


Abstract
Cockayne syndrome (CS) is a segmental premature aging syndrome caused primarily by defects in the CSA or CSB genes. In addition to premature aging, CS patients typically exhibit microcephaly, progressive mental and sensorial retardation and cutaneous photosensitivity. Defects in the CSB gene were initially thought to primarily impair transcription-coupled nucleotide excision repair (TC-NER), predicting a relatively consistent phenotype among CS patients. In contrast, the phenotypes of CS patients are pleiotropic and variable. The latter is consistent with recent work that implicates CSB in multiple cellular systems and pathways, including DNA base excision repair, interstrand cross-link repair, transcription, chromatin remodeling, RNAPII processing, nucleolin regulation, rDNA transcription, redox homeostasis, and mitochondrial function. The discovery of additional functions for CSB could potentially explain the many clinical phenotypes of CSB patients. This review focuses on the diverse roles played by CSB in cellular pathways that enhance genome stability, providing insight into the molecular features of this complex premature aging disease.
INTRODUCTION
Normal aging is believed to be a manifestation of molecular damage that gradually accumulates in cells and organisms over time, leading to increasing dysfunction and eventually death. A complex interplay of genetic, epigenetic and environmental factors determine the course of aging for any individual/organism. Cockayne syndrome (CS) is a rare autosomal recessive genetic disorder characterized by premature aging, and one of the most studied segmental progeroid syndromes.
CS patients are genetically heterogeneous, carrying mutations in genes encoding Cockayne syndrome B (CSB), excision repair cross-complementing protein group 6 (ERCC6) on chromosome 10q11, and Cockayne syndrome A (CSA), ERCC8 gene on chromosome 5q11. Mutations in the ERCC3 gene on chromosome 2q21 [xeroderma pigmentosum complementation group B (XPB)], ERCC4 gene on chromosome 16p13 (XPF), and ERCC5 on chromosome 13q33 (XPG) show some overlap with features of CS (1–5). Approximately 70% of CS-affected individuals have a mutation in CSB, and the majority of the remaining cases harbor CSA mutations, with relatively few from the other listed genes. A total of 108 distinct mutations are documented in CS patients. In general, CS group A patients, carrying mutations in CSA, present with less severe phenotypes than CS group B patients (6,7). CS has a prevalence of 2–3 per million globally, including the US, although the incidence is greater in some European countries (7–9). Based on the severity and the age of onset, CS is classified into 3 types: type I includes early-onset CS, where patients typically show clinical signs in the first year after birth; type II CS includes early-onset cases with more severe symptoms; and type III CS includes patients with a late-onset, mild clinical presentation (8,10,11).
PROTEIN BIOCHEMISTRY
CSA is a 44-kDa protein, consisting of 396 amino acids. CSA lacks any detectable enzymatic function, belonging to the ‘‘Trp-Asp (WD) 40 repeat’’ family of structural and regulatory proteins (12). The WD40 domain of CSA consists of a helix-loop-helix motif and seven well ordered WD40 β-propeller structures (13). CSA is part of a multi-subunit E3 ubiquitin ligase complex (CRL4), comprised of Cullin 4A (CUL4A), a regulator of cullins-1 (Rbx1/ROC1) and DNA damage binding protein 1 (DDB1) (14,15). Stalled RNA polymerases activate CSA in a CSB-dependent manner, which in turn targets several proteins for ubiquitination, including CSB and RNA polymerase II (RNAPII) (14,16–18). Mutations of CSA predominantly cause type I CS (7).
CSB is a 168-kDa protein with 1493 amino acids. The N-terminal domain is followed by an acidic stretch, a glycine-rich region, a central helicase domain, two putative nuclear localization signal sequences, and a number of serine phosphorylation sites (Figure 1). CSB belongs to the SWI2/SNF2-family of DNA-dependent ATPases and contains the highly-conserved canonical seven ATPase motifs in the helicase region, characteristic of DNA and RNA helicases (19). The ATPase activity, but not the acidic region, of CSB is important for its role in DNA repair (20). CSB does not appear to possess helicase activity, but does possess ATP-dependent chromatin remodeling activity (21–23). CSB has ssDNA strand annealing and exchange activities (24). A ubiquitin binding domain (UBD) is part of the larger winged-helix domain (WHD) located in C-terminus and is important for recruitment of CSB to double strand breaks (DSBs) (25) and TC-NER (26). CSB interacts with CSA through its CSA-interaction motif (CIM) upstream of the UBD, to recruit CSA to DNA damage-stalled RNAPII (27) (Figure 1). CSB functions as a homodimer (28), where each CSB subunit wraps around the DNA helix, altering DNA conformation, which in turn alters protein-DNA interactions in the affected DNA region (29).
Figure 1.

Schematic representation of the CSB protein. Some of the important mutations of the CSB are depicted here. AD represents acidic domain, blue color is glycine stretch, NLS is nuclear localization signal, NBF is nucleotide binding fold, CIM is CSA-interaction motif and UBD is ubiquitin binding domain.
CSB FUNCTIONS
A broad range of DNA lesions are caused by endogenous cellular molecules, such as metabolism-driven reactive oxygen species (ROS) as well as exogenous agents or compounds, including ionizing radiation (IR) and ultraviolet (UV) light. UV induces the formation of cyclobutane pyrimidine dimers (CPDs) and 6-pyrimidine-4-pyrimidone products (6,4-PP) in DNA. Sensitivity to UV light is a characteristic feature of CS. Many DNA lesions in the template strand of actively transcribed regions of the genome are preferentially repaired by TC-NER, a subpathway of NER (30,31). CS cells are deficient in TC-NER and demonstrate increased sensitivity towards UV irradiation (32). However, many clinical phenotypes of CS cannot be explained solely based on the TC-NER defect. Evidence demonstrating that CSA and CSB play roles in multiple cellular pathways, as opposed to a singular role in TC-NER, is presented below.
Although this review is primarily focused on CSB, a brief discussion of current understanding of the biological roles of CSA is needed. For example, a better understanding of CSA-CSB interaction would provide insight into the function of this complex and the underlying reason for the similarity between CSA and CSB human phenotypes. At the molecular level, CSA is a cofactor of an E3 ubiquitin ligase complex, which plays an important role in the ubiquitination (direct and indirect) and degradation of proteins involved in TC-NER. Further, CSA directly promotes UV-dependent proteasome-mediated degradation of CSB (16). Besides TC-NER, CSA is involved in transcription and ribosomal biogenesis (33–35). CSA forms a complex with XPA-binding protein 2 (XAB2) (36) and promotes interaction between CSB and stalled RNAPII (17,27). A patient with mild UV-sensitive syndrome (UVSS) was diagnosed with a novel amino acid substitution mutation (W361C) in CSA. The cellular phenotype associated with CSA W361C involves sensitivity to UV but not to oxidative damage (37). In this case, the retention of effective repair of oxidative damage could possibly explain a mild CS phenotype.
CSA is recruited to the RNAPII-CSB complex by the CIM in CSB (27). Once recruited, CSA promotes association of UVSSA and stalled RNAPII through ubiquitination and helps recruit the transcription initiation factor IIH (TFIIH) complex (17,27). A number of proteins, including XAB2, nucleolin and UVSSA, are shared interacting partners of CSA and CSB and these interactions are thought to help maintain cellular homeostasis (17,27,36,38,39). Given the emerging diverse roles of CSA and CSB, the neuropathological features of CS may be more tightly linked to defects in these roles than to their roles in TC-NER.
Transcription coupled nucleotide excision repair (TC-NER)
CS has traditionally been associated with and linked to defects in TC-NER (Figure 2) (40,41). Helix-distorting lesions in active genes can physically block or stall RNAPII progression, which initiates TC-NER to rescue stalled RNAPII. The region within the DNA footprint of stalled RNAPII, ∼35 nucleotides, can no longer be accessed by DNA repair enzymes (42). Stalled RNAPII recruits CSB to the lesion site, leading to CSB-dependent deployment of the CSA/E3 ubiquitin ligase complex (CRL4), and recruitment of other NER factors and TC-NER specific proteins such as UVSSA, ubiquitin-specific processing protease 7 (USP7), and XAB2 (36,43). TFIIH is involved in DNA opening and lesion verification (44–46). XPA coordinates with TFIIH and the pre-incision complex allowing the XPF-ERCC1 complex to incise 5′ of the lesion site to initiate DNA repair synthesis. Subsequently, XPG generates a 5′ phosphate group 3′ to the incision, which is required for ligation (44,46,47). Following the removal of the damaged oligonucleotides, DNA polymerases δ and ϵ, with the accessory proteins proliferating cell nuclear antigen (PCNA) and replication factor C (RFC), fill the gap and DNA LIG1 or LIG3α ligate the nick (48–50).
Figure 2.
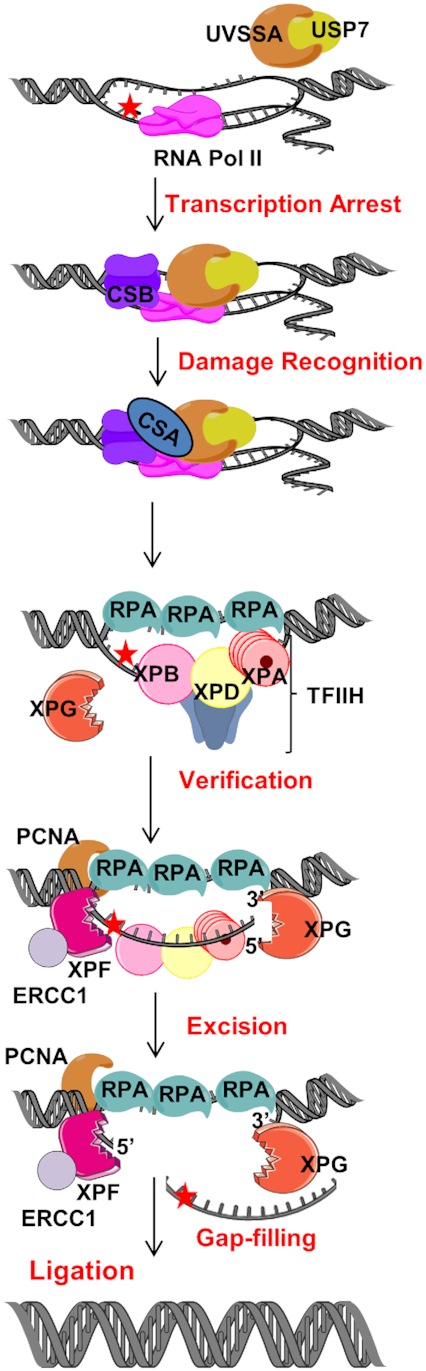
The role of CSB in TC-NER. In TC-NER, blocking lesions (red star) are identified by stalled RNA polymerase. Persistent stalled RNA polymerase leads to recruitment of the TC-repair factors, CSB and CSA. Following lesion recognition, it is verified by TFIIH and excision is performed by XPG and XPF-ERCC1. Nick is sealed by ligase after gap-filling. Figures were created using artwork of Servier Medical Art and Chemdraw.
