

Abstract
The QT interval, a global index of ventricular repolarization, varies among individuals and is influenced by diverse physiologic and pathophysiologic stimuli such as gender, age, heart rate, electrolyte concentrations, concomitant cardiac disease, and other diseases such as diabetes. Many drugs produce a small but reproducible effect on QT interval but in rare instances this is exaggerated and marked QT prolongation can provoke the polymorphic ventricular tachycardia ‘torsades de pointes’, which can cause syncope or sudden cardiac death. The generally accepted common mechanism whereby drugs prolong QT is block of a key repolarizing potassium current in heart, IKr, generated by expression of KCNH2, also known as HERG. Thus, evaluation of the potential that a new drug entity may cause torsades de pointes has relied on exposure of normal volunteers or patients to drug at usual and high concentrations, and on assessment of IKr block in vitro. More recent work, focusing on anticancer drugs with QT prolonging liability, is defining new pathways whereby drugs can prolong QT. Notably, the in vitro effects of some tyrosine kinase inhibitors to prolong cardiac action potentials (the cellular correlate of QT) can be rescued by intracellular phosphatidylinositol 3,4,5-trisphosphate, the downstream effector of phosphoinositide 3-kinase. This finding supports a role for inhibition of this enzyme, either directly or by inhibition of upstream kinases, to prolong QT through mechanisms that are being worked out, but include enhanced inward ‘late’ sodium current during the plateau of the action potential. The definition of non-IKr-dependent pathways to QT prolongation will be important for assessing risk, not only with anticancer therapies but also with other QT prolonging drugs and for generating a refined understanding how variable activity of intracellular signalling systems can modulate QT and associated arrhythmia risk.
Keywords: Cardio-oncology, QT Prolongation, Arrhythmia, Torsades de pointes, PI3 Kinase
This article is part of the Spotlight Issue on Cardio-oncology.
1. Introduction
Many drugs prolong the QT interval on the electrocardiogram a small amount but in some patients this prolongation can be exaggerated and provoke the morphologically distinctive polymorphic ventricular tachycardia, ‘torsades de pointes’ (TdP; Figure 1). Symptoms associated with TdP include syncope and sudden cardiac death (SCD) if the arrhythmia is prolonged or degenerates to ventricular fibrillation; exaggerated QT prolongation and TdP constitute the syndrome of drug-induced long QT syndrome (diLQTS). TdP is also characteristic of a rare set of familial arrhythmia syndromes, collectively termed the congenital long QT syndromes (cLQTS), in which marked QT interval prolongation is associated with a risk of TdP and SCD. The congenital and drug-induced forms of LQTS share clinical features and likely arise from similar mechanisms.
Figure 1.

(A) Two rhythm strips obtained 5 min apart in a 25-year-old woman with sepsis, hypokalaemia, and therapy with ondansetron and a fluoroquinolone. The top rhythm strip shows ventricular bigeminy with a remarkable U wave (orange arrow) and prolonged repolarization. When the U wave amplitude is large, it is included in the QT measurement; in this case, the end of the QT-U complex is not seen and the QT-U exceeds 600 ms. The bottom strip shows a post-ectopic pause (red star), a sinus beat with a long QT-U, and an episode of the polymorphic ventricular tachycardia torsades de pointes (black line). QT interval was entirely normal without U waves after recovery. (B) Rhythm strip in a 60-year-old woman with Philadelphia chromosome positive acute leukaemia being treated with dasatinib and who developed cytokine storm when blinatumomab was added. The pauses, U wave, and TdP are indicated as above. QT interval was entirely normal pre-dasatinib, and prolonged to 500 ms with drug.
2. A brief history
Jervell and Lange-Nielsen described the first cLQTS in the late 1950s as an autosomal recessive condition, associated with marked QT interval prolongation, congenital sensorineural deafness, and a high risk of SCD in childhood. Subsequently, autosomal dominant forms of the disease, termed the ‘Romano-Ward’ syndrome were described in the early 1960s. Mutations have now been described in over a dozen cLQTS disease genes1 and in the vast majority of instances the disease is autosomal dominant and there are no extracardiac features. Occasional subtypes have been associated with neuromuscular symptoms or with syndactyly and neurodevelopmental delay, but these are rare and will not be discussed further here. Further, the frequency of polymorphisms in long QT (and all other) genes ranges from very common to ultra-rare; the latter are termed mutations when clearly associated with a disease phenotype, but the role of other polymorphisms as contributors to or modifiers of the long QT phenotype is only now being explored.
2.1 Risk factors for diLQTS
The idea that drugs could provoke polymorphic ventricular tachycardia was first reported in the 1960s and the term torsades de pointes was coined to describe a polymorphic ventricular tachycardia occurring in the setting of advanced atrioventricular block in an elderly woman (who, in fact, was not receiving any QT prolonging drugs). The repolarization time of the sinus beat immediately preceding an episode of TdP is generally >500 ms, and often includes a prominent U wave (Figure 1). Heart rate modulates QT interval, so a variety of formulae have been proposed to ‘rate-correct’ QT to allow comparisons [e.g. baseline vs. on drug electrocardiogram (ECGs)] obtained at different rates. However, such comparisons may be problematic for several reasons. First, if a drug has important effects on both heart rate and QT (e.g. if a drug has autonomic effects that can influence both), dissecting QT effects alone may be difficult. Second, robust methods are not in place to measure rate-corrected QT in the presence of certain conditions, notably bundle branch block or atrial fibrillation (AF). Thus, comparisons between baseline and on-drug ECGs cannot be viewed as reliable, especially if these confounders are present on one tracing but not another. Third, TdP occurs when the absolute value of the QT (not the rate-corrected value) is long (Figure 1).
