

Abstract
Motivation
Isoforms are mRNAs produced from the same gene locus by alternative splicing and may have different functions. Although gene functions have been studied extensively, little is known about the specific functions of isoforms. Recently, some computational approaches based on multiple instance learning have been proposed to predict isoform functions from annotated gene functions and expression data, but their performance is far from being desirable primarily due to the lack of labeled training data. To improve the performance on this problem, we propose a novel deep learning method, DeepIsoFun, that combines multiple instance learning with domain adaptation. The latter technique helps to transfer the knowledge of gene functions to the prediction of isoform functions and provides additional labeled training data. Our model is trained on a deep neural network architecture so that it can adapt to different expression distributions associated with different gene ontology terms.
Results
We evaluated the performance of DeepIsoFun on three expression datasets of human and mouse collected from SRA studies at different times. On each dataset, DeepIsoFun performed significantly better than the existing methods. In terms of area under the receiver operating characteristics curve, our method acquired at least 26% improvement and in terms of area under the precision-recall curve, it acquired at least 10% improvement over the state-of-the-art methods. In addition, we also study the divergence of the functions predicted by our method for isoforms from the same gene and the overall correlation between expression similarity and the similarity of predicted functions.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
In eukaryotes, the mechanism of alternative splicing produces multiple isoforms from the same gene. Studies in Pan et al. (2008) and Wang et al. (2008) reveal that more than 95% of human multi-exon genes undergo alternative splicing. Though the changes in the sequences of the isoforms of the same gene are very small, they may have a systematic impact on cell functions and regulation (Gallego-Paez et al., 2017). It has been widely reported that isoforms from the same gene sometimes have distinct or even opposite functions (Himeji et al., 2002; Melamud and Moult, 2009; Mittendorf et al., 2012). For example, among the two isoforms, l-KlCpo and s-KlCpo, of gene KlHEM13 that use different transcription start sites, only s-KlCpo is involved in the growth of Kluyveromyces lactis Δhem13 mutants (Vázquez et al., 2011). There is also evidence that alternative splicing plays an important role in the evolutionary process (Gueroussov et al., 2015). For example, the absence of exon 9 in one of the isoforms of gene PTBP1 expressed in the brains of mammals amplifies the evolutionary difference between mammals and the other vertebrates (Gueroussov et al., 2015). Many studies have found that alternative splicing is critical in human health and diseases. For example, to escape from cell death in tumorigenesis, gene BCL2L1 produces two isoforms with opposite functions, where BCL-XS is pro-apoptosis but BCL-XL is anti-apoptosis (Revil et al., 2007). Similarly, gene CASP3 has two isoforms, with CASP3-L being pro-apoptosis and CASP3-S anti-apoptosis (Végran et al., 2006). An isoform of gene TNR6 that skips exon 6 may initiate cell death (Bouillet and O’Reilly, 2009). Among the two isoforms of gene PKM, PKM1 and PKM2 that skip exons 9 and 10, respectively, only PKM2 is widely expressed in cancer cells (Mazurek et al., 2005). Besides these examples, the results in Himeji et al. (2002), Melamud and Moult (2009), Oberwinkler et al. (2005) and Pickrell et al. (2010) offer more interesting stories of isoforms with dissimilar functions and hence motivate the study of specific functions of isoforms.
There is rich literature concerning the prediction of gene functions (Barutcuoglu et al., 2006; Mi et al., 2012; Schietgat et al., 2010; Vinayagam et al., 2004; Yang et al., 2015). In particular, the UniProt Gene Ontology (GO) database has been widely used as a standard reference for gene function annotation (Ashburner et al., 2000; Barrell et al., 2009). It is organized as a directed acyclic graph where the nodes represent functional terms (referred to as GO terms) and edges indicate how a term is subdivided into more detailed functional concepts. The directed acyclic graph is comprised of three main branches, i.e. Biological Process (BP), Molecular Function (MF) and Cellular Component (CC) (Ashburner et al., 2000), representing three distinct classes of functional concepts. The functions of a gene are then represented by mapping the gene to all relevant terms in GO. In contrast, very little systematic study has been done about the specific functions of isoforms and there is no central database that provides annotated isoform functions. Recently, several machine learning approaches were proposed to predict isoform functions from GO and RNA-Seq expression data (Eksi et al., 2013; Li et al., 2014, 2015; Luo et al., 2017; Panwar et al., 2016). In other words, these methods attempt to distribute the annotated functions of a gene to its isoforms based on their expression profiles. Since labeled training data were generally unavailable, Eksi et al. (2013) (see also Panwar et al., 2016), Li et al. (2014) and Luo et al. (2017) solved the problem by using a semi-supervised learning technique called multiple instance learning (MIL). However, the experimental performance of their methods was quite poor. For example, on their respective datasets, the best areas under the receiver operating characteristics curve (or AUCs) achieved by the methods were only 0.681, 0.671 and 0.677, respectively. We believe that a primary cause of the poor performance was due to the lack of labeled training data.
In this paper, we propose a novel method, DeepIsoFun, for predicting isoform functions from GO and RNA-Seq expression data. It directly addresses the challenge from the lack of labeled training data by combining MIL with the domain adaptation (DA) technique. The two techniques are somewhat complementary to each other since while MIL takes advantage of the gene-isoform relationship, DA helps to transfer the existing knowledge of gene functions to the prediction of isoform functions. More precisely, we consider each gene as a bag and each isoform as an instance in the context of MIL where the labels (i.e. functions) of the instances in each bag are given as a set (Dietterich et al., 1997). The goal of MIL is to assign the labels (i.e. functions) of each bag (i.e. gene) to its instances (i.e. isoforms) with the constraint that each label is assigned to at least one instance in the bag and no instance is assigned a label that does not belong to its bag (Andrews et al., 2003; Wang et al., 2017). To apply the DA technique, we take advantage of the fact that genes actually have expression data and thus can be considered as instances in another domain (i.e. the gene domain). In other words, a gene can be regarded as both an instance in the gene domain as well as a bag in the isoform domain. Since gene functions are known in GO, the DA technique can be used to transfer knowledge (i.e. the relationship between expression and function) from the gene domain (called the source domain) into the isoform domain (called the target domain) (Ganin and Lempitsky, 2015; Long et al., 2015; Pan et al., 2011; Tzeng et al., 2014). Hence, the gene domain helps provide the much needed labeled training data.
