

Abstract
The COVID-19 pandemic has affected more than 20 million people worldwide with mortality exceeding 800,000 patients. Risk factors associated with severe disease and mortality include advanced age, hypertension, diabetes, and obesity. Each of these risk factors pathologically disrupts the lipidome, including immunomodulatory eicosanoids and docosanoids lipid mediators (LM). We hypothesized that dysregulation of LMs may be a defining feature of the severity of COVID-19. By examining LMs and polyunsaturated fatty acid precursor lipids in serum from hospitalized COVID-19 patients, we demonstrate that moderate and severe disease are separated by specific differences in abundance of immune-regulatory and pro-inflammatory LMs. This difference in LM balance corresponded with decreased LM products of ALOX12 and COX2 and an increase LMs products of ALOX5 and cytochrome p450. Given the important immune regulatory role of LMs, these data provide mechanistic insight into an immuno-lipidomic imbalance in severe COVID-19.
Introduction
Lipids function in disease to rearrange cellular signaling structures, modify metabolic processes, absorb reactive species, and act directly as both autocrine and endocrine ligands in the regulation of the immune system. In the context of an immune insult, eicosanoid and docosanoid polyunsaturated fatty acids (PUFA) are liberated from glycerolipids and converted via enzymatic hydroxylation to immune regulating lipid mediators (LM) (1–7). LMs function as inflammatory, immune regulatory, or pro-resolving immune signals during chronic and acute immune responses (1, 8). Previous studies have demonstrated that co-morbidities associated with severe COVID-19 including obesity, hypertension, diabetes and heart disease feature pathological disruption of the lipidome including altered baseline levels of LMs and the PUFA-containing LM precursors in absence of infection (9–13). Several studies have examined systemic metabolic correlates of COVID-19 including shifts in the lipidome (14–17) (Troisi, J, et. al., Research Square, 2020, https://doi.org/10.21203/rs.3.rs-34085/v1). Collectively, these studies demonstrated that severe COVID-19 is marked by a systemic dysregulation of metabolism and widespread changes in the lipidome suggesting the potential for dysregulation of LMs. However, the specific behavior of LMs and their implications for disease severity remains unexplored in the context of COVID-19.
Based on these previous results, we hypothesized that severe COVID-19 is associated with a disrupted balance of LMs potentially as a result of the associated metabolic comorbidities. Herein we characterize a dysregulation of the LM lipidome, including the PUFA-containing LM precursors, in severe COVID-19 and identify new potential targets for modulating the aberrant immune response that results in morbidity.
Materials and Methods
Ethics Statement
This study was approved by Yale Human Research Protection Program Institutional Review Boards (FWA00002571, Protocol ID. 2000027690). Informed consents were obtained from all enrolled patients. Samples from healthy patients were from frozen banked donations which were obtained under the protocol (CPIRT, HIC 0901004619) before the onset of COVID-19 outbreak.
Patient cohort and serum collection
COVID-19 patients were recruited among those who were admitted to the Yale-New Haven Hospital between March 18th and May 9th, 2020 and were positive for SARS-CoV-2 by RT-PCR from nasopharyngeal and/or oropharyngeal swabs and displayed respiratory symptoms. Patients in this study were enrolled through the IMPACT biorepository study after obtaining informed consent. Samples were collected an average of 3 ± 1 and 4 ± 2 days post admission for moderate and severe groups, respectively (supplemental table 1). Control biospecimens are from healthy subjects without known inflammatory or lung disease or co-morbidities from our Yale CPIRT biorepository. Basic demographics and clinical information of study participants were obtained and shown in Supplemental table 1. Following collection, all samples were immediately frozen at −80°C. Prior to thawing, all samples including healthy controls were gamma-irradiated (2 MRad) to inactivate potential infectious virus.
Sample processing for aqueous, organic, and lipid mediator extraction
LCMS grade solvents were used for all LCMS experiments. Sample order was randomized for each extraction. For aqueous and organic metabolites samples were extracted using a modified Bligh and Dyer extractions with details in Schwarz, B, et. al., medRxiv, 2020 (https://doi.org/10.1101/2020.07.09.20149849) (18).
Lipid mediators sample processing and extraction
Lipid mediators were extracted from patient serum as previously described with modifications detailed in Schwarz, B, et. al. medRxiv, 2020 (https://doi.org/10.1101/2020.07.09.20149849) (19).
LC-MS/MS analysis
Aqueous metabolite, lipid, and lipid mediator samples were analyzed using a series of targeted multiple-reaction monitoring (MRM) methods. All samples were separated using a Sciex ExionLC™ AC system and analyzed using a Sciex 5500 QTRAP® mass spectrometer. All methods were derived from previously published methodology with modifications detailed in Schwarz, B, et. al. medRxiv, 2020 (https://doi.org/10.1101/2020.07.09.20149849) (19, 20).
Single cell RNA sequencing analysis
The published single cell RNA sequencing dataset from Wilk, et al Nature Medicine 2020 was interrogated using Seurat v3.0 (21).
Statistical Analysis
Demographic data is presented as either counts and percentages (for categorical data) or means and standard deviations (for continuous data). To investigate the difference in the control, moderate and severe groups, GraphPad Prism (version 8.4.2) was used. The results were compared using the chi-square test or Fisher’s exact test for categorical variables and one-way analysis of variance (ANOVA) or unpaired t test was used for continuous variables with significance set at p ≤ 0.05.