Isolated cases, and subsequently small case series, defined the clinical features of diLQTS.2 Well-recognized risk factors include female gender, hypokalaemia and hypomagnesaemia, and heart rate slowing, which is most often due to a post-ectopic pause or underlying bradyarrhythmia (see Figure 1), just prior to the onset of the arrhythmia, and certain types of heart disease such as left ventricular hypertrophy or heart failure. Other clinical features that have been associated with high risk include the several hours following cardioversion of AF,3 advanced age,4 inflammatory states,5 auto-immune conditions associated with antibodies that appear to have QT prolonging activity,6 and testosterone deficiency often generated by prostate cancer therapy.7 In most instances, risk is increased by high drug doses or elevated drug concentrations due to suicide attempts or drug interactions. An exception is quinidine, which in the 1970s and 1980s was probably the most commonly implicated drug. In this instance, risk was seen even at ‘subtherapeutic’ quinidine concentrations, and in the absence of accumulation of active metabolites. Patients with diLQTS often have exposure to multiple potential culprit drugs and display multiple risk factors (Figure 1).8
2.2 Enter the regulators
Despite increasing recognition, diLQTS remained an electrophysiologic curiosity until its impact on risk vs. benefit considerations in the development of new drugs came into sharp focus. This was due to a single case report9 that described an episode of marked QT interval prolongation and TdP in an otherwise healthy young woman receiving the widely-used antihistamine terfenadine in combination with the antifungal agent ketoconazole. Terfenadine was one of the first non-sedating antihistamines brought to market and was so widely used that it was considered for over-the-counter status. It was subsequently recognized that terfenadine undergoes near-complete first-pass hepatic metabolism, mediated by CYP3A4, to fexofenadine, also a non-sedating antihistamine (now marked as Allegra).10 Terfenadine itself is a potent IKr blocker (discussed further below) and markedly prolongs QT interval, while fexofenadine lacks this effect. Ketoconazole is a potent CYP3A4 inhibitor, and thus prevents terfenadine biotransformation resulting in much higher systemic concentrations than usual and diLQTS. As the mechanisms underlying terfenadine-induced diLQTS were being defined, case series and mechanistic studies reported a relationship between other widely used drugs and diLQTS, which then became a leading cause of drug relabelling and drug withdrawal in the United States in the 1990s. Drugs withdrawn included terfenadine, a second antihistamine (astemizole), the gastric motility drug cisapride, and sparfloxacin and other newer antibiotics.2 As discussed further below, regulatory agencies promulgated guidelines for evaluating risk of QT prolongation for new drug entities and these have become standard parts of the workup of any drug.11 More recently, regulatory agencies in the United States, Europe, Canada, and Japan have proposed newer methods for evaluating diLQTS risk for new drug entities and these are currently being finalized.12
3. From the ECG to individual ion currents: mechanisms of QT prolongation
The single cell correlate of the QT interval is action potential duration in individual heart cells (Figure 2). Early studies demonstrated that experimental conditions mimicking those predisposing to TdP, including slow drive rates, low extracellular potassium, and exposure to QT prolonging drugs, could provoke marked action potential prolongation, discontinuities in the trajectory of terminal repolarization termed early afterdepolarizations (EADs), and triggered secondary upstrokes arising from EADs (Figure 3).13,14 EADs were most readily elicited in the cardiac conduction system (Purkinje cells) where action potentials are among the longest in the heart and are thought to play a key role in initiating and possibly maintaining TdP. One concept is that TdP is generated by repetitive EADs propagated from the conduction system into myocardium and that the polymorphic nature of the tachycardia reflects action potential prolongation and beat-to-beat functional block of conducted EADs. An alternate formulation posits that EAD(s) serve as a trigger for a re-entrant rhythm whose activation pathway varies from beat to beat because of local heterogeneities in action potential duration and arcs of functional block. The latter concept has been popularized in studies of a ‘wedge’ of perfused myocardium (most commonly from dogs) in which marked action potential heterogeneities can be observed at baseline and these are exaggerated in the presence of QT prolonging drugs.15 These studies have not only defined differences in action potential duration and configuration between epicardial and endocardial cells, but have also defined a group of mid-myocardial cells (‘M cells’) with unusually long action potential durations, which may therefore be the site of local conduction block during TdP. Early studies drew a sharp distinction between mechanisms underlying EADs and those underlying delayed afterdepolarizations (DADs), typically seen with rapid drive rates and elevated intracellular calcium concentration in genetic disorders of intracellular calcium regulation (such as catecholaminergic polymorphic ventricular tachycardia, CPVT) and digitalis intoxication. However, perturbed intracellular calcium control has been implicated as a contributor to arrhythmogenic afterdepolarizations (EADs or DADs) under a range of conditions including diLQTS.
Figure 2.
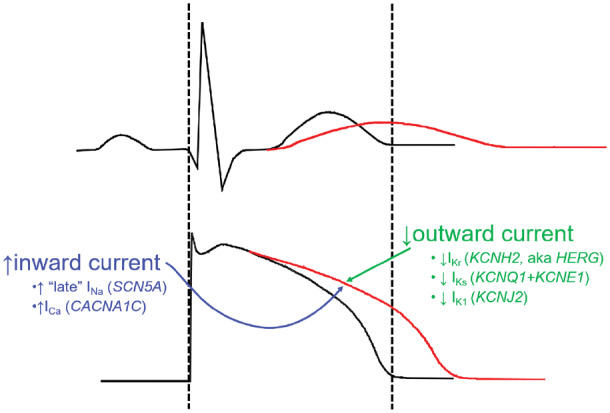
The normal QT interval (black, top; delineated by the dashed lines) is a rough indicator of the duration of action potentials in the ventricle (bottom). When the QT is prolonged (red), action potentials in at least some cells in the ventricle must be prolonged, and this can arise from increased inward current through sodium or potassium channels or decreased outward current through potassium channels. The genes encoding these key channels are also shown.
