

SUMMARY
Stromal-interaction molecules (STIM1/2) sense endoplasmic reticulum (ER) Ca2+ depletion and activate Orai channels. However, the choreography of interactions between native STIM/Orai proteins under physiological agonist stimulation is unknown. We show that the five STIM1/2 and Orai1/2/3 proteins are non-redundant and function together to ensure the graded diversity of mammalian Ca2+ signaling. Physiological Ca2+ signaling requires functional interactions between STIM1/2, Orai1/2/3, and IP3Rs, ensuring that receptor-mediated Ca2+ release is tailored to Ca2+ entry and nuclear factor of activated T cells (NFAT) activation. The N-terminal Ca2+-binding ER-luminal domains of unactivated STIM1/2 inhibit IP3R-evoked Ca2+ release. A gradual increase in agonist intensity and STIM1/2 activation relieves IP3R inhibition. Concomitantly, activated STIM1/2 C termini differentially interact with Orai1/2/3 as agonist intensity increases. Thus, coordinated and omnitemporal functions of all five STIM/Orai and IP3Rs translate the strength of agonist stimulation to precise levels of Ca2+ signaling and NFAT induction, ensuring the fidelity of complex mammalian Ca2+ signaling.
Graphical Abstract
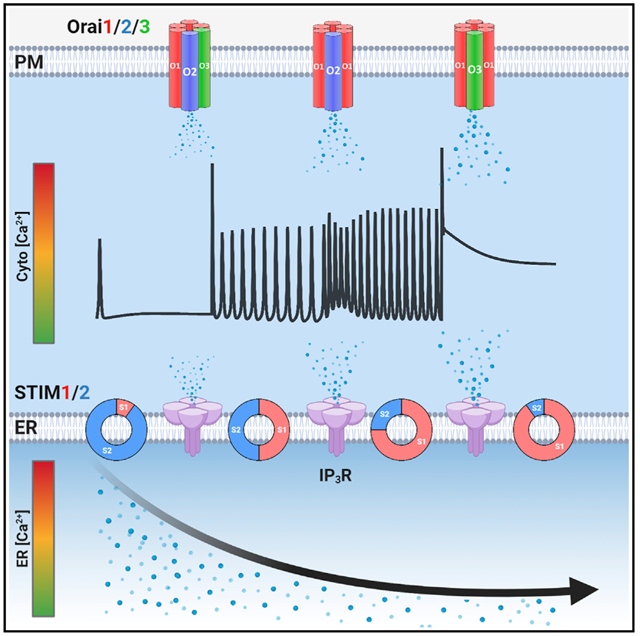
In brief
Ca2+ signals are crucial for cell function. Emrich et al. show that the five store-operated STIM/Orai proteins are non-redundant. Physiological store-operated channel activities are omnitemporal choreographies of all five STIM1/2 and Orai1/2/3 proteins and their functional interactions with IP3Rs, ensuring the fidelity and diversity of Ca2+ signaling and NFAT activation.
INTRODUCTION
Ca2+ is a universal second messenger that regulates a myriad of physiological responses, including immune function, exocrine secretion, and muscle contraction (Ahuja et al., 2020; Berridge, 2016; Trebak and Kinet, 2019). The endoplasmic reticulum (ER) luminal-free Ca2+ is 10,000-fold higher than the cytosol (Bagur and Hajnóczky, 2017). ER-free Ca2+ is tightly controlled through the opposing functions of the inositol 1,4,5-trisphosphate receptor (IP3R) channels, which release Ca2+ into the cytosol and the sarcoplasmic/ER Ca2+-ATPases (SERCA), which pump Ca2+ from the cytosol into the ER (Bagur and Hajnóczky, 2017; Berridge, 2016; Yule, 2001). Agonist stimulation of plasma membrane (PM) receptors coupled to phospholipase C (PLC) produces diffusible IP3, leading to activation of IP3Rs and depletion of ER Ca2+ stores (Putney and Tomita, 2012). Store depletion triggers the activation of ER resident stromal-interaction molecule (STIM) proteins to couple with and gate PM Orai channels and drive Ca2+ entry through the ubiquitous store-operated Ca2+ entry (SOCE) pathway (Hogan and Rao, 2015; Prakriya and Lewis, 2015; Trebak and Putney, 2017), mediated by the Ca2+ release-activated Ca2+ (CRAC) current (Hoth and Penner, 1992). SOCE plays a critical dual role in the acute refilling of ER Ca2+ to support Ca2+ release and in generating local Ca2+ signals that activate Ca2+-dependent nuclear factor of activated T cells (NFAT) transcription factors (Gwack et al., 2007; Trebak and Putney, 2017). Dysregulation of SOCE contributes to the pathogenesis of multiple immune, muscle, exocrine, and metabolic disorders (Feske, 2019).
Stimulation of cells with either SERCA blockers (e.g., thapsigargin) or high-agonist concentrations induces massive depletion of ER Ca2+ and robust cytosolic Ca2+ plateaus. Under these conditions, STIM1 and Orai1 have emerged as the major participants in SOCE. However, maximal activation of SOCE represents only the extreme end of diverse and graded SOCE activities in response to the full range of agonist concentrations. Stimulation of mammalian cells with relatively low physiological concentrations of agonists results in the generation of cytosolic Ca2+ oscillations (Dupont et al., 2011). In lymphocytes, Ca2+ oscillations are driven by oscillating SOCE activity independently of IP3Rs (Dolmetsch and Lewis, 1994; Lewis and Cahalan, 1989). In most cells, however, Ca2+ oscillations are initiated by IP3R-mediated Ca2+ release and maintained by ER Ca2+ refilling through SOCE (Bird and Putney, 2005). We recently demonstrated that Orai1 is dispensable for sustaining regenerative Ca2+ oscillations in HEK293 cells (Yoast et al., 2020b). Surprisingly, endogenous Orai2 and Orai3, which contribute an almost undetectable fraction of SOCE, support oscillations, suggesting that miniscule amounts of SOCE are enough to support sustained Ca2+ oscillations (Yoast et al., 2020b).
Although structurally similar, STIM1 and STIM2 are activated by distinct levels of ER Ca2+ depletion (Brandman et al., 2007; Liou et al., 2005). By comparison to STIM1, STIM2 is a weak activator of Orai channels and is triggered by more modest levels of ER Ca2+ depletion (Brandman et al., 2007; Wang et al., 2014; Zheng et al., 2018a). The contributions and precise choreographies of native STIM1 and STIM2 translocation to ER-PM junctions and interactions with Orai1/2/3 over the range of physiological agonist concentrations are unknown. A number of studies have used knockdown to investigate STIM1/2 in regulating Ca2+ oscillations (Ong et al., 2015; Thiel et al., 2013; Wedel et al., 2007) but with conflicting results. Earlier reports suggested that STIM1 is solely required for Ca2+ oscillations (Bird et al., 2009; Wedel et al., 2007), while more recent studies suggested that STIM2 is the key regulator of Ca2+ oscillations (Ong et al., 2015; Thiel et al., 2013). Further complicating this picture, evidence has emerged that STIM2 actively recruits STIM1 to ER-PM junctions under conditions of modest ER Ca2+ depletion (Ong et al., 2015; Son et al., 2020; Subedi et al., 2018), highlighting the complex function of STIM1/2 in Ca2+ signaling.
Here, we show that, in response to various agonist concentrations, both STIM isoforms are always required together to ensure the fidelity of Ca2+ signaling. The distinct C termini of STIM1 and STIM2 coordinate the wide range of Ca2+ oscillation frequencies in response to low- and mid-range agonist concentrations, while the C terminus of STIM1 exclusively drives sustained Ca2+ plateaus in response to high-agonist concentrations. STIM1/STIM2 cooperation is required for optimal nuclear translocation of NFAT1 and NFAT4. The Ca2+ binding N-terminal EF hand of unactivated (resting) STIMs is required for STIM1/2-mediated negative regulation of IP3Rs, likely setting a threshold for IP3 activation of IP3Rs. As the ratio of activated/unactivated STIMs increases with increasing agonist concentrations, IP3R inhibition by STIMs is relieved concomitantly with increased CRAC activity, effectively matching Ca2+ release to Ca2+ entry. This ensures that graded Ca2+ signals and NFAT activities are tailored to their matching physiological agonist intensities. Our data challenge the current oversimplified model of mammalian CRAC channel activation involving either STIM2/Orai1 or STIM1/Orai1 interactions under basal and agonist stimulation, respectively. Physiological CRAC channel activities are Omni tempore choreographies of all five STIM/Orai proteins and their functional interaction with IP3Rs.
RESULTS
Both native STIM1 and STIM2 support Ca2+ oscillations
We generated several clones of HEK293 cells lacking STIM1 (STIM1-KO), STIM2 (STIM2-KO), or both (STIM1/2-KO) using CRISPR/Cas9 (Figure 1A). Western blots showed no compensatory change in either STIM protein (Figure 1A) or the three isoforms of IP3Rs (Figures S1D–S1G) in these knockout cells; HEK293 cells in which all three isoforms of IP3R were knocked out were used as controls (Alzayady et al., 2016). There was no difference in Ca2+ release between parental wild-type (WT)-HEK293 and STIM-KO cells in response to store depletion by 2 μM thapsigargin (Figure 1B; see also Figure 4F for ER measurements). As expected, however, SOCE was slightly reduced in STIM2-KO cells, mostly reduced in STIM1-KO cells, and completely abrogated in STIM1/2-KO cells (Figures 1B and 1C). Orai1/2/3 triple-knockout cells (Orai-TKO), which show complete abrogation of SOCE (Yoast et al., 2020b), were included as controls. Previously, we determined that 10 μM of the muscarinic agonist carbachol (CCh) mostly induces Ca2+ oscillations (Yoast et al., 2020b). For each condition, five representative cells are shown (Figures 1D–1H), and data from several independent experiments with two independent CRISPR/Cas9-KO clones per condition are statistically analyzed (Figures 1I–1L), including Ca2+ oscillations frequency, the percentage of oscillating cells, cells with Ca2+ plateaus, and non-responding cells (either 0 or 1 spike) as defined in Figures S1A–S1C.
Figure 1. Native STIM1/STIM2 mediate unique Ca2+ oscillation profiles and agonist sensitivities.
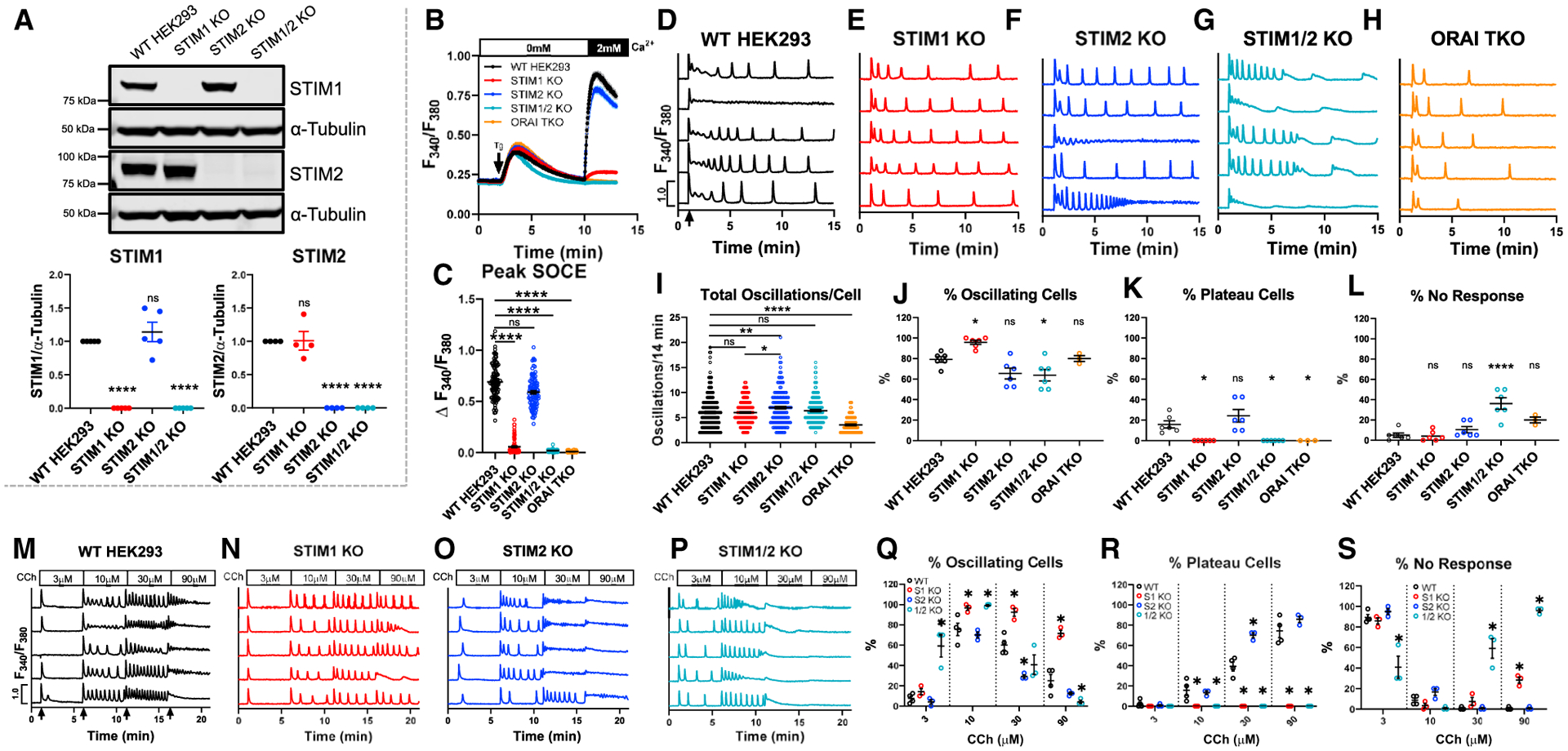
(A) Top: western blot of STIM1 and STIM2 in WT-HEK293 and STIM-KO cells. Bottom: quantification (mean ± SEM) of STIM1/2 band intensities normalized to α-tubulin. Each point is a biological replicate (STIM1, n = 5; STIM2, n = 4).
(B) Fura2 Ca2+ measurements upon store depletion with 2 μM thapsigargin in 0 mM Ca2+ and re-addition of 2 mM Ca2+.
(C) Quantification (mean ± SEM) of SOCE for (B). From left to right n = 100, 101, 100, 99, and 120 cells.
(D–H) Representative Ca2+ oscillation traces from 5 cells/condition upon stimulation with 10 μM CCh at 1 min (arrow in D) in 2 mM Ca2+.
(I) Quantification (mean ± SEM) of total oscillations in 14 min. From left to right n = 189, 230, 156, 152, and 96 cells.