Univariate and multivariate analysis was performed in MarkerView® Software 1.3.1. The aqueous dataset and the combined lipid mediators/cytokine dataset data were autoscaled prior to multivariate analysis in order to visualize the contribution of low ionization efficiency species. Lipid datasets were pareto scaled to avoid overrepresenting low abundance signals within each lipid class. For all datasets a missing value cut-off of 50 % and a CV of 30 % were utilized as filters. For all univariate analysis an unpaired t-test was used and a Benjamini-Hochberg correction with a false discovery of 10 % was utilized on the combined polar metabolite and lipidomic datasets to correct for multiple comparisons. Adjusted p-value cut-off levels were moderate vs. healthy: 0.050, severe vs. healthy: 0.032, severe vs. moderate: 0.017.
Results and Discussion
To measure differences in LM profiles between moderate and severe cases of COVID-19 and gain mechanistic insights into how these changes may drive disease severity, we used serum draws from 19 healthy patients (healthy), 18 hospitalized COVID-19 patients with respiratory symptoms who did not require ICU admission (moderate), and 20 patients with respiratory symptoms of a severity that required ICU admission (severe). Serum draws from healthy patients without known co-morbidities were collected prior to the COVID-19 pandemic, to avoid any contribution of asymptomatic or convalescent cases in this data set. The demographics, preexisting conditions, and treatment details of these patients are indicated in Supplemental Table 1. To ensure recovery of both lipids and polar metabolites a modified chloroform extraction was used and measurements were made using a series of targeted LC-MS/MS methods providing high-confidence feature identification (20, 22, 23).
Changes in primarily polar metabolites, including methanol-soluble polar lipids, among COVID-19 patient cohorts from China, Italy, and France have been reported (14–17). In agreement with those studies, we observed a dysregulation of amino acids and lactate and dysregulation of nucleotide catabolic products such as xanthine, hypoxanthine, and urate (Sup. Fig. 1A) (14). Amongst lipid species, separation of disease from healthy and severe from moderate disease was marked by changes in sphingomyelins, lyso-phospholipids, cholesterol esters and multiple classes of glycerophospholipids in agreement with previous studies (Fig. 1A&B, Sup. Fig. 1B–M)(14–17).
Figure 1. Mobilization of plasmalogen-derived PUFAs correlates with the disease severity in COVID-19.
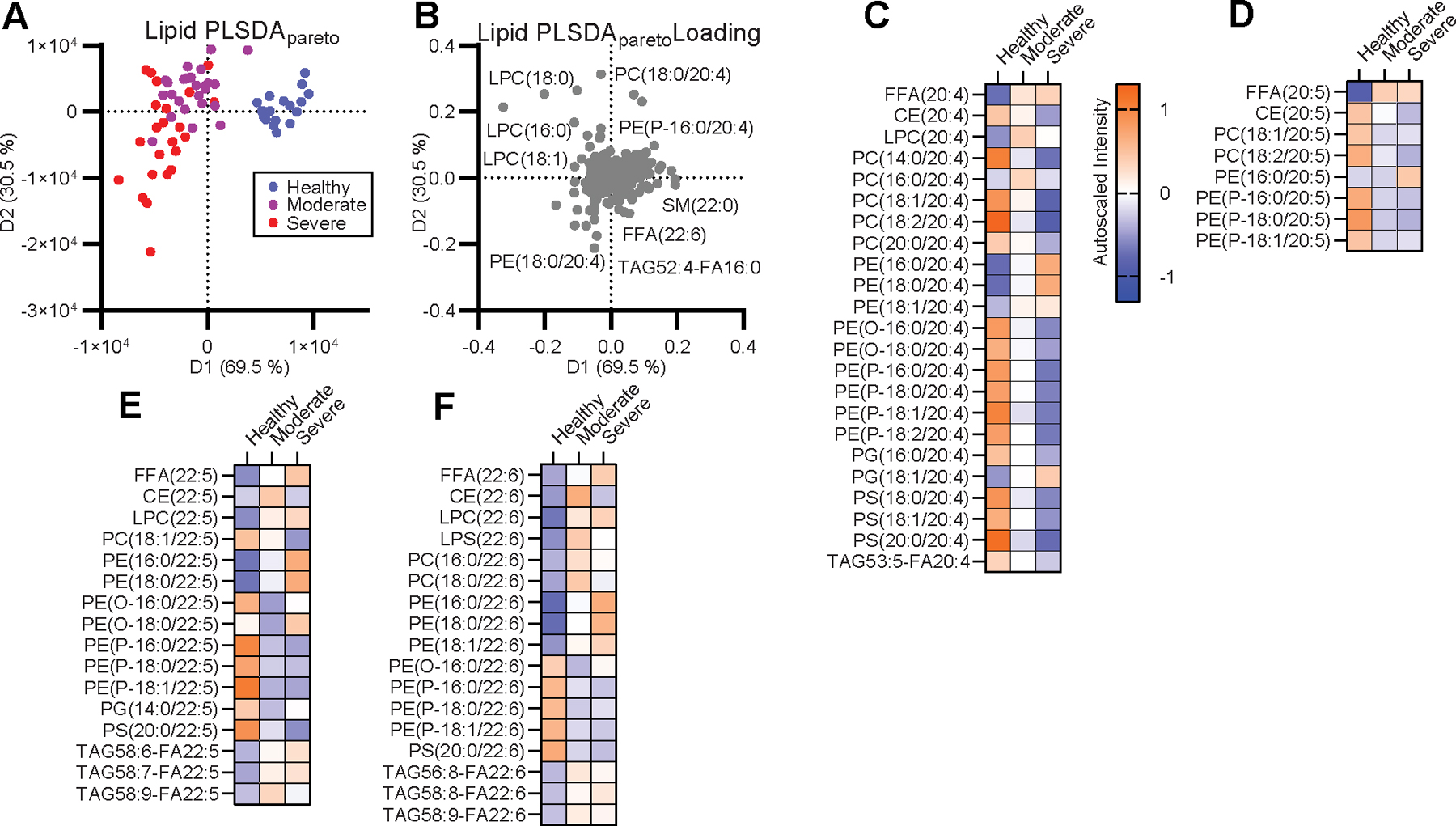
(A) Supervised PLSDA analysis of the healthy, moderate and severe disease groups and (B) the corresponding feature loading plot. (C-F) Heatmap of the autoscaled mean intensity of each patient group for significantly varied lipids containing C20:4 (C), C20:5 (D), C22:5 (E), and C22:6 (F) between one or more groups adjusted for a false discovery rate (FDR) of 10 % due to multiple comparisons. Color scale is consistent for (C-F).
