

Abstract
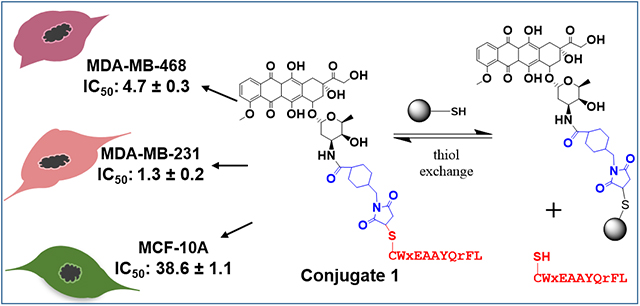
In this study, we have designed and synthesized two novel peptide-drug conjugates (PDCs) where the drug, doxorubicin (Dox), is linked to the peptide via a succinimidyl thioether bond or a hydrazone linker. A highly specific and proteolytically stable breast cancer cell targeting peptide (WxEAAYQrFL) is conjugated to Dox to synthesize peptide-Dox thioether (1) or hydrazone (2) conjugate. The evaluation of the stability in water, media, and human serum showed that the conjugate 1 with the succinimidyl thioether linkage is more stable compared to the acid-sensitive hydrazone containing conjugate 2. The cytotoxicity studies showed that the two PDCs were as toxic as free Dox toward the triple negative breast cancer (TNBC) cells and were 7–30 times less toxic (IC50 1.2–4.7 μM for TNBC cells versus 15–39 μM for non-cancerous cells) toward the non-cancerous breast cells compared to the free doxorubicin (IC50 0.35–1.5 μM for TNBC cells versus 0.24 μM for non-cancerous cells). The results from the comparative study of the two PDCs suggest that both may have translational potential for TNBC treatment.
Keywords: Triple negative breast cancer (TNBC), Breast cancer cell targeting peptide, Peptide-doxorubicin conjugate, Succinimidyl thioether linker, Conjugate stability, Cellular toxicity
Graphical Abstract
INTRODUCTION
Cancer treatment is hampered by the non-specific uptake of the chemotherapeutic agents by the peripheral tissues. Several approaches have been used to enhance the uptake of chemotherapeutic agents by the cancer cells in tumors sparing peripheral healthy cells and tissues. In this regard, targeting ligands like engineered antibodies and tumor homing peptides have gained attention as these target specific receptors on cancer cells.1–4 Targeted antibodies or peptides are conjugated to chemotherapeutic agents to provide antibody-drug conjugates (ADCs) or peptide-drug conjugates (PDCs), respectively, that deliver chemotherapeutic specifically to cancer cells.5–8 Ado-trastuzumab emtansine (Kadcyla) and brentuximab vedotin (Adcetris) are two ADCs, approved by FDA, that are used clinically for the treatment of metastatic breast cancer and refractory Hodgkin’s lymphoma, respectively.9, 10 ADCs, however, face several limitations like high manufacturing costs, issues with production methods leading to heterogenous mixtures with different number of chemotherapeutic drugs conjugated to an antibody, and poor penetration into tumor tissue.1, 2 PDCs being much smaller in size have several advantages over ADCs. PDCs can be easily synthesized to homogeneity, are economical and can penetrate deep into the tumors. Design of PDCs involves selection of a proper targeting peptide, linker that allows sufficient circulation time and is mainly cleaved at the tumor site, and a highly toxic chemotherapeutic drug.
Triple negative breast cancer (TNBC), an aggressive subtype of breast cancer, is difficult to target as this breast cancer subtype lacks expression of hormone receptors (estrogen or progesterone) as well as lacks overexpression of human epidermal growth factor receptor 2 (HER2).11, 12 Chemotherapy is the mainstay treatment for TNBC.11 Several cancer targeting peptides have been proposed for targeting overexpressed receptors in breast cancer.6, 13–18 For instance, peptide 18–4 binds keratin 1 (KRT1) and peptide GE11 binds epidermal growth factor receptor (EGFR or ErbB1) overexpressed on breast cancer cells, and both keratin 1 and EGFR are suggested to play key roles in TNBC.15, 18 The linker is an important component of the PDC and allows timely release of the drug after it is internalized. Linkers like esters, amides, disulfides, and acid-labile hydrazones have been explored that get cleaved in the intracellular tumor environment.5–7, 19–21 In addition, thioether linker has been used where the drug is released from the peptide or antibody by proteolytic degradation (intracellular) of the peptide/antibody. The succinimidyl thioether linker is gaining attention as it is present in two clinically used ADCs, Kadcyla and Adcetris.2 The succinimidyl thioether linkage shows superior in vivo efficacy compared to disulfide bonded ADCs.5 For the cytotoxic agent, it is recommended to use a highly toxic agent in the PDC with IC50 values in the subnanomolar range such as drugs like maytansine derivatives, auristatin and doxorubicin (Dox).1
In this study, we have explored two different chemistries for covalently conjugating peptide 18–4 to Dox to obtain conjugates 1 and 2 (Figure 1) to deliver Dox to TNBC specifically. In conjugate 1, Dox is conjugated to the peptide via a succinimidyl thioether linkage whereas in conjugate 2, an acid-labile hydrazone linker along with succinimidyl thioether is employed. The stability of the conjugates in different environments is studied, followed by in vitro cytotoxicity using TNBC and normal breast tissue-derived cells. The results show that conjugate 1 is more stable compared to conjugate 2 in human serum, and both conjugates display much higher cytotoxicity toward TNBC cells compared to normal breast cells.
Figure 1.
