

Abstract
In the decades since the discovery of angiotensin-converting enzyme 2 (ACE2), its protective role in terms of antagonizing activation of the classical renin-angiotensin system (RAS) axis has been recognized in clinical and experimental studies on kidney and cardiovascular diseases. The effects of ACE inhibitor/angiotensin type 1 receptor blockers (ACEi/ARBs) on ACE2-angiotensin-(1-7) (Ang-(1-7))-Mas receptor (MasR) axis activation has encouraged the use of such blockers in patients with kidney and cardiovascular diseases, until the emergence of coronavirus disease 2019 (COVID-19). The previously unchallenged functions of the ACE2-Ang-(1-7)-MasR axis and ACEi/ARBs are being re-evaluated in the era of COVID-19; the hypothesis is that ACEi/ARBs may increase the risk of severe acute respiratory syndrome coronavirus 2 infection by upregulating the human ACE2 receptor expression level. In this review, we examine ACE2 molecular structure, function (as an enzyme of the RAS), and distribution. We explore the roles played by ACE2 in kidney, cardiovascular, and pulmonary diseases, highlighting studies that defined the benefits imparted when ACEi/ARBs activated the local ACE2-Ang-(1-7)-MasR axis. Finally, the question of whether ACEi/ARBs therapies should be stopped in COVID-19-infected patients will be reviewed by reference to the available evidence.
Keywords: Angiotensin converting enzyme 2, Cardiovascular diseases, COVID-19, Kidney diseases, Severe acute respiratory syndrome coronavirus 2
INTRODUCTION
Over the two decades since its discovery in 2000 [1,2], angiotensin-converting enzyme 2 (ACE2) has been shown to protect against certain actions of the classical renin-angiotensin system (RAS) (Fig. 1). The RAS is physiologically essential, but contributes to the pathogenesis of many diseases. The RAS is composed of an ACE, angiotensin II (Ang II) and the angiotensin type 1 receptor (AT1R). Suppression of the ACE-Ang II-AT1R axis by ACE inhibitors/AT1R blockers (ACEi/ARBs) has become the “dogma” of management for patients with kidney and cardiovascular diseases [3-6]. In contrast, the ACE2-angiotensin-(1-7) (Ang-(1-7))-Mas receptor (MasR) axis largely mitigates the consequences of AT1R activation, thus counterbalancing activation of the classical RAS [7]. Thus, blockade of the classical RAS via concurrent activation of the ACE2-Ang-(1-7)-MasR axis has emerged as an attractive therapeutic strategy [8-11]. The evidence indicates that ACEi/ARBs enhance ACE2-Ang-(1-7)-MasR axis activity [12-15], although the agents do not directly target ACE2, Ang-(1-7), or MasR, further suggesting that ACEi/ARBs are valuable for patients with kidney or cardiovascular diseases.
Figure 1.

A schematic of the renin-angiotensin system (RAS) components and their modes of action. Renin converts angiotensinogen to angiotensin I (Ang I), which is subsequently cleaved by angiotensin-converting enzyme (ACE) to form Ang II. Ang II binds to its cognate G-protein-coupled receptor, angiotensin II type 1 receptor (AT1R), playing as a major effector molecule of classic RAS such as water and salt retention, vasoconstriction, and proliferative, proinflammatory, and profibrotic processes. ACE2 hydrolyzes Ang I and Ang II to Ang 1–9 and Ang-(1–7), respectively, although the enzyme efficacy for Ang II is 400-fold greater for Ang I. Angiotensin-converting enzyme 2 (ACE2) cleaves Ang II to generate Ang-(1-7), which binds to another G-protein-coupled receptor, Mas receptor (MasR). The activation of MasR is associated with abrogation of pathogenic processes medicated by AT1R, in large, counterbalances the classic RAS activation to prevent target organ damage. Ang-(1-7) is also a substrate of ACE, which is converted to an inactive metabolite. Both Ang II and Ang-(1-7) are reported to activate angiotensin II type 2 receptor (AT2R), resulting in the effect similar to MasR activation. While ACEi/angiotensin II receptor blockers (ARBs) blocks ACE and AT1R, respectively, either of ACE2, MasR, or AT2R is not inhibited by conventional inhibitors of RAS.
This dogma, however, is being threatened by the viral pandemic coronavirus disease 2019 (COVID-19). Structural studies [16-18] have shown that the causative virus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), utilizes airway epithelial ACE2 as the receptor for entry into the human host. It has been suggested that ACEi/ARBs may increase vulnerability to SARS-CoV-2 infection by upregulating the viral ACE2 receptor [19-21], triggering an intense debate as to whether ACEi/ARBs should be continued or stopped in patients with COVID-19. Indeed, clinicians have been forced to continue or discontinue ACEi/ARBs in patients with COVID-19 and underlying kidney and/or cardiovascular diseases, without reliable evidence for such decisions.
In this review, the molecular structure, function (as an enzyme of the RAS), and distribution of ACE2 will be discussed. We will explore the role played by ACE2 in kidney diseases, highlighting studies that have demonstrated the benefits afforded when ACEi/ARBs activate the local ACE2-Ang-(1-7)-MasR axis. The roles played by ACE2 in cardiovascular and pulmonary diseases will also be summarized. Finally, the differing opinions on the use or disuse of ACEi/ARBs in patients with COVID-19 will be reviewed by drawing on the evidence that has accumulated to date; decision-making must be reasonable.
ACE2 STRUCTURE, FUNCTION, AND DISTRIBUTION
More than a century after the discovery of renin, ACE2 was near-simultaneously discovered by two research groups in 2000 [1,2]. Its enzymic properties were soon established [22] and the ACE2-Ang-(1-7)-MasR axis defined [23]. The human ACE2 gene is located in chromosome Xp22 and is 40 kb in size. The gene features 18 exons, most of which resemble those of ACE [2]. As is true of ACE, the N-terminus of ACE2 is a zinc metalloprotease domain exposed to the extracellular surface. Structurally, ACE exhibits two enzymatically active sites, whereas ACE2 has only one (Fig. 2). A major difference between ACE and ACE2 lies in the distinct substrate specificities of the N-terminal domains. ACE cleaves C-terminal dipeptide residues from susceptible substrates (and is thus a peptidyl dipeptidase), converting Ang I to Ang II [22,24] and bradykinin to inactive metabolites [25]. Ang-(1-7) is also a substrate of ACE, and is converted to an inactive metabolite, Ang-(1-5), by the enzyme [26,27]. In contrast, ACE2 is a simple carboxypeptidase that hydrolyzes Ang I and Ang II to Ang 1-9 and Ang-(1-7), respectively, although the affinity for Ang II is 400-fold greater than that for Ang I [24]. ACE2 does not cleave bradykinin. Conventional inhibitors of ACE, including ramipril, block the enzymatic activity, but ACE2 is insensitive to this class of agents [22]. The C-terminus of ACE2 is a transmembrane domain with a cytosolic tail lacking any similarity to ACE. This is termed the collectrin-like domain, being a homolog of collectrin, a protein expressed in the kidney, and regulates the trafficking of amino acid transporters to the cell surface, conferring a unique function on ACE2. Unexpectedly, ACE2 serves as the host receptor for coronavirus. Interaction between the receptor-binding domain of the viral spike protein and the protease domain of the host ACE2 was shown, in the early 2000s, to be essential for entry of the SARS-CoV virus and, more recently, SARS-CoV-2 [16,17,28,29]. Compared to SARS-CoV, several mutations in amino acid residues in the interface between SARS-CoV-2 and ACE2 are evident; some may strengthen the interactions between SARS-CoV-2 and ACE2 but others may reduce the affinity by negatively affecting hydrophobic interactions and salt bridge formation [17], although the overall binding affinity of SARS-CoV-2 to ACE2 is 10- to 20-fold higher than that of SARS-CoV [29], which may partly explain the persistence of COVID-19.
