

Abstract
Identifying nonnative, trimeric forms of SOD1 trimers as the toxic species, rather than large aggregates revolutionizes our understanding of ALS pathophysiology. Large protein aggregates, what was previously thought as the central cause of neurodegeneration, play protective role and are not responsible for neuronal death. SOD1 trimers are implicated at the molecular, cellular, and organismal level. Understanding the formation of the nonnative trimer and its role in the cell, leading to cell death, holds the key to developing a new standard of therapeutics for ALS and for other neurodegenerative diseases. This review highlights recent advances of knowledge for the role of SOD1 oligomers in ALS.
Keywords: SOD1 oligomers, SOD1 timer, ALS, neurodegeneration
Graphical Abstract
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a progressive and ultimately fatal neurodegenerative disease. ALS is characterized by the loss of motor neurons in the brain, brainstem, and spinal cord leading to extra-motor symptoms such as difficulty in speech, swallowing, and limb paralysis [1]. The most feared complication of the disease is the loss of voluntary respiratory muscle function resulting in death [2]. The varied clinical manifestations of ALS suggest that the disease is complex and most likely caused by several different mechanisms [3,4]. Much of our insight into the molecular mechanisms of ALS comes from the study of familial ALS. About 20% of ALS patients have a family history of ALS or frontotemporal dementia [5–7] and 70% of the familial ALS cases are accounted for by mutations in the following genes: superoxide dismutase 1 (SOD1), C9orf72, TAR DNA binding protein, and FUS RNA binding protein [8]. SOD1 was the first gene identified in familial ALS [9] and is linked to the greatest number of pathophysiological mechanisms. A few mechanisms of disease implicated with SOD1 mutations are protein misfolding, proteasome impairment, oxidative stress, oligodendrocyte degeneration, and mitochondrial dysfunction [2,4]. The variety of mechanisms associated with SOD1 illustrates the complicated nature of ALS and uncovering how SOD1 causes disease opens a door for understanding a multitude of mechanisms in neurodegeneration.
Recent work from Proctor et al. [10] puts a spot light on oligomers, specifically trimeric forms, of SOD1 as the neurotoxic species. This work demonstrated that SOD1 mutants which promote trimerization increased neuronal cell death. They further showed a direct link between misfolded oligomers and neuron death by correlating cytotoxicity with trimer stability. The finding that trimeric forms of SOD1 is responsible for toxicity in cells is evidence against previous theories of large aggregates, a hallmark in neurodegenerative diseases, as the causative agent of the disease. Additional studies show that large aggregates are not toxic and play a neuroprotective role [11,12]. The work described above repositions the central cause of ALS and of neurodegenerative disease away from large aggregates and directly to specific oligomers. It also demands further investigation for understanding the etiology of ALS. In this review, we summarize prospective disease mechanisms of oligomeric SOD1 in ALS and oligomeric SOD1-associated therapeutic strategies.
Body
SOD1 trimer formation, structure, and stability
The formation, structure, and stability of the SOD1 trimer provides evidence for the basis of the trimer’s potential to disrupt cellular functions lead to cell death. Recent work studied the formation of SOD1 trimers and oligomers computationally and experimentally [10,13–17]. Structural studies comparing wild-type SOD1 and oligomeric SOD1 hypothesize that an unmodified Trp32 crosslink in the native dimer leads to the formation of SOD1 oligomers and trimers [15]. Furthermore, in vivo studies show that superoxide exposure in cell culture can initiate the formation of SOD1 trimers [16] and SOD1 maturation minimizes copper wastage and reduces the production of toxic SOD1 species [17]. The exact mechanism for the formation of the non-native trimer is unknown due to the fact that SOD1 is very stable, but in order to form trimers, it must dissociate into monomers [18]. The main issue with understanding non-native trimer formation begs the question; how do SOD1 dimers fall into monomers? Exogenous factors such as oxidative stress [19] or BMMA exposure [20,21] may play a role. Currently, the two main hypotheses for trimer formation are that trimer formation occurs on-pathway from the native dimer to larger aggregates or off-pathway meaning it is separate to the formation of large aggregates (Figure 1).
Figure 1: SOD1 trimer formation: on- or off-pathway.