(J–L) Mean ± SEM of percentage of (J) oscillating, (K) plateau, and (L) non-responding cells for (D)–(H). From left to right n = 6, 6, 6, 6, and 3 independent experiments.
(M–P) Representative Ca2+ traces in response to increasing CCh concentrations in 2 mM Ca2+ for 5 min each (arrows in M).
(Q–S) Mean ± SEM of percentage of (Q) oscillating, (R) plateau, and (S) non-responding cells for (M)–(P). From left to right n = 4, 3, 3, and 3 independent experiments. For all figures, *p < 0.05, **p < 0.01, ****p < 0.0001.
Figure 4. N-terminal EF-hands of STIM1/2 regulate receptor-mediated Ca2+ release.
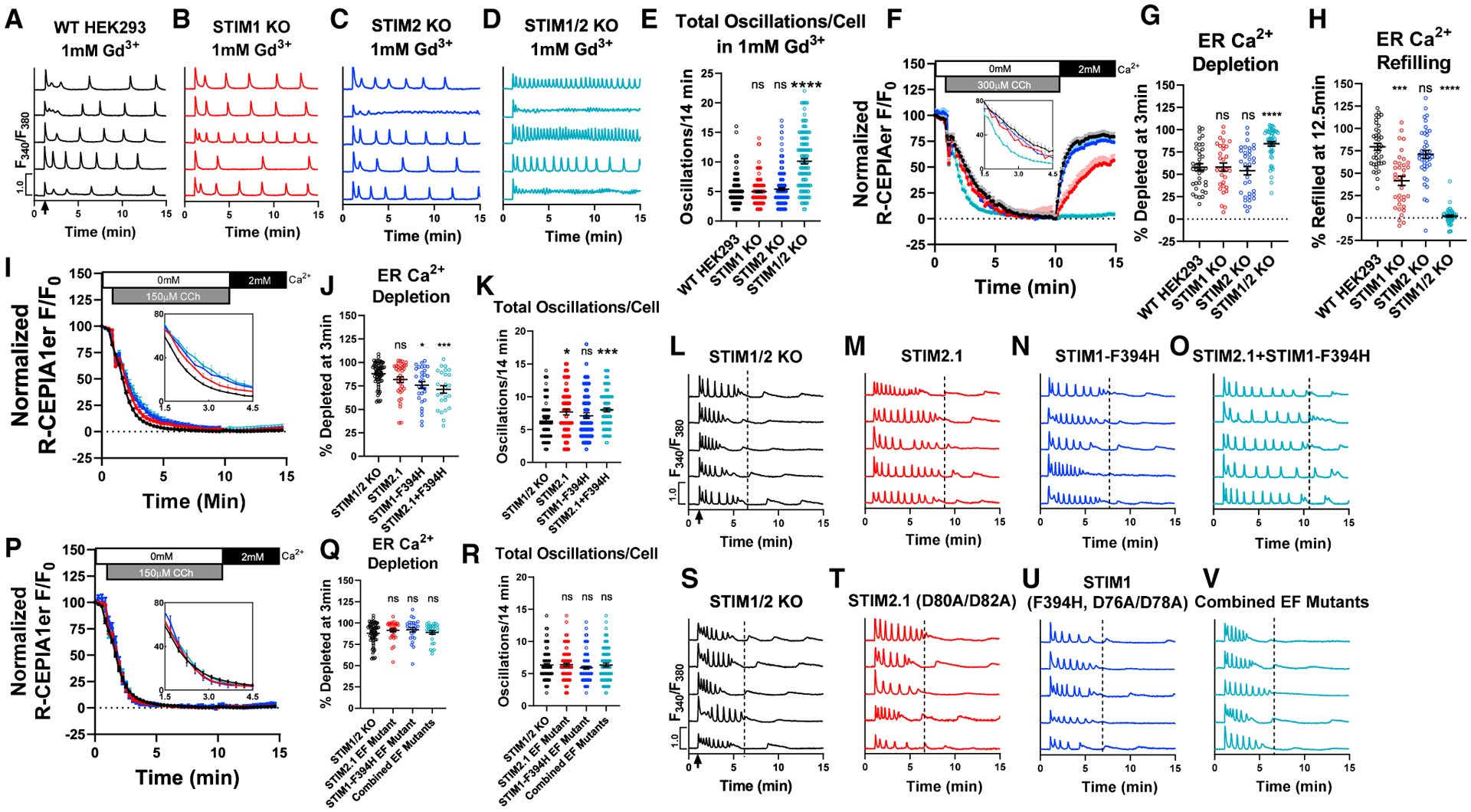
(A–D) Representative Ca2+ traces from 5 cells/condition with 10 μM CCh stimulation at 1 min (arrow in A) with 1 mM Gd3+ and 2 mM Ca2+ in bath.
(E) Total oscillations/14 min (mean ± SEM) from (A)–(D). Left to right, n = 173, 191, 166, and 123 cells.
(F) ER Ca2+ measurements in cells using R-CEPIA1er. Cells were stimulated with 300 μM CCh in 0 mM Ca2+ followed by 2 mM Ca2+ at 10 min (without CCh). Inset: zoomed in from 1.5–4.5 min.
(G and H) ER Ca2+ depletion rate (G; mean ± SEM) in (F) at 3 min after CCh in 0 mM Ca2+. (H) ER Ca2+ refilling rate (mean ± SEM) in (F) at 12.5 min in 2 mM Ca2+. Left to right n = 37, 35, 41, and 53 cells.
(I and J) ER Ca2+ measurements in STIM1/2-KO or STIM1/2-KO cells rescued with YFP-STIM2.1, STIM1-F394H-CFP, or STIM2.1 + STIM1-F394H (I). Inset: zoomed in from 1.5–4.5 min. (J) ER Ca2+ depletion rate (mean ± SEM) in (I) at 3 min after CCh stimulation in 0 mM Ca2+. Left to right n = 67, 36, 30, and 25 cells.
(K) Total oscillations/14 min (mean ± SEM) from STIM1/2-KO or STIM1/2-KO cells rescued with YFP-STIM2.1, STIM1-F394H-CFP, or combined STIM2.1 + STIM1-F394H. Cells were stimulated with 10 μM CCh in 2 mM Ca2+. Left to right n = 81, 64, 67, and 88 cells.
(L–O) Representative Ca2+ traces from 5 cells/condition from (K).
(P) ER Ca2+ measurements in STIM1/2-KO or STIM1/2-KO cells rescued with YFP-STIM2.1-(D80A/D82A), STIM1-(F394H/D76A/D78A)-CFP, or STIM2.1+STIM1-F394H EF-hand mutants. Inset: zoomed in from 1.5–4.5 min.
(Q) ER Ca2+ depletion rate (mean ± SEM) in (P) at 3 min after CCh stimulation in 0 mM Ca2+. Left to right n = 67, 31, 26, and 29 cells.
(R) Total oscillations/14 min (mean ± SEM) from (Q) after stimulation with 10 μM CCh in 2 mM Ca2+. Left to right n = 108, 97, 101, and 93 cells.
(S–V) Ca2+ traces from 5 cells/condition from (R). For all figures, *p < 0.05, ***p < 0.001, ****p < 0.0001.
Stimulation of WT-HEK293 cells with 10 μM CCh in the presence of 2 mM Ca2+ produced an average of 6 oscillations/14 min (Figure 1I). At this concentration, WT-HEK293 cells demonstrated an average of 79% oscillating cells, 16% plateau cells, and 5% non-responders (Figures 1J–1L). While STIM1-KO cells demonstrated a similar oscillation frequency compared to WT-HEK293 (Figure 1I), 95% of those cells responded with Ca2+ oscillations and 0% with plateaus (Figures 1J and 1K). STIM2-KO cells produced an increased average of ~6.9 oscillations/14 min, as well as an increased percentage of plateau cells (24%) compared to WT-HEK293 (Figures 1J–1L). These results suggest that, while both STIM1 and STIM2 can independently support Ca2+ oscillations, Ca2+ plateaus strictly require STIM1 and that STIM2 negatively regulates plateau responses.
Unexpectedly, STIM1/2-KO cells showed rapid oscillations in the first 5 min of CCh stimulation, followed by a progressive decrease in peak amplitude and subsequent rundown (Figure 1G). STIM1/2-KO cells showed a significantly increased percentage of non-responding cells (36%) and no cells eliciting a plateau response (Figures 1J–1L). The rapid oscillatory phenotype of STIM1/2-KO cells was unrelated to Ca2+ entry as inhibition of SOCE with 5 μM Gd3+ (Trebak et al., 2002) reduced the frequency of Ca2+ oscillations in WT-HEK293 but not in STIM1/2-KO cells (Figures S1L–S1P). Furthermore, Orai-TKO cells have a significantly reduced frequency of oscillations with only ~3 oscillations/14 min (Figures 1H and 1I; see also Yoast et al., 2020b). These findings suggest that STIM proteins exert control over ER Ca2+ release through IP3Rs. This behavior of STIM1/2-KO cells will be addressed further below.
Interestingly, we also observed a qualitative change in the behavior of Ca2+ oscillations between WT-HEK293 cells and STIM-KO cells as summarized in Figures S1H–S1K. A significant portion of WT-HEK293 and STIM2-KO cells typically responded as in Figures S1H and S1J, respectively, with 44.6% of WT-HEK293 cells stimulated with 10 μM CCh showing a rapid oscillatory response at first, which gradually transitioned into sustained oscillations that return to baseline (Figure S1H). 21.3% of STIM2 KO cells showed a reversal of this phenotype with oscillations that become rapid and tetanize into moderate plateaus (Figure S1J); only 1.6% of WT-HEK293 cells responded as in Figure S1J, and only 0.8% of STIM2-KO cells responded as in Figure S1H. STIM1-KO and STIM1/2-KO cells showed consistent behavior throughout recordings (Figures S1I and S1K). Thus, while endogenous STIM1 and STIM2 can independently support Ca2+ oscillations, they likely function together under physiological conditions. Quantification of oscillation data from two independent CRISPR/Cas9 knockout clones per condition is presented in Figures S2A–S2H.
STIM1/STIM2 cooperate across the range of agonist concentrations
Although STIM1 and STIM2 are highly homologous, the EF-hand domain of STIM2 has lower affinity for Ca2+ (Zheng et al., 2018a). Current models suggest that basal unstimulated SOCE activity is controlled by STIM2, while STIM1 is increasingly recruited with increasing agonist concentrations (Ong et al., 2015; Thiel et al., 2013). We measured Ca2+ signals to increasing CCh concentrations ranging from 3 to 90 μM. At 3 μM CCh, WT-HEK293, STIM1-KO, and STIM2-KO cells were almost entirely refractory, while nearly 60% of STIM1/2-KO cells demonstrated Ca2+ oscillations (Figures 1M–1S). At 10 μM CCh, WT-HEK293 and STIM2-KO cells showed 75% and 70% of oscillating cells, respectively, with 100% of STIM1-KO and STIM1/2-KO cells oscillating. Interestingly, STIM2-KO cells demonstrated increased sensitivity by exhibiting plateaus at 30 μM CCh (70%) compared to WT-HEK293 cells (40%), suggesting that STIM2 negatively regulates STIM1 and SOCE, in agreement with an earlier study (Soboloff et al., 2006). At 90 μM CCh, WT-HEK293 and STIM2-KO cells reached comparable percentages of cells responding with plateaus (~80%). Importantly, STIM1-KO cells failed to support plateaus at all CCh concentrations (Figures 1Q–1S), further documenting that plateaus are exclusively mediated by STIM1. These data suggest that STIM1 and STIM2 work collaboratively across the range of stimulus intensities to tailor Ca2+ responses to their respective agonist concentrations.
STIM C termini determine the diversity of receptor-activated Ca2+ signaling events
We utilized STIM1/2-KO cells to determine whether STIM1 and STIM2 re-expression can rescue the phenotype of STIM1/2-KO cells. To achieve near endogenous protein levels, we expressed YFP-tagged STIM constructs driven by the relatively weak thymidine kinase (tk) promoter (Korzeniowski et al., 2010) (Figure S3A shows re-expression of STIM1/2 to near endogenous levels). Both tk-STIM1 and tk-STIM2 rescued SOCE activated by store depletion with 2 μM thapsigargin. As expected, tk-STIM1 supported significantly larger SOCE compared to tk-STIM2 (Figures 2F and 2H). Furthermore, 10 μM CCh induced sustained oscillations and plateaus in STIM1/2-KO cells expressing tk-STIM1 with a similar percentage to those of STIM2-KO cells (Figures 2A–2C and 2J–2L). Similarly, tk-STIM2 expression in STIM1/2-KO cells supported oscillations with a percentage similar to that of STIM1-KO cells, with 0% of cells displaying plateaus (Figures 2A–2C and 2J–2L). Frequencies of oscillations were generally higher in these rescue experiments with tk-STIM1 and tk-STIM2 supporting ~12.6 and 9 oscillations/14 min, respectively (Figure 2I), suggesting that, while average tk-STIM1 and tk-STIM2 protein levels within cell populations is similar to endogenous STIM levels (Figure S3A), tk-STIM1 and tk-STIM2 protein levels in individual cells are likely higher compared to native levels.
Figure 2. STIM1/STIM2 have distinct interactions with Orai1/2/3.