The model of DeepIsoFun consists of three classifiers. The first attempts to correctly label the functions of each gene. The second attempts to correctly label the functions of each isoform (via bags). The third tries to make sure that instances from the source and target domains are indistinguishable so knowledge can be transferred. To implement the model, we use a neural network (NN) auto-encoder to extract features from expression data that are both domain-invariant and discriminative for functional prediction, inspired by the work in Ajakan et al. (2014) and Ganin and Lempitsky (2015). The three classifiers are also implemented as parallel NNs and connected to the auto-encoder NN to form a deep feed-forward network. The NNs involve mostly standard hidden layers and loss functions and can be trained for each GO term sequentially using a standard back-propagation algorithm based on stochastic gradient descent, but we also incorporated the gradient reversal layer to facilitate the DA method as introduced in Ganin and Lempitsky (2015) and take advantage of the hierarchical structure of GO in training. In particular, we traverse GO starting at the leaf nodes and make sure that the model is trained for all child nodes before it is trained for a parent node so the training for the parent node can benefit from earlier trainings. This also helps maintain the prediction consistency throughout GO.
To evaluate the performance of DeepIsoFun, we use three RNA-Seq expression datasets of human and mouse collected from the NCBI Reference Sequence Archive (SRA) at different times. The first is a new (also the largest) dataset that we extracted from the SRA recently. The other two were studied in Eksi et al. (2013) and Li et al. (2014). To measure the prediction accuracy, we use both AUC and area under the precision-recall curve (AUPRC) against specific baselines [measured at the gene level, as done in Eksi et al. (2013), Li et al. (2014) and Luo et al. (2017)]. Our experimental results consist of two parts. In the first part, we analyze various properties of DeepIsoFun such as the effect of DA on its performance, impact of the frequency of a GO term in genes on its performance, difference in performance across the three main branches of GO, divergence of the functions predicted for the isoforms of a gene, and correlation between the similarity of expression profiles and the similarity of predicted functions. In the second part, we compare the performance of DeepIsoFun with the methods introduced in Eksi et al. (2013), Li et al. (2014), Luo et al. (2017) and Panwar et al. (2016), Multiple Instance SVM (mi-SVM), Multiple Instance Label Propagation (iMILP) and Weighted Logistic Regression Method (WLRM), based on support vector machines (SVMs), label propagation and weighted logistic regression, respectively. On our new dataset, DeepIsoFun outperformed these mi-SVM, iMILP and WLRM methods by 31, 64 and 23% (against baseline 0.5) in AUC, respectively. In terms of AUPRC, DeepIsoFun outperformed them by 59, 11 and 63%, respectively, against baseline 0.1. Similar improvements on the other two datasets were also observed. We believe that besides the deep learning framework, the DA technique also played an important role in these significant improvements.
The rest of the paper is organized as follows. In Section 2, we describe the proposed method and its NN implementation in more detail. Section 3 shows how to determine the key parameters in the NN, the construction of experimental datasets and all computational results on these datasets. Some possible future work is briefly outlined in the Discussion section.
2 Materials and methods
In this section, we detail our proposed method, DeepIsoFun, for predicting isoform functions from GO and RNA-Seq data. As outlined above, our learning framework consists of two domains, the gene domain (denoted as yd = 0) and the isoform domain (denoted as yd = 1), where yd represents a domain class label. In the isoform domain, the isoforms of each gene form a bag in the context of MIL. The gene domain will be considered as the source domain and the isoform domain as the target domain in the context of DA. Suppose that there are n genes and m isoforms. Hence, the isoforms are divided into n bags in the isoform domain. Suppose that the expression profiles consist of r experiments.
Given a GO term, the data in the gene (or source) domain are denoted as a pair (xs, ys), where xs is an n × r feature matrix representing the expression profiles of all n genes over the r experiments and ys is an n-dimensional binary vector (called gene class labels) indicating whether each gene has the functional term or not. Similarly, the data in the isoform (or target) domain is denoted as a pair (xt, yt), where xt is an m × r feature matrix representing the expression profile of all isoforms and yt is an m-dimensional binary vector (called isoform class labels) indicating whether each isoform has the functional term or not. The data for each bag of the isoform domain are also denoted as a pair (XT, YT), where XT is a binary matrix representing the membership of isoforms in each bag (or gene) and YT is an n-dimensional binary vector (called bag class labels) indicating whether the isoforms in each bag collectively have the functional term or not. Observe that YT = ys.
As mentioned above, our method combines the MIL and DA techniques and uses three classifiers to classify isoforms with respect to each GO term. It is implemented on a deep NN architecture with four modules: an auto-encoder, a gene function predictor in the gene domain, an isoform function predictor in the isoform domain and a domain label predictor, as illustrated in Figure 1. The input gene and isoform expression features (xs and xt) are mapped by the auto-encoder to obtain an encoded feature matrix xf. We denote the training weights used in this mapping as wf.
Fig. 1.

The proposed NN architecture. It includes an auto-encoder, a gene class label predictor, an isoform class label predictor and a domain label predictor. These four modules jointly form a feed-forward NN, where the auto-encoder consists of two hidden layers and other three components consist of one hidden layer each. The NN is trained using a standard cross-validation so that the auto-encoder extracts features from the input expression profiles to minimize the loss in gene class label prediction loss, minimize the loss in bag class label prediction and maximize the loss in domain classification so that knowledge can be transferred from the gene domain to the isoform domain. In the figure, the rectangle boxes represent the input data and extracted features. Particularly, xs is the input gene expression data, xt is the input isoform expression data, the encoded gene feature data and the encoded isoform feature data. Each variable and represents a predicted gene class label vector, a predicted isoform class label vector and a predicted domain class label vector, respectively. The notation ys represents the true gene class label vector that is used to calculate Ls, i.e. gene class label loss. The notation XT represents the membership of isoforms in the bags and YT=ys is the true bag class label vector that is used to calculate Lt, i.e. bag class label loss via a multiple instance loss procedure. The notation yd is the true domain class label vector that is used for calculating Ld, i.e. domain class label loss. Forward arrows represent forward propagation and backward arrows show how losses are backpropagated to allow for the adjustment of the weights wf, ws, wt and wd used in the auto-encoder, gene class label predictor, isoform class label predictor and domain label predictor, respectively
Our goal for the auto-encoder is to generate new feature vectors that will reduce the loss of predicted gene class label, reduce the loss of predicted bag class label and at the same time, increase the loss of predicted domain class label. This will hopefully force the auto-encoder to generate domain-invariant features and hence realize the transfer of knowledge from the gene domain into the isoform domain. The new (encoded) feature vectors in the matrix xf are then partitioned into encoded gene feature vectors and encoded isoform feature vectors . Each former vector is mapped by the gene class label predictor to predict a label in the gene domain and we denote the weights in this mapping as ws. Each latter vector is mapped by the isoform class label predictor to predict a label in the target domain and we denote the weights in this mapping as wt. See Figure 1 for the detailed NN architecture.