To pursue our hypothesis that dysregulation of PUFA-containing lipids and immune-regulatory LMs may contribute to the inflammatory progression of severe COVID-19, we specifically interrogated patterns of LM-precursors amongst the portion of lipids that significantly varied between any two groups in the cohort. Disease groups contained lowered levels of PUFA-containing phosphatidylcholine (PC), PUFA-containing phosphatidylserine (PS), and PUFA-containing phosphatidylethanolamine-plasmalogen and increased levels of PUFA free fatty acids (FFA) as well as PUFA-containing phosphatidylethanolamine (PE), PUFA-containing lyso-phospholipids, and PUFA-containing triacylglycerols (TAG) compared to healthy controls (Fig. 1C–F). These PUFA patterns were exacerbated in severe patients compared to moderate patients. Of these PUFA-containing lipids, plasmalogen was of particular interest as a primary pool of PUFAs for the generation of LMs in both immune and structural cells (24, 25). This shift in PUFA pools strongly suggested differential mobilization of PUFAs for the generation of LMs between moderate and severe COVID-19.
To assess LMs directly, we targeted 67 LMs species using LC-MS/MS with comparison to standards and available spectral libraries (https://serhanlab.bwh.harvard.edu/). Principle component analysis (PCA) of the LM lipidome showed separation of infected and healthy cohorts and between moderate and severe patients (Fig. 2A). Nearly all LMs measured were positively correlated with infection and moderate and severe disease were characterized by unique profiles of LMs (Fig. 2B, Sup. Fig. 1N–P). Of particular interest were LMs present at lower levels in the severe group compared to the moderate group as these molecules behave counter to the pattern of increased immune activity expected in the severe disease. Specifically, moderate disease was characterized by significantly higher levels of the pro-resolving LM resolvin E3 (RvE3) and a trend toward increased presence of the prostaglandin family members, particularly PGE2 (p= 0.051), PGD2 (p= 0.220) and PGF2a (p= 0.242). In contrast, severe disease was characterized by a significant increase in free PUFAs levels, mono-hydroxylated species, and AA-derived dihydroxylated species (Supp. Fig. 1P). Multiple DHA- and AA-derived pro-resolving LMs also trend upward in severe disease (RvD3, p = 0.063) but do not reach significance.
Figure 2: A unique milieu of LMs defines moderate and severe COVID-19 disease.
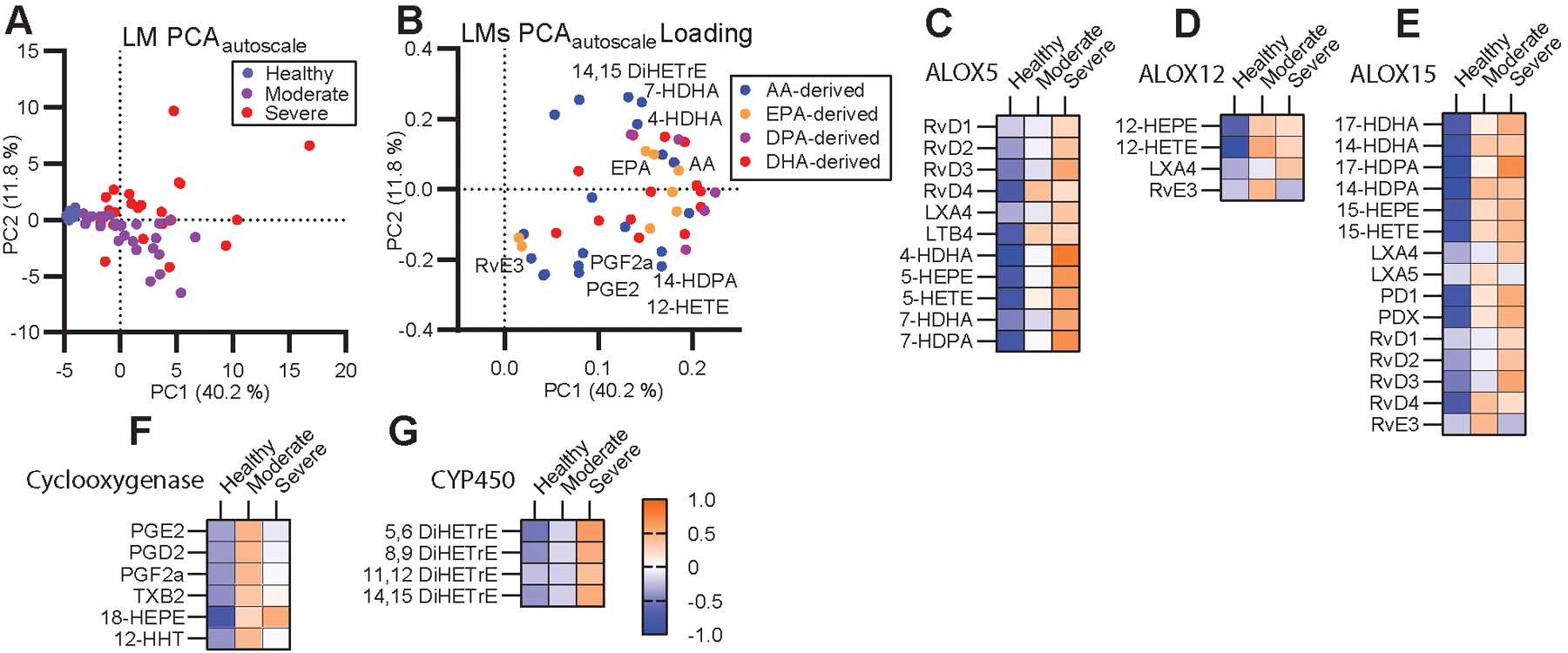
(A) Unsupervised PCA of autoscaled lipid mediators and (B) corresponding feature loading plot. (C-G) Heatmaps of the autoscaled mean for each patient group across molecules synthesized by ALOX5 (C), ALOX12 (D), ALOX15 (E), Cyclooxygenases (F) or cytochrome P450 (G). Color scale is consistent across (C-G).