Figure 2.
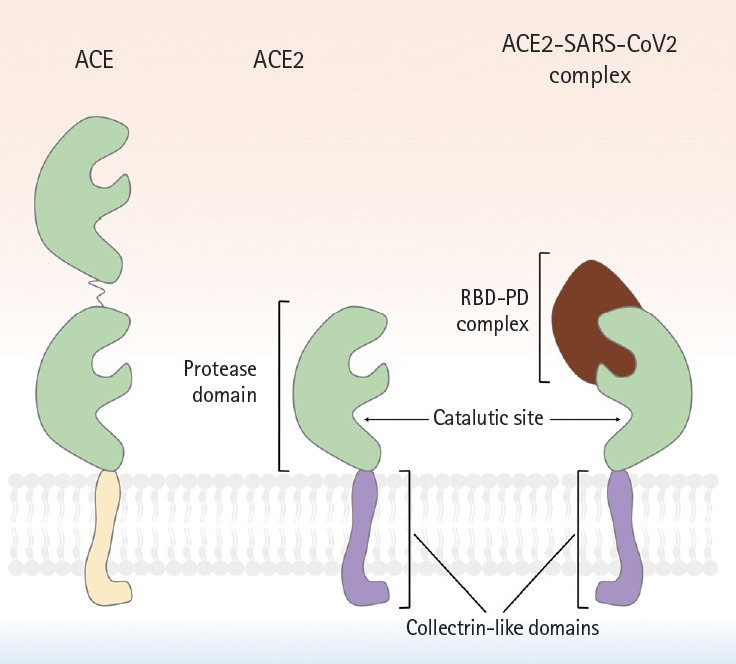
A schematic showing the molecular structures of angiotensin-converting enzyme (ACE), angiotensin-converting enzyme 2 (ACE2), and the ACE2-severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) complex. Although ACE2 is homologous to ACE. But, ACE2 has only a single active site, whereas ACE possesses 2 enzymatically active sites. Similar to ACE, the N-terminus of ACE2 is a protease domain (PD, colored in green) that is exposed to extracellular surfaces, acting as a zinc metalloprotease. The C-terminus of ACE2 is a transmembrane domain with a cytosolic tail that has no similarity with ACE. It is referred to as collectrin-like domain (colored in purple), as it is a homolog of collectrin, a protein expressed in the kidney. The interaction between receptor binding domain of viral spike protein and PD of host ACE2 is known to be crucial for viral entry of SARS-CoV in early 2000s, and more recently, SARS-CoV-2. Compared to SARS-CoV, several mutations in amino acid residues in the interface between SARS-CoV-2 and ACE2 were reported, resulting in the increase of binding affinity. RBD, receptor binding domain.
Renin converts angiotensinogen to Ang I, which is subsequently cleaved by ACE to form Ang II (Fig. 1). Ang II is a major effector molecule of the classic RAS, and binds to its cognate G-protein-coupled receptor (GPCR), termed AT1R. As AT1R activation mediates vasoconstrictive, proliferative, proinflammatory, and profibrotic processes, receptor antagonists such as ACEi/ARBs have become the cornerstone of kidney and cardiovascular disease therapeutics [4-6,30]. ACE2 cleaves Ang II to Ang-(1-7), which binds to another GPCR (MasR) [7,23,31]. As MasR activation abrogates the pathogenic processes mediated by AT1R, the ACE2-Ang-(1-7)-MasR axis essentially counterbalances the actions of the classical RAS, preventing the organ damage that will be discussed later. Conversely, RAS blockade by ACEi/ARBs upregulates ACE2 expression, although the precise mechanism was long elusive despite robust evidence from several animal studies [15,32-34]. However, the role played by the TNF-α converting enzyme (TACE) in cleavage of the ACE2 ectodomain was then discovered [35]. Using p47phox-/- mice, in which the p47phox subunit of nicotinamide adenine dinucleotide phosphate plays a crucial role in the superoxide generation induced by Ang II, it was found that oxidative stress followed by RAS activation enhanced TACE expression/activity via phosphorylation of p38 mitogen-activated protein kinase, to cleave ACE2 from cardiomyocytes (Fig. 3). Specific deletion of Tace from the myocardium prevented ACE2 shedding despite Ang II infusion [35].
Figure 3.

A schematic of proteolytic angiotensin-converting enzyme 2 (ACE2) ectodomain shedding after angiotensin II (Ang II)-induced TNF-α converting enzyme (TACE) activation. Activation of angiotensin II type 1 receptor (AT1R) by Ang II leads to superoxide generation, which in turn enhances phosphorylation of p38-mitogen-activated protein kinase (MAPK). Phosphorylated p38 MAPK is critical for the activation of TACE, via phosphorylation of a cytosolic residue. Activated TACE cleaved ACE2 from extracelluar surface, resuling in the shedding of ACE2 ectodomain. Ang-(1-7), angiotensin-(1-7); MasR, Mas receptor; ROS, reactive oxygen species.
The role of the angiotensin type 2 receptor (AT2R) is related to that of the ACE2-Ang-(1-7)-MasR axis, but is poorly understood (Fig. 1). AT2R is a receptor for Ang II, but the consequences of AT2R activation are opposite to those of AT1R activation. The anti-inflammatory vasoprotective effect of AT1R blockade by valsartan was significantly attenuated in AT2R knockout (KO) mice, implying that AT2R stimulation after AT1 blockade is important in terms of vascular protection [36]. Furthermore, Ang-(1-7) seems to act as an AT2R agonist, as best illustrated by the report that the anti-atherosclerotic effect of Ang-(1-7) in ApoE-/- mice was abolished by chemical inhibition of AT2R. AT2R blockade also revealed that the anti-hypertensive effect of Ang-(1-7) was largely mediated by AT2R rather than MasR [37,38]. More recently, an organ-specific protective role for ATR2 has been implied by a series of studies using a non-peptide AT2R agonist [39-42]. AT2R activation in the proximal tubules prevented sodium retention via internalization/inactivation of the major sodium transporters, and reduced the blood pressure of hypertensive rats [42]. As AT2R also exerts anti-inflammatory effects in the kidney [41], AT2R stimulation significantly ameliorated renal pathology in a rodent model of type 1 diabetes mellitus (T1DM). Thus, together with MasR, AT2R seems to be the principal receptor of an alternative, counterbalancing arm of the RAS. Organ-specific functions by the site of AT2R expression, and AT2R functions that differ from those of MasR, require further study.
Northern blotting initially implied that ACE2 expression was restricted to the heart, kidney, and testis [2]. However, later studies expanded the anatomical distribution. Lung type 2 pneumocytes and the endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) of various organs express ACE2 [43]. The epithelial cells of intestinal villi also express ACE2, which regulates amino acid transport [44]. ACE2 is normally expressed by the hepatocytes of healthy humans, and also in bile duct epithelial cells and sinusoidal ECs of patients with cirrhotic liver disease [45]. In contrast, direct evidence of ACE2 expression in the human brain remains lacking, although ACE2 expression was reported in a subset of mouse paraventricular neurons [46]. Most importantly, with the emergence of COVID-19, data based on single-cell transcriptome analysis of ACE2 expression are growing explosively [47-49], yielding many high-quality insights that will revolutionize our knowledge of ACE2 expression.