We train the NN by following a 5-fold cross-validation procedure in the isoform domain and use the annotated GO terms of genes to evaluate its performance, similar to Eksi et al. (2013), Li et al. (2014) and Panwar et al. (2016). The data is partitioned by genes instead of isoforms to avoid potential data leak, as done in Panwar et al. (2016). Note that since isoforms from homologous genes of the human genome (i.e. paralogs) do not generally share similar expression profiles (Li et al., 2014), it is unlikely for them to cause data leak in expression-based prediction of isoforms as demonstrated in Eksi et al. (2013). All data from the gene domain is always applied to enable DA, but the single-isoform genes (SIGs) in the isoform domain are left out of training to avoid overfitting. Before the training is started, the variable for isoform i is initialized as follows:
| (1) |
The model is then trained for each GO term separately. To take advantage of the hierarchical structure of GO, we traverse GO starting from the leaf nodes and train the model on a parent node only after all its children have been considered. This allows the training for a parent node to benefit from the knowledge learned from its children, as sketched schematically in Supplementary Figure S1 as well as help make the predicted labels more consistent between parents and children.
The weights wf, wd, ws, wt are determined during training to minimize the following objective function:
| (2) |
where denotes the loss in gene class label prediction at the ith gene, the loss in bag class label prediction at the ith bag and the loss in domain class label prediction at the ith gene or isoform (see Fig. 1). More precisely, for a fixed gene (or bag or isoform/gene) i, these loss functions are:
| (3) |
We now show how the loss function Lt[i] is derived. Given the predicted class labels of the isoforms in bag i, we can estimate the class label of the bag using the method proposed in Wang et al. (2015) for dealing with multiple instance loss as shown in Equation (4). Clearly, if at least one instance of the bag is positive, the bag will be predicted as positive; otherwise, it will be considered as negative.
| (4) |
| (5) |
| (6) |
Here, i denotes a bag and j an isoform. The predicted isoform class label for isoform j is calculated by the sigmoid function in Equation (5). The encoded feature vector of isoform j, , is calculated by another sigmoid function given in Equation (6). The weights wf(t) represent the part of wf derived from the isoform data. The other part of wf, denoted as wf(s), represents the weights derived from the gene data. Similar sigmoid functions are used to derive the values of and used in Equation (3).
As mentioned above, we would like to seek the values of wf, ws, wt, wd to achieve a saddle point of Equation (2) such that
| (7) |
At the saddle point, the weights wd of the domain label predictor maximize the loss in domain classification while the weights ws and wt of the class label predictors minimize the loss in functional prediction in both domains. The feature mapping weights wf help minimize the class label prediction loss while maximizing the domain classification loss. A saddle point of Equation (7) can be found as a stationary point by using the following stochastic updates as suggested in Ganin and Lempitsky (2015):
where the parameters λ1 and λ2 control the relative contributions of the predictors during learning and α denotes the learning rate in this process.
3 Experimental evaluation
In this section, we describe in detail how to choose the key parameters in the NN model, how the test data is collected, and how the computational experiments are performed as well as what are their results.
3.1 The deep NN parameters
DeepIsoFun has been implemented in Caffe (Jia et al., 2014). In our NN architecture, the auto-encoder consists of two fully connected layers to extract common features of the gene and isoform domains. The first fully connected layer consists of 600 neurons and the second fully connected layer consists of 200 neurons. The number of hidden layers and size of each layer (i.e. number of neurons in the layer) were optimized by a standard grid search method (Bergstra et al., 2011; Bergstra and Bengio, 2012). The gene class label predictor and isoform class label predictor modules are both output layers, and hence have only a single output neuron each. The domain label predictor module uses a fully connected layer with 300 neurons and an output layer with a domain output neuron. We used a standard stochastic gradient descent optimization method to minimize the training error as represented by the loss function given in Equation (2) that involves two parameters λ1 and λ2. Both parameters were tuned experimentally by following suggestions in the literature (Bergstra and Bengio, 2012; Snoek et al., 2012). In particular, the parameter λ2 weighting the contribution from domain label prediction was set by using the following formula:
By adjusting , we gradually tuned λ2 so that noise from the domain label predictor is minimized at early training stages. The isoform domain data were partitioned in the 5-fold cross-validation procedure to produce the training and test data. The batch size used in stochastic training of the NN model was 200. In other words, 200 source samples (genes) and 200 target samples (isoforms) are merged to create a batch. At the initial training stage, the leaning rate was set as α = 0.001. As training progresses, we update the learning rate by using the standard step decay procedure (Sutskever et al., 2013) implemented in Caffe. We also checked if the learning was diverging (e.g. very large loss values were observed), and dropped the initial learning rate by a factor 10 until convergence has been achieved.
3.2 Collection of datasets
Manually reviewed mRNA isoform sequences and gene sequences of human were collected from the NCBI RefSeq (Pruitt et al., 2005). To collect the expression profiles of these isoforms, we took an initial set of 4643 RNA-Seq experiments from the NCBI SRA database (Leinonen et al., 2011), and selected datasets with 50–100 million reads. These experiments represented different physiological and cell conditions but were not involved in population studies. Such a diverse set of expression data may reflect many complex characteristics of the isoforms. The tool Kallisto (Bray et al., 2016) with Sleuth (Pimentel et al., 2016) was used to generate isoform expression data measured in Transcripts Per Million. The expression level of a gene in a dataset was estimated by summing up the expression levels of all its isoforms. Experiments with the pseudo-alignment ratio <0.7 were discarded to ensure data quality. We also filtered out poorly covered genes and their corresponding isoforms in these experiments. Finally, the expression data of 19 532 genes and 47 393 isoforms from 1735 RNA-Seq experiments formed our first dataset (simply called Dataset#1). Out of these genes, 9039 have only one isoform and are called SIGs and 10 313 have more than one isoform and are called multiple-isoform genes (MIGs). The distribution of isoforms over genes is shown in Supplementary Figure S2. UniProt genes were mapped to RefSeq genes by using the UniProt ID mapping file. The UniProt GO database was used to annotate the functions of each RefSeq gene, where GO functions inferred from electronic annotation evidence code were discarded as done in Li et al. (2014). In other words, only manually curated functions were used for the final annotation. The number of genes associated with a GO term is referred to as the GO term size. Intuitively, GO terms with small sizes are computationally difficult to learn since its data are highly skewed (i.e. mostly negative). In particular, it was assumed in Eksi et al. (2013) that a GO term with size <5 might be very specific to certain genes and thus not very useful in the cross-validation training procedure. We hence did not consider such infrequent GO terms in our experiments. The basic version of GO was used to generate the parent–child relationship between GO terms (Ashburner et al., 2000). Out of all 44 612 GO terms, 14 563 appear in human annotations. After the above filtration, 4272 GO terms were kept for our experimental evaluation work. In addition to Dataset#1, we also used the datasets with their respective GO annotations introduced in Eksi et al. (2013) and Li et al. (2014) (called Dataset#2 and Dataset#3, respectively) to ensure our comparison results are unbiased, where Dataset#2 was generated from 116 SRA mouse studies consisting of 365 experiments and Dataset#3 was generated from 29 SRA human studies consisting of 455 experiments with the requirement that each study had more than 6 experiments.
