

Abstract
Histological and molecular analyses of urothelial carcinoma often reveal intratumoural and intertumoural heterogeneity at the genomic, transcriptional and cellular levels. Despite the clonal initiation of the tumour, progression and metastasis often arise from subclones that can develop naturally or during therapy, resulting in molecular alterations with a heterogeneous distribution. Variant histologies in tumour tissues that have developed distinct morphological characteristics divergent from urothelial carcinoma are extreme examples of tumour heterogeneity. Ultimately, heterogeneity contributes to drug resistance and relapse after therapy, resulting in poor survival outcomes. Mutation profile differences between patients with muscle-invasive and metastatic urothelial cancer (interpatient heterogeneity) probably contribute to variability in response to chemotherapy and immunotherapy as first-line treatments. Heterogeneity can occur on multiple levels and averaging or normalizing these alterations is crucial for clinical trial and drug design to enable appropriate therapeutic targeting. Identification of the extent of heterogeneity might shape the choice of monotherapy or additional combination treatments to target different drivers and genetic events. Identification of the lethal tumour cell clones is required to improve survival of patients with urothelial carcinoma.
Heterogeneity resulting from clonal expansion of individual mutations, genomic alterations and variability of gene expression between tumour regions and, in patients with metastasis, between the primary tumour and metastases, forms the basis of the complexity of cancer1. Tumour heterogeneity is recognized as a hallmark of urothelial carcinoma and is potentially related to high mutational burden that can change cellular differentiation over time with each cell division2,3. Next-generation sequencing enables identification and characterization of urothelial carcinoma heterogeneity at the genomic and transcriptomic levels and provides the opportunity to associate alterations with tumour morphology and clinical outcome4. Nevertheless, tumour heterogeneity is a considerable obstacle for both scientists and clinicians when developing new agents or choosing therapeutic strategies for treatment of patients with urothelial carcinoma5. For example, treatments directed at individual genomic targets are likely to result in expansion of non-responding clones that do not harbour these targets, and less-targeted therapies (for example, chemotherapy and immunotherapy) might fundamentally alter the clonal and/or transcriptional subtypes of an individual tumour6.
Evidence for the critical role of tumour heterogeneity was identified in lung cancer7, renal cell carcinoma8 and colorectal cancer9,10. In these detailed studies of multiple tumour sites, parental driver alterations are shared, but new clones result over time. For example, in lung cancer, driver mutations in TP53, MET, EGFR and BRAF were often clonal, whereas alterations in PIK3CA, NF1 and DNA damage repair and chromatin-regulatory genes were more heterogeneous and occurred as later alterations7. New methods and model systems for assessing and studying the effects of tumour heterogeneity on phenotype and clinical behaviour will facilitate an improved understanding of this cancer hallmark in urothelial carcinoma11.
Bladder cancer heterogeneity occurs on multiple levels and directly affects clinical care. Patients with bladder cancer usually die from muscle-invasive disease and much research is focused on this disease stage. However, 75% of patients are diagnosed with non-muscle-invasive disease12 and tumour heterogeneity is likely to have a role in the management of these patients, as it might affect the selection of non-muscle-invasive bladder cancer (NMIBC) risk groups, surveillance monitoring strategies, intravesical therapies and early application of radical therapy13. Unfortunately, few data exist to suggest that substantial tumour heterogeneity is present and a driver of treatment resistance in NMIBC. In addition, the total mutation burden reported for NMIBC is lower than that of muscle-invasive bladder cancer (MIBC)14, and data on the application of tumour subtypes to NMIBC are limited. Overall, tumour heterogeneity affects several major aspects of bladder cancer management: molecular profiling of MIBC to assess risk of relapse, selection of aggressive tumours (NMIBC or MIBC) for radical treatment, and use of urine and blood biomarkers to identify aggressive tumours, apply early radical therapy and identify relapse.
In this Review, we describe the multiple levels of heterogeneity in bladder cancer and how they affect tumour biology and clinical care. We summarize common definitions of tumour heterogeneity and discuss the link between heterogeneity and tumour evolution, as well as the influence of treatments and molecular drivers. We then describe current knowledge of genomic heterogeneity at the DNA level and the expression level, resulting in different tumour subtypes and the morphological heterogeneity seen in variant bladder cancer histology. Finally, we discuss the influence of heterogeneity on treatment decision-making, drug development and clinical trial design.
Definitions of tumour heterogeneity
Improved technology to characterize the heterogeneity of tumours at the morphological, genomic and transcriptional levels led to an appreciation by clinicians that neoplastic disease is inherently unstable, characterized by heterogeneous tumour composition and evolving morphological changes that occur in the disease course. With this recognition, scientists and clinicians have worked to develop a framework to understand the heterogeneity of urothelial carcinoma between tumours, within a tumour and over time (BOX 1; FIG. 1).
Box 1 |. Definitions of heterogeneity.
Heterogeneity: the state of comprising diverse or dissimilar elements
Interpatient heterogeneity: differences between tumours of patients diagnosed with the same type of primary cancer
Intratumoural heterogeneity: differences between spatial regions of the primary tumour of the same patient
Intertumoural heterogeneity: differences between multiple primary tumours in the same patient, the primary tumour and metastatic deposits or multiple metastatic sites
Temporal heterogeneity: molecular or genetic changes in the tumour over time and/or during treatment of the same patient
Circulation heterogeneity: differences between circulating tumour markers and tissue-based tumour markers
Fig. 1 |. Different types of heterogeneity found in bladder cancer.
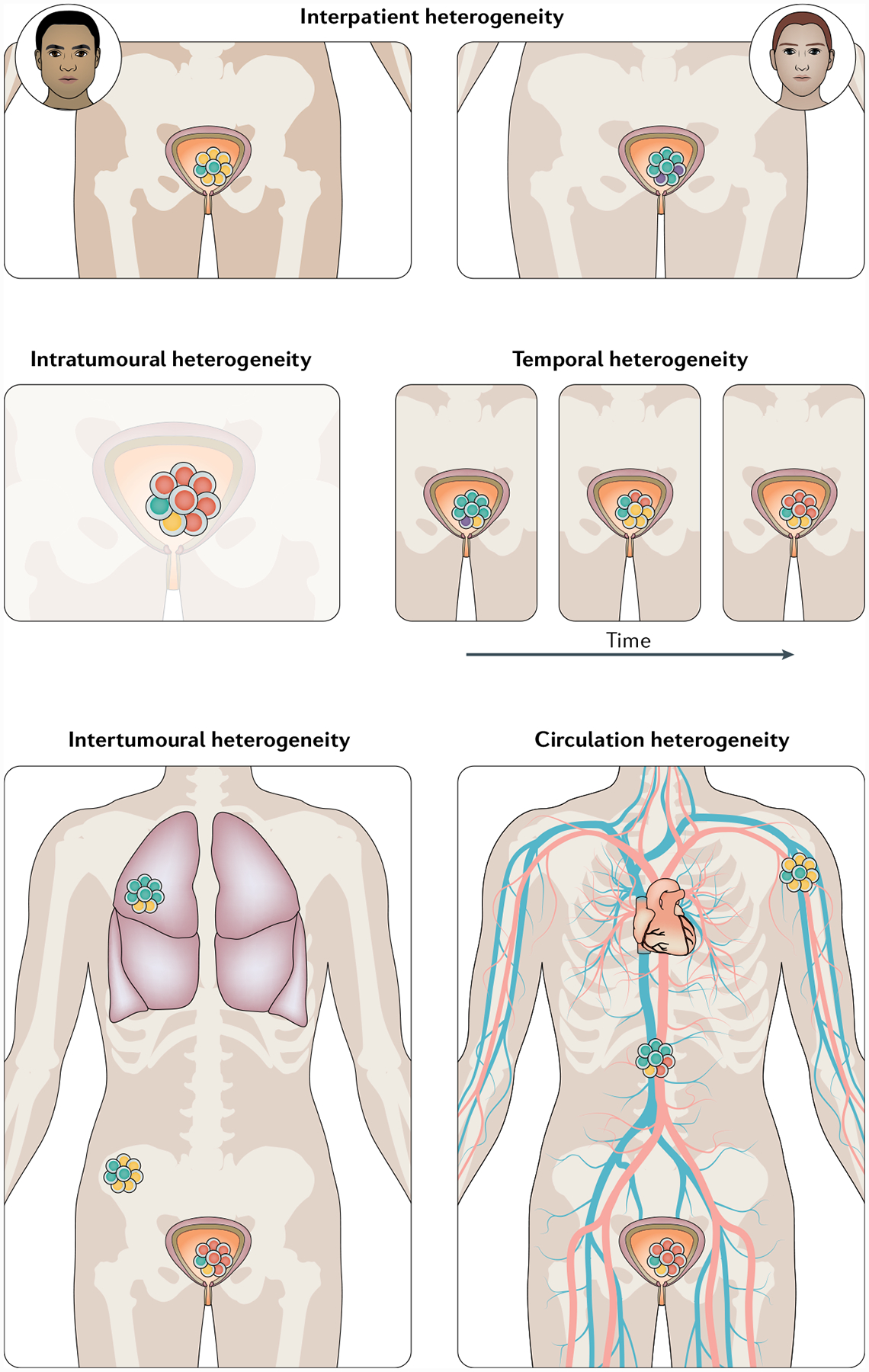
Bladder tumours can vary in morphology, gene expression profile and mutations. This heterogeneity exists not only between patients (interpatient heterogeneity) but also within the same patient, where subclassifications can be made. Intratumoural heterogeneity describes variations between regions of one tumour and can be affected or caused by clonality, immune cell infiltration and the tumour microenvironment. Differences between multiple tumours and/or metastases within one patient are termed intertumoural heterogeneity. Heterogeneity can also change over time and during treatment (temporal heterogeneity). Finally, differences can also exist between tissue-based tumour markers and circulating markers (circulation heterogeneity) and can be assessed by comparing data from tumour deposits with those from liquid biopsy approaches.