LMs are generated by a single or a series of oxygenase mediated conversions of the parent PUFA. To examine the potential contribution of each oxygenase enzyme to the severe disease phenotype, we grouped LMs according to synthesis pathway (Fig. 2C–G). Several LMs are shared between multiple enzyme groups as they require sequential stereospecific hydroxylations by multiple enzymes. This grouping revealed that moderate disease was characterized by higher levels of LMs that require cyclooxygenase activity (COX) as well as certain EPA products of ALOX12 while severe disease was characterized by LMs that require activity of ALOX5 and cytochrome p450 (CYP) enzymes. This is in good agreement with previous observations from influenza, which associated symptom severity with ALOX5 activity (26).
Elevation of ALOX5- and CYP-dependent LMs in severe COVID-19 patient sera suggested upregulation of these pathways but provided no data as to the origin of these LMs. To begin to examine potential cellular origins of these enzymes in COVID-19 patients, we interrogated a published single-cell RNAseq dataset of severe COVID-19 patient PBMCs (Fig 3A) for expression of ALOX and CYP genes (21). CYP gene expression was not detected (data not shown) while ALOX5 expression was detected in multiple cell types (Fig 3B). ALOX5 was significantly increased in neutrophils and trended upward in CD14 monocytes, CD16 monocytes, and developing neutrophils (a population found almost exclusively in diseased individuals) from severe COVID-19 patients compared to healthy controls (Fig 3C). While expression is important, ALOX enzymes are primarily regulated by subcellular location and pathways involving LM production are intercellular highlighting the importance of ALOX expressing cell numbers and not just expression levels in these pathways(27, 28). Interestingly, severe COVID-19 is characterized by elevated ALOX5 expressing monocyte/macrophage population and depletion of lymphocyte populations (21, 29, 30). The absence of CYP gene expression in peripheral blood is consistent with the primarily hepatic localization of these enzymes (31). Taken together, these data support a systemic increase in ALOX5 activity in severe COVID-19 and provide a plausible foundation for the data here wherein ALOX5 products separate moderate and severe disease (32).
Figure 3. Human PBMCs from COVID-19 patients are enriched for ALOX5 expressing cells and express higher levels of ALOX5.
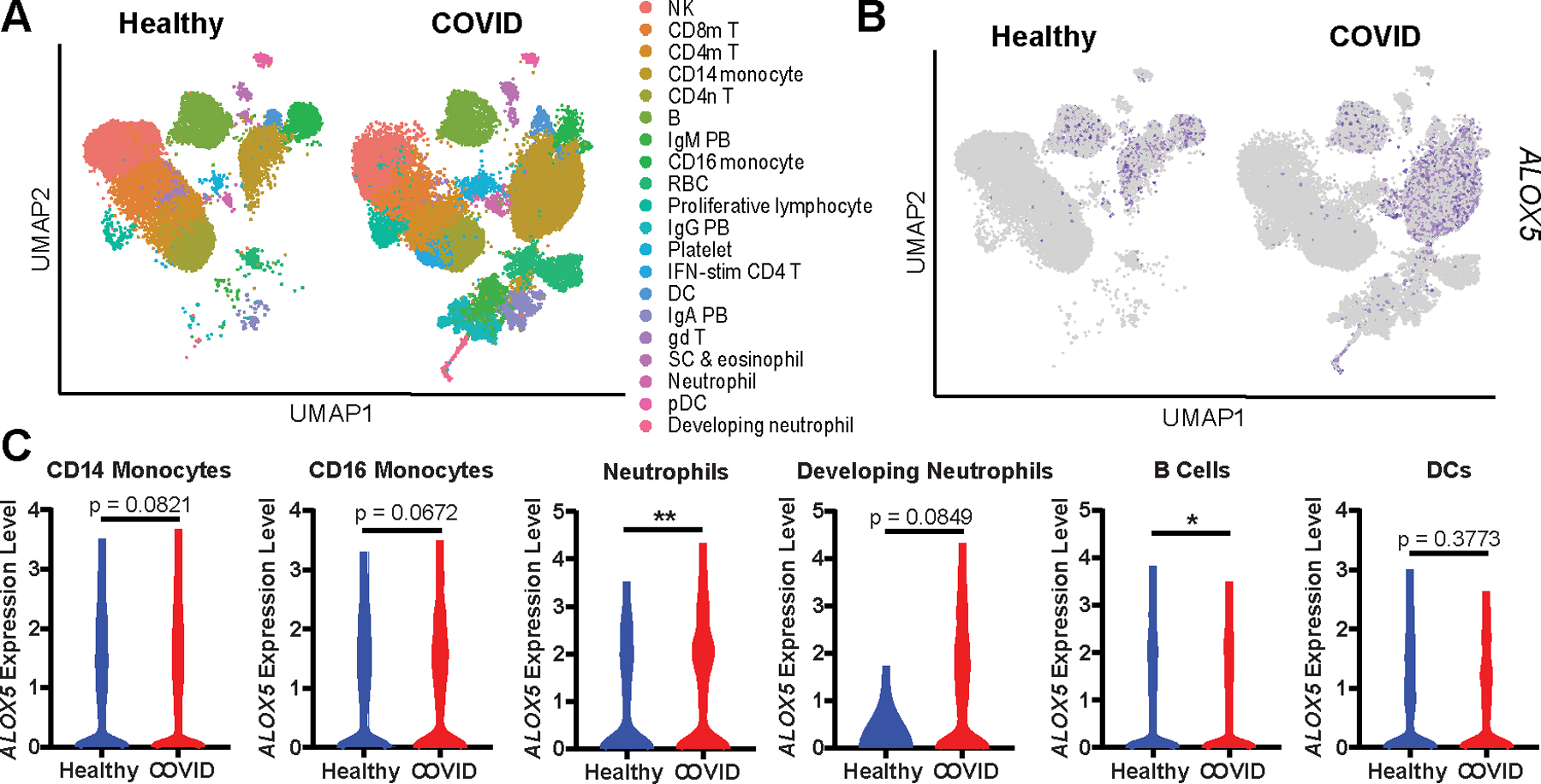
(A) UMAP dimensionality reduction plot of a published human PBMC single-cell RNA Seq dataset (21) identifying twenty cell types. (B) UMAP depicting ALOX5 expressing cells in blue. (C) Violin plots indicating ALOX5 expression levels within specific cellular populations in healthy (blue) or COVID (red) PBMCs. Statistical significance was determined by a Mann-Whitney test; * p < 0.05. ** p< 0.01.