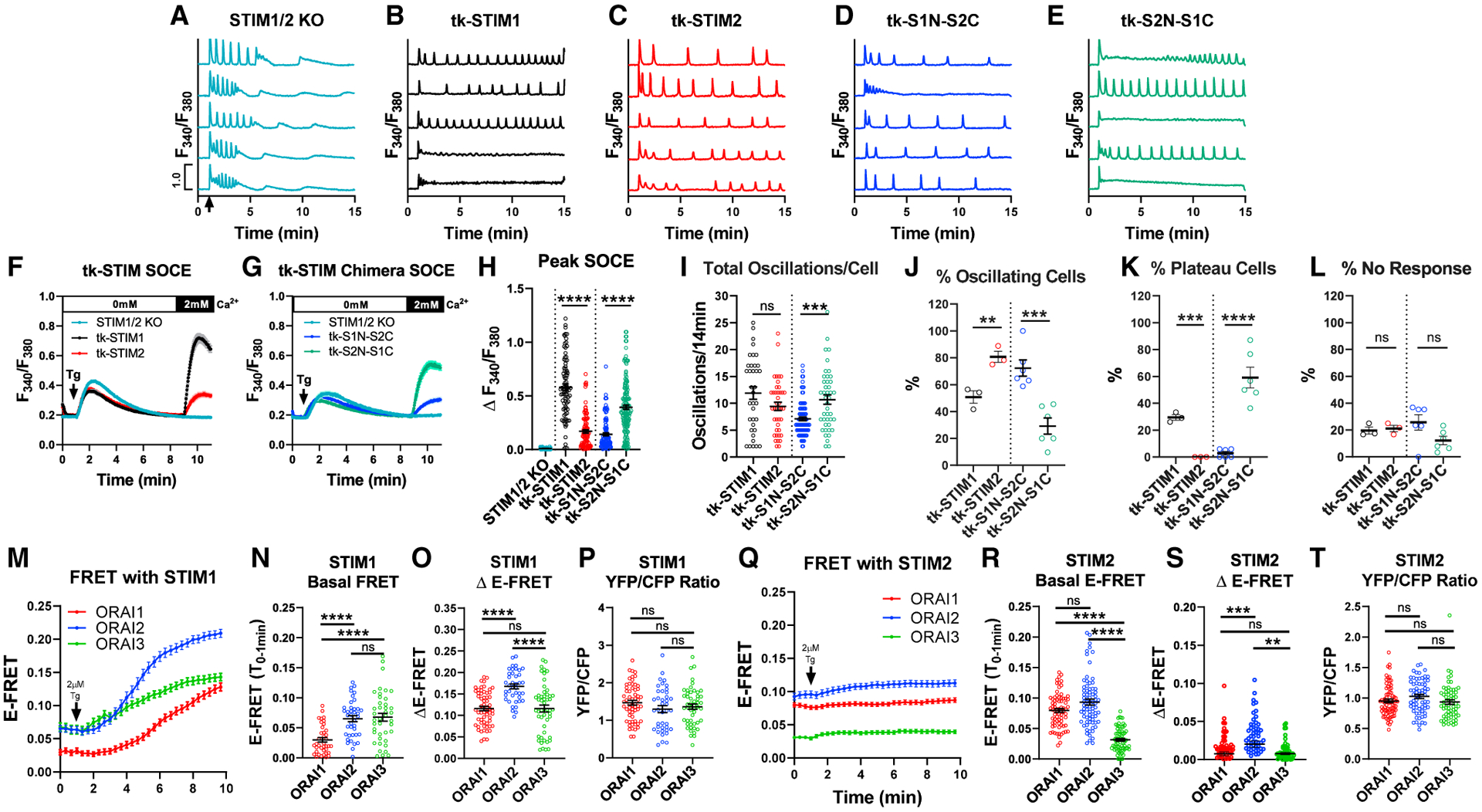
(A–E) Ca2+ traces of (A) non-transfected STIM1/2-KO or STIM1/2-KO cells individually rescued with (B) tk-YFP-STIM1, (C) tk-YFP-STIM2, (D) tk-YFP-S1N-S2C, and (E) tk-YFP-S2N-S1C from 5 representative cells/condition upon stimulation with 10 μM CCh at 1 min (arrow in A) in 2 mM Ca2+.
(F) SOCE in response to 2 μM thapsigargin in Ca2+-free followed by re-addition of 2 mM Ca2+ in STIM1/2-KO or STIM1/2-KO cells rescued with tk-YFP-STIM1 or tk-YFP-STIM2.
(G) SOCE in STIM1/2-KO cells rescued with tk-YFP-S1N-S2C or tk-YFP-S2N-S1C.
(H) Maximal SOCE (mean ± SEM) for (F) and (G). From left to right n = 90, 88, 96, 116, and 154 cells.
(I) Total oscillations (mean ± SEM) in 14 min for (A)–(E). From left to right n = 36, 41, 87, and 43 cells.
(J–L) Percentage (mean ± SEM) of (J) oscillating, (K) plateau, and (L) non-responding cells for (B)–(E); from left to right n = 3, 3, 6, and 6 independent experiments.
(M) E-FRET plotted as mean ± SEM between STIM1-YFP and each CFP-Orai expressed in Orai-TKO cells at baseline and after 2 μM thapsigargin in 2 mM Ca2+.
(N–P) Scatterblots (mean ± SEM) quantifying basal E-FRET, (O) change in E-FRET after thapsigargin, and (P) YFP/CFP fluorescence ratios from data in (M). From left to right n = 60 (Orai1), 40 (Orai2), and 51 (Orai3) cells.
(Q) E-FRET presented as mean ± SEM between STIM2-YFP and each CFP-Orai expressed in Orai-TKO cells before and after thapsigargin.
(R–T) Scatterblots (mean ± SEM) quantifying basal E-FRET (R), change in E-FRET after thapsigargin (S), and YFP/CFP fluorescence ratios (T) from data in (Q). From left to right n = 71 (Orai1), 67 (Orai2), and 64 (Orai3) cells. For all figures, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we utilized two STIM chimeric constructs that we recently developed (Emrich et al., 2019). The first chimera consists of the N-terminal and transmembrane domains of STIM1 combined with the C-terminus of STIM2 (tk-S1N-S2C), while the second is the N-terminal and transmembrane domains of STIM2 combined with the C-terminus of STIM1 (tk-S2N-S1C). Both STIM chimeras rescued SOCE triggered by thapsigargin when expressed in STIM1/2-KO cells with tk-S2N-S1C demonstrating significantly higher Ca2+ entry compared to tk-S1N-S2C (Figures 2G and 2H). Whole-cell patch-clamp recordings in STIM1/2-KO cells co-expressing Orai1 with either STIM chimera (driven by the robust CMV promoter) demonstrated that S2N-S1C supported a larger CRAC current density compared to S1N-S2C (Figures S3C and S3D). In terms of frequency of Ca2+ oscillations, percentage of oscillating cells, plateau cells, and non-responding cells, the behavior of these chimeras was solely determined by their C termini, with tk-S2N-S1C and tk-S1N-S2C supporting similar responses to those of tk-STIM1 and tk-STIM2, respectively (Figures 2I–2L). These experiments further demonstrate that STIM1 and STIM2 can independently support Ca2+ oscillations and that the C -terminus of STIM1 is the primary driver of plateau responses.
STIM/Orai isoform interactions shape the diversity of Ca2+ signaling events
We recently proposed that native CRAC channels are heteromers of all three Orai isoforms. Without Orai2/3, physiological Ca2+ oscillations in response to low/mid-range agonist concentrations are mostly replaced with Orai1-driven Ca2+ plateaus (Yoast et al., 2020b). Here, we performed side-by-side Förster resonance energy transfer (FRET) experiments with either STIM1-YFP or STIM2-YFP co-expressed with each CFP-Orai isoform in Orai-TKO cells (Figures 2M–2T). STIM1 exhibited significant interaction under basal conditions with Orai2 and Orai3 in comparison to Orai1 (Figures 2M and 2N), and the interaction between STIM1 and Orai isoforms was enhanced after store depletion (Figures 2M–2P). In contrast, STIM2 demonstrated significant basal interactions with Orai1 and Orai2 compared to Orai3 (Figures 2Q and 2R), and the interaction of STIM2 with each Orai isoform only marginally increased after store depletion (Figures 2Q–2T). One potential interpretation of these data is that STIM2 interaction with Orai1 at rest mediates SOCE at low-agonist concentrations, while interactions of STIM1 with Orai3 dampens SOCE at high-agonist concentrations. The interaction of both STIM1 and STIM2 with Orai2 likely fine-tunes SOCE across the full spectrum of physiological agonist concentrations.
We used CRISPR/Cas9 to generate cells expressing combinations of one native Orai and one native STIM. We generated six different combinations consisting of the double Orai knockout (Orai2,3-KO, Orai1,2-KO, and Orai1,3-KO), in which we knocked out either STIM1 or STIM2 (Figure S3B). STIM2 knockout in Orai2,3-KO cells caused a slight reduction in SOCE, while STIM1 knockout caused a near complete loss of SOCE (Figures 3A and 3B). In agreement with our previous data (Yoast et al., 2020b), relieving the negative regulatory effects of Orai2 and Orai3 on native Orai1 results in enhanced percentage (~70%) of Orai2,3-KO cells responding with Ca2+ plateaus upon stimulation with 10 μM CCh (Figures 3C and 3Q–3S; cf. to 16% of WT-HEK293 cells in Figure 1K). Importantly, in the absence of STIM1, 100% of Orai2,3-STIM1-KO cells responded with Ca2+ oscillations (Figures 3D and 3Q–3S). Interestingly, Orai2,3-STIM2-KO cells presented an intermediary phenotype with ~40% of cells responding with plateaus and ~60% of cells responding with Ca2+ oscillations (Figures 3E and 3Q–3S). Within oscillating cells, deletion of either STIM1 or STIM2 did not affect the frequency of Ca2+ oscillations per cell (Figure 3P).
Figure 3. Coordinated STIM/Orai interactions enhance the diversity of Ca2+ signaling events.
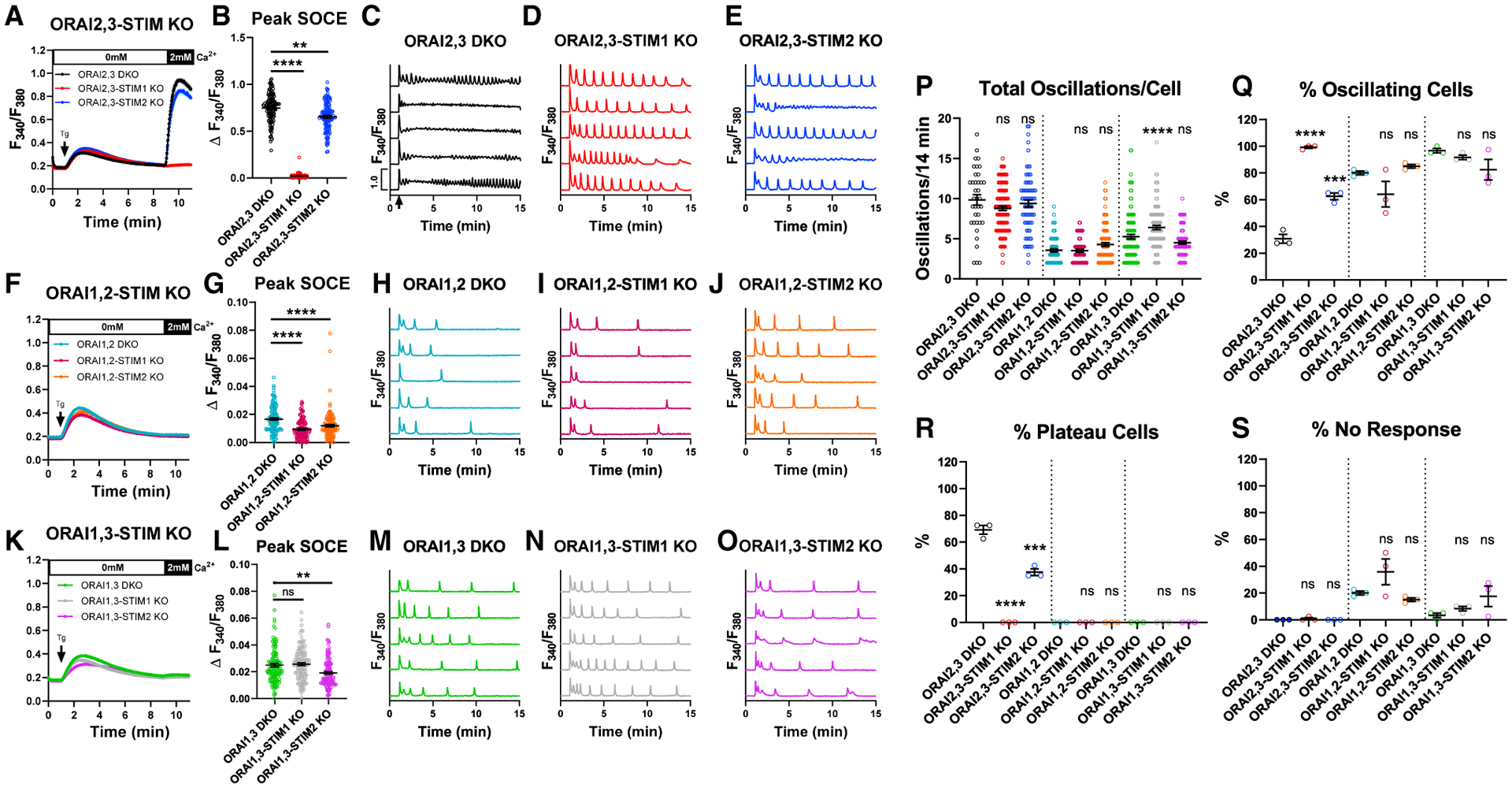
(A) SOCE measurement upon store depletion with 2 μM thapsigargin in 0 mM Ca2+ followed by re-addition of 2 mM Ca2+ in Orai2,3-STIM KO cells.
(B) Quantification (mean ± SEM) of maximal SOCE for (A). From left to right n = 120, 120, and 120 cells.
(C–E) Representative Ca2+ traces from 5 cells/condition upon stimulation with 10 μM CCh at 1 min (arrow in C) in 2 mM Ca2+.
(F and G) SOCE in Orai1,2-STIM KO cells (F). (G) SOCE quantification (mean ± SEM) from (F). From left to right n = 120, 120, and 120 cells.
(H–J) Representative Ca2+ traces of Orai1,2-STIM KO cells as in (C)–(E).
(K and L) SOCE in Orai1,3-STIM KO cells (K). (L) SOCE quantification (mean ± SEM) from (K). From left to right n = 120, 121, and 120 cells.
(M–O) Representative Ca2+ oscillation in Orai1,3-STIM KO cells as in (C)–(E).
(P) Quantification (mean ± SEM) of oscillations/14 min for (C)–(E), (H)–(J), and (M)–(O). From left to right n = 37, 119, 75, 96, 76, 102, 116, 110, and 99 cells.
(Q–S) Quantification (mean ± SEM) of percentage of (Q) oscillating, (R) plateau, and (S) non-responding cells for (C)–(E), (H)–(J), and (M)–(O). n = 3 independent experiments. For all figures, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Orai1,2-KO cells have an almost complete abrogation of SOCE activated by thapsigargin (Figure 3F). Yet, knockout of either STIM1 or STIM2 in these Orai1,2-KO cells demonstrated an additional very small reduction in SOCE, which only became evident after analyzing many cells (Figures 3F and 3G). Consistent with Ca2+ plateaus requiring Orai1 (Yoast et al., 2020b), Orai1,2-KO cells lacking either STIM isoform or neither responded strictly with Ca2+ oscillations upon stimulation with 10 μM CCh (Figures 3H–3J and 3Q–3S). Orai1,2-STIM1-KO cells showed an increased percentage of non-responding cells compared to the parental Orai1,2-KO cells, although this increase was not significant (Figure 3S). While Orai1,2-KO and Orai1,2-STIM1-KO cells displayed a similar oscillation frequency (~3.5 oscillations/14 min), oscillations in Orai1,2-STIM2-KO cells were slightly enhanced (~4.2 oscillations/14 min), although this did not reach significance (Figure 3P).