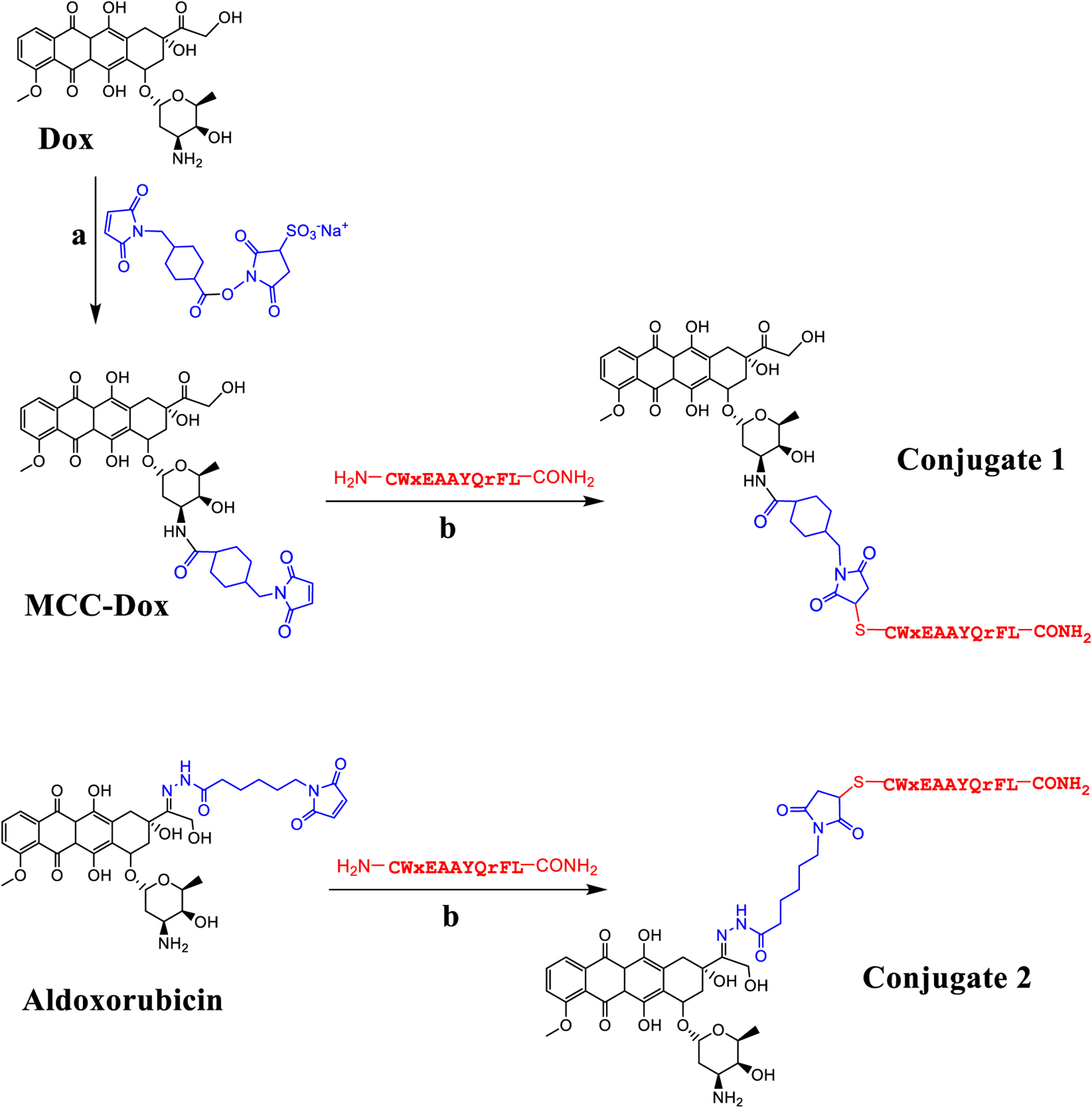
The chemical synthesis of PDCs 1 and 2 starting with Dox or aldoxorubicin, respectively. The reagents used were (a) sulfo-SMCC, DMF/water and (b) peptide (H2N-CWxEAAYQrFL-CONH2), DMF/water. Lower case r equals D-arginine and x equals D-norleucine.
RESULTS AND DISCUSSION
Synthesis and Characterization of Peptide-Dox Conjugates.
PDCs 1 and 2 were synthesized utilizing separate cross-linkers but with similar conjugation chemistries (maleimide-thiol). For conjugate 1, Dox was conjugated to a sulfo-SMCC linker, whereas for the conjugate 2, a prodrug of Dox was used where Dox was already conjugated to a linker (Figure 1). In both cases, it is the maleimide group that reacts with a thiolated peptide to give the respective conjugate. To obtain thiolated peptide, an extra cysteine residue was inserted at the N-terminal of the previously reported decapeptide 18–4. The peptide (CWxEAAYQrFL) was synthesized using standard Fmoc SPPS on Rink amide resin to give C-terminally amidated peptide, that prevented side reactions during conjugation to Dox.7, 22
Conjugate 1 was synthesized in two steps (Figure 1). In the first step, Dox was reacted with the sulfo-SMCC linker to obtain MCC-Dox. Sulfo-SMCC is a bifunctional crosslinker that allows covalent conjugation of molecules containing amines and sulfhydryls through its N-hydroxysuccinimide (NHS) ester and maleimide groups, respectively.23 Dox has a primary amine which reacts with the NHS ester of sulfo-SMCC at ~ pH 7.6 to form an amide bond. The reaction was done in aqueous DMF (DMF/water; 1:1) at r.t. and the pH was maintained by PBS. Sulfo-SMCC is a water-soluble molecule, whereas, Dox is highly lipophilic therefore a mix of DMF and water was used for solubilization. MCC-Dox that formed, was purified using RP-HPLC. Pure MCC-Dox was characterized using NMR spectroscopy (SI, Figure S1), analytical RP-HPLC and mass spectrometry (Figure S2a). It was obtained in good yield (91%) with 98% purity.
In the second step, the peptide with a free sulfhydryl group on Cys residue was reacted with the maleimide group of pure MCC-Dox to deliver the peptide-Dox conjugate 1. The crude conjugate was purified and characterized using RP-HPLC (Figure S3a) and was obtained in 63.5% yield (98% purity). The mass analysis of the pure conjugate using Q-TOF mass spectrometry showed the desired mass as a single peak (Figure S3b). The MALDI-TOF mass spectrometry showed a peak for the conjugate 1 at 2161.5 ([M+H]+ calc 2161.9) (Figure S3c). In addition, it showed a peak at 1765.1 which is most likely fragmentation of the conjugate by the acidic matrix (α-cyano-4-hydroxycinnamic acid and trifluoroacetic acid) of MALDI-TOF.
Peptide-Dox conjugate 2 was synthesized by reacting the peptide with the commercially available prodrug aldoxorubicin or (6-maleimidocaproyl)hydrazone of doxorubicin.24–26 The reaction was run in DMF/water (pH 7.4) with aldoxorubicin in 3-fold excess at 37 °C. At the end, precipitation of the crude reaction mixture with cold acetonitrile gave pure conjugate 2 in 70% yield (Figure S4). It was used as is for the stability and cell cytotoxicity studies.
Stability of Peptide-Dox Conjugate 1.
We evaluated the stability of conjugate 1 in aqueous conditions, aqueous acidic conditions and in the cell culture media. While the conjugate was mostly stable in aqueous conditions (pH 7.0) when incubated for up to 48 hours (>80% intact), the stability in the aqueous acidic conditions (pH 5) and cell culture medium was much less (Figure 2a). A solution of conjugate 1 at pH 5 or in cell culture media (DMEM/F12 containing HEPES, 100 μM) was incubated at 37 °C, and aliquots were analyzed by RP-HPLC. The HPLC peak (area under the curve) for the conjugate slowly decreased over time with a half-life of 24 hours at pH 5 and ~48 hours in media. The loss of conjugate under acidic conditions is most likely due to the hydrolysis of the acetal group present in Dox (Figure 2). This is supported by the appearance of acetal hydrolysis fragment with a mass of 1764.4 (calculated mass 1764.7) in aqueous acidic conditions (Figure S5).
Figure 2.