COVID-19 comorbidities are known to pathologically disrupt the homeostatic lipidome and may predispose patients to the different signatures of LMs seen here. To examine this potential association further, we correlated the LM dataset with the severity of disease, comorbidities, and demographics (Fig. 4A & B). Thirteen LMs were significantly (p ≤ 0.05) correlated with disease severity and several families of LMs group together in the positive and negative direction including COX2 and ALOX12 products in the negative direction and ALOX15 and CYP in the positive direction despite not all members reaching significance. Among comorbidities, BMI most closely reflected the LM pattern of disease severity with all but 3 LMs sharing a similar coefficient direction and magnitude as disease severity (Fig. 4A). Hypertension and heart disease reflected the pattern of disease severity but not to the degree of BMI. Of note, prostaglandins (PGE2, PGD2 and PGF2a) and RvE3 were negatively correlated with disease severity, BMI, hypertension, and heart disease. Neither age, gender, nor presence of diabetes correlated well with the pattern of LM dysregulation observed in severe cases of COVID-19. This correlative analysis supports the association of the specific LM dysregulation in severe disease with known comorbidities, in particular high BMI.
Figure 4. Correlation of comorbidities with LMs and severe COVID-19.
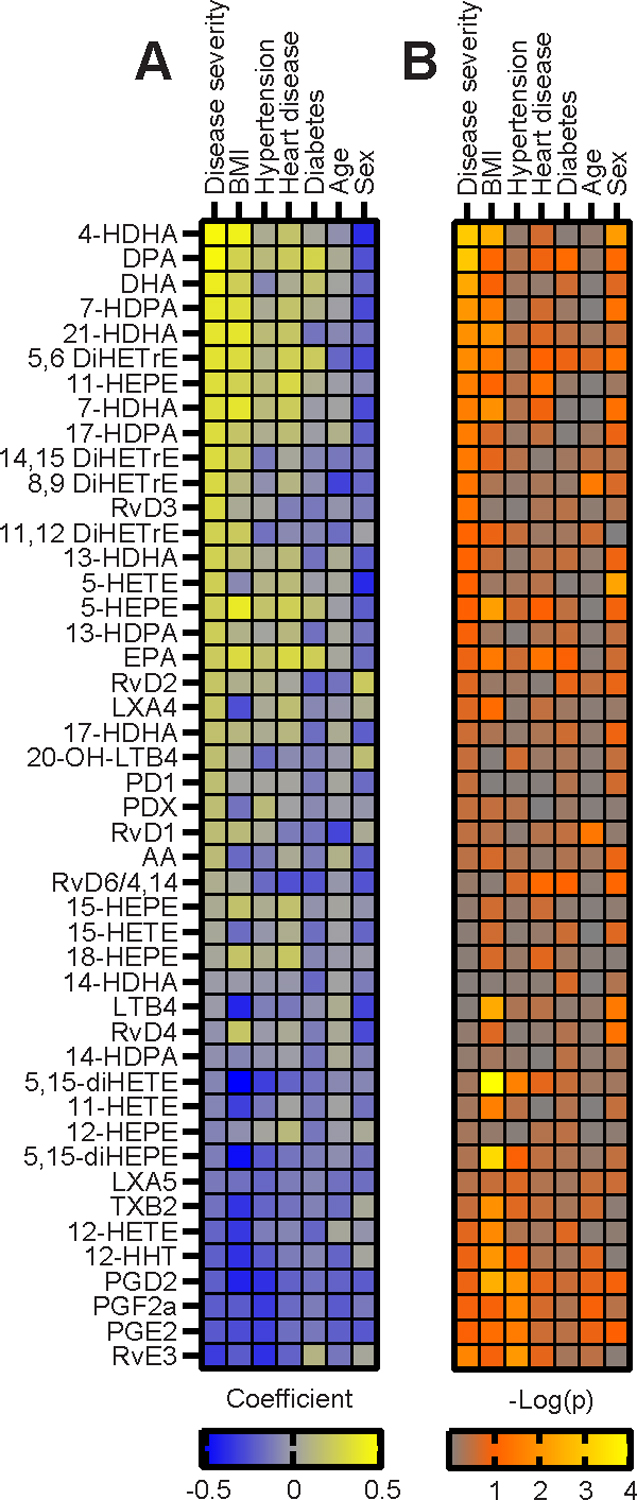
(A) Correlation plot of spearman (BMI, age) or point biserial pearson (disease severity, hypertension, heart disease, diabetes, gender) correlation coefficient of each LM with disease severity or patient demographics and comorbidities. LMs are ordered by the strength of correlation with disease severity. For gender a positive correlation is correlated with male. (B) Corresponding p-values of each LM-patient condition correlation displayed as -Log(p).
The downward trend of prostaglandins in severe disease and the strong negative correlation of this family of LMs with COVID-19 comorbidities suggested a role for these molecules in the differential immune response between moderate and severe disease. Products of COX, including the prostaglandins, have been shown to play complex roles in regulating initiation and suppression of elements of inflammation in viral infection (33). In the context of COVID-19, the potentially negative effects of both inhibitors of COX and the COX product thromboxane (TXB2) in severe disease and death have been discussed, albeit inconclusively, further highlighting the complexity and multifunctional nature of this pathway (34, 35). Herein, lower levels of prostaglandins, in the context of hospitalized COVID-19 patients, were associated with higher disease severity, but further investigation is needed to understand the mechanistic role prostaglandins play in COVID-19 progression.