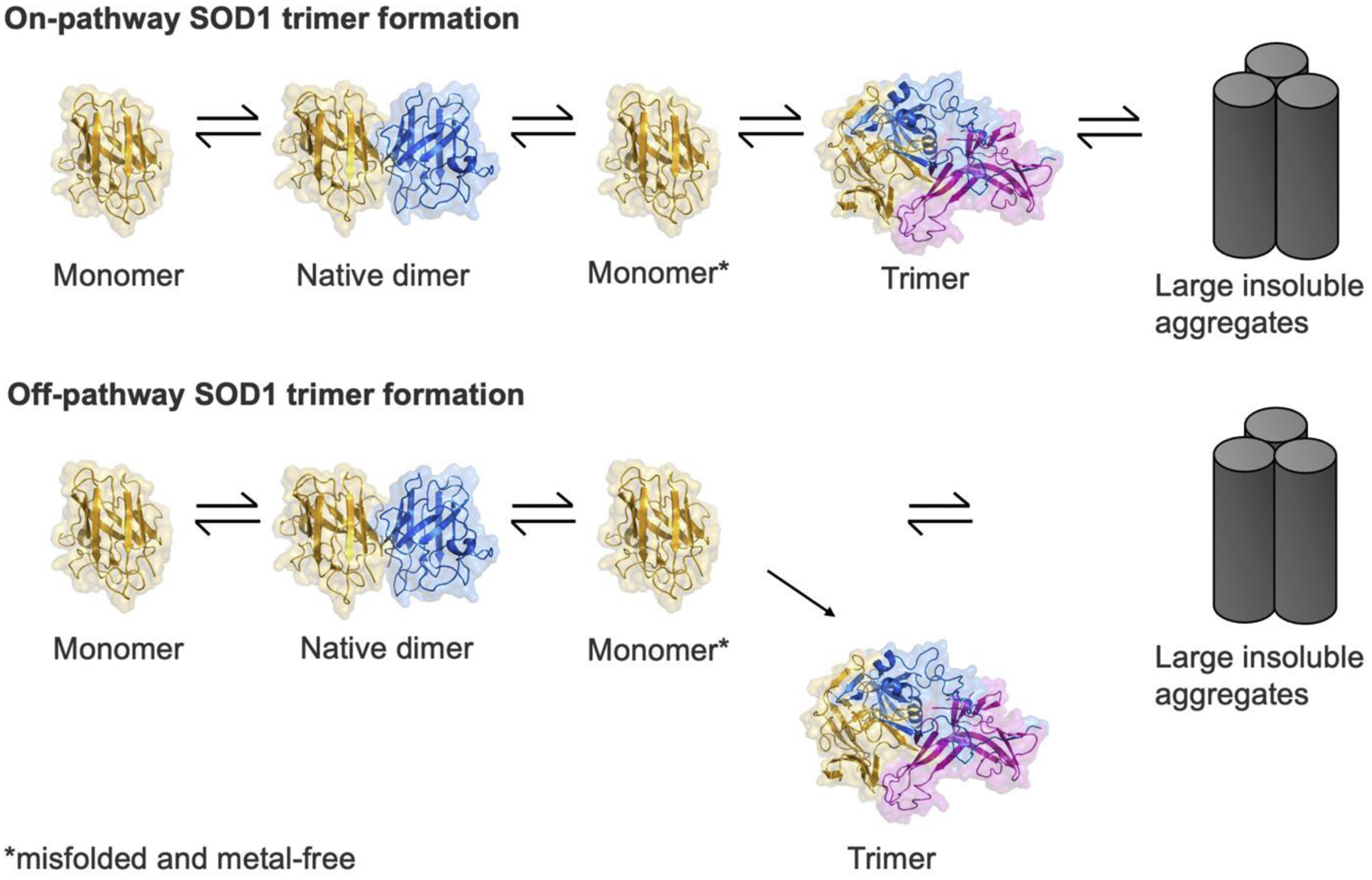
The formation of SOD1 trimers is unclear. One hypothesis is that the SOD1 trimer forms on-pathway from monomers, to native dimers (with the native dimer dissociating into monomers), to trimers, and then to large aggregates. The other hypothesis is that the formation of the trimer is off-pathway meaning it does not lead to the formation of large aggregates.
Structures of SOD1 trimers were solved with stabilizing and destabilizing mutations [10]. The structures of various SOD1 trimers affect nucleation of aggregation and the degree of toxicity. Specifically, trimeric SOD1 with a clinically potent mutation, A4V [22], exhibits a decreased number of water molecules surrounding the site of oxidative modification. The difference in water molecules surrounding the trimer compared to wild type affects the potentiation of aggregation [23]. In addition, superoxide exposure initiated the formation of SOD1 trimers and increased aggregation propensity in cells [16]. Altering superoxide exposure in cells to only halt trimer formation may yield an increase in cell viability since trimers are the neurotoxic species [10] and several studies have shown that large aggregates play a protective role [11,12]. Understanding the structure of the trimer is critical for determining changes in normal cell function such as protein homeostasis and provides insight into potential disease mechanisms discussed later in the review.