Several studies have reported CSB and RNAPII interaction in various ways. In some observations, this interaction was via a complex of proteins, whereas others showed direct interaction of CSB with RNAPII. The interaction between CSB and RNAPII is independent of DNA, and forms under multiple conditions (51–53). A recent study of the Saccharomyces cerevisiae Pol II-Rad26 (the yeast homologue of human CSB) complex solved by cryo-electron microscopy demonstrated that Rad26 alters the RNAPII path by binding DNA upstream of it. The core ATPase domain of Rad26 promotes the forward translocation of RNAPII, which suggests a role of CSB (Rad26) in TC-NER and transcription elongation (54). The other domains of CSB also play important roles in regulating its functions. Under normal growth conditions, the N-terminal region prevents stable CSB interaction with chromatin whereas the C-terminal region stabilizes CSB chromatin interaction in the presence of lesion-stalled transcription. The central ATPase domain interacts with the N- and C-terminal regions of CSB (55,56). The N-terminal region of CSB negatively regulates chromatin association of CSB by hiding a DNA-binding region within the C-terminal region of CSB. Thus, ATP hydrolysis is dispensable for chromatin binding under normal growth conditions (56). However, following UV-induced DNA damage, the C-terminal region of CSB disengages from the ATPase domain of CSB. The C-terminal of CSB contains the WHD and UBD. Both are involved in interaction with the CSB ATPase domain (55). Possibly, ATP hydrolysis drives a conformational change in CSB thereby disrupting engagement of the WHD and UBD domains with the ATPase domain, which exposes a DNA-binding region within the C-terminal region of CSB for stable chromatin association (55,56). Thus, it helps overcoming autorepression of CSB association to chromatin through its N-terminal region in an ATP-dependent process (56). This CSB-RNAPII interaction initiates TC-NER. CSA gets recruited to CSB and to the stalled RNAPII complex by direct interaction with CSB through the CIM region of CSB (27). CSB dynamically associates with RNAPII under normal conditions and this association is stabilized upon UV irradiation (56,57). Hence, CSA association to the CSB-RNAPII complex becomes part of a CRL4CSA complex (14,58). UVSSA recruitment to the lesion stalled RNAPII is dependent on both CS proteins (27). CSA helps recruiting UVSSA to the stalled RNAPII complex by interacting with a N-terminal VHS domain of UVSSA and this interaction is then stabilized by CSB (27). Some studies suggest that recruitment of UVSSA to DNA damage-stalled RNAPII is independent of the CS proteins (43,59). However, UVSSA was found to be involved in stabilization of CSB in TC-NER and facilitates ubiquitination of stalled RNAPII at DNA damage sites (60,61). Damage induced ubiquitination of RNAPII occurs before recruitment of UVSSA and TFIIH but UVSSA-K414 ubiquitination is required for the efficient transfer of TFIIH from UVSSA to the stalled RNAPII (17,27). CSB enhances UV induced RNAPII ubiquitination but delays its turnover (62). A single ubiquitylation site in RPB1 (K1268) (a larger subunit of RNAPII) was found to regulate DNA damage-induced degradation of RNAPII in human cells. This ubiquitylation affects TC-NER, the global transcription recovery to UV irradiation and the RNAPII pool in cells (17,63). RPB1-K1268 ubiquitination does not affect the association of CS protein with stalled RNAPII, but rather CS protein facilitates RPB1-K1268 ubiquitination (17). These findings explain the role of CSB in recognition of stalled RNAPII and modulating its fate (processing) and downstream TC-NER process. This area of CSB function has been widely covered in recent reviews (64–68) and here we will focus on emerging areas of roles for CSB in other repair pathways.
Base excision repair (BER)
DNA base excision repair (BER) acts selectively on small, non-helix distorting lesions caused primarily by oxidative DNA damage. The biochemistry of BER is well studied and has been extensively reviewed (69–71). Briefly, in the first steps of BER, a DNA glycosylase scans the genome and searches for damaged and inappropriate bases (Figure 3). The DNA glycosylase removes the damaged base forming an abasic (AP) site, while leaving the DNA backbone intact. Mammalian cells express 11 different damage-specific DNA glycosylases, seven of which are also expressed in the mitochondria. After a DNA glycosylase removes the damaged base, bifunctional glycosylases cleave the sugar-phosphate backbone. When a DNA lesion is incised by a monofunctional glycosylase, the next step is AP endonuclease APE1-mediated cleavage of the DNA backbone, to generate a gapped intermediate. After AP site cleavage, repair proceeds via either a ‘short-patch’ (single-nucleotide) BER or ‘long-patch’ (multi-nucleotide) BER. Gap-filling is performed by a DNA polymerase and the nick is sealed by a DNA ligase (72–74).
Figure 3.

CSB roles in BER, transcription and chromatin remodeling. The classical BER (nuclear) is initiated by damaged base removal (blue star) which creates an AP site (blue circle). AP site is processed by AP lyase and induce incision at AP site. Following incision, the incision is processed by either short or long patch repair depending upon nature of the substrate and cellular environment and subsequently nick is sealed by ligase. CSB interacts with some of the integral player of BER like OGG1, NEIL1, APE1 and PARP1. Similar kinds of BER events also take place in response to damage in mitochondria (mitochondrial BER). In response to UV induced DNA damage, firstly RNAPII is ubiquitinated and degraded which leads to transcription repression then ATF3 recruites to inactive promoters. ATF3 is ubiquitinated in presence of CSB and leads to its proteasomal degradation. This allows recruitment of RNAPII and transcription reactivation. The transcription factor like c-Jun targets CSB to TREs in normal condition. This allows CSB to regulate transcription by ATP dependent chromatin remodeling which results in recruitment of other transcription regulators (see text for detail).
Several lines of evidence support a role for CSB in the repair of oxidative DNA damage. First, CSB-deficient cell lines and patients incise 8-oxoguanine (8-oxoG) less efficiently and express lower levels of OGG1, the glycosylase that removes this lesion from DNA, than corresponding normal control cells (75,76). Next, primary fibroblasts from 11 different CS patients accumulate significant levels of 8-oxoG and 8-oxoadenine (8-oxoA) after exposure to IR, while six normal control fibroblast cultures do not accumulate detectable levels of these DNA lesions (75–77), which are induced by oxidative stress and repaired by BER. Consistent with this, the putative helicase activity of CSB, especially functions encoded by helicase motifs V and VI, has been shown to play an important role in repair of 8-oxoG and 8-oxoA (77–80). OGG1 is present and functional in both the nucleus and mitochondria. Although mitochondrial BER is independent from nuclear BER, the proteins responsible for mtBER are encoded by nuclear genes. The mitochondrial extracts of CSB-deficient cells and CSB-knockout mouse liver cells exhibited reduced 8-oxoG incision activity, consistent with CSB’s role in transcription of OGG1 (81).
In addition to OGG1, CSB functionally interacts with several other BER proteins (38), one of which is NEIL1, a glycosylase that excises formamidopyrimidine DNA lesions including Fapy-G, Fapy-A, and 5-hydroxycytosine. Consistent with this, CSB-deficient mouse cells showed elevated levels of Fapy-G and Fapy-A in DNA (82). CSB stimulates NEIL1 incision activity and AP lyase activity in a dose-dependent manner through its N-terminal domain (82). APE1 plays an important role in processing repair intermediates produced by OGG1 and other glycosylases. (83). The N-terminal domain of APE1 interacts with CSB, which also interacts with X-ray cross-complementing protein 1 (XRCC1) (84). CSB deficiency leads to a three-fold hypersensitivity to methyl methane sulfonate (MMS), which produces primarily N7-methylguanine and N3-methyadenine DNA adducts repaired by BER. Because CSB stimulates APE1 activity in vitro, defects in CSB may decrease BER efficiency, and increase sensitivity to alkylating agents such as MMS (85).
Poly (ADP-ribose) polymerase-1 (PARP1) is a molecular sensor of DNA damage that binds single-strand DNA breaks (SSB) with high affinity. Upon binding to SSBs, PARP1 covalently modifies its target proteins, adding linear or branched chains of ADP-ribose (PARylation) using the cofactor nicotinamide adenine dinucleotide (NAD+). In addition to auto-PARylation, PARP1 substrates include core histones and CSB (86,87). In general, PARylation alters the binding and function of the modified protein (86).
The N-terminal domain of CSB interacts with unmodified and PARylated PARP-1 in the presence and absence of oxidative stress. PARylation of CSB by PARP1 inhibits its ATPase activity, making cells sensitive to oxidative stress. In addition, formation of CSB foci is inhibited by PARP inhibition, suggesting that PARP-1 and/or PARylation promotes formation of CSB foci (88). Under conditions of oxidative stress, the CSB/PARP-1 complex recognizes and binds to DNA lesions and promotes repair (89). When CSB-proficient cells are treated with PARP inhibitors, the repair of oxidative lesions is decreased, suggesting that PARP stimulates BER in a CSB-dependent manner (90). CSB plays an important role in displacing activated PARP1 from DNA damage. Therefore, CSB-deficient cells have higher levels of PAR and more PARP-1 foci (91). These observations indicate that CSB plays a critical role in regulating PARP1-dependent DNA repair.
The 70-kDa XRCC1 protein acts as a non-enzymatic scaffold that recruits other proteins to SSBs generated as BER-intermediates, and promotes processing of the nicked DNA (84). Binding of XRCC1 to SSBs is stimulated by CSB and transcription and tightly linked to BER, although XRCC1 binds SSBs generated by pathways other than BER is independent of CSB and transcription (92). Taken together, these findings establish a role for CSB in facilitating BER at several types of DNA lesions and promoting their efficient repair.
DNA Double-strand break repair
Double-strand DNA breaks (DSBs) are generated directly by IR or through processing events (referred to as two-ended DSBs) or as intermediates when the replication fork encounters a persistent SSB or other bulky lesion (referred to as one-ended DSBs). Based on the phase of the cell cycle and on the cell type, DSBs are resolved by either nonhomologous end joining (NHEJ) or homologous recombination (HR). In S and G2 phases, HR is favored over NHEJ (30). NHEJ repairs broken ends in the absence of sequence homology and is often error prone. HR repairs DSBs using sister chromatid homology in S/G2 phases and is considered to be almost error free. Ataxia-telangiectasia mutated (ATM) is an important signaling molecule in double strand break repair. It recognizes DSBs and phosphorylates hundreds of DNA damage response proteins including breast cancer 1 (BRCA1) (93). During HR repair, resection of DNA occurs at DSBs to produce 3′ single-stranded DNA (ssDNA). Initially, this ssDNA is protected by replication protein A (RPA), and later replaced by RAD51. This filamentous RAD51-ssDNA is important for homology search and strand invasion (94). The MRN complex (MRE11, DNA repair protein RAD50 and Nibrin/NBS1), along with its cofactor CtIP, is involved in DNA resection. BRCA1 binds phosphorylated CtIP and forms a complex with MRN-CtIP. This BRCA1- MRN-CtIP complex performs DNA end resection through the endonuclease and exonucleolytic activities of MRN (95–98). UV exposure stimulates interaction between BRCA1 and CSB. BRCA1 polyubiquitinates CSB, leading to CSB degradation (99). ATM phosphorylates 53BP1, promoting RIF1 recruitment to DSBs, preventing DNA end-resection, which in turn favors the error-prone NHEJ pathway. CSB is recruited to FokI nuclease-induced DSBs through its interaction with RIF1, an effector of 53BP1, via its WHD during S phase. Phosphorylation on S10 and S158 amino acids of CSB serve as molecular signals and governs chromatin remodeling activity of CSB at DSBs. CSB can remove histones from damaged chromatin, promote efficient HR-dependent DSB repair, and can restrict RIF1-mediated NHEJ (100,101). Thus, CSB regulates DSB repair pathway choice and check point activation (102).