It remains unclear whether these LM patterns represent drivers of disease, arise from preexisting conditions, or COVID-19 related treatment. LMs operate as part of an expansive immune signaling network and broadly deconvoluting their role from other immune ligands will require controlled experimentation. Baseline PUFA and LM levels are known to be dysregulated in several of the comorbidities examined here(11–13). Thus, while the LM levels here reflect the COVID-19 disease state, the differences between disease severity groups is likely to be affected by different starting levels of both LMs and LM precursors. Differences in LM levels could also arise from treatment differences. Unfortunately, in this cohort, therapies were either overrepresented or underrepresented to an extent that prevented correlative analysis and these therapies remain potentially confounding factors.
Together, the results presented here provide the first detailed description of the behavior of LMs in COVID-19 disease progression (26, 36). We provide evidence that a systemic lipid network consisting of liberation of PUFAs from plasmalogen and their subsequent conversion to LMs, capable of modulating inflammatory responses, characterizes both the onset and severity of COVID-19. Specifically, the loss of the immune regulatory prostaglandins and RvE3 and the increased products of ALOX5 and cytochrome P450 provides both a measure of disease severity and a potentially mechanistic understanding of the immune balance allowing for patient recovery (37). Importantly, these pathways are directly targetable with drugs previously approved for use in other inflammatory conditions and, thus, provide new therapeutic opportunities to control severe COVID-19 (2, 5).
Yale IMPACT Team
Kelly Anastasio, Michael H. Askenase, Maria Batsu, Sean Bickerton, Kristina Brower, Molly L. Bucklin, Staci Cahill, Yiyun Cao, Edward Courchaine, Giuseppe DeIuliis, Bertie Geng, Laura Glick, Akiko Iwasaki, Nathan Grubaugh, Chaney Kalinich, William Khoury-Hanold, Daniel Kim, Lynda Knaggs, Maxine Kuang, Eriko Kudo, Joseph Lim, Melissa Linehan, Alice Lu-Culligan, Anjelica Martin, Irene Matos, David McDonald, Maksym Minasyan, M. Catherine Muenker, Nida Naushad, Allison Nelson, Jessica Nouws, Abeer Obaid, Camilla Odio, Saad Omer, Isabel Ott, Annsea Park, Hong-Jai Park, Mary Petrone, Sarah Prophet, Harold Rahming, Tyler Rice, Kadi-Ann Rose, Lorenzo Sewanan, Denise Shepard, Erin Silva, Michael Simonov, Mikhail Smolgovsky, Nicole Sonnert, Yvette Strong, Codruta Todeasa, Jordan Valdez, Sofia Velazquez, Pavithra Vijayakumar, Annie Watkins, Elizabeth B. White, Yexin Yang
Supplementary Material
Key Points.
Systemic lipidomic changes distinguish COVID-19 disease severity in humans.
Lipidomic changes correspond to different levels of immune lipid mediators.
Lipid mediators differences correlate with BMI, hypertension and heart-disease.