ACE2 IN KIDNEY DISEASES
Results from animal studies
The role of ACE2 in kidney diseases has been best-established in animal models of diabetic nephropathy. It is now widely accepted that intra-renal RAS activation is of particular importance in terms of the pathogenesis of such nephropathy [50]. Paradoxically, systemic RAS components are downregulated despite the robust activation of intra-renal RAS components in patients with diabetic nephropathy; this has spawned a great deal of debate [51]. T1DM induction with streptozotocin (STZ) downregulates ACE2 expression in mouse proximal tubular epithelial cells [52]. Compared to wild-type T1DM mice, deletion of Ace2 in STZ-induced T1DM mice accelerated the decline in renal function and increased the extents of glomerular and tubulointerstitial damage in a time-dependent manner [53]. Loss of Ace2 was associated with aggravated albuminuria and blood pressure elevation [52]; the responsiveness to perindopril was markedly attenuated by genetic deletion of Ace2 or treatment with an ACE2 inhibitor. Exacerbation of renal histology and albuminuria after Ace2 gene deletion has also been demonstrated in Akita mice, another animal model of T1DM [46].
The results from animal models of type 2 diabetes mellitus (T2DM) are rather complicated. Compared to wildtype mice, ACE2 expression was significantly higher in the kidney of db/db mice, a model of T2DM, with concurrent elevation of the ACE2 level in urine but not plasma [54,55]. This may reflect TACE-mediated shedding of the ACE2 ectodomain [56], which is activated by high glucose levels in various cells, including kidney, proximal tubular epithelial cells [57,58]. TACE, also known as a disintegrin and metalloproteinase 17 (ADAM17), is a metalloproteinase that can shed the ACE2 ectodomain upregulated in the kidneys of diabetic mice when its endogenous inhibitor, tissue inhibitor of metalloproteinase 3, is downregulated [55]. Together with observations from mouse models of T1DM, it has been speculated that upregulation of tubular ACE2 expression and activity may be an early event during the natural course of disease, but these features seem to decay on TACE upregulation as diabetic nephropathy progresses, although further studies are required to reveal the precise link between time-dependent glucose signaling and intra-renal regulation of ACE2. Changes in ACE2 expression have also been reported in Col4a3-/- mice, an animal model of Alport syndrome [59], characterized by a genetic defect in the glomerular basement membrane. The mice are normal at birth, but soon develop proteinuria and progressive, glomerular tubulointerstitial injuries. ACE2 expression is inversely correlated with the progression of renal injury in such mice, with a resultant rise in the Ang II level and a decline in the Ang-(1-7) level.
The effect of ACE2 administration has been examined in several animal models of kidney injury. Recombinant human ACE2 (rhACE2) effectively attenuated Ang II-mediated hypertension and renal injury [10,11], delayed progression of diabetic nephropathy in Akita mice (with a reduction in albuminuria) [60], and suppressed tubulointerstitial fibrosis in ApoE-/- animals (a model of atherosclerosis with progressive kidney lesions) by abrogating phosphorylation of AKT [61]. Recombinant murine ACE2 (rmACE2) has been given to mice with STZ-induced diabetic nephropathy, thus db/db mice and Col4a3-/-mice [62,63]. Surprisingly, a protective effect of rmACE2 was evident only in Col4a3-/- mice [62,63], regardless of the route of delivery; rmACE2 was delivered by an osmotic mini-pump in one study [62] and Ace2 minicircle DNA was injected once in the other study [63]. Thus, the differences in rmACE2 treatment efficacy may be primarily attributable to the ACE2 level/activity in the kidney tissue or urine, as urinary ACE2 activity increased significantly only in Col4a3-/- mice, despite striking rises in the serum activities of all STZ-injected mice, db/db mice, and Col4a3-/-mice [63]. One possible explanation is that rmACE2 can pass through the glomerular filtration barrier only when overt proteinuria is in play (e.g., in Col4a3-/- mice), and thus, the efficacy of systemic ACE2 delivery may be limited in mice with mild proteinuria (e.g., STZ-injected mice, db/db mice) [63].
Based on the role played by ACE2 in kidney homeostasis, several pharmacological interventions seeking to upregulate ACE2 expression have been tested in various disease models; the most consistent results are those of studies using ACEi/ARBs. One study on normotensive rats reported that lisinopril or losartan significantly augmented ACE2 activity in the renal cortex, and increased urinary excretion of Ang-(1-7) [34]. A 2-week telmisartan treatment of mice upregulated ACE2 and downregulated ACE expression in the tunica media and endothelial layer of the kidney arterioles, respectively [64]. In db/db mice given candesartan, renal tubular damage and albuminuria were ameliorated; the expression levels of ACE2, AT2R, and MasR increased; and ACE2 activity enhanced, with a reduction in extracellular signal-regulated protein kinase (ERK) 1/2 phosphorylation [12], although ultra-high doses of candesartan promoted renal injury and increased renal ERK1/2 activation. Olmesartan treatment of Col4a3-/-mice ameliorated both the glomerular and tubulointerstitial (pathological) histology, with upregulation of ACE2 expression and subsequent activation of the ACE2-Ang-(1-7)-MasR axis, despite persistence of the genetic defect [65].
Other pharmacological interventions seeking to activate the ACE2-Ang-(1-7)-MasR axis have yielded less consistent results or require further validation [66-72]. The results of treatment with diminazene aceturate (DIZE), a known ACE2 activator, are somewhat conflicting, and seem to depend on the experimental model chosen [66,67]. DIZE restored glomerular ACE2 expression and normalized whole-kidney Ang II and Ang-(1–7) levels in STZ-induced diabetic rats [66], but had no effect on blood pressure in indole-3-carbinol-induced Cyp1a1-Ren-2 transgenic rats with malignant hypertension, despite significant induction of kidney ACE2 activity and Ang-(1-7) expression [67]. This may mean that the genetic overexpression of renin was not overcome via pharmacological activation of ACE2. The fact that vitamin D is a negative regulator of renin transcription [69] has encouraged investigations of the effects of active vitamin D on kidney ACE2 expression. Calcitriol upregulated ACE2, but downregulated ACE expression in the kidney of STZ-induced diabetic rats; kidney phosphorylation of p38 and ERK was mitigated [70]. However, paricalcitol alone or in combination with aliskiren, a direct renin inhibitor, did not reduce urinary albumin excretion in non-obese diabetic mice, despite a reduction in serum ACE2 activity and enhanced cortical ACE2 expression [71]. Conversely, a role for fibroblast growth factor 23 (FGF23) in negative modulation of ACE2 expression has been suggested [73-75]. Indeed, combination FGF23/losartan compromised the effect of losartan on Ace2 mRNA upregulation in the contralateral kidney of the unilateral ureter obstruction model [72]; FGF23 alone did not affect the Ace2 mRNA level.
Together, the experimental evidence indicates that ACE2 plays a protective role in several kidney disease models, especially when activation is local rather than systemic. Pharmacological interventions enhance local ACE2 expression and activity. ACEi/ARBs have shown promising, but not fully consistent, results. Further studies on the local actions of ACE2 in kidney diseases should focus on context-dependent tailoring of therapeutics.