Interstrand crosslink (ICL) repair
Interstrand crosslinks (ICLs) are DNA lesions that covalently link opposite strands of dsDNA. Many chemotherapeutic agents including cisplatin and mitomycin C, as well as natural processes like lipid peroxidation cause ICLs. ICLs are repaired by multiple repair pathways including NER and the Fanconi anemia (FA) pathway. The FA pathway involves at least 14 gene products (103). CSA- or CSB-deficient cells have increased sensitivity towards cisplatin and mitomycin C (104–106). CSB-deficient cells repair ICLs less efficiently than control cells during G1 (107). CSB is rapidly and robustly recruited to ICLs (108). CSB interacts with 5′ to 3′ exonuclease DNA crosslink repair 1A (DCLRE1A) (also known as sensitive to nitrogen mustard 1A (SNM1A)). CSB stimulates the SNM1A exonuclease activity, recruits it to ICLs and facilitates ICL repair. Additional studies will be needed, before we understand CSB’s role in ICL repair at the molecular level (109).
Chromatin structure/remodeling
CSB is a SWI/SNF – protein with DNA-dependent ATPase activity. The central ATPase domain of CSB mediates CSB homodimerization, which is essential for its chromatin remodeling activity (Figure 3) (23,28). Mutations in the CSB ATPase domain range from the E646Q mutation in motif II, which shows no ATPase activity at all to the T912/913V and Q942E mutations in motifs V and VI, respectively, which have low but measurable ATPase activity. CSB ATPase activity is also regulated by post-translational modification (110). Phosphorylation by casein kinase or Abelson murine leukemia viral oncogene homolog 1 (ABL1) and PARylation by PARP1 negatively regulate CSB ATPase (88,111). The ATPase domain also regulates UV-induced apoptosis in CSB-deficient cells. Cells harboring the E646Q ATPase mutation or a null allele of CSB die by apoptosis at similar rates (112). Both CSB-deficient and the Q942E mutant CSB cells express low levels of OGG1 protein and correspondingly low OGG1 incision activity (113).
In contrast, the phenotype of cells with deletions in the acidic domain of CSB was relatively mild (112), with nearly normal levels of gene-specific repair of CPDs and PCNA relocation after UV irradiation (114). Although CSB is unlikely to directly recruit PCNA as it interacts directly with upstream NER proteins (CSA, UVSSA and TFIIH), PCNA complex formation was reduced in CSB deficient cells in response to oxidative- and UV-induced DNA damage (115,116). In general, the acidic domain of CSB is dispensable, with the exception of its role in facilitating protein–protein interactions (114). An interesting hypothesis is that the protein-protein interacting acidic region may regulate proteins that control chromatin structure, potentially by modulating the rate of transcription and/or activation of a subset of target genes. More evidence is needed to validate this hypothesis.
CSB ATPase and its chromatin remodeling activity (23,117), are required for CSB’s contribution to transcription and DNA repair. The CSB nucleosome remodeling activity is 10-fold less active than the well characterized human ATP-dependent chromatin assembly factor (ACF) remodeling complex. However, CSBs interaction with nucleosome assembly protein 1(NAP1)-like histone chaperones (NAP1L1 or NAP1L4) enhances its nucleosome remodeling activity to a level comparable to ACF, potentially by weakening the interaction between CSB and undamaged DNA. Under conditions of cellular stress, activation and translocation of CSB increase, which in turn stimulates TC-NER (117,118). Recent studies show that CSB-deficient cells downregulate H3K9me3-specific methyltransferases SUV39H1 and SETDB1. SETDB1 expression is thought to be regulated by CSB through activating transcription factor 3 (ATF3). The downregulation of methyltransferases in CSB-deficient cells results in loss of heterochromatin and increased PARylation in highly-transcribed regions. Increased PARylation by PARP-1 depletes cellular NAD+, leading to mitochondrial dysfunction (see below) (119).
Hypoxia-inducible factor-1 (HIF-1) induces CSB to compete with p53 for histone acetyltransferase (HAT) p300 in response to hypoxia (120). Highly condensed and packed (‘closed’) chromatin sterically hinders access to and repair of genomic DNA. In contrast, CSB-mediated chromatin remodeling promotes an ‘open’ chromatin conformation, which modulates association of HAT p300 and HMGN1 to enhance unwind/relax chromatin structure and facilitate DNA repair (120–123).
Transcription regulation
It has been known for >20 years that CSB-deficient cells have a nearly 50% lower rate of transcription than some control cells (124,125). Similarly, transcriptome analysis of CSB-deficient human fibroblasts showed dysregulation of thousands of genes including neuronal genes (126). In one study of CSB-deficient cells under conditions of oxidative stress, global transcription appears to be impaired. For example, 122 genes (∼1.8% of all genes analyzed in this study) were differentially-expressed in CS cells (127). Interestingly, CSB ATPase domain was found to play an important role in regulating transcription (127). These results suggest that CSB may regulate transcription in cells with or without DNA damage.
The tumor suppressor p53 is a master regulator of the transcriptional response to genotoxic stress, which regulates cell cycle progression and the initiation of apoptosis. MDM2, an E3 ubiquitin ligase, promotes polyubiquitination and degradation of p53 in unstressed cells, while p53 is phosphorylated, stabilized and more abundant under conditions of cellular stress. Several post-translational modifications regulate p53, modulating its binding to the promoters of a variety of genes including genes involved in cell cycle and apoptosis (128). Early studies suggest a direct physical interaction between p53 and CSB’s C-terminal region (129–131). In CS cells under conditions of stress, the duration of the p53-induced transcriptional response and the frequency of apoptosis increase (112,132,133). CSB-deficient primary fibroblasts cells express a higher level of p53 than control cells, potentially due to decreased expression of MDM2, which in turn would limit the rate of p53 degradation (134). Both CSA and CSB proteins play an important role in polyubiquitination-mediated p53 degradation (134). Thus, CSB indirectly regulates the abundance of p53 in the cell.
Following in vitro reconstitution with purified proteins, CSB interacts with RNAPII and the transcription elongation complex, resulting in a 3-fold stimulation of transcription elongation (51,53,135). Furthermore, chromatin immunoprecipitated sequencing (ChIP-seq) using anti-CSB antibody revealed higher occupancy of CSB at promoter and enhancer sites which suggests that CSB plays a direct role in transcription initiation (136). This study showed that CSB was enriched at 12-O-tetradecanoylphorbol-13-acetate (TPA) response elements (TREs) that contain binding motifs for the activator protein 1 (AP-1), which belongs to a family of bZIP transcription factors, including c-Jun (Figure 3). c-Jun plays an important role in cell cycle progression and has an anti-apoptotic activity. The transcription factor (c-Jun) can recruit CSB to TRE-containing sites to regulate gene expression at transcription initiation stage (136).
Similarly, in response to oxidative stress, CSB regulates genes involved in stress response, cell cycle, transcription, and translation (127). Another study of cells with menadione-induced oxidative DNA damage showed that CSB occupancy at target promoters was 6-fold (11%) higher than in unexposed cells (2%). For example, the occupancy of transcriptional regulator CCCTC-binding factor (CTCF) went from 1% in normal cells to 11% in menadione-treated cells. CTCF is involved in long-range chromatin interactions (137,138). In vitro protein interaction studies showed that CSB and CTCF interact with each other and that this interaction is stimulated by oxidative stress (137). Hence, these studies suggest that CSB may associate with CTCF and that it regulates transcription in response to oxidative stress. Nevertheless, the effect of oxidative stress on TREs is not dependent on CSB, indicating a distinct regulatory pathway for basal TRE transcription.
CSB interacts with RNAPII resulting in sequential recruitment of CSA, UVSSA and transcription factor TFIIH to promote transcription recovery after UV (17,27). The first evidence suggesting a role for CSB in transcription was the inability of UV-irradiated CS cells to restore global RNA synthesis (139). This led to two hypotheses: that CS played a general role in transcription, or alternatively, that CSB played a role in transcription restart after UV exposure. The fact that CSA and CSB could be depleted in cells without transcriptional failure and that antibodies against CSA or CSB did not upset basal transcription favor the latter hypothesis (53,140). However, later studies demonstrated that CSB-deficient cells were unable to restore RNA synthesis after UV in both damaged and undamaged regions of the genome, suggesting a more global role for CSB in transcription in addition to TC-NER (141,142). Indeed, several proposed models linking CSB and CSA to general dysregulation of transcription in cells exposed to UV have emerged (17,63).
Upon UV irradiation, many genes are repressed. The promoters of UV-repressed genes bind and are repressed by ATF3 in response to UV stress (Figure 3) (143,144). In normal cells exposed to UV, ATF3 reaches its maximum expression ∼8 h after UV irradiation, and then gene expression is maximally repressed. By 12–24 h, ATF3 is degraded, RNAPII is recruited and transcripts begins to resume. In contrast, in CSB-deficient cells, ATF3 remains bound to promoters for extended periods of time, leading to persistent transcriptional arrest and downregulation of ∼85% of all genes (143,144). CSB and CSA promote ubiquitin-mediated proteasomal degradation of ATF3, restoring transcription after UV-induced damage (143). In fact, this effect is so pronounced in CS cells that it has been suggested that ATF3-responsive genes could be used as markers for the diagnosis of CS (145). However, ATF3 is activated in response to other genotoxins such as IR, but CS cells are not as sensitive to these genotoxins as to UV irradiation (146,147). This suggests that CSB-dependent ubiquitin-mediated proteasomal degradation of ATF3 may not be the principal mechanism for transcription recovery from exposure to UV. Therefore, other roles for CSB in UV-exposed cells should be explored in future studies. Indeed, an alternative mechanism involving the influence of CSB on RNAPII stability was recently proposed (17,63).