Figure 3.

The concept of reduced repolarization reserve. (A) Computed (in silico) action potentials at baseline (black) are shown. The top panel is a normal action potential with the dashed line indicating the end of the action potential, and the bottom panel shows the effects of a 15% reduction in IKs, leading to a minimal prolongation of baseline action potential evident at the blue arrow. When IKr block is superimposed, the expected action potential prolongation (seen in the normal situation; green) is highly exaggerated in the presence of the minimal IKs lesion, and an early afterdepolarization is elicited (red arrow). (B) The effect on the electrocardiogram is shown. At baseline, the QT in the presence of the IKs lesion is minimally prolonged (blue arrow), but with drug exposure, there is minimal QT prolongation in the normal situation (top), and marked QT prolongation with development of a long, low-amplitude bifid T wave often seen in diLQTS.
4. From ion currents to the genes whose expression generates ion channel proteins
Having established that action potential prolongation is the proximate mechanism underlying generation of EADs in single cells and TdP in the intact heart, a next obvious question is the mechanism(s) underlying such action potential prolongation. The earliest studies of individual ion currents in mammalian ventricular myocytes defined the rapid inward movement of sodium through sodium channels (INa) as the initiator of the action potential by generating the rapid initial upstroke, and the long plateau phase as a ‘balance’ between inward depolarizing current through calcium channels and outward repolarizing potassium current, generically termed ‘IK’. Subsequent work defined two distinct components of IK: a rapidly-activating current (IKr) blocked by many QT prolonging drugs and a slowly-activating current (IKs).16
A major advance in electrophysiology was the cloning of genes whose expression generates or modulates IKr, IKs, INa, and other critical components of the action potential. This cloning effort was enabled by studies in families with cLQTS and highlights the first principle that prolonged cardiac repolarization must reflect increased inward current and/or decreased outward current during the plateau of the action potential (Figure 2). The vast majority of cases of cLQTS arise as a result of loss-of-function variants the genes that encode IKs and IKr: KCNQ1 and KCNE1 (whose co-expression results in IKs) and KCNH2 (also known as HERG, whose expression results in IKr). The third common form of cLQTS arises from mutations in SCN5A, whose expression underlies INa: these mutations (often termed ‘gain-of-function’) destabilize fast inactivation and thus increase a persistent or ‘late’ sodium current during the plateau of the action potential.17 Mutations in a number of other genes that underlie other key components of action potential control (such as the L-type calcium channel or the inward delayed rectifier IK1 that accomplishes terminal repolarization) or that interact with sodium or potassium channels to modify their function have also been described in cLQTS.1 Many are quite rare, and the evidence supporting their role in causality is sometimes less firm than for the three major forms of cLQTS.
4.1 Currents underlying phases of the action potential
The rapid initial upstroke of a typical ventricular cardiomyocyte is generated by opening of cardiac sodium channels and rapid inward movement of sodium ions along their electrochemical gradient. After opening, sodium channels undergo fast inactivation that rapidly decreases sodium current back to baseline within several milliseconds. The rapid initial depolarization initiates the opening of potassium channels and of calcium channels whose balance generates the plateau of the action potential. In some conditions, notably Type 3 cLQTS as well as abnormalities of intracellular signalling described further below, fast inactivation of the sodium channel is unstable resulting in a residual inward current through sodium channels during the plateau, producing action potential prolongation.
A waning (inactivating) calcium current and increasing IKr and IKs during the plateau ultimately return the voltage towards its resting state; terminal repolarization is further accelerated by IK1. When the action potential plateau is prolonged, EADs are thought to arise during delayed repolarization because of reactivation of inward current through L-type calcium channels or sodium channels, or via sodium-calcium exchange.18IKr displays unusual inactivation properties which markedly increase current amplitude late during the plateau, thereby accelerating rapid repolarization.19 In contrast, IKs amplitude is low at baseline in human ventricular myocytes in the absence of beta-adrenergic stimulation.20 Beta adrenergic stimulation not only increases IKs but also increases L-type calcium current and heart rate, which, if unopposed, would result in marked action potential prolongation at faster rates. Thus, it is thought that a major role of IKs is to limit this action potential prolongation to prevent marked action potential prolongation and EADs.21,22 Consistent with this idea is the observation that cLQTS mutations that decrease IKs are typically manifest as arrhythmias occurring with adrenergic stimulation.
4.2 How do drugs prolong action potentials and QT intervals?
While enhanced inward current or block of any of the repolarizing potassium currents could prolong action potentials, the predominant mechanism among drugs in clinical use associated with diLQTS is IKr block. Examples of drugs that appear to prolong action potential through block of IKs or enhanced calcium current have been reported, but appear rare. Interestingly, the canonical sodium channel blocking antiarrhythmic quinidine is actually a much more potent IKr blocker, which likely explains the development of diLQTS at low drug concentrations. The intracellular face of the of the channel encoded by KCNH2/HERG displays two unusual features that are thought to account for the ‘promiscuity’ of the channel with respect to drug block.23 It is unusually large, allowing drugs to access the potassium-permeating pathway, and it is lined with a series of aromatic residues that are thought to enable binding of high affinity methanesulfonanilide drugs such as dofetilide. A recent cryo-EM structure of the channel also highlights potential drug-binding ‘pouches’ within this intracellular domain.24,25 While drug block of IKr is the most commonly implicated mechanism, other studies have suggested drug-induced changes in IKr amplitude, for example due to inflammatory mediators26 or to reduced cell surface expression,27 may also play a role.