Although Orai1,3-KO cells showed an almost complete abrogation of SOCE activated by thapsigargin, the miniscule remaining SOCE was significantly larger than in Orai1,2-KO cells (Figures 3K and 3L), consistent with Orai2 displaying higher channel activity than Orai3 (Yoast et al., 2020b). STIM2 knockout in Orai1,3-KO cells caused an appreciable decrease of this miniscule SOCE, while STIM1 knockout did not (Figure 3L). Orai1,3-KO cells lacking either STIM isoform or neither responded strictly with Ca2+ oscillations upon 10 μM CCh stimulation (Figures 3M–3O and 3Q–3S). Orai1,3-STIM2-KO cells showed an increased in percentage of non-responding cells compared to the parental Orai1,3-KO cells, although this was not significant (Figure 3S). The oscillatory frequency of Orai1,3-KO cells was significantly higher than Orai1,2-KO cells (Figure 3P). STIM1 knockout in Orai1,3-KO cells significantly enhanced the oscillatory frequency (from ~5.25 to ~6.4 oscillations/14 min), while STIM2 knockout caused a small decrease (to ~4.5 oscillations/14 min) that was not significant (Figure 3P).
Unactivated STIM inhibit IP3R-mediated Ca2+ release
The rapid oscillatory response of STIM1/2-KO cells (Figures 1G and 1P) was not observed in Orai-TKO cells (Figure 1H), arguing that this is independent of the loss of SOCE. The frequency of Ca2+ oscillations in STIM1/2-KO cells was unchanged in the presence of 5 μM Gd3+ (Figures S2A–S2E), which blocks all Orai channel isoforms (Yoast et al., 2020b; Zhang et al., 2020), ruling out contributions from STIM-independent Ca2+ influx through Orai channels. Blockade of Ca2+ entry and Ca2+ extrusion by 1 mM Gd3+, termed lanthanide insulation (Wedel et al., 2007), did not alter the Ca2+ oscillatory frequencies of WT-HEK293, STIM1-KO, and STIM2-KO cells in response to 10 μM CCh (Figures 4A–4C and 4E; cf. Figure 4E to Figure 1I). Importantly, although the Ca2+ oscillations of STIM1/2-KO cells were sustained due to the lanthanide insulation, their frequency remained rapid (Figures 4D and 4E), suggesting that the rapid Ca2+ oscillations of STIM1/2-KO cells is independent of both Ca2+ entry and Ca2+ extrusion and is likely the result of enhanced ER Ca2+ release. Of note, while 10 μM CCh stimulation of STIM1/2-KO cells failed to produce Ca2+ plateaus (Figure 1K), when the lanthanide insulation protocol was used ~40% of STIM1/2-KO cells responded with plateaus (Figures S4A–S4C), suggesting that the combination of enhanced ER Ca2+ release and abrogation of Ca2+ extrusion increase cytosolic Ca2+ enough to produce Ca2+ plateaus in STIM1/2-KO cells.
We performed direct ER Ca2+ measurements using the genetically encoded R-CEPIA1er. WT-HEK293 cells and their STIM-KO counterparts were stimulated with a supramaximal concentration of CCh (>100 μM) in 0 mM external Ca2+ to deplete ER Ca2+, followed by restoration of 2 mM Ca2+ without CCh to allow ER refilling (Figure 4F). Interestingly, both STIM1-KO and STIM2-KO cells displayed a slight increase in the rate of ER Ca2+ depletion compared to WT-HEK293 that was not significant. However, we observed a significant increase in the rate of ER Ca2+ depletion in STIM1/2-KO cells (Figure 4G). The rate of ER Ca2+ refilling was significantly reduced in STIM1-KO cells while only marginally reduced in STIM2-KO cells (Figure 4H). As expected, acute ER Ca2+ refilling was essentially abrogated in STIM1/2-KO cells (Figures 4F and 4H). While these data suggest that acute ER refilling at the timescale of minutes is STIM dependent in agreement with previous work (Zheng et al., 2018b), STIM1/2-KO cells eventually refill their ER Ca2+ as documented by the state of their stores at the beginning of recordings. This STIM-independent refilling likely involves alternative, and perhaps slower, Ca2+ entry routes.
We utilized two variants of STIM1 and STIM2 that are incapable of activating Orai channels and mediating SOCE. The first is STIM1 with a point mutation (STIM1-F394H) (Wang et al., 2014) within the C-terminal STIM-Orai activating region (SOAR) (Park et al., 2009; Yuan et al., 2009). The second is a naturally occurring STIM2 splice variant called STIM2.1, which does not activate Orai channels (Miederer et al., 2015; Rana et al., 2015; Zhou et al., 2018). STIM1-F394H tagged with CFP and STIM2.1 tagged with YFP were expressed individually and together in STIM1/2-KO cells, and ER Ca2+ depletion and replenishment were analyzed. Interestingly, expression of either STIM1-F394H or STIM2.1 reduced the rate of ER Ca2+ depletion compared to STIM1/2-KO cells, and this effect was further enhanced with co-expression of both STIM variants (Figures 4I and 4J). As expected, expression of these STIM variants, which are unable to engage and activate Orai proteins, individually or together, failed to support acute ER refilling (Figure 4I). In response to 10 μM CCh, STIM1/2-KO cells rapidly oscillate and then cease this activity by ~5 min. Strikingly, rescue with either STIM1-F394H or STIM2.1 prolonged the time of rundown, thereby increasing total oscillation number (Figures 4K–4N). This effect was further enhanced when STIM1-F394H and STIM2.1 were co-expressed in STIM1/2-KO cells (Figures 4K and 4O). Obviously, because these cells lack both functional Orai-activating STIM1/2, Ca2+ oscillations eventually cease. Rescue with either STIM variant, or both, did not substantially alter the percentage of non-responders, plateau, or oscillating cells (Figures S4J–S4L). Under lanthanide insulation, co-expression of STIM1-F394H and STIM2.1 in STIM1/2-KO cells caused a reduction in the frequency of oscillations and a decrease in the percentage of plateau cells (Figures S4D–S4I), strongly arguing that STIM1/2 N termini negatively regulate Ca2+ release from the ER.
We mutated key residues within the EF-hand of STIM1-F394H (D76A/D78A) and STIM2.1 (D80A/D82A) to abolish Ca2+ binding and mimic STIM activation (Brandman et al., 2007; Liou et al., 2005). The expression of these EF-hand mutants in STIM1/2-KO cells either individually or together failed to alter the rate of ER Ca2+ depletion in response to supramaximal concentration of CCh (Figures 4P and 4Q) and failed to alter the oscillatory profile in response to 10 μM CCh (Figures 4R–4V), suggesting that unactivated STIMs inhibit Ca2+ release. Rescue with either STIM1-F394H (D76A/D78A) and STIM2.1 (D80A/D82A) EF-hand mutants or both did not substantially alter the percentage of non-responders, plateau, or oscillating cells (Figures S4M–S4O). STIM EF-hand domains are not simply regulating Ca2+ release through buffering of ER Ca2+. Indeed, ER Ca2+ measurements after passive store depletion with the reversible SERCA pump blocker cyclopiazonic acid (CPA, 25 μM) showed that the rate of ER Ca2+ depletion by CPA, but not refilling, was identical among WT-HEK293, STIM1-KO, STIM2-KO, and STIM1/2-KO cells (Figures S5A–S5C).
We then measured the elementary Ca2+ signals generated by IP3Rs (called Ca2+ puffs) in WT-HEK293 cells and STIM1/2-KO cells using total internal reflection fluorescence (TIRF) microscopy. Cells were loaded with caged IP3 (ci-IP3/PM), and Ca2+ puffs were recorded after photolysis of ci-IP3 for 60 s at a rate of 166 frames/s. Ca2+ puffs were detected following photolysis of ci-IP3 in both WT-HEK293 and STIM1/2-KO cells (Figures 5A and 5B). The number of puffs and number of puff sites were significantly higher in STIM1/2-KO cells compared to WT-HEK293 cells (Figures 5C and 5D; Videos S1 and S2). The increased number of Ca2+ puffs and number of puff sites are consistent with the rapid oscillatory behavior and the increased sensitivity of STIM1/2-KO cells to low CCh concentrations (Figures 1G and 1P). Further, Ca2+ signals globalized in ~33% of STIM1/2-KO cells compared to ~6% of WT-HEK293 cells within 60 s (Figure 5E), consistent with increased sensitivity of IP3R in STIM1/2-KO cells. The mean rise (r) and fall (f) time of the Ca2+ puffs was similar between WT-HEK293 and STIM1/2-KO cells (Figure 5F), indicating that the fundamental biophysical properties of IP3R clusters were unaltered. The amplitude distribution of puffs did not differ between WT-HEK293 and STIM1/2-KO cells with most puffs ranging from 0.3 to 0.9 peak amplitude (Figures 5G and 5H). Similar results were obtained when Ca2+ puffs were evoked by 1 μM CCh (Figures S5D–S5I). The number of puffs and number of puff sites were significantly greater in STIM1/2-KO as compared to WT-HEK293 cells. While Ca2+ signals globalized rarely within 90 s in WT cells (~9%), ~45% of STIM1/2-KO cells globalized within this period. The amplitude distribution and mean rise and fall time of Ca2+ puffs were similar between WT-HEK293 and STIM1/2-KO cells (Figures S5G–S5I).
Figure 5. STIM1/STIM2 regulate IP3R-mediated Ca2+ puffs in response to IP3 uncaging.
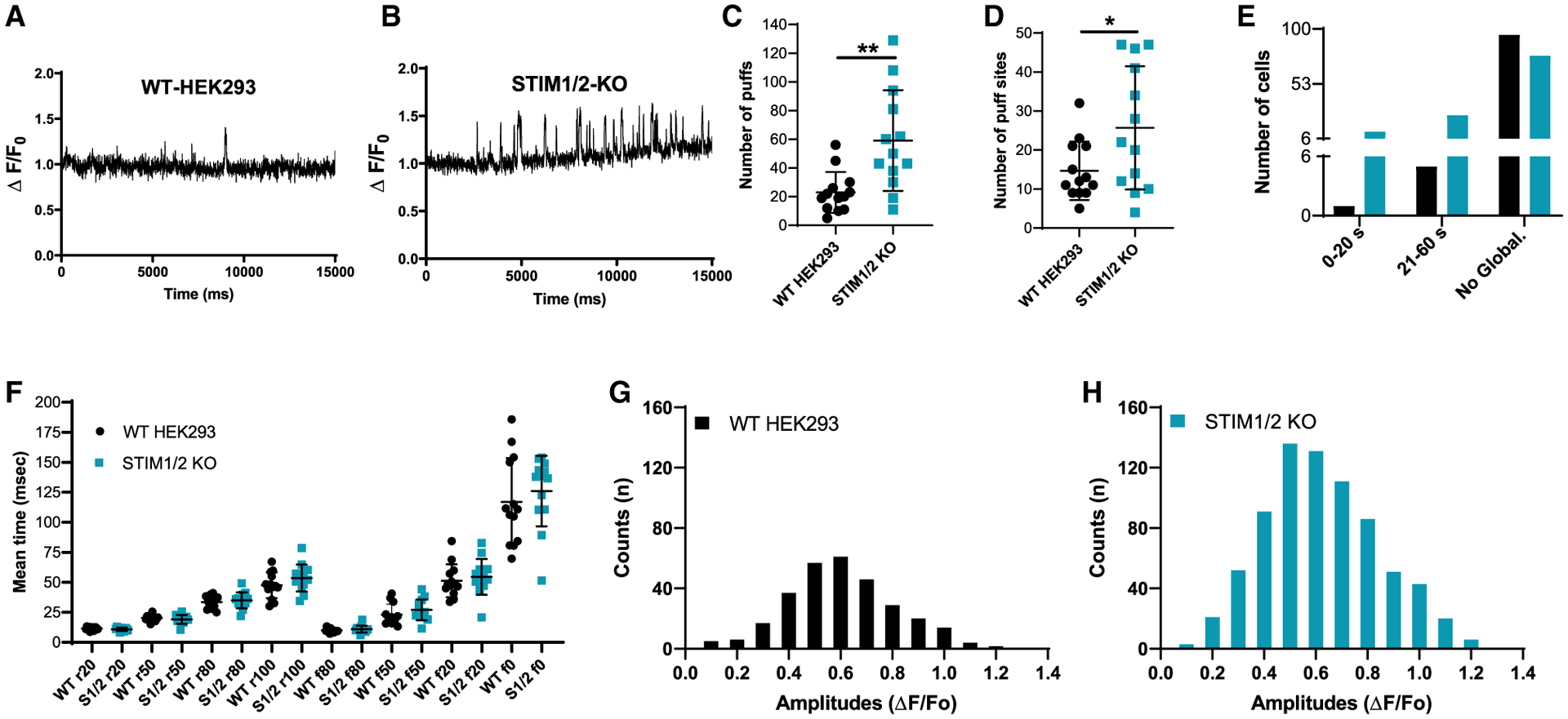
(A and B) Representative traces of Cal-520 fluorescence ratios (ΔF/F0) from the center of a single puff site (1.3 × 1.3 μm) evoked by photolysis of ci-IP3 at 3 s in WT-HEK293 (n = 12 experiments, 101 cells) and STIM1/2-KO (n = 10 experiments, 115 cells), respectively.
(C and D) 13 cells from each condition were randomly selected to quantify the number of puffs (C) and puff sites (D). (C) n = 768 puffs in 13 STIM1/2-KO cells when compared to n = 298 puffs in 13 HEK293 cells. (D) n = 334 puff sites in 13 STIM1/2-KO cells when compared to n = 191 puff sites in 13 HEK293 cells.
(E) From a total of 101 HEK293 cells and 115 STIM1/2-KO cells, a bar graph shows the proportions of WT-HEK293 and STIM1/2-KO cells in which the Ca2+ signals either did not globalize or globalized within 0–20 or 21–60 s.
(F) Mean rise and decay times of fluorescence of Ca2+ puffs when it increases or decreases to 20%, 50%, 80%, and 100% from 13 cells each of HEK293 and STIM1/2-KO.
(G and H) Amplitudes distribution of the Ca2+ puffs in WT-HEK293 (G) and STIM1/2-KO cells (H). For all figures, *p < 0.05, **p < 0.01.