3.3 Experimental results
Since isoform functions are generally unavailable, we evaluated the performance of DeepIsoFun using gene level functional annotations by considering SIGs and MIGs either together or separately, as done similarly in Eksi et al. (2013) and Li et al. (2014). Because each SIG contains only one isoform, its functional annotation can be used to directly validate the predicted functions of the involved isoform. For a MIG, we can only check if the set of the predicted functions of its isoforms is consistent with its annotated GO terms (Eksi et al., 2013). We also estimated the functional divergence achieved by the isoforms of the same gene by calculating the semantic dissimilarity for each of the three main branches of GO (i.e. CC, BP and MF). The tool GOssTo (Caniza et al., 2014) was used to perform this estimation because it was able to take into account the hierarchical structure of GO. Moreover, we analyzed how the DA technique really helped the performance of our method, how the size of a GO term impacted the performance and the correlation between expression similarity and predicted function similarity for isoforms. Finally, we compared our method with the methods in Eksi et al. (2013), Li et al. (2014), Luo et al. (2017) and Panwar et al. (2016) in terms of AUC and AUPRC against specific baselines by focusing on a small set of GO terms (i.e. GO Slim with 117 terms) that have been widely used in the literature (Ashburner et al., 2000). Here, a baseline represents the performance of a random (untrained) classifier (Saito and Rehmsmeier, 2015). While the baseline in an AUC estimation is always 0.5 (Fawcett, 2006; Metz, 1978), the baseline in an AUPRC estimation depends on data imbalance and equals the proportion of positive instances (Saito and Rehmsmeier, 2015). The latter measure is known to be more suitable for imbalanced data. Note that for highly imbalanced data (like ours), AUPRC values are often quite low (Davis and Goadrich, 2006; Saito and Rehmsmeier, 2015). However, we may still use them to compare the relative performance of different methods on various datasets, taking into account actual baselines.
3.3.1. Performance on the three main branches of GO
Since the three main branches carry very different meanings in gene functions and are often treated separately in the literature, we compared the performance of DeepIsoFun on them. Out of the 4272 GO terms, 699 belong to CC, 2178 BP and 1395 MF. The distributions of GO term sizes on the three branches are similar. The average AUC values on BP, CC and MF are 0.735, 0.728 and 0.722, respectively (see Fig. 2a), and the average AUPRC values are 0.301, 0.279 and 0.294, respectively (see Fig. 2b). This robust performance of DeepIsoFun on the three main branches of GO shows that the terms on the branches probably follow similar distributions (as already observed on the distributions of their sizes).
Fig. 2.
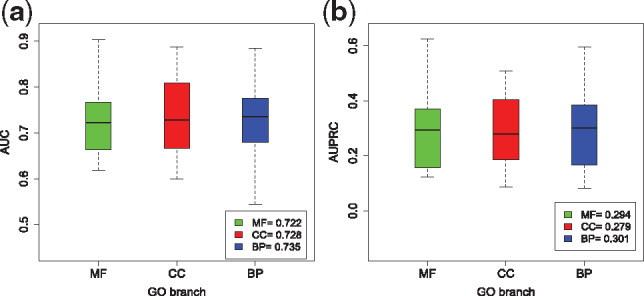
Comparison of performance on the three main branches of GO. (a) The average AUC values on the three branches. (b) The average AUPRC values on the three branches
3.3.2. Impact of the size of a GO term on performance
Some GO terms are very specific to certain genes while the others are more general. To test how the size (or popularity) of a GO term would impact the performance of DeepIsoFun, we divided the GO terms into four groups based on size. The four groups consist of GO terms of sizes in ranges [5–10], [11–20], [21–50] and [51–1000], respectively. The performance of DeepIsoFun on these groups is given in Figure 3a. DeepIsoFun performed better on GO terms with smaller sizes in general. This pattern seems to contradict intuition, but it is consistent with the findings in Li et al. (2014) and can perhaps be explained by the large the amount of (annotation) noise in large size GO terms. To confirm this, we further analyzed the correlation between expression similarity and functional similarity with respect to GO terms in each of the four groups. The results in Supplementary Figure S3 suggest that the correlation decreases as the GO term size increases. The weak correlations shown in the figure also partially explain why the AUC and AUPRC values obtained in Figure 2 are not very high.
Fig. 3.