Results from patients with kidney diseases
Most studies on ACE2 in humans with kidney diseases are observational. In the human kidney, ACE2 has been observed in proximal tubular epithelial cells and, to a lesser degree, in glomeruli, where concurrent ACE2 downregulation and ACE upregulation have been reported in patients with T2DM and overt diabetic nephropathy [76,77], suggestive of changes in the ACE/ACE2 ratios. ACE2 downregulation in kidney tissue has been consistently reported in other studies on patients with T2DM and nodular glomerulosclerosis [78], although ACE was also downregulated; the ACE/ACE2 ratio was not measured. Analysis of urinary ACE2 protein level/activity is non-invasive and has provided valuable insights into intra-renal ACE2-Ang-(1-7)-MasR axis activity. Urinary ACE2 levels independently predict the risk of microalbuminuria, and reflect the stages and progression of chronic kidney disease (CKD) [79]. Diabetes further increases urinary ACE2 levels in such patients [80]. Intriguingly, urinary ACE2 protein excretion and activity were elevated in adolescents with uncomplicated T1DM compared to healthy controls, correlating with higher hemoglobin A1c levels, but not with the estimated glomerular filtration rate, blood pressure, or albuminuria [81], strongly suggesting that urinary ACE2 may be an early (and sensitive) biomarker of diabetic nephropathy, thus reflecting the severity of renal injury, given the mechanism of ACE2 shedding by proximal tubular epithelial cells after high-glucose exposure [56-58,82].
ACE2 IN CARDIOVASCULAR AND PULMONARY DISEASES
ACE2 in cardiovascular diseases
ACE2 is normally found in the cardiomyocytes, fibroblasts, epicardial adipocytes, and ECs of coronary vessels [35,83,84]; Ang-(1-7)/MasR is expressed in the cardiomyocytes, fibroblasts, ECs, and VSMCs of coronary vessels [85-88]. The role of the ACE2-Ang-(1-7)-MasR axis has been intensively explored in various animal models of cardiovascular disease as well as in humans. Immunohistochemistry for ACE2 in human and rat hearts revealed that ischemic injury upregulates ACE expression, principally in the vascular endothelium and smooth muscle, and less so in cardiomyocytes [89]. This seems to be a compensatory response to ischemia, rather than a mediator of tissue injury, as loss of Ace2 further accelerates maladaptive, left ventricular remodeling after myocardial infarction (MI), which was prevented by treatment with an ARB [90,91]. Overexpression of ACE2 [92] or systemic administration of Ang-(1-7) [93] also preserved cardiac function and attenuated inflammation after MI. A study of patients with idiopathic dilated cardiomyopathy revealed that heterozygotic loss of ACE2 was sufficient to promote adverse myocardial remodeling in response to pressure overload [94], implying a protective role for ACE2 in heart failure (HF). HF with a preserved ejection fraction is closely linked to obesity, and is characterized by inflammation of epicardial adipose tissue [84,95,96], which is further augmented by loss of ACE2 and increased macrophage polarization to the pro-inflammatory M1 phenotype. Ang-(1-7) attenuated M1 macrophage polarization in epicardial adipose tissue of obese Ace2 KO mice, preventing HF progression [84,97].
Activation of the ACE2-Ang-(1-7)-MasR axis assists blood pressure control. Renal Ace2 mRNA levels decreased in spontaneously hypertensive rats (SHRs) and stroke-prone SHRs [98], and lentiviral overexpression of ACE2 [99,100] or pretreatment with rhACE2 [10] attenuated blood pressure elevation in SHRs and Ang II-induced hypertensive mice, respectively. Diabetic retinopathy is another pro-inflammatory condition that is curtailed by activation of the ACE2-Ang-(1-7)-MasR axis [101,102]. Of note, ACE2 mRNA levels strongly predict microvascular disease in diabetic patients; such patients who remained free of retinopathy despite > 40 years of poor glycemic control exhibited higher levels of mRNAs transcribed from the genes of the ACE2-Ang-(1-7)-MasR axis than did age-, sex-, and glycemia-matched diabetics with retinopathy [103]. Specifically, Ang-(1-7) treatment restored the in vivo function of CD34+ bone marrow-derived vascular reparative cells and the circulating angiogenic cells that are dysfunctional in diabetics, preventing vascular injury inflicted by oxidative stress [103]. Intraocular administration of AAV-ACE2 or Ang-(1–7) reduced diabetes-induced retinal vascular leakage and inflammation, thus preventing retinopathy [102].
The efficacy of various ACEi/ARBs in terms of enhancing ACE2 expression/activity in the heart and vessels has been intensely examined, yielding quite promising and consistent results [13-15,80,104-106]. For example, olmesartan upregulated ACE2 and Ang-(1-7) expression in the aorta of SHRs [104]. Blood pressure was controlled by either lisinopril or losartan, accompanied by an increase in cardiac Ace2 mRNA levels [105]. It seems likely that the effects of ACEi/ARBs on the ACE2-Ang-(1-7)-MasR axis will further reinforce the appropriateness of ACEi/ARBs for patients with cardiovascular diseases.
ACE2 in pulmonary diseases
ACE2 is abundantly expressed by the alveolar and bronchiolar epithelium, the endothelium, and smooth muscle cells of the pulmonary vessels of rats, but not in bronchiolar smooth muscle cells [90]. ACE2 expression falls dramatically with aging in both sexes; female rats retain more ACE2 expression than do males. Acute lung injury induced by smoking downregulates lung ACE2 and upregulates ACE [107]. A precise role for ACE2 during acute lung injury has been implied by studies on Ace2 KO mice. Lung injury in such mice was induced by acid inhalation or sepsis [108], SARS-CoV infection [109], and bleomycin [110], and was more severe than in wild-type mice. rhACE2 ameliorated the histological and lung function changes in sepsis-induced [108] and pulmonary hypertension (PH) models of lung injury, and fibrosis in a bleomycin-induced model [110]. Notably, intravenous rhACE2 injection into patients with PH improved pulmonary hemodynamics and reduced the levels of oxidative and inflammatory markers [111]. Eleven patients with heritable or idiopathic PH exhibited lower ACE2 activity than healthy controls. This was the first study to explore the therapeutic efficacy of rhACE2 in humans, emphasizing the potential utility of ACE2 as a novel therapeutic. DIZE [112] and losartan [108,109,113] attenuated mouse lung injury with preservation of ACE2 expression [113], although their efficacies have not been proven in humans with pulmonary diseases.
ACEi/ARBs IN THE ERA OF COVID-19
The SARS-CoV-2 was first identified in late 2019, but lies on a continuum shared by two other highly pathogenic human coronaviruses (CoVs) described during the past two decades, thus SARS-CoV and Middle East respiratory syndrome (MERS)-CoV [114]. During the first SARS-CoV epidemic, human ACE2 was identified as the receptor for the surface spike protein (S protein) of SARS-CoV [108,109]. It remains unclear whether SARS-CoV-2 infection changes ACE2 expression; recent studies found that ACE2 expression increased after infection [115], and suggested that this was triggered by induction of interferons when the cell detected viral entry [49]. Any concern that ACEi/ARBs use might increase vulnerability to SARS-CoV by upregulating the viral ACE2 receptor was not marked prior to the emergence of COVID-19, probably because most studies on the effects of ACEi/ARBs on ACE2 expression were reported thereafter.
Initial reports indicate that comorbidities are very common in patients infected with COVID-19; these include hypertension, diabetes, coronary artery disease, and CKD [116-118]. Moreover, the frequency of pre-existing underlying conditions is considerably higher in patients exhibiting more severe clinical courses of COVID-19 infection, compared to patients with mild clinical courses [119]. Soon after identification of ACE2 as the receptor for SARS-CoV-2 [16-18], it was suggested that continued use of ACEi/ARBs in patients with underlying diseases would facilitate SARS-CoV-2 infection and increase the risk of severe disease and a fatal outcome [19]. Others later raised similar concerns [20,21,120], triggering an intense debate on continued use or cessation of ACEi/ARBs in patients with COVID-19 and underlying cardiovascular or kidney diseases. In fact, a recent letter reported that many patients from South America, Central America, and Spain, have already stopped or intend to interrupt their treatments with such drugs [121].