RNAPII processing
Some DNA lesions stall RNAPII, which initiates DNA repair to remove the damage and also triggers global transcriptional shutdown to avoid an aberrant expression of genes. However, the mechanism of transcriptional arrest is not clear. A newly identified ubiquitination site (K1268) on RBP1 may provide insight into this question, as it plays a key role in both DNA damage and the transcriptional response to UV irradiation (17,63). Mechanistically, K1268 ubiquitination induces the binding of the TFIIH core complex with the stalled RNAPII in a process also involving UVSSA mono-ubiquitination. When K1268 ubiquitination is impaired, two main consequences have been noted: (i) stalled RNAPII is not effectively processed/degraded, preventing global transcription shutdown and leading to dysregulation in transcription, (ii) TC-NER is impaired as UVSSA and TFIIH fail to interact with RNAPII. The role of CSA or CSB in K1268 ubiquitination, on the other hand, is less well understood. After the insult, CSA and CSB are both recruited to the stalled RNAPII and this process does not require RNAPII K1268 ubiquitination. However, the subsequent recruitment of UVSSA and TFIIH to the stalled RNAPII in the process of TC-NER depends on CSA and CSB. K1268 ubiquitination on RBP1 is mediated by cullin-ring type E3 ligases (CRLs) as suppression of CRLs completely abolishes UV-induced RBP1 ubiquitination (17). Despite CSA being a part of the CRL4 E3 ubiquitin ligase complex, it is not clear whether CSA is directly involved in the K1268 ubiquitination process as there are many types of CRLs. However, CSA and CSB contribute to the K1268 ubiquitination of RNAPII, potentially enhancing processing and degradation of stalled RNAPII (17,61). Indeed, mice expressing ubiquitination-resistant RBP1 K1268R in a NER-compromised background displayed CS-like phenotypes such as growth retardation and neurodegeneration (17). These results suggest a model that CSB promotes degradation of stalled RNAPII, which is also supported by other reports (61,148). However, in contrast, there are other findings showing lower levels of RNAPII in UV-treated CSB- deficient cells, resulting in sustained depletion of RNAPII and defective post-UV recovery of global transcription (63). Indeed, the introduction of non-degradable RBP1 partially restores transcription restart in CSB-deficient cells, suggesting that the lack of transcription recovery after UV irradiation is due to excessive degradation and depletion of RNAPII (63). Further studies will be needed to elucidate the mechanism by which CSA or CSB regulates K1268 ubiquitination and RNAPII stability.
rDNA transcription
In general, RNA translation depends upon rRNA and ribosome biosynthesis. rDNA genes encode rRNA, which are present in the nucleolus in multiple copies and are transcribed by RNAPI and RNAPIII. A subset of these genes are transcriptionally active at any given time to support cell growth (149). CSB is localized in the nucleolus and is part of a complex that includes RNAPI, TFIIH, XPG and TIF-1B (150,151). Both CSB and TFIIH are required for RNAPI transcription (151). Chromatin remodeling and epigenetic changes regulate rRNA transcription (152). CSB regulates both these activities. Transcription termination factor 1 (TTF-1) binds to the promoter-proximal terminator site and helps recruit chromatin remodeling factors that facilitate RNAPI transcription. Co-immunoprecipitation experiments revealed that TTF-1, CSB and RNAPI were part of the same complex. A direct physical interaction between CSB and TTF-1 has been observed, but direct interaction between CSB and RNAPI has not been demonstrated. The CSB-TTF-1 complex competes with other factors of the chromatin remodeling complex to regulate rDNA transcription. Furthermore, G9a, a methyl transferase, epigenetically regulates RNAPI transcription by mono- and di-methylating histone 3 lysine 9 (H3K9). In the same study, the direct interaction between CSB and G9a was established using coimmunoprecipitation and a role for this interaction in regulating RNAPI transcription is suggested (153,154). H3K9 modification promotes binding of the heterochromatin protein 1 gamma (HP1γ), which regulates rRNA synthesis initiation and elongation. Furthermore, CSB ATPase activity is required for formation of H3K9me by G9a (153).
Recent studies demonstrate that CSA and CSB deficiency cause defects in ribosomal DNA transcription (155). CSB’s UBD domain interacts with the most abundant nucleolar protein, nucleolin, which plays a key role in rDNA transcription and pre-rRNA synthesis. Indeed, mutations in the CSB UBD results in inhibition of the synthesis of pre-rRNA. More detailed mechanistic studies showed that CSA, as a part of the E3 ubiquitin ligase complex, promotes the ubiquitination of nucleolin and enhances the interaction of CSB with nucleolin (39). Nucleolin is present at the dense fibrillar and the granular center compartment of the nucleolus. rDNA transcription, pre-rRNA processing, and ribosome subunit assembly occur predominantly in these nucleolar compartments. CSB and CSA increase RNAPI loading to the coding region of rDNA, which is dependent on nucleolin. Thus, CSB and its interaction with nucleolin regulate rRNA transcription and ribosome biogenesis (39). G4 structures occur at a high frequency in rDNA genes and play a role in regulation of both replication and transcription. CSB promotes resolution of G4 structures via its helicase activity and also reduces transcriptional pausing at G4 structures (155).
Mitochondria
There is significant interest and a body of recent evidence supporting a role for mitochondrial dysfunction in many diseases as well as normal human aging. CS patients experience mitochondrial disease and neurological dysfunction (156–158). A recent cross-species transcriptomic analysis suggested that mitochondrial dysfunction is a common feature found in CS postmortem brain tissue, CS mice and CS nematodes (159). The mitochondrial theory of aging is based on that ROS, predominantly produced in mitochondria, damage various macromolecules including mitochondrial DNA (mtDNA), leading to mitochondrial dysfunction (160). Lack of abundant protective histones and fewer DNA repair pathways have been documented in mitochondria than in the nucleus (161–163); however, mitochondrial transcription factor TFAM binds strongly to DNA and may substitute functionally for nuclear histones.
Mitochondria are a major source of endogenous ROS and CSB deficiency might lead to accumulation of mutations in mtDNA. This idea is supported by the observation that CSB interacts with the mitochondrial transcription protein TFAM in vivo (164). Mitodb.com is a bioinformatics tool that collects and analyzes clinical and genetic information, using clustering algorithms, to determine whether a disease clusters with known mitochondrial diseases, and the probability that it is linked to mitochondrial dysfunction. The Mitodb algorithms show that CS clusters tightly with known mitochondrial diseases (165). As described below, CSB-associated mitochondrial dysfunction might reflect a direct role for CSB in mitochondria or might reflect an indirect role, where CSBs nuclear role in DNA repair propagates through nuclear to mitochondrial signaling. CSB does not have a classic mitochondrial localization signal, but it may utilize TOM20 or similar transport system. Mitochondrial dysfunction in CSB cells could potentially explain many poorly understood clinical features of CSB, including hearing loss, severe neurological deficiencies, dysfunction in skeletal muscle and heart, as well as premature aging (81,166–168).
BER plays a prominent role in protecting the nervous system against oxidative DNA damage. Increased mtDNA damage and defective nuclear or mitochondrial BER correlate with neurodegeneration and aging (169,170). BER in mitochondria is associated with the inner mitochondrial membrane (Figure 3) (171,172). Menadione-induced oxidative stress induces CSB mitochondrial localization. In the absence of CSB, BER activity at the mitochondrial membrane is decreased, resulting in an increased mutation rate (164). CSB-deficient cells and mice have increased mitochondrial content and higher overall ROS, perhaps due to dysregulation of mitochondrial BER. This could cause damaged mitochondria to accumulate, which then must be removed via mitophagy (105,132,173,174). CSB-deficient cells have increased oxygen consumption rates and FCCP-uncoupled respiration, probably due to increased ATP demand. FCCP is a protonophore which mimics physiological energy demand to investigate the role of mitochondria in cellular function and CSB-deficient cells showed increased mitochondrial respiration to meet energy demand. The NIX/PINK1-Parkin-mediated mitophagy pathway recycles dysfunctional mitochondria. Damaged, ubiquitinated mitochondria recruit the scaffolding protein, P62, which facilitates mitophagy by fusion with an autophagosome coated by the linkage protein LC-3B isoform II. In response to stress, CSB-deficient cells demonstrated decreased colocalization of LC3, P62 and ubiquitin to the mitochondria, resulting in decreased mitophagy (173,175).
CSB’s role in relation to nuclear transcription is described above. CSB also plays a role in mitochondrial transcription. Mitochondrial RNA polymerase (mtRNAP) is a single subunit enzyme similar to the bacteriophage T7 RNA polymerase (176). In vitro experiments showed that CSB stimulates mtRNAP elongation similarly to RNAPII. Furthermore, transcription from the mitochondrial heavy strand promoter was lower in CSB-deficient cells than in control cells (177). Although the exact mechanism of this regulation by CSB is still unknown, the DNA annealing and translocation activities of CSB were required for this regulation (178). The majority of mitochondrial proteins are encoded in the nucleus and translocated to mitochondria in response to environmental stimuli. Some studies demonstrate nuclear-mitochondrial crosstalk through various proteins in the mitochondria (179). This crosstalk, from the mitochondria to the nucleus or nucleus to mitochondria, is essential for mitochondrial and cellular health (180). Thus, changes in the cellular environment, abundance of NAD+, and response to stress play roles in this type of crosstalk, ultimately regulating mitochondrial homeostasis.
Nicotinamide adenine dinucleotide (NAD) and its oxidized (NAD+) and reduced (NADH) forms are major metabolites and coenzymes involved in critical pathways including glycolysis, the electron transport chain (ETC), the tricarboxylic acid (TCA) cycle, and DNA repair pathways. NAD+ abundance in various cellular compartments plays an important role regulating several metabolic pathways (Figure 4). For example, the NAD+–SIRT1–PGC1α axis regulates nucleus to mitochondria signaling. PARP1 utilizes NAD+ as its substrate to PARylate itself and other target repair proteins in response to DNA damage. There are reports that PARP1 localizes to mitochondria, though there is debate over the functional significance of this observation (181–183). The sirtuin family of proteins (SIRT1–7) also uses NAD+ as a substrate, primarily for deacetylase activities. Sirtuins localize to the mitochondrial or nuclear compartments and serve important roles in DNA damage response and genomic stability (184–188). Of the seven SIRT proteins, SIRT1 is the most studied and has a well validated role in mitochondrial biogenesis through deacetylation of the transcription factor, PGC-1α (189,190). In instances of limiting NAD+, the activity of all seven sirtuins is inhibited. As discussed earlier, CSB interacts with PARP1 and both are present in mitochondria. PARP1 PARylates its substrates in response to DNA damage, whereas CSB mechanically displaces PARP1 to limit its activity (91). However, in the absence of CSB, this regulation of PARP1 is lost, leading to persistent PARP1 activity and subsequent NAD+ deficiency. It was observed that NAD+ supplementation activates SIRT1 and rescues CS-associated phenotypes in mice and cells (91,191). AMP-activated protein kinase (AMPK), which acts as an energy sensor of cells, upon activation can phosphorylate a number of proteins including SIRT1, PGC-1α, and HIF-1α (192,193). Interestingly, lower AMPK activity was observed in CS patient-derived brain samples and restored with NAD+ augmentation in CSB deficient cells. AMPK plays an important role in mitochondrial biogenesis and maintains mitochondrial homeostasis through NAD+ (194). This influences NAD+ consumption and the interaction between these proteins helps to regulate nuclear to mitochondrial crosstalk (Figure 4). Based on our studies in other premature aging and DNA repair-deficient syndromes including XPA, ATM, and Werner syndrome, we speculate that NAD+ depletion due to dysregulated PARylation may be the most significant cause of mitochondrial dysfunction in CS cells (185,195,196). Partial mitochondrial dysfunction in CS cells could result from defective CSB in the mitochondria. However, mitochondrial dysfunction also occurs in CSA- and XPA-deficient cells (185,197), which are not reported in the mitochondria. Thus, it is likely that the major cause of mitochondrial dysfunction in CS is defective nuclear-mitochondrial crosstalk in response to DNA damage.
Figure 4.

NAD+ supplementation as a therapeutic approach to CS. CSB play important role in regulation of repair and transcription under stressed and normal conditions through chromatin remodeling and maintains normal cellular homeostasis. In CSB-deficient cells, in response to endogenous and exogenous DNA damage, cells are unable to process DNA damage-stalled RNAPII which lead to prolonged transcription arrest and dysregulation of number of proteins. PARP1 gets activated and persist at damage sites and PARylates number of proteins utilizing NAD+. Eventually this leads to decreased level of NAD+ which affects number of NAD+ dependent proteins like sirtuins. Inactivation of sirtuins causes increase in mitochondrial reactive oxygen species in response to decreased downstream activation of mitochondrial factors like AMPK, HIF1α and PGC1α resulting in mitochondrial dysfunction. Decreased repair, stalled transcription and mitochondrial dysfunction all together contribute to neurodegeneration in CSB deficient patient. Thus, impaired cellular metabolite concentration can be replenished by NAD+ precursors such as nicotinamide riboside (NR) to overcome some of premature aging phenotypes (see text).