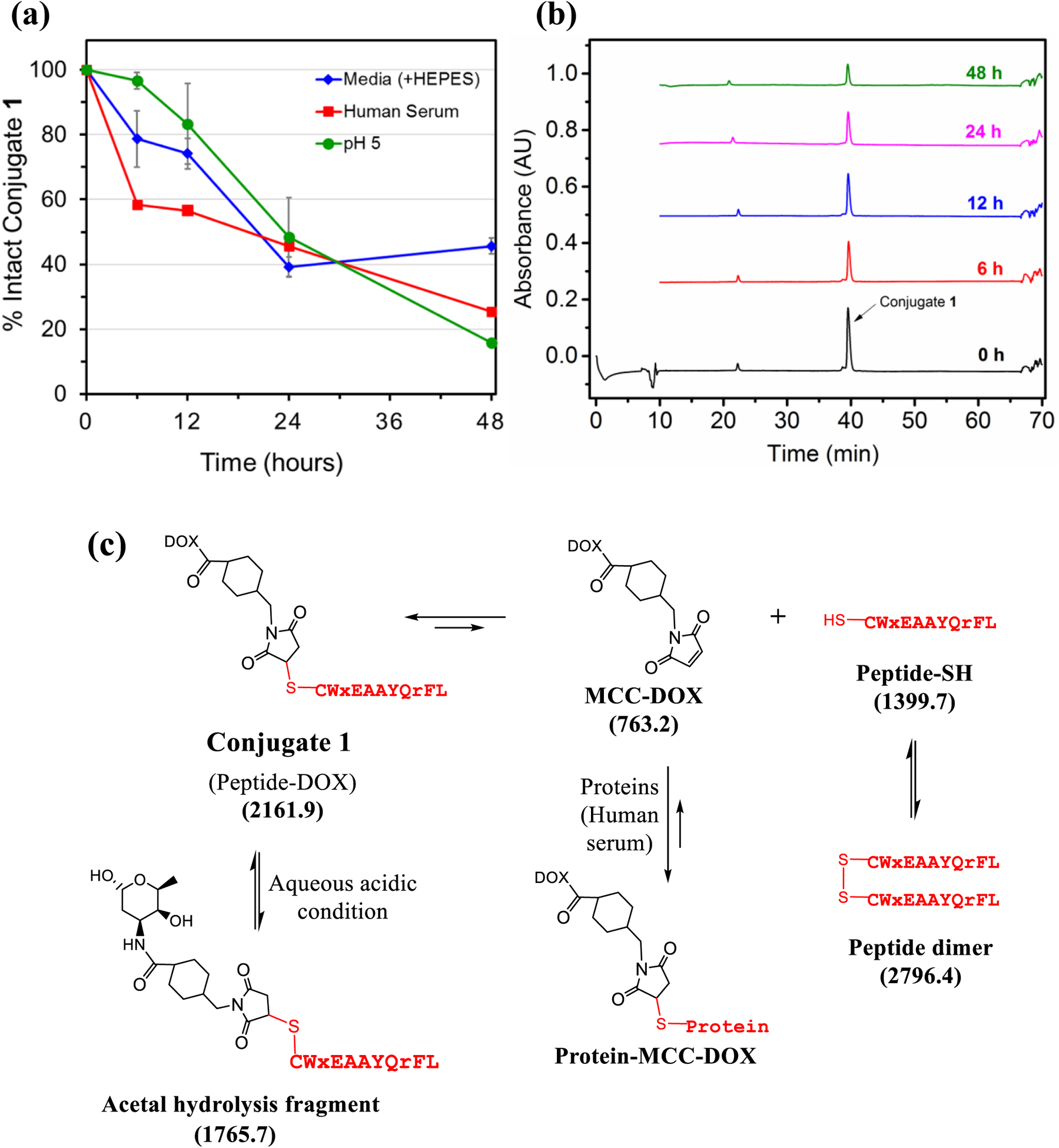
(a) The stability of conjugate 1 in aqueous solution, cell culture media, and human serum at 37 °C. Each experiment was repeated twice and error bars show standard deviation. (b) Analysis of different moieties present in solution during incubation of conjugate 1 with human serum using RP-HPLC. (c) Structures of the possible molecules/adducts when conjugate 1 is incubated in human serum (due to the reversible thiol-maleimide reaction) or in acidic conditions. The numbers in parenthesis are the calculated mass [M+H]+.
Next, the stability of conjugate 1 in human serum was conducted by incubating conjugate (100 μM) with human serum at 37 °C. Aliquots from the incubation mixture were taken at regular intervals and subjected to precipitation by adding methanol to remove serum proteins. The supernatant was analyzed by HPLC and mass spectrometry. As shown in Figures 2a and 2b, the conjugate was fairly stable in human serum (t1/2 ~ 18 hours), and 25% of the conjugate was still intact at 48 hours. The depletion of conjugate 1 under different conditions is apparent as it is a succinimidyl thioether that is produced by Michael addition reaction of a thiolate with the double bond of the MCC-Dox maleimide.27, 28 This reaction is reversible, and therefore can undergo the retro-Michael reaction to give back the free thiol and the MCC-Dox maleimide (Figure 2c). In human serum, the presence of other proteins can enhance the formation of other protein-MCC-Dox adducts and over time, the conjugate 1 is depleted. In addition, the formation of peptide dimer (disulfide formation) is also observed when the conjugate is incubated with the human serum (37 °C). Peptide dimer was observed ([M+H]+ 2797.3) in the precipitate fraction when the aliquots at 6, 12, and 24 hours were precipitated with methanol (Figure S6). The succinimidyl thioether conjugates have become popular over the past few years with the approval of ADCs like brentuximab vedotin (Adcetris) and adotrastuzumab emtansine (Kadcyla) that utilize the same chemistry.1, 2 These conjugates function well over a short period of time (1–2 days), however, may not be the best when a prolonged drug circulation is required.28 Over a long time these conjugates will undergo either thiol exchange reaction or a stabilizing succinimide ring opening reaction. Several strategies to synthesize the ring opened conjugates with the hydrolysis of the succinimide ring have been proposed.27–29
Stability of Peptide-Dox Conjugate 2.
The conjugate 2 contains an acid-sensitive hydrazone linker which is expected to release Dox in acidic conditions. A solution of conjugate at pH 5 (37 °C) was completely hydrolyzed over 24 hours (Figure 3a). MALDI-TOF analysis showed two main peaks, one for the conjugate and the other for the peptide-linker portion without the Dox ([M+H]+1 1624.0) (Figure S7a). Conjugate 2 in acetonitrile/water (−20 °C) was found to be stable for a month. The MALDI-TOF analysis showed that the conjugate was completely intact at days 1, 14 and 30 (Figure S7b). In order to evaluate the stability of conjugate 2 during the MTT cell cytotoxicity assay, the conjugate was incubated with DMEM/F12 media (± HEPES) and aliquots at various times were analyzed as before. Conjugate 2 was completely hydrolyzed in DMEM/F12 media with HEPES in <3 hours. On the other hand, conjugate 2 was stable (near 100% intact) in DMEM/F12 media without HEPES for at least 24 hours (Figure 3a). HEPES is a zwitterionic sulfonic acid buffer that, when removed from the media led to completely stable conjugate 2.
Figure 3.