It should be emphasized; however, that, although these concerns have been (mostly) expressed by experts, they are grounded on very weak scientific evidence (Table 1) [122-129]. To date, no clinical evidence strongly recommends either the cessation or continued use of ACEi/ARBs in patients infected with COVID-19 [28,130-132]. Rather, the evidence that is available favors continued use of ACEi/ARBs in patients with COVID-19 (Table 2) [133-135], although one study suggested that ACEi/ARBs increased the risk of acute kidney injury in patients with severe COVID-19 infections [136]. For example, inhibition of AT1R by losartan attenuated SARS-CoV-induced acute lung injury in mice [108], and SARS-CoV infection downregulated ACE2 expression in mouse lungs and cultured cells, implying that uncontrolled RAS activation plays a crucial role in the pathogenesis of SARS-CoV-induced acute lung injury. One study enrolling a small number of COVID-19 patients reported that Ang II plasma levels were linearly associated with the viral load and extent of lung injury, and the authors even suggested that ARBs should be repurposed to treat COVID-19 [137]. More recently, a large retrospective multicenter study including 1,128 COVID-19 patients with hypertension on ACEi/ARBs showed that inpatient use by COVID-19 patients lowered all-cause mortality [133]. Despite potential biases attributable to extrapolation of data from mice infected with SARS-CoV, those of studies with limited numbers of patients, and those of retrospective analyses, the evidence that ACEi/ARB therapies are dangerous seems to be balanced by evidence for the opposite view. The fears are groundless. Appropriately, learned societies have recommended continuation of RAS antagonist therapies in COVID-19 patients with underlying conditions such as HF, hypertension, ischemic heart disease, or kidney disease. Unless concrete evidence supporting ACEi/ARB cessation is available; treatment should be individualized by reference to patient hemodynamic status and clinical presentation [131,132].
Table 1.
A summary of the results of recent studies revealing no clear association between ACEi/ARB use and COVID-19 infection status
| Study | Date of release | Study design | Population | Key findings |
|---|---|---|---|---|
| Mehta et al. [123] | May 5, 2020 | Retrospective cohort study | 18,472 Patients tested for COVID-19. | No association between ACEi/ARB use and COVID-19 test positivity |
| Jung et al. [124] | May 22, 2020 | Nationwide population-based cohort study | 5,179 Confirmed COVID-19 cases | Prior use of RAAS inhibitors was not independently associated with mortality among COVID-19 patients in Korea. |
| Raisi-Estabragh et al. [125] | July 14, 2020 | Prospective cohort study | 7,099 Participants from the UK Biobank tested for COVID-19 | ACE/ARB use did not associate with COVID-19 status. |
| De Spiegeleer et al. [126] | Jul 18, 2020 | Retrospective multicenter cohort study | 154 COVID-19-positive subjects | No statistically significant association between ACEi/ARB and asymptomatic status or serious clinical outcome |
| Zhang et al. [127] | August 4, 2020 | Multicenter retrospective study | 13,981 Patients with COVID-19 in Hubei Province, China | No significant association between ACEi/ARB therapy and 28-day mortality in individuals with hypertension and statin treatment |
| Bean et al. [128] | June 2, 2020 | Multicenter retrospective study | 1,200 Acute inpatients with COVID-19 | No evidence for increased severity of COVID-19 in hospitalized patients on chronic treatment with ACEi/ARBs |
| Fosbol et al. [129] | June 19, 2020 | Retrospective cohort study | 4,480 Patients with COVID-19 | Taking ACEi/ARBs did not result in more diagnoses of COVID-19, nor did they have a higher mortality rate. |
| Mackey et al. [122] | August 4, 2020 | Meta-analysis | NA | High-certainty evidence suggests that ACEi/ARB use is not associated with more severe COVID-19 disease. |
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin type 1 receptor blocker; COVID-19, coronavirus disease 2019; RAAS, renin-angiotensin-aldosterone system; NA, not applicable.
Table 2.
A summary of the results of recent studies revealing favorable outcomes of patients with COVID-19 infections who continued to use ACEi/ARBs
| Study | Date of release | Study design | Population | Key findings |
|---|---|---|---|---|
| Zhang et al. [133] | June 5, 2020 | Retrospective, multi-center study | 1,128 Adult patients with HTN diagnosed with COVID-19 | Inpatient use of ACEi/ARB was associated with lower risk of all-cause mortality compared with ACEi/ARB nonusers. |
| Grover et al. [134] | June 15, 2020 | Meta analysis | NA | ACEi/ARBs should be continued in COVID-19 patients. ACE2 polymorphisms which might confer higher risk of adverse outcomes. |
| Lam et al. [135] | July 23, 2020 | Retrospective single-center study | 614 Hypertensive laboratory-confirmed COVID-19 patients | Continued ACEi/ARB use in hypertensive COVID-19 patients yields better clinical outcomes. |
COVID-19, coronavirus disease 2019; ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin type 1 receptor blocker; HTN, hypertension; NA, not applicable; ACE2, angiotensin-converting enzyme 2.
CONCLUSIONS
In the time since ACE2 was discovered, our understanding of how the RAS affects kidney and cardiovascular disease progression has expanded. Over the last two decades, the protective role played by ACE2 (in terms of antagonizing activation of the classical RAS axis) has been demonstrated in many clinical and experimental settings. The positive effects of ACEi/ARBs on ACE2-Ang-(1-7)-MasR axis activation encouraged their prescription for patients with kidney and cardiovascular diseases, until COVID-19 appeared. The human virus receptor is ACE2; the previously unchallenged utilities of ACEi/ARBs have thus been challenged. It has been suggested that ACEi/ARB use may increase the risk of SARS-CoV-2 infection by upregulation of the ACE2 receptor; however, the scientific evidence is minimal. Much accumulated evidence to date indicates that SARS-CoV-2 infection does not imply that ACEi/ARB therapy should cease in patients conventionally indicated for such drugs. Planned directed trials [122,138] will soon guide clinical decision-making in the ACEi/ARB context in COVID-19-infected patients.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by Korean government (MIST) (2020R1A2C2005620, NRF-2019R1A2C1003971, NRF-2017M3A9E8023001 & NRF-2020R1F1A1074001), and by Chonnam National University Hospital Biomedical Research Institute Grant (BCRI 20025&20076).