4.3 A genetic basis for diLQTS?
Studies in kindreds with cLQTS, using family tree structures or genotyping, demonstrate that some mutation carriers do not display QT prolongation at baseline.28,29 This observation suggests the hypothesis that subclinical cLQTS contributes to susceptibility to the drug-induced form of the syndrome. Sequencing cLQTS disease genes identifies cLQTS-related rare variants (mutations) in approximately ∼10–20% of cases of diLQTS (Figure 4).30–33 This is consistent with the idea that genetic variants could remain subclinical until repolarization is further compromised by superimposition of an IKr blocker, when marked action potential prolongation and afterdepolarizations can then occur. Causes for such ‘reduced repolarization reserve’34 (Figure 3) could include not only altered ion current amplitude or gating due to genetic variants in ion channels or other proteins critical for action potential control, but also changes in the extracellular environment (such as hypokalaemia), disease-related changes in ion channel expression or function, or alterations in intracellular signalling systems that modulate channel function.
Figure 4.
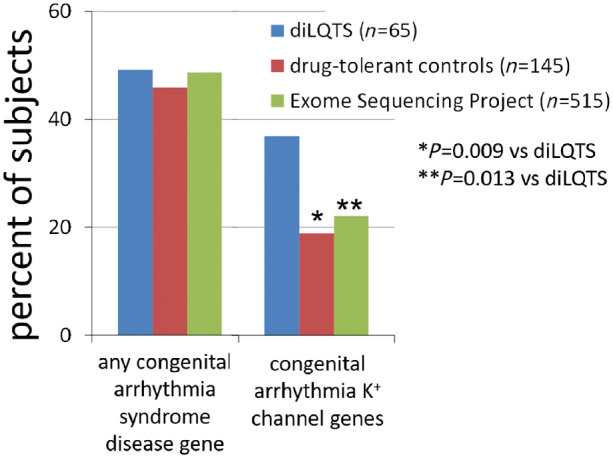
Frequency of rare missense variants in congenital arrhythmia syndrome disease genes in patients with drug-induced long QT syndrome (diLQTS), drug-tolerant controls, and population controls from the Exome Sequencing Project. There was a high (∼50%) frequency of rare variants across all three groups, but far more patients with diLQTS had rare variants in arrhythmia genes encoding potassium channel subunits compared with the two control groups.33
A large candidate gene survey implicated a common variant in KCNE1, resulting in D85N as a risk allele for diLQTS, likely by subtly altering IKs amplitude and gating.35 This variant occurs in 0.9–2.5% of the general population (depending on ancestry36) and was found in up to 9% of cases of diLQTS, resulting in an odds ratio of approximately 10. A genome-wide association study (GWAS) did not identify other loci with large effect sizes modulating risk for diLQTS.37 However, a genetic risk score, derived from 60 SNPs known to modulate baseline QT interval, did effectively (P = 10−7) distinguish cases of diTdP from controls (Figure 5).38 This result suggests that the genetic architecture of the QT interval at baseline, determined by the expression of multiple genes and captured by the genetic risk score, can contribute to risk when IKr block is superimposed. That is, some ‘normal’ QT intervals may be more susceptible than others to marked QT prolongation when IKr block is superimposed, consistent with the ‘reduced repolarization reserve’ hypothesis.
Figure 5.
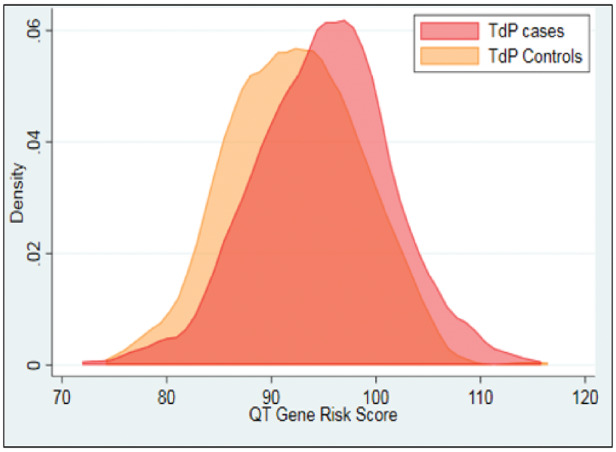
Distribution of genetic risk scores derived from a large genome-wide association study of baseline QT intervals. The distribution is significantly (P = 10−7) skewed to higher risk scores in subjects with drug-induced torsades de pointes (TdP cases) compared with drug exposed controls not developing TdP.38
5. Identifying drugs at risk for causing torsades de pointes
5.1 The thorough QT study
After recognition that high concentrations of terfenadine could provoke QT prolongation, a double-blind study evaluated the effects of the drug on the rate corrected QT interval (QTc) in normal volunteers and in patients with recognized heart disease.39 At baseline, average QTc was 407 ms in the volunteers and 417 ms in those with heart disease and 60 mg terfenadine twice daily (the ‘usual’ dose) provoked a mean QTc increase of 6 ms in normals and 12 ms in those with heart disease. With dosing at 180 mg twice daily, the increases in QTc were correspondingly larger, 19 ms and 26 ms, respectively. The protocol also included evaluation of 300 mg twice daily, but this arm was suspended after an elderly patient with heart disease, who had tolerated the lower doses, died suddenly (off a cardiac monitor) on Day 4 of dosing. A second patient developed prominent U waves.
This study suggested that systematic examination of the QT interval during exposure to therapeutic and supratherapeutic doses of new drug molecules could expose TdP liability, and laid the groundwork for the regulatory recommendation that new drug entities be evaluated in a ‘thorough QT study’ along these lines.11 In this design, a supratherapeutic dosage could also be evaluated by a usual drug dosage in combination along with a metabolic inhibitor (e.g. terfenadine + ketoconazole40). The thorough QT study design also generally includes a set of subjects exposed to placebo and subjects exposed to a positive control, usually the antibiotic moxifloxacin which consistently produces a 5–10 ms increase in QTc.