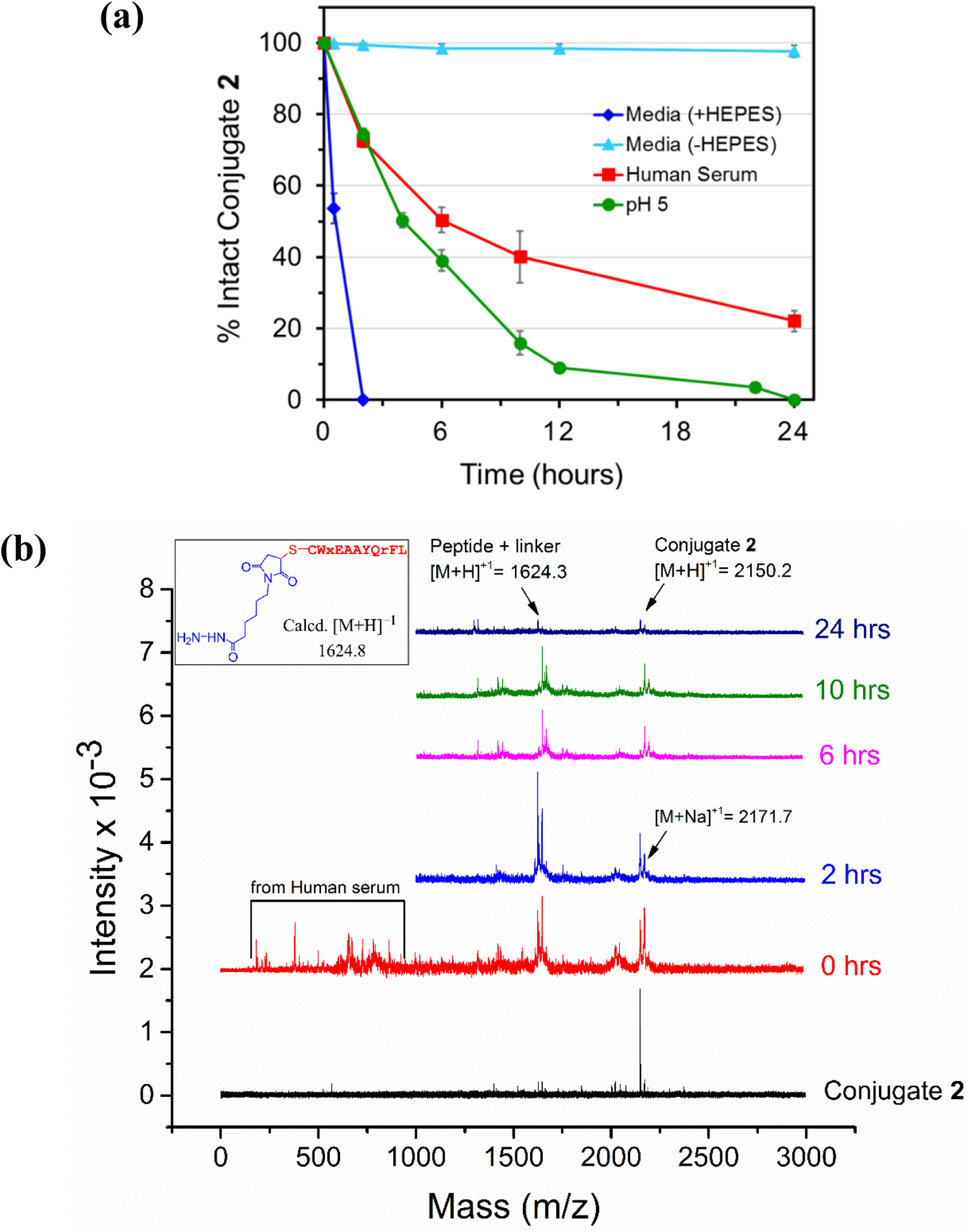
(a) The stability of conjugate 2 in aqueous acidic solution (pH 5), cell culture media (+ and − HEPES), and human serum at 37 °C. Each experiment was repeated at least once. Error bars show standard deviation. (b) Analysis of different moieties present in solution during incubation of conjugate 2 with human serum using MALDI-TOF mass spectrometry. Inset shows the structure of the peptide-linker fragment released upon acid hydrolysis of conjugate 2.
The stability of conjugate 2 in human serum (37 °C) was examined by the mass analysis of the aliquots from the incubated mixture. It showed that over time conjugate 2 hydrolyzes at the acid sensitive part (hydrazone) of the linker with an estimated half-life of 6 hours. The percent intact for conjugate 2 decreased with an increase in incubation time with about 22% intact conjugate remaining at 24 hours (Figure 3b). Although MALDI-TOF analysis showed only the peak for the hydrolysis at the acid sensitive linker portion (1624.3), it is notable that conjugate 2 is also a succinimidyl thioether conjugate like the conjugate 1. Consequently, a reversible retro-Michael reaction to give back free thiol and the prodrug aldoxorubicin which then can react with other moieties from serum is very likely. Altogether, these mechanisms explain why conjugate 1 is more stable compared to conjugate 2.
Cytotoxicity of Conjugates 1 and 2.
The main treatment for TNBC is chemotherapy.11 Therefore, to enhance the therapeutic efficacy of Dox two conjugates were prepared and their cell cytotoxicity was evaluated using two TNBC cell lines (MDA-MB-231 and MDA-MB-468) and one noncancerous mammary epithelial cell line (MCF-10A). To assess the cell viability via MTT assay and compare the IC50 values, cells in serum-free media were used during the assay to prevent early release of Dox from the conjugates (Figure 4).
Figure 4.
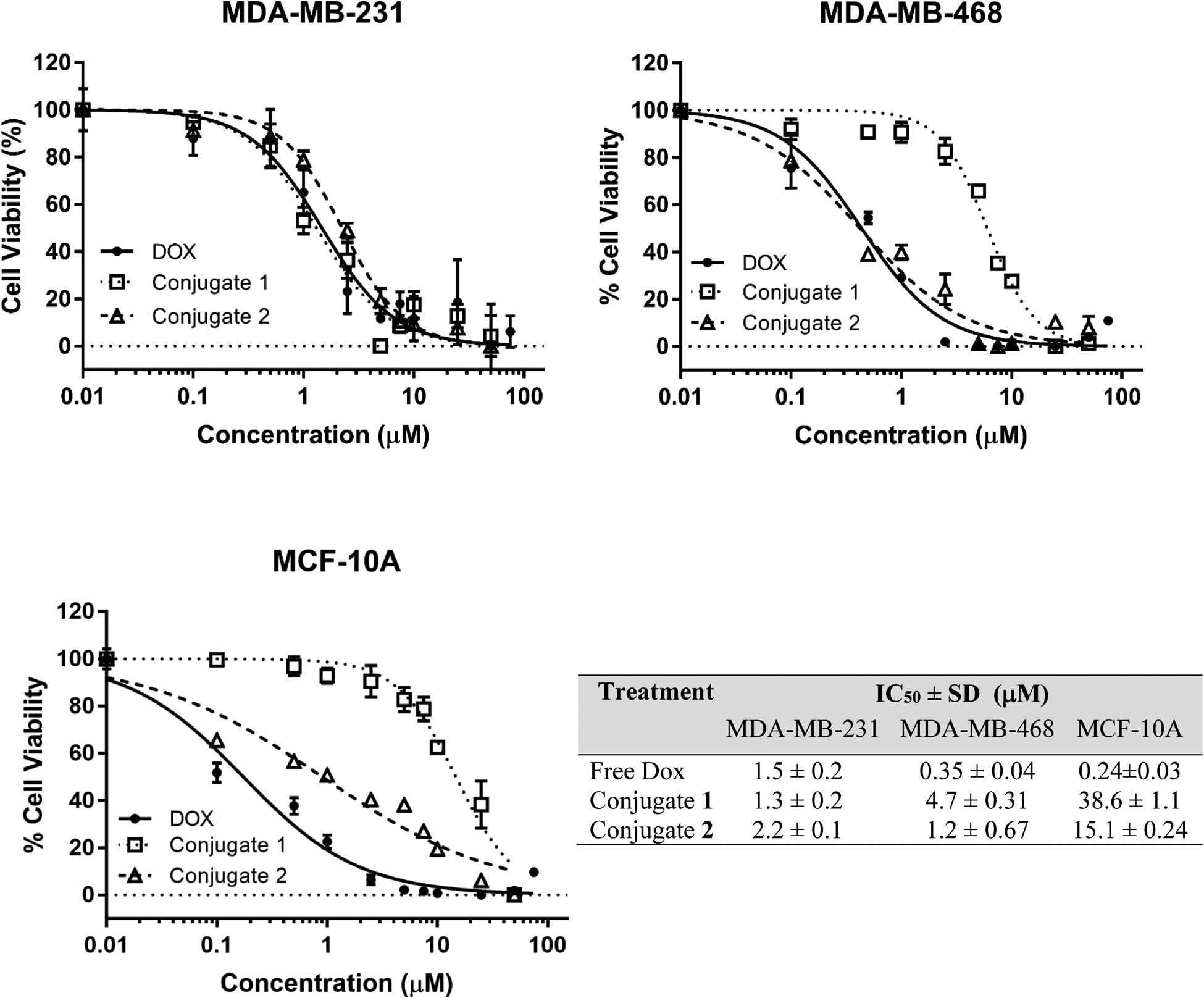
Cytotoxicity of PDCs 1 and 2 using MTT assay. The percent cell viability is plotted against conjugate or Dox (control) concentration and the data are fitted using non-linear fit of normalized data to obtain IC50 values using GraphPad Prism 7.04. The plots show a representative experiment for each cell line used, namely, MDA-MB-231, MDA-MB-468, and MCF-10A. The table lists corresponding mean IC50 values and the standard deviation for Dox, conjugate 1 and conjugate 2 for the three cell lines. Each experimental point was done in triplicates and the experiment was repeated once. The horizontal dotted line at 0% cell viability serves as a baseline.