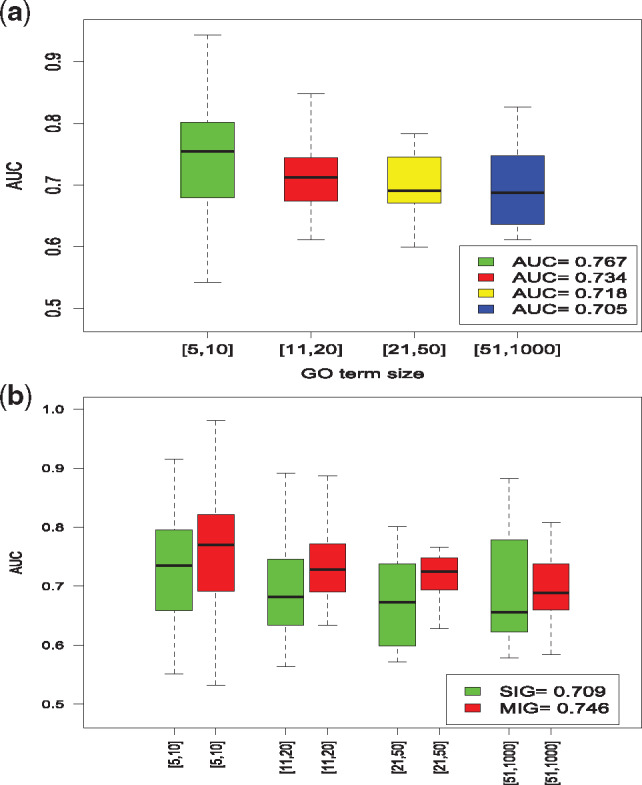
Comparison of AUCs achieved in four groups of GO terms from CC with different sizes. The four groups contain terms with sizes in ranges [5–10], [11–20], [21–75] and [76–1000], respectively. (a) The average AUC values achieved by the terms in the four groups are 0.767, 0.734, 0.718 and 0.705, respectively. The plot shows that generally as the size of a GO term increases, its achieved AUC actually decreases. (b) DeepIsoFun consistently performed better on MIGs over SIGs. The average AUC values on MIGs achieved in the four groups are 0.79, 0.748, 0.745 and 0.709, respectively. The average AUC values on SIGs achieved by in four groups are 0.755, 0.702, 0.693 and 0.675, respectively
3.3.3. Performance on MIGs versus SIGs
In the previous section, we considered the performance of DeepIsoFun on all genes, including both SIGs and MIGs. Since our ultimate goal is to dissect functions of different functions of the same gene, we would like to compare the performance on MIGs with that on SIGs in this subsection. As shown in Figure 3b, the performance increases as term size decreases. Moreover, the performance on MIGs achieved in these groups is consistently better than the performance on SIGs. More precisely, the performance on MIGs was 14, 23, 27 and 19 better (against the baseline 0.5) than that on SIGs in the four groups, respectively. Hence, DeepIsoFun was more effective in predicting functions for genes with multiple isoforms than genes with a single isoform, probably because of the functional diversity usually acquired by the former. Another plausible cause is that, since most (95%) human genes are expected to be MIGs, many SIGs could represent poorly annotated genes that have large numbers of undiscovered isoforms. Therefore, we also analyzed the performance of DeepIsoFun on MIGs with a certain number of isoforms. As shown in Supplementary Figure S4, the AUC performance of DeepIsoFun increases (slightly) as more isoforms are found in a MIG.
3.3.4. Dissimilarity among the predicted functions of isoforms
Since our ultimate goal is to dissect the functions of isoforms, we estimate the functional divergence of the isoforms of the same gene. For each GO term, the gene-wise method simGIC of GOssTo (Caniza et al., 2014; Pesquita et al., 2007) was used to calculate the semantic similarity score in the range of [0, 1] for each gene based on the predicted functions of its isoforms. The dissimilarity score was simply defined as one minus the similarity score (Li et al., 2014). Again, the three main branches of GO (i.e. CC, BP and MF) were considered separately. Out of the 10 313 MIGs, 4310 genes appear in CC, 5224 appear in BP and 3217 appear in MF (a gene may contain functions from multiple branches). For each branch, the functional divergence of a gene is calculated as the average dissimilarity scores over all terms on the branch. Figure 4 shows the distribution of functional dissimilarity scores among the isoforms of each gene. As observed in the literature (Li et al., 2014; Schlicker et al., 2006), many genes exhibited low average dissimilarity scores. More precisely, about 24% (1033) of the genes that appear in CC showed average dissimilarity scores<0.1. For BP and MF, this percentage rose to 46% (2405 genes) and 39% (1280 genes). On the other hand, about 7, 3 and 4% of the genes have average dissimilarity scores >0.3 on the three branches, respectively. These results are consistent with the fact that the isoforms of the same genes have very similar sequences, which lead them to perform mostly similar functions, but some isoforms may still have very different functions due to large changes in promoters and/or composition of coding exons.
Fig. 4.

Functional dissimilarity distributions on the three main branches of GO
3.3.5. Effectiveness of DA
A main novelty in DeepIsoFun is the use of DA to create labeled training data and transfer knowledge from the gene domain to the isoform domain. To test the effectiveness of DA in the experiments, we compared DeepIsoFun with a version without DA where the third part of the objective function in Equation (2) is disabled. Compared with the average AUC of 0.695 achieved by the restricted DeepIsoFun without DA, DeepIsoFun with DA performed 18% better against the baseline 0.5 as shown in Figure 5. We then further compared the two versions on the four GO term groups based on term sizes and found that the DA technique always made a significant difference. More specifically, it helped DeepIsoFun to achieve 19, 19, 17, and 20% better AUC (against the baseline 0.5) in the four groups, respectively.
Fig. 5.
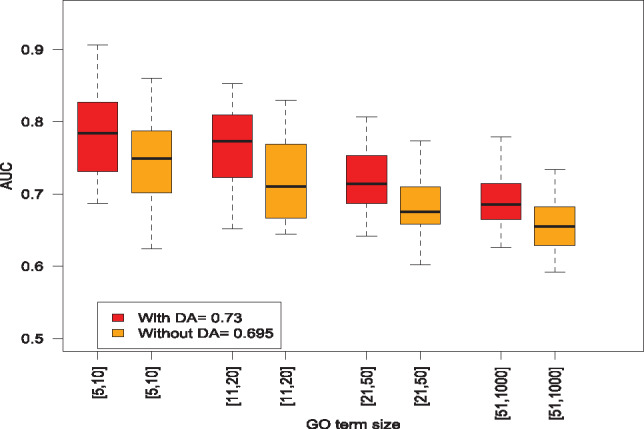
Comparison of AUCs achieved in the four groups of GO terms by DeepIsoFun with and without the DA technique. The average AUC value achieved by DeepIsoFun with DA in all four groups is 0.730 and the corresponding AUC achieved by DeepIsoFun without DA is 0.695. The benefit of DA is also clearly shown in the comparison over individual groups
We also tested if the DA technique was actually able to mix the two domains (so knowledge can be transferred). The plots in Supplementary Figure S5 made by using t-SNE (Maaten and Hinton, 2008) show clearly that the extracted features from the two domains became indistinguishable with the help of DA. This makes it possible to transfer knowledge (i.e. the relationship between expression profiles and functions) from the gene domain to the isoform domain and is a key to the improved performance of DeepIsoFun.