Stabilization studies are critical for understanding the role of proteins in the cell and predicting cascading events of the nonnative protein. The stability of the trimer affects the degree of aggregation that occurs [17,20,24]. Amino acid substitutions resulting in destabilization promotes SOD1 aggregation and neurotoxicity [20]. Destabilization of SOD1 in monomeric form in a protein crowded environment showed residue specific interactions of SOD1 with crowder proteins and chemical shift perturbations [25]. This implies that the stability of the trimer affects cell function by direct interaction with other proteins as a chaperone for folding or a marker for degradation. At the clinical level, human spinal cord homogenates from familial ALS patients induce more protein aggregation than tissue homogenates from sporadic ALS patients [26]. This finding was further supported by experiments showing attenuation of aggregation by small molecules or antibodies specific to misfolded SOD1. This result may root from differences in presence of stable and non-stable trimers in familial and sporadic ALS. Stabilizing mutations in A4V mutant SOD1 dimer show disturbances to protein aggregation and aids in regaining protein structural conformations [24]. Taken together, we can conclude that the stability of dimer decreases cytotoxicity while stabilizing the trimer increases cytotoxicity.
SOD1 trimer mechanisms of toxicity
SOD1 trimers are responsible for cytotoxicity [10] and large aggregates do not impact cell viability [11,12]. The exact mechanism of cytotoxicity mediated by SOD1 trimers is not well understood but a range of studies from the atomic to clinical levels provide clues into potential cellular pathways and functions central to the trimer’s mode of toxicity (Figure 2). At the atomic level, the crosslink of unmodified Trp32 discussed above in the native SOD1 dimer also hinders proteolytic mechanisms [15]. The inability for proteolysis to occur implies that the SOD1 oligomer will persist intracellularly and carry the potential to aggregate leading to the classically seen insoluble protein aggregates in ALS histology. Prion-like propagation of SOD1 is highly discussed in the literature [27–29]. At the cellular level, in motor neuron-like cells, misfolded SOD1 exposure in the presence of a strong binder to mutant SOD1 prevented the prion-like propagation of SOD1 [30]. Propagation of misfolded SOD1 in cell culture was interrupted by direct blocking of SOD1 from cell to cell. Protein folding and processing interactions are important for cellular homeostasis. SOD1-chaperone interactions impact neuronal cell loss in ALS [31]. At the organismal level, disaggregation of SOD1 proteins occurs by heat shock proteins, specifically by Heat Shock Protein 104, in an ATP-dependent manner. This action restored mobility in mice, but produced trimers and other oligomers rather than the native dimer [32]. Stress granules are also a cellular component of interest in aging since the granules act as seeds for disease-related protein aggregation [33–35]. In mice, SOD1 inclusion studies showed that SOD1 localizes to RNA-rich structures, such as stress granules, but did not show evidence of direct SOD1 binding to RNA [36]. Da Ros et al. [36] acknowledge the potential role of trimers due to SOD1 trimers detected in their work. Other evidence of trimers in mouse models of ALS includes increased levels of soluble misfolded subfractions of SOD1 in spinal cords correlated to decreased life span [37]. At the clinical level, central nervous system regions of ALS patients least affected by the disease had the highest levels of aggregated SOD1 while those most affected by disease had lower levels of misfolded soluble SOD1. The correlation of high misfolded soluble SOD1 and disease burden indicates that SOD1 oligomers are more likely the disease drivers compared to large aggregates [12]. They concluded, in support of work from Zhu et al. [11], that aggregated SOD1 serves to sequester the toxic oligomeric SOD1 species and are neuroprotective.
Figure 2: Atomic to organism evidence of SOD1 trimer toxicity.