Interpatient heterogeneity.
Patients with cancer have traditionally been characterized clinically by the location of their primary tumour and its histology. Clinicians treat eligible patients with metastatic urothelial carcinoma using cisplatin-based chemotherapy; some of these patients will initially respond very well to this therapy and others will not respond at all3. Currently, the molecular features associated with response to chemotherapy are not fully understood but might include tumour immune cell invasion (for example, indicated by PDL1 status), total mutation burden (a surrogate for neoantigen load), DNA damage response defects and tumour morphology6. As genomic and molecular characterization become routinely utilized, this information can help to evaluate interpatient heterogeneity in patients with the same tumour type, providing new data to characterize patients’ tumours beyond their histological information and determine their optimal treatment plan.
Intratumoural heterogeneity.
Perhaps the most commonly referenced type of heterogeneity for urothelial carcinoma is intratumoural heterogeneity, which describes differences among regions of the primary tumour that might have discreet genomic and functional alterations during tumour evolution7,8,15. Intratumoural heterogeneity has long been recognized clinically by histological differences in distinct tumour areas. For example, in prostate cancer, the Gleason Score is calculated by adding the two most prominent grades from different tumour areas to give a sum score16. Specific clinical considerations for patients with urothelial carcinoma include whether multiple areas of the tumour should be biopsied; how the molecular information might influence care and whether different molecular targets exist; and — when multiple histologies are present in the same tumour — whether the more aggressive histology and its relative representation can be identified.
Intertumoural heterogeneity.
This type of heterogeneity refers to differences between the primary tumour and metastases, different metastatic sites or multiple tumours found in the same primary location. Research from patients with metastases suggests that very few genomic alterations are shared between the primary tumour and the metastases17. Several investigations have demonstrated heterogeneous genetic findings of metastatic lesions from the same patient: the genetic makeup of some of the lesions represented that of a subclonal population in the primary tumour, whereas others represented distinct mutations found only in the metastatic lesions8,18,19. The key consideration for the clinician is when to biopsy a metastatic deposit and how to use the information gained from genetic analysis. The identification of a new or different mutation in a metastasis might indicate the need to change therapy, add additional therapy or treat a rogue metastatic lesion with local therapy if it is believed to be an isolated event in a patient with otherwise adequate systemic disease control.
Temporal heterogeneity.
Temporal heterogeneity describes changes in the tumour over time. Urothelial carcinomas are inherently genetically unstable and new mutational events occur frequently, which can accumulate over time20,21. Temporal heterogeneity is more likely to affect patients with metastatic cancer22. One practical consideration for clinicians and when planning a clinical trial is to determine when archival tissue is acceptable or when a new biopsy is needed. Clinically, the answer is sometimes self-evident, for example, in a patient with a tissue sample from many years ago who now has recurrent disease a biopsy is likely to be part of the assessment of new drivers in the metastases. In trials that involve evaluation of genomic alterations, archival tissue for analysis is commonly allowed if available from within a certain time period (for example, from the past 12 months)23. Clinicians are hesitant to pursue re-biopsy owing to the potential medical risks, discomfort to the patient and cost24. The rapidly expanding availability and utility of blood-based and urine-based liquid biopsies might obviate this problem25,26.
Circulation heterogeneity.
Circulation heterogeneity refers to differential genomic profiles of circulating DNA compared with tissue from the primary or metastatic site27,28. Liquid biopsy is the measurement of the cell-free DNA in the blood29 but can be extended to include circulating tumour cells30. The utility of liquid biopsies is rapidly improving as technological advances are made and they are increasingly used in both clinical and trial settings31. Several important questions regarding circulation heterogeneity in urothelial carcinoma currently remain unanswered. For example, whether direct comparison of genomic alterations and allele frequencies between tissue, urine and blood samples is possible; which platforms, tumour types and tumour burdens are best evaluated by blood assays; and whether integrating data from multiple tumour sites with divergent evolution is meaningful. Much work is being done to address these questions and developments in blood-based assays now provide the opportunity for longitudinal and frequent assessment, especially of specific mutational events that can be targeted with therapy at progression32.
Heterogeneity and branching evolution
Urothelial carcinoma is characterized by a high total mutational burden of >7 mutations per Mb — only exceeded by lung and skin cancer33. This high mutation rate is believed to fuel tumour heterogeneity and tumour evolution (FIG. 2). Several studies have examined the evolutionary dynamics of urothelial carcinoma2,17,34–36. Mutational analyses of early-late tumour pairs identified the existence of a single ancestral origin within each assessed patient demonstrated by identical mutations at a high cellular prevalence in the primary and invasive tumour pairs. Furthermore, subclonal mutations that were specific to the individual tumours were identified34. Branched evolution was also found as an early event in the natural history of urothelial carcinoma with metastasis. Phylogenetic analysis of 21 sets of matched early and late tumours showed that the ancestral clone gave rise to multiple cell populations that evolved in parallel during the early stages of tumour evolution17. A high level of intertumoural heterogeneity between primary tumours and metastases was also seen in another study37. Evaluation of molecular features of metachronous tumours from 29 patients initially diagnosed with early-stage bladder cancer revealed a common origin of the metachronous tumours that developed years later. Tumours from patients with progressive disease had a higher variation in the intrapatient mutational spectrum and a higher frequency of APOBEC-related mutations than those from patients with non-progressive disease2. Genomic studies have shown a significant difference in the number and frequency of individual mutations and rearrangements between ancestral and progressive clones4. Frequent mutations of tumour suppressors and oncogenes, including in KDM6A, TP53, PIK3CA and FGFR3, were found in the ancestral clone, whereas mutations in TP53, MLL3, FBXW7 and SETD2 were found in progressing clones2,34. Non-aggressive subclones in early tumours harboured mutations (in FGFR3, AFDN and H3F3A) that were absent in invasive clones. Fibroblast growth factor (FGF) signalling was enriched in ancestral clones of early invasive urothelial carcinoma with PIK3CA mutations, whereas DNA damage checkpoint regulation signalling was enriched in progressing subpopulations34. The differences in genes altered before and after progression suggest that tumour evolution continues as a function of time.
Fig. 2 |. Tumour evolution with emergence of distinct tumour subtypes.