5.2 Screening for IKr block in drug development
A complementary approach to evaluating diLQTS liability of new drug entities is to establish the extent to which they block IKr (generally assessed in cells transiently or stably transfected with KCNH2), the concentrations required to achieve this effect, the relationship of those concentrations to those achieved clinically, and other ion channel blocking actions that may blunt the effects of IKr block.11 For example, the L-type calcium channel blocker, verapamil also blocks IKr, but the calcium channel effect predominates and QT interval prolongation is nearly unheard of. In silico methods that have potential to predict IKr block from using in silico structural analysis are in development.41
5.3 Other approaches to risk evaluation
Almost two decades of experience with the thorough QT study design has suggested to the drug development and regulatory communities that compounds with potential therapeutic utility and low TdP liability may be dropped from development because of an early ‘QT signal’ detected by the thorough QT study. As a result, an alternate scheme to more effectively screen for dangerous drugs has been proposed by the ‘comprehensive in vitro proarrhythmia assay’ (CiPA) initiative. The goal is to replace the thorough QT study and with a set of assays including in assessment of drug effects on individual ion currents (including IKr) and prediction of global drug effects on repolarization and potential proarrhythmia using in silico modelling and studies in cardiomyocytes from human induced pluripotent stem cells (iPSC-CMs).12
5.4 A range of effects on QT
In some cases, QT effects are well documented, occur in a substantial portion of patients, and are not disputed. For example, the potent IKr-blocking antiarrhythmic dofetilide produces TdP in 1.7% of patients treated for AF even when strict dosing guidelines (in patient initiation with QT monitoring, dose adjustment for excessive QT prolongation or for renal dysfunction) are incorporated into dosing.42 In other cases, risk evaluated by IKr block in vitro or from clinical reports is more modest; the antibiotic moxifloxacin, for example, blocks IKr but reports of TdP are exceedingly rare. The website crediblemeds.org43 summarizes and continually updates the spectrum of risks across drugs, based ongoing analyses of large public spontaneous adverse drug event reporting databases and in vitro literature. The site is particularly useful for patients with congenital long QT syndrome in whom high-risk drugs such as methadone or dofetilide should be avoided entirely and lower risk drugs used only with caution, if at all.
Some drugs labelled as known or possible risk for TdP at crediblemeds.org have special relevance to treatment of cancer itself or symptoms such as pain or nausea for which cancer patients are often treated (Table 1). These include antibiotics, antiemetics, and pain medications, as well as anticancer drugs. In some cases, alternate therapies may be available. In others, such as arsenic trioxide, the drug is first line (in this case for promyelocytic leukaemia), the benefits of treatment are thought to outweigh the risks and alternate treatments are not available.
Table 1.
Selected drugs with QT liability listed at crediblemeds.org
| Anticancer agents | Other drugs with potential use in cancer and ‘known TdP risk’ |
|---|---|
| Anti-infective agents | |
| ‘Known TdP risk’ | Azithromycin |
| Aclarubicina | Chloroquine |
| Anagrelide | Ciprofloxacin |
| Arsenic trioxide | Clarithromycin |
| Oxaliplatin | Erythromycin |
| Vantenatib | Fluconazole |
| Halofantrinea | |
| ‘Possible TdP risk’ | Levofloxacin |
| Abarelixa | Moxifloxacin |
| Apalutemide | Pentamidine |
| Bendamustine | Roxithromycina |
| Bortezomid | |
| Bosutinib | Antidepressants |
| Cabozantinib | Citalopram |
| Capecitabine | Escitalopram |
| Ceritinib | |
| Crizotinib | Antiemetics |
| Dafratinib | Domperidonea |
| Epirubicin | Droperidol |
| Fluorouracil | Ondansetron |
| Inotuzumab ozogamicin | |
| Lapatinib | Pain management |
| Leuprolide | Methadone |
| Midostaurin | |
| Necitumumab | |
| Nilotinib | |
| Oimertinib | |
| Panobinostat | |
| Pazopanib | |
| Ribociclib | |
| Romidepsin | |
| Sorafenib | |
| Suntinib | |
| Tamoxifen | |
| Tipiracil + Trifluridine | |
| Toremifene | |
| Vemurafinib | |
| Vorinostat |
Not marketed in the United States.
6. A new mechanism in diLQTS: relevance to anticancer therapy
The IKr-based paradigm described above was disrupted when Lu et al.44 reported an evaluation of the effects of a series of tyrosine kinase inhibitors on action potential duration in canine cardiac ventricular myocytes. The Bcr-Abl kinase inhibitor nilotinib, which carries a QT ‘black box’ warning on its FDA label, did not acutely block IKr but prolonged drug exposure (2 h) did generate action potential prolongation, and this was reversed by including phosphatidylinositol 3,4,5-trisphosphate (PIP3) in the pipette solution (Figure 6). PIP3 is the downstream effector of phosphoinositide 3-kinase (PI3K), a key intracellular signalling system modulating cell growth and, in cardiomyocytes, hypertrophy. Similar effects were seen with the multi-kinase inhibitors dasatinib and sunitinib; all three drugs are classified as ‘possible’ TdP risk at crediblemeds.org. PI3K inhibition could be a direct effect or could reflect inhibition of upstream receptor tyrosine kinases and the group reported similar effects with direct inhibitors of PI3K. Subsequent studies have identified the alpha isoform of PI3-kinase as the major target that mediates action potential prolongation through these pathways.45
Figure 6.

Role of phosphoinositide 3-kinase (PI3K) in ion channel signalling. As discussed in the text, occupancy of receptor tyrosine kinases (RTKs) activates PI3K (the α isoform in cardiomyocytes) which then generates its downstream effector phosphatidylinositol 3,4,5-trisphosphate (PIP3). Drug block of RTKs, or decreased transcription, results in enhanced late INa and decreased IKr and IKs, all of which (green) act to prolong action potential and QT, as well as decreased L-type calcium current and peak INa (orange) which would be expected to shorten QT. As described in the text, this effect on repolarization has now been reported with RTK inhibitors used in cancer, with other QT prolonging drugs formerly thought to act via IKr alone, and in diabetes. In each instance, the electrophysiologic abnormality is corrected by adding PIP3 to the intracellular solution.