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 μM, respectively), as well as the free Dox (IC50 = 1.5 μM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 μM) was slightly more toxic compared to conjugates 1 (4.7 μM) and 2 (1.2 μM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 μM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 μM, respectively). The conjugates were 13–30 times less toxic toward non-cancerous breast cells compared to the breast cancer cells, while free Dox showed either the same or more toxicity towards noncancerous cells compared to the breast cancer cells. The variability in toxicity of both conjugates to different cell lines could be attributed to the differential expression of keratin 1 among the cell lines, which is the target receptor for peptide 18–4.18 This, however, needs to be further verified as the surface expression levels of keratin 1 in different breast cancer cell lines is not reported yet.
CONCLUSIONS
The use of succinimidyl thioether linker for the synthesis of PDC gave conjugate 1 which has optimal characteristics such as decent stability and high selectivity for targeted delivery of Dox to TNBC cells. The cause of instability of conjugate 1 was that it is a reversible maleimide-thiol adduct which can convert back to the free thiolated peptide and maleimide-Dox overtime. For instance, when conjugate 1 was incubated with human serum, only 45% intact conjugate was left after 24 hours (Figure 2). While conjugate 2 also contains the same thioether linkage as conjugate 1, its instability was pronounced due to the acid-labile hydrazone linker (Figure 3). The peptide 18–4 (WxEAAYQrFL) component of the conjugate is stable in human serum as the peptide has been engineered to be proteolytically stable by exchange of the labile amino acids with unnatural d-amino acids.22 Furthermore, the targeting capability of the peptide 18–4, and the uptake of the peptide and peptide-Dox amide/ester conjugates via specific receptor-mediated endocytosis in breast cancer cells is reported previously.7, 18, 22 Peptide 18–4 binds keratin 1 on the breast cancer cell surface18 and keratin 1 portrays an important role in breast30 and other cancers or disease conditions.31–34 The purpose here was to develop linker chemistries for the conjugation of peptide to Dox to obtain PDCs with superior in vitro and in vivo efficacy. The maleimide-thiol reaction which forms succinimidyl thioether linker has been used previously for FDA approved ADCs,1 and with the promising in vitro results presented here with the new PDCs (1 and 2) that use similar linker chemistry, we conjecture that these conjugates will lead to improved efficacy and lesser side effects for TNBC treatment. The comparative in vitro study of the two novel PDCs that use different linker chemistries suggest that they may serve as potential anticancer agents, especially for TNBC treatment. The in vivo efficacy experiments with conjugates 1 and 2 are currently in progress using MDA-MB-231 subcutaneous xenograft in NOD-SCID mice.
EXPERIMENTAL SECTION
Materials.
Doxorubicin hydrochloride (Dox.HCl) salt and aldoxorubicin were bought from LC Laboratories (MA, USA) and MedChem express (NJ, USA), respectively. Fmoc-Leu TentaGel S RAM resin (loading 0.21 mmol/g) was bought from Rapp Polymere GmbH (Germany). The coupling agent 2-(6-chloro-1H-benzotriazole-1-yl)- 1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU), and the Fmoc-amino acids with the following side chain protections: tert-butyl in tyrosine, tert-butoxy in glutamic acid, trityl (Trt) in glutamine and cysteine, tert-butoxycarbonyl (Boc) in lysine and tryptophan, and pentamethyl dihydrobenzofuran (Pbf) in d-arginine were purchased from Fischer Scientific (IL, USA). N, N-dimethylformamide (DMF), N-methylmorpholine (NMM), trifluoroacetic acid (TFA), triisoproylsilane (TIS), piperidine, and all other reagents were bought from Sigma-Aldrich. Sulfo SMCC (sulfosuccinimidyl 4-[N-maleimidomethyl] cyclohexane-1-carboxylate) was purchased from Thermofisher Scientific (NY, USA). α-Cyano-4-hydroxycinnamic acid (CHCA) matrix was bought from Fluka. Ultra-pure water was from Milli-Q system. MTT (98%) and IGEPAL®CA-630 (or NP40) were purchased from Sigma-Aldrich.
Human breast cancer cell lines (MDA-MB-231 and MDA-MB-468) and normal breast cell line (MCF-10A) were purchased from ATCC (USA). All cells were maintained in a 5% CO2 incubator (37 °C). MDA-MB-231 was cultured in DMEM/F-12 (cat # 11330–032, Corning, Virginia, USA), 100 IU/mL penicillin, and 100 IU/mL streptomycin supplemented with FBS, whereas, MDA-MB-468 was cultured in DMEM (1x) + GlutaMAX™-l media (10567–014, Gibco) containing 100 IU/mL penicillin, and 100 IU/mL streptomycin supplemented with FBS. MCF-10A was cultured in DMEM/F-12 with HEPES (cat # 10–092-CV) and without HEPES (cat # 10-090-CV, Corning, Virginia, USA), 100 IU/mL penicillin, and 100 IU/mL streptomycin supplemented with horse serum. Human serum was purchased from Sigma-Aldrich.