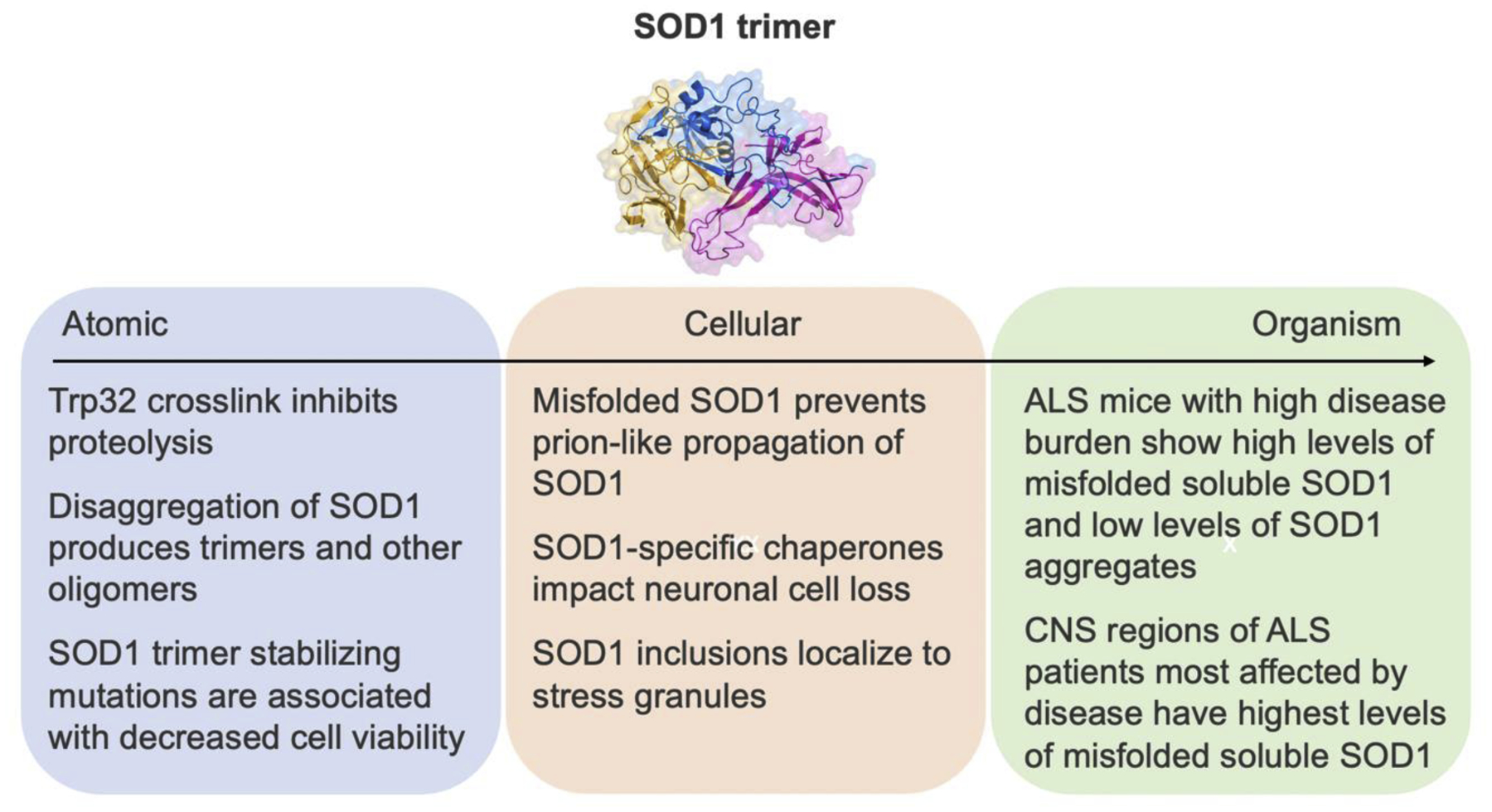
The mechanism of toxicity mediated by the SOD1 trimer is unknown. This figure illustrates evidence from the atomic to organismal level of SOD1 trimer implications.
SOD1 trimer-associated therapeutic strategies
A wide range of potential therapeutics for ALS are currently undergoing clinical trials [4], but there is still no cure. Treatment of ALS relies of multi-disciplinary symptomatic management [38,39] while patients only survive 3–5 years after diagnosis [2,40]. Only two drugs, riluzole [41] and edaravone [42], are FDA-approved and they possess modest efficacy [43] in symptomatic treatment only. Based on the work described above, we discuss SOD1 trimer-associated therapeutic strategies (Figure 3).
Figure 3: SOD1 trimer-specific therapeutic strategies.