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 2.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 3.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2007;112:417–428. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 4.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001 Sep;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 5.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 6.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 7.Kostenis E, Milligan G, Christopoulos A, et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–1813. doi: 10.1161/01.CIR.0000160867.23556.7D. [DOI] [PubMed] [Google Scholar]
- 8.Culver S, Li C, Siragy HM. Intrarenal angiotensin-converting enzyme: the old and the new. Curr Hypertens Rep. 2017;19:80. doi: 10.1007/s11906-017-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams VR, Scholey JW. Angiotensin-converting enzyme 2 and renal disease. Curr Opin Nephrol Hypertens. 2018;27:35–41. doi: 10.1097/MNH.0000000000000378. [DOI] [PubMed] [Google Scholar]
- 10.Wysocki J, Ye M, Rodriguez E, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong J, Guo D, Chen CB, et al. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension. 2011;57:314–322. doi: 10.1161/HYPERTENSIONAHA.110.164244. [DOI] [PubMed] [Google Scholar]
- 12.Callera GE, Antunes TT, Correa JW, et al. Differential renal effects of candesartan at high and ultra-high doses in diabetic mice-potential role of the ACE2/AT2R/Mas axis. Biosci Rep. 2016;36:e00398. doi: 10.1042/BSR20160344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111:2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- 14.Ocaranza MP, Godoy I, Jalil JE, et al. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension. 2006;48:572–578. doi: 10.1161/01.HYP.0000237862.94083.45. [DOI] [PubMed] [Google Scholar]
- 15.Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–976. doi: 10.1161/01.HYP.0000124667.34652.1a. [DOI] [PubMed] [Google Scholar]
- 16.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94:e00127. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–481. doi: 10.1016/S2213-2600(20)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz JH. Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med. 2020;27:taaa041. doi: 10.1093/jtm/taaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esler M, Esler D. Can angiotensin receptor-blocking drugs perhaps be harmful in the COVID-19 pandemic? J Hypertens. 2020;38:781–782. doi: 10.1097/HJH.0000000000002450. [DOI] [PubMed] [Google Scholar]
- 22.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt 1):45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos RA, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 25.Georgiadis D, Beau F, Czarny B, Cotton J, Yiotakis A, Dive V. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ Res. 2003;93:148–154. doi: 10.1161/01.RES.0000081593.33848.FC. [DOI] [PubMed] [Google Scholar]
- 26.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension. 1998;31(1 Pt 2):362–367. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- 27.Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdos EG. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1-7) and keto-ACE. Hypertension. 1998;31:912–917. doi: 10.1161/01.hyp.31.4.912. [DOI] [PubMed] [Google Scholar]
- 28.Aronson JK, Ferner RE. Drugs and the renin-angiotensin system in covid-19. BMJ. 2020;369:m1313. doi: 10.1136/bmj.m1313. [DOI] [PubMed] [Google Scholar]
- 29.Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruiz-Ortega M, Ruperez M, Esteban V, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant. 2006;21:16–20. doi: 10.1093/ndt/gfi265. [DOI] [PubMed] [Google Scholar]
- 31.Pinheiro SVB, Ferreira AJ, Kitten GT, et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009;75:1184–1193. doi: 10.1038/ki.2009.61. [DOI] [PubMed] [Google Scholar]
- 32.Tikellis C, Johnston CI, Forbes JM, et al. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension. 2003;41:392–397. doi: 10.1161/01.HYP.0000060689.38912.CB. [DOI] [PubMed] [Google Scholar]
- 33.Agata J, Ura N, Yoshida H, et al. Olmesartan is an angiotensin II receptor blocker with an inhibitory effect on angiotensin-converting enzyme. Hypertens Res. 2006;29:865–874. doi: 10.1291/hypres.29.865. [DOI] [PubMed] [Google Scholar]
- 34.Ferrario CM, Jessup J, Gallagher PE, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005;68:2189–2196. doi: 10.1111/j.1523-1755.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 35.Patel VB, Clarke N, Wang Z, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–176. doi: 10.1016/j.yjmcc.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Wu L, Iwai M, Nakagami H, et al. Roles of angiotensin II type 2 receptor stimulation associated with selective angiotensin II type 1 receptor blockade with valsartan in the improvement of inflammation-induced vascular injury. Circulation. 2001;104:2716–2721. doi: 10.1161/hc4601.099404. [DOI] [PubMed] [Google Scholar]
- 37.Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45:960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- 38.Bosnyak S, Widdop RE, Denton KM, Jones ES. Differential mechanisms of ang (1-7)-mediated vasodepressor effect in adult and aged candesartan-treated rats. Int J Hypertens. 2012;2012:192567. doi: 10.1155/2012/192567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koulis C, Chow BS, McKelvey M, et al. AT2R agonist, compound 21, is reno-protective against type 1 diabetic nephropathy. Hypertension. 2015;65:1073–1081. doi: 10.1161/HYPERTENSIONAHA.115.05204. [DOI] [PubMed] [Google Scholar]
- 40.Matavelli LC, Zatz R, Siragy HM. A nonpeptide angiotensin II type 2 receptor agonist prevents renal inflammation in early diabetes. J Cardiovasc Pharmacol. 2015;65:371–376. doi: 10.1097/FJC.0000000000000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhande I, Ali Q, Hussain T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: role in renoprotection in obese rats. Hypertension. 2013;61:1218–1226. doi: 10.1161/HYPERTENSIONAHA.111.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kemp BA, Howell NL, Keller SR, Gildea JJ, Padia SH, Carey RM. AT2 receptor activation prevents sodium retention and reduces blood pressure in angiotensin II-dependent hypertension. Circ Res. 2016;119:532–543. doi: 10.1161/CIRCRESAHA.116.308384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vuille-dit-Bille RN, Camargo SM, Emmenegger L, et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids. 2015;47:693–705. doi: 10.1007/s00726-014-1889-6. [DOI] [PubMed] [Google Scholar]
- 45.Paizis G, Tikellis C, Cooper ME, et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790–1796. doi: 10.1136/gut.2004.062398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong DW, Oudit GY, Reich H, et al. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol. 2007;171:438–451. doi: 10.2353/ajpath.2007.060977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. 2020;116:1097–1100. doi: 10.1093/cvr/cvaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14:185–192. doi: 10.1007/s11684-020-0754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181:1016–1035. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 51.Price DA, Porter LE, Gordon M, et al. The paradox of the low-renin state in diabetic nephropathy. J Am Soc Nephrol. 1999;10:2382–2391. doi: 10.1681/ASN.V10112382. [DOI] [PubMed] [Google Scholar]
- 52.Tikellis C, Bialkowski K, Pete J, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 53.Shiota A, Yamamoto K, Ohishi M, et al. Loss of ACE2 accelerates time-dependent glomerular and tubulointerstitial damage in streptozotocin-induced diabetic mice. Hypertens Res. 2010;33:298–307. doi: 10.1038/hr.2009.231. [DOI] [PubMed] [Google Scholar]