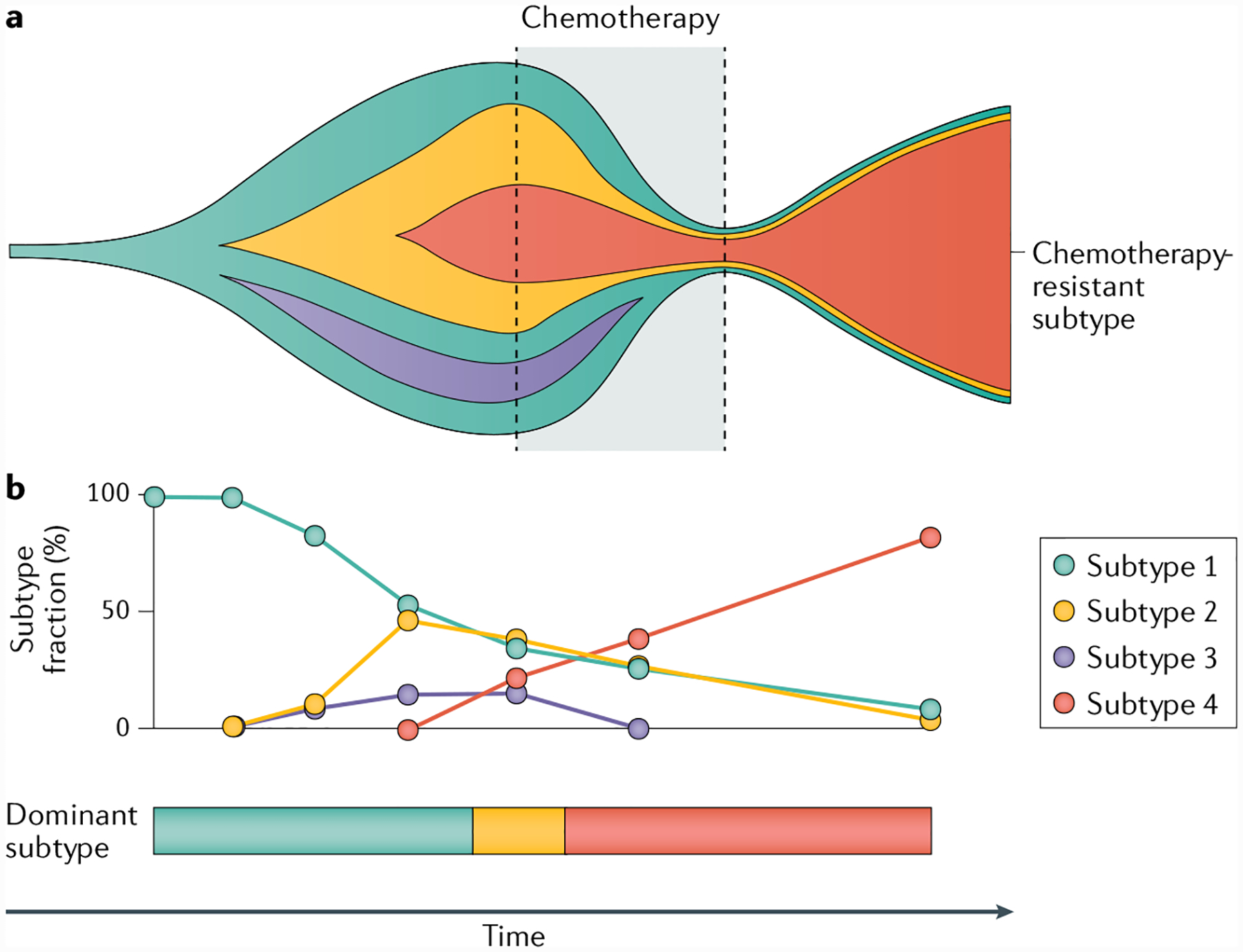
a | Schematic of the occurrence of tumour subclones over time. Genomic differences between subclones might result in different expression patterns and thereby different tumour subtypes. Chemotherapy can cause contraction of subtypes and isolation of a specific subtype, which becomes the dominant clone after chemotherapy (subtype 4 in this example). b | The fractions of different subtypes can vary over time and under the selection pressure of treatments, resulting in inconsistent subtype calling even if the entire tumour is analysed. Sampling of only parts of a tumour would be expected to further complicate consistent subtype calling.
Chemotherapy-driven clonal evolution.
The effect of systemic therapies on the evolutionary trajectory of urothelial carcinoma (FIG. 2) was studied by comparing the genomic profiles of samples from matched untreated and chemotherapy-resistant tumours from individual patients17. Whole-exome sequencing and clonality were estimated in tumour analyses of 16 matched chemotherapy-naive and cisplatin-treated tumours. Only one-third of the mutations were shared within the tumour pairs, demonstrating mutational heterogeneity for each pair. Reconstructing the phylogenetic relationship of each patient’s samples revealed early branching evolution occurring in successive waves of clonal expansion. The observed increase in the clonality of mutations found in post-chemotherapy tumours suggested that chemotherapy restricted the mutational landscape of the tumour17. Chemotherapy-resistant tumours were enriched in genes involved in integrin signalling, which is linked to cell-adhesion-mediated survival and drug resistance38. Increased activity of integrin signalling pathways is a possible shared link between drug resistance and metastatic spread of urothelial cells. These findings are consistent with mathematical models showing that even a small advantage in a single cell under selective pressure from chemotherapy can enable the descendants of this resistant cell to replace the precursor tumour mass, thereby increasing clonality and restricting mutational heterogeneity39. A study of the mutational patterns in chemotherapy-resistant muscle-invasive urothelial carcinoma using whole-exome sequencing of matched samples from 30 patients before and after neo-adjuvant cisplatin-based chemotherapy identified a new cisplatin mutation signature, which was linked to 14% of mutations in treated tumours, supporting the idea that chemotherapy shapes the mutational landscape of urothelial carcinoma6. The cisplatin mutation signature is enriched in T>A and C>A mutations compared with other mutational signatures, such as the APOBEC (C>T) and mutation signature 5 (comparatively flat signature with minimal signature peaks). Collectively, these data suggest that systemic chemotherapy for urothelial carcinoma affects the evolution of a cancer, constraining the clonality, and ultimately leading to treatment resistance.
APOBEC3-related mutagenesis.
The true initiating steps of bladder cancer are unknown, but the development of genomic mutations is likely to have a fundamental role. Compared with other solid tumours, the high mutational burden in urothelial carcinoma might be partly driven by enzymatic activity. The DNA-editing enzyme apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC3) family consists of seven enzymes34,40. These enzymes are members of a super family of evolutionarily conserved deaminases, including activation-induced cytidine deaminase, APOBEC1, APOBEC2 and APOBEC4 (REF.41). APOBEC3 enzymes are known for their role in restricting viruses by editing viral DNA42. APOBEC3-induced DNA editing is caused by the deamination of cytidines (C) to uridines (U), which are repaired to guanines (G) or thymidines (T)41. Each of the seven human APOBEC3 para-logues has a preferred cytidine-harbouring motif. For instance, APOBEC3B preferentially deaminates cytidines in a TCW motif (in which W can be A or T)43. APOBEC3-induced mutational signatures are prevalent in bladder, cervical, breast, head and neck, and lung cancers44. Analysis of gene expression data and mutation patterns, distributions and loads of 19 different cancer types showed that APOBEC3B-catalysed genomic uracil lesions are responsible for a large proportion of mutations in urothelial carcinoma45.
The frequency of APOBEC mutational signatures found at all stages of bladder cancer provides evidence for a role of APOBEC in this disease40. APOBEC3 mutational signatures become enriched during progression from early-stage to muscle-invasive urothelial tumours2. These signatures have been specifically identified in high-risk NMIBC and potentially fuel tumour progression and evolution, even in these early stages46. Analysis of the mutational signatures from The Cancer Genome Atlas (TCGA) muscle-invasive urothelial carcinoma patient cohort show that both APOBEC3A and APOBEC3B signatures accounted for 67% of single nucleotide variations40. Patients with APOBEC-enriched tumours had a better prognosis (median survival 50 months versus not reached, 1.5 × 10−4)40. Whether this improved survival reflects better response to treatment is not known.
APOBEC3A and APOBEC3B expression levels also correlate with APOBEC3-associated mutational load47. Several studies suggest that APOBEC3-associated mutations have a role in shaping urothelial carcinoma evolution. More than 40% of clonal mutations in cancer driver genes of several tumour types, including urothelial carcinoma, were found to have APOBEC-signature enrichment1. In urothelial carcinoma, 62% and 75% of mutations associated with APOBEC3A and APOBEC3B are clonal, respectively, suggesting that the majority of APOBEC3 signature mutations occur early in urothelial carcinoma evolution40. Mutational signatures associated with APOBEC3A and APOBEC3B were enriched in urothelial carcinoma after cisplatin-based chemotherapy17. APOBEC3 activity is enriched in lagging DNA strands in early-replicating, gene-dense and active chromatin regions and it is plausible that conditions that increase the abundance of single-strand DNA, such as chemotherapy, could increase the substrate availability for APOBEC3-induced mutagenesis43,48–53. These data suggest a potential interaction between chemotherapy and APOBEC3-induced mutagenesis in shaping the evolutionary landscape of urothelial carcinoma. In addition, previous findings in breast cancer models suggest that APOBEC3B promotes tamoxifen resistance54. Whether APOBEC3 enzymes have similar roles in treatment resistance in urothelial cancer remains to be determined.
Gene expression heterogeneity
The substantial intertumoural heterogeneity identified in urothelial carcinoma might be driven by variations in cell cycle activity and cellular differentiation programmes between patients55. To classify the differences in gene expression between urothelial carcinomas, a system of molecular subtypes was proposed, with distinct molecular characteristics and associations with pathological findings and clinical outcome40,46,55. The characteristics and evolution of subtypes in NMIBC and MIBC have been previously described13,56 and, in MIBC, a consensus classification of six subtypes is emerging, which includes luminal papillary, luminal non-specific, luminal unstable, stroma-rich, basal squamous, neuronal-like subtypes57. Some studies have observed molecular subtypes to be associated with response to therapeutic treatment, but conflicting results have been reported and no consensus exists58–62.
Interpatient heterogeneity is likely to be caused by a range of underlying DNA changes (mutations, rearrangements, insertions or deletions, long non-coding RNAs and methylations) accumulated during the evolution of each cancer, but observed differences might also be a product of varying cell-type compositions in the analysed tissue sample. The constant evolution of the cancer genome generates new genomic subclones63 that might give rise to differences in gene expression patterns within the tumour. However, most analyses of molecular subtypes are based on the assumption that no intratumoural heterogeneity exists and try to assign a single subtype to each tumour. Multiple subtype classification studies have reported unclassified samples and varying classification strengths (for example, silhouette width measures64), suggesting that substantial intratumoural heterogeneity in gene expression exists. This heterogeneity might also be caused by undiscovered subtypes, but, on the basis of current knowledge of genomic evolution, several subtypes are likely to coexist in single tumours.