Lu et al. described multiple mechanisms that can explain how direct or indirect PI3Kα inhibition prolongs action potentials: the drugs reduce IKr and IKs amplitude (which would prolong action potential), and reduce peak calcium and sodium current which would, if anything, shorten action potentials. Another prominent effect was to enhance late sodium current, and this was also seen (in the absence of drug) in mice in which PI3Kα was knocked out. QT interval prolongation is well recognized in diabetes and in genetic mouse models (Akita mice deficient in insulin and db/db mice with insulin resistance) marked action potential prolongation and increased late sodium current were observed. These abnormalities were all reversed by intracellular PIP3,46 suggesting a role for decreased insulin receptor signalling to PI3K.
Drugs designated as ‘pure’ IKr blockers, such as dofetilide and moxifloxacin, have widely divergent risks for diLQTS. Accordingly, we examined the effect of prolonged exposure to a range of IKr blockers in adult mouse cardiomyocytes (which lack IKr), human iPSC-CMs, and on late sodium current in heterologous expression systems.47 Dofetilide and other potent IKr blockers also enhanced late sodium current on prolonged (≥2 h) drug exposure whereas moxifloxacin and verapamil, IKr blockers rarely if ever associated with TdP, did not. Interestingly, Lu et al.44 reported that prolonged exposure to terfenadine produced marked action potential prolongation in canine cardiac myocytes (which do express IKr), and that this was largely, but not completely, reversed by intracellular PIP3. This result supports the contention that potent ‘high risk’ IKr-blocking drugs, such as terfenadine and dofetilide, may produce marked action potential prolongation due to combined IKr block and enhanced late INa.
The effects of prolonged infusion of dofetilide have also been examined in intact dogs.48 Prolonged infusion inhibited Akt phosphorylation, a marker of PI3K activity. Dofetilide prolonged QT interval acutely, and with continued infusion QT interval continued to prolong, whereas with moxifloxacin, QT interval prolongation was observed acutely, but did not prolong with continued infusion. Administration of lidocaine, a blocker of the late sodium current, shortened QT more after prolonged dofetilide infusion than after acute exposure, also consistent with a delayed effect on late sodium current.
These initial studies are prompting a re-evaluation of the mechanisms underlying diLQTS especially in the setting of emerging anticancer therapies that target multiple tyrosine kinases coupled to downstream PI3K signalling as well as those directly targeting PI3Kα itself. In each case, risk will depend on the potency of inhibition of this pathway in cardiomyocytes, other electrophysiologic properties of such as block of IKr or calcium current, and other pharmacologic actions such as testosterone inhibition that may affect QT.7 In some instances (a minority), these other actions have been addressed, but for most drugs only IKr block has been studied and the fuller spectrum of basic mechanisms that might generate a QT signal is not fully worked out; hence, ECG monitoring during development of the drug may be a first indication of this issue.
There are now emerging examples in each of these domains. Lapatinib and vandetanib can prolong QT and both block IKr; thus, while other mechanisms could contribute, they need not be invoked.49,50 Sporadic case reports suggest the HDAC inhibitor vorinostat can rarely produce TdP.51 However, follow-up studies indicated no effect on late INa52 or on IKr53; altered transcription of key genes involved in transport of ion channels to the cell surface was implicated.53 After preliminary studies suggested increased late INa could be generated by decreased platelet derived growth factor (PDGF) signalling acting to inhibit PI3K, we showed that the PDGF inhibitor crenolanib markedly prolonged action potentials in human iPSC-CMs.54 There is a high incidence of QT interval prolongation with the cyclin-dependent kinase 4/6 inhibitor ribociclib although the mechanism has not been worked out.55
Interestingly, late INa is also enhanced by reactive oxygen species acting via calcium/calmodulin-dependent kinase (CaM kinase) delta.56 Thus, clinical conditions that modulate reactive oxygen, PI3K, or CaM kinase signalling in myocytes may, in turn, contribute to variability to day-to-day or patient-to-patient variability in the extent to which drugs such as dofetilide prolong the QT interval.
7. Summary
Initial lessons learned from sporadic cases of drug exposure that appear to phenocopy a congenital arrhythmia syndrome have had profound implications for the drug development community. For several decades, the major focus has been on IKr block and this is a predominant mechanism of QT prolongation and drug-induced TdP risk. More recent study, focusing on anticancer drugs, but also extending to other classes, emphasize the potential of PI3-kinase alpha inhibition as an additional mechanism controlling QT prolongation/TdP risk.
Conflict of interest: none declared.
Funding
This work was supported in part by P50 GM115305.