Peptide synthesis was done employing an automated peptide synthesizer Tribute from Protein Technologies (Protein Technology, Inc., Arizona, USA). Purification and HPLC analysis were conducted on a RP-HPLC system Prominence-i (Shimadzu Corp., Kyoto, Japan) using C18 semi-preparative (10 mm × 250 mm, 5 μm) and analytical (4.6 mm × 250 mm, 5 μm) columns. For mass spectra, either an autoflex speed MALDI-TOF mass spectrometer (Bruker, USA) or EVOQ Triple Quadrupole LC-TQ Mass Spectrometer (Bruker, USA) was used. UV-Vis spectrophotometer (UV-2600, Shimadzu) was used to obtain an absorption spectrum. NMR experiments were conducted on an Ascend 400 MHz NMR spectrometer (Bruker BioSpin Corporation, Billerica, Massachusetts, USA).
Peptide Synthesis.
We synthesized the 11-mer peptide (NH2-CWxEAAYQrFL-CONH2) on Rink amide resin (0.1 mmol scale) pre-loaded with Fmoc-Leu residue (Fmoc-Leu Rink amide resin, loading 0.21 mmol/g) using automated SPPS.17, 22, 35 The activation and coupling at each step were carried out for 2 hours using HCTU and NMM in DMF. After each coupling step, Fmoc deprotection was done using piperidine/DMF (2:8). After complete assembly of the peptide sequence, the peptide was released from the resin by treating the resin with a cleavage mixture of TFA/TIS/water (10 mL, 90:5:5) for 2 h.17, 22 Crude peptide was precipitated by adding diethyl ether (20 mL, chilled) to the filtered TFA cocktail which was then collected by centrifugation (10 min). The crude peptide was characterized using MALDI-TOF and RP-HPLC. The purity of the peptide was assessed using an analytical C18 column. The peptide eluted with 35% acetonitrile at 31 min (tR) (method used: 10–100% with 0.05% TFA, flow rate = 1 mL/min, 100 min run time at 220 nm) and was found to be ~99% pure (Figure S2b). MALDI-TOF [M+H]+ found 1399.2 (calc. 1399.7).
Synthesis of Peptide-Dox Conjugate 1.
The synthesis of conjugate 1 involved two steps, Dox conjugation to sulfo-SMCC linker to give MCC-Dox and MCC-Dox reaction with the peptide. Briefly, to a solution of sulfo-SMCC (5 mg, 11.5 μmol) in 1.2 mL of DMF, water, and PBS (100 mM, pH 7.6) at ratio 1:1:0.4, Dox-HCl (4.3 mg, 7.7 μmol) in DMF/water (1 mL, 1:1) was added. The reaction mixture was stirred (r.t.) under nitrogen for 4 h. The reaction was monitored every hour using mass spectrometry and it was determined all free Dox was used up after 4 hours. The purification of the resulting crude product (MCC-Dox) was carried out using a semi-preparative RP-HPLC (method used: 10–70% acetonitrile/water with 0.05% TFA at 1 mL/min in100 min). MCC-Dox peak that eluted at 36 min (tR) with 48% acetonitrile in RP-HPLC was collected and combined and dried under a rotary evaporator to obtain pure MCC-Dox as a red powder with 91% (5.3 mg, 98% purity) yield. Pure MCC-Dox was characterized using 1H and 13C NMR spectroscopy (Figure S1), MALDI-TOF mass spectrometry and analytical HPLC (Figure S2a). For NMR, MCC-Dox was dissolved in 100% deuterium oxide (Cambridge Isotope Laboratories Inc., Massachusetts, USA). 1D (1H and 13C) and 2D (1H-13C-HSQC and HMBC) NMR experiments were run at 25 °C. The spectra were analyzed using Topspin 3.7 software (Bruker BioSpin Corporation, Billerica, Massachusetts, USA). Complete chemical shift assignments (1H and 13C) were made using the HSQC and HMBC experiments. MALDI-TOF [M+Na]+ calc. 785.2, found 785.4.
A solution of peptide (1.1 mg, 0.78 μmol) in DMF (0.25 mL) was added to the MCC-Dox (0.8 mg, 1 μmol) in DMF (0.5 mL), and additional DMF (1.25 mL) was added. Finally, a catalytic amount of DIPEA (1 wt%) was added, and the reaction mixture was stirred at r.t. under nitrogen. The reaction progress was monitored using RP-HPLC, and after 4 hours the conjugate formation halted. The DMF was removed by rotary evaporator and the mixture was diluted with acetonitrile/water and injected into RP-HPLC for purification. The conjugate eluted at 27 min (38% acetonitrile) using RP-HPLC (30–45% acetonitrile/water containing 0.05% TFA, 1 mL/min flow rate, 65 min run time, tR= 27 min) to give pure peptide-Dox conjugate 1 as TFA salt with 63.5% yield and 98% purity (Figure S3). Q-TOF found 1081.43 charge +2 therefore unprotonated mass found (1081.43–1)*2 = 2160.86, calc. mass = 2160.9; MALDI-TOF [M+H]+ found 2161.5, calc. 2161.9. Until used, the conjugate was stored as a dry powder at −20 °C.
Synthesis of Peptide-Dox Conjugate 2.
Peptide (1.68 mg, 1.2 μmol) dissolved in DMF and PBS (pH 7.4) (1:9, v/v, 0.6 mL) was added to aldoxorubicin (2.7 mg, 3.6 μmol, 3 equiv.) in DMF/water (0.6 mL, 1:1), and the reaction mixture was left stirring for 3 h at 37 °C. The progress of the reaction was monitored by RP-HPLC. After about 3 hours, the major peak observed with the HPLC was the conjugate eluting at 34 min (39% acetonitrile) and no peak for the peptide was observed suggesting that all the peptide was consumed. The conjugate was collected as a precipitate by adding cold acetonitrile (6 mL) to the reaction mixture.36 The precipitate was dried and stored at −20 °C. A small amount of precipitate (product) was dissolved in 10% acetonitrile in water and analyzed using analytical RP-HPLC (method used: acetonitrile/water 10–50% with 0.01% TFA and flow rate 1 mL/min at 495 nm, absorbance maxima for aldoxorubicin). The conjugate 2 eluted at 34 min (tR) and was found to be 98% pure with a yield of 70% (1.81 mg) (Figure S4). MALDI-TOF [M+H]+ calc. 2149.7, found 2149.9. Conjugate 2 has an acid-sensitive hydrazone linker, therefore, it was not subjected to HPLC purification.
Stability of the Conjugates in Aqueous and Aqueous Acidic Conditions.
The stability of the conjugates 1 or 2 in aqueous solution was assessed by dissolving the pure conjugates (100 μM) in water and acetonitrile (9:1, v/v, pH 7.0) and keeping at 4 °C or −20 °C, respectively. At different time intervals aliquots were taken and injected into RP-HPLC. The stability of the conjugate was monitored using the peak (area under the curve) from HPLC and the MALDI-TOF mass analysis of the HPLC peak. The stability was also evaluated in acidic aqueous media at ~ pH 5. A solution of the conjugate (1 or 2) in PBS pH 5 (100 μM) was incubated at 37 °C, and aliquots removed over time were examined with MALDI-TOF mass spectrometry. Stock solution concentrations for conjugates 1 and 2 were obtained using Quick drop spectrophotometer at 481 nm (ε = 10410 L mol−1 cm−1)37 and 495 nm (ε = 9250 L mol−1 cm−1)38 wavelengths, respectively.
Stability of the Conjugates in Cell Culture Media.