Studies of temporal intratumoural heterogeneity have shown differences in tumour classifications. In a study from 2005 of array-based gene expression analysis in metachronous tumours, most paired tumour samples had similar expression patterns but several exceptions were found65. In 9 of 14 patients, tumours at presentation had gene expression similar to tumours at progression. Across all patients, gene expression of early-stage and late-stage tumours from an individual patient was more similar than that of tumours at the same stage across patients. New studies of synchronous and metachronous tumours had similar results2,46,66. A study in 57 primary MIBCs and 28 matched lymph node metastases found only 18% discordance (12 of 67 pairs) in subtype classification between primary tumours and metastases overall but 58% discordance (7 of 12 pairs) in the basal/squamous-like subtype67. In 6 of these 12 discordant pairs, the primary tumour showed substantial intratumoural heterogeneity, including mesenchymal, genomically unstable and small-cell neuroendocrine subtype regions. Thus, discordance might be caused by intratumoural heterogeneity in half of these pairs and the rest might reflect subtype plasticity.
In a study of spatial intratumoural heterogeneity in four patients68, the authors identified both luminal-like and basal-like gene expression subtypes in laser-microdissected tumour tissue from cystectomy specimens68. Importantly, different subtype classifications were only observed in the two patients with multifocal tumours. In one patient, basal-like expression patterns were observed in a muscle-invasive tumour and luminal-like expression patterns in synchronous Ta tumours. However, in the second patient with multifocal MIBC, the luminal-like and basal-like expression subtypes were intermixed in different areas of the individual tumours. Overall, the gene expression differences mirrored genomic alterations in tumour biopsy samples, suggesting that gene expression patterns might be founded in DNA alterations. In TCGA MIBC data set, associations of mutation patterns specifically with a tumour subtype were limited, although luminal papillary tumours were enriched in FGFR3 alterations56. Histone regulation might have a role in subtype development, as luminal tumours have an increased frequency of KDM6A mutations, which are also found in low-grade (papillary) tumours. Thus, subtypes might be affected by genetic and epigenetic mechanisms of regulation.
Collectively, intratumoural heterogeneity complicates gene expression subtyping of a subset of bladder tumours, and average bulk tumour estimates might confound subtype analysis (for example, for therapy response estimation), and more advanced approaches to subtyping might be required. In colorectal cancer, a meta-analysis of expression subtypes documented evidence for a continuous subtype score instead of the traditional subtype association approach69. The high mutation rate and intratumoural heterogeneity observed in bladder cancer suggests that differences in gene expression patterns are likely to arise within the tumour during its development; hence, a similar approach for subtyping of bladder tumours might be useful. However, further studies are required to evaluate whether continuous scores might provide a more clinically relevant classification of bladder tumours than the current subtype likelihood scores.
The rapid developments in single-cell sequencing approaches might enable better delineations and more granular definitions of subtypes than those from bulk tumour analyses. No large studies of single cell RNA sequencing (RNA-seq) have been published for bladder cancer, but, in colorectal cancer, specific T cells, identified by single-cell analysis, were found to be preferentially enriched in patients with microsatellite-instable tumours, which might explain favourable responses to immune checkpoint blockade in patients with these tumours70. In melanoma, single-cell RNA-seq revealed a resistance programme in malignant cells that is associated with T cell exclusion and immune evasion71. Specific cell subpopulations will probably not be identified from bulk tumour analysis and the high-resolution cellular maps of tumours generated from single-cell analysis might be crucial for an improved understanding of gene expression heterogeneity and subtype differences and for identification of better predictive biomarkers.
Morphology of heterogeneity
The genomic and transcriptional drivers that cause intratumoural heterogeneity are ultimately reflected in the varied histological morphologies of urothelial carcinoma. This heterogeneity can be observed in tumours that include more than one histological appearance within the same tumour. These morphologies reveal the wide spectrum of morphological heterogeneity in urothelial cancer and ultimately affect how a tumour responds to treatment72–75. Variants of urothelial carcinoma are divided on the basis of their microscopic morphological features, but increasing knowledge of the genetic and transcriptional attributes of histological variants have led to a better understanding of the molecular features associated with a subset of these lesions. The morphological spectrum of urothelial carcinoma includes divergent differentiation, such as squamous and glandular, as well as variant histologies such as nested, plasmacytoid, micropapillary, sarcomatoid and small-cell (neuroendocrine) carcinoma. Rarely, tumours of non-urothelial histology can develop in the bladder, such as squamous cell carcinoma and adenocarcinoma (FIG. 3).
Fig. 3 |. Variant histology of urothelial carcinoma.
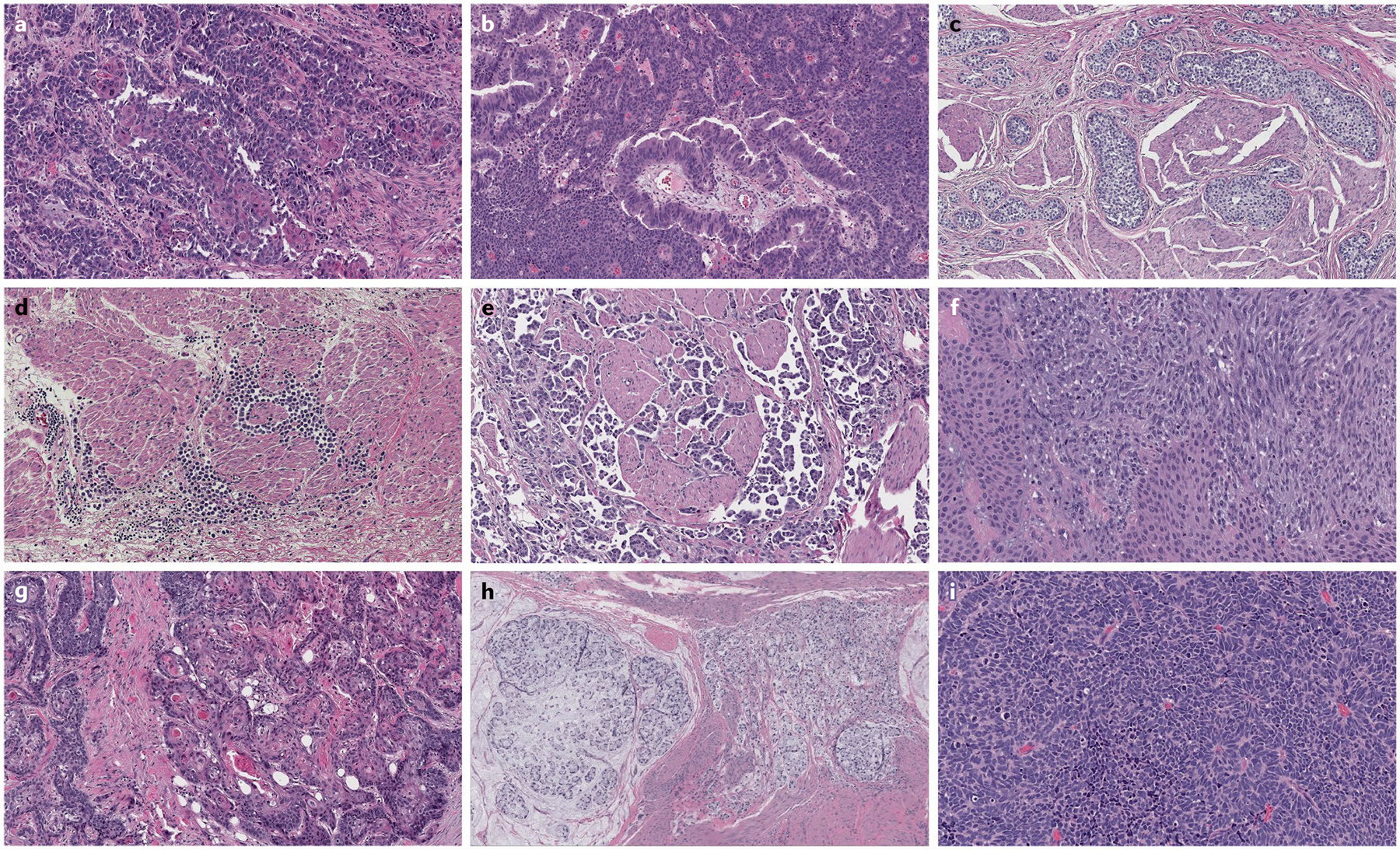
Tumour heterogeneity is most pronounced at the morphological level when comparing urothelial carcinomas with variant histology. The morphological spectrum of urothelial carcinoma includes divergent differentiation, such as squamous differentiation (part a) and glandular differentiation (part b). Variant histologies of urothelial carcinoma include nested variant (part c), plasmacytoid variant (part d), micropapillary variant (part e) and sarcomatoid variant (part f). Primary tumours of non-urothelial histology can also develop in the bladder, including squamous cell carcinoma (part g), mucinous adenocarcinoma with signet ring cells (part h) and small-cell or neuroendocrine carcinoma (part i). Haematoxylin and eosin staining in all images, magnification ×50.
Urothelial carcinoma with divergent differentiation.