Acknowledgements
We are deeply indebted to the patients and families of patients for their contribution to this study. Prof. Charles N. Serhan and his group including, K. Boyle, A. Shay, C. Jouvene, X. de la Rosa, S. Libreros, and N. Chiang generously provided methodology, consultation and extensive training for the assessment of lipid mediators by LC-MS/MS. AB Sciex in particular M. Pearson, P. Norris and P. Baker (currently Avanti Polar Lipids) provided LC-MS/MS consultation and methods.
Abbreviations
- LM
eicosanoid and docosanoid lipid mediators
- PE
phosphatidylethanolamine
- LPE
lyso-PE
- PC
phosphatidylcholine
- LPC
lyso-PC
- PS
phosphatidylserine
- PE(O)
plasmanyl plasmalogen
- PE(P)
plasmenyl plasmalogen
- TAG
triacylglycerol
- DAG
diacylglycerol
- MAG
monoacylglycerol
- CE
cholesterol ester
- Cer
ceramide
- DCer
dihydroceramide
- HCer
hexosylceramide
- LCer
lactosylceramide
- SM
sphingomyelin
- FAC
frees fatty acid
- Rv
resolving
- LX
lipoxin
- LT
leukotriene
- HETE
hydroxyeicosatetraenoic acid
- HEPE
hydroxyeicosapentaenoic acid
- HDHA
hydroxydocosahexaenoic acid
- HDPA
hydroxydocosapentaenoic acid
- PG
prostaglandin
- PD
D-series protectin
- TxB2
Thromboxane B2
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- PCA
principle component analysis
- PLSDA
partial least square discriminant analysis
1 Footnotes
Lokesh Sharma is supported by Parker B Francis Fellowship. Charles Dela Cruz is supported by Veterans affairs Merit Grant (BX004661) and Department of Defense grant (PR181442). Albert Ko and Charles Dela Cruz are supported by a U19 supplement for this work (AI089992-09S2). This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases. This work was supported by the Department of Internal Medicine at the Yale School of Medicine, Yale School of Public Health, and the Beatrice Kleinberg Neuwirth Fund.
Competing Interests
The authors declare no competing interests
References
- 1.Serhan CN, and Haeggstrom J 2010. Lipid mediators in acute inflammation and resolution: Eicosanoids, PAF, resolvins, and protectins. Fundamentals of Inflammation. Serhan CN, PA Ward, DW Gilroy, and SS Ayoub, editors. Cambridge University Press, Cambridge: 153–174. [Google Scholar]
- 2.Bitto A, Minutoli L, David A, Irrera N, Rinaldi M, Venuti FS, Squadrito F, and Altavilla D 2012. Flavocoxid, a dual inhibitor of COX-2 and 5-LOX of natural origin, attenuates the inflammatory response and protects mice from sepsis. Critical Care 16: doi: 10.1186/1364-8535-1116-R1132. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Dalli J, and Serhan CN 2012. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120: e60–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dennis EA, Cao J, Hsu Y-H, Magrioti V, and Kokotos G 2011. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chemical reviews 111: 6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis EA, and Norris PC 2015. Eicosanoid storm in infection and inflammation. Nature Reviews Immunology 15: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norris PC, Gosselin D, Reichart D, Glass CK, and Dennis EA 2014. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proceedings of the National Academy of Sciences 111: 12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lämmermann T, Afonso PV, Angermann BR, Wang JM, Kastenmüller W, Parent CA, and Germain RN 2013. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowal‐Bielecka O, Kowal K, Distler O, Rojewska J, Bodzenta‐Lukaszyk A, Michel BA, Gay RE, Gay S, and Sierakowski S 2005. Cyclooxygenase‐and lipoxygenase‐derived eicosanoids in bronchoalveolar lavage fluid from patients with scleroderma lung disease: An imbalance between proinflammatory and antiinflammatory lipid mediators. Arthritis & Rheumatism 52: 3783–3791. [DOI] [PubMed] [Google Scholar]
- 9.Bellis C, Kulkarni H, Mamtani M, Kent JW Jr, Wong G, Weir JM, Barlow CK, Diego V, Almeida M, and Dyer TD 2014. Human plasma lipidome is pleiotropically associated with cardiovascular risk factors and death. Circulation: Cardiovascular Genetics 7: 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holland WL, Knotts TA, Chavez JA, Wang L-P, Hoehn KL, and Summers SA 2007. Lipid mediators of insulin resistance. Nutrition reviews 65: S39–S46. [DOI] [PubMed] [Google Scholar]
- 11.Neuhofer A, Zeyda M, Mascher D, Itariu BK, Murano I, Leitner L, Hochbrugger EE, Fraisl P, Cinti S, and Serhan CN 2013. Impaired local production of proresolving lipid mediators in obesity and 17-HDHA as a potential treatment for obesity-associated inflammation. Diabetes 62: 1945–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross DJ, Hough G, Hama S, Aboulhosn J, Belperio JA, Saggar R, Van Lenten BJ, Ardehali A, Eghbali M, and Reddy S 2015. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulmonary circulation 5: 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spite M, Clària J, and Serhan CN 2014. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell metabolism 19: 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, Ge W, Liu W, Liang S, Chen H, Zhang Y, Li J, Xu J, He Z, Chen B, Wang J, Yan H, Zheng Y, Wang D, Zhu J, Kong Z, Kang Z, Liang X, Ding X, Ruan G, Xiang N, Cai X, Gao H, Li L, Li S, Xiao Q, Lu T, Zhu Y, Liu H, Chen H, and Guo T 2020. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 182: 59–72 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song JW, Lam SM, Fan X, Cao WJ, Wang SY, Tian H, Chua GH, Zhang C, Meng FP, Xu Z, Fu JL, Huang L, Xia P, Yang T, Zhang S, Li B, Jiang TJ, Wang R, Wang Z, Shi M, Zhang JY, Wang FS, and Shui G 2020. Omics-Driven Systems Interrogation of Metabolic Dysregulation in COVID-19 Pathogenesis. Cell Metab 32: 188–202 e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blasco H, Bessy C, Plantier L, Lefevre A, Piver E, Bernard L, Marlet J, Stefic K, Benz-de Bretagne I, Cannet P, Lumbu H, Morel T, Boulard P, Andres CR, Vourc’h P, Hérault O, Guillon A, and Emond P 2020. The specific metabolome profiling of patients infected by SARS-COV-2 supports the key role of tryptophan-nicotinamide pathway and cytosine metabolism. Scientific Reports 10: doi: 10.1038/s41598-41020-73966-41595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu D, Shu T, Yang X, Song J-X, Zhang M, Yao C, Liu W, Huang M, Yu Y, Yang Q, Zhu T, Xu J, Mu J, Wang Y, Wang H, Tang T, Ren Y, Wu Y, Lin S-H, Qiu Y, Zhang D-Y, Shang Y, and Zhou X 2020. Plasma metabolomic and lipidomic alterations associated with COVID-19. National Science Review 7: 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bligh EG, and Dyer WJ 1959. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology 37: 911–917. [DOI] [PubMed] [Google Scholar]
- 19.English JT, Norris PC, Hodges RR, Dartt DA, and Serhan CN 2017. Identification and profiling of specialized pro-resolving mediators in human tears by lipid mediator metabolomics. Prostaglandins, Leukotrienes and Essential Fatty Acids 117: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCloskey D, Gangoiti JA, Palsson BO, and Feist AM 2015. A pH and solvent optimized reverse-phase ion-paring-LC–MS/MS method that leverages multiple scan-types for targeted absolute quantification of intracellular metabolites. Metabolomics 11: 1338–1350. [Google Scholar]
- 21.Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, Simpson LJ, Grant P, Subramanian A, Rogers AJ, and Blish CA 2020. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 26: 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis A, Rudnitskaya A, Blackburn GJ, Fauzi NM, Pitt AR, and Spickett CM 2013. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. Journal of lipid research 54: 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyagi RK, Azrad A, Degani H, and Salomon Y 1996. Simultaneous extraction of cellular lipids and water‐soluble metabolites: evaluation by NMR spectroscopy. Magnetic resonance in medicine 35: 194–200. [DOI] [PubMed] [Google Scholar]
- 24.Braverman NE, and Moser AB 2012. Functions of plasmalogen lipids in health and disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1822: 1442–1452. [DOI] [PubMed] [Google Scholar]
- 25.Lebrero P, Astudillo AM, Rubio JM, Fernández-Caballero L, Kokotos G, Balboa MA, and Balsinde J 2019. Cellular plasmalogen content does not influence arachidonic acid levels or distribution in macrophages: A role for cytosolic phospholipase A2γ in phospholipid remodeling. Cells 8: 799–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, Thomas PG, Dennis EA, and Aderem A 2013. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell 154: 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flamand N, Lefebvre J, Surette ME, Picard S, and Borgeat P 2006. Arachidonic acid regulates the translocation of 5-lipoxygenase to the nuclear membranes in human neutrophils. Journal of Biological Chemistry 281: 129–136. [DOI] [PubMed] [Google Scholar]
- 28.Lehmann C, Homann J, Ball AK, Blöcher R, Kleinschmidt TK, Basavarajappa D, Angioni C, Ferreirós N, Häfner AK, and Rådmark O 2015. Lipoxin and resolvin biosynthesis is dependent on 5‐lipoxygenase activating protein. The FASEB Journal 29: 5029–5043. [DOI] [PubMed] [Google Scholar]
- 29.Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, and Wang F 2020. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nature medicine: 1–3. [DOI] [PubMed] [Google Scholar]
- 30.Merad M, and Martin JC 2020. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nature Reviews Immunology: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renaud HJ, Cui JY, Khan M, and Klaassen CD 2011. Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicological sciences 124: 261–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, and Gu X 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The lancet 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sander WJ, O’Neill HG, and Pohl CH 2017. Prostaglandin E2 as a modulator of viral infections. Frontiers in Physiology 8: doi: 10.3389/fphys.2017.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, Hochman J, and Berger JS 2020. Platelet and Vascular Biomarkers Associate With Thrombosis and Death in Coronavirus Disease. Circulation Research 127: doi: 10.1161/CIRCRESAHA.1120.317803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Little P 2020. Non-steroidal anti-inflammatory drugs and covid-19. BMJ 368: doi: 10.1136/bmj.m1185. [DOI] [PubMed] [Google Scholar]
- 36.Titz B, Luettich K, Leroy P, Boue S, Vuillaume G, Vihervaara T, Ekroos K, Martin F, Peitsch MC, and Hoeng J 2016. Alterations in serum polyunsaturated fatty acids and eicosanoids in patients with mild to moderate chronic obstructive pulmonary disease (COPD). International journal of molecular sciences 17: doi: 10.3390/ijms17091583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalinski P 2012. Regulation of immune responses by prostaglandin E2. The Journal of Immunology 188: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.