Conjugate 1 or 2 was dissolved in DMEM/F12 (+ HEPES) media and the resulting solution (100 μM) was incubated at 37 °C for up to 48 hours. Aliquots, removed at different time intervals, were assessed using mass spectrometry analysis. Following a similar procedure, the stability of conjugate 2 was also evaluated in DMEM/F12 media (− HEPES).
Stability of the Conjugates in Human Serum.
A stock solution of conjugate 1 at 1 mM concentration was prepared in sterile water. Human serum was thawed at r.t. Human serum (250 μL) was diluted by adding sterile water (650 μL) and conjugate stock solution (100 μL) to give a sample with 25% human serum and 100 μM conjugate.7 The sample was kept at 37°C in an incubator to imitate human body temperature. At regular intervals, aliquots (100 μL) were taken and diluted with methanol (200 μL). After centrifugation (500 g) for 15 min, the supernatant was analyzed by HPLC at 495 nm, and the major HPLC peaks were analyzed by mass spectrometry. The mobile phase used was acetonitrile/water with 0.05% TFA with a gradient of 30–55% with a flow rate of 1 mL/min in 100 min. The intact conjugate (relative percentage) was plotted against incubation time. HPLC and MALDI-TOF mass analysis were used to confirm degradation products, and t1/2 was calculated as the time needed to hydrolyze half of the initial conjugate in human serum.
The percent intact for conjugate 2 in human serum was monitored with MALDI-TOF MS only as this conjugate is not stable in acidic conditions of the RP-HPLC (solvents). Conjugate 2 was incubated with human serum at the same concentrations as used above for conjugate 1. Aliquots (20 μL) were added to methanol (40 μL) to quench the reaction at different times (0 – 24). The conjugate 2 precipitated along with human serum proteins, therefore the precipitate was used to analyze the percent intact for conjugate 2. The relative percent intact for conjugate 2 was plotted against time in hours.
In Vitro Cytotoxicity.
The cellular toxicity of the conjugates and free Dox were assessed for three cell lines (MDA-MB-231, MDA-MB-468, and MCF-10A) using MTT assay. After seeding the cells in 96-well microtiter plates (5×103 cells/well) and growing in complete media, the next day the media was substituted with fresh serum-free media (200 μL) containing different concentrations of the Dox or conjugates (0–100 μM). It was incubated for 48 h at 37 °C. The negative and positive controls used were cells in serum-free medium (without any drug) and cells with Dox, respectively. Cell survival assay was performed by adding MTT solution (20 μL, 5 mg/mL) to each well and the plates were further incubated for 2 h. A sterile MTT solution was prepared by dissolving MTT (60 mg) in PBS 1X (12 mL) and the resulting solution was filtered using EZFlow syringe filter (diameter 13 mm, pore size 0.22 μm) with Foxx CA membrane (Foxx Life Sciences, Salem, NH, USA). To solubilize the formazan pellets that formed after adding MTT solution to the wells, a solubilizing solution (4 mM HCl, 0.1% NP40, 2-propanol, 1:1:1, v/v/v) was added to each well (100 μL/well). The plate was placed on a shaker for 10 mins and was read using plate reader (SpectraMax M5 UV VIZ plate reader) at 570 nm. The percent cell viability was calculated for each well by taking the absorbance ratio of treated cells to untreated cells and the average of the triplicates was plotted. The percent cell viability was plotted against the log drug (Dox or conjugate) concentrations and fitted using a non-linear fit of normalized data to obtain IC50 values using GraphPad Prism 7.04.
Supplementary Material
ACKNOWLEDGEMENT
The National Cancer Institute of the National Institutes of Health (Award Number R15CA208656) supported this research. The core labs at the Chapman University School of Pharmacy are also acknowledged. The authors thank Basir Syed, Hanieh Hossein-Nejad-Ariani, Shirley Fong, and Mona Alas for assistance with the techniques used in this study.
ABBREVIATIONS
- ADC
antibody-drug conjugate
- Boc
tert-butoxycarbonyl
- CHCA
α-Cyano-4-hydroxycinnamic acid
- DIPEA
N,N-diisopropylethylamine
- DMF
N, N,-dimethylformamide
- Dox
doxorubicin
- EGFR
epidermal growth factor receptor
- HCTU
2-(6-chloro-1H-benzotriazole-1-yl)- 1,1,3,3-tetramethylaminium hexafluorophosphate
- HMBC
heteronuclear multiple bond correlation
- HSQC
heteronuclear single quantum coherence spectroscopy
- KRT1
keratin 1
- MALDI-TOF MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NHS
N-hydroxy succinimide
- NMM
N-methylmorpholine
- Pbf
pentamethyl dihydrobenzofuran
- PDC
peptide-drug conjugate
- r
d-arginine
- RP-HPLC
reversed-phase high performance liquid chromatography
- r.t.
room temperature
- SPPS
solid-phase peptide synthesis
- Sulfo-SMCC
(sulfosuccinimidyl 4-[N-maleimidomethyl] cyclohexane-1-carboxylate)
- TFA
trifluoroacetic acid
- TIS
triisoproylsilane
- TNBC
triple negative breast cancer
- Trt
trityl
- x
d-norleucine
Footnotes
Supporting Information. Figures S1-S7 with the characterization of MCC-Dox, peptide, PDC 1 and PDC 2 as well as stability experiments for the two conjugates. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- [1].Thomas A, Teicher BA, and Hassan R (2016) Antibody-drug conjugates for cancer therapy, Lancet Oncol 17, e254–e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trail PA, Dubowchik GM, and Lowinger TB (2018) Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol. Ther 181, 126–142. [DOI] [PubMed] [Google Scholar]
- [3].Vrettos EI, Mezo G, and Tzakos AG (2018) On the design principles of peptide-drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem 14, 930–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang Y, Cheetham AG, Angacian G, Su H, Xie L, and Cui H (2017) Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev 110–111, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RV, Lutz RJ, et al. (2008) Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290. [DOI] [PubMed] [Google Scholar]
- [6].Salem AF, Wang S, Billet S, Chen JF, Udompholkul P, Gambini L, Baggio C, Tseng HR, Posadas EM, Bhowmick NA, and Pellecchia M (2018) Reduction of Circulating Cancer Cells and Metastases in Breast-Cancer Models by a Potent EphA2-Agonistic Peptide-Drug Conjugate. J. Med. Chem 61, 2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Soudy R, Chen C, and Kaur K (2013) Novel peptide-doxorubucin conjugates for targeting breast cancer cells including the multidrug resistant cells. J. Med. Chem 56, 7564–7573. [DOI] [PubMed] [Google Scholar]
- [8].Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, Qiu Q, Wu R, Hong E, Bogalhas M, et al. (2017) Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug. Chem 28, 1371–1381. [DOI] [PubMed] [Google Scholar]