Neurodegeneration is a major clinical feature of CS, but its etiology and mechanism are not fully understood. However, it is clear that defects in TC-NER can not fully account for neurodegenerative features in CS, because patients with UV-sensitive syndrome and defective TC-NER manifest mild UV-sensitivity and lack the neuropathological features of CS (198). Recent studies show that mitochondrial dysfunction contributes significantly to neurodegeneration. In a Caenorhabditis elegans model of CS, CSB mutant worms showed mitochondrial defects and compromised respiratory activity and neurodegeneration (199). Intriguingly, as mentioned above, features of mitochondrial diseases and CS strongly overlap. Indeed, mitochondrial dysfunction due to persistent PARylation leading to NAD+ depletion is observed in CS cells. Notably, NAD+ augmentation corrected many aspects of mitochondrial abnormalities in CS and prevented the progression of sensorineural hearing loss in CS mice. However, it is not yet know whether NAD+ supplementation alleviates other CS neurological features, such as progressive retinal degeneration or neuropathy (200). Nevertheless, these findings suggest CSB’s role in mitochondrial function as a new emerging mechanism linking CSB deficiency to neurodegeneration.
Closing remarks
CS is a multisystem progeroid disorder, exhibiting some features of normal human aging. This review provides a broad picture of the current understanding of CS focusing on emerging roles for CSB. This knowledge has expanded our understanding and role of CSA in TC-NER. One third of CS cases are associated with CSA mutations and two thirds of these CSA patients are classified as type I CS (7). No obvious genotype-phenotype correlation can be drawn between CSA and CSB mutations and clinical phenotypes due to overlapping symptoms. Even though there are no specific clinical symptoms that differentiate CSA from CSB, there are subtle differences in these phenotypes, as shown in Table 1 (201). Since both patient types have similar clinical features, it is likely that CSA and CSB regulate some of the same process(es) in the cell. Research demonstrating differences between the other phenotype of XP patients deficient in TC-NER and CS patients makes clear that other DNA repair pathways are important for CS deficiencies. More research is needed to understand the relationship between CSB and especially CSA in those pathways.
Table 1.
Comparison of clinical features in CSA and CSB
| Clinical features | |
|---|---|
| Similar prevalence (in CSA & CSB mutations) | Variable prevalence (in CSA & CSB mutations) |
| Growth failure | Low birth weight |
| Cachexia | Cataract |
| Mental retardation | Microphthalmia |
| Microcephaly | Hearing loss |
| Retinal degeneration | Dental anomalies |
| Photosensitivity | |
Initially, CSB was only considered as a TC-NER protein, but the CS- deficient TC-NER defect is not sufficient to explain many of the clinical features of CS. The developmental defect of CS can be explained by the transcriptional role of CSB whereas neurodegeneration is more likely attributed to defective oxidative DNA damage processing. CSA and CSB mouse models are considered mild CS models as they only have few features of the patients. The severe CS phenotype is precipitated only when these genes are inactivated in XPC or XPA –null background (202). Therefore, the role of CS proteins must be more than DNA damage repair. Thus, it is important to understand the principle molecular functions of CS proteins and their contribution and correlation to different clinical pathologies. CSB participates in several critical aspects of DNA repair, RNAPII processing, transcription, signaling, chromatin structure and cellular bioenergetics (e.g. mitochondrial health). Thus, patient-to-patient variation in CS severity may reflect CSBs diverse functions manifesting at different stages of development.
CSB is involved in both RNAPI and RNAPII transcription. CS cells demonstrate BER deficiency (166), which may reflect reduced transcription of BER proteins or perhaps indicate that CSB directly contributes to BER. The potential role of CSB in regulating mitochondrial transcription is intriguing and in need of further research.
More details about the regulation of CSB itself are also needed. CSB is phosphorylated in response to UV and also contains a C-terminal UBD and CIM. CSB binds to CSA through CIM in response to UV induced DNA damage due to a conformational change triggered by association of CSB with lesion stalled RNAPII (27). Protein-protein interaction of CSB with other transcription factors or repair proteins might also be involved in the activation and regulation of CSB. A study on yeast Rad26 using cryo-electron microscopy shed some light on its role in TC-NER and transcription elongation (54). There is a need for further structure-function studies to understand CSB regulation.
Mitochondrial dysfunction is another area where research on CSB’s role is emerging and advancing. A study showed the presence of CSA inside mitochondria and its involvement with mtBER upon oxidative stress (203). It is intriguing that generally the NER proteins are not present in the mitochondria, yet CS patients demonstrate mitochondrial disease phenotypes. Clearly, in the case of CSB, we can speculate that the loss of mtBER activity and resulting increase in mtDNA mutations contributes to this mitochondrial phenotype. However, how this relates directly to CS cases caused by CSA mutations is a subject of active research.
Finally, therapeutic intervention is another goal of our research. Previously, the use of antioxidants (204) and pharmacological chaperones (205) have been suggested. There are also some drugs currently under clinical trials (Prodarsan and Sirolimus). Our research indicates that NAD+ supplementation demonstrates promise for restoring mitochondrial homeostasis, promoting DNA repair and energy balance, and reversing CS-associated hearing loss (Figure 4) (157,167,175,185). There is tremendous scientific interest in the potential of NAD+ supplementation as a therapeutic approach for CS, and for understanding how NAD+ abundance regulates multiple cellular pathways, including DNA repair, transcription, chromatin structure and mitochondrial bioenergetics.
ACKNOWLEDGEMENTS
We thank Drs Deborah Croteau, Burcin Duan Şahbaz and Seoyun Choi (NIA) for their helpful comments on the article.
Contributor Information
Vinod Tiwari, Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA.
Beverly A Baptiste, Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA.
Mustafa N Okur, Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA.
Vilhelm A Bohr, Laboratory of Molecular Gerontology, National Institute on Aging, National Institutes of Health, Baltimore, MD 21224, USA.
FUNDING
Intramural Research Program of the NIH; National Institute on Aging. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cleaver J.E., Thompson L.H., Richardson A.S., States J.C.. A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum. Mutat. 1999; 14:9–22. [DOI] [PubMed] [Google Scholar]
- 2. Mallery D.L., Tanganelli B., Colella S., Steingrimsdottir H., van Gool A.J., Troelstra C., Stefanini M., Lehmann A.R.. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am. J. Hum. Genet. 1998; 62:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weeda G., van Ham R.C., Vermeulen W., Bootsma D., van der Eb A.J., Hoeijmakers J.H.. A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell. 1990; 62:777–791. [DOI] [PubMed] [Google Scholar]
- 4. Vermeulen W., Jaeken J., Jaspers N.G., Bootsma D., Hoeijmakers J.H.. Xeroderma pigmentosum complementation group G associated with Cockayne syndrome. Am. J. Hum. Genet. 1993; 53:185–192. [PMC free article] [PubMed] [Google Scholar]
- 5. Kashiyama K., Nakazawa Y., Pilz D.T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J.F., Lewin S.O., Carr L.et al.. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013; 92:807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao H., Williams C., Carter M., Hegele R.A.. CKN1 (MIM 216400): mutations in Cockayne syndrome type A and a new common polymorphism. J. Hum. Genet. 2004; 49:61–63. [DOI] [PubMed] [Google Scholar]
- 7. Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013; 134:161–170. [DOI] [PubMed] [Google Scholar]
- 8. Nance M.A., Berry S.A.. Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 1992; 42:68–84. [DOI] [PubMed] [Google Scholar]
- 9. Kleijer W.J., Laugel V., Berneburg M., Nardo T., Fawcett H., Gratchev A., Jaspers N.G., Sarasin A., Stefanini M., Lehmann A.R.. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst.). 2008; 7:744–750. [DOI] [PubMed] [Google Scholar]
- 10. Wilson B.T., Stark Z., Sutton R.E., Danda S., Ekbote A.V., Elsayed S.M., Gibson L., Goodship J.A., Jackson A.P., Keng W.T.et al.. The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. Genet. Med. 2016; 18:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kubota M., Ohta S., Ando A., Koyama A., Terashima H., Kashii H., Hoshino H., Sugita K., Hayashi M.. Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. Pediatr. Int. 2015; 57:339–347. [DOI] [PubMed] [Google Scholar]
- 12. Henning K.A., Li L., Iyer N., McDaniel L.D., Reagan M.S., Legerski R., Schultz R.A., Stefanini M., Lehmann A.R., Mayne L.V.et al.. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995; 82:555–564. [DOI] [PubMed] [Google Scholar]
- 13. Xu C., Min J.. Structure and function of WD40 domain proteins. Protein Cell. 2011; 2:202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer E.S., Scrima A., Böhm K., Matsumoto S., Lingaraju G.M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F.et al.. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011; 147:1024–1039. [DOI] [PubMed] [Google Scholar]
- 15. Vermeulen W., Fousteri M.. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Groisman R., Kuraoka I., Chevallier O., Gaye N., Magnaldo T., Tanaka K., Kisselev A.F., Harel-Bellan A., Nakatani Y.. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006; 20:1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakazawa Y., Hara Y., Oka Y., Komine O., van den Heuvel D., Guo C., Daigaku Y., Isono M., He Y., Shimada M.et al.. Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell. 2020; 180:1228–1244. [DOI] [PubMed] [Google Scholar]
- 18. Liebelt F., Schimmel J., Verlaan de Vries M., Klemann E., van Royen M.E., van der Weegen Y., Luijsterburg M.S., Mullenders L.H., Pines A., Vermeulen W.et al.. Transcription-coupled nucleotide excision repair is coordinated by ubiquitin and SUMO in response to ultraviolet irradiation. Nucleic Acids Res. 2020; 48:231–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Troelstra C., Odijk H., de Wit J., Westerveld A., Thompson L.H., Bootsma D., Hoeijmakers J.H.. Molecular cloning of the human DNA excision repair gene ERCC-6. Mol. Cell. Biol. 1990; 10:5806–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brosh R.M. Jr, Balajee A.S., Selzer R.R., Sunesen M., Proietti De Santis L., Bohr V.A. The ATPase domain but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair. Mol. Biol. Cell. 1999; 10:3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pazin M.J., Kadonaga J.T.. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions. Cell. 1997; 88:737–740. [DOI] [PubMed] [Google Scholar]
- 22. Selby C.P., Sancar A.. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 1997; 272:1885–1890. [DOI] [PubMed] [Google Scholar]
- 23. Citterio E., Van Den Boom V., Schnitzler G., Kanaar R., Bonte E., Kingston R.E., Hoeijmakers J.H., Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 2000; 20:7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muftuoglu M., Sharma S., Thorslund T., Stevnsner T., Soerensen M.M., Brosh R.M. Jr, Bohr V.A. Cockayne syndrome group B protein has novel strand annealing and exchange activities. Nucleic Acids Res. 2006; 34:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi T.S., Sato Y., Yamagata A., Goto-Ito S., Saijo M., Fukai S.. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res. 2019; 47:3784–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anindya R., Mari P.O., Kristensen U., Kool H., Giglia-Mari G., Mullenders L.H., Fousteri M., Vermeulen W., Egly J.M., Svejstrup J.Q.. A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol. Cell. 2010; 38:637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van der Weegen Y., Golan-Berman H., Mevissen T.E.T., Apelt K., González-Prieto R., Goedhart J., Heilbrun E.E., Vertegaal A.C.O., van den Heuvel D., Walter J.C.et al.. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020; 11:2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christiansen M., Thorslund T., Jochimsen B., Bohr V.A., Stevnsner T.. The Cockayne syndrome group B protein is a functional dimer. FEBS J. 2005; 272:4306–4314. [DOI] [PubMed] [Google Scholar]