3.3.6. Correlation between expression similarity and the similarity of predicted functions
Given the difficulty in testing the performance of DeepIsoFun directly due to the lack of isoform function benchmark, we tested how the predicted isoform functions are correlated with their expression profiles. After all, this was the original hypothesis behind the design of DeepIsoFun. We performed a hierarchical clustering of the isoforms based on the expression data and Euclidean distance by using a standard tool (hclust) in the R Stats package. Eight clusters were defined from the clustering tree using the same tool. Then, the average distance between the expression profiles of the isoforms within each cluster was calculated and normalized to the range of [0, 1]. The same thing was done to estimate the average distance between the predicted GO terms of the isoforms within each cluster. The distributions of the average distances over the clusters are shown in Figure 6. Clearly, isoforms with similar expression profiles resulted in similar predicted functions.
Fig. 6.
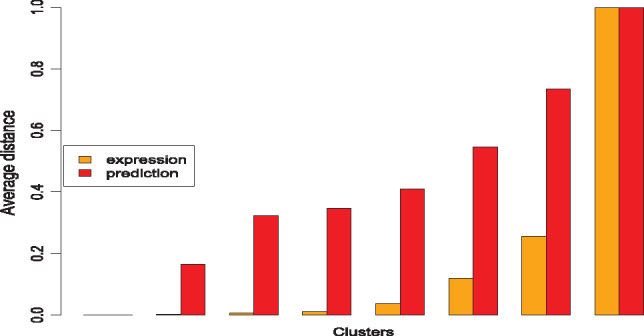
Comparison between the distribution of expression similarity and the distribution of similarity concerning predicted functions. The plot shows a clear positive correlation between the two distributions
3.3.7. Comparison with the existing methods
We compared the performance of DeepIsoFun with three existing methods, iterative iMILP (Li et al., 2014), mi-SVM (Eksi et al., 2013; Panwar et al., 2016) and the WLRM (Luo et al., 2017). Here, iMILP is the iterative version of MILP where a feature selection wrapper method is run over MILP to achieve better performance (Li et al., 2014). For completeness, we will also include MILP in the comparison. Note that WLRM was compared in Luo et al. (2017) with two recent methods for solving MIL, namely miFV (Wei et al., 2014) and miVLAD (Wei et al., 2017), and found to perform better in the prediction of isoform functions. In addition to Dataset#1 analyzed above, we also considered the two expression datasets introduced in Eksi et al. (2013) and Li et al. (2014), Dataset#2 and Dataset#3, respectively. Since mi-SVM and WLRM follow a 2-class classification framework but MILP/iMILP adapt a 3-class classification framework, different benchmarks were used to create functional labels for training and testing in Eksi et al. (2013), Li et al. (2014), Luo et al. (2017) and Panwar et al. (2016). In a 2-class classification framework, an isoform is classified as either positive or negative with respect to each GO term, while in a 3-class classification framework, an isoform is classified as positive, negative or unknown. Hence, we present the comparison between DeepIsoFun and MILP/iMILP in Table 1 and the comparison among DeepIsoFun, mi-SVM and WLRM in Table 2. Note that the values in the two tables are not directly comparable. On all three datasets, DeepIsoFun performed significantly better than the other methods. The average AUC values achieved by DeepIsoFun on all three datasets are 0.742, 0.734 and 0.720 with respect to the first benchmark (Table 1), and 0.735, 0.729 and 0.704 with respect to the second benchmark (Table 1). The corresponding values of AUPRC are 0.368 (baseline 0.1), 0.270 (baseline 0.08) and 0.331 (baseline 0.11) with respect to the first benchmark, and 0.292 (baseline 0.1), 0.246 (baseline 0.08) and 0.234 (baseline 0.11) with respect to the second benchmark. Note that although the AUPRC values are lower, they still represent quite decent performance when compared to the baseline values. The best performance achieved on Dataset#1 is perhaps due to the quality of data (since it was collected most recently and processed with updated tools) and its diversity across different tissue conditions. On this dataset, compared to iMILP, MILP, mi-SVM and WLRM, the AUC of DeepIsoFun increased 64, 102, 31 and 23% against the baseline 0.5, respectively. Similarly, on Dataset#2 (or Dataset#3), the improvements are 73, 216, 37 and 45% (or 26, 450, 43 and 24%) against the baseline, respectively. Since our labeled data were imbalanced, we also compared the performance in AUPRC and observed similar improvements. On Dataset#1 (Dataset#2 and Dataset#3), DeepIsoFun performed 59% (29 and 41%, respectively) better than mi-SVM in AUPRC against respective baselines, 11% (23 and 10%, respectively) better than iMILP, 57% (32 and 20%, respectively) better than MILP and 63% (62 and 85%, respectively) better than WLRM. We think that these significant improvements in performance over the existing methods on several human and mouse datasets demonstrate the success of the DA technique as well as the power of deep learning.
Table 1.
Comparison between DeepIsoFun and MILP/iMILP on different expression datasets in terms of AUC and AUPRC values
| AUC |
AUPRC |
|||||
|---|---|---|---|---|---|---|
| Dataset method | DeepIsoFun | MILP | iMILP | DeepIsoFun | MILP | iMILP |
| Dataset#1 | 0.742 | 0.620 | 0.648 | 0.368 | 0.271 | 0.342 |
| Dataset#2 | 0.734 | 0.574 | 0.635 | 0.270 | 0.224 | 0.235 |
| Dataset#3 | 0.720 | 0.540 | 0.674 | 0.331 | 0.294 | 0.311 |
Note: Dataset#1 was generated from 1735 RNA-Seq experiments by using Kallisto (Bray et al., 2016). Dataset#2 and Dataset#3 were obtained from Eksi et al. (2013) and Li et al. (2014), respectively. The benchmark positive and negative instances of each GO term used in testing were defined by following the procedure in Li et al. (2014). The unlabeled instances were ignored in testing. Both Dataset#1 and Dataset#2 were divided based on read length to create different ‘study groups’. There are 24, 24 and 29 study groups in Dataset#1, Dataset#2 and Dataset#3, respectively. On the average, each study group consists of 71, 16 and 17 SRA experiments in Dataset#1, Dataset#2 and Dataset#3, respectively. As done in Li et al. (2014), a selection algorithm was employed by iMILP to choose a subset of study groups on each dataset optimize its performance.
Table 2.