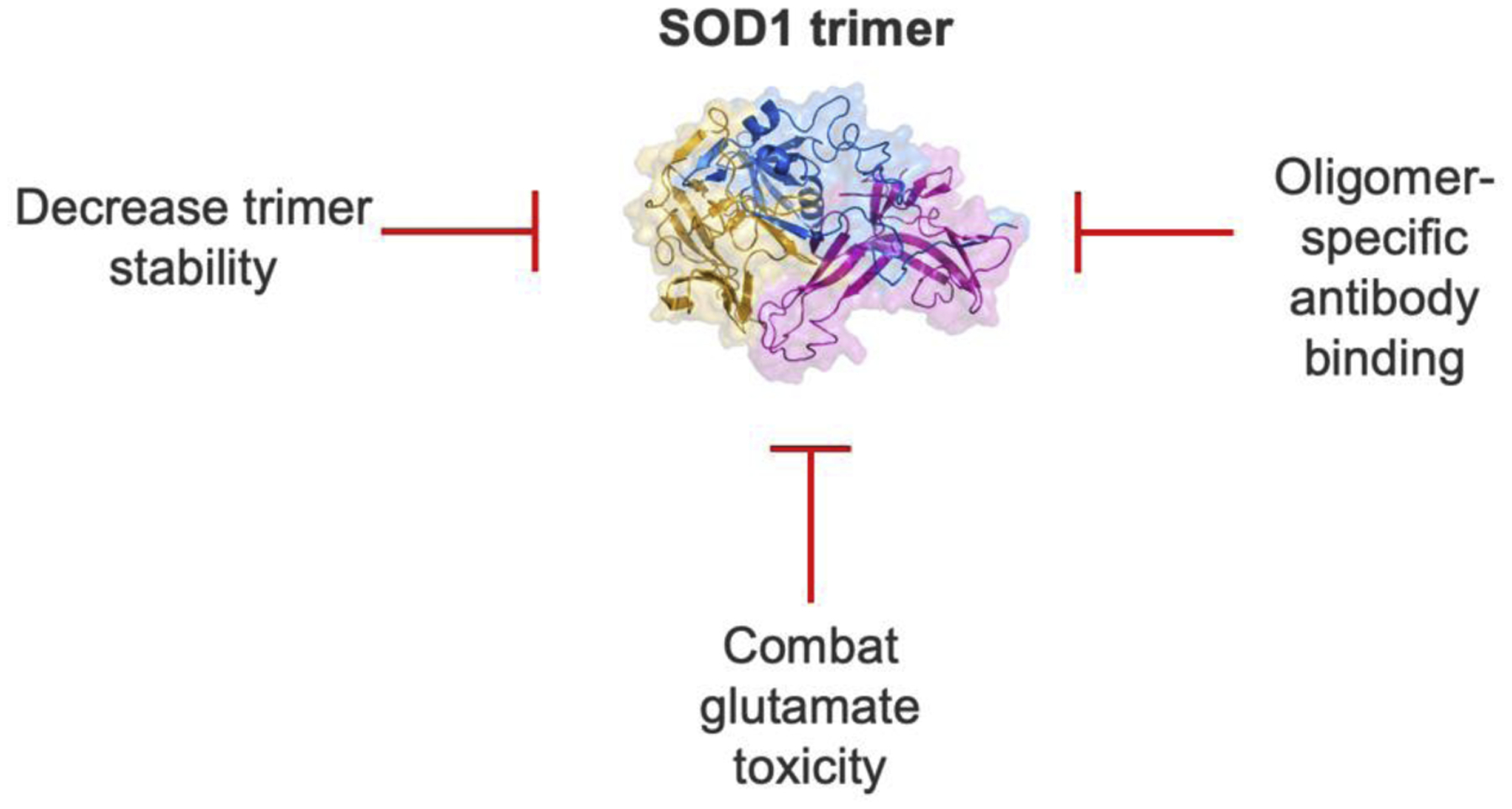
Targeting SOD1 timers show therapeutic promise. Several proposed mechanisms discussed in this review are decreasing trimer stability, addressing glutamate toxicity due to trimers, and direct binding to sequester SOD1 oligomers with specific antibodies.
First, as discussed earlier in this review, SOD1 dimer stability decreases toxicity [24,44] while trimer stabilization increases toxicity [10,45]. A potential therapeutic approach is to manipulate the cellular environment or to use direct SOD1 binders to stabilize the native dimer or decrease timer stability. Direct mutagenesis or binding to modify the stability of the dimer and trimer is a possible mode of action. Sequestering the toxic species into larger aggregates plays a protective role [11,12] and thus, enhancing trimer clearance through protein tagging for specific degradation may benefit cell viability. An example of this approach is to enhance chaperone protein’s role in clearing misfolded SOD1. Macrophage-migration inhibitory factor (MIF) inhibits accumulation of misfolded SOD1 [31,46]. Increasing MIF expression and activity in the cell may show enhanced survival. Next, glutamate toxicity is the basis for one of the FDA-approved drugs, riluzole. Toxicity by glutamate is mechanistically linked to the conversion of mutant SOD1. Disease onset was delayed ALS mice models with reduced calcium permeability in a type of glutamate receptors [47]. Alternative approaches to combat glutamate toxicity from riluzole use changes in expression of glutamate to modulate its toxic levels of glutamate. Trimeric SOD1 may play a role in glutamate homeostasis by direct binding of regulator proteins. Enzymatic activity assays or binding affinity studies investigating trimeric SOD1 are needed to elucidate glutamate toxicity. Finally, oligomer-specific antibodies show promise as a therapeutic for ALS. Misfolded SOD1-specific antibodies provide insight into SOD1 misfolding [48]. Antibody, W20 [49], is an example of an SOD1 oligomer-specific antibody with efficacy at low doses improving motor neuron survival and motor performance in SOD1G93A mice [50]. To summarize, preventing binding interactions of trimeric SOD1 is the key to preventing cytotoxicity and preserving neuronal function.