The two most common variants of divergent differentiation in urothelial carcinoma are squamous and/or glandular differentiation76. Squamous differentiation is the most common variant, occurring in up to 30% of high-grade and/or high-stage urothelial carcinomas. In urothelial carcinoma with squamous differentiation, expression profile analysis of separate areas of urothelial and squamous morphology in the same tumour classified urothelial areas as luminal and squamous areas as basal/squamous in a subset of cases4,77. An association of squamous differentiation with human papillomavirus infection has been explored, but little genomic information exists to support a viral origin72,75. However, rarely, such as in patients with neurogenic bladder dysfunction or those requiring repeated catheterization, a viral aetiology has been identified, supported by p16 expression and human papillomavirus in situ hybridization78,79.
The presence of glandular differentiation in urothelial carcinoma is less common than squamous differentiation (8–18% of high-grade tumours)80. Morphologically, the glandular component resembles adenocarcinomas of other organs, most commonly showing features of enteric or colonic adenocarcinoma, but can also rarely resemble mucinous or various mixed types of glandular morphology. One analysis of the molecular features of glandular differentiation in urothelial carcinoma has revealed high rates of hotspot mutations in the TERT promoter region, similar to urothelial carcinoma ithout glandular differentiation81.
Nested urothelial carcinoma.
This rare variant of urothelial carcinoma belongs to a group of morphologically deceptively bland tumours that can be associated with an aggressive clinical course82. The morphological characteristics of nested urothelial carcinoma include the presence of invasive clusters of tumour cells without considerable morphological atypia or generally not associated with a stromal reaction. Identifying this variant can be challenging, as it is shares features with benign conditions, such as proliferative cystitis, von Brunn nest hyperplasia, nephrogenic adenoma or inverted papilloma83,84. To date, a high rate of TERT promoter mutations, which was not found in benign mimickers, was the only molecular finding in this tumour type85, suggesting that it has molecular alterations similar to those of urothelial carcinoma in general.
Plasmacytoid urothelial carcinoma.
The plasmacytoid variant of urothelial carcinoma is another rare but aggressive tumour primarily composed of discohesive, infiltrating cells that resemble plasma cells, mostly admixed with other cells that contain intracytoplasmic vacuoles, giving the appearance of signet ring cells86–88. Patients with this tumour typically present at an advanced stage and have low survival. This tumour is also associated with high relapse rates and frequent peritoneal carcinomatosis, although patients might initially respond to chemotherapy86–90.
This tumour shares immunohistochemical and genetic features with urothelial carcinoma. It frequently expresses markers of urothelial differentiation, such as CK7, p63, GATA3 and uroplakins, and generally has genetic alterations similar to those of urothelial carcinoma, such as mutations in TP53, RB1, KMT2D and ARID1A86–90. However, in contrast to urothelial carcinoma, loss-of-function mutations in CDH1, and less commonly promoter hypermethylation of CDH1, drive the development of this variant and contribute to its aggressive biology86. Targeted next-generation sequencing of macrodissected areas of plasmacytoid and urothelial histologies from the same tumour revealed shared mutations, suggesting that both arose from the same origin, but CDH1 mutation was limited to the plasmacytoid component, supporting the role of CDH1 loss in the development of this variant histology86.
Micropapillary urothelial carcinoma.
The designation of micropapillary carcinoma requires the presence of small tight tumour clusters without true fibrovascular cores located within clear spaces, which is the result of reverse cellular orientation or polarization91–93 and a lack of cohesion between tumour and stroma. This tumour type is generally associated with high rates of ERBB2 alterations, mostly amplification94 and less commonly mutations95. Morphological intratumoural heterogeneity in micropapillary carcinoma is common, as most of these tumours also contain a component of not otherwise specified (NOS) urothelial carcinoma72. In addition, intratumoural heterogeneity of ERBB2 amplification is common. In tumours with mixed micropapillary and NOS urothelial carcinoma, ERBB2 amplification was more common in micropapillary than NOS urothelial carcinoma components96. Additionally, the rate of ERBB2 amplification in the NOS urothelial carcinoma component associated with micropapillary components was much higher than that in NOS urothelial carcinoma not mixed with micropapillary components40,97,98.
Sarcomatoid urothelial carcinoma.
When a component of urothelial carcinoma takes the form of a mesenchymal neoplasm, the tumour is designated sarcomatoid urothelial carcinoma, which is a rare form of bladder cancer that is typically associated with an advanced stage and overall poor prognosis99. The presence of a sarcomatous component in urothelial carcinoma does not exclude other variant histologies, as tumours with urothelial, glandular, squamous and/or small-cell or neuroendocrine differentiation have been reported72. The most common morphology of the sarcomatous component is that of spindle-cell proliferation, but it can also include myxoid, pseudoangiosarcomatous, undifferentiated pleomorphic sarcoma-like morphology, and true heterologous elements in the form of cartilaginous, osseous and other elements100. The sarcomatous and urothelial components within the same tumour have been reported to share a common clonal origin101. A comprehensive study of sarcomatoid urothelial carcinoma showed that this tumour type is enriched with mutations in TP53, RB1 and PIK3CA and is associated with dysregulation of the epithelial-mesenchymal transition pathway and overexpression of epithelial-mesenchymal transition markers100,102.
Small-cell (neuroendocrine) carcinoma.
Small-cell carcinoma is a rare form of urothelial carcinoma and can be admixed with a urothelial (invasive or non-invasive), glandular, squamous of sarcomatous component72. Its morphological appearance is similar to small-cell carcinomas of other organs; similarly, it commonly harbours co-alterations in both TP53 and RB1 (REFS97,103,104). However, TP53 and RB1 mutations are insufficient to explain development of small-cell carcinomas of the bladder, as these genetic alterations also often occur in urothelial carcinoma that does not exhibit features of small-cell or neuroendocrine differentiation. Furthermore, other alterations detected in urothelial carcinoma are also found in small-cell carcinoma of the bladder, in contrast to small-cell carcinoma of other organs. These aberrations include TERT promoter mutations in ~95% of samples and truncating alterations within chromatin remodelling genes, such as CREBBP, EP300, ARID1A and KMT2D, in nearly 75% of samples104. A high level of chromosomal instability is observed in bladder small-cell carcinoma, including whole genome duplication in 72% of tumours. Similar to MIBC, the APOBEC mutation signature is present in 95% of small-cell carcinoma of the bladder20, which contrasts with small-cell carcinoma of the lung, whose mutation signature is typically associated with tobacco exposure104. These findings suggest that bladder small-cell carcinoma develops through transdifferentiation from urothelial carcinoma, but the exact molecular mechanisms for this transition are not yet clear20,97,105. In contrast to, for example, prostate cancer, where neuroendocrine differentiation almost always develops in the setting of androgen receptor-directed therapy106, neuroendocrine differentiation in the bladder seems to develop de novo. Studies based on RNA and immunohistochemical expression profiling have identified a subtype of bladder cancer that is enriched in neuroendocrine markers but does not have the microscopic appearance of a true small-cell or neuroendocrine carcinoma107,108. This subtype has been referred to as “neuronal subtype” by TCGA classification40 and “neuroendocrine-like” by a consensus clustering recommendation of the Bladder Cancer Molecular Taxonomy Group57. Whether this subtype represents an early step in the development of frank neuroendocrine carcinoma is yet to be determined.
Adenocarcinoma.
The presence of pure glandular morphology is required for the diagnosis of primary adenocarcinoma of the bladder, which can develop anywhere in the bladder. If such a tumour develops in the bladder dome and is associated with a urachal remnant, it will be designated as urachal adenocarcinoma109. Most of these tumours resemble colorectal adenocarcinomas, but they can also resemble adenocarcinoma of any other organs. Genetically, adenocarcinomas are different from urothelial carcinoma, as they generally lack mutations in chromatin-modifying genes and the TERT promoter region and resemble a subset of colorectal adenocarcinoma that is enriched in mutations in TP53, KRAS and SMAD4, as well as a small subset with EGFR and ERBB2 amplification110–112.
Heterogeneity, systemic therapy and drug design
Tumour heterogeneity complicates systemic therapy, drug development and delivery owing to the potential variable response to therapeutics in different tumour regions8,113. Relapse is often associated with the emergence of resistance and resistance can occur because of intratumoural heterogeneity, and not only because of drug resistance mechanisms or the presence of natively resistant populations, such as cancer stem cells114,115. Subtyping of muscle-invasive urothelial carcinoma on the basis of gene expression profiles provides an opportunity for personalized medicine4,55,58. Similarly, expression profiles of NMIBC might enable subclassification of these tumours, but the therapeutic implications have not yet been explored46. Histological variants of MIBC, such as the micropapillary variant, might have distinct genetic profiles (for example, ERBB2 overexpression), but whether these correlate with response to certain therapeutics (for example, HER2-targeted agents) has not been determined116. Defining which subtypes represent well-delineated groups, either natively or after therapy, remains a priority in the field.
Neoadjuvant chemotherapy and chemoradiotherapy.