Mathematical modeling supports STIM1/2, Orai1/2/3, and IP3Rs shape receptor-evoked Ca2+ signaling
We constructed a simplified model of how SOCE is controlled by the interaction of STIMs (defined as STIMi where i = 1,2) with Orai dimers (defined as Oraij,k where j,k = 1,2,3). This Orai model was refined from our recent study (Yoast et al., 2020b) by the addition of STIM1 and STIM2 interactions with Oraij,k. The model makes five assumptions that are described in the STAR Methods. The model qualitatively reproduces the range of Ca2+ oscillations and plateaus in response to increasing concentrations of agonist (represented by “p”; Figures 6A–6D; solid graphs). Specifically, the model predicts the following experimental data: (1) both STIM1 and STIM2 support oscillations and are both involved across the full range of agonist concentrations. However, in the absence of STIM1, even high-agonist concentrations do not lead to sustained plateaus (Figures 6A–6C; solid graphs). (2) High sensitivity to agonist and faster oscillatory frequency of STIM1/2-KO cells (Figure 6D; solid graphs), in support of the negative regulation of IP3Rs by unactivated STIM proteins. (3) The Gd3+ insulation protocol (i.e., closed cell; Figures S6A–S6D): STIM1/2-KO cells readily respond by Ca2+ plateaus when Ca2+ extrusion is abrogated in the closed cell condition (Figure S6D). (4) The ER Ca2+ measurements (Figures 6E and 6F; depicting normalized and absolute ER Ca2+ levels, respectively). (5) Data obtained with Orai double-knockout/STIM single-knockout cells with either thapsigargin stimulation, first in 0 mM Ca2+ and then in Ca2+-containing external solution or with application of increasing concentrations of agonist in Ca2+-containing external solution (Figures S7A–S7C). Native STIM1 and Orai1 are essential for mediating Ca2+ plateaus in response to high-agonist concentrations with STIM2, Orai2, and Orai3 regulating SOCE across the full range of agonist concentrations but have negligible contributions to plateaus on their own (Figures S7A–S7C). Consistent with these results, model simulations show that, in WT-HEK293 cells, at all agonist concentrations, STIM1 interaction with the Orai1,1 homodimer mediates the great majority of SOCE activity (Figure S7D). Finally, the model reproduces the dynamic change of oscillation properties in response to agonist application in WT-HEK293 cells, as the oscillations initially start faster but gradually change to slower oscillations on a lower baseline (see, e.g., Figure S1H), a known feature of Ca2+ oscillation models (Sneyd et al., 2004, 2017).
Figure 6. Mathematical modeling supports coordinated STIM1/STIM2 functions over the diverse range of Ca2+ signaling events.
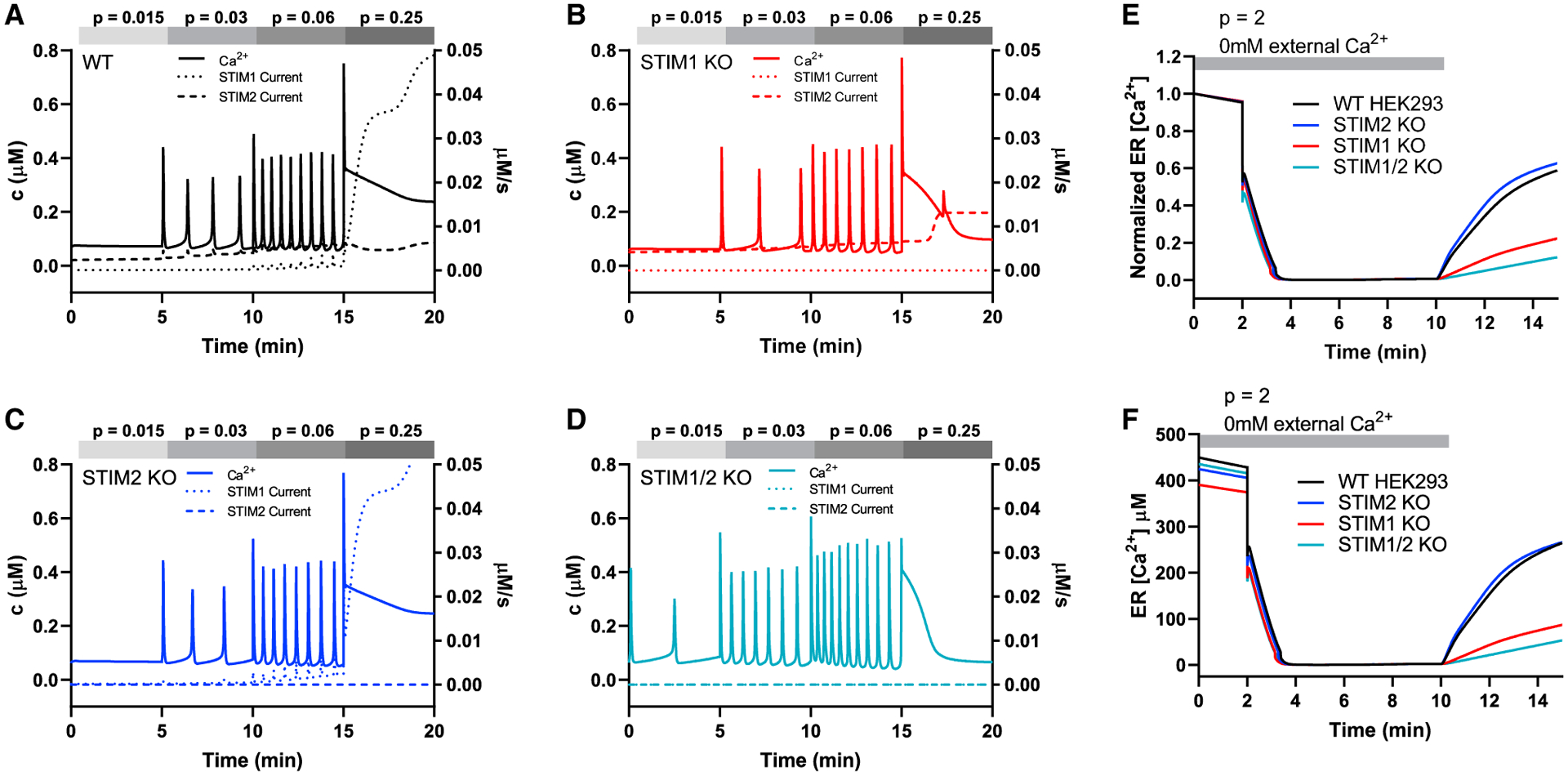
(A–D) Simulation of responses to different agonist concentrations (p = 0.015, p = 0.03, p = 0.06, p = 0.25) applied sequentially in that order, each for 5 min, to WT- and STIM-KO cells in 2 mM Ca2+. Solid lines are Ca2+ concentrations plotted against the left axis, while the dotted and dashed lines are STIM-mediated currents, which are plotted against the right axis.
(E and F) Simulation of normalized (E) and absolute (F) ER [Ca2+] in response to high-agonist concentration (p = 2), in the absence of external Ca2+, followed (at 10 min) by restoration of external Ca2+ and removal of agonist. In (F), the ER starts at different concentrations due to changes in the leak current (called α0 in the model).
The experimental data show that, at the beginning of recordings, ER Ca2+ stores of STIM1/2-KO cells are full (Figures 4F, 4I, and 4P; Figures 5E and 5F; see also Zheng et al., 2018b), suggesting the presence of an unknown alternative pathway that refills ER Ca2+ in hours or days. As such, the model predicts that, under basal conditions (i.e., without agonist), only 28% of the Ca2+ influx into the cell is through STIM/Orai channels; 7% (of this 28%) of STIM/Orai-mediated current is mediated by STIM1, with the remaining 93% mediated by STIM2 (Figure 6; dotted graphs). For low-agonist concentrations, this proportion is maintained. At higher agonist concentrations, 92% of the Ca2+ influx into the cell is mediated by STIM/Orai channels, and, of that, 93% is mediated by STIM1 and 7% by STIM2.
Coordinated STIM1/STIM2 functions tailor receptor activation to NFAT induction
SOCE is a critical regulator of NFAT isoform nuclear translocation (Feske et al., 2006; Gwack et al., 2007; Rao, 2009; Smedler and Uhlén, 2014; Tomida et al., 2003; Zhang et al., 2019). The nuclear translocation of NFAT1 is relatively slow and requires robust SOCE activity, while NFAT4 nuclear translocation is more rapid and requires relatively low SOCE activity (Srikanth et al., 2017; Srinivasan and Frauwirth, 2007; Yissachar et al., 2013; Yoast et al., 2020b; Zhang et al., 2019). We expressed GFP-tagged NFAT1 and NFAT4 reporter constructs in WT-HEK293 and STIM-KO cells. These four cell types were then stimulated with either low (10 μM) or high (300 μM) concentrations of CCh and NFAT nuclear translocation was monitored. In WT-HEK293 cells, stimulation with 10 μM CCh caused a slow and gradual accumulation of NFAT1 in the nucleus (Figures 7A and 7E). NFAT1 nuclear translocation was significantly reduced in STIM1-KO and STIM2-KO cells and essentially abolished in STIM1/2-KO cells (Figures 7A, 7B, and 7E). When WT-HEK293 cells were stimulated with 300 μM CCh, NFAT1 nuclear translocation was more robust (Figure 7C). While NFAT1 nuclear translocation was completely inhibited in STIM1/2-KO cells, it was only slightly inhibited in STIM2-KO cells and substantially reduced in STIM1-KO cells (Figures 7C, 7D, and 7F). The nuclear translocation of NFAT4 in WT-HEK293 cells was significantly faster by comparison to that of NFAT1 at both agonist concentrations (Figures 7G–7L). NFAT4 nuclear translocation in response to either 10 or 300 μM of CCh was significantly reduced in STIM1-KO and STIM2-KO cells and essentially abrogated in STIM1/2-KO cells (Figures 7G–7L). These results demonstrate that both STIM1 and STIM2 are required for optimal NFAT nuclear translocation under both low- and high-agonist stimulus intensities.
Figure 7. Both STIM1 and STIM2 are required for optimal NFAT1/4 activation.
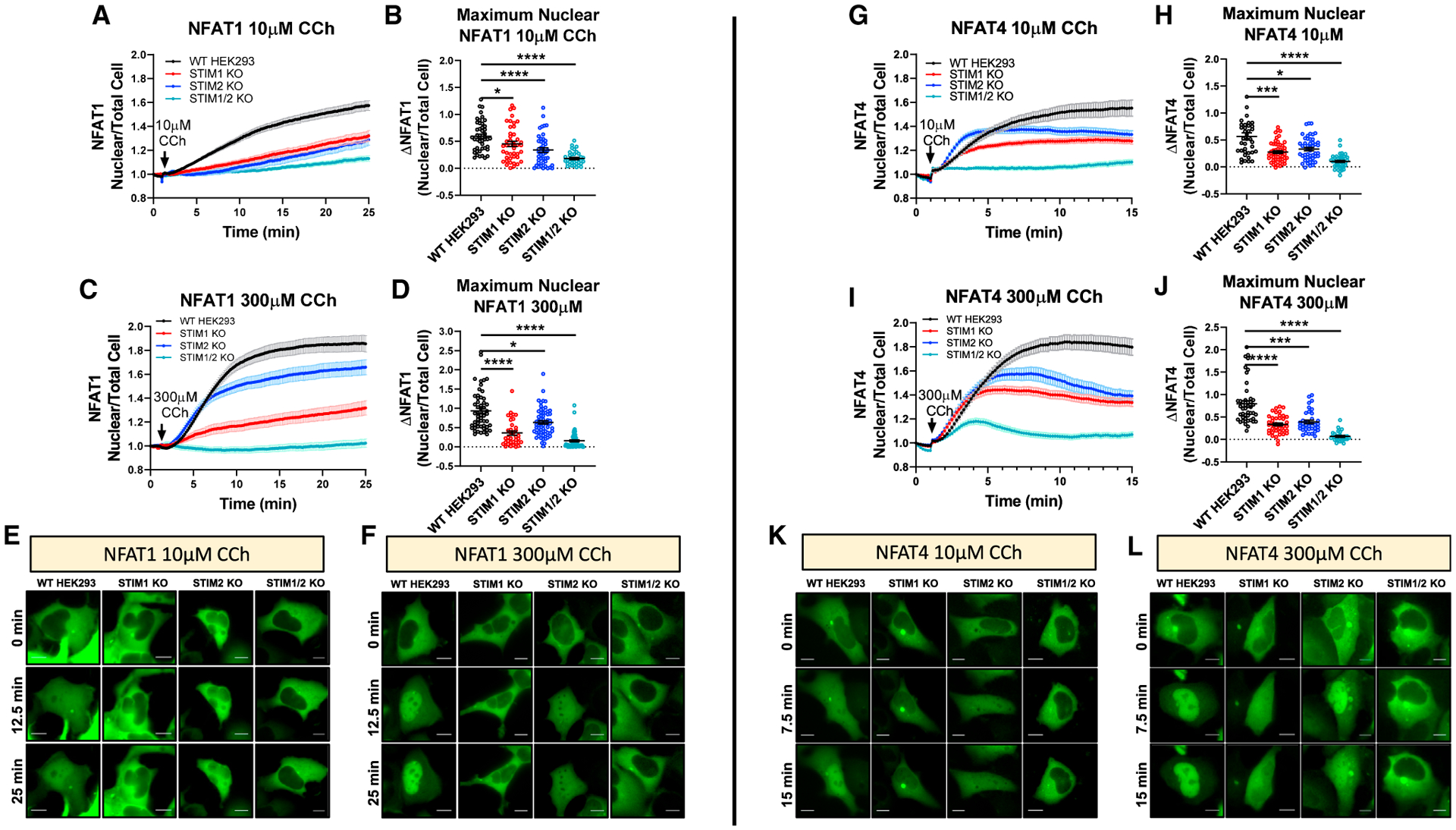
(A) Time-lapse quantification of NFAT1-GFP nuclear translocation in response to 10 μM CCh in 2 mM Ca2+ as determined by the ratio of nuclear/total GFP fluorescence.
(B) Mean ± SEM of nuclear NFAT1 fluorescence for (A) at 25 min. From left to right n = 46, 43, 44, and 36 cells.
(C) Mean ± SEM of NFAT1-GFP nuclear translocation in response to 300 μM CCh.
(D) Mean ± SEM of nuclear NFAT1 fluorescence from (C) at 25 min. From left to right n = 55, 36, 62, and 59 cells.
(E and F) Representative time-lapse images of NFAT1-GFP-expressing cells after 10 μM CCh (E) or 300 μM CCh (F).
(G) Same as in (A) but utilizing NFAT4-GFP-expressing cells.
(H) Mean ± SEM of nuclear NFAT4 fluorescence for (G) at 15 min. From left to right n = 39, 53, 49, and 40 cells.
(I) Mean ± SEM of NFAT4-GFP nuclear translocation in response to 300 μM CCh.
(J) Mean ± SEM of nuclear NFAT4 fluorescence from (I) at 15 min. From left to right n = 46, 40, 36, and 29 cells.