Conclusion
Here, we discuss recent advances in understanding the role of trimeric SOD1 in ALS pathophysiology. SOD1 trimers are a novel discovery in ALS pathophysiology. The trimers are responsible for toxicity instead of as previously assumed, large aggregates, [10,11] and show promise as a therapeutic target. The structure and formation of the SOD1 trimer is not fully understood; however, we are beginning to understand the importance of the trimer based on cell viability assays with stability studies and in animal models. Trimers are either formed on- or off-pathway in SOD1 aggregation and better understanding their origin will shed light on their mechanism of toxicity. Working hypotheses about SOD1 trimers show that direct interaction of the trimer impairs cell function such as protein homeostasis. Currently, riluzole and edaravone are the only FDA-approved therapeutics for ALS with modest effects and more effective therapeutics are needed [51]. Guiding the development of therapeutics based on recent discoveries regarding SOD1 trimers may be a viable strategy moving forward in mitigating neurotoxicity in ALS. ALS is a fatal and complicated disease and cracking the code on a mode of a cell death mechanism through SOD1 trimers will revolutionize the field of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease since many phenotypic features are shared and no cure exists.
Highlights.
SOD1 trimers are the toxic species responsible for cell death rather than large aggregates.
Stability of the native SOD1 dimer decreases cellular toxicity whereas increased SOD1 trimer stability increases cellular toxicity.
SOD1 trimer formation is not fully understood, but direct interaction of the trimers affects cellular function.
Inhibition of SOD1 trimer interactions hold promise as a therapeutic approach.
Acknowledgements
We thank B.H., C.S., J.R, and J.W. for proofreading the manuscript. We acknowledge support from the National Institutes for Health (1R35 GM134864 to N.V.D.) and the Passananti Foundation. The project described was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare no conflict of interest.
References
- 1.Grad LI, Rouleau GA, Ravits J, Cashman NR: Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb Perspect Med 2017, 7:a024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH: Amyotrophic lateral sclerosis. Nat Rev Dis Prim 2017, 3:17071. [DOI] [PubMed] [Google Scholar]
- 3.Al-Chalabi A, Hardiman O: The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 2013, 9:617–628. [DOI] [PubMed] [Google Scholar]
- 4.Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA: ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?. Front Neurosci 2019, 13:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC: Amyotrophic lateral sclerosis. Lancet 2011, 377:942–955. [DOI] [PubMed] [Google Scholar]
- 6.Brenner D, Weishaupt JH: Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol 2019, 32:735–739. [DOI] [PubMed] [Google Scholar]
- 7.Ghasemi M, Brown RH: Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med 2018, 8:a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiò A, Battistini S, Calvo A, Caponnetto C, Conforti FL, Corbo M, Giannini F, Mandrioli J, Mora G, Sabatelli M: Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 2014, 85:478–485. [DOI] [PubMed] [Google Scholar]
- 9.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362:59–62. [DOI] [PubMed] [Google Scholar]
- 10.Proctor EA, Fee L, Tao Y, Redler RL, Fay JM, Zhang Y, Lv Z, Mercer IP, Deshmukh M, Lyubchenko YL, et al. : Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci 2016, 113:614 LP–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.**.Zhu C, Beck MV, Griffith JD, Deshmukh M, Dokholyan NV: Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci 2018, 115:4661 LP–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]; Large SOD1 aggregates are not the cause of cellular toxicity as was previously thought in the field of ALS and the large aggregates may be protective. This work supports previous studies that SOD1 trimers are the toxic species and not large aggregates.
- 12.**.Gill C, Phelan JP, Hatzipetros T, Kidd JD, Tassinari VR, Levine B, Wang MZ, Moreno A, Thompson K, Maier M: SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci Rep 2019, 9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; CNS regions show inversely correlated levels of aggregated SOD1 and disease progression. The CNS regions affected most by disease show high levels of misfolded to soluble total SOD1 meaning that soluble SOD1 are disease drivers in ALS.
