

GREB1 is an inducible cytoplasmic glycosyltransferase.
Abstract
What covalent modifications control the temporal ubiquitination of ERα and hence the duration of its transcriptional activity remain poorly understood. We show that GREB1, an ERα-inducible enzyme, catalyzes O-GlcNAcylation of ERα at residues T553/S554, which stabilizes ERα protein by inhibiting association with the ubiquitin ligase ZNF598. Loss of GREB1-mediated glycosylation of ERα results in reduced cellular ERα levels and insensitivity to estrogen. Higher GREB1 expression in ERα+ve breast cancer is associated with greater survival in response to tamoxifen, an ERα agonist. Mice lacking Greb1 exhibit growth and fertility defects reminiscent of phenotypes in ERα-null mice. In summary, this study identifies GREB1, a protein with an evolutionarily conserved domain related to DNA-modifying glycosyltransferases of bacteriophages and kinetoplastids, as the first inducible and the only other (apart from OGT) O-GlcNAc glycosyltransferase in mammalian cytoplasm and ERα as its first substrate.
INTRODUCTION
Transcription factors, including hormone receptors, are master regulators of gene expression programs that maintain homeostasis (1). Because misregulations of transcription factor activities often lead to diseases, their levels are tightly controlled by a myriad of mechanisms, including posttranslational modifications that regulate temporal finetuning, as seen with nuclear factor κB (NF-κB) (2) or p53 (3) pathways. The levels and hence activity of hormone receptor estrogen receptor α (ERα), key for reproduction (4) and cancer development (5), are temporally regulated by ubiquitination and phosphorylation upon ligand binding (6). Nonetheless, unlike with NF-κB or p53 pathways, what controls the temporal ubiquitination of ERα and hence the duration of its transcriptional activity remains poorly understood. O-GlcNAcylation is a posttranslational modification that has been implicated in regulating protein stability in mammals (7, 8). However, unlike enzymes mediating other covalent modifications, enzymes that inducibly control O-GlcNAcylation are not known. Moreover, while O-GlcNAcylation of ERα has been reported (9), it is unclear how ERα glycosylation is regulated and what effect it has on ERα levels and signaling.
In eukaryotes, most proteins are glycosylated with glycans added to the side chains of either asparagine on the carbonyl nitrogen (N-linked) or serine/threonine on the alcohol oxygen (O-linked). Dedicated glycosylation systems mediate the decoration of secreted or cell surface proteins with mono- or polysaccharide tags concurrent with their maturation in the endoplasmic reticulum (10, 11). In the nucleus and other intracellular compartments, the O-linked glycosylation of serines/threonines in proteins with N-acetylglucosamine (O-GlcNAcylation), catalyzed by OGT enzyme, plays important regulatory roles (12, 13). O-GlcNAcylation of histones H2A, H2B, and H4 is believed to have an epigenetic role (14, 15). Likewise, OGT regulates the metabolism of cells in normal and disease states: For instance, O-GlcNAcylation of the pyruvate kinase isoform PKM2 leads to a metabolic reconfiguring characteristic of the Warburg effect seen in cancer cells (16). However, it remains unknown whether enzymes other than OGT play a role in intracellular glycosylation events of physiological relevance. Furthermore, OGT does not associate with all the cytoplasmic proteins, like ERα, and is constitutively active, raising the question: What enzymes O-GlcNAcylate substrates like ERα in a temporal manner?
Computational sequence analysis of growth regulation by estrogen in breast cancer 1 (GREB1), a top ERα target gene, indicated that it might be a putative glycosyltransferase (GT) (17). GREB1 encodes a 1949–amino acid protein that contains an N-terminal Zn-binding domain followed by a circularly permuted catalytically inactive superfamily II helicase domain and a C-terminal Fringe-like GT domain. The GT domain is related to the DNA-base GTs found in bacteriophages and kinetoplastids (17) that catalyze the transfer of sugars to 5-hydroxymethylated cytosine or thymine. This homology raised the question of whether the GT domain of GREB1 might be part of a previously unidentified inducible glycosylation apparatus distinct from the previously characterized glycosylation system centered on OGT. GREB1 is implicated in hormone-dependent cancers, such as ERαve+ subtype of breast cancer (18) and ovarian and prostate cancer (19, 20), and as a risk factor in endometriosis and variations in bone mineral density (21–24). In turn, GREB1 has been postulated to function as a coactivator of ERα as it was documented to co-occupy ERα-bound chromatin (25). We thought that it was very likely that GREB1 had other roles than merely an adaptor for ERα-mediated transcription, as GREB1 itself evolved much earlier in eukaryotes than ERα (17, 26). Here, we identify GREB1 as the first inducible O-GlcNAc transferase in mammals. GREB1 uses sugars produced by the hexosamine biosynthetic pathway (HBP) driven by another ERα target gene XBP1 and temporally modifies ERα. GREB1-mediated glycosylation of ERα blocks its degradation mediated by ZNF598 ubiquitin E3 ligase. This study explains how site-specific O-GlcNAc modifications of ERα are achieved in an inducible and temporal fashion by two of its target genes, XBP1 and GREB1. GREB1 is the first inducible O-GlcNAc GT in the cytoplasm to be identified in mammals, and these findings have lessons for expanding inducible protein modifications in health and disease.
RESULTS
GREB1 regulates the proliferation of ERα+ve breast cancer cells
The predicted conserved domains of GREB1 protein are schematically illustrated in Fig. 1A. In accordance with previous studies (18, 27), expression of GREB1 mRNA was detected in ERα+ve but not in ERα−ve breast cancer lines (fig. S1A). In the human breast adenocarcinoma cell line MCF7, GREB1 was among the top 10 mRNA transcripts induced by estradiol (E2) treatment (Fig. 1B), suggesting that it may play a key role in ERα signaling. We used CRISPR-Cas9 technology to target a region downstream of the start codon and created GREB1 knockout (KO) MCF7 cell lines (GREB1-KO) numbered 517, 417, and 441, which showed disruption of gene sequences (fig. S1B) and consequent loss of GREB1 protein (fig. S1C). Proliferation of GREB1-KO clones was significantly impaired, as measured by fluorometric viability assays (fig. S1D) and colony formation assays (fig. S1E). These in vitro differences in cell proliferation were also evident in vivo; when implanted into immunosuppressed nonobese diabetic (NOD)/severe combined immunodeficient (SCID) mice supplemented with human E2, MCF7–wild-type (WT)–derived tumors grew significantly faster than did their GREB1-KO counterparts (fig. S1, F and G). Knocking out GREB1 led to a loss of estrogen-responsive genes, e.g., GREB1 itself and X-box protein 1 (XBP1) (Fig. 1C). CRISPR-mediated removal of GREB1 in other ERα+ve breast cancer lines, BT474 and T47D, also led to a loss of GREB1 protein and dampened cell growth (fig. S1, H to J), suggesting the universality of our observations.
Fig. 1. GREB1 regulates ERα signaling through stabilization of ERα.
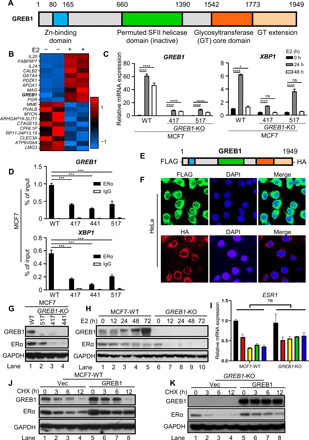
(A) Schematic diagram of predicted GREB1 domains (17). (B) Heatmap illustrating the top 10 up-regulated and down-regulated genes from RNA sequencing of MCF7-WT cells with or without estradiol (E2) stimulation. (C) MCF7-WT and GREB1-KO cells were treated with E2 and analyzed by qPCR. n = 3; ns, not significant, *P < 0.05, ***P < 0.001, and ****P < 0.0001 by two-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test. (D) ERα recruitment at indicated promoters in MCF7-WT and GREB1-KO cells were analyzed by ChIP followed by qPCR. n = 2; ***P < 0.001 and ****P < 0.0001 by one-way ANOVA with Holm-Sidak’s multiple comparisons test. (E) Schematic illustration of the GREB1 construct used for immunofluorescence. (F) GREB1 localization was analyzed by immunofluorescence microscopy in HeLa cells expressing the tagged GREB1 construct. Nuclei were stained with DAPI. (G) In MCF7-WT and GREB1-KO cells, levels of indicated proteins were analyzed by Western blot. (H and I) After E2 treatment in MCF7-WT and GREB1-KO cells, protein levels (H) and ESR1 mRNA expression (I) were analyzed. n = 2; by ANOVA two-way test. (J) MCF7-WT cells were transduced with empty vector (Vec) or GREB1 construct and subsequently treated with CHX. (K) GREB1-KO cells were transduced with empty Vec or GREB1 construct and subsequently treated with CHX.
Ectopic expression of GREB1 through lentiviral transduction (fig. S2A) restored ERα target gene XBP1 expression (fig. S2B) and proliferation (fig. S2C) in GREB1-KO cells. Notably, ERα (ESR1) mRNA levels remained unchanged with or without GREB1 (fig. S2B), suggesting that GREB1 does not affect transcriptional levels of ERα. These experiments collectively show that GREB1 regulates proliferation in ERα+ve breast cancer cell lines and its expression is needed for normal induction of ERα target genes like XBP1 following estrogen treatment.
GREB1 regulates ERα by direct interaction via its ligand-binding domain
To elucidate how GREB1 modulates ERα signaling, we performed chromatin immunoprecipitation (ChIP), which revealed that ERα recruitment to the promoters of estrogen-responsive genes GREB1 and XBP1 was reduced in GREB1-KO cells (Fig. 1D). This loss of ERα recruitment to GREB1 and XBP1 promoters in GREB1-KO cells could be restored by reintroducing GREB1 (fig. S2, D and E). GREB1 protein localization is key to understand its function. However, there had been no consensus of GREB1 localization (25, 28, 29). Our immunofluorescence microscopy of HeLa cells expressing an N-terminal FLAG-tagged and C-terminal hemagglutinin (HA)–tagged GREB1 (Fig. 1E) suggested a predominantly cytoplasmic localization (Fig. 1F). To rule out the possibility of artifact due to ectopic expression, we biochemically fractionalized cytoplasmic and nuclear lysates from a panel of human cell lines. Evidently, GREB1 was only highly expressed in ERα+ve breast cancer cell lines MCF7 and T47D and predominantly cytoplasmic with a detectable nuclear presence (fig. S2F). Notably, ERα itself was present in both the cytoplasm and the nucleus, while histone H3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as nuclear and cytoplasmic markers, respectively, demonstrating the rigidity of our fractionation conditions.