Cisplatin-based regimens that comprise multiple agents are the most effective chemotherapy in advanced urothelial carcinoma117. Following evidence of improved survival of patients with metastatic bladder cancer treated with cisplatin chemotherapy, prospective randomized trials have demonstrated improved survival of patients with MIBC treated with neoadjuvant cisplatin-based chemotherapy118,119. Neoadjuvant chemotherapy is the current standard of care, but positive responses to immune checkpoint inhibitors (ICIs) in patients with MIBC and metastatic urothelial carcinoma have altered the neoadjuvant landscape to include immunotherapy120. Thus, determining the response of each tumour subtype to neoadjuvant chemotherapy and/or immunotherapy will be critical in determining the most efficacious precision therapy. For example, patients with basal-subtype tumours have the most improved survival benefit from neoadjuvant chemotherapy (3-year overall survival without neoadjuvant chemotherapy 49.2% versus 77.8% with neoadjuvant chemotherapy)59, whereas those with luminal tumours treated without systemic therapy have the lowest rate of upstaging compared with those with nonluminal tumours (34% versus 51%)121. Subtype assignment is usually considered absolute, but some subtypes are more ‘stable’ than others, meaning that repeat bio-informatic analysis of tumours with these designated subtypes is likely to result in the same subtype assignment. By contrast, subtypes such as luminal infiltrated tumours contain varying amounts of stroma and immune cells and are ‘unstable, with an increased likelihood of being designated as other subtypes in repeat clustering4,68. Thus, complexity at the cellular level is likely to affect subtype membership, which might have the greatest influence on the choice of neoadjuvant systemic therapy.
Targeted therapy.
Relatively few MIBCs are driven by single-gene drivers, with the exception of FGFR3, RAS and PPARG, which are predominantly found in luminal-subtype tumours40. BLC2001, a phase II dose-escalation study of the FGFR3-targeted agent erdafitinib in 99 patients, demonstrated an overall response rate of 34% and a median duration of response of 5.5 months in patients with metastatic urothelial cancer that harbours FGFR3 mutations and overexpression122,123. On the basis of these results, erdafitinib was approved by the FDA in 2019 for the treatment of patients with locally advanced or metastatic bladder cancer with FGFR3 or FGFR2 alterations and has progressed following platinum-containing chemotherapy122. In addition, in a phase II trial, the pan-FGFR inhibitor infigratinib demonstrated a 25.4% response rate and a 38.8% disease stabilization rate in patients with metastatic urothelial carcinoma and FGFR alterations31. Response to small molecular therapies targeting mutations or activation of FGFR might depend on intratumoural heterogeneity. For example, an evaluation of 27 MIBCs found FGFR3 alterations in ~30%, but only ~15% had FGFR3 alterations at deep tissue levels124. In addition, activating mutations of PPARG are common in MIBC (up to 15% of patients)125. Targeting of PPARG in urothelial carcinoma cell lines showed that inverse agonism of PPARG reduced proliferation rates of PPARG-mutant cells but not PPARG wild-type cells125, pointing to another strategy by which patients with luminal tumours might benefit from targeted therapy in addition to established benefit from chemotherapy125.
Response to immunotherapy.
Immunotherapy is contingent upon T cell infiltration in response to neoantigen expression61,126. Neoadjuvant immunotherapy in melanoma indicates that ICIs alter intratumoural heterogeneity, in part through selection of specific populations with a low antigen load, and might be more efficacious in regions with high immunogenicity127. In the phase II trial PURE-01 in patients with MIBC, treatment with the ICI pembrolizumab before cystectomy resulted in a reduction in total mutation burden or neoantigens in matched tumour samples, indicating removal of tumours with high mutational burden128. Thus, one conceivable mechanism of developed resistance to ICIs is a change in the neoantigen burden and the type of alterations found across a tumour after ICI treatment.
Clinical trial considerations
As our understanding of the molecular features that differentiate types of bladder cancer improves, biomarkers are likely to have an expanded role in future clinical trials. To improve their accuracy, the reliability of both prognostic and predictive biomarkers in the setting of tumour heterogeneity is being investigated. Clinical decisions are made on the presence or absence of a biomarker and the accuracy of this biomarker to represent a treatment response of the tumour is critically dependent on the heterogeneity of the tested sample. One contemporary example in bladder cancer is the SWOG S1314 (co-expression extrapolation) study129. This randomized phase II trial includes 167 evaluable patients with non-metastatic MIBC and uses an Affymetrix gene expression model. Two separate models were tested (one for gemcitabine plus cisplatin (GC) and one for a dose-dense combination of methotrexate, vinblastine, doxorubicin and cisplatin (ddMVAC)) as prognostic and predictive biomarkers in these patients randomized to GC or ddMVAC chemotherapy regimens. Early data suggest that the individual GC and ddMVAC biomarkers do not predict response in their individual treatment arms but that the GC biomarker predicts response when applied to the larger, combined cohort of all patients (HR 2.33, 95% CI 1.11–4.89; P = 0.02)130. Future studies are essential to validate these results.
The S1314 study identified the challenges associated with heterogeneity that need to be addressed to optimize future biomarker-oriented bladder cancer clinical trials in the neoadjuvant setting. First, adequate tumour sampling, including depth of biopsy and multiple regions, will be required to evaluate the biomarker across multiple tumour sites. Depending on the tumour stage, a minimum tumour size or specifying the amount of viable tumour might be necessary. This might be easier for MIBC than for NMIBC, owing to the tumour burden, whereas the challenge in metastatic disease will be obtaining adequate tissue from needle biopsies. In multicentre trials, engaging local pathologists might also be advantageous, for example, to enable separate verification of an adequate amount of viable tissue as part of determining eligibility for inclusion in the trial. Second, the use of archival tissues versus the requirement for new tissue biopsy is an important consideration. Biologically, current assessment is desirable, as it minimizes aspects of temporal heterogeneity. However, in practice, requiring a new tissue biopsy of a metastatic site might put patients at increased risk and potential discomfort, making the patient less likely to participate in the trial.
The use of blood-based biopsies in clinical trials is quickly emerging as a new option131. This approach has the potential to obviate many concerns around the assessment of temporal heterogeneity, as contemporary sampling is easier than with tissue biopsies. In addition, the utility of urine analysis for tumour DNA continues to be investigated. This approach might enable direct measurement of tumour burden and identify specific dominant clones. For example, in patients with NMIBC, serial evaluation of urine specimens might identify drivers of persistence and recurrence during intravesical therapy. The utility of this strategy depends on the identification of cancer drivers and heterogeneity in NMIBC. For example, the currently limited genomic evaluation of Ta and T1 tumours suggests that FGFR3 alterations are a dominant driver, but the clonality of this mutation during selective intravesical treatment remains unclear132. However, the handling of urine specimens requires special methods to avoid lysis of white blood cells and preferentially sampling urine, for example, in the morning to avoid increased dilution of cell-free DNA. Furthermore, identification of novel actionable targets through liquid biopsy analysis has been demonstrated in patients with metastatic bladder cancer and could complement current patient selection procedures for clinical trials. A study in 68 patients with MIBC demonstrated an association between circulating tumour DNA fluctuations and chemotherapy response, which might enable more efficient treatment response evaluations in clinical trials than the current standard of radiographical imaging133. Thus, blood-based evaluation of circulating DNA might be a dynamic method of capturing a snapshot of tumour heterogeneity and inform changes in treatment owing to tumour evolution.
Conclusions
Our current understanding of the development and progression of urothelial carcinoma at the genetic and molecular levels indicates that single-agent therapy is unlikely to be successful, predominantly owing to intratumoural and intertumoural heterogeneity. This heterogeneity occurs at multiple levels and is best demonstrated in tumours with variant histology. Therapeutic targeting of the primary and metastatic lesions might require analysis of solid tumour or liquid biopsy samples to identify the evolving landscape of clones. Metastatic cells might have different subtypes and therapy might need to be further tailored as we begin to understand these populations. New therapeutics are still needed as response rates to agents currently in clinical trials remain suboptimal. As new drug candidates are being developed, designing trials that include those patients who are most likely to benefit and developing biomarkers to define these patients will be imperative.
Key points.
Bladder cancers have a high total mutational burden and considerable intratumoural and intertumoural heterogeneity at the genomic, transcriptional and cellular levels that remain difficult to quantify.
Heterogeneity might be driven by genomic events initiated by APoBEC enzymes and selection pressure from therapeutic interventions, which both drive tumour evolution.
Bladder tumours can be categorized into different subtypes on the basis of gene expression signatures, but these molecular subtypes might be unstable and different subtypes can occur within the same tumour causing intratumoural heterogeneity.
variant tumour histologies are the morphological extreme of tumour heterogeneity and include glandular, squamous, nested, plasmacytoid, micropapillary, sarcomatoid and small-cell carcinoma.
Tumour heterogeneity might affect treatment efficacy, for example, of neoadjuvant chemotherapy and immune checkpoint inhibitors, as well as targeted therapy, for example, when individual actionable mutations only occur in a fraction of the tumour.
Biomarkers to select personalized treatments in precision medicine approaches will likely shape future clinical trial design, but their validity might be affected by heterogeneity.