(K and L) Representative time-lapse images of NFAT4-GFP-expressing cells after 10 μM CCh (K) or 300 μM CCh (L). Scale bar, 10 μm. For all figures, *p < 0.05, ***p < 0.001, ****p < 0.0001.
DISCUSSION
SOCE requires STIM1 and Orai1, and the molecular mechanisms underlying their coupling have been intensively investigated (Fahrner et al., 2017; Lewis, 2020; Yeung et al., 2020; Zhou et al., 2017, 2019). STIM2 is a weaker SOCE activator and its recruitment is triggered under basal conditions, when ER Ca2+ depletion is modest (Brandman et al., 2007). Recently, we showed that all three Orai isoforms oligomerize to form native CRAC channels, mediating the diversity of Ca2+ signaling events in response to the full range of stimulus intensities (Yoast et al., 2020a, 2020b). Here, we addressed the choreography of interactions between native STIM1/2 and Orai1/2/3. Multiple caveats have hindered the understanding of the contributions of each native STIM/Orai to Ca2+ signaling. First, ectopic expression has been performed on a wild-type background, such that the interference of endogenous SOCE is unknown. Ectopic expression typically produces higher protein levels than native levels (Zhang et al., 2019), altering stoichiometry and channel activity. In most of these overexpression studies, only STIM1 and Orai1 were considered. Second, measurements of native SOCE activity have been mostly performed upon maximal store depletion. Under physiological conditions, stimulation of cells with relatively low or moderate agonist concentrations induces concomitant IP3R-mediated Ca2+ oscillations and ER Ca2+ replenishment through SOCE, resulting in only minor and transient fluctuations of ER Ca2+. Third, prior reports investigating native STIM1 and STIM2 in Ca2+ oscillations have relied on small interfering RNA (siRNA) knockdown (Bird et al., 2009; Ong et al., 2015; Thiel et al., 2013; Wedel et al., 2007), producing conflicting results with variations in knockdown efficiencies between studies as likely confounding factors.
To overcome these limitations, we studied multiple clones of HEK293 cells devoid of STIM1 or STIM2 proteins individually and in combination. Both STIM1-KO and STIM2-KO cells maintain oscillation frequencies like WT-HEK293 cells. However, loss of STIM1 completely abolished Ca2+ plateaus. STIM2-KO cells exhibit plateaus at lower concentrations of agonist in comparison to WT-HEK293, suggesting that STIM2 negatively regulates SOCE activity and is required for enhancing the repertoire of Ca2+ signaling events. Ca2+ plateaus are driven exclusively by the C terminus of STIM1. Alterations in Ca2+ oscillation profiles are predicted to have important downstream functional consequences, particularly in the activation of the Ca2+-dependent transcription factors, NFAT (Gwack et al., 2007; Vaeth and Feske, 2018; Zhang et al., 2019). By investigating the activation of NFAT1 and NFAT4, which have distinct nuclear translocation requirements (Srikanth et al., 2017; Yissachar et al., 2013), we demonstrate that loss of either STIM1 or STIM2 impairs NFAT1 and NFAT4 translocation in response to both low- and high-agonist concentrations, suggesting that the presence of both native STIM1 and STIM2 is always required for optimal activation of both NFAT1 and NFAT4 at all agonist concentrations. The combined functions of STIM1 and STIM2 are required to tailor the Ca2+ signal to the strength of agonist stimulation and NFAT transcriptional activity. STIM1 plays the major role for NFAT1 nuclear translocation in response to high concentrations of agonist. This is consistent with the known properties of NFAT1, which requires robust Ca2+ concentrations for activation and with STIM1 function described herein as the exclusive driver of Ca2+ plateaus. We recently revealed that both endogenous STIM1 and STIM2 have major contributions to SOCE in the colorectal cancer cell line HCT116 (Emrich et al., 2019). Recent studies proposed that STIM2 enhances Ca2+ signals by recruiting STIM1 to Orai1 channels under conditions of modest store depletion (Ong et al., 2015; Son et al., 2020; Subedi et al., 2018), in agreement with our findings. Whether STIM1/2 function as homodimers or heterodimers requires further investigations. Native CRAC channels are likely heterohexamers of all three Orais. STIM1 displays stronger basal interactions with Orai2 and Orai3 compared to Orai1 (Yoast et al., 2020b; Zhang et al., 2013). Interestingly, STIM2 shows stronger basal interaction with Orai1 and Orai2 compared to Orai3. Combinations of single STIM-KO/double Orai-KO and modeling showed that Ca2+ plateaus strictly require the STIM1/Orai1 duo, which also mediate the diversity of Ca2+ oscillation frequencies in combination with the other five STIM/Orai duos.
Although STIM1/2-KO cells completely lack SOCE, their oscillation profile does not phenocopy that of Orai-TKO cells. STIM1/2-KO cells are more sensitive to IP3-mediated Ca2+ release, and we show that STIM1/2 negatively regulate IP3R channel activity. We failed to detect physical interactions between STIM and IP3R proteins with co-immunoprecipitations assays (data not shown). While the mechanisms of this IP3R regulation by STIMs require further experimentation, our mutagenesis, ER Ca2+ measurements, and modeling data suggest it is the unactivated STIMs that inhibit IP3R channels, likely setting the threshold of activation by IP3. As STIM proteins unfold and become increasingly activated (a state mimicked by EF-hand mutants), the inhibition of IP3Rs is lifted with concomitant and equivalent CRAC channel activation, thus precisely coordinating the levels of Ca2+ release and Ca2+ entry. Taken together, our work clarifies the complex and dynamic choreography of native interactions between IP3Rs, two STIM, and three Orai proteins and how this set of proteins provides the mammalian cell with an effective toolkit to enhance the diversity of Ca2+ signaling events and subsequent transcriptional control in response to the full spectrum of agonist intensities.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for resources and reagents should be directed and fulfilled by the Lead Contact, Dr. Mohamed Trebak (mtrebak@psu.edu).
Materials availability
All unique/stable reagents generated in the study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
Raw data, unprocessed micrographs, and statistics of all data included in the manuscript are available here: https://data.mendeley.com/datasets/9dtvhhkzdz/draft?a=c43ec1d3-e142-4fd0-b29f-86660a9ecbe4
This study did not generate any unique software or code sets.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
All HEK293 cell lines were cultured in high glucose (4.5g/L) Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1X Antibiotic-Antimycotic (Thermo Scientific), and cultured at 37°C with 5% CO2. For CMV-driven STIM and Orai constructs, HEK293 cells were transfected using an Amaxa Nucleofector II (Amaxa Biosystems) with the Nucleofector Kit V (VVCA-1003) and program Q-001. For TK-driven and NFAT reporter constructs, cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Following each transfection cells were plated on 25mm glass coverslips and incubated for 24 hours before use in experiments.
Generation of STIM and Orai CRISPR/Cas9 knockout cell lines
HEK293 Orai and STIM knockout cell lines were generated as previously described (Emrich et al., 2019; Yoast et al., 2020b; Zhang et al., 2019). Briefly, sequence specific guide RNAs (gRNA) targeting STIM1, STIM2, Orai1, Orai2, or Orai3 were cloned into one of two fluorescent vectors (pSpCas9(BB)-2A-GFP, Addgene #48138; pU6-(BbsI)_CBh-Cas9-T2A-mCherry, Addgene #64324). For genes with commercially available validated antibodies (STIM1, STIM2, and Orai1) a single gRNA was utilized to reduce off target effects. For genes without validated antibodies (Orai2 and Orai3), two gRNA sequences were used to excise the entire gene. Parental HEK293 cell lines were transfected with each fluorescent CRISPR vector and individual single or double GFP/mCherry positive cells were FACS sorted (BD FACS Aria SORP) into 96-well cell culture plates 24 hours after transfection. Individual knockout clones were validated using western blot analysis, Sanger sequencing, and functional Ca2+ imaging experiments. Each gRNA sequence used to generate single and double Orai and STIM knockout cell lines was as follows:
Orai1 g1: 5′-GTTGCTCACCGCCTCGATGT-3′
Orai2 g1: 5′-ACGACAGGGCCTGTACCGAG-3′
Orai2 g3: 5′-CTCATGCGGGGACTCGCTGA-3′
Orai3 g2: 5′-GTTCGTGCACCGCGGCTACC-3′
Orai3 g4: 5′-CCAGAGACTGCACCGCTACG-3′
STIM1 g3: 5′-TGATGAGCTTATCCTCACCA-3′
STIM2 g1: 5′-AGATGGTGGAATTGAAGTAG-3′
METHOD DETAILS
DNA constructs
All novel tk-driven constructs (tk-STIM1, tk-STIM2, tk-S1N-S2C, tk-S2N-S1C) and STIM1/STIM2 EF-hand mutants were generated with primers compatible with the In-Fusion cloning system (Takara Bio Inc.). Primers used to create the D80A/D82A substitutions in YFP-STIM2.1 were 5′-ACAAATGGCTGATGCCAAAGATGGTGGAATTG-3′ and 5′-TGGCATCAGCCATTTGTTTATGTATTGTTTGAAG-3′. Primers used to create the D76A/D78A substitutions in STIM1-F394H-YFP were 5′-ACTGATGGCCGATGCTGCCAATGGTGATGTGG-3′ and 5′-CAGCATCGGCCATCAGTTTGTGGATGTTACGG-3′. CMV-CFP-Orai1, CMV-CFP-Orai2, CMV-CFP-Orai3, STIM1-YFP, STIM2-YFP, YFP-STIM2.1, and STIM1-F394H-YFP were from Dr. Don Gill. HA-NFAT1(4–460)-GFP (Addgene #11107) and HA-NFAT4(3–407)-GFP (Addgene #21664) were from Dr. Anjana Rao. pCMV R-CEPIA1er was from Dr. Masamitsu Iino (Addgene #58216).
Fluorescence imaging
HEK293 cells (1.5×105) were plated onto 25mm glass coverslips and allowed to attach overnight. For cytosolic Ca2+ measurements, coverslips were mounted in Attofluor cell chambers (Thermo Scientific) and cells were incubated in complete DMEM containing 2μM Fura-2AM (Molecular Probes) for 30 minutes at 37°C. Following Fura-2 loading, cells were washed 3 times with a HEPES-buffered saline solution (HBSS) containing the following components: 120mM NaCl, 5.4mM KCl, 0.8mM MgCl2, 2mM CaCl2, 20mM HEPES, 10mM D-glucose, and adjusted to pH 7.4 with NaOH. Chambers were mounted on a Leica DMi8 fluorescence microscope and Fura-2 fluorescence was measured every 2 s by excitation at 340nm and 380nm using a fast shutter wheel and the emission at 510nm was collected through a 20X fluorescence objective. Fluorescence data was collected from individual cells on a pixel by pixel basis and processed using Leica Application Suite X. All cytosolic Ca2+ concentrations are presented as the ratio of F340/F380 and plotted as means ± SEM from at least three independent experiments.
For ERCa2+measurements, HEK293 cell lines were transfected by Nucleofection with 1μg of R-CEPIA1er and imaged 24 hours later on a Leica TCS SP8 confocal microscope with a 40X oil objective. RFP fluorescence was excited with the 552nm laser at 1% output at room temperature. Images were collected every 10–15 s and traces were plotted as means ± SEM after normalization to the first time point. Prior to the start of each recording, cells were switched from HBSS containing 2mM CaCl2 to a HBSS solution nominally free of CaCl2.
For NFAT nuclear translocation, HEK293 cell lines were transfected with 1μg of NFAT1-GFP or NFAT4-GFP reporter constructs using Lipofectamine 2000. Cells were imaged 24 hours post transfection using a Leica DMi8 fluorescence microscope equipped with a Hamamatsu Flash 4 camera with a 40X oil objective. GFP fluorescence from the whole cell and nucleus was collected every 10 s using a 488nm fast filter wheel with emission captured through a corresponding GFP filter cube. NFAT1 and NFAT4 nuclear translocation was calculated using the equation and plotted as a function of time in response to carbachol stimulation.
Förster resonance energy transfer (FRET) measurements
Measurement of FRET between STIM1 and STIM2 with each Orai isoform was performed as previously described(Yoast et al., 2020b). Briefly, Orai-TKO cells were transfected by Nucleofection with 2μg STIM1-YFP in combination with 3μg CFP-Orai1, 4μg CFP-Orai2 or 5μg CFP-Orai3 and 2μg STIM2-YFP together with 3μg CFP-Orai1, 4μg CFP-Orai2, or 5μg CFP-Orai3. Imaging was performed on a Leica DMI 6000B inverted automated fluorescence microscope equipped with a 40X oil objective with simultaneous collection of CFP, YFP, and FRET images every 20 s. Exposure times for STIM1 and STIM2 FRET were 1000ms (CFP), 250ms (YFP), and 1000mM (FRET). Three channel FRET was calculated with SlideBook 6.0 software (Intelligent Imaging Innovations) using the formula where, IDD, IAA, and IDA were the respective fluorescent intensities of CFP, YFP, and FRET channels after taking individual CFP and YFP background correction images. Calculation of E-FRET was performed as previously described using the formula (Zal and Gascoigne, 2004).
Detection and analysis of Ca2+ puffs using TIRF microscopy
Parental WT-HEK293 cells or STIM1/2-KO cells were grown on 15-mm glass coverslips coated with poly-D-lysine (100 μg/ml) in a 35-mm dish for 2 days. Prior to imaging, the cells were washed three times with imaging buffer (137 mM NaCl, 5.5 mM glucose, 0.56 mM MgCl2, 4.7 mM KCl, 1.26 mM CaCl2, 10 mM HEPES, 1 mM Na2HPO4 at pH 7.4). The cells were subsequently incubated with Cal520-AM (5 μM; AAT Bioquest #21130) and 6-O-[(4,5-Dimethoxy-2-nitrophenyl)methyl]-2,3-O-(1-methylethylidene)-D-myo-Inositol 1,4,5-tris[bis[(1-oxopropoxy)methyl]phosphate] (ci-IP3/PM; 1 μM, Tocris #6210) in imaging buffer with 0.01% BSA in the dark at room temperature. After 1 hour incubation, the cells were washed three times with imaging buffer and incubated in imaging buffer containing EGTA-AM (5 μM, Invitrogen #E1219). After 45 minutes incubation, the media was replaced with fresh imaging buffer and incubated for additional 30 minutes at room temperature to allow for de-esterification of loaded reagents(Lock et al., 2015).