To elucidate the interaction between ERα and GREB1, we cloned N-terminal FLAG-tagged ERα constructs encoding (i) full-length (FL) ERα and (ii) truncation mutants, including combinations of known ERα domains (fig. S2G). After transfection in MCF7 cells, FLAG IP revealed that GREB1 predominantly interacted with the ligand-binding domain of ERα, which alone coprecipitated a similar amount of endogenous GREB1 as did the FL ERα (fig. S2H)
GREB1 regulates the posttranslational stability of ERα protein
The basal level of ERα protein is a driving factor in ERα signaling in response to estrogen, as observed in some breast cancers (6). Unexpectedly, GREB1-KO cells had lower levels of ERα protein compared to parental MCF7 cells under basal conditions (Fig. 1G). Following E2 treatment, in MCF7-WT cells, GREB1 accumulated as expected of an estrogen-responsive gene, whereas ERα slowly decreased before stabilizing at 48-hour time point (Fig. 1H, lanes 1 to 5). In contrast, in the GREB1-KO cells, the absence of GREB1 correlated with a marked loss of ERα protein from 12 hours after treatment (Fig. 1H, lanes 6 to 10). Thus, both under basal conditions and following E2 stimulation, the presence of GREB1 is associated with higher ERα protein levels. Deletion of GREB1 in other breast ERα+ve cancer cell lines BT474 and T47D also led to lower ERα levels (fig. S1, H and I), suggesting the universality of GREB1-mediated control of ERα protein levels. Because ESR1 transcript levels are not notably affected by the presence or absence of GREB1 (fig. S2B and Fig. 1I), GREB1 likely increases either posttranslational stability or translation efficiency of ERα mRNA. ERα protein turnover rates were assessed when protein synthesis was inhibited by cycloheximide (CHX) treatment. In cells transduced with the empty vector, ERα degraded much faster in GREB1-KO cells than in MCF7-WT cells (Fig. 1, J and K). However, the expression of GREB1 slowed the rate of ERα degradation in both MCF7-WT and GREB1-KO cells (Fig. 1, J and K). Together, these results suggest that GREB1 stabilizes ERα protein.
GREB1 catalyzes O-GlcNAcylation of ERα
Glycosylation is a common mediator of the stability of cellular proteins (7, 30, 31). Comparing the amino acid sequences of GREB1 homologs and the GREB1-related gene family members to other characterized GTs, including the J-base GT and the TET/JBP-associated GT (TAGT) in phages, we observed strong alignment and conservation at key residues in the GT domain (fig. S3A). While GREB1 GT domain is related to DNA-modifying GTs, there is currently no evidence for DNA glycosylation in mammals. Moreover, predominantly cytoplasmic localization of GREB1 (Fig. 1F and fig. S2F) suggests the possibility that GREB1 might function as protein GT rather than nucleic acid GT and ERα itself might be a substrate for GREB1. To date, OGT is the only cytoplasmic GT whose modification of serine and threonine residues with O-GlcNAc (O-GlcNAcylation) regulates target protein function and stability (32–35). Immunoprecipitated FLAG-ERα from GREB1-KO cells had much lower O-GlcNAcylation than that from MCF7-WT cells (Fig. 2A, lanes 3 and 4). To address whether the predicted GT domain of GREB1 was responsible for the differences in ERα O-GlcNAcylation between MCF7-WT and GREB1-KO cells, we created a catalytically dead GREB1 mutant (GT-Mut) construct by mutating residues predicted to be involved in substrate binding (Fig. 2B and fig. S3A). Unlike GT-WT reconstituted GREB1-KO cells, GT-Mut reconstituted GREB1-KO cells continued to display reduced growth rate in vitro (Fig. 2C) or in vivo in xenograft experiments using NOD/SCID mice (Fig. 2, D and E). Reconstitution of GREB1-KO cells with GT-WT, but not GT-Mut, rescued the stability of ERα protein (Fig. 2F), suggesting that the GT domain of GREB1 is important in GREB1-mediated cell growth via ERα stability.
Fig. 2. GREB1 GT activity is important in stabilizing ERα.
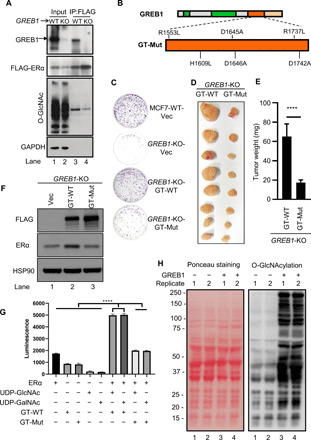
(A) FLAG pulldown was analyzed by Western blot in MCF7-WT or GREB1-KO cells expressing FLAG-ERα. (B) Catalytically dead mutant GREB1 (GT-Mut) was generated by mutating indicated residues in GREB1’s GT domain. (C) The growth of GREB1-KO cells transduced with Vec, GT-WT, or GT-Mut was assessed by colony formation assay with crystal violet staining. Photo credit: Sultan Abda Neja, Institute of Cell and Molecular Biology, A*STAR. (D and E) GREB1-KO cells transduced with GT-WT or GT-Mut were subcutaneously injected to the flanks of the same NOD/SCID mice, each cell line on one side. The resulting tumors in these mice were harvested (D) and weighted (E). n = 8; ****P < 0.0001 by unpaired t test. Photo credit: Anandhkumar Raju, Institute of Molecular and Cell Biology, A*STAR. (F) GREB1-KO cells transduced with Vec, GT-WT, or GT-Mut. (G) ERα, GT-WT, and GT-Mut were individually purified from 293T. In vitro assay using a UDP-Glo Glycosyltransferase Assay kit was performed as indicated. n = 3. (H) S. cerevisiae yeast was transfected with either Vec or FLAG-conjugated GREB1 construct.
To assess GREB1’s GT activity, the luminescence UDP-Glo assay was used to measure the hydrolysis of donor substrate sugars catalyzed by GT-WT and GT-Mut purified from human embryonic kidney (HEK) 293T. We used uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) or UDP N-acetylgalactosamine (UDP-GalNAc) as donor substrate sugars and ERα as the acceptor. GT-WT protein catalyzed the hydrolysis of UDP-GlcNAc and UDP-GalNAc when ERα was present in the reaction, shown by the increase in luminescence compared to controls and conditions with GT-Mut (Fig. 2G), suggesting that GREB1 GT domain can catalyze the glycosylation of ERα in vitro. However, the absolute value of the luminescence was low, and the assay did not show specificity for a sugar donor that is typically observed for other GTs. Availability of full-length purified GREB1 together with its associated cofactor(s) and optimal conditions in the future would be necessary to design a reliable in vitro assay wherein GREB1-mediated glycosylation of its physiological substrates can be robustly measured. In the interim, we used budding yeast, which was shown to be a reliable system to test for O-GlcNAcylation (36), as it lacks the only other known eukaryotic cytoplasmic GT, OGT. Although there was some background labeling, ectopic expression of codon-optimized FLAG-GREB1 in yeast, similar to OGT ectopic expression in yeast (36), increased O-GlcNAcylation of proteins from total lysates (Fig. 2H). There was no difference in O-GalNAcylation as probed by GalNAc lectin VLL under the same conditions (fig. S3B). Furthermore, while GREB1 is a strong interactor of ERα, no interaction of OGT with GREB1 or ERα was seen (fig. S3C), which was in line with the results using the state-of-the-art rapid immunoprecipitation mass spectrometry of endogenous protein (RIME) method designed to identify ERα interactors (25). Together, these assays suggest that GREB1 has a GT activity, and it directly transfers O-GlcNAc to ERα and possibly other yet to be identified cellular substrates.
GREB1 glycosylates ERα at T553 and S554
To address whether GREB1 increases the overall O-GlcNAcylation of ERα or through a site-specific modification, as observed with several other protein modifications (37–39), we performed mass spectrometry (MS). FLAG-tagged ERα was ectopically expressed in HEK293T cells alongside with either an empty vector (Vec), GT-WT, or GT-Mut (Fig. 3A). Equal amounts of FLAG-ERα were immunoprecipitated from the described conditions (Fig. 3B) and analyzed by liquid chromatography–MS/MS (LC-MS/MS). To improve the mapping of labile O-linked sugar-modified ERα peptides, we used β-elimination followed by Michael addition of dithiothreitol (40) after alkaline phosphatase treatment for removal of phosphorylation. Most of the threonine and serine residues on ERα were either unmodified or exhibited low levels of O-linked glycosylation (fig. S4A), except for T553 and S554, which were highly glycosylated only when ERα was coexpressed with GT-WT (Fig. 3, C to F). The modified residues are located C-terminal to the globular core of ERα ligand-binding domain, which interacts with GREB1 (fig. S2, G and H). Glycosylation at S10, an O-GlcNAc site previously shown in murine ERα (41), remained very low across all conditions (Fig. 3G). Considering the fact that OGT is not found to associate with ERα in vivo (fig. S3C) (25), these results strongly suggest that GREB1 specifically glycosylates ERα on T553 and S554 rather than merely being a general chaperone that recruits OGT to ERα.
Fig. 3. GREB1 glycosylates ERα on T553 and S554.
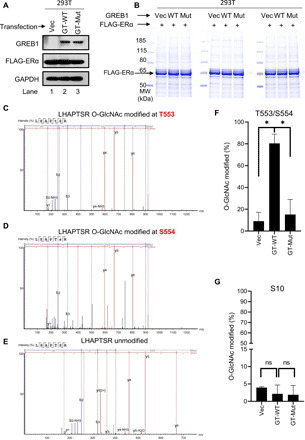
(A) HEK293T cells were transfected with FLAG-ERα together with either Vec, GT-WT, or GT-Mut. (B) FLAG pulldown was performed in HEK293T cells described in (A). Total protein eluates were analyzed by Coomassie blue staining. (C to E) O-GlcNAcylation of gel-extracted ERα from the corresponding band from (B) was labeled using BEMAD [β-elimination and Michael addition with dithiothreitol (DTT)] methodology and analyzed by LC-MS/MS. Spectra of ERα tryptic peptide LHAPTSR with modifications at T553 (C) and S554 (D) and with no modification (E) are illustrated. (F) Quantification of the fraction of peptides that were glycosylated at T553 and S554 in the indicated samples. n = 2; *P < 0.05 by one-way ANOVA with Holm-Sidak’s multiple comparisons test. (G) Quantification of the fraction of peptides that were glycosylated at S10 in the indicated samples. n = 2; by one-way ANOVA with Holm-Sidak’s multiple comparisons test.
GREB1-mediated O-GlcNAcylation of T553 and S554 regulates ERα stability
To assess the significance of glycosylation on T553 and S554 residues, we created FLAG-tagged constructs for WT ERα (ERα-WT) and mutant ERα (ERα-2M), wherein T553 and S554 residues are substituted by alanine (Fig. 4A). With an equal amount of protein (Fig. 4B, left), comparable binding to DNA oligonucleotides encoding estrogen response elements (EREs) (Fig. 4B, right) by FLAG-tagged ERα-WT and ERα-2M suggests that these mutations do not cause structural changes affecting basic ERα function. After MCF7-WT or GREB1-KO cells were transduced with either empty vector, ERα-WT, or ERα-2M, ERα stability was monitored using CHX chase assay. In MCF7-WT cells, ERα-2M protein levels dropped more markedly than did ERα-WT protein levels (Fig. 4C and fig. S4B). However, in the absence of GREB1, ERα-WT levels reduced at a similar rate as ERα-2M (Fig. 4D and fig. S4C), indicating that T553 and S554 residues are important for stabilization of ERα mediated by GREB1.