References
- 1. Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013;62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roden DM. Drug-induced prolongation of the QT Interval. N Engl J Med 2004;350:1013–1022. [DOI] [PubMed] [Google Scholar]
- 3. Choy AM, Darbar D, Dell'Orto S, Roden DM. Exaggerated QT prolongation after cardioversion of atrial fibrillation. J Am Coll Cardiol 1999;34:396–401. [DOI] [PubMed] [Google Scholar]
- 4. Astrom-Lilja C, Odeberg JM, Ekman E, Hagg S. Drug-induced torsades de pointes: a review of the Swedish pharmacovigilance database. Pharmacoepidemiol drug safety 2008;17:587–592. [DOI] [PubMed] [Google Scholar]
- 5. Lazzerini PE, Laghi-Pasini F, Bertolozzi I, Morozzi G, Lorenzini S, Simpatico A, Selvi E, Bacarelli MR, Finizola F, Vanni F, Lazaro D, Aromolaran A, El Sherif N, Boutjdir M, Capecchi PL. Systemic inflammation as a novel QT-prolonging risk factor in patients with torsades de pointes. Heart 2017;103:1821–1829. [DOI] [PubMed] [Google Scholar]
- 6. Yue Y, Castrichini M, Srivastava U, Fabris F, Shah K, Li Z, Qu Y, El-Sherif N, Zhou Z, January C, Hussain MM, Jiang X-C, Sobie EA, Wahren-Herlenius M, Chahine M, Capecchi P-L, Laghi-Pasini F, Lazzerini P-E, Boutjdir M. Pathogenesis of the novel autoimmune-associated long QT syndrome. Circulation 2015;132:230–240. [DOI] [PubMed] [Google Scholar]
- 7. Salem JE, Waintraub X, Courtillot C, Shaffer CM, Gandjbakhch E, Maupain C, Moslehi JJ, Badilini F, Haroche J, Gougis P, Fressart V, Glazer AM, Hidden-Lucet F, Touraine P, Lebrun-Vignes B, Roden DM, Bachelot A, Funck-Brentano C. Hypogonadism as a reversible cause of torsades de pointes in men. Circulation 2018;138:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, Philippides GJ, Roden DM, Zareba W. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. J Am Coll Cardiol 2010;55:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monahan BP, Ferguson CL, Killeavy ES, Lloyd BK, Troy J, Cantilena LR Jr. Torsades de pointes occurring in association with terfenadine use. J Am Med Assoc 1990;264:2788–2790. [PubMed] [Google Scholar]
- 10. Woosley RL, Chen Y, Freiman JP, Gillis RA. Mechanism of the cardiotoxic actions of terfenadine. J Am Med Assoc 1993;269:1532–1536. [PubMed] [Google Scholar]
- 11. Shah R. Drugs, QTc interval prolongation and final ICH E14 guideline. Drug Saf 2005;28:1009–1028. [DOI] [PubMed] [Google Scholar]
- 12. Colatsky T, Fermini B, Gintant G, Pierson JB, Sager P, Sekino Y, Strauss DG, Stockbridge N. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—update on progress. J Pharmacol Toxicol Methods 2016;81:15–20. [DOI] [PubMed] [Google Scholar]
- 13. Roden DM, Hoffman BF. Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Relationship to potassium and cycle length. Circ Res 1985;56:857–867. [DOI] [PubMed] [Google Scholar]
- 14. Strauss HC, Bigger JT, Hoffman BF. Electrophysiological and beta-receptor blocking effects of MJ 1999 on dog and rabbit cardiac tissue. Circ Res 1970;26:661–678. [DOI] [PubMed] [Google Scholar]
- 15. Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu DW. Heterogeneity within the ventricular wall: electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res 1991;69:1427–1449. [DOI] [PubMed] [Google Scholar]
- 16. Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K + current: differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 1990;96:195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995;376:683–685. [DOI] [PubMed] [Google Scholar]
- 18. Wit AL. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin Electrophysiol 2018;doi:10.1111/pace.13419. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 19. Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 1996;379:833–836. [DOI] [PubMed] [Google Scholar]
- 20. Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, Bogats G, Lathrop DA, Papp JG, Varro A. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 2005;112:1392–1399. [DOI] [PubMed] [Google Scholar]
- 21. Xie Y, Grandi E, Puglisi JL, Sato D, Bers DM. beta-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J Mol Cell Cardiol 2013;58:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation 2005;112:1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 2000;97:12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vandenberg JI, Perozo E, Allen TW. Towards a structural view of drug binding to hERG K(+) channels. Trends Pharmacol Sci 2017;38:899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang W, MacKinnon R. Cryo-EM structure of the open human ether-a-go-go-related K(+) channel hERG. Cell 2017;169:422–430.e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sordillo PP, Sordillo DC, Review HL. The prolonged QT interval: role of pro-inflammatory cytokines, reactive oxygen species and the ceramide and sphingosine-1 phosphate pathways. In Vivo 2015;29:619–636. [PubMed] [Google Scholar]
- 27. Ficker E, Kuryshev YA, Dennis AT, Obejero-Paz C, Wang L, Hawryluk P, Wible BA, Brown AM. Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol Pharmacol 2004;66:33–44. [DOI] [PubMed] [Google Scholar]
- 28. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation 1999;99:529–533. [DOI] [PubMed] [Google Scholar]
- 29. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–811. [DOI] [PubMed] [Google Scholar]
- 30. Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, Schulze-Bahr E, Haverkamp W, Breithardt G, Cohen N, Aerssens J. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med 2004;82:182–188. [DOI] [PubMed] [Google Scholar]
- 31. Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton M, Murray KT, Norris K, George AL Jr, Roden DM. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 2002;105:1943–1948. [DOI] [PubMed] [Google Scholar]