Acknowledgements
The authors acknowledge the Leo and Anne Albert Institute of Bladder Cancer Research. J.J.M. is supported by a VA Merit Award (BX003692-01) and a Department of Defense Grant (W81XWH-18-0257). J.J.M and L.D. are supported by an award from the Leo and Anne Albert Institute of Bladder Cancer Research. H.A. is supported by a Sloan Kettering Institute for Cancer Research Cancer Center Support Grant P30CA008748 and SPORE in Bladder Cancer P50CA221745. B.M.F. is supported by the Department of Defense CDMRP Career Development Award (grant CA160212). D.J.D. is supported in part by RSG 17-233-01-TBE from the American Cancer Society. B.L.W. is supported by an American Urological Association Research Scholar Award.
Footnotes
Competing interests
B.M.F. received grant/research support from Eli Lilly and Company and participated in an Advisory Board for Immunomedics. J.J.M. received research support from Epizyme, Tesaro and Abbvie, is a consultant for Ferring and AstraZeneca, and participated in advisory boards for Cold Genesys and Janssen. H.A. is a consultant for AstraZeneca/MedImmune, Bristol-Myers Squibb and EMD Serono. L.D. received research support from Ferring and Natera, and is a consultant for Ferring.
References
- 1.McGranahan N & Swanton C Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Lamy P et al. Paired exome analysis reveals clonal evolution and potential therapeutic targets in urothelial carcinoma. Cancer Res. 76, 5894–5906 (2016). [DOI] [PubMed] [Google Scholar]
- 3.da Costa JB et al. Molecular tumor heterogeneity in muscle invasive bladder cancer: biomarkers, subtypes, and implications for therapy. Urol. Oncol 10.1016/j.urolonc.2018.11.015 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Warrick JI et al. Intratumoral heterogeneity of bladder cancer by molecular subtypes and histologic variants. Eur. Urol 75, 18–22 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Ma G et al. Precision medicine and bladder cancer heterogeneity. Bull. Cancer 105, 925–931 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Liu D et al. Mutational patterns in chemotherapy resistant muscle-invasive bladder cancer. Nat. Commun 8, 2193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jamal-Hanjani M et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Gerlinger M et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo M et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. CancerDiscov. 6, 147–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks E, Rizvi SM, Sarwani N, Yang Z & El-Deiry WS A case of heterogeneous sensitivity to panitumumab in cetuximab-refractory colorectal adenocarcinoma metastases. Cancer Biol. Ther 16, 377–382 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pauli C et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 7, 462–477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babjuk M et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2016. Eur. Urol 71, 447–461 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Dyrskjøt L & Ingersoll MA Biology of nonmuscle-invasive bladder cancer: pathology, genomic implications, and immunology. Curr. Opin. Urol 28, 598–603 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Hurst CD & Knowles MA Mutational landscape of non-muscle-invasive bladder cancer. Urol. Oncol 10.1016/j.urolonc.2018.10.015 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Gerlinger M et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet 46, 225–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epstein JI et al. The 2014 International Society of Urological Pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma: definition of grading patterns and proposal for a new grading system. Am. J. Surg. Pathol 40, 244–252 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Faltas BM et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet 48, 1490–1499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yachida S et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPherson A et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet 48, 758–767 (2016). [DOI] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Findlay JM et al. Differential clonal evolution in oesophageal cancers in response to neo-adjuvant chemotherapy. Nat. Commun 7, 11111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McQuerry JA, Chang JT, Bowtell DDL, Cohen A & Bild AH Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J. Mol. Med 95, 1167–1178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meeks JJ, Goldkorn A, Aparicio AM & McConkey DJ Development of a translational medicine protocol for an NCTN genitourinary clinical trial: critical steps, common pitfalls and a basic guide to translational clinical research. Urol. Oncol 37, 313–317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma M, Gogia A, Deo SSV & Mathur S Role of rebiopsy in metastatic breast cancer at progression. Curr. Probl. Cancer 43, 438–442 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Hench IB, Hench J & Tolnay M Liquid biopsy in clinical management of breast, lung, and colorectal cancer. Front. Med 5, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen E et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol 37, 1547–1557 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Thierry AR et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann. Oncol 28, 2149–2159 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Agarwal N et al. Characterization of metastatic urothelial carcinoma via comprehensive genomic profiling of circulating tumor DNA. Cancer 124, 2115–2124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paweletz CP et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin. Cancer Res 22, 915–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barata PC et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: a pilot assessment of concordance. Ann. Oncol 28, 2458–2463 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Pal SK et al. Efficacy of BGJ398, a fibroblast growth factor receptor 1–3 Inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. CancerDiscov. 8, 812–821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oellerich M et al. Using circulating cell-free DNA to monitor personalized cancer therapy. Crit. Rev Clin. Lab. Sci 54, 205–218 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nordentoft I et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 7, 1649–1663 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Cha EK et al. Branched evolution and intratumor heterogeneity of urothelial carcinoma of the bladder. J. Clin. Oncol 32, 293–293 (2014). [Google Scholar]
- 36.Prandi D et al. Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol. 15, 439 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomsen MBH et al. Spatial and temporal clonal evolution during development of metastatic urothelial carcinoma. Mol. Oncol 10, 1450–1460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dentro SC, Wedge DC & Van Loo P Principles of reconstructing the subclonal architecture of cancers. Cold SpringHarb. Perspect. Med 10.1101/cshperspect.a026625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waclaw B et al. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 525, 261–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson AG et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540–556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moris A, Murray S & Cardinaud S AID and APOBECs span the gap between innate and adaptive immunity. Front. Microbiol 5, 534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swanton C, McGranahan N, Starrett GJ & Harris RS APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 5, 704–712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salter JD, Bennett RP & Smith HC The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci 41, 578–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts SA et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet 45, 970–976 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burns MB, Temiz NA & Harris RS Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet 45, 977–983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedegaard J et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell 30, 27–42 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Blanes A, Rubio J, Sanchez-Carrillo JJ & Diaz-Cano SJ Coexistent intraurothelial carcinoma and muscle-invasive urothelial carcinoma of the bladder: clonality and somatic down-regulation of DNA mismatch repair. Hum. Pathol 40, 988–997 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Kazanov MD et al. APOBEC-induced cancer mutations are uniquely enriched in early-replicating, gene-dense, and active chromatin regions. Cell Rep. 13, 1103–1109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoopes JI et al. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 14, 1273–1282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seplyarskiy VB et al. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 26, 174–182 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nowarski R & Kotler M APOBEC3 cytidine deaminases in double-strand DNA break repair and cancer promotion. Cancer Res. 73, 3494–3498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okeoma CM, Lovsin N, Peterlin BM & Ross SR APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445, 927–930 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Smith HC, Bennett RP, Kizilyer A, McDougall WM & Prohaska KM Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol 23, 258–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Law EK et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv 2, e1601737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sjödahl G et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res 18, 3377–3386 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Choi W et al. Genetic alterations in the molecular subtypes of bladder cancer: illustration in the cancer genome atlas dataset. Eur. Urol 72, 354–365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamoun A et al. A consensus molecular classification of muscle-invasive bladder cancer. Eur. Urol 77, 420–433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi W et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25, 152–165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seiler R et al. Impact of molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol. 72, 544–554 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Mariathasan S et al. TGFß attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenberg JE et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma P et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Greaves M & Maley CC Clonal evolution in cancer. Nature 481, 306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rousseeuw PJ Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math 20, 53–65 (1987). [Google Scholar]