Fig. 4. Glycosylation on T553 and S554 is important for GREB1 stabilization of ERα.
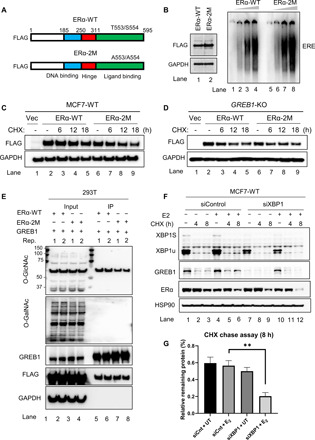
(A) Schematic of ESR1 WT construct (ERα-WT) or mutated construct (ERα-2M). (B) ERα-WT and ERα-2M expressed by HEK293T (left) were analyzed for DNA binding capability by electrophoretic mobility shift assay (EMSA) (right panel). (C and D) MCF7-WT cells (C) and GREB1-KO cells (D) were transduced with Vec, ERα-WT, or ERα-2M construct and subsequently treated with CHX. (E) HEK293T cells were transfected with GREB1 alongside either ERα-WT or ERα-2M constructs, and FLAG pulldown was performed. Two replicates (Rep.) were analyzed together on Western blot. (F and G) MCF7 cells were transfected with either control siRNA (siControl) or XBP1 siRNA (siXBP1). Cells were treated with or without E2 for 24 hours and subsequently subjected to a CHX chase assay. Levels of indicated proteins were analyzed by Western blot (F), and the amount of the remaining ERα after CHX treatment was analyzed by densitometry quantification (G). n = 3, **P < 0.01 by t test.
However, even with the interaction study by co-IP assay (fig. S3C), it is still impossible to completely rule out the possibility of transient interactions between ERα and OGT. Therefore, to disentangle the possible overlapping activities of GREB1 and OGT, OGT was knocked down in MCF7 cells using small interfering RNA (siRNA) technology (fig. S4, D and E). However, in this condition, O-GlcNAcylation of ERα remained unaltered (fig. S4F), and the difference in stability between ERα-WT and ERα-2M remained unchanged (fig. S4G). These observations in cells with appreciable depletion of OGT are opposite to that in GREB1 null cells, wherein O-GlcNAcylation of ERα decreases (Fig. 2A) and the difference in degradation rates between ERα-WT and ERα-2M is blunt (Fig. 4D and fig. S4C). Furthermore, the degradation rates of ERα-WT and ERα-2M in cells with OGT match that in cells with appreciable depletion of OGT (fig. S4H), indicating that OGT does not have a role in either ERα-WT or ERα-2M stability levels.
When the two ERα constructs were coexpressed with GREB1, there was no GalNAcylation on ERα under any of the conditions (Fig. 4E), while O-GlcNAcylation was more abundant on ERα-WT (Fig. 4E, lanes 5 and 6, replicates) than on ERα-2M (Fig. 4E, lanes 7 and 8, replicates), confirming that GREB1 modifies ERα with O-GlcNAc. Thus, knocking down XBP1, which positively regulates the HBP that produces the UDP-GlcNAc in cells (fig. S4, I and J) (42), should compromise GREB1’s ability to stabilize ERα. In conjunction with estrogen and CHX treatments, XBP1 was knocked down by siRNA in MCF7-WT cells to assess ERα protein stability (Fig. 4F). Where the GREB1 levels were low (without E2), knocking down XPB1 slightly affected ERα stability (Fig. 4F, lanes 1 to 3 versus lanes 7 to 9, quantified in Fig. 4G). In contrast, when GREB1 was induced by E2, XBP1 knockdown decreased ERα protein stability more sharply after CHX treatment (Fig. 4F, lanes 10 to 12 versus lanes 4 to 6, quantified in Fig. 4G). Moreover, in cells with XBP1 knocked down (fig. S4K), O-GlcNAcylation of ERα decreased (fig. S4L, lanes 7 and 8 in comparison with lanes 5 and 6; fig. S4M), further reiterating a role for XBP1 in regulating the glycosylation of ERα. Together, these results show that GREB1-mediated O-GlcNAcylation of ERα residues T553 and S554 occurs in co-operation with the HBP mediated by XBP1, and together, these two ERα target genes are responsible for ERα stabilization in cells upon ligand stimulation.
GREB1-mediated glycosylation of T553 and S554 regulates ERα stability by limiting binding of the ubiquitin ligase ZNF598
To test whether GREB1-mediated glycosylation at T553 and S554 would lead to a reduced association between ERα and its cognate ubiquitin ligases, FLAG-ERα-WT ectopically expressed in MCF7-WT and GREB1-KO cells was immunoprecipitated for analysis of protein interactions by MS. By designating the FLAG beads as the limiting factor, we obtained equal amounts of FLAG-ERα from MCF7-WT and GREB1-KO cells (Fig. 5A) to compare peptide association stoichiometry per molecule of ERα. As expected, GREB1 co-immunoprecipitated with ERα from MCF7 but not from GREB1-KO cells (Fig. 5B). More ubiquitin was found associated with ERα in the absence of GREB1 (Fig. 5C). Most ERα interaction partners (336) were the same in both cell lines; only 33 and 81 were specific to MCF7-WT and GREB1-KO cells, respectively (Fig. 5D). Of these, of note was the interaction between ERα and ZNF598, specifically in GREB1-KO cells (Fig. 5E). IP of FLAG-tagged ERα-WT and ERα-2M ectopically expressed in MCF7-WT or GREB1-KO cells showed that ZNF598 associates more with ERα-2M (Fig. 5, F and G, lane 6) than with ERα-WT (Fig. 5, F and G, lane 5) in MCF7-WT. In addition, in the absence of GREB1, ZNF598 interacted more with both ERα-WT and ERα-2M (Fig. 5, F and G; comparing lanes 7 and 8 with lane 5). These data correlate well with the ERα stability (Fig. 1, G to K) and indicate that both GREB1 and T553/S554 are necessary to avoid the accumulation of the ubiquitin ligase ZNF598 on ERα. Notably, we did not detect OGT as an ERα interactor in our MS result (table S1), which corroborates our biochemistry assay analyzed by Western blot (fig. S3C) and ERα interactors identified by the RIME method (25).
Fig. 5. GREB1 stabilizes ERα by limiting binding of the ubiquitin ligase ZNF598.
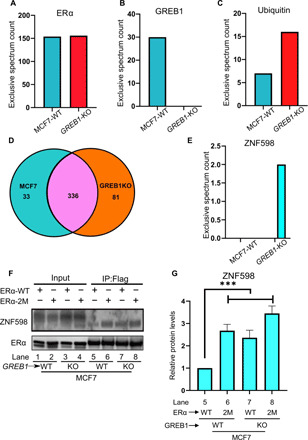
(A to E) MCF7-WT or GREB1-KO cells were transduced with FLAG-ERα, and FLAG pulldown was performed. Eluates were analyzed by MS. The number of peptide reads for ERα (A) and GREB1 (B) was examined as controls. The number of peptide reads for ubiquitin (C), unique or common interactors (D), and ZNF598 (E) from MCF7-WT and GREB1-KO was illustrated. (F) FLAG pulldown was performed to MCF7-WT or GREB1-KO cells transduced with ERα-WT or ERα-2M. (G) Densitometry analysis of ZNF598 bands from the Western blot in (F) for corresponding lanes. n = 3; ***P < 0.005 by one-way ANOVA.
Greb1 KO phenocopies partial loss of ERα signaling
Greb1-KO mice were generated according to the strategy shown in Fig. 6A. Using allele-specific primers, we genotyped these mice using polymerase chain reaction (PCR) and confirmed Greb1-KO (Fig. 6B); this was corroborated by the reduced expression of Greb1 mRNA in heterozygous mice (Het) and the lack of Greb1 mRNA expression in the ovaries of Greb1-KO mice (Fig. 6C). Greb1-KO mice were smaller than their WT counterparts (Fig. 6, D to F), consistent with the reported smaller stature for ERα-KO mice (43). Across multiple mating pairings, Greb1-KO females have lower rates of conception (Fig. 6G) and fewer pups per litter (Fig. 6H) compared to WT or Het females. These results reiterate that Greb1-KO mice phenocopy a partial loss of ERα function. This fertility defect was caused by weaker ERα signaling in Greb1-KO mice, as the expression of the progesterone receptor (Pgr), an ERα target gene that functions in fertility, was much lower in Greb1-KO mice than in either Het or WT mice (Fig. 6I). The Esr1 transcript levels were comparable across WT, Het, and Greb1-KO mice (Fig. 6J), reiterating that the reduction in ERα function in Greb1-KO mice is likely due to changes in its protein stability. Together, these in vivo data demonstrate a growth and fertility phenotype in mice lacking GREB1 and postulate that these effects are partial phenocopy of ERα loss, reflective of reduction, but not a total loss of ERα protein in the absence of GREB1.
Fig. 6. Greb1 KO reduces growth and fertility in female mice.
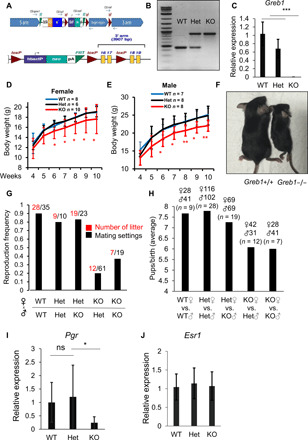
(A) Schematic presentation of the generation of Greb1-KO mice by inserting a trapping cassette “SA-βgeo-pA” upstream exons 16 and 17 of Greb1 and corresponding allele expressing GREB1-WT when the trapping cassette is removed by Flp-recombinase targeting FRT sites flanking the SA-βgeo-pA cassette. (B) Representative genotyping for WT, Het, and Greb1-KO mice by PCR. (C) Mouse ovaries were harvested from WT, Het, or Greb1-KO mice. mRNA level of Greb1 was evaluated by qRT-PCR. n = 3, ***P < 0.001 by t test. (D) Body weight (in grams) of WT or Greb1-KO female mice. *P < 0.05 and **P < 0.01 by t test. (E) Body weight (in grams) of WT or Greb1-KO male mice. (F) Representative photo image of age-matched WT and Greb1-KO female mice at 25 days. Photo credit: Chia Yi Liu, Institute of Molecular and Cell Biology, A*STAR. (G and H) A total of six rounds of mattings were performed with multiple mating cages, one male and two female mice per mating cage, for each pairing consisting of indicated genotypes. In different mating combinations of WT, Het, and Greb1-KO mice, the frequency of conceptions was calculated as the ratio between the number of litter conceived per number of mating cages (G), and the average number of pups per litter from different mating settings of WT and Greb1-KO mice was calculated (H). (I) Ovary samples from WT, Het, and Greb1-KO mice were collected and analyzed for mouse progesterone receptor (Pgr) expression at mRNA levels. (J) Ovary samples from WT, Het, and Greb1-KO mice were collected and analyzed for mouse estrogen receptor α (Esr1) expression at mRNA levels. n = 3, *P < 0.05 by t test.