Comparison among DeepIsoFun, mi-SVM and WLRM on different expression datasets in terms of AUC and AUPRC values
| AUC |
AUPRC |
|||||
|---|---|---|---|---|---|---|
| Dataset method | DeepIsoFun | mi- SVM | WLRM | DeepIsoFun | mi- SVM | WLRM |
| Dataset#1 | 0.735 | 0.679 | 0.691 | 0.292 | 0.221 | 0.218 |
| Dataset#2 | 0.729 | 0.667 | 0.658 | 0.246 | 0.209 | 0.182 |
| Dataset#3 | 0.704 | 0.643 | 0.664 | 0.234 | 0.198 | 0.177 |
Note: The benchmark positive and negative instances of each GO term used in testing were defined by following the procedure in Eksi et al. (2013).
Some comparisons of the methods in terms of divergence of predicted isoform functions and time efficiency are given in the Supplementary Material (Supplementary Fig. S6 and Table S1). We also compared the performance of the methods on two additional datasets concerning Arabidopsis thaliana and Drosophila melanogaster (i.e. fruit fly) and summarize the comparison results in Supplementary Tables S2 and S3. As the tables show, DeepIsoFun consistently performed better than the other methods in both AUC and AUPRC. The performance of the methods on all five datasets with respect to different GO term sizes is given in Supplementary Tables S4 and S5.
3.3.8. Validation of some predicted isoform functions
As mentioned before, there has been little systematic study on isoform functions in the literature, and not many specific experimentally verified functions of isoforms have been reported. Some of the reported functions concern differential regulatory behaviors of isoforms in important processes such as the ‘regulation of apoptosis process’ (GO: 0042981). Apoptosis refers to programed cell death. This GO term has two children with opposite functions, i.e. the ‘positive regulation of apoptosis process’ or pro-apoptosis (GO: 0043065) and the ‘negative regulation of apoptosis process’ or anti-apoptosis (GO: 0043066). For MIGs with both pro-apoptosis and anti-apoptosis functions, it would be interesting to know if it has some isoforms that are pro-apoptosis but not anti-apoptosis and some other isoforms that are anti-apoptosis but not pro-apoptosis. In other words, we would like to know if the pro- and anti-apoptosis functions of the gene are differentiated among its isoforms. To investigate such MIGs, we searched for all genes that have multiple isoforms and are annotated with both pro-apoptosis and anti-apoptosis functions. Totally, 18 such genes were found (see Supplementary Table S6). The number of isoforms in each of these genes ranges from 2 to 17. Supplementary Tables S6–S9 show the performance of DeepIsoFun, iMILP, mi-SVM and WLRM, respectively, in predicting the apoptosis regulatory, pro-apoptosis and anti-apoptosis functions, measured at the gene level. DeepIsoFun was able to predict the apoptosis regulatory function for the isoforms of 17 out of the 18 genes (94.4% recall), the pro-apoptosis function for the isoforms of 13 genes (72.2% recall) and the anti-apoptosis function for the isoforms of 14 genes (77.7% recall). In contrast, iMILP achieved recalls 77.7, 55.6 and 61.1%, mi-SVM achieved recalls 83.3, 66.7 and 61.1% and WLRM achieved recalls 77.7, 61.1 and 72.2% in predicting the three functions, respectively. Futhermore, the tables show that DeepIsoFun was able to differentiate the pro- and anti-apoptosis functions among isoforms for 8 of the 18 genes while iMILP, mi-SVM and WLRM were only able to do it for 5, 4 and 3 genes, respectively. Although we do not know exactly how many of these genes have differentiated pro- and anti-apoptosis functions among their isoforms, it is perhaps reasonable to conjecture that most of these genes do possess this property.
4 Discussion
Although DeepIsoFun achieved significant improvement over the existing methods in isoform function prediction, its performance as measured by AUC and AUPRC in our experiments still remained less than desirable. The prediction of isoform functions is challenging not only because of the lack of labeled training data (i.e. specific functions are known for very few isoforms) and noisy GO annotation, but also because the data were very imbalanced. That is, most GO terms are only associated with a small number of genes and hence the negative examples are far more than the positive examples. This makes the situation especially bad when the performance is measured in AUPRC since the number of false positive examples tends to be high and thus the precision tends to be low. We dealt with the problem by leaving out infrequent GO terms that are associated with fewer than five genes, although such terms often represent specific functions and could be biologically the most relevant. On the other hand, most functions of genes are yet to be discovered. Hence, three-class classification was proposed in Li et al. (2014) as a way to address the data imbalance issue. However, such an approach often leads to conservative predictions and may fail to predict many isoform specific functions. We plan to study machine learning (including unsupervised learning) techniques that can help produce meaningful predictions for infrequent GO terms.
Another challenge we faced was the heterogeneity of the expression data. While a large dataset covering many tissues and conditions (such as Dataset#1) provides rich information about isoform functions, it also contains a lot of noise that makes the extraction of informative features difficult. Li et al. (2014) solved this problem by using an elaborate search procedure to identify the best subset of RNA-Seq experiments in the input data. However, the search consumes a lot of time, especially when the number of input RNA-Seq experiments is large. We plan to apply DeepIsoFun to tissue-specific data to see how its performance will be affected as well as if some tissue-specific isoform functions can be discovered.
Supplementary Material
Acknowledgements
We would like to thank the authors of Eksi et al. (2013), Li et al. (2014) and Luo et al. (2017) for sharing their data and code with us.
Funding
This research was partially supported by National Science Foundation grant [IIS-1646333]; the Natural Science Foundation of China grant [61772197] and the Natioanl Key Research and Development Program of China grant 2018YFC0910404.
Conflict of Interest: none declared.
References
- Ajakan H. et al. (2014) Domain-adversarial neural networks. arXiv, 1412, 4446. [Google Scholar]
- Andrews S. et al. (2003) Support vector machines for multiple-instance learning. In: Advances in Neural Information Processing Systems, pp. 577–584.
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell D. et al. (2009) The GOA database in 2009 an integrated Gene Ontology Annotation resource. Nucleic Acids Res., 37, D396–D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutcuoglu Z. et al. (2006) Hierarchical multi-label prediction of gene function. Bioinformatics, 22, 830–836. [DOI] [PubMed] [Google Scholar]
- Bergstra J.S., Bengio Y. (2012) Random search for hyper-parameter optimization. J. Mach. Learn. Res., 13, 281–305. [Google Scholar]
- Bergstra J.S. et al. (2011) Algorithms for hyper-parameter optimization. In: Advances in Neural Information Processing Systems, pp. 2546–2554.