Following loading, the coverslip was mounted in a chamber and imaged using an inverted Olympus IX83 total internal reflection fluorescence microscope (TIRFM) equipped with an oil-immersion PLAPO OTIRFM 60x objective lens (1.45 numerical aperture). The cells were illuminated using a 488 nm laser to excite Cal-520 and the emitted fluorescence was collected through a bandpass filter by a Hamamatsu ORCA-Fusion CMOS camera. The angle of the excitation beam was adjusted to achieve TIRF with a penetration depth of ~140 nm. Images were captured from a field of view by directly streaming into RAM. To photorelease IP3, UV light from a 405 nm laser was introduced to uniformly illuminate the field of view. Both the intensity of the UV flash (~200 μW) and the duration (1000 msec) for uncaging IP3 were optimized to prevent spontaneous puff activity in the absence of loaded ci-IP3. TIRF images were captured using 4 × 4 pixel binning (433.333 nm/pixel) from equal field of views for WT-HEK293 and STIM1/2-KO cells at a rate of ~166 frames per second. After visualizing images with the cellSens [Ver.2.3] life science imaging software (Olympus), images were exported as vsi files. Images, 10 s before and 60 s after flash photolysis of ci-IP3, were captured.
The vsi files were converted to tif files using Image J (Fiji) and further processed using FLIKA, a Python programming based tool for image processing(Ellefsen et al., 2014). From each recording, 500 frames (~3 s) before photolysis of ci-IP3 were averaged to obtain a ratio image stack (F/Fo) and standard deviation for each pixel for recording up to 20 s following photolysis. The image stack was Gaussian-filtered, and pixels that exceeded a critical value (1.0 for our analysis) were located. The ‘Detect-puffs’ plug-in was utilized to detect the number of clusters (puff sites), number of events (number of puffs), amplitudes and durations of localized Ca2+ signals from individual cells. All the puffs identified automatically by the algorithm were manually confirmed before analysis(Lock et al., 2018; Mataragka and Taylor, 2018). The results from FLIKA were saved as excel and graphs were plotted using GraphPad Prism 8.
A similar protocol for loading was followed to test the effect of CCh on Ca2+ puffs. In this case, the cells were not loaded with ci-IP3. Cells were stimulated by perfusing 1 μM CCh to generate IP3-induced Ca2+ release.
Western blot analysis
HEK293 cell lines were harvested and lysed for 15 minutes in ice cold RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0; Sigma) containing 1X Halt protease/phosphatase inhibitors (Thermo Scientific). Following lysis, samples were clarified by centrifugation at 15,000×g for 10 minutes at 4°C. Supernatants were collected and protein concentration was determined using the Pierce Rapid Gold BCA Protein Assay Kit (Thermo Scientific). Equal concentrations of protein extract were loaded into 4%–12% NuPAGE BisTris gels (Life Technologies) and transferred to PVDF membranes utilizing the Transblot Turbo Transfer System (Bio-Rad). Membranes were blocked for 1 hour at room temperature in Odyssey Blocking Buffer in TBS (LI-COR) and incubated overnight at 4°C with primary antibody. The following antibodies and dilutions were used: STIM1 (1:2000; 4916, Cell Signaling Technology), STIM2 (1:1000; 4917, Cell Signaling Technology), α-tubulin (1:5000; 3873, Cell Signaling Technology), GAPDH (1:5000; MAB374, Sigma), IP3R-1 (1:1000, Yule Lab), IP3R-2 (1:1000, Yule Lab), and IP3R-3 (1:1000; 610313, BD Biosciences). Membranes were washed with TBST and incubated for 1 hour at room temperature with the following secondary antibodies: IRDye 680RD goat anti-mouse (1:10,000 LI-COR) or IRDye 800RD donkey anti-rabbit (1:10,000 LI-COR). Membranes were imaged on an Odyssey CLx Imaging System (LI-COR) and analysis performed in Image Studio Lite version 5.2 (LI-COR) and ImageJ.
Patch clamp electrophysiology
For measurement of Icrac, STIM1/2-KO cells were transfected by Nucleofection with 3.5μg of each YFP-tagged STIM chimera (YFP-S1N-S2C or YFP-S2N-S1C) along with 1.5μg of CFP-Orai1. Cells were seeded onto 30mm glass coverslips and patch clamp recordings were performed 24 hours after transfection. Patch-clamp recordings were performed using an Axopatch 200B along with a Digidata 1440A (Molecular Devices). Pipettes were pulled from borosilicate glass capillaries using a P-1000 Flaming/Brown micro-pipette puller (Sutter Instrument Company) and a DMF1000 (World Precision Instruments) was used for polishing. Glass electrodes with a resistance of 2 to 4 MΩ were utilized. Recordings were only performed when a > 16 GΩ seal was formed and when whole cell series resistance was < 8 MΩ. Patch Master (HEKA) was used for data acquisition and analysis. For recording of CRAC currents, cells were maintained at a +30mV holding potential and reverse voltage ramps from +100 to −140mV for 250ms at 2 s intervals were performed. After establishing the whole-cell configuration, intracellular stores were depleted using 20mM BAPTA. The following pipette and bath solutions were utilized:
Pipette solution: 145 mM cesium methanesulfonate, 20 mM BAPTA, 10 mM HEPES, 8 mM MgCl2, and 25 μM IP3 (pH to 7.2 with CsOH).
Bath solution: 115 mM Na-methanesulfonate, 10 mM CsCl, 1.2 mM MgSO4, 10 mM HEPES, 20 mM CaCl2, and 10 mM glucose (pH 7.4 with NaOH).
Mathematical modeling
The majority of the model parameters remain unchanged from (Yoast et al., 2020b). The only additional parameters are the equilibrium constants of the STIMi-Oraij,k interactions and the unitary current through a single STIMi-Oraij,k complex.
The following model assumptions are important for obtaining the correct qualitative behavior. There is flexibility in the precise numbers used, but the qualitative trends are critical.
There is a background STIM-independent influx of Ca2+ into the cell, which we call a leak. Knockout of STIM changes the size of this leak, which in turn sets the resting ER Ca2+ concentration. However, changes in the leak are not always sufficient to maintain 100% of the basal ER Ca2+ concentration when STIM1 or STIM2 are knocked out. STIM2-KO, STIM1-KO and STIM1,2-KO cells have resting ER Ca2+ of, respectively, 94%, 86% and 97% of basal. We propose no mechanism for how the leak changes. However, the counterintuitive assumption that STIM1,2-DKO cells have a higher resting ER Ca2+ than the STIM1-KO cells is consistent with the findings of Zheng et al. (Zheng et al., 2018b).
Unactivated STIM1 and STIM2 inhibit IP3R. This is modeled by a phenomenological function (see Modeling Methods below) that increases IP3R activity as the amount of unactivated STIM1 and/or STIM2 decreases, as a result of activation or knockout of STIM. This is a positive feedback loop (agonist stimulation decreases ER Ca2+, which activates STIM, which decreases the amount of unactivated STIM, which removes inhibition from the IP3R, which lowers ER Ca2+ further), but plays no significant role in the generation of single oscillations. Its main function is to increase oscillation frequency in a STIM1,2 DKO cell that has been closed by 1mM Gd3+ (see Figure 4D).
Binding ratios to the Orai homodimers (for example, the ratio of STIM1/Orai1,1 to STIM1/Orai2,2 binding, and so on) are determined from the FRET data in Figures 2M and 2Q; see details below in Modeling methods.
There is three times more STIM1 than STIM2.
In most cases, the unitary current through a STIM2-Oraij,k complex is about 1/5 of the current through the analogous STIM1-Oraij,k complex. The two exceptions are the unitary currents through STIM2-Orai2,2 and STIM2-Orai3,3 which are ten times the current through STIM1-Orai2,2 and STIM1-Orai3,3 respectively.
Modeling methods
The models of Sneyd et al. (2017) and Yoast et al. (2020b) are combined with a simple STIM model. Let S1 denote the concentration of STIM1 and let denote the concentration of activated STIM1, with analogous notation for STIM2. Assuming that S1 is activated in the reaction
it follows that, at steady state,
where is the maximal concentration of STIM1, and where . Similarly,
The STIM bind to the various Orai dimers as follows,
and thus, at steady state,
for some constants pij and qij, which determine how well each STIM binds to the Orai dimers. Each pij and qij is a combination of the equilibrium binding constant of the relevant Orai and STIM and the ratio of .
The ratios of the equilibrium binding constants are estimated from the data in Figures 3M and 3Q. This gives , , , . In other words, STIM1 binds Orai2 twice as well as it binds Orai1, and binds Orai3 with an intermediate efficacy. Similarly STIM2 binds Orai1 twice as well as it binds Orai3, and binds Orai2 better still. For want of a better choice, each binding constant pij with i≠j was set to (pii + pjj)/2. The relative sizes of pij and qij are then adjusted by choice of . We chose , in order to get qualitative agreement with the data presented here.
The inward Ca current, Jin, is given by
for some constants αij and βij which control the amount of currrent through each open STIM-Oraii,j complex. The αij were taken from (Yoast et al., 2020b), and the βij were chosen to obtain qualitative agreement with the data presented here. Reasonable agreement was obtained by setting βij = αij/5, with the exceptions β22 = 10α22 and β33 = 10α33: Hence, the current through STIM2-Oraii,j is generally only 20% of the current through the analogous STIM1-Oraii,j, with the exception of Orai2,2 and Orai3,3. The parameter α0 is the STIM-independent leak.
We also have a conservation equation for Orai1,
with analogous conservation equations for Orai2 and Orai3. Given known values of ce, pij, qij, αij, βij the conservation equations can be solved to determine Oij, and thus determine Jin. This needs to be done for every time step of the differential equations for the calcium concentration, which are taken from (Sneyd et al., 2017).
To model the inhibition of the IP3R by unactivated STIM, we multiply the usual IP3R current (Sneyd et al., 2017) by the factor
where K = 0.005 and n = 6, and where Si denotes the normalized concentration of activated STIMi. Under basal conditions, S1 and S2 are much less than 1, and F is thus close to 1. As S1 and S2 increase, F also increases, to a maximum value of 3. Thus, in the absence of any unactivated STIM, the current through the IP3R is three times greater than the current would be in the absence of feedback from STIM.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were conducted using GraphPad Prism 8 and data presented as mean ± SEM. N numbers are included in each figure legends. An unpaired, nonparametric Mann-Whitney t test was used when comparing two sample groups. For comparisons of greater than two groups, the Kruskal-Wallis test with Dunn’s multiple comparison was used. Specifically, for Figure 1. Data analyzed with the Kruskal-Wallis one-way ANOVA with multiple comparisons to WT-HEK293 cells. For Figure 2. (H) and (I) are analyzed with the Mann-Whitney U test. (J-L) are analyzed with an unpaired Student’s t test. (N-P) and (R-T) are analyzed with the Kruskal-Wallis one-way ANOVA with multiple comparisons. For Figure 3. (B, G, L) and (P) are analyzed with the Kruskal-Wallis one-way ANOVA with multiple comparisons to the respective parental cell line. (Q-S) are analyzed with one-way ANOVA with multiple comparisons to the respective parental cell line. For Figure 4. Data analyzed with the Kruskal-Wallis one-way ANOVA with multiple comparisons to WT-HEK293 (E, G, H) or non-transfected STIM1/2-KO controls (J, K, Q, R). For Figure 5. Data analyzed with Student’s t test (unpaired, two-tailed). For Figure 7. Data analyzed with the Kruskal-Wallis one-way ANOVA with multiple comparisons to WT-HEK293. For representation of statistical significance, *, **, ***, and **** indicate p values of < 0.05, < 0.01, < 0.001, and < 0.0001 respectively. ns; not significant. Exact p values for all comparisons performed in the study along with all raw data are posted online on Mendeley Data.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GAPDH | MilliporeSigma | Cat# MAB374; RRID: AB_2107445 |
| Rabbit monoclonal anti-STIM1 | Cell Signaling Technologies | Cat# 5668; RRID: AB_10828699 |
| Rabbit polyclonal anti-STIM2 | Cell Signaling Technologies | Cat# 4917; RRID: AB_2198021 |
| α-IP3R1 | Yule Lab | N/A |
| α-IP3R2 | Yule Lab | N/A |
| Anti-IP3R-3 | Becton, Dickinson (BD) | Cat# 610313; RRID: AB_397705 |
| α-Tubulin (DM1A) Mouse mAb | Cell Signaling Technologies | Cat# 3873S; RRID: AB_1904178 |
| 680 CW goat anti-Mouse | LI-COR | Cat# 926–68070; RRID: AB_10956588 |
| 800 CW donkey anti-Rabbit | LI-COR | Cat# 926–32213; RRID: AB_621848 |
| Chemicals, peptides, and recombinant proteins | ||
| Thapsigargin | Calbiochem | Cat#586005 |
| Carbachol | MilliporeSigma | Cat#212385 |
| Fura-2 AM | Invitrogen | Cat#F1201 |
| BAPTA-tetracesium Salt | Santa Cruz Biotechnology | Cat#480436-84-8 |
| Intercept® (TBS) Blocking Buffer | LI-COR | Cat#927–60001 |
| Halt Protease and Phosphatase Inhibitor | Thermo Scientific | Cat#PI78443 |
| RIPA Buffer | Sigma-Aldrich | Cat#R0278–50ML |
| Gadolinium(III) Chloride | ACROS Organics | Cat#AC383560050 |
| Cyclopiazonic Acid | Alomone Labs | Cat#C-750 |
| Lipofectamine 2000 Transfection Reagent | Thermo Scientific | Cat#11668027 |
| Cal-520, potassium salt | AAT Bioquest | Cat#21140 |
| ci-IP3/PM | Tocris | Cat#6210 |
| EGTA, AM | Invitrogen | Cat#E1219 |
| Critical commercial assays | ||
| Guide-it Mutation Detection Kit | Clontech Laboratories | Cat#631443 |
| Cell line Nucleofector Kit | Lonza | Cat#VCA-1003 |
| In-Fusion® HD EcoDry Cloning System | Clontech Laboratories | Cat#639688 |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo Scientific | Cat#A53225 |
| Deposited data | ||
| http://data.mendeley.com/login?redirectPath=/datasets/9dtvhhkzdz/draft?a=c43ec1d3-e142-4fd0-b29f-86660a9ecbe4 | Trebak Lab, this paper | N/A |
| Experimental models: cell lines | ||
| HEK293 cells | ATCC | Cat#CRL-1573 |
| STIM1 KO cells | Trebak Lab (Emrich et al., 2019) | N/A |
| STIM2 KO cells | Trebak Lab (Emrich et al., 2019) | N/A |
| STIM1/2 KO cells | Trebak Lab (Emrich et al., 2019) | N/A |
| Orai2,3 DKO cells | Trebak Lab (Yoast et al., 2020b) | N/A |
| Orai1,3 DKO cells | Trebak Lab (Yoast et al., 2020b) | N/A |
| Orai1,2 DKO cells | Trebak Lab (Yoast et al., 2020b) | N/A |
| Orai TKO cells | Trebak Lab (Yoast et al., 2020b) | N/A |
| Orai2,3-STIM1 KO cells | Trebak Lab, this paper | N/A |
| Orai2,3-STIM2 KO cells | Trebak Lab, this paper | N/A |
| Orai1,3-STIM1 KO cells | Trebak Lab, this paper | N/A |
| Orai1,3-STIM2 KO cells | Trebak Lab, this paper | N/A |
| Orai1,2-STIM1 KO cells | Trebak Lab, this paper | N/A |