GREB1 expression is a robust biomarker for the effectiveness of endocrine treatment in ERα+ve breast cancer
A major limitation of tamoxifen therapy, a common treatment for ERα+ve breast cancers, is the development of resistance. In a cohort of breast cancer patients from a curated public dataset (44), the level of GREB1 mRNA expression strongly predicted overall patient survival, with prognosis power similar to the expression level of ESR1 (fig. S5, A and B), which is currently considered one of the most reliable prognostic biomarkers in breast cancer. On the basis of our results, the loss of GREB1 in ERα+ve breast cancer would hypothetically lead to down-regulation of ERα, the primary targets of endocrine therapy, resulting in refractory to tamoxifen treatment and eventual disease recurrence by the activation of other proliferative pathways. In line with this idea, in patients treated with endocrine therapy (fig. S5, C and D) or tamoxifen alone (Fig. 7, A and B), low GREB1 expression correlated with lower overall survival. ESR1 mRNA expression loses its prognostic capability in patients treated with endocrine therapy and tamoxifen (Fig. 7A and fig. S5C). This is consistent with the in vitro biochemistry results above, showing that GREB1 regulates ERα at the protein stability level rather than its transcription level (Fig. 7C). Hence, GREB1 expression levels are more indicative of ERα signaling than ESR1 mRNA transcript level. These results suggest that GREB1 has great potential as a biologically relevant biomarker to predict the chance of cancer recurrence in these patients.
Fig. 7. GREB1 role in tamoxifen response in breast cancer.
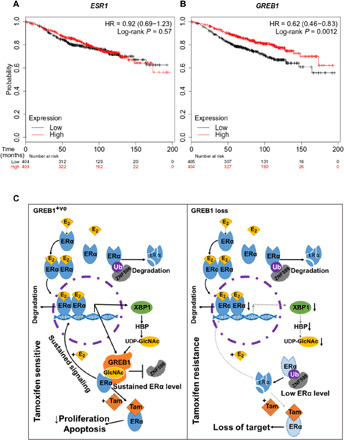
(A and B) Kaplan-Meier plots in a cohort of tamoxifen-treated breast cancer patients, plotting tumor’s ESR1 (A) and GREB1 (B) mRNA expression as a function of overall patient survival. (C) Graphical abstracts of GREB1 function in ER+ve breast cancer and drug response. When cells express GREB1, ERα is stabilized by glycosylation and imposes ERα signaling transcription signature, which is vulnerable to tamoxifen, an ERα agonist. For cells that transcriptionally repress GREB1, ERα protein and its transcriptional profile are lost. Because of this loss of target, these cells are resistance to tamoxifen.
DISCUSSION
In this study, we uncover and characterize the GT activity of GREB1 and the role of GREB1-mediated glycosylation of ERα using assays including O-GlcNAcylation–specific antibodies, luminescence assays, and MS. GREB1 is induced by ERα and it, in turn, catalyzes the O-GlcNAcylation of ERα specifically at T553 and S554, which is important in maintaining ERα stability by occluding its association with the ubiquitin ligase ZNF598. KO of GREB1, mutation of its catalytic active site, mutation of the glycosylation sites on ERα, and reducing levels of UDP-GlcNAc due to XBP1 loss result in reduced stability of ERα. GREB1 not only is an ERα adapter in regulating transcription but also plays a key role in feedforward autoregulation of ERα stability and hence function. Here, we provide compelling evidence for the role of the ERα-GREB1-XBP1 axis in linking together hormonal signaling, nutrient sensing, and cell proliferation. The loss of GREB1 stabilization of ERα in vivo phenocopies ERα-KO mice, displaying a profound reduction in growth and fertility in female Greb1-KO mice. These findings also offer an insight into breast cancer recurrence in ERα+ve tumors and identify GREB1 as a potential biomarker for tamoxifen treatment in breast cancer.
Previous studies looking at the interaction between GREB1 and ERα suggested that GREB1 works as a transcriptional coactivator of ERα, binding alongside ERα to a subset of estrogen-responsive elements and enhancing the binding of the chromatin regulators p300 and CBP at these GREB1-bound sites (25). Because our assays show that GREB1 has some nuclear presence in addition to its cytoplasmic localization, it is plausible that GREB1 is a multifunctional protein capable of protein glycosylation in the cytoplasm, in addition to having a distinct nuclear function. This is consistent with the presence of the catalytically inactive circular permuted SF-2 helicase domain in GREB1 (Fig. 1A), which could potentially bind nucleic acids.
GATA3 and ERα are master transcription factors in mammary tissue development and are implicated in breast cancer (45). Expressions of GATA3 and ERα are strongly correlated, and these transcription factors have been shown to directly cross–up-regulate each other’s transcription levels (46). However, there are conflicting views on GATA3 as a prognosis marker in breast cancer (47, 48). ERα protein loss overwhelmingly correlates to poor response to tamoxifen in recurrent tumors (49). GATA3 protein labeling is maintained in all ERα loss metastasis (50). Our study now explains how GATA3 and ERα levels can be decoupled by the loss of GREB1, leading to the loss of ERα protein levels, independent from its mRNA levels.
In ERα+ve breast cancers, GREB1 plays an important role in maintaining ERα protein levels and transcriptional signaling, resulting in susceptibility to tamoxifen and hormonal therapy. However, there are breast cancers and other cancers in which GREB1 is expressed without ERα (25, 28). Which transcriptional factors activate GREB1 and how GREB1 interacts and functions in these pathways are not well understood. So far, liver receptor homolog 1 (LRH-1) (51), androgen receptor (52), and progesterone receptor (22) have been shown to activate transcription of GREB1. When considering only ERα−ve cases in the same patient cohort analyzed in Fig. 7, GREB1 correlates with lower survival (fig. S5E), suggesting that context-specific cooperation of transcription factors that co-regulate GREB1 may drive the ultimate response and this code must be evaluated in future studies to better predict response to hormone therapy or responsiveness.
This study also biochemically demonstrates a temporally regulated and context-specific O-GlcNAcylation paradigm mediated by GREB1. In contrast, the only known cytoplasmic O-GlcNAc GT OGT is constitutively expressed. Previously, the mechanism by which OGT targets and selects an enormous number of its cytoplasmic substrates remained very poorly understood. While O-GlcNAcylation is ubiquitous and has far-reaching regulation in cellular signaling, the specific effect on each substrate remains unknown. Previous studies have reported that the increase in cellular O-GlcNAcylation levels by knocking down O-GlcNAcase (OGA) or using dominant-negative mutant OGA (53) or using OGA inhibitor (54) all led to a decrease in ERα protein and transcript levels. Because our MS also detects other O-GlcNAc sites on ERα, which are not regulated by GREB1, it is likely that general sites glycosylated by OGT are distinct and have different functions from those regulated by GREB1. However, OGT does not regulate O-GlcNAcylation at T553/S554 and GREB1 sites and consequentially does not regulate ERα stability controlled by these sites (fig. S4, D to H). Identification of the precise sites on ERα, which are glycosylated and which positively regulate its stability, is a crucial knowledge generated by our study.
While our results suggest that GREB1 is a GT, it is essential to discuss the limitations of our claims. In this study, we specifically study the role of GREB1 in the context of signaling mediated by ERα, a major hormone receptor and transcription factor. Using molecular, biochemical, and genetic evidence, we suggest that GREB1 regulates ERα function by controlling its O-GlcNAcylation and hence stability. However, our experiments were performed in cells that constitutively expressed OGT, which raises the question of whether GREB1 is a previously unknown O-GlcNAc GT or an auxiliary protein for OGT to catalyze O-GlcNAcylation of ERα. Although evidence from our studies and those from others (25) suggest that GREB1 and OGT do not interact, it would have been useful to use OGT-deficient cell lines to make a watertight case. However, in mammals, OGT is essential for embryonic development (55) and cellular functions. Given that the ERα+ve breast cancer cells required for these studies must divide for our ability to make sufficient reagents for biochemical assays, it is technically challenging to create OGT-null ERα+ve cells in sufficient amounts. An added layer of technical complication in carrying out these experiments is that the cells without OGT must also be responsive to estrogen to perform the biochemical experiments necessary for making unambiguous conclusions that OGT is not a surrogate for GREB1 in some of our assays. To fortify the confidence in our findings, we have assessed GREB1’s GT activity in yeast and performed OGT knockdown experiments. Saccharomyces cerevisiae does not contain an OGT paralog or homolog (36), and hence, no OGT enzymatic activity is seen in bakers’ yeasts. Therefore, it has been used by many scientists in the field as a model to study OGT’s activity and substrate interactions (36, 56–58). Ectopic expression of GREB1 in S. cerevisiae leads to O-GlcNAcylation of yeast cellular proteins (Fig. 2H), much like when OGT is ectopically expressed in yeast (36, 56–58). Furthermore, knockdown of OGT to the degree that has been documented to have effects on O-GlcNAcylation of its other substrates in the literature (59–61) does not affect ERα’s O-GlcNAcylation in our assays (fig. S4F). Thus, in addition to Western blots, in vitro experiment, and MS, these results strongly indicate that GREB1 O-GlcNAcylates ERα independent of OGT.
ERα is a critical regulatory transcription factor in hormonal response and breast cancer proliferation and thus is subjected to myriad levels of regulations. Many posttranslational modifications have been reported to modulate ERα activity. In addition to the well-known ubiquitination and proteasome-mediated degradation when ERα is liganded with estrogen (62), many ubiquitin ligases or deubiquitinases have been reported to regulate ERα, such as BRCA1 (63), OTUB1 (64), USP7 (65), and ChIP (66). However, not all ubiquitinations on ERα are the same. For example, RNF31 protects ERα by mono-ubiquitination (67), and TRIM56 increases ERα stability through k63-linked ubiquitination (68). Similarly, many phosphorylation events were demonstrated to regulate ERα in a site-specific manner, as has been reviewed elsewhere (69). Here, we further unravel the posttranslational modification complexity of ERα with O-GlcNAcylation of its ligand-binding domain by GREB1. S554 was previously identified as a phosphosite with an unknown function (70). Because O-GlcNAcylation and phosphorylation extensively cross-talk either through competition of occupying the same residue or sterically hindrance of nearby residues (71), as a consequence, GREB1 integrates glycosylation, ubiquitination, and phosphorylation to regulate ERα. Hence, GREB1, unlike OGT, may be a major integrator and rate-limiting enzyme in these cross-talks via these covalent modifications in a stimulus- and context-dependent manner, as its levels and activity are not constitutive like OGT.