- 29. Beerens N., Hoeijmakers J.H., Kanaar R., Vermeulen W., Wyman C.. The CSB protein actively wraps DNA. J. Biol. Chem. 2005; 280:4722–4729. [DOI] [PubMed] [Google Scholar]
- 30. Tiwari V., Wilson D.M. 3rd. DNA damage and associated DNA repair defects in disease and premature aging. Am. J. Hum. Genet. 2019; 105:237–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vessoni A.T., Guerra C.C.C., Kajitani G.S., Nascimento L.L.S., Garcia C.C.M.. Cockayne syndrome: the many challenges and approaches to understand a multifaceted disease. Genet Mol Biol. 2020; 43:e20190085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Venema J., Mullenders L.H., Natarajan A.T., van Zeeland A.A., Mayne L.V.. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aamann M.D., Hvitby C., Popuri V., Muftuoglu M., Lemminger L., Skeby C.K., Keijzers G., Ahn B., Bjørås M., Bohr V.A.et al.. Cockayne syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech. Ageing Dev. 2014; 135:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koch S., Garcia Gonzalez O., Assfalg R., Schelling A., Schäfer P., Scharffetter-Kochanek K., Iben S.. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle. 2014; 13:2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu S., Chen L., Ye L., Fei L., Tang W., Tian Y., Geng Q., Yi X., Xie J.. Identification of two missense mutations of ERCC6 in three Chinese sisters with Cockayne syndrome by whole exome sequencing. PLoS One. 2014; 9:e113914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakatsu Y., Asahina H., Citterio E., Rademakers S., Vermeulen W., Kamiuchi S., Yeo J.P., Khaw M.C., Saijo M., Kodo N.et al.. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J. Biol. Chem. 2000; 275:34931–34937. [DOI] [PubMed] [Google Scholar]
- 37. Nardo T., Oneda R., Spivak G., Vaz B., Mortier L., Thomas P., Orioli D., Laugel V., Stary A., Hanawalt P.C.et al.. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:6209–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aamann M.D., Muftuoglu M., Bohr V.A., Stevnsner T.. Multiple interaction partners for Cockayne syndrome proteins: implications for genome and transcriptome maintenance. Mech. Ageing Dev. 2013; 134:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Okur M.N., Lee J.H., Osmani W., Kimura R., Demarest T.G., Croteau D.L., Bohr V.A.. Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 2020; 48:2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Troelstra C., van Gool A., de Wit J., Vermeulen W., Bootsma D., Hoeijmakers J.H.. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992; 71:939–953. [DOI] [PubMed] [Google Scholar]
- 41. Balajee A.S., Bohr V.A.. Genomic heterogeneity of nucleotide excision repair. Gene. 2000; 250:15–30. [DOI] [PubMed] [Google Scholar]
- 42. Kresge N., Simoni R.D., Hill R.L.. Discovery and characterization of DNA excision repair pathways: the work of Philip Courtland Hanawalt. J. Biol. Chem. 2010; 285:e9–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schwertman P., Lagarou A., Dekkers D.H., Raams A., van der Hoek A.C., Laffeber C., Hoeijmakers J.H., Demmers J.A., Fousteri M., Vermeulen W.et al.. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012; 44:598–602. [DOI] [PubMed] [Google Scholar]
- 44. Volker M., Moné M.J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J.H., van Driel R., van Zeeland A.A., Mullenders L.H.. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell. 2001; 8:213–224. [DOI] [PubMed] [Google Scholar]
- 45. Yokoi M., Masutani C., Maekawa T., Sugasawa K., Ohkuma Y., Hanaoka F.. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem. 2000; 275:9870–9875. [DOI] [PubMed] [Google Scholar]
- 46. Riedl T., Hanaoka F., Egly J.M.. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003; 22:5293–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wakasugi M., Sancar A.. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:6669–6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu Q., Wani A.A.. Nucleotide excision repair: finely tuned molecular orchestra of early pre-incision events. Photochem. Photobiol. 2017; 93:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marteijn J.A., Lans H., Vermeulen W., Hoeijmakers J.H.. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014; 15:465–481. [DOI] [PubMed] [Google Scholar]
- 50. Shivji M.K., Podust V.N., Hübscher U., Wood R.D.. Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. Biochemistry. 1995; 34:5011–5017. [DOI] [PubMed] [Google Scholar]
- 51. Tantin D., Kansal A., Carey M.. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol. 1997; 17:6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sarker A.H., Tsutakawa S.E., Kostek S., Ng C., Shin D.S., Peris M., Campeau E., Tainer J.A., Nogales E., Cooper P.K.. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol. Cell. 2005; 20:187–198. [DOI] [PubMed] [Google Scholar]
- 53. van Gool A.J., Citterio E., Rademakers S., van Os R., Vermeulen W., Constantinou A., Egly J.M., Bootsma D., Hoeijmakers J.H.. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997; 16:5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu J., Lahiri I., Wang W., Wier A., Cianfrocco M.A., Chong J., Hare A.A., Dervan P.B., DiMaio F., Leschziner A.E.et al.. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature. 2017; 551:653–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Batenburg N.L., Qin J., Walker J.R., Zhu X.D.. Efficient UV repair requires disengagement of the CSB winged helix domain from the CSB ATPase domain. DNA Repair (Amst.). 2018; 68:58–67. [DOI] [PubMed] [Google Scholar]
- 56. Lake R.J., Geyko A., Hemashettar G., Zhao Y., Fan H.Y.. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol. Cell. 2010; 37:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van den Boom V., Citterio E., Hoogstraten D., Zotter A., Egly J.M., van Cappellen W.A., Hoeijmakers J.H., Houtsmuller A.B., Vermeulen W.. DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J. Cell Biol. 2004; 166:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A.F., Tanaka K., Nakatani Y.. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003; 113:357–367. [DOI] [PubMed] [Google Scholar]
- 59. Wienholz F., Zhou D., Turkyilmaz Y., Schwertman P., Tresini M., Pines A., van Toorn M., Bezstarosti K., Demmers J.A.A., Marteijn J.A.. FACT subunit Spt16 controls UVSSA recruitment to lesion-stalled RNA Pol II and stimulates TC-NER. Nucleic Acids Res. 2019; 47:4011–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang X., Horibata K., Saijo M., Ishigami C., Ukai A., Kanno S., Tahara H., Neilan E.G., Honma M., Nohmi T.et al.. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 2012; 44:593–597. [DOI] [PubMed] [Google Scholar]
- 61. Nakazawa Y., Sasaki K., Mitsutake N., Matsuse M., Shimada M., Nardo T., Takahashi Y., Ohyama K., Ito K., Mishima H.et al.. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 2012; 44:586–592. [DOI] [PubMed] [Google Scholar]
- 62. He J., Zhu Q., Wani G., Wani A.A.. UV-induced proteolysis of RNA polymerase II is mediated by VCP/p97 segregase and timely orchestration by Cockayne syndrome B protein. Oncotarget. 2017; 8:11004–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tufegdžić Vidaković A., Mitter R., Kelly G.P., Neumann M., Harreman M., Rodríguez-Martínez M., Herlihy A., Weems J.C., Boeing S., Encheva V.et al.. Regulation of the RNAPII pool is integral to the DNA damage response. Cell. 2020; 180:1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oh J., Xu J., Chong J., Wang D. Molecular basis of transcriptional pausing, stalling, and transcription-coupled repair initiation. Biochim. Biophys. Acta Gene Regul. Mech. 2021; 1864:194659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lans H., Hoeijmakers J.H.J., Vermeulen W., Marteijn J.A.. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019; 20:766–784. [DOI] [PubMed] [Google Scholar]
- 66. Puget N., Miller K.M., Legube G.. Non-canonical DNA/RNA structures during transcription-coupled double-strand break repair: roadblocks or bona fide repair intermediates. DNA Repair (Amst.). 2019; 81:102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gregersen L.H., Svejstrup J.Q.. The cellular response to transcription-blocking DNA damage. Trends Biochem. Sci. 2018; 43:327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Strick T.R., Portman J.R.. Transcription-coupled repair: from cells to single molecules and back again. J. Mol. Biol. 2019; 431:4093–4102. [DOI] [PubMed] [Google Scholar]
- 69. Karahalil B., Bohr V.A., Wilson D.M. 3rd. Impact of DNA polymorphisms in key DNA base excision repair proteins on cancer risk. Hum. Exp. Toxicol. 2012; 31:981–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Howard M.J., Wilson S.H.. DNA scanning by base excision repair enzymes and implications for pathway coordination. DNA Repair (Amst.). 2018; 71:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mullins E.A., Rodriguez A.A., Bradley N.P., Eichman B.F.. Emerging roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci. 2019; 44:765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krokan H.E., Bjørås M.. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dizdaroglu M., Coskun E., Jaruga P.. Repair of oxidatively induced DNA damage by DNA glycosylases: mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. 2017; 771:99–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li M., Wilson D.M. 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox. Signal. 2014; 20:678–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dianov G., Bischoff C., Sunesen M., Bohr V.A.. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999; 27:1365–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tuo J., Jaruga P., Rodriguez H., Bohr V.A., Dizdaroglu M.. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003; 17:668–674. [DOI] [PubMed] [Google Scholar]
- 77. Tuo J., Jaruga P., Rodriguez H., Dizdaroglu M., Bohr V.A.. The cockayne syndrome group B gene product is involved in cellular repair of 8-hydroxyadenine in DNA. J. Biol. Chem. 2002; 277:30832–30837. [DOI] [PubMed] [Google Scholar]
- 78. Tuo J., Müftüoglu M., Chen C., Jaruga P., Selzer R.R., Brosh R.M., Rodriguez H., Dizdaroglu M., Bohr V.A.. The Cockayne syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J. Biol. Chem. 2001; 276:45772–45779. [DOI] [PubMed] [Google Scholar]
- 79. Muftuoglu M., Selzer R., Tuo J., Brosh R.M. Jr, Bohr V.A. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene. 2002; 283:27–40. [DOI] [PubMed] [Google Scholar]
- 80. Selzer R.R., Nyaga S., Tuo J., May A., Muftuoglu M., Christiansen M., Citterio E., Brosh R.M. Jr, Bohr V.A.. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 2002; 30:782–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stevnsner T., Nyaga S., de Souza-Pinto N.C., van der Horst G.T., Gorgels T.G., Hogue B.A., Thorslund T., Bohr V.A.. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002; 21:8675–8682. [DOI] [PubMed] [Google Scholar]
