- Sterner T, Jansen M, Hou YM. 1995. Structural and functional accommodation of nucleotide variations at a conserved tRNA tertiary base pair. RNA 1: 841–851. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Nagao A, Suzuki T. 2011. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet 45: 299–329. 10.1146/annurev-genet-110410-132531 [DOI] [PubMed] [Google Scholar]
- Sweeney MG, Bundey S, Brockington M, Poulton KR, Winer JB, Harding AE. 1993. Mitochondrial myopathy associated with sudden death in young adults and a novel mutation in the mitochondrial DNA leucine transfer RNAUUR gene. Q J Med 86: 709–713. [PubMed] [Google Scholar]
- Taniike M, Fukushima H, Yanagihara I, Tsukamoto H, Tanaka J, Fujimura H, Nagai T, Sano T, Yamaoka K, Inui K, et al. 1992. Mitochondrial tRNAIle mutation in fatal cardiomyopathy. Biochem Biophys Res Commun 186: 47–53. 10.1016/S0006-291X(05)80773-9 [DOI] [PubMed] [Google Scholar]
- Teramoto T, Kaitany KJ, Kakuta Y, Kimura M, Fierke CA, Hall TMT. 2020. Pentatricopeptide repeats of protein-only RNase P use a distinct mode to recognize conserved bases and structural elements of pre-tRNA. Nucleic Acids Res 48: 11815–11826. 10.1093/nar/gkaa627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Rossmanith W. 2015. Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res 43: 6649. 10.1093/nar/gkv658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W. 2012. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res 40: 11583–11593. 10.1093/nar/gks910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li R, Fettermann A, Li Z, Qian Y, Liu Y, Wang X, Zhou A, Mo JQ, Yang L, et al. 2011. Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res 108: 862–870. 10.1161/CIRCRESAHA.110.231811 [DOI] [PubMed] [Google Scholar]
- Wang M, Zhou XL, Liu RJ, Fang ZP, Zhou M, Eriani G, Wang ED. 2013. Multilevel functional and structural defects induced by two pathogenic mitochondrial tRNA mutations. Biochem J 453: 455–465. 10.1042/BJ20130294 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zeng QY, Zheng WQ, Ji QQ, Zhou XL, Wang ED. 2018. A natural non-Watson-Crick base pair in human mitochondrial tRNAThr causes structural and functional susceptibility to local mutations. Nucleic Acids Res 46: 4662–4676. 10.1093/nar/gky243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenhagen LM, Kelley SO. 2003. Impact of disease-related mitochondrial mutations on tRNA structure and function. Trends Biochem Sci 28: 605–611. 10.1016/j.tibs.2003.09.006 [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Champion MP, van den Heuvel L, Barth H, Trutnau B, Craig K, Lammens M, Schreuder MF, Taylor RW, Smeitink JA, et al. 2012. Mitochondrial DNA m.3242G>A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction? Eur J Med Genet 55: 552–556. 10.1016/j.ejmg.2012.06.002 [DOI] [PubMed] [Google Scholar]
- Yan H, Zareen N, Levinger L. 2006. Naturally occurring mutations in human mitochondrial pre-tRNASer(UCN) can affect the transfer ribonuclease Z cleavage site, processing kinetics, and substrate secondary structure. J Biol Chem 281: 3926–3935. 10.1074/jbc.M509822200 [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Hino N, Suzuki T, Watanabe K, Ueda T, Ohta S. 2000. A pathogenic point mutation reduces stability of mitochondrial mutant tRNAIle. Nucleic Acids Res 28: 3779–3784. 10.1093/nar/28.19.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HY, Wang SW, Liu L, Chen R, Wang L, Gong XL, Zhang ML. 2009a. Genetic variants in mitochondrial tRNA genes are associated with essential hypertension in a Chinese Han population. Clin Chim Acta 410: 64–69. 10.1016/j.cca.2009.09.023 [DOI] [PubMed] [Google Scholar]
- Zhu HY, Wang SW, Liu L, Li YH, Chen R, Wang L, Holliman CJ. 2009b. A mitochondrial mutation A4401G is involved in the pathogenesis of left ventricular hypertrophy in Chinese hypertensives. Eur J Hum Genet 17: 172–178. 10.1038/ejhg.2008.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.