GREB1-like proteins are widely conserved across eukaryotes and are also found in organisms outside of the animal lineage, such as Choanoflagellatea, Ichthyosporea, Mycetozoa, Cryptomonada, Haptophyta, Heterokonta, and Naegleria-like Heterolobosea that lack ERα and related proteins. This work also opens the field of O-GlcNAcylation catalyzed by GREB1 and possibly its paralog GREB1L, indicating that there are inducible posttranslational protein O-GlcNAcylations beyond those catalyzed by OGT. The wide distribution of GREB1 clade proteins in eukaryotes suggests that these modifications might have a more general significance in cancer beyond the stabilization of ERα reported here. GREB1 is the first inducible O-GlcNAc GT in the cytoplasm to be identified in mammals, and these findings have lessons for expanding inducible modifications and temporal control of signaling in health and disease.
MATERIALS AND METHODS
Cell lines and reagents
MCF7, T47D, MDA-MB-231, PC-3, U2OS, HeLa, and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Merck) and penicillin-streptomycin (50 U/ml). OV-CAR-3 cells were cultured in DMEM supplemented with 20% FBS and penicillin-streptomycin (50 U/ml). BT474, BT549, A2780, and LNCaP cells were cultured in RPMI 1640 with l-glutamine supplemented with FBS and penicillin-streptomycin (50 U/ml). Cells were grown at 37°C with 5% CO2 in a humidified incubator.
For E2 stimulation assays, MCF7, T47D, and BT474 cells were first grown in culture medium without phenol red and supplemented with 10% charcoal-stripped FBS. E2 (10 μM) was added to the culture medium after 24 hours. Plates of cells were harvested at the indicated time. For drug treatment, the following drugs and concentrations were used: CHX (10 μg/ml, Sigma-Aldrich) and proteasome inhibitor MG132 (10 μM in dimethyl sulfoxide, Abcam). Biotinylated Vicia Villosa Lectin (VVL; Vector Laboratories Inc., catalog no. B-1235) was provided by F. Bard’s laboratory [Institute of Molecular and Cell Biology (IMCB), A*STAR, Singapore].
Generation of GREB1-KO cell lines
CRISPR-Cas9 guide sequences were designed using the Zhang laboratory guide design tool at http://crispr.mit.edu/. A pair of guide RNA spacer sequences, which target genomic locus about 100 base pairs (bp) downstream of GREB1’s start codon, was selected: gRNA1, 5′-GAAGACGACACGCTTTGAAG-3′ and gRNA2, 5′-ATCCCTGCGGTCCAACAACC-3′. Spacer sequences were cloned into pX330 according to the Zhang laboratory general cloning protocol (72). Plasmids were transiently transfected into MCF7, T47D, or BT474 cells using Lipofectamine LTX reagent (Thermo Fisher Scientific) and single cell–sorted into 96-well plates.
Colony formation and cell growth assays
Colony formation assays were performed by seeding 400 cells into poly-l-lysine–coated six-well plates. Culture medium was refreshed every 3 and 4 days. After 14 days, cells were fixed and stained in 75% ethanol with 0.2% crystal violet for 10 min. Cell growth assays were performed by seeding 3000 cells into 96-well plates. Cell growth was measured at 1-day intervals using Cell Counting Kit-8 (Dojindo), measuring absorbance at 450 nm with a reference wavelength of 620 nm using a Spark M10 plate reader (Tecan).
Plasmids
All plasmids used in this study were generated in V.T.’s laboratory except for the virus packaging plasmids or information indicated with provider. MCF7 RNA was used for complementary DNA (cDNA)–based cloning for GREB1 and ESR1. For GREB1 cloning, GREB1 was first cloned into Escherichia coli expression vectors pSY5 and pSY7 using ligation-independent cloning method. pSY5 and pSY7 plasmids were provided by B. Robinson’s laboratory (IMCB, A*STAR, Singapore). Using pLenti vector backbone, we cloned no-tag-GREB1, 3xFLAG-GREB1-WT, 1xFLAG-GREB1-WT, no-tag-GT-Mut, 3xFLAG-GT-Mut, no-tag-ESR1, 3xFLAG-ESR1, 1xFLAG-ESR1, 3xFLAG-ESR1-2M, 1xFLAG-GREB1-D1, and 1xFLAG-GREB1-D2 mammalian expression constructs. Using pBOBI vector backbone, we cloned ERα-FL, 1xFLAG-ERα-FL, 1xFLAG-ERα-Del-1, 1xFLAG-ERα-Del-2, 1xFLAG-ERα-Del-3, 1xFLAG-ERα-Del-4, and 1xFLAG-ERα-Del-5 mammalian expression constructs. For lentiviral packaging and transduction, GREB1 gene is cloned from MCF7 cDNA into pLenti-CMV-zeo-GFP (green fluorescent protein) vector. The GFP in the vector was removed for cloning purposes using Bam HI and Sal I restriction enzymes. A silent mutation in the sequence of both GT-WT and GT-Mut was made to remove a Bam HI site for cloning purposes.
Site-directed mutagenesis
Site-directed mutagenesis of plasmids containing GREB1 and ESR1 was performed using the QuikChange II Site-Directed Mutagenesis Kit (Agilent). Primers were designed using a QuikChange Primer Design program (Agilent). GREB1 was mutated at R1553 to leucine (R1553L), H1609 to leucine (H1609D), D1645 to alanine (D1645A), D1646 to alanine (D1646A), R1737 to leucine (R1737L), and D1742 to alanine (D1742A) in GT-Mut GREB. ESR1 was mutated at T553 and S554 to alanine. Mutagenesis was confirmed by Sanger sequencing.
Western blot analysis
Total cellular extracts were prepared by lysis of cells in Totex lysis buffer [20 mM Hepes (pH 7.9), 150 mM NaCl, 20% glycerol, 1% NP-40, 1 mM MgCl2, 0.5 mM EDTA, and protease inhibitor cocktail (Roche)] for 30 min at 4°C followed by sonication using Bioruptor UCD-200 (Diagenode) for 5 min. Soluble proteins were obtained by taking the supernatant after centrifugation of the lysate at 18,000g for 15 min at 4°C. Protein quantification was performed by a Bradford assay (Bio-Rad), and equal amounts of lysate were loaded onto 4 to 12% bis-tris polyacrylamide gels (Thermo Fisher Scientific) for electrophoretic separation. Separated proteins were transferred onto a methanol-activated polyvinylidene difluoride membrane (Bio-Rad). Blocking was performed in 5% skim milk dissolved in PBS-T (phosphate-buffered saline, 0.1% Tween 20) for 30 min at room temperature. Primary antibody staining was performed overnight at 4°C. Secondary antibody staining was performed for 1 hour at room temperature. Washes with PBS-T were performed after antibody staining. Proteins were detected using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). The following antibodies were used: anti-GREB1 (Millipore, catalog no. MABS62, RRID:AB_10806474), anti-ERα (Santa Cruz Biotechnology, catalog no. sc-8002, RRID:AB_627558), anti-HSP90α/β (Santa Cruz Biotechnology, catalog no. sc-13119, RRID:AB_675659), anti–XBP-1 (Santa Cruz Biotechnology, catalog no. sc-8015, RRID:AB_628449), anti-GAPDH (Santa Cruz Biotechnology, catalog no. sc-32233, RRID:AB_627679), anti-FLAG (Sigma-Aldrich, catalog no. F1804, RRID:AB_262044), anti-IκBα (Santa Cruz Biotechnology, catalog no. sc-371, RRID:AB_2235952), anti–histone H3 (Cell Signaling Technology, catalog no. 9715, RRID:AB_331563), anti–O-GlcNAc (Cell Signaling Technology, catalog no. 9875, RRID:AB_10950973), anti–O-GlcNAc (Cell Signaling Technology, catalog no. 82332, RRID:AB_2799991), anti–O-GlcNAc (Abcam, catalog no. ab2739, RRID:AB_303264), ZNF598 antibody (GeneTex, catalog no. GTX119245, RRID:AB_10619017), anti-ubiquitin (Santa Cruz Biotechnology, catalog no. sc-8017 AC, RRID:AB_2762364), anti-Cdc2 p34 (Santa Cruz Biotechnology, catalog no. sc-53, RRID:AB_2074908), β-actin (Santa Cruz Biotechnology, catalog no. sc-47778 HRP, RRID:AB_2714189), rabbit anti-mouse immunoglobulin G (IgG)–horseradish peroxidase (HRP) (Santa Cruz Biotechnology, catalog no. sc-358914, RRID:AB_10915700), goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, catalog no. sc-2004, RRID:AB_631746), and streptavidin-HRP (Cell Signaling Technology, catalog no. 3999, RRID:AB_10830897).
Immunoprecipitation
Total cellular extracts were prepared by lysing cells in IP lysis buffer [70 mM NaCl, 50 mM tris-HCl (pH 8.0), 5% glycerol, 1% Triton X-100, 0.5% NP-40, 1 mM EDTA, protease inhibitor cocktail (Roche)] for 1 hour on ice followed by sonication using Bioruptor UCD-200 (Diagenode) for 5 min. Soluble proteins were obtained by taking the supernatant after centrifugation of the lysate at 18,000g for 15 min at 4°C. Lysate and antibody were incubated overnight at 4°C. Antibody-antigen complex was captured by the addition of PureProteome Protein A magnetic beads (Merck) for 2 hours at 4°C. After washes, elution was performed by boiling the beads in 2× Laemmli buffer for 6 min. We performed FLAG pulldown with anti-FLAG M2 magnetic beads (Sigma-Aldrich, catalog no. M8823, RRID:AB_2637089) for 8 hours at 4°C. FLAG-tagged proteins were eluted by incubation with an excess amount of FLAG peptide (Merck) at 4°C for 30 min. The following antibodies were used: anti-ERα (Santa Cruz Biotechnology, catalog no. sc-8002, RRID:AB_627558) and anti-FLAG (Sigma-Aldrich, catalog no. F1804, RRID:AB_262044).
Cytoplasmic and nucleic protein fractionation Western blot analysis
The cytoplasmic membrane was lysed in hypotonic buffer [10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.025% NP-40, protease inhibitor cocktail (Roche)] for 3 min on ice. The cytoplasmic fraction contained in the supernatant was separated by centrifugation at 2000g for 3 min at 4°C. The nuclear membrane was lysed in nuclear lysis buffer [170 mM NaCl, 10 mM tris-HCl (pH 8.0), 0.5% NP-40, protease inhibitor cocktail (Roche)] for 15 min at 4°C. The nuclear fraction contained in the supernatant was separated by centrifugation at 18,000g for 15 min at 4°C.