- 13.Shirvanyants D, Redler RL, Tandon A, Kim DN, Dagliyan O, Proctor EA, Dokholyan NV, Kota P, Ramachandran S, Ding F: Computational approaches to understanding protein aggregation in neurodegeneration. J Mol Cell Biol 2014, 6:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redler RL, Dokholyan NV: The complex molecular biology of amyotrophic lateral sclerosis (ALS). In Progress in molecular biology and translational science. Elsevier; 2012:215–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medinas DB, Gozzo FC, Santos LFA, Iglesias AH, Augusto O: A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic Biol Med 2010, 49:1046–1053. [DOI] [PubMed] [Google Scholar]
- 16.*.Workman A: Nucleation and kinetics of SOD1 aggregation in human cells for ALS1. Mol Cell Biochem 2020, 466:117–128. [DOI] [PubMed] [Google Scholar]; Cells have increased aggregation propensity with SOD1 trimers formed by superoxide exposure. The kinetic formation of SOD1 show that superoxide may initiate its radical polymerization.
- 17.*.Sala FA, Wright GSA, Antonyuk SV, Garratt RC, Hasnain SS: Molecular recognition and maturation of SOD1 by its evolutionarily destabilised cognate chaperone hCCS. PLoS Biol 2019, 17:e3000141. [DOI] [PMC free article] [PubMed] [Google Scholar]; Destabilizing mutations at the chaperone and SOD1 interface reduced homodimer affinity and icreased interaction with immarture SOD1. This work implicates copper chaperones in producing toxic SOD1 species.
- 18.Khare SD, Caplow M, Dokholyan NV: The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2004, 101:15094 LP–15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilcox KC, Zhou L, Jordon JK, Huang Y, Yu Y, Redler RL, Chen X, Caplow M, Dokholyan NV: Modifications of superoxide dismutase (sod1) in human erythrocytes a possible role in amyotrophic lateral sclerosis. J Biol Chem 2009, 284:13940–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.*.Proctor EA, Mowrey DD, Dokholyan NV: β-Methylamino-L-alanine substitution of serine in SOD1 suggests a direct role in ALS etiology. PLOS Comput Biol 2019, 15:e1007225. [DOI] [PMC free article] [PubMed] [Google Scholar]; Computational evidence of SOD1 structural destabilization promotes SOD1 aggregation and neurotoxicity.
- 21.Petrozziello T, Secondo A, Tedeschi V, Esposito A, Sisalli M, Scorziello A, Di Renzo G, Annunziato L: ApoSOD1 lacking dismutase activity neuroprotects motor neurons exposed to beta-methylamino-L-alanine through the Ca2+/Akt/ERK1/2 prosurvival pathway. Cell Death Differ 2017, 24:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur SJ, McKeown SR, Rashid S: Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016, 577:109–118. [DOI] [PubMed] [Google Scholar]
- 23.*.Chao W-C, Lu J-F, Wang J-S, Chiang T-H, Lin L-J, Lee Y-L, Chou P-T: Unveiling the structural features of nonnative trimers of human superoxide dismutase 1. Biochim Biophys Acta (BBA)-General Subj 2020, 1864:129483. [DOI] [PubMed] [Google Scholar]; Trimeric SOD1 exhibits less water molecules surrounding the site of oxidative modification. It also is a potentiator of aggregation compared to WT providing insight into how structural features of trimers in ALS cause disease phenotypes.
- 24.Srinivasan E, Rajasekaran R: Cysteine to serine conversion at 111th position renders the disaggregation and retains the stabilization of detrimental SOD1 A4V mutant against amyotrophic lateral sclerosis in human—a discrete molecular dynamics study. Cell Biochem Biophys 2018, 76:231–241. [DOI] [PubMed] [Google Scholar]
- 25.**.Bille A, Jensen KS, Mohanty S, Akke M, Irbäck A: Stability and local unfolding of SOD1 in the presence of protein crowders. J Phys Chem B 2019, 123:1920–1930. [DOI] [PubMed] [Google Scholar]; SOD1 interactions with crodwer proteins occur in a residue-specific manner. SOD1 stability is lost in apo monomeric form when in a protein crowded environment meaning that SOD1 unfolding and binding are affected by neighboring proteins.