- 82. Muftuoglu M., de Souza-Pinto N.C., Dogan A., Aamann M., Stevnsner T., Rybanska I., Kirkali G., Dizdaroglu M., Bohr V.A.. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 2009; 284:9270–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim Y.J., Wilson D.M. 3rd. Overview of base excision repair biochemistry. Curr Mol Pharmacol. 2012; 5:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vidal A.E., Boiteux S., Hickson I.D., Radicella J.P.. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001; 20:6530–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wong H.K., Muftuoglu M., Beck G., Imam S.Z., Bohr V.A., Wilson D.M. 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007; 35:4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alemasova E.E., Lavrik O.I.. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019; 47:3811–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ray Chaudhuri A., Nussenzweig A.. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017; 18:610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Thorslund T., von Kobbe C., Harrigan J.A., Indig F.E., Christiansen M., Stevnsner T., Bohr V.A.. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell. Biol. 2005; 25:7625–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Boetefuer E.L., Lake R.J., Dreval K., Fan H.Y.. Poly(ADP-ribose) polymerase 1 (PARP1) promotes oxidative stress-induced association of Cockayne syndrome group B protein with chromatin. J. Biol. Chem. 2018; 293:17863–17874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Flohr C., Bürkle A., Radicella J.P., Epe B.. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003; 31:5332–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Scheibye-Knudsen M., Mitchell S.J., Fang E.F., Iyama T., Ward T., Wang J., Dunn C.A., Singh N., Veith S., Hasan-Olive M.M.et al.. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014; 20:840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Menoni H., Wienholz F., Theil A.F., Janssens R.C., Lans H., Campalans A., Radicella J.P., Marteijn J.A., Vermeulen W.. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res. 2018; 46:7747–7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Clouaire T., Marnef A., Legube G.. Taming Tricky DSBs: ATM on duty. DNA Repair (Amst.). 2017; 56:84–91. [DOI] [PubMed] [Google Scholar]
- 94. Symington L.S., Gautier J.. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011; 45:247–271. [DOI] [PubMed] [Google Scholar]
- 95. Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P.. Human CtIP promotes DNA end resection. Nature. 2007; 450:509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen L., Nievera C.J., Lee A.Y., Wu X.. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008; 283:7713–7720. [DOI] [PubMed] [Google Scholar]
- 97. Aparicio T., Gautier J.. BRCA1-CtIP interaction in the repair of DNA double-strand breaks. Mol Cell Oncol. 2016; 3:e1169343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Syed A., Tainer J.A.. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018; 87:263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wei L., Lan L., Yasui A., Tanaka K., Saijo M., Matsuzawa A., Kashiwagi R., Maseki E., Hu Y., Parvin J.D.et al.. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011; 102:1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Batenburg N.L., Walker J.R., Noordermeer S.M., Moatti N., Durocher D., Zhu X.D.. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017; 8:1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Batenburg N.L., Walker J.R., Coulombe Y., Sherker A., Masson J.Y., Zhu X.D.. CSB interacts with BRCA1 in late S/G2 to promote MRN- and CtIP-mediated DNA end resection. Nucleic Acids Res. 2019; 47:10678–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Batenburg N.L., Thompson E.L., Hendrickson E.A., Zhu X.D.. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015; 34:1399–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Deans A.J., West S.C.. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011; 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Furuta T., Ueda T., Aune G., Sarasin A., Kraemer K.H., Pommier Y.. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002; 62:4899–4902. [PubMed] [Google Scholar]
- 105. McKay B.C., Becerril C., Ljungman M.. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene. 2001; 20:6805–6808. [DOI] [PubMed] [Google Scholar]
- 106. Enoiu M., Jiricny J., Schärer O.D.. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012; 40:8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Richards S., Liu S.T., Majumdar A., Liu J.L., Nairn R.S., Bernier M., Maher V., Seidman M.M.. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways. Nucleic Acids Res. 2005; 33:5382–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Iyama T., Wilson D.M. 3rd. Elements that regulate the DNA damage response of proteins defective in cockayne syndrome. J. Mol. Biol. 2016; 428:62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Iyama T., Lee S.Y., Berquist B.R., Gileadi O., Bohr V.A., Seidman M.M., McHugh P.J., Wilson D.M.. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015; 43:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Christiansen M., Stevnsner T., Modin C., Martensen P.M., Brosh R.M. Jr, Bohr V.A.. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003; 31:963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Imam S.Z., Indig F.E., Cheng W.H., Saxena S.P., Stevnsner T., Kufe D., Bohr V.A.. Cockayne syndrome protein B interacts with and is phosphorylated by c-Abl tyrosine kinase. Nucleic Acids Res. 2007; 35:4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Balajee A.S., Proietti De Santis L., Brosh R.M. Jr, Selzer R., Bohr V.A.. Role of the ATPase domain of the Cockayne syndrome group B protein in UV induced apoptosis. Oncogene. 2000; 19:477–489. [DOI] [PubMed] [Google Scholar]
- 113. Tuo J., Chen C., Zeng X., Christiansen M., Bohr V.A.. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst.). 2002; 1:913–927. [DOI] [PubMed] [Google Scholar]
- 114. Sunesen M., Selzer R.R., Brosh R.M. Jr, Balajee A.S., Stevnsner T., Bohr V.A.. Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acids Res. 2000; 28:3151–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Balajee A.S., May A., Dianova I., Bohr V.A.. Efficient PCNA complex formation is dependent upon both transcription coupled repair and genome overall repair. Mutat. Res. 1998; 409:135–146. [DOI] [PubMed] [Google Scholar]
- 116. Balajee A.S., Dianova I., Bohr V.A.. Oxidative damage-induced PCNA complex formation is efficient in xeroderma pigmentosum group A but reduced in Cockayne syndrome group B cells. Nucleic Acids Res. 1999; 27:4476–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cho I., Tsai P.F., Lake R.J., Basheer A., Fan H.Y.. ATP-dependent chromatin remodeling by Cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLoS Genet. 2013; 9:e1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee J.Y., Lake R.J., Kirk J., Bohr V.A., Fan H.Y., Hohng S.. NAP1L1 accelerates activation and decreases pausing to enhance nucleosome remodeling by CSB. Nucleic Acids Res. 2017; 45:4696–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lee J.H., Demarest T.G., Babbar M., Kim E.W., Okur M.N., De S., Croteau D.L., Bohr V.A.. Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging. Nucleic Acids Res. 2019; 47:8548–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ye X.W., Zhang X.P., Liu F.. CSB modulates the competition between HIF-1 and p53 upon hypoxia. Math Biosci Eng. 2019; 16:5247–5262. [DOI] [PubMed] [Google Scholar]
- 121. Birger Y., West K.L., Postnikov Y.V., Lim J.H., Furusawa T., Wagner J.P., Laufer C.S., Kraemer K.H., Bustin M.. Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. EMBO J. 2003; 22:1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Filippi S., Latini P., Frontini M., Palitti F., Egly J.M., Proietti-De-Santis L.. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J. 2008; 27:2545–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Newman J.C., Bailey A.D., Weiner A.M.. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Balajee A.S., May A., Dianov G.L., Friedberg E.C., Bohr V.A.. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dianov G.L., Houle J.F., Iyer N., Bohr V.A., Friedberg E.C.. Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 1997; 25:3636–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang Y., Chakravarty P., Ranes M., Kelly G., Brooks P.J., Neilan E., Stewart A., Schiavo G., Svejstrup J.Q.. Dysregulation of gene expression as a cause of Cockayne syndrome neurological disease. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:14454–14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kyng K.J., May A., Brosh R.M. Jr, Cheng W.H., Chen C., Becker K.G., Bohr V.A.. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene. 2003; 22:1135–1149. [DOI] [PubMed] [Google Scholar]
- 128. Rodier F., Campisi J., Bhaumik D.. Two faces of p53: aging and tumor suppression. Nucleic Acids Res. 2007; 35:7475–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang X.W., Yeh H., Schaeffer L., Roy R., Moncollin V., Egly J.M., Wang Z., Freidberg E.C., Evans M.K., Taffe B.G.et al.. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat. Genet. 1995; 10:188–195. [DOI] [PubMed] [Google Scholar]
- 130. Frontini M., Proietti-De-Santis L.. Cockayne syndrome B protein (CSB): linking p53, HIF-1 and p300 to robustness, lifespan, cancer and cell fate decisions. Cell Cycle. 2009; 8:693–696. [DOI] [PubMed] [Google Scholar]
- 131. Frontini M., Proietti-De-Santis L.. Interaction between the Cockayne syndrome B and p53 proteins: implications for aging. Aging (Albany NY). 2012; 4:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Laposa R.R., Huang E.J., Cleaver J.E.. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Proietti De Santis L., Garcia C.L., Balajee A.S., Latini P., Pichierri P., Nikaido O., Stefanini M., Palitti F.. Transcription coupled repair efficiency determines the cell cycle progression and apoptosis after UV exposure in hamster cells. DNA Repair (Amst.). 2002; 1:209–223. [DOI] [PubMed] [Google Scholar]
- 134. Latini P., Frontini M., Caputo M., Gregan J., Cipak L., Filippi S., Kumar V., Vélez-Cruz R., Stefanini M., Proietti-De-Santis L.. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle. 2011; 10:3719–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Selby C.P., Sancar A.. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11205–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lake R.J., Boetefuer E.L., Tsai P.F., Jeong J., Choi I., Won K.J., Fan H.Y.. The sequence-specific transcription factor c-Jun targets Cockayne syndrome protein B to regulate transcription and chromatin structure. PLoS Genet. 2014; 10:e1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lake R.J., Boetefuer E.L., Won K.J., Fan H.Y.. The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res. 2016; 44:2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lee B.K., Iyer V.R.. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J. Biol. Chem. 2012; 287:30906–30913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mayne L.V., Lehmann A.R.. Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res. 1982; 42:1473–1478. [PubMed] [Google Scholar]
- 140. van Gool A.J., van der Horst G.T., Citterio E., Hoeijmakers J.H.. Cockayne syndrome: defective repair of transcription. EMBO J. 1997; 16:4155–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Proietti-De-Santis L., Drané P., Egly J.M.. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006; 25:1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rockx D.A., Mason R., van Hoffen A., Barton M.C., Citterio E., Bregman D.B., van Zeeland A.A., Vrieling H., Mullenders L.H.. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:10503–10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Epanchintsev A., Costanzo F., Rauschendorf M.A., Caputo M., Ye T., Donnio L.M., Proietti-de-Santis L., Coin F., Laugel V., Egly J.M.. Cockayne's Syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol. Cell. 2017; 68:1054–1066. [DOI] [PubMed] [Google Scholar]
- 144. Kristensen U., Epanchintsev A., Rauschendorf M.A., Laugel V., Stevnsner T., Bohr V.A., Coin F., Egly J.M.. Regulatory interplay of Cockayne syndrome B ATPase and stress-response gene ATF3 following genotoxic stress. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E2261–E2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Epanchintsev A., Rauschendorf M.A., Costanzo F., Calmels N., Obringer C., Sarasin A., Coin F., Laugel V., Egly J.M.. Defective transcription of ATF3 responsive genes, a marker for Cockayne Syndrome. Sci. Rep. 2020; 10:1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wei L., Nakajima S., Böhm S., Bernstein K.A., Shen Z., Tsang M., Levine A.S., Lan L.. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3495–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kool J., Hamdi M., Cornelissen-Steijger P., van der Eb A.J., Terleth C., van Dam H.. Induction of ATF3 by ionizing radiation is mediated via a signaling pathway that includes ATM, Nibrin1, stress-induced MAPkinases and ATF-2. Oncogene. 2003; 22:4235–4242. [DOI] [PubMed] [Google Scholar]