Electrophoretic mobility shift assay
The ERE sequence for DNA binding was labeled, and electrophoretic mobility shift assay (EMSA) was performed as described previously (73). Briefly, for nuclear protein lysis, cytoplasmic proteins were first isolated using buffer A [10 mM Hepes (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonyl fluoride (PMSF)] containing protease inhibitors and spun down at 3000g for 5 min at 4°C. After washing pellet with cold PBS two times, nucleic proteins were further lysed from pellet in buffer C lysis [20 mM Hepes (pH 7.9), 400 mM NaCl, 1 mM EDTA, and 1 mM EGTA] with 1 mM DTT and 0.5 mM PMSF, 0.1% aprotinin, or 1× protease inhibitor mix using occasional vortex. ERE consensus sequence used is 5′-GGATCTAGGTCACTGTGACCTCGGATC-3′. EMSA images were scanned using Typhoon FLA 7000 (GE Life Science).
Protein in-gel digestion, peptide dephosphorylation, and β-elimination and Michael addition with DTT derivatization
As previously described (74), the immunoprecipitated protein was separated by SDS–polyacrylamide gel electrophoresis, and protein bands corresponding to ESR1 and GREB1 were excised and digested in-gel with Trypsin Gold, Mass Spectrometry Grade (Promega) after reduction and alkylation. Peptides were extracted and analyzed directly for LC-MS/MS or treated with calf intestine alkaline phosphatase (CIAP) (New England Biolabs) in CIAP buffer [50 mM tris-HCl (pH 9.3), 1 mM MgCl2, 0.1 mM ZnCl2, and 1 mM spermidine], with 10 pmol MassPrep phosphopeptide standard spiked as a dephosphorylation quality control. The dephosphorylated peptides were then desalted with Pierce C18 Spin Tips (Thermo Fisher Scientific) and subjected to BEMAD (β-elimination and Michael addition with DTT) treatment as reported (40) with modifications. Briefly, the peptides were dissolved in BEMAD buffer [1.5% (v/v) triethylamine, 20 mM DTT, pH adjusted to 12.5 with 0.15% NaOH] and incubated at 52°C for 3 hours with gentle shaking. The reactions were quenched by final concentration of 2% trifluoroacetic acid, and the peptides were desalted as described above and reconstituted in 2% (v/v) acetonitrile–1% (v/v) formic acid before LC–electrospray ionization–MS/MS.
LC-MS/MS and data analysis
MS analysis was performed on the LTQ-Orbitrap Elite ETD Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with nanoACQUITY UPLC system (Waters, Milford, MA) and Thermo Xcalibur 3.063 and LTQ Tune Plus 2.7.0.1112 SP2 instrument control. Five microliters of peptides was trapped in Symmetry C18 trapping column, 180 μm × 20 mm, 5 μm (Waters) for 5 min with 1% (v/v) acetonitrile in mobile phase A [0.1% (v/v) formic acid in water] at 8 μl/min. The eluted peptides were separated online by nanoACQUITY UPLC BEH130 1.7-μm C18 column, 75 μm × 200 mm, at a flow rate of 300 nl/min at 35°C over 90-min linear gradient from 5 to 40% mobile phase B [0.1% (v/v) formic acid in acetonitrile], followed by column washed with 90% buffer B and reequilibrated to 5% B, while mobile phase A was composed of 0.1% (v/v) formic acid in water. Spectra were obtained in the positive ion mode using data-dependent scanning MS/MS, in which one full MS scans at 120,000 resolution from 350 to 1600 m/z (mass/charge ratio) was followed by HCD Orbitrap MS/MS scans of the 15 most intense peptide ions with normalized collision energy of 35.0% at a resolution of 15,000.
MS/MS data were analyzed with PEAKS Studio 10 software (Bioinformatics Solutions Inc.) by database search, quantification, and MS/MS spectra annotation such as GlcNAc or BEMAD modification. The protein database was UniProtKB/Swiss-Prot Release 2017_07, which includes Homo sapiens protein entries of 20213. The peptide and fragment ion mass tolerances used were ±10 ppm (parts per million) and ±0.02 Da, respectively. Carbamidomethylation of cysteine was included as a fixed modification, oxidation of methionine, phosphorylation, and O-linked HexNAc (O-GlcNAc or O-GalNAc) (+203.08 Da) of serine, threonine, and BEMAD modification (+136 Da) at serine and threonine (+136 Da) were selected as dynamic and variable modification. One missed cleavage was allowed for searching the data.
The percentage of the modified peptide area (total area of the features associated with the corresponding BEMAD-modified peptide) over sum area of modified and unmodified peptide features) was calculated. Tandem T553 and S554 in the same tryptic peptide were combined for percentage calculation due to the challenge of accurate residual assignment for the O-GlcNAc modification, two independent biological replicates were analyzed, and the results were reproducible.
RNA isolation, reverse transcription, and real-time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) and Nucleospin RNA columns (Macherey-Nagel). cDNA was synthesized using a SuperScript VILO cDNA Synthesis kit (Thermo Fisher Scientific). Reverse transcription PCR (RT-PCR) was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on the CFX96 Real-Time System (Bio-Rad). Readings were first normalized to ACTB in each sample, followed by a second normalization step to one sample within the set of samples. Primer sequences are as follows: GREB1, 5′-AAGAGGAAGAAGAGGGAGAAGGAG-3′ (forward) and 5′-GTTGGAAATGGAGACAAGGCG-3′ (reverse); XBP1, 5′-CCTGGTTGCTGAAGAGGAGG-3′ (forward) and 5′-CCATGGGGAGATGTTCTGGAG-3′ (reverse); ESR1, 5′-CAGGTGCCCTACTACCTGGA-3′ (forward) and 5′-ACTGGCCAATCTTTCTCTGC-3′ (reverse); ACTB, 5′-GCCAACCGCGAGAAGATGA-3′ (forward) and 5′-CCATCACGATGCCAGTGGTA-3′ (reverse); Greb1, 5′-AGGGTGACATTGACATTTTGCTGGAC (forward) and 5′-ATCGCAGATTCCAGCACCCAGAATCTGGAC (reverse); Pgr, 5′-CTCCGGGACCGAACAGAGT (forward) and 5′-ACAACAACCCTTTGGTAGCAG (reverse) (PrimerBank ID:26331756a1); Esr1, 5′-CCTCCCGCCTTCTACAGGT (forward) and 5′-CACACGGCACAGTAGCGAG (reverse) (PrimerBank ID: 6678695a1); Actb, 5′-GGCTGTATTCCCCTCCATCG (forward) and 5′-CCAGTTGGTAACAATGCCATGT (reverse) (PrimerBank ID: 6671509a1) (PrimerBank, https://pga.mgh.harvard.edu/primerbank/).
Chromatin immunoprecipitation
Formaldehyde was directly added to MCF7 cells to a final concentration of 1% for 10 min at room temperature to fix cells. Then, cells were harvested and lysed in SDS buffer [100 mM NaCl, 50 mM tris-HCl (pH 8.0), 5 mM EDTA, 0.5% SDS, protease inhibitor cocktail (Roche)]. The cell lysate was sonicated using Bioruptor UCD-200 (Diagenode) to generate DNA fragments 200 to 500 bp in length. Anti-ERα (HC-20; sc-543, Santa Cruz Biotechnology) or control IgG (normal rabbit IgG, sc-2027, Santa Cruz Biotechnology) antibodies were added overnight at 4°C. Antibody-bound chromatin was pulled down using PureProteome Protein A magnetic beads (Merck) for 2 hours at 4°C. After washing and elution, cross-links were reversed and DNA was purified. Quantitative PCR (qPCR) was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on the CFX96 Real-Time System (Bio-Rad). Readings were normalized to input samples. The following primers were used: GREB1, 5′-AGCAGTGAAAAAAAGTGTGGCAACTGGG-3′ and 5′-CGACCCACAGAAATGAAAAGGCAGCAAACT-3′; XBP1, 5′-ATACTTGGCAGCCTGTGACC-3′ and 5′-GGTCCACAAAGCAGGAAAAA-3′.
RNA interference
For siRNA-mediated knockdown of human XBP1, ESR1, and OGT, MCF7-WT cells were transfected with 100 nM of either the targeting or control siRNA using Lipofectamine RNAiMAX Reagent (Invitrogen) for 48 hours. Two independent duplexes of siRNAs for ESR1 knockdown and the siRNA pools of three duplexes were used to target XBP1 and OGT (synthesized by Integrated DNA Technologies) knockdown. The sequences of siRNA targeting human ESR1 are as follows: (i) sense: 5′-CAAGCUACCUUGUCAUGUAUACAGT-3′; antisense: 5′-ACUGUAUACAUGACAAGGUAGCUUGaa-3′; (ii) sense: 5′-GAUGAUGGGCUUACUGACCAACCTG-3′; antisense: 5′-CAGGUUGGUCAGUAAGCCCAUCAUCga-3′. The sequences of siRNA targeting human OGT are as follows: (i) sense: 5′-AAGCAUUUCUUGAUAGUCUACCAGA-3′; antisense: 5′-UCUGGUAGACUAUCAAGAAAUGCUUug-3′; (ii) sense: 5′-GUGGUACUGUCAUUGAUAAUAAUAT-3′; antisense: 5′-AUAUUAUUAUCAAUGACAGUACCACaa-3′; (iii) sense: 5′-GUCUUGAGCUUAUUGCUAAAAACAG-3′; antisense: 5′-CUGUUUUUAGCAAUAAGCUCAAGACaa-3′. The sequences of siRNA targeting human XBP1 are as follows: (i) sense: 5′-GUCCAAGGUAUUGAGACAUAUUACT-3′; antisense: 5′-AGUAAUAUGUCUCAAUACCUUGGACug-3′; (ii) sense: 5′- UAGAAAAUCAGCUUUUACGAGAGAA-3′; antisense: 5′- UUCUCUCGUAAAAGCUGAUUUUCUAgc-3′; (iii) sense: 5′- CAACUUGGACCCAGUCAUGUUCUTC-3′; antisense: 5′- GAAGAACAUGACUGGGUCCAAGUUGuc-3′.
Immunofluorescence microscopy
Construct expressing GREB1 with N-terminal FLAG-tagged and C-terminal HA-tagged was transfected in 2 × 104 of HeLa cells for 24 hours. Transfected cells were washed two times with PBS, fixed using 4% paraformaldehyde for 15 min at room temperature, and then permeabilized using 0.1% Triton X-100 in PBS for 15 min on ice. After blocking with 5% bovine serum albumin (BSA) in PBS at room temperature with shaking, fixed cells were incubated with anti-FLAG (F3156, Sigma-Aldrich) and anti-HA overnight at 4°C. After primary antibody incubation, cells were washed three times with PBS followed by incubation in secondary antibody (1:2000 dilution, 5% BSA in PBS) for 2 hours at room temperature in the dark. Prolong Gold Antifade Mount with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, P36935) was added. Slides were imaged using an LSM800 (Zeiss) confocal microscope.