- 65.Dyrskjøt L et al. A molecular signature in superficial bladder carcinoma predicts clinical outcome. Clin. Cancer Res 11, 4029–4036 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Dyrskjøt L et al. Prognostic impact of a 12-gene progression score in non-muscle-invasive bladder cancer: a prospective multicentre validation study. Eur Urol. 72, 461–469 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Sjödahl G et al. Discordant molecular subtype classification in the basal-squamous subtype of bladder tumors and matched lymph-node metastases. Mod. Pathol 31, 1869–1881 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Thomsen MBH et al. Comprehensive multiregional analysis of molecular heterogeneity in bladder cancer. Sci. Rep 7, 11702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma S et al. Continuity of transcriptomes among colorectal cancer subtypes based on meta-analysis. Genome Biol. 19, 142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amin MB Histological variants of urothelial carcinoma: diagnostic, therapeutic and prognostic implications. Mod. Pathol 22, S96–S118 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Linder BJ et al. The impact of histological reclassification during pathology re-review — evidence of a Will Rogers effect in bladder cancer? J. Urol 190, 1692–1696 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Shah RB, Montgomery JS, Montie JE & Kunju LP Variant (divergent) histologic differentiation in urothelial carcinoma is under-recognized in community practice: impact of mandatory central pathology review at a large referral hospital. Urol. Oncol 31, 1650–1655 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Moch H, Humphrey PA, Ulbright TM & Reuter VE WHO Classification of Tumours of the Urinary System and Male Genital Organs Vol. 4, 77–133 (International Agency for Research on Cancer, 2016). [Google Scholar]
- 76.Humphrey PA, Moch H, Cubilla AL, Ulbright TM & Reuter VE The 2016 WHO classification of tumours of the urinary system and male genital organs — part B: prostate and bladder tumours. Eur. Urol 70, 106–119 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Hovelson DH et al. Targeted DNA and RNA sequencing of paired urothelial and squamous bladder cancers reveals discordant genomic and transcriptomic events and unique therapeutic implications. Eur. Urol 74, 741–753 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Blochin EB, Park KJ, Tickoo SK, Reuter VE & Al-Ahmadie H Urothelial carcinoma with prominent squamous differentiation in the setting of neurogenic bladder: role of human papillomavirus infection. Mod. Pathol 25, 1534–1542 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Chapman-Fredricks JR et al. High-risk human papillomavirus DNA detected in primary squamous cell carcinoma of urinary bladder. Arch. Pathol. Lab. Med 137, 1088–1093 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Solomon JP et al. Challenges in the diagnosis of urothelial carcinoma variants: can emerging molecular data complement pathology review? Urology 102, 7–16 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Vail E et al. Telomerase reverse transcriptase promoter mutations in glandular lesions of the urinary bladder. Ann. Diagn. Pathol 19, 301–305 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Lopez-Beltran A et al. Variants and new entities of bladder cancer. Histopathology 74, 77–96 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Dhall D, Al-Ahmadie H & Olgac S Nested variant of urothelial carcinoma. Arch. Pathol. Lab. Med 131, 1725–1727 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Volmar KE, Chan TY, De Marzo AM & Epstein JI Florid von Brunn nests mimicking urothelial carcinoma: a morphologic and immunohistochemical comparison to the nested variant of urothelial carcinoma. Am. J. Surg. Pathol 27, 1243–1252 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Zhong M et al. Distinguishing nested variants of urothelial carcinoma from benign mimickers by TERT promoter mutation. Am. J. Surg. Pathol 39, 127–131 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Al-Ahmadie HA et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat. Genet 48, 356–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Keck B et al. The plasmacytoid carcinoma of the bladder-rare variant of aggressive urothelial carcinoma. Int. J. Cancer 129, 346–354 (2011). [DOI] [PubMed] [Google Scholar]
- 88.Nigwekar P et al. Plasmacytoid urothelial carcinoma: detailed analysis of morphology with clinicopathologic correlation in 17 cases. Am. J. Surg. Pathol 33, 417–424 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Dayyani F et al. Plasmacytoid urothelial carcinoma, a chemosensitive cancer with poor prognosis, and peritoneal carcinomatosis. J. Urol 189, 1656–1661 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaimakliotis HZ et al. Plasmacytoid bladder cancer: variant histology with aggressive behavior and a new mode of invasion along fascial planes. Urology 83, 1112–1116 (2014). [DOI] [PubMed] [Google Scholar]
- 91.Amin MB et al. Micropapillary variant of transitional cell carcinoma of the urinary bladder. Histologic pattern resembling ovarian papillary serous carcinoma. Am. J. Surg. Pathol 18, 1224–1232 (1994). [DOI] [PubMed] [Google Scholar]
- 92.Nassar H et al. Pathogenesis of invasive micropapillary carcinoma: role of MUC1 glycoprotein. Mod. Pathol 17, 1045–1050 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Luna-More S, Gonzalez B, Acedo C, Rodrigo I & Luna C Invasive micropapillary carcinoma of the breast. A new special type of invasive mammary carcinoma. Pathol. Res. Pract 190, 668–674 (1994). [DOI] [PubMed] [Google Scholar]
- 94.Ching CB et al. HER2 gene amplification occurs frequently in the micropapillary variant of urothelial carcinoma: analysis by dual-color in situ hybridization. Mod. Pathol 24, 1111–1119 (2011). [DOI] [PubMed] [Google Scholar]
- 95.Ross JS et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin. Cancer Res 20, 68–75 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Isharwal S et al. Intratumoral heterogeneity of ERBB2 amplification and HER2 expression in micropapillary urothelial carcinoma. Hum. Pathol 77, 63–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iyer G et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J. Clin. Oncol 31, 3133–3140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fleischmann A, Rotzer D, Seiler R, Studer UE & Thalmann GN Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur. Urol 60, 350–357 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Lopez-Beltran A et al. Carcinosarcoma and sarcomatoid carcinoma of the bladder: clinicopathological study of 41 cases. J. Urol 159, 1497–1503 (1998). [DOI] [PubMed] [Google Scholar]
- 100.Sanfrancesco J et al. Sarcomatoid urothelial carcinoma of the bladder: analysis of 28 cases with emphasis on clinicopathologic features and markers of epithelial-to-mesenchymal transition. Arch. Pathol. Lab. Med 140, 543–551 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Sung MT et al. Histogenesis of sarcomatoid urothelial carcinoma of the urinary bladder: evidence for a common clonal origin with divergent differentiation. J. Pathol 211, 420–430 (2007). [DOI] [PubMed] [Google Scholar]
- 102.Guo CC et al. Dysregulation of EMT drives the progression to clinically aggressive sarcomatoid bladder cancer. Cell Rep. 27, 1781–1793.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.George J et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chang MT et al. Small cell carcinomas of the bladder and lung are characterized by a convergent but distinct pathogenesis. Clin. Cancer Res 24, 1965–1973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim PH et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur. Urol 67, 198–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sjödahl G, Eriksson P, Liedberg F & Höglund M Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J. Pathol 242, 113–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Batista da Costa J et al. Molecular characterization of neuroendocrine-like bladder cancer. Clin. Cancer Res 25, 3908–3920 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Gopalan A et al. Urachal carcinoma: a clinicopathologic analysis of 24 cases with outcome correlation. Am. J. Surg. Pathol 33, 659–668 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jordan E et al. Assessment of genomic alterations in bladder adenocarcinoma and urachal adenocarcinoma. Eur. J. Cancer 51, S530–S530 (2015). [Google Scholar]
- 111.Collazo-Lorduy A et al. Urachal carcinoma shares genomic alterations with colorectal carcinoma and may respond to epidermal growth factor inhibition. Eur. Urol 70, 771–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reis H et al. Pathogenic and targetable genetic alterations in 70 urachal adenocarcinomas. Int. J. Cancer 143, 1764–1773 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Saunders NA et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol. Med 4, 675–684 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Garraway LA & Janne PA Circumventing cancer drug resistance in the era of personalized medicine. CancerDiscov. 2, 214–226 (2012). [DOI] [PubMed] [Google Scholar]
- 115.Steinbichler TB et al. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol 53, 156–167 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Guo CC et al. Gene expression profile of the clinically aggressive micropapillary variant of bladder cancer. Eur. Urol 70, 611–620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meeks JJ et al. A systematic review of neoadjuvant and adjuvant chemotherapy for muscle-invasive bladder cancer. Eur. Urol 62, 523–533 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Grossman HB et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N. Engl. J. Med 349, 859–866 (2003). [DOI] [PubMed] [Google Scholar]
- 119.Plimack ER et al. Accelerated methotrexate, vinblastine, doxorubicin, and cisplatin is safe, effective, and efficient neoadjuvant treatment for muscle-invasive bladder cancer: results of a multicenter phase II study with molecular correlates of response and toxicity. J. Clin. Oncol 32, 1895–1901 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kamat AM et al. Bladder cancer. Lancet 388, 2796–2810 (2016). [DOI] [PubMed] [Google Scholar]
- 121.Lotan Y et al. Molecular subtyping of clinically localized urothelial carcinoma reveals lower rates of pathological upstaging at radical cystectomy among luminal tumors. Eur. Urol 76, 200–206 (2019). [DOI] [PubMed] [Google Scholar]
- 122.The ASCO Post. FDA grants accelerated approval to erdafitinib for metastatic urothelial carcinoma. The ASCO Post https://www.ascopost.com/News/59934 (2019). [Google Scholar]
- 123.Loriot Y et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med 381, 338–348 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Pouessel D et al. Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: implications for perioperative anti-FGFR3 treatment. Ann. Oncol 27, 1311–1316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goldstein JT et al. Genomic activation of PPARG reveals a candidate therapeutic axis in bladder cancer. Cancer Res. 77, 6987–6998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Siefker-Radtke A & Curti B Immunotherapy in metastatic urothelial carcinoma: focus on immune checkpoint inhibition. Nat. Rev. Urol 15, 112–124 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Riaz N et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Necchi A et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm, phase II study. J. Clin. Oncol 6, 3353–3360 (2018). [DOI] [PubMed] [Google Scholar]
- 129.US National Library of Medicine. ClinicalTrials.gov http://www.clinicaltrials.gov/ct2/show/NCT02177695 (2019). [DOI] [PubMed]
- 130.Flaig TW et al. SWOG S1314: a randomized phase II study of co-expression extrapolation (COXEN) with neoadjuvant chemotherapy for localized, muscle-invasive bladder cancer. J. Clin. Oncol 37, 4506–4506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Christensen E et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol 37, 1547–1557 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Hurst CD et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 32, 701–715.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vandekerkhove G et al. Circulating tumor DNA reveals clinically actionable somatic genome of metastatic bladder cancer. Clin. Cancer Res 23, 6487–6497 (2017). [DOI] [PubMed] [Google Scholar]