- 54.Wysocki J, Garcia-Halpin L, Ye M, et al. Regulation of urinary ACE2 in diabetic mice. Am J Physiol Renal Physiol. 2013;305:F600–F611. doi: 10.1152/ajprenal.00600.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chodavarapu H, Grobe N, Somineni HK, Salem ES, Madhu M, Elased KM. Rosiglitazone treatment of type 2 diabetic db/db mice attenuates urinary albumin and angiotensin converting enzyme 2 excretion. PLoS One. 2013;8:e62833. doi: 10.1371/journal.pone.0062833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lambert DW, Yarski M, Warner FJ, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J Biol Chem. 2005;280:30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy AB, Ramana KV, Srivastava S, Bhatnagar A, Srivastava SK. Aldose reductase regulates high glucose-induced ectodomain shedding of tumor necrosis factor (TNF)-alpha via protein kinase C-delta and TNF-alpha converting enzyme in vascular smooth muscle cells. Endocrinology. 2009;150:63–74. doi: 10.1210/en.2008-0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao F, Zimpelmann J, Burger D, Kennedy C, Hebert RL, Burns KD. Protein kinase C-δ mediates shedding of angiotensin-converting enzyme 2 from proximal tubular cells. Front Pharmacol. 2016;7:146. doi: 10.3389/fphar.2016.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bae EH, Konvalinka A, Fang F, et al. Characterization of the intrarenal renin-angiotensin system in experimental alport syndrome. Am J Pathol. 2015;185:1423–1435. doi: 10.1016/j.ajpath.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 60.Oudit GY, Liu GC, Zhong J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen LJ, Xu YL, Song B, et al. Angiotensin-converting enzyme 2 ameliorates renal fibrosis by blocking the activation of mTOR/ERK signaling in apolipoprotein E-deficient mice. Peptides. 2016;79:49–57. doi: 10.1016/j.peptides.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 62.Bae EH, Fang F, Williams VR, et al. Murine recombinant angiotensin-converting enzyme 2 attenuates kidney injury in experimental Alport syndrome. Kidney Int. 2017;91:1347–1361. doi: 10.1016/j.kint.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 63.Wysocki J, Ye M, Khattab AM, et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int. 2017;91:1336–1346. doi: 10.1016/j.kint.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soler MJ, Ye M, Wysocki J, William J, Lloveras J, Batlle D. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am J Physiol Renal Physiol. 2009;296:F398–F405. doi: 10.1152/ajprenal.90488.2008. [DOI] [PubMed] [Google Scholar]
- 65.Suh SH, Choi HS, Kim CS, et al. Olmesartan attenuates kidney fibrosis in a murine model of alport syndrome by suppressing tubular expression of TGFβ. Int J Mol Sci. 2019;20:3843. doi: 10.3390/ijms20153843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goru SK, Kadakol A, Malek V, Pandey A, Sharma N, Gaikwad AB. Diminazene aceturate prevents nephropathy by increasing glomerular ACE2 and AT2 receptor expression in a rat model of type 1 diabetes. Br J Pharmacol. 2017;174:3118–3130. doi: 10.1111/bph.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huskova Z, Kopkan L, Cervenkova L, et al. Intrarenal alterations of the angiotensin-converting enzyme type 2/angiotensin 1-7 complex of the renin-angiotensin system do not alter the course of malignant hypertension in Cyp1a1-Ren-2 transgenic rats. Clin Exp Pharmacol Physiol. 2016;43:438–449. doi: 10.1111/1440-1681.12553. [DOI] [PubMed] [Google Scholar]
- 68.Suh SH, Choi HS, Kim CS, et al. CG200745, a novel HDAC inhibitor, attenuates kidney fibrosis in a murine model of Alport syndrome. Int J Mol Sci. 2020;21:1473. doi: 10.3390/ijms21041473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan W, Pan W, Kong J, et al. 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem. 2007;282:29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 70.Lin M, Gao P, Zhao T, et al. Calcitriol regulates angiotensin-converting enzyme and angiotensin converting-enzyme 2 in diabetic kidney disease. Mol Biol Rep. 2016;43:397–406. doi: 10.1007/s11033-016-3971-5. [DOI] [PubMed] [Google Scholar]
- 71.Riera M, Anguiano L, Clotet S, et al. Paricalcitol modulates ACE2 shedding and renal ADAM17 in NOD mice beyond proteinuria. Am J Physiol Renal Physiol. 2016;310:F534–F546. doi: 10.1152/ajprenal.00082.2015. [DOI] [PubMed] [Google Scholar]
- 72.de Jong MA, Mirkovic K, Mencke R, et al. Fibroblast growth factor 23 modifies the pharmacological effects of angiotensin receptor blockade in experimental renal fibrosis. Nephrol Dial Transplant. 2017;32:73–80. doi: 10.1093/ndt/gfw105. [DOI] [PubMed] [Google Scholar]
- 73.de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22:1603–1609. doi: 10.1681/ASN.2010121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Humalda JK, Lambers Heerspink HJ, Kwakernaak AJ, et al. Fibroblast growth factor 23 and the antiproteinuric response to dietary sodium restriction during renin-angiotensin-aldosterone system blockade. Am J Kidney Dis. 2015;65:259–266. doi: 10.1053/j.ajkd.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 75.Zoccali C, Ruggenenti P, Perna A, et al. Phosphate may promote CKD progression and attenuate renoprotective effect of ACE inhibition. J Am Soc Nephrol. 2011;22:1923–1930. doi: 10.1681/ASN.2011020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizuiri S, Hemmi H, Arita M, et al. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am J Kidney Dis. 2008;51:613–623. doi: 10.1053/j.ajkd.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 77.Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008;74:1610–1616. doi: 10.1038/ki.2008.497. [DOI] [PubMed] [Google Scholar]
- 78.Wang M, Zhang X, Song X, et al. Nodular glomerulosclerosis and renin angiotensin system in Chinese patients with type 2 diabetes. Mol Cell Endocrinol. 2016;427:92–100. doi: 10.1016/j.mce.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 79.Mizuiri S, Aoki T, Hemmi H, Arita M, Sakai K, Aikawa A. Urinary angiotensin-converting enzyme 2 in patients with CKD. Nephrology (Carlton) 2011;16:567–572. doi: 10.1111/j.1440-1797.2011.01467.x. [DOI] [PubMed] [Google Scholar]
- 80.Rigatto K, Casali KR, Shenoy V, Katovich MJ, Raizada MK. Diminazene aceturate improves autonomic modulation in pulmonary hypertension. Eur J Pharmacol. 2013;713:89–93. doi: 10.1016/j.ejphar.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burns KD, Lytvyn Y, Mahmud FH, et al. The relationship between urinary renin-angiotensin system markers, renal function, and blood pressure in adolescents with type 1 diabetes. Am J Physiol Renal Physiol. 2017;312:F335–F342. doi: 10.1152/ajprenal.00438.2016. [DOI] [PubMed] [Google Scholar]
- 82.Basu R, Lee J, Wang Z, et al. Loss of TIMP3 selectively exacerbates diabetic nephropathy. Am J Physiol Renal Physiol. 2012;303:F1341–F1352. doi: 10.1152/ajprenal.00349.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patel VB, Zhong JC, Fan D, et al. Angiotensin-converting enzyme 2 is a critical determinant of angiotensin II-induced loss of vascular smooth muscle cells and adverse vascular remodeling. Hypertension. 2014;64:157–164. doi: 10.1161/HYPERTENSIONAHA.114.03388. [DOI] [PubMed] [Google Scholar]
- 84.Patel VB, Mori J, McLean BA, et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes. 2016;65:85–95. doi: 10.2337/db15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 2007;49:185–192. doi: 10.1161/01.HYP.0000251865.35728.2f. [DOI] [PubMed] [Google Scholar]
- 86.Santos RA, Castro CH, Gava E, et al. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension. 2006;47:996–1002. doi: 10.1161/01.HYP.0000215289.51180.5c. [DOI] [PubMed] [Google Scholar]
- 87.Sampaio WO, Henrique de Castro C, Santos RA, Schiffrin EL, Touyz RM. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50:1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- 88.Iwata M, Cowling RT, Gurantz D, et al. Angiotensin-(1-7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol. 2005;289:H2356–H2363. doi: 10.1152/ajpheart.00317.2005. [DOI] [PubMed] [Google Scholar]
- 89.Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26:369–75. doi: 10.1093/eurheartj/ehi114. [DOI] [PubMed] [Google Scholar]
- 90.Xie X, Chen J, Wang X, Zhang F, Liu Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006;78:2166–2171. doi: 10.1016/j.lfs.2005.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kassiri Z, Zhong J, Guo D, et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2:446–455. doi: 10.1161/CIRCHEARTFAILURE.108.840124. [DOI] [PubMed] [Google Scholar]
- 92.Qi YF, Zhang J, Wang L, et al. Angiotensin-converting enzyme 2 inhibits high-mobility group box 1 and attenuates cardiac dysfunction post-myocardial ischemia. J Mol Med (Berl) 2016;94:37–49. doi: 10.1007/s00109-015-1356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Qian C, Roks AJ, et al. Circulating rather than cardiac angiotensin-(1-7) stimulates cardioprotection after myocardial infarction. Circ Heart Fail. 2010;3:286–293. doi: 10.1161/CIRCHEARTFAILURE.109.905968. [DOI] [PubMed] [Google Scholar]