- Bouillet P., O’Reilly LA. (2009) CD95, BIM and T cell homeostasis. Nat. Rev. Immunol., 9, 514–519. [DOI] [PubMed] [Google Scholar]
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Caniza H. et al. (2014) GOssTO: a stand-alone application and a web tool for calculating semantic similarities on the Gene Ontology. Bioinformatics, 30, 2235–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J., Goadrich M. (2006) The relationship between Precision-Recall and ROC curves. In: Proceedings of the 23rd International Conference on Machine Learning, ACM, pp. 233–240.
- Dietterich T.G. et al. (1997) Solving the multiple instance problem with axis-parallel rectangles. Artif. Intell., 89, 31–71. [Google Scholar]
- Eksi R. et al. (2013) Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data. PLoS Comput. Biol., 9, e1003314.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. (2006) An introduction to ROC analysis. Pattern Recognit. Lett., 27, 861–874. [Google Scholar]
- Gallego-Paez L. et al. (2017) Alternative splicing: the pledge, the turn, and the prestige. Hum. Genet., 136, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganin Y., Lempitsky V. (2015) Unsupervised domain adaptation by backpropagation. In: International Conference on Machine Learning, pp. 1180–1189.
- Gueroussov S. et al. (2015) An alternative splicing event amplifies evolutionary differences between vertebrates. Science, 349, 868–873. [DOI] [PubMed] [Google Scholar]
- Himeji D. et al. (2002) Characterization of caspase-8l: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood, 99, 4070–4078. [DOI] [PubMed] [Google Scholar]
- Jia Y. et al. (2014) Caffe: convolutional architecture for fast feature embedding. In: Proceedings of the 22nd ACM International Conference on Multimedia, ACM, pp. 675–678.
- Leinonen R. et al. (2011) The sequence read archive. Nucleic Acids Res., 39, D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.D. et al. (2015) MIsoMine: a genome-scale high-resolution data portal of expression, function and networks at the splice isoform level in the mouse. Database, 2015, bav045.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. et al. (2014) High-resolution functional annotation of human transcriptome: predicting isoform functions by a novel multiple instance-based label propagation method. Nucleic Acids Res., 42, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. et al. (2015) Learning transferable features with deep adaptation networks. In: International Conference on Machine Learning, pp. 97–105.
- Luo T. et al. (2017) Functional annotation of human protein coding isoforms via non-convex multi-instance learning. In: Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, pp. 345–354.
- Maaten L.V.D., Hinton G. (2008) Visualizing data using t-SNE. J. Mach. Learn. Res., 9, 2579–2605. [Google Scholar]
- Mazurek S. et al. (2005) Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol., 15, 300–308. [DOI] [PubMed] [Google Scholar]
- Melamud E., Moult J. (2009) Stochastic noise in splicing machinery. Nucleic Acids Res., 37, 4873–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz C.E. (1978) Basic principles of ROC analysis. Semin. Nucl. Med., 8, 283–298. [DOI] [PubMed] [Google Scholar]
- Mi H. et al. (2012) Panther in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res., 41, D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittendorf K.F. et al. (2012) Tailoring of membrane proteins by alternative splicing of pre-mRNA. Biochemistry, 51, 5541.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberwinkler J. et al. (2005) Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J. Biol. Chem., 280, 22540–22548. [DOI] [PubMed] [Google Scholar]
- Pan Q. et al. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet., 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Pan S.J. et al. (2011) Domain adaptation via transfer component analysis. IEEE Trans. Neural Netw., 22, 199–210. [DOI] [PubMed] [Google Scholar]
- Panwar B. et al. (2016) Genome-wide functional annotation of human protein-coding splice variants using multiple instance learning. J. Proteome Res., 15, 1747–1753. [DOI] [PubMed] [Google Scholar]
- Pesquita C. et al. (2007) Evaluating GO-based semantic similarity measures. In Proceedings of 10th Annual Bio-Ontologies Meeting, Vol. 37, pp. 38. [Google Scholar]
- Pickrell J.K. et al. (2010) Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet., 6, e1001236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H.J. et al. (2016) Differential analysis of RNA-seq incorporating quantification uncertainty. biorxiv 058164. [DOI] [PubMed] [Google Scholar]
- Pruitt K.D. et al. (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res., 33, D501–D504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revil T. et al. (2007) Protein kinase C-dependent control of Bcl-x alternative splicing. Mol. Cell. Biol., 27, 8431–8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Rehmsmeier M. (2015) The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One, 10, e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietgat L. et al. (2010) Predicting gene function using hierarchical multi-label decision tree ensembles. BMC Bioinformatics, 11, 2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker A. et al. (2006) A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics, 7, 302.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoek J. et al. (2012) Practical Bayesian optimization of machine learning algorithms. In: Advances in Neural Information Processing Systems, pp. 2951–2959.
- Sutskever I. et al. (2013) On the importance of initialization and momentum in deep learning. In: International conference on machine learning, pp. 1139–1147.
- Tzeng E. et al. (2014) Deep domain confusion: maximizing for domain invariance. arXiv, 1412, 3474. [Google Scholar]
- Vázquez Á.V. et al. (2011) Two proteins with different functions are derived from the KIHEM13 gene. Eukaryot. Cell, 10, 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Végran F. et al. (2006) Overexpression of caspase-3s splice variant in locally advanced breast carcinoma is associated with poor response to neoadjuvant chemotherapy. Clin. Cancer Res., 12, 5794–5800. [DOI] [PubMed] [Google Scholar]
- Vinayagam A. et al. (2004) Applying support vector machines for Gene Ontology based gene function prediction. BMC Bioinformatics, 5, 116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T. et al. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature, 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (2017) Multiple-instance learning via an RBF kernel-based extreme learning machine. J. Intell. Syst., 26, 185–195. [Google Scholar]
- Wang X. et al. (2015) Relaxed multiple-instance SVM with application to object discovery. In: Proceedings of the IEEE International Conference on Computer Vision, pp. 1224–1232.
- Wei X.S. et al. (2014) Scalable multi-instance learning. In: 2014 IEEE International Conference on Data Mining (ICDM), IEEE, pp. 1037–1042.
- Wei X.S. et al. (2017) Scalable algorithms for multi-instance learning. IEEE Trans. Neural Netw. Learn. Syst., 28, 975–987. [DOI] [PubMed] [Google Scholar]
- Yang J. et al. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods, 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.