- [9].Peddi PF, and Hurvitz SA (2013) Trastuzumab emtansine: the first targeted chemotherapy for treatment of breast cancer. Future Oncol. 9, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Senter PD, and Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol 30, 631–637. [DOI] [PubMed] [Google Scholar]
- [11].Irvin WJ Jr., and Carey LA (2008) What is triple-negative breast cancer? Eur. J. Cancer 44, 2799–2805. [DOI] [PubMed] [Google Scholar]
- [12].Parise C, and Caggiano V (2018) The influence of marital status and race/ethnicity on risk of mortality for triple negative breast cancer. PLoS One 13, e0196134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arap W, Pasqualini R, and Ruoslahti E (1998) Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279, 377–380. [DOI] [PubMed] [Google Scholar]
- [14].Askoxylakis V, Zitzmann S, Mier W, Graham K, Kramer S, von Wegner F, Fink RH, Schwab M, Eisenhut M, and Haberkorn U (2005) Preclinical evaluation of the breast cancer cell-binding peptide, p160. Clin. Cancer Res 11, 6705–6712. [DOI] [PubMed] [Google Scholar]
- [15].Hossein-Nejad-Ariani H, Althagafi E, and Kaur K (2019) Small Peptide Ligands for Targeting EGFR in Triple Negative Breast Cancer Cells. Sci. Rep 9, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hu D, Mezghrani O, Zhang L, Chen Y, Ke X, and Ci T (2016) GE11 peptide modified and reduction-responsive hyaluronic acid-based nanoparticles induced higher efficacy of doxorubicin for breast carcinoma therapy. Int. J. Nanomedicine 11, 5125–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Raghuwanshi Y, Etayash H, Soudy R, Paiva I, Lavasanifar A, and Kaur K (2017) Proteolytically Stable Cyclic Decapeptide for Breast Cancer Cell Targeting. J. Med. Chem 60, 4893–4903. [DOI] [PubMed] [Google Scholar]
- [18].Soudy R, Etayash H, Bahadorani K, Lavasanifar A, and Kaur K (2017) Breast Cancer Targeting Peptide Binds Keratin 1: A New Molecular Marker for Targeted Drug Delivery to Breast Cancer. Mol. Pharm 14, 593–604. [DOI] [PubMed] [Google Scholar]
- [19].Fan M, Yang D, Liang X, Ao J, Li Z, Wang H, and Shi B (2015) Design and biological activity of epidermal growth factor receptor-targeted peptide doxorubicin conjugate. Biomed. Pharmacother 70, 268–273. [DOI] [PubMed] [Google Scholar]
- [20].Krasnovskaya OO, Malinnikov VM, Dashkova NS, Gerasimov VM, Grishina IV, Kireev II, Lavrushkina SV, Panchenko PA, Zakharko MA, Ignatov PA, et al. (2019) Thiourea Modified Doxorubicin: A Perspective pH-Sensitive Prodrug, Bioconjug. Chem 30, 741–750. [DOI] [PubMed] [Google Scholar]
- [21].Mollaev M, Gorokhovets N, Nikolskaya E, Faustova M, Zabolotsky A, Zhunina O, Sokol M, Zamulaeva I, Severin E, and Yabbarov N (2019) Type of pH sensitive linker reveals different time-dependent intracellular localization, in vitro and in vivo efficiency in alpha-fetoprotein receptor targeted doxorubicin conjugate. Int. J. Pharm 559, 138–146. [DOI] [PubMed] [Google Scholar]
- [22].Soudy R, Gill A, Sprules T, Lavasanifar A, and Kaur K (2011) Proteolytically stable cancer targeting peptides with high affinity for breast cancer cells. J. Med. Chem 54, 7523–7534. [DOI] [PubMed] [Google Scholar]
- [23].Bae S, Ma K, Kim TH, Lee ES, Oh KT, Park ES, Lee KC, and Youn YS (2012) Doxorubicin-loaded human serum albumin nanoparticles surface-modified with TNF-related apoptosis-inducing ligand and transferrin for targeting multiple tumor types. Biomaterials 33, 1536–1546. [DOI] [PubMed] [Google Scholar]
- [24].Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, and Kratz F (2010) INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest. New Drugs 28, 14–19. [DOI] [PubMed] [Google Scholar]
- [25].Kratz F (2007) DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin. Investig. Drugs 16, 855–866. [DOI] [PubMed] [Google Scholar]
- [26].Kruger M, Beyer U, Schumacher P, Unger C, Zahn H, and Kratz F (1997) Synthesis and Stability of Four Maleimide Derivatives of the Anticancer Drug Doxorubicin for the Preparation of Chemoimmunoconjugates. Chem. Pharm. Bull 45, 399–401. [Google Scholar]
- [27].Fontaine SD, Reid R, Robinson L, Ashley GW, and Santi DV (2015) Long-term stabilization of maleimide-thiol conjugates. Bioconjug. Chem 26, 145–152. [DOI] [PubMed] [Google Scholar]
- [28].Lyon RP, Setter JR, Bovee TD, Doronina SO, Hunter JH, Anderson ME, Balasubramanian CL, Duniho SM, Leiske CI, Li F, and Senter PD (2014) Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol 32, 1059–1062. [DOI] [PubMed] [Google Scholar]
- [29].Tumey LN, Charati M, He T, Sousa E, Ma D, Han X, Clark T, Casavant J, Loganzo F, Barletta F, Lucas J, and Graziani EI (2014) Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjug. Chem 25, 1871–1880. [DOI] [PubMed] [Google Scholar]
- [30].DeAngelis JT, Li Y, Mitchell N, Wilson L, Kim H, and Tollefsbol TO (2011) 2D difference gel electrophoresis analysis of different time points during the course of neoplastic transformation of human mammary epithelial cells. J. Proteome Res 10, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chuang NN, and Huang CC (2007) Interaction of integrin beta1 with cytokeratin 1 in neuroblastoma NMB7 cells. Biochem. Soc. Trans 35, 1292. [DOI] [PubMed] [Google Scholar]
- [32].Collard CD, Montalto MC, Reenstra WR, Buras JA, and Stahl GL (2001) Endothelial oxidative stress activates the lectin complement pathway: role of cytokeratin 1. Am. J. Pathol 159, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tang S, Huang W, Zhong M, Yin L, Jiang H, Hou S, Gan P, and Yuan Y (2012) Identification Keratin 1 as a cDDP-resistant protein in nasopharyngeal carcinoma cell lines. J. Proteomics 75, 2352–2360. [DOI] [PubMed] [Google Scholar]
- [34].Toivola DM, Boor P, Alam C, and Strnad P (2015) Keratins in health and disease. Curr. Opin. Cell Biol 32, 73–81. [DOI] [PubMed] [Google Scholar]
- [35].Ahmed S, Mathews AS, Byeon N, Lavasanifar A, and Kaur K (2010) Peptide arrays for screening cancer specific peptides. Anal. Chem 82, 7533–7541. [DOI] [PubMed] [Google Scholar]
- [36].Sheng Y, You Y, and Chen Y (2016) Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int. J. Pharm 512, 1–13. [DOI] [PubMed] [Google Scholar]
- [37].Yabbarov NG, Posypanova GA, Vorontsov EA, Popova ON, and Severin ES (2013) Targeted delivery of doxorubicin: drug delivery system based on PAMAM dendrimers. Biochemistry (Mosc) 78, 884–894. [DOI] [PubMed] [Google Scholar]
- [38].Moktan S, Perkins E, Kratz F, and Raucher D (2012) Thermal targeting of an acid-sensitive doxorubicin conjugate of elastin-like polypeptide enhances the therapeutic efficacy compared with the parent compound in vivo. Mol. Cancer Ther 11, 1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.