| Orai1,2-STIM2 KO cells | Trebak Lab, this paper | N/A |
| IP3R TKO cells | Yule Lab(Alzayady et al., 2016) | N/A |
| Oligonucleotides | ||
| Forward primer for CMV-YFP-STIM2.1 (D80A/D82A) 5′-ACAAATGGCTGATGCCAAAGATGGTGGAATTG-3′ | Trebak Lab, this paper | N/A |
| Reverse Primer for CMV-YFP-STIM2.1 (D80A/D82A) 5′-TGGCATCAGCCATTTGTTTATGTATTGTTTGAAG-3′ | Trebak Lab, this paper | N/A |
| Forward primer for CMV-STIM1-F394H (D76A/D78A)-YFP 5′-ACTGATGGCCGATGCTGCCAATGGTGATGTGG-3′ | Trebak Lab, this paper | N/A |
| Reverse primer for CMV-STIM1-F394H (D76A/D78A)-YFP 5′-CAGCATCGGCCATCAGTTTGTGGATGTTACGG-3′ | Trebak Lab, this paper | N/A |
| ORAI1 g1 CRISPR/Cas9 Guide RNA 5′-GTTGCTCACCGCCTCGATGT-3′ | Trebak Lab, this paper | N/A |
| ORAI2 g1 CRISPR/Cas9 Guide RNA 5′-ACGACAGGGCCTGTACCGAG-3′ | Trebak Lab, this paper | N/A |
| ORAI2 g3 CRISPR/Cas9 Guide RNA 5′-CTCATGCGGGGACTCGCTGA-3′ | Trebak Lab, this paper | N/A |
| ORAI3 g2 CRISPR/Cas9 Guide RNA 5′-GTTCGTGCACCGCGGCTACC-3′ | Trebak Lab, this paper | N/A |
| ORAI3 g4 CRISPR/Cas9 Guide RNA 5′-CCAGAGACTGCACCGCTACG-3′ | Trebak Lab, this paper | N/A |
| STIM1 g3 CRISPR/Cas9 Guide RNA 5′-TGATGAGCTTATCCTCACCA-3′ | Trebak Lab, this paper | N/A |
| STIM2 g1 CRISPR/Cas9 Guide RNA 5′-AGATGGTGGAATTGAAGTAG-3′ | Trebak Lab, this paper | N/A |
| Recombinant DNA | ||
| HA-NFAT1(4–460)-GFP | Addgene | Cat#11107 |
| HA-NFAT4(3–407)-GFP | Addgene | Cat#21664 |
| LentiCRISPR v2 | Addgene | Cat#52961 |
| pU6-(Bbs1)-mCherry | Addgene | Cat#64324 |
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat#48138 |
| pCMV R-CEPIA1er | Addgene | Cat#58216 |
| CMV-CFP-ORAI1 | Gill Lab | NA |
| CMV-CFP-ORAI2 | Gill Lab | NA |
| CMV-CFP-ORAI3 | Gill Lab | NA |
| CMV-STIM1-YFP | Gill Lab | NA |
| CMV-STIM2-YFP | Gill Lab | NA |
| CMV-YFP-S1N-S2C | Trebak Lab(Emrich et al., 2019) | NA |
| CMV-YFP-S2N-S1C | Trebak Lab(Emrich et al., 2019) | NA |
| TK-YFP-S1N-S2C | Trebak Lab, this paper | NA |
| TK-YFP-S2N-S1C | Trebak Lab, this paper | NA |
| TK-STIM1-YFP | Trebak Lab, this paper | NA |
| TK-STIM2-YFP | Trebak Lab, this paper | NA |
| CMV-YFP-STIM2.1 | Gill Lab | NA |
| CMV-STIM1-F394H-YFP | Gill Lab | NA |
| CMV-YFP-STIM2.1 (D80A/D82A) | Trebak Lab, this paper | NA |
| CMV-STIM1-F394H (D76A/D78A)-YFP | Trebak Lab, this paper | NA |
| Software and algorithms | ||
| Graphpad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Clampfit 10.3 | Molecular Devices, LLC. | https://www.moleculardevices.com/ |
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| LAS X | Leica | https://www.leica-microsystems.com/ |
| SlideBook 6.0 | Intelligent Imaging Innovations, Inc. | https://www.intelligent-imaging.com/slidebook |
| MAXCHELATOR | Chris Patton: cpatton@stanford.edu | https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/ |
| Image Studio Lite | LI-COR | https://www.licor.com/bio/image-studio-lite/download |
Highlights.
All five STIM/Orai proteins are always required together during Ca2+ signaling
Unactivated STIM1/2 inhibit IP3R and activated STIM1/2 cooperatively activate Orai1/2/3
STIM1 contribution increases and that of STIM2 decreases as agonist intensifies
Graded IP3R disinhibition and Orai activation tailor agonist strength to NFAT activity
ACKNOWLEDGMENTS
We are grateful to Drs. Jeff Lock and Ian Parker (University of California, Irvine) for their advice and help with the Ca2+ puff measurements using TIRF microscopy and with data analysis. This work was supported by NIH/NHLBI (R35-HL150778 to M.T.), NIH/NHLBI (R01 HL137852 to Scott Earley and M.T.), NIH NIDCR (R01 DE019245 to D.I.Y. and J.S.), NIH/NIGMS (1R35 GM131916 to D.L.G.), and NIH/NCI (R01 CA242021 to N.H.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108760.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahuja M, Chung WY, Lin WY, McNally BA, and Muallem S (2020). Ca2+ Signaling in Exocrine Cells. Cold Spring Harb. Perspect. Biol 12, a035279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzayady KJ, Wang L, Chandrasekhar R, Wagner LE 2nd, Van Petegem F, and Yule DI (2016). Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci. Signal 9, ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagur R, and Hajnóczky G (2017). Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 66, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (2016). The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev 96, 1261–1296. [DOI] [PubMed] [Google Scholar]
- Bird GS, and Putney JW Jr. (2005). Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J. Physiol 562, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR, and Putney JW Jr. (2009). STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol 19, 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Liou J, Park WS, and Meyer T (2007). STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, and Lewis RS (1994). Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol 103, 365–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont G, Combettes L, Bird GS, and Putney JW (2011). Calcium oscillations. Cold Spring Harb. Perspect. Biol 3, a004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefsen KL, Settle B, Parker I, and Smith IF (2014). An algorithm for automated detection, localization and measurement of local calcium signals from camera-based imaging. Cell Calcium 56, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emrich SM, Yoast RE, Xin P, Zhang X, Pathak T, Nwokonko R, Gueguinou MF, Subedi KP, Zhou Y, Ambudkar IS, et al. (2019). Cross-talk between N-terminal and C-terminal domains in stromal interaction molecule 2 (STIM2) determines enhanced STIM2 sensitivity. J. Biol. Chem 294, 6318–6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner M, Schindl R, Muik M, Derler I, and Romanin C (2017). The STIM-Orai Pathway: The Interactions Between STIM and Orai. Adv. Exp. Med. Biol 993, 59–81. [DOI] [PubMed] [Google Scholar]
- Feske S (2019). CRAC channels and disease - From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 80, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, and Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Feske S, Srikanth S, Hogan PG, and Rao A (2007). Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 42, 145–156. [DOI] [PubMed] [Google Scholar]
- Hogan PG, and Rao A (2015). Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun 460, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, and Penner R (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356. [DOI] [PubMed] [Google Scholar]
- Korzeniowski MK, Manjarrés IM, Varnai P, and Balla T (2010). Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal 3, ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS (2020). Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol 12, a035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, and Cahalan MD (1989). Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., and Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JT, Ellefsen KL, Settle B, Parker I, and Smith IF (2015). Imaging local Ca2+ signals in cultured mammalian cells. J. Vis. Exp 3, 52516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JT, Alzayady KJ, Yule DI, and Parker I (2018). All three IP3 receptor isoforms generate Ca2+ puffs that display similar characteristics. Sci. Signal 11, eaau0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataragka S, and Taylor CW (2018). All three IP3 receptor subtypes generate Ca2+ puffs, the universal building blocks of IP3-evoked Ca2+ signals. J. Cell Sci 131, jcs220848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miederer AM, Alansary D, Schwär G, Lee PH, Jung M, Helms V, and Niemeyer BA (2015). A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun 6, 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S, and Ambudkar IS (2015). STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal 8, ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, and Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, and Lewis RS (2015). Store-Operated Calcium Channels. Physiol. Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW, and Tomita T (2012). Phospholipase C signaling and calcium influx. Adv. Biol. Regul 52, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Yen M, Sadaghiani AM, Malmersjö S, Park CY, Dolmetsch RE, and Lewis RS (2015). Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol 209, 653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A (2009). Signaling to gene expression: calcium, calcineurin and NFAT. Nat. Immunol 10, 3–5. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of Image Analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedler E, and Uhlén P (2014). Frequency decoding of calcium oscillations. Biochim. Biophys. Acta 1840, 964–969. [DOI] [PubMed] [Google Scholar]
- Sneyd J, Tsaneva-Atanasova K, Yule DI, Thompson JL, and Shuttle-worth TJ (2004). Control of calcium oscillations by membrane fluxes. Proc. Natl. Acad. Sci. USA 101, 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneyd J, Han JM, Wang L, Chen J, Yang X, Tanimura A, Sanderson MJ, Kirk V, and Yule DI (2017). On the dynamical structure of calcium oscillations. Proc. Natl. Acad. Sci. USA 114, 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, and Gill DL (2006). STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol 16, 1465–1470. [DOI] [PubMed] [Google Scholar]
- Son GY, Subedi KP, Ong HL, Noyer L, Saadi H, Zheng C, Bhardwaj R, Feske S, and Ambudkar IS (2020). STIM2 targets Orai1/STIM1 to the AKAP79 signaling complex and confers coupling of Ca2+ entry with NFAT1 activation. Proc. Natl. Acad. Sci. USA 117, 16638–16648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Woo JS, Sun Z, and Gwack Y (2017). Immunological Disorders: Regulation of Ca2+ Signaling in T Lymphocytes. Adv. Exp. Med. Biol 993, 397–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, and Frauwirth KA (2007). Reciprocal NFAT1 and NFAT2 nuclear localization in CD8+ anergic T cells is regulated by suboptimal calcium signaling. J. Immunol 179, 3734–3741. [DOI] [PubMed] [Google Scholar]
- Subedi KP, Ong HL, Son GY, Liu X, and Ambudkar IS (2018). STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 23, 522–534. [DOI] [PubMed] [Google Scholar]
- Thiel M, Lis A, and Penner R (2013). STIM2 drives Ca2+ oscillations through store-operated Ca2+ entry caused by mild store depletion. J. Physiol 591, 1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida T, Hirose K, Takizawa A, Shibasaki F, and Iino M (2003). NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 22, 3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, and Kinet JP (2019). Calcium signalling in T cells. Nat. Rev. Immunol 19, 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, and Putney JW Jr. (2017). ORAI Calcium Channels. Physiology (Bethesda) 32, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, Bird GS, McKay RR, and Putney JW Jr. (2002). Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J. Biol. Chem 277, 21617–21623. [DOI] [PubMed] [Google Scholar]
- Vaeth M, and Feske S (2018). NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res. 7, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, and Gill DL (2014). Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun 5, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel B, Boyles RR, Putney JW Jr., and Bird GS (2007). Role of the store-operated calcium entry proteins Stim1 and Orai1 in muscarinic cholinergic receptor-stimulated calcium oscillations in human embryonic kidney cells. J. Physiol 579, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung PS, Yamashita M, and Prakriya M (2020). Molecular basis of allosteric Orai1 channel activation by STIM1. J. Physiol 598, 1707–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yissachar N, Sharar Fischler T, Cohen AA, Reich-Zeliger S, Russ D, Shifrut E, Porat Z, and Friedman N (2013). Dynamic response diversity of NFAT isoforms in individual living cells. Mol. Cell 49, 322–330. [DOI] [PubMed] [Google Scholar]
- Yoast RE, Emrich SM, and Trebak M (2020a). The anatomy of native CRAC channel(s). Curr. Opin. Physiol 17, 89–95. [PMC free article] [PubMed] [Google Scholar]
- Yoast RE, Emrich SM, Zhang X, Xin P, Johnson MT, Fike AJ, Walter V, Hempel N, Yule DI, Sneyd J, et al. (2020b). The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun 11, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, and Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yule DI (2001). Subtype-specific regulation of inositol 1,4,5-trisphosphate receptors: controlling calcium signals in time and space. J. Gen. Physiol 117, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zal T, and Gascoigne NR (2004). Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J 86, 3923–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, González-Cobos JC, Schindl R, Muik M, Ruhle B, Motiani RK, Bisaillon JM, Zhang W, Fahrner M, Barroso M, et al. (2013). Mechanisms of STIM1 activation of store-independent leukotriene C4-regulated Ca2+ channels. Mol. Cell. Biol 33, 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Pathak T, Yoast R, Emrich S, Xin P, Nwokonko RM, Johnson M, Wu S, Delierneux C, Gueguinou M, et al. (2019). A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun 10, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xin P, Yoast RE, Emrich SM, Johnson MT, Pathak T, Benson JC, Azimi I, Gill DL, Monteith GR, and Trebak M (2020). Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 91, 102281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Ma G, He L, Zhang T, Li J, Yuan X, Nguyen NT, Huang Y, Zhang X, Gao P, et al. (2018a). Identification of molecular determinants that govern distinct STIM2 activation dynamics. PLoS Biol. 16, e2006898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Zhou L, Ma G, Zhang T, Liu J, Li J, Nguyen NT, Zhang X, Li W, Nwokonko R, et al. (2018b). Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflugers Arch. 470, 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Nwokonko RM, Loktionova NA, Wang Y, and Gill DL (2017). The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium 63, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nwokonko RM, Cai X, Loktionova NA, Abdulqadir R, Xin P, Niemeyer BA, Wang Y, Trebak M, and Gill DL (2018). Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 115, E3398–E3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nwokonko RM, Baraniak JH Jr., Trebak M, Lee KPK, and Gill DL (2019). The remote allosteric control of Orai channel gating. PLoS Biol. 17, e3000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data, unprocessed micrographs, and statistics of all data included in the manuscript are available here: https://data.mendeley.com/datasets/9dtvhhkzdz/draft?a=c43ec1d3-e142-4fd0-b29f-86660a9ecbe4
This study did not generate any unique software or code sets.