- 32. Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, Priori SG, Roden DM, George AL Jr, Goldstein SA. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl. Acad Sci USA 2000;97:10613–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weeke P, Mosley JD, Hanna D, Delaney JT, Shaffer C, Wells QS, Van Driest S, Karnes JH, Ingram C, Guo Y, Shyr Y, Norris K, Kannankeril PJ, Ramirez AH, Smith JD, Mardis ER, Nickerson D, George AL Jr, Roden DM. Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug-induced long QT interval syndrome. J Am Coll Cardiol 2014;63:1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roden DM. Taking the idio out of idiosyncratic - predicting torsades de pointes. Pacing Clin Electrophysiol 1998;21:1029–1034. [DOI] [PubMed] [Google Scholar]
- 35. Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, Meitinger T, Peters A, Wichmann HE, Ingram C, Bradford Y, Carter S, Norris K, Ritchie MD, George AL Jr, Roden DM. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet 2012;5:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roden DM, Glazer AM, Kroncke B. Arrhythmia genetics: not dark and lite, but 50 shades of gray. Heart Rhythm 2018;15:1231–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Behr ER, Ritchie MD, Tanaka T, Kaab S, Crawford DC, Nicoletti P, Floratos A, Sinner MF, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Zumhagen S, Guicheney P, Bishopric NH, Marshall V, Shakir S, Dalageorgou C, Bevan S, Jamshidi Y, Bastiaenen R, Myerburg RJ, Schott JJ, Camm AJ, Steinbeck G, Norris K, Altman RB, Tatonetti NP, Jeffery S, Kubo M, Nakamura Y, Shen Y, George AL Jr, Roden DM. Genome wide analysis of drug-induced torsades de pointes: lack of common variants with large effect sizes. PloS One 2013;8:e78511.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, Roden DM, Woosley R, Kosova G, Rosenberg MA, Newton-Cheh C. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation 2017;135:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pratt CM, Ruberg S, Morganroth J, McNutt B, Woodward J, Harris S, Ruskin J, Moye L. Dose-response relation between terfenadine (Seldane) and the QTc interval on the scalar electrocardiogram: distinguishing a drug effect from spontaneous variability. Am Heart J 1996;131:472–480. [DOI] [PubMed] [Google Scholar]
- 40. Honig PK, Wortham DC, Zamani K, Conner DP, Mullin JC, Cantilena LR. Terfenadine-ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. J Am Med Assoc 1993;269:1513–1518. [PubMed] [Google Scholar]
- 41. Munawar S, Windley MJ, Tse EG, Todd MH, Hill AP, Vandenberg JI, Jabeen I. Experimentally validated pharmacoinformatics approach to predict hERG inhibition potential of new chemical entities. Front Pharmacol 2018;9:1035.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abraham JM, Saliba WI, Vekstein C, Lawrence D, Bhargava M, Bassiouny M, Janiszewski D, Lindsay BD, Militello M, Nissen SE, Poe S, Tanaka-Esposito C, Wolski K, Wilkoff BL. Safety of oral dofetilide for rhythm control of atrial fibrillation and atrial flutter. Circ Arrhythm Electrophysiol 2015;8:772–776. [DOI] [PubMed] [Google Scholar]
- 43. Woosley RL, Black K, Heise CW, Romero K. CredibleMeds.org: what does it offer? Trends Cardiovasc Med 2018;28:94–99. [DOI] [PubMed] [Google Scholar]
- 44. Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS, Lin RZ. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med 2012;4:131ra150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang T, Meoli DF, Moslehi J, Roden DM. Inhibition of the alpha-subunit of phosphoinositide 3-kinase in heart increases late sodium current and is arrhythmogenic. J Pharmacol Exp Ther 2018;365:460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu Z, Jiang YP, Wu CY, Ballou LM, Liu S, Carpenter ES, Rosen MR, Cohen IS, Lin RZ. Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes 2013;62:4257–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C, Roden DM. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation 2014;130:224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qiu XS, Chauveau S, Anyukhovsky EP, Rahim T, Jiang YP, Harleton E, Feinmark SJ, Lin RZ, Coronel R, Janse MJ, Opthof T, Rosen TS, Cohen IS, Rosen MR. Increased late sodium current contributes to the electrophysiological effects of chronic, but not acute, dofetilide administration. Circ Arrhythm Electrophysiol 2016;9:e003655.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shah RR, Morganroth J, Shah DR. Cardiovascular safety of tyrosine kinase inhibitors: with a special focus on cardiac repolarisation (QT interval). Drug Saf 2013;36:295–316. [DOI] [PubMed] [Google Scholar]
- 50. Lee HA, Kim EJ, Hyun SA, Park SG, Kim KS. Electrophysiological effects of the anti-cancer drug lapatinib on cardiac repolarization. Basic Clin Pharmacol Toxicol 2010;107:614–618. [DOI] [PubMed] [Google Scholar]
- 51. Lynch DR Jr, Washam JB, Newby LK. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: a case report and review of the literature. Cardiol J 2012;19:434–438. [PubMed] [Google Scholar]
- 52. Xu Q, Patel D, Zhang X, Veenstra RD. Changes in cardiac Nav1.5 expression, function, and acetylation by pan-histone deacetylase inhibitors. Am J Physiol Heart Circ Physiol 2016;311:H1139–h1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spence S, Deurinck M, Ju H, Traebert M, McLean L, Marlowe J, Emotte C, Tritto E, Tseng M, Shultz M, Friedrichs GS. Histone deacetylase inhibitors prolong cardiac repolarization through transcriptional mechanisms. Toxicol Sci 2016;153:39–54. [DOI] [PubMed] [Google Scholar]
- 54. Bersell KR, Yang T, Mosley JD, Glazer AM, Brown JD, Campbell CM, Hong CC, Wells QS, Johnson AN, Short L, Blair MA, Behr ER, Petropoulou E, Jamshidi Y, Gerszten R, Benson MD, Vasan RS, Yang Q, Jarvik GP, Namjou B, Shaffer CM, Parikh S, Sheng Q, Kannankeril PJ, Wang TJ, Knollmann BC, Roden DM. Abstract 14477: Integrating patient-specific cardiomyocyte function with population multi-omics identifies a novel arrhythmia pathway (**Katz Prize finalist**). Circulation 2017;136:A14477–A14477. [Google Scholar]
- 55. Infante JR, Cassier PA, Gerecitano JF, Witteveen PO, Chugh R, Ribrag V, Chakraborty A, Matano A, Dobson JR, Crystal AS, Parasuraman S, Shapiro GI. A phase I study of the cyclin-dependent kinase 4/6 inhibitor ribociclib (LEE011) in patients with advanced solid tumors and lymphomas. Clin Cancer Res 2016;22:5696–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]