RNA sequencing
MCF7 cells were treated with either ethanol or E2 (10 nM) for 12 hours before total RNA was extracted. The prepared library mix was sequenced at A*STAR GIS sequencing facility, Singapore. After the qualities of sequenced results were checked, RNA-sequencing reads were aligned to the Gencode version 19 splice junctions mapped to hg19/GRCh37 build of the human genome using STAR (version 2.4a) (75). Subsequent quantification of Gencode version 19 genes and isoform expression levels was done using RSEM (version 1.2.20) (76). Differential genes and isoform expression estimation were performed using EdgeR (version 3.10.5 with limma 3.24.15) (77) according to log fold change (LogFC) at ±2, and statistical significance P value (>0.05) was used as a cutoff value to determine the significance of differential gene expression. Sequencing results were further analyzed on MS Excel by sorting the differentially expressed genes, and vane diagram, functional grouping, and pathway analysis of the selected genes were made accordingly. The R software package was also used to make a heatmap based on the read per kilobase of transcript per million mapped read value of the differentially expressed genes.
Stable transduction and transient transfection
Stable transfection was made using pLenti-GFP-Zeocin or pBOBI vector plasmid and other third-generation lentiviral packaging plasmid. Plasmids were cotransfected into HEK293T cells for virus productions. Virions are harvested and filtered from HEK293T medium supernatant. Polybrene was added to the virion solution for transduction.
All transient transfection was made using X-tremeGENE DNA Transfection Reagent (Roche, Basel, Switzerland) in Opti-MEM medium (without FBS). The transfection media were changed 8 hours after transfection.
GT activity assay
The UDP-Glo Glycosyltransferase Assay Kit (Promega) was used to determine the GT activity of GREB1. From the MS of ERα, we identified the sugar to be a hexosamine, which narrows possible GREB1 sugar donors to UDP-GalNAc and UDP-GlcNAc; thus, these two sugar donors were used in our assay. For the GT reaction, purified GREB1, ERα, and UDP-GalNAc or GlcNAc were incubated in 25 μl of GT reaction buffer (50 mM tris, 50 mM NaCl, and 10 mM MnCl2) for 1 hour. For detection, UDP-Glo solution was prepared according to the manual and added to every GT reaction at a 1:1 (v/v) ratio. After 1-hour incubation, 22 μl of each final reaction mix was transferred to a solid white bottom, 96-well plate for the reading of luminescence.
S. cerevisiae cell culture and protein purification
S. cerevisiae strain BY4741 (MATa his3del1 leu2del0 met15del0 ura3del0) was grown in yeast extract peptone dextrose (YLPD) [yeast extract (1.1%), bactopeptone (2.2%), and glucose (2%)] or yeast synthetic tryptophan dropout medium [yeast nitrogen base without amino acid (6.7 g/liter), 2% glucose, and tryptophan dropout amino acid supplement (0.87 g/liter)] overnight with shaking. YEplac112 vector (TRP1) was used for ectopic expression and selection of positive clones.
Yeast GREB1 expression and Western blot analysis
Codons of 5′-3xFLAG–tagged human GREB1 gene was optimized for expression in yeast and cloned into YEplac112+Tryptopan vector with TEF1 promoter sequence (Bio Basic Canada Inc.). YEplac112 vector and GREB1-YEplac112 were transformed in Trp− S. cerevisiae and selected by plating and picking clones from culturing plates without tryptophan (Trp−). To extract protein for Western analysis, yeast cells were grown overnight until the OD600 (optical density at 600 nm) measurement reached 1. Cells were harvested and lysed in 20 mM Hepes (pH 8.0), 9 M urea, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, and 1 mM ß-glycerophosphate with glass beads.
Mouse tumor xenograft model
All animal studies were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) guidelines approved by the Biological Resource Centre (BRC; Biomedical Sciences Institute, A∗STAR). Immunosuppressed NSG mice (NOD/SCID female mice, 6 to 8 weeks old), supplied by InVivsos Pte Ltd., Singapore, were subcutaneously implanted with slow-releasing 17β-estradiol hormone pellets (catalog no. NE-121, 0.72 mg/pellet, Innovative Research of America, Sarasota, FL, USA) in the subscapular region. After 1 week, 5 × 106 cells per mouse were subcutaneously injected onto the right and left flanks. Tumor growth was monitored over a period of 4 weeks (28 days) or until tumor volume reached at least 1000 mm3, whichever is earlier. Tumor diameter was measured using a Vernier caliper, and tumor volume was calculated as V = a × b2 × 0.52, where a is the largest and b is the smallest diameter of the tumor.
Generation of GREB1-KO mice
Greb1-null mouse was generated by using a targeting vector (HTGRS01001_A_G07) in embryonic stem (ES) cell clone (EPD0165_2_B02, parental cell line, JM8.N4) and genetic background (C57BL/6N). The vector, ES cell(s), and/or mouse strain used for this research project were generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org). Strain ID: Greb1tm1a(KOMP)Wtsi.
The following primers were used for genotyping: WT forward (F1): 5′-ATAGAGTGAGTGACAGAGGGATGG (467 bp); KO forward (NeoF1): 5′-TCGCCTTCTATCGCCTTCTTGAC (902 bp); and reverse (ttR2, common for F1 and NeoF1): 5′-CCTCTCAGACATCTTCCTCTTCA.
Supplementary Material
Acknowledgments
Culturing protocol, yeast cells, and Vec expression construct were provided by U. Surana (IMCB). Histological processing and analysis done in this study were performed with the help of Advanced Molecular Pathology Laboratory (IMCB). Funding: The V.T. laboratory is funded by the National Research Foundation, Singapore (NRF-CRP17-2017-02). V.T.H. is supported by SINGA program. L.M.I. and L.A. were supported by the Intramural Research Program of the NIH, National Library of Medicine. Author contributions: Conceptualization: V.T.; software: L.M.I. and L.A.; investigation: V.T.H., E.M.S., S.A.N., C.Y.L., A.R., K.T., J.G., and X.B.; writing (original draft): V.T.H. and V.T.; writing (review and editing): V.T.H., V.T., E.M.S., L.M.I., and L.A.; supervision: V.T. and N.S.T. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All unique/stable reagents generated in this study are available from the lead contact (vinayt@imcb.a-star.edu.sg) without restriction.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe2470/DC1
REFERENCES AND NOTES
- 1.Lee T. I., Young R. A., Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayden M. S., Ghosh S., Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Tavana O., Gu W., p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 11, 564–577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hewitt S. C., Korach K. S., Oestrogen receptor knockout mice: Roles for oestrogen receptors alpha and beta in reproductive tissues. Reproduction 125, 143–149 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Tang Z.-R., Zhang R., Lian Z.-X., Deng S.-L., Yu K., Estrogen-receptor expression and function in female reproductive disease. Cells 8, 1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tecalco-Cruz A. C., Ramírez-Jarquín J. O., Mechanisms that increase stability of estrogen receptor alpha in breast cancer. Clin. Breast Cancer 17, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Lo P.-W., Shie J.-J., Chen C.-H., Wu C.-Y., Hsu T.-L., Wong C.-H., O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 115, 7302–7307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng X., Hart G. W., Alternative O-glycosylation/O-phosphorylation of serine-16 in murine estrogen receptor β: Post-translational regulation of turnover and transactivation activity. J. Biol. Chem. 276, 10570–10575 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Jiang M.-S., Hart G. W., A subpopulation of estrogen receptors are modified by O-linked N-acetylglucosamine. J. Biol. Chem. 272, 2421–2428 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Gludovacz E., Maresch D., de Carvalho L. L., Puxbaum V., Baier L. J., Sützl L., Guédez G., Grünwald-Gruber C., Ulm B., Pils S., Ristl R., Altmann F., Jilma B., Salminen T. A., Borth N., Boehm T., Oligomannosidic glycans at Asn-110 are essential for secretion of human diamine oxidase. J. Biol. Chem. 293, 1070–1087 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Li S., Wang J., Chen S., Sun X.-L., Wu Q., N-glycosylation in the protease domain of trypsin-like serine proteases mediates calnexin-assisted protein folding. eLife 7, e35672 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hart G. W., Housley M. P., Slawson C., Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O., Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q., Chen Y., Bian C., Fujiki R., Yu X., TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deplus R., Delatte B., Schwinn M. K., Defrance M., Méndez J., Murphy N., Dawson M. A., Volkmar M., Putmans P., Calonne E., Shih A. H., Levine R. L., Bernard O., Mercher T., Solary E., Urh M., Daniels D. L., Fuks F., TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Liu J., Jin X., Zhang D., Li D., Hao F., Feng Y., Gu S., Meng F., Tian M., Zheng Y., Xin L., Zhang X., Han X., Aravind L., Wei M., O-GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 114, 13732–13737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer L. M., Zhang D., Burroughs A. M., Aravind L., Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 41, 7635–7655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh M. G., Thompson D. A., Weigel R. J., PDZK1 and GREB1 are estrogen-regulated genes expressed in hormone-responsive breast cancer. Cancer Res. 60, 6367–6375 (2000). [PubMed] [Google Scholar]
- 19.Hodgkinson K., Forrest L. A., Vuong N., Garson K., Djordjevic B., Vanderhyden B. C., GREB1 is an estrogen receptor-regulated tumour promoter that is frequently expressed in ovarian cancer. Oncogene 37, 5873–5886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee E., Wongvipat J., Choi D., Wang P., Lee Y. S., Zheng D., Watson P. A., Gopalan A., Sawyers C. L., GREB1 amplifies androgen receptor output in human prostate cancer and contributes to antiandrogen resistance. eLife 8, e41913 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fung J. N., Holdsworth-Carson S. J., Sapkota Y., Zhao Z. Z., Jones L., Girling J. E., Paiva P., Healey M., Nyholt D. R., Rogers P. A. W., Montgomery G. W., Functional evaluation of genetic variants associated with endometriosis near GREB1. Hum. Reprod. 30, 1263–1275 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Camden A. J., Szwarc M. M., Chadchan S. B., DeMayo F. J., O’Malley B. W., Lydon J. P., Kommagani R., Growth regulation by estrogen in breast cancer 1 (GREB1) is a novel progesterone-responsive gene required for human endometrial stromal decidualization. Mol. Hum. Reprod. 23, 646–653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegarty K. G., Drummond F. J., Daly M., Shanahan F., Molloy M. G., GREB1 genetic variants are associated with bone mineral density in Caucasians. J. Bone Miner. Metab. 36, 189–199 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Nyholt D. R., Low S.-K., Anderson C. A., Painter J. N., Uno S., Morris A. P., Gregor S. M., Gordon S. D., Henders A. K., Martin N. G., Attia J., Holliday E. G., Evoy M. M., Scott R. J., Kennedy S. H., Treloar S. A., Missmer S. A., Adachi S., Tanaka K., Nakamura Y., Zondervan K. T., Zembutsu H., Montgomery G. W., Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat. Genet. 44, 1355–1359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammed H., D’Santos C., Serandour A. A., Ali H. R., Brown G. D., Atkins A., Rueda O. M., Holmes K. A., Theodorou V., Robinson J. L. L., Zwart W., Saadi A., Ross-Innes C. S., Chin S.