- 94.Wang W, Patel VB, Parajuli N, et al. Heterozygote loss of ACE2 is sufficient to increase the susceptibility to heart disease. J Mol Med (Berl) 2014;92:847–858. doi: 10.1007/s00109-014-1149-y. [DOI] [PubMed] [Google Scholar]
- 95.Patel VB, Shah S, Verma S, Oudit GY. Epicardial adipose tissue as a metabolic transducer: role in heart failure and coronary artery disease. Heart Fail Rev. 2017;22:889–902. doi: 10.1007/s10741-017-9644-1. [DOI] [PubMed] [Google Scholar]
- 96.Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol. 2018;71:2360–2372. doi: 10.1016/j.jacc.2018.03.509. [DOI] [PubMed] [Google Scholar]
- 97.Patel VB, Basu R, Oudit GY. ACE2/Ang 1-7 axis: a critical regulator of epicardial adipose tissue inflammation and cardiac dysfunction in obesity. Adipocyte. 2016;5:306–311. doi: 10.1080/21623945.2015.1131881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 99.Diez-Freire C, Vazquez J, Correa de Adjounian MF, et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics. 2006;27:12–19. doi: 10.1152/physiolgenomics.00312.2005. [DOI] [PubMed] [Google Scholar]
- 100.Rentzsch B, Todiras M, Iliescu R, et al. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52:967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 101.Verma A, Shan Z, Lei B, et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012;20:28–36. doi: 10.1038/mt.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dominguez JM, 2nd, Hu P, Caballero S, et al. Adeno-associated virus overexpression of angiotensin-converting enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am J Pathol. 2016;186:1688–1700. doi: 10.1016/j.ajpath.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jarajapu YP, Bhatwadekar AD, Caballero S, et al. Activation of the ACE2/angiotensin-(1-7)/Mas receptor axis enhances the reparative function of dysfunctional diabetic endothelial progenitors. Diabetes. 2013;62:1258–1269. doi: 10.2337/db12-0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H1013–H1019. doi: 10.1152/ajpheart.00068.2005. [DOI] [PubMed] [Google Scholar]
- 105.Jessup JA, Gallagher PE, Averill DB, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol. 2006;291:H2166–H2172. doi: 10.1152/ajpheart.00061.2006. [DOI] [PubMed] [Google Scholar]
- 106.Jin HY, Song B, Oudit GY, et al. ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS One. 2012;7:e38502. doi: 10.1371/journal.pone.0038502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yilin Z, Yandong N, Faguang J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns. 2015;41:1468–1477. doi: 10.1016/j.burns.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rey-Parra GJ, Vadivel A, Coltan L, et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J Mol Med (Berl) 2012;90:637–647. doi: 10.1007/s00109-012-0859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hemnes AR, Rathinasabapathy A, Austin EA, et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51:1702638. doi: 10.1183/13993003.02638-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fang Y, Gao F, Liu Z. Angiotensin-converting enzyme 2 attenuates inflammatory response and oxidative stress in hyperoxic lung injury by regulating NF-κB and Nrf2 pathways. QJM. 2019;112:914–924. doi: 10.1093/qjmed/hcz206. [DOI] [PubMed] [Google Scholar]
- 113.Yuan YM, Luo L, Guo Z, Yang M, Ye RS, Luo C. Activation of renin-angiotensin-aldosterone system (RAAS) in the lung of smoking-induced pulmonary arterial hypertension (PAH) rats. J Renin Angiotensin Aldosterone Syst. 2015;16:249–253. doi: 10.1177/1470320315576256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li G, He X, Zhang L, et al. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun. 2020;112:102463. doi: 10.1016/j.jaut.2020.102463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McMichael TM, Currie DW, Clark S, et al. Epidemiology of COVID-19 in a long-term care facility in King County, Washington. N Engl J Med. 2020;382:2005–2011. doi: 10.1056/NEJMoa2005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li G, Hu R, Zhang X. Antihypertensive treatment with ACEI/ARB of patients with COVID-19 complicated by hypertension. Hypertens Res. 2020;43:588–590. doi: 10.1038/s41440-020-0433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marin GH. Facts and reflections on COVID-19 and anti-hypertensives drugs. Drug Discov Ther. 2020;14:105–106. doi: 10.5582/ddt.2020.01017. [DOI] [PubMed] [Google Scholar]
- 122.Mackey K, King VJ, Gurley S, et al. Risks and impact of angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers on SARS-CoV-2 infection in adults: a living systematic review. Ann Intern Med. 2020;173:195–203. doi: 10.7326/M20-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mehta N, Kalra A, Nowacki AS, et al. Association of use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with testing positive for coronavirus disease 2019 (COVID-19) JAMA Cardiol. 2020;5:1020–1026. doi: 10.1001/jamacardio.2020.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jung SY, Choi JC, You SH, Kim WY. Association of renin-angiotensin-aldosterone system inhibitors with COVID-19-related outcomes in Korea: a nationwide population-based cohort study. Clin Infect Dis. 2020;71:2121–2128. doi: 10.1093/cid/ciaa624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Raisi-Estabragh Z, McCracken C, Ardissino M, et al. Renin-angiotensin-aldosterone system blockers are not associated with coronavirus disease 2019 (COVID-19) hospitalization: study of 1,439 UK Biobank cases. Front Cardiovasc Med. 2020;7:138. doi: 10.3389/fcvm.2020.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Spiegeleer A, Bronselaer A, Teo JT, et al. The effects of ARBs, ACEis, and statins on clinical outcomes of COVID-19 infection among nursing home residents. J Am Med Dir Assoc. 2020;21:909–914. doi: 10.1016/j.jamda.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang XJ, Qin JJ, Cheng X, et al. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab. 2020;32:176–187. doi: 10.1016/j.cmet.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bean DM, Kraljevic Z, Searle T, et al. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers are not associated with severe COVID-19 infection in a multi-site UK acute hospital trust. Eur J Heart Fail. 2020;22:967–974. doi: 10.1002/ejhf.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fosbol EL, Butt JH, Ostergaard L, et al. Association of angiotensin-converting enzyme inhibitor or angiotensin receptor blocker use with COVID-19 diagnosis and mortality. JAMA. 2020;324:168–177. doi: 10.1001/jama.2020.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382:1653–1659. doi: 10.1056/NEJMsr2005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bavishi C, Maddox TM, Messerli FH. Coronavirus disease 2019 (COVID-19) infection and renin angiotensin system blockers. JAMA Cardiol. 2020;5:745–747. doi: 10.1001/jamacardio.2020.1282. [DOI] [PubMed] [Google Scholar]
- 132.South AM, Tomlinson L, Edmonston D, Hiremath S, Sparks MA. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat Rev Nephrol. 2020;16:305–307. doi: 10.1038/s41581-020-0279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang P, Zhu L, Cai J, et al. Association of inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020;126:1671–1681. doi: 10.1161/CIRCRESAHA.120.317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Grover A, Oberoi M. A systematic review and meta-analysis to evaluate the clinical outcomes in COVID-19 patients on angiotensin-converting enzyme inhibitors or angiotensin receptor blockers. Eur Heart J Cardiovasc Pharmacother. 2020 Jun 15; doi: 10.1093/ehjcvp/pvaa064. [Epub]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lam KW, Chow KW, Vo J, et al. Continued in-hospital angiotensin-converting enzyme inhibitor and angiotensin ii receptor blocker use in hypertensive COVID-19 patients is associated with positive clinical outcome. J Infect Dis. 2020;222:1256–1264. doi: 10.1093/infdis/jiaa447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. doi: 10.1093/cid/ciaa677. [Epub]. [DOI] [Google Scholar]
- 137.Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lopes RD, Macedo AVS, de Barros E Silva PGM, et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The BRACE CORONA Trial. Am Heart J. 2020;226:49–59. doi: 10.1016/j.ahj.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]