- 26.Pokrishevsky E, Hong RH, Mackenzie IR, Cashman NR: Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS One 2017, 12:e0184384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Münch C, O’Brien J, Bertolotti A: Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci 2011, 108:3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAlary L, Plotkin SS, Yerbury JJ, Cashman N: Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci 2019, 12:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayers JI, Fromholt SE, O’Neal VM, Diamond JH, Borchelt DR: Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol 2016, 131:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dagan B, Oren O, Banerjee V, Taube R, Papo N: A hyperthermophilic protein G variant engineered via directed evolution prevents the formation of toxic SOD1 oligomers. Proteins Struct Funct Bioinforma 2019, 87:738–747. [DOI] [PubMed] [Google Scholar]
- 31.Wright GSA: Molecular and pharmacological chaperones for SOD1. Biochem Soc Trans 2020, 48:1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim Y, Park J-H, Jang J-Y, Rhim H, Kang S: Characterization and Hsp104-induced artificial clearance of familial ALS-related SOD1 aggregates. Biochem Biophys Res Commun 2013, 434:521–526. [DOI] [PubMed] [Google Scholar]
- 33.Wolozin B, Ivanov P: Stress granules and neurodegeneration. Nat Rev Neurosci 2019, 20:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandes N, Eshleman N, Buchan JR: Stress granules and ALS: a case of causation or correlation? In RNA Metabolism in Neurodegenerative Diseases. Springer; 2018:173–212. [DOI] [PubMed] [Google Scholar]
- 35.Lee D-Y, Jeon GS, Sung J-J: ALS-Linked Mutant SOD1 Associates with TIA-1 and Alters Stress Granule Dynamics. Neurochem Res 2020, doi: 10.1007/s11064-020-03137-5. [DOI] [PubMed] [Google Scholar]
- 36.Da Ros M, Deol HK, Savard A, Guo H, Meiering EM, Gibbings D: Wild-type and mutant SOD1 localizes to RNA-rich structures in cells and mice but does not bind RNA. J Neurochem 2020, [DOI] [PubMed] [Google Scholar]
- 37.Zetterström P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brännström T, Oliveberg M, Marklund SL: Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci 2007, 104:14157–14162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholson K, Murphy A, McDonnell E, Shapiro J, Simpson E, Glass J, Mitsumoto H, Forshew D, Miller R, Atassi N: Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve 2018, 57:20–24. [DOI] [PubMed] [Google Scholar]
- 39.Niedermeyer S, Murn M, Choi PJ: Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155:401–408. [DOI] [PubMed] [Google Scholar]
- 40.Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG, Consortium OB of the E: Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler 2009, 10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bensimon G, Lacomblez L, Meininger V: A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N Engl J Med 1994, 330:585–591. [DOI] [PubMed] [Google Scholar]
- 42.Rothstein JD: Edaravone: a new drug approved for ALS. Cell 2017, 171:725. [DOI] [PubMed] [Google Scholar]
- 43.Jaiswal MK: Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev 2019, 39:733–748. [DOI] [PubMed] [Google Scholar]
- 44.Fay JM, Zhu C, Proctor EA, Tao Y, Cui W, Ke H, Dokholyan NV: A phosphomimetic mutation stabilizes SOD1 and rescues cell viability in the context of an ALS-associated mutation. Structure 2016, 24:1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redler RL, Fee L, Fay JM, Caplow M, Dokholyan NV: Non-native Soluble Oligomers of Cu/Zn Superoxide Dismutase (SOD1) Contain a Conformational Epitope Linked to Cytotoxicity in Amyotrophic Lateral Sclerosis (ALS). Biochemistry 2014, 53:2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.**.Shvil N, Banerjee V, Zoltsman G, Shani T, Kahn J, Abu-Hamad S, Papo N, Engel S, Bernhagen J, Israelson A: MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: implications for familial ALS. Cell Death Dis 2018, 9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]; Macrophage-migration inhibitory factor inhibits accumulation of misfolded SOD1 and can be a therapeutic target.
- 47.Tateno M, Sadakata H, Tanaka M, Itohara S, Shin R-M, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H: Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum Mol Genet 2004, 13:2183–2196. [DOI] [PubMed] [Google Scholar]
- 48.*.Atlasi RS, Malik R, Corrales CI, Tzeplaeff L, Whitelegge JP, Cashman NR, Bitan G: Investigation of anti-SOD1 antibodies yields new structural insight into SOD1 misfolding and surprising behavior of the antibodies themselves. ACS Chem Biol 2018, 13:2794–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]; Misfolded SOD1-specific antibodies are described in this work and give insight into SOD1 misfolding.
- 49.Wang X, Zhang J, Wang Y, Feng Y, Zhang X, Sun X, Li J, Du X, Lambert MP, Yang S: Conformation-dependent single-chain variable fragment antibodies specifically recognize beta-amyloid oligomers. FEBS Lett 2009, 583:579–584. [DOI] [PubMed] [Google Scholar]
- 50.**.Dong Q, Zhu J, Liu S, Yu X, Liu R: An oligomer-specific antibody improved motor function and attenuated neuropathology in the SOD1-G93A transgenic mouse model of ALS. Int Immunopharmacol 2018, 65:413–421. [DOI] [PubMed] [Google Scholar]; W20 is a SOD1 oligomer-specific antibody and a promising therapeutic for ALS due to W20’s ability to reduce SOD1 oligomer levels in the spinal cord and brainstem as well as improing motor neuron survival and motor performance in ALS mouse models.
- 51.Petrov D, Mansfield C, Moussy A, Hermine O: ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci 2017, 9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]