-F., Menon S., Stingl J., Palmieri C., Caldas C., Carroll J. S., Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 3, 342–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker M. E., Steroid receptors and vertebrate evolution. Mol. Cell. Endocrinol. 496, 110526 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Rae J. M., Johnson M. D., Scheys J. O., Cordero K. E., Larios J. M., Lippman M. E., GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res. Treat. 92, 141–149 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Hnatyszyn H. J., Liu M., Hilger A., Herbert L., Gomez-Fernandez C. R., Jorda M., Thomas D., Rae J. M., El-Ashry D., Lippman M. E., Correlation of GREB1 mRNA with protein expression in breast cancer: Validation of a novel GREB1 monoclonal antibody. Breast Cancer Res. Treat. 122, 371–380 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Haines C. N., Braunreiter K. M., Mo X. M., Burd C. J., GREB1 isoforms regulate proliferation independent of ERα co-regulator activities in breast cancer. Endocr. Relat. Cancer 25, 735–746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itkonen H. M., Minner S., Guldvik I. J., Sandmann M. J., Tsourlakis M. C., Berge V., Svindland A., Schlomm T., Mills I. G., O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 73, 5277–5287 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Culyba E. K., Price J. L., Hanson S. R., Dhar A., Wong C.-H., Gruebele M., Powers E. T., Kelly J. W., Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science 331, 571–575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., Qian K., Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang F., Su K., Yang X., Bowe D. B., Paterson A. J., Kudlow J. E., O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115, 715–725 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Whisenhunt T. R., Yang X., Bowe D. B., Paterson A. J., van Tine B. A., Kudlow J. E., Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology 16, 551–563 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Yang X., Ongusaha P. P., Miles P. D., Havstad J. C., Zhang F., So W. V., Kudlow J. E., Michell R. H., Olefsky J. M., Field S. J., Evans R. M., Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451, 964–969 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi H., Li F., Han B., Arai S., Gao X.-D., Yeast cells as an assay system for in vivo O-GlcNAc modification. Biochim. Biophys. Acta 1861, 1159–1167 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Shi J., Gu J.-H., Dai C.-L., Gu J., Jin X., Sun J., Iqbal K., Liu F., Gong C.-X., O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci. Rep. 5, 14500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han C., Gu Y., Shan H., Mi W., Sun J., Shi M., Zhang X., Lu X., Han F., Gong Q., Yu W., O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nat. Commun. 8, 1491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu C.-S., Lo P.-W., Yeh Y.-H., Hsu P.-H., Peng S.-H., Teng Y.-C., Kang M.-L., Wong C.-H., Juan L.-J., O-GlcNAcylation regulates EZH2 protein stability and function. Proc. Natl. Acad. Sci. U.S.A. 111, 1355–1360 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wells L., Vosseller K., Cole R. N., Cronshaw J. M., Matunis M. J., Hart G. W., Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol. Cell. Proteomics 1, 791–804 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Cheng X., Hart G. W., Glycosylation of the murine estrogen receptor-α. J. Steroid Biochem. Mol. Biol. 75, 147–158 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Wang Z. V., Deng Y., Gao N., Pedrozo Z., Li D. L., Morales C. R., Criollo A., Luo X., Tan W., Jiang N., Lehrman M. A., Rothermel B. A., Lee A.-H., Lavandero S., Mammen P. P. A., Ferdous A., Gillette T. G., Scherer P. E., Hill J. A., Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 156, 1179–1192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker V. R., Korach K. S., Estrogen receptor knockout mice as a model for endocrine research. ILAR J. 45, 455–461 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Györffy B., Lanczky A., Eklund A. C., Denkert C., Budczies J., Li Q., Szallasi Z., An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Cohen H., Ben-Hamo R., Gidoni M., Yitzhaki I., Kozol R., Zilberberg A., Efroni S., Shift in GATA3 functions, and GATA3 mutations, control progression and clinical presentation in breast cancer. Breast Cancer Res. 16, 464 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eeckhoute J., Keeton E. K., Lupien M., Krum S. A., Carroll J. S., Brown M., Positive cross-regulatory loop ties GATA-3 to estrogen receptor α expression in breast cancer. Cancer Res. 67, 6477–6483 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Mehra R., Varambally S., Ding L., Shen R., Sabel M. S., Ghosh D., Chinnaiyan A. M., Kleer C. G., Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 65, 11259–11264 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Voduc D., Cheang M., Nielsen T., GATA-3 expression in breast cancer has a strong association with estrogen receptor but lacks independent prognostic value. Cancer Epidemiol. Biomarkers Prev. 17, 365–373 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Kuukasjärvi T., Kononen J., Helin H., Holli K., Isola J., Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 14, 2584–2589 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Cimino-Mathews A., Subhawong A. P., Illei P. B., Sharma R., Halushka M. K., Vang R., Fetting J. H., Park B. H., Argani P., GATA3 expression in breast carcinoma: Utility in triple-negative, sarcomatoid, and metastatic carcinomas. Hum. Pathol. 44, 1341–1349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chand A. L., Wijayakumara D. D., Knower K. C., Herridge K. A., Howard T. L., Lazarus K. A., Clyne C. D., The orphan nuclear receptor LRH-1 and ERα activate GREB1 expression to induce breast cancer cell proliferation. PLOS ONE 7, e31593 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rae J. M., Johnson M. D., Cordero K. E., Scheys J. O., Larios J. M., Gottardis M. M., Pienta K. J., Lippman M. E., GREB1 is a novel androgen-regulated gene required for prostate cancer growth. Prostate 66, 886–894 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Bowe D. B., Sadlonova A., Toleman C. A., Novak Z., Hu Y., Huang P., Mukherjee S., Whitsett T., Frost A. R., Paterson A. J., Kudlow J. E., O-GlcNAc integrates the proteasome and transcriptome to regulate nuclear hormone receptors. Mol. Cell. Biol. 26, 8539–8550 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanwal S., Fardini Y., Pagesy P., N’Tumba-Byn T., Pierre-Eugène C., Masson E., Hampe C., Issad T., O-GlcNAcylation-inducing treatments inhibit estrogen receptor α expression and confer resistance to 4-OH-tamoxifen in human breast cancer-derived MCF-7 cells. PLOS ONE 8, e69150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Donnell N., Zachara N. E., Hart G. W., Marth J. D., Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol. 24, 1680–1690 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iyer S. P. N., Akimoto Y., Hart G. W., Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J. Biol. Chem. 278, 5399–5409 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Oh H. J., Moon H. Y., Cheon S. A., Hahn Y., Kang H. A., Functional analysis of recombinant human and Yarrowia lipolytica O-GlcNAc transferases expressed in Saccharomyces cerevisiae. J. Microbiol. 54, 667–674 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Li F., Yang G., Tachikawa H., Shao K., Yang Y., Gao X.-D., Nakanishi H., Identification of novel O-GlcNAc transferase substrates using yeast cells expressing OGT. J. Gen. Appl. Microbiol., 2020.04.002 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Peng C., Zhu Y., Zhang W., Liao Q., Chen Y., Zhao X., Guo Q., Shen P., Zhen B., Qian X., Yang D., Zhang J.-S., Xiao D., Qin W., Pei H., Regulation of the hippo-YAP pathway by glucose sensor O-GlcNAcylation. Mol. Cell 68, 591–604. e5 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Pyo K. E., Kim C. R., Lee M., Kim J.-S., Kim K. I., Baek S. H., ULK1 O-GlcNAcylation is crucial for activating VPS34 via ATG14L during autophagy initiation. Cell Rep. 25, 2878–2890.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Ali A., Kim S. H., Kim M. J., Choi M. Y., Kang S. S., Cho G. J., Kim Y. S., Choi J.-Y., Choi W. S., O-GlcNAcylation of NF-κB promotes lung metastasis of cervical cancer cells via upregulation of CXCR4 expression. Mol. Cells 40, 476–484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lonard D. M., Nawaz Z., Smith C. L., O’Malley B. W., The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol. Cell 5, 939–948 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Eakin C. M., MacCoss M. J., Finney G. L., Klevit R. E., Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 104, 5794–5799 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stanišić V., Malovannaya A., Qin J., Lonard D. M., O’Malley B. W., OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) deubiquitinates estrogen receptor (ER) α and affects ERα transcriptional activity. J. Biol. Chem. 284, 16135–16145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xia X., Liao Y., Huang C., Liu Y., He J., Shao Z., Jiang L., Dou Q. P., Liu J., Huang H., Deubiquitination and stabilization of estrogen receptor α by ubiquitin-specific protease 7 promotes breast tumorigenesis. Cancer Lett. 465, 118–128 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Tateishi Y., Kawabe Y.-i., Chiba T., Murata S., Ichikawa K., Murayama A., Tanaka K., Baba T., Kato S., Yanagisawa J., Ligand-dependent switching of ubiquitin–proteasome pathways for estrogen receptor. EMBO J. 23, 4813–4823 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu J., Zhao C., Kharman-Biz A., Zhuang T., Jonsson P., Liang N., Williams C., Lin C.-Y., Qiao Y., Zendehdel K., Strömblad S., Treuter E., Dahlman-Wright K., The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor α and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene 33, 4340–4351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xue M., Zhang K., Mu K., Xu J., Yang H., Liu Y., Wang B., Wang Z., Li Z., Kong Q., Li X., Wang H., Zhu J., Zhuang T., Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis 8, 30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murphy L. C., Seekallu S. V., Watson P. H., Clinical significance of estrogen receptor phosphorylation. Endocr. Relat. Cancer 18, R1–R14 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Atsriku C., Britton D. J., Held J. M., Schilling B., Scott G. K., Gibson B. W., Benz C. C., Baldwin M. A., Systematic mapping of posttranslational modifications in human estrogen receptor-α with emphasis on novel phosphorylation sites. Mol. Cell. Proteomics 8, 467–480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z., Gucek M., Hart G. W., Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A. 105, 13793–13798 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shin E. M., Sin Hay H., Lee M. H., Goh J. N., Tan T. Z., Sen Y. P., Lim S. W., Yousef E. M., Ong H. T., Thike A. A., Kong X., Wu Z., Mendoz E., Sun W., Salto-Tellez M., Lim C. T., Lobie P. E., Lim Y. P., Yap C. T., Zeng Q., Sethi G., Lee M. B., Tan P., Goh B. C., Miller L. D., Thiery J. P., Zhu T., Gaboury L., Tan P. H., Hui K. M., Yip G. W.-C., Miyamoto S., Kumar A. P., Tergaonkar V., DEAD-box helicase DP103 defines metastatic potential of human breast cancers. J. Clin. Invest. 124, 3807–3824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee K.-G., Kim S. S.-Y., Kui L., Voon D. C.-C., Mauduit M., Bist P., Bi X., Pereira N. A., Liu C., Sukumaran B., Rénia L., Ito Y., Lam K.-P., Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep. 10, 1055–1065 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe2470/DC1