- 148. Anindya R., Aygün O., Svejstrup J.Q.. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell. 2007; 28:386–397. [DOI] [PubMed] [Google Scholar]
- 149. Grummt I., Ladurner A.G.. A metabolic throttle regulates the epigenetic state of rDNA. Cell. 2008; 133:577–580. [DOI] [PubMed] [Google Scholar]
- 150. Iyama T., Okur M.N., Golato T., McNeill D.R., Lu H., Hamilton R., Raja A., Bohr V.A., Wilson D.M. 3rd. Regulation of the Intranuclear Distribution of the Cockayne Syndrome Proteins. Sci. Rep. 2018; 8:17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bradsher J., Auriol J., Proietti de Santis L., Iben S., Vonesch J.L., Grummt I., Egly J.M.. CSB is a component of RNA pol I transcription. Mol. Cell. 2002; 10:819–829. [DOI] [PubMed] [Google Scholar]
- 152. McStay B., Grummt I.. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 2008; 24:131–157. [DOI] [PubMed] [Google Scholar]
- 153. Yuan X., Feng W., Imhof A., Grummt I., Zhou Y.. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol. Cell. 2007; 27:585–595. [DOI] [PubMed] [Google Scholar]
- 154. Strohner R., Nemeth A., Jansa P., Hofmann-Rohrer U., Santoro R., Längst G., Grummt I.. NoRC–a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001; 20:4892–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Scheibye-Knudsen M., Tseng A., Borch Jensen M., Scheibye-Alsing K., Fang E.F., Iyama T., Bharti S.K., Marosi K., Froetscher L., Kassahun H.et al.. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12502–12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Licht C.L., Stevnsner T., Bohr V.A.. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 2003; 73:1217–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Babbar M., Basu S., Yang B., Croteau D.L., Bohr V.A.. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020; 186:111207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Scheibye-Knudsen M., Croteau D.L., Bohr V.A.. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013; 134:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Okur M.N., Fang E.F., Fivenson E.M., Tiwari V., Croteau D.L., Bohr V.A.. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD+ signaling. Aging Cell. 2020; 19:e13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Stevnsner T., Thorslund T., de Souza-Pinto N.C., Bohr V.A.. Mitochondrial repair of 8-oxoguanine and changes with aging. Exp. Gerontol. 2002; 37:1189–1196. [DOI] [PubMed] [Google Scholar]
- 161. Kauppila T.E.S., Kauppila J.H.K., Larsson N.G.. Mammalian mitochondria and aging: an update. Cell Metab. 2017; 25:57–71. [DOI] [PubMed] [Google Scholar]
- 162. Dianov G.L., Souza-Pinto N., Nyaga S.G., Thybo T., Stevnsner T., Bohr V.A.. Base excision repair in nuclear and mitochondrial DNA. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:285–297. [DOI] [PubMed] [Google Scholar]
- 163. López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The hallmarks of aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Aamann M.D., Sorensen M.M., Hvitby C., Berquist B.R., Muftuoglu M., Tian J., de Souza-Pinto N.C., Scheibye-Knudsen M., Wilson D.M. 3rd, Stevnsner T.et al.. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010; 24:2334–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Scheibye-Knudsen M., Scheibye-Alsing K., Canugovi C., Croteau D.L., Bohr V.A.. A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging (Albany NY). 2013; 5:192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Stevnsner T., Muftuoglu M., Aamann M.D., Bohr V.A.. The role of Cockayne syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008; 129:441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Okur M.N., Mao B., Kimura R., Haraczy S., Fitzgerald T., Edwards-Hollingsworth K., Tian J., Osmani W., Croteau D.L., Kelley M.W.et al.. Short-term NAD(+) supplementation prevents hearing loss in mouse models of Cockayne syndrome. NPJ Aging Mech Dis. 2020; 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Scheibye-Knudsen M., Fang E.F., Croteau D.L., Bohr V.A.. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy. 2014; 10:1468–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. de Souza-Pinto N.C., Wilson D.M. 3rd, Stevnsner T.V., Bohr V.A.. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair (Amst.). 2008; 7:1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Maynard S., Schurman S.H., Harboe C., de Souza-Pinto N.C., Bohr V.A.. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009; 30:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Thorslund T., Sunesen M., Bohr V.A., Stevnsner T.. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair (Amst.). 2002; 1:261–273. [DOI] [PubMed] [Google Scholar]
- 172. Stuart J.A., Mayard S., Hashiguchi K., Souza-Pinto N.C., Bohr V.A.. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 2005; 33:3722–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Scheibye-Knudsen M., Ramamoorthy M., Sykora P., Maynard S., Lin P.C., Minor R.K., Wilson D.M., Cooper M., Spencer R., de Cabo R.et al.. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012; 209:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Pascucci B., Lemma T., Iorio E., Giovannini S., Vaz B., Iavarone I., Calcagnile A., Narciso L., Degan P., Podo F.et al.. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell. 2012; 11:520–529. [DOI] [PubMed] [Google Scholar]
- 175. Fakouri N.B., Hou Y., Demarest T.G., Christiansen L.S., Okur M.N., Mohanty J.G., Croteau D.L., Bohr V.A.. Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 2019; 286:1058–1073. [DOI] [PubMed] [Google Scholar]
- 176. Arnold J.J., Smidansky E.D., Moustafa I.M., Cameron C.E.. Human mitochondrial RNA polymerase: structure-function, mechanism and inhibition. Biochim. Biophys. Acta. 2012; 1819:948–960. [DOI] [PubMed] [Google Scholar]
- 177. Berquist B.R., Canugovi C., Sykora P., Wilson D.M. 3rd, Bohr V.A.. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012; 40:8392–8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Berquist B.R., Wilson D.M. 3rd. Nucleic acid binding activity of human Cockayne syndrome B protein and identification of Ca(2+) as a novel metal cofactor. J. Mol. Biol. 2009; 391:820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Fang E.F., Scheibye-Knudsen M., Chua K.F., Mattson M.P., Croteau D.L., Bohr V.A.. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016; 17:308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Saki M., Prakash A.. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017; 107:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hocsak E., Szabo V., Kalman N., Antus C., Cseh A., Sumegi K., Eros K., Hegedus Z., Gallyas F. Jr, Sumegi B.et al.. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic. Biol. Med. 2017; 108:770–784. [DOI] [PubMed] [Google Scholar]
- 182. Rossi M.N., Carbone M., Mostocotto C., Mancone C., Tripodi M., Maione R., Amati P.. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009; 284:31616–31624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Szczesny B., Brunyanszki A., Olah G., Mitra S., Szabo C.. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res. 2014; 42:13161–13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Joo H.Y., Yun M., Jeong J., Park E.R., Shin H.J., Woo S.R., Jung J.K., Kim Y.M., Park J.J., Kim J.et al.. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem. Biophys. Res. Commun. 2015; 462:294–300. [DOI] [PubMed] [Google Scholar]
- 185. Fang E.F., Scheibye-Knudsen M., Brace L.E., Kassahun H., SenGupta T., Nilsen H., Mitchell J.R., Croteau D.L., Bohr V.A.. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014; 157:882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Serrano L., Martínez-Redondo P., Marazuela-Duque A., Vazquez B.N., Dooley S.J., Voigt P., Beck D.B., Kane-Goldsmith N., Tong Q., Rabanal R.M.et al.. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013; 27:639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Finley L.W., Haigis M.C.. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol. Med. 2012; 18:516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Mostoslavsky R., Chua K.F., Lombard D.B., Pang W.W., Fischer M.R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M.M.et al.. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006; 124:315–329. [DOI] [PubMed] [Google Scholar]
- 189. Cohen H.Y., Miller C., Bitterman K.J., Wall N.R., Hekking B., Kessler B., Howitz K.T., Gorospe M., de Cabo R., Sinclair D.A.. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004; 305:390–392. [DOI] [PubMed] [Google Scholar]
- 190. Cantó C., Auwerx J.. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009; 20:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Luna A., Aladjem M.I., Kohn K.W.. SIRT1/PARP1 crosstalk: connecting DNA damage and metabolism. Genome Integr. 2013; 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jäger S., Handschin C., St-Pierre J., Spiegelman B.M.. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ruderman N.B., Xu X.J., Nelson L., Cacicedo J.M., Saha A.K., Lan F., Ido Y.. AMPK and SIRT1: a long-standing partnership. Am. J. Physiol. Endocrinol. Metab. 2010; 298:E751–E760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Okur M.N., Fang E.F., Fivenson E.M., Tiwari V., Croteau D.L., Bohr V.A.. Cockayne syndrome proteins CSA and CSB maintain mitochondrial homeostasis through NAD(+) signaling. Aging Cell. 2020; 19:e13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Fang E.F., Kassahun H., Croteau D.L., Scheibye-Knudsen M., Marosi K., Lu H., Shamanna R.A., Kalyanasundaram S., Bollineni R.C., Wilson M.A.et al.. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016; 24:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Fang E.F., Hou Y., Lautrup S., Jensen M.B., Yang B., SenGupta T., Caponio D., Khezri R., Demarest T.G., Aman Y.et al.. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019; 10:5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Pascucci B., D’Errico M., Romagnoli A., De Nuccio C., Savino M., Pietraforte D., Lanzafame M., Calcagnile A.S., Fortini P., Baccarini S.et al.. Overexpression of parkin rescues the defective mitochondrial phenotype and the increased apoptosis of Cockayne syndrome A cells. Oncotarget. 2017; 8:102852–102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Brooks P.J. Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair (Amst.). 2013; 12:656–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Lopes A.F.C., Bozek K., Herholz M., Trifunovic A., Rieckher M., Schumacher B.. A C. elegans model for neurodegeneration in Cockayne syndrome. Nucleic Acids Res. 2020; 48:10973–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Calaza K.C., Kam J.H., Hogg C., Jeffery G.. Mitochondrial decline precedes phenotype development in the complement factor H mouse model of retinal degeneration but can be corrected by near infrared light. Neurobiol. Aging. 2015; 36:2869–2876. [DOI] [PubMed] [Google Scholar]
- 201. Calmels N., Botta E., Jia N., Fawcett H., Nardo T., Nakazawa Y., Lanzafame M., Moriwaki S., Sugita K., Kubota M.et al.. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 2018; 55:329–343. [DOI] [PubMed] [Google Scholar]
- 202. Jaarsma D., van der Pluijm I., van der Horst G.T., Hoeijmakers J.H.. Cockayne syndrome pathogenesis: lessons from mouse models. Mech. Ageing Dev. 2013; 134:180–195. [DOI] [PubMed] [Google Scholar]
- 203. Kamenisch Y., Fousteri M., Knoch J., von Thaler A.K., Fehrenbacher B., Kato H., Becker T., Dollé M.E., Kuiper R., Majora M.et al.. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010; 207:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cleaver J.E., Revet I.. Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech. Ageing Dev. 2008; 129:492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Alupei M.C., Maity P., Esser P.R., Krikki I., Tuorto F., Parlato R., Penzo M., Schelling A., Laugel V., Montanaro L.et al.. Loss of proteostasis is a pathomechanism in Cockayne syndrome. Cell Rep. 2018; 23:1612–1619. [DOI] [PubMed] [Google Scholar]