

Abstract
From a clinical, morphological and molecular perspective, prostate cancer is a heterogeneous disease. Primary prostate cancers are often multifocal, having topographically and morphologically distinct tumour foci. Sequencing studies have revealed that individual tumour foci can arise as clonally distinct lesions with no shared driver gene alterations. This finding demonstrates that multiple genomically and phenotypically distinct primary prostate cancers can be present in an individual patient. Lethal metastatic prostate cancer seems to arise from a single clone in the primary tumour but can exhibit subclonal heterogeneity at the genomic, epigenetic and phenotypic levels. Collectively, this complex heterogeneous constellation of molecular alterations poses obstacles for the diagnosis and treatment of prostate cancer. However, advances in our understanding of intra-tumoural heterogeneity and the development of novel technologies will allow us to navigate these challenges, refine approaches for translational research and ultimately improve patient care.
Cancer cells show differences in many measurable traits, including proliferation, metastatic potential and therapeutic resistance. Such differences are determined by heritable genetic and epigenetic alterations1,2. Every patient’s cancer has unique genomic and phenotypic changes, but over the past few years an increasing number of studies have highlighted the high level of genetic and phenotypic heterogeneity within a given tumour2–4. These insights have been driven by advances in sequencing technology and molecular pathology approaches that have enabled the study of genomic and molecular alterations at an unprecedented resolution5–7. Applying methods adapted from evolutionary biology to such sequencing data has demonstrated the complex relationship of different lesions in primary tumours and metastases and provided evidence for independent clonal evolution of tumour cell subpopulations8,9.
Prostate cancer is the most common non-skin malignancy diagnosed in men in the Western world and has long been known to be a heterogeneous disease10. The clinical presentation of prostate cancer can range from localized indolent to a rapidly progressing lethal metastatic disease11–13. Although the majority of men are diagnosed with organ-confined disease, long-term oncological outcomes can vary greatly14–16. Furthermore, histomorphological and molecular tumour characteristics show substantial diversity between different patients and within a given tumour. This poses diagnostic challenges and has major implications for clinical management4,17,18.
Much of the complexity of primary prostate cancer diagnosis is rooted in the multifocal nature of the disease. Numerous reports have documented that >80% of primary prostate cancers show multiple topographically and histomorphologically distinct tumour foci19–22. Sequencing efforts have demonstrated a high level of genomic diversity between different patients (inter-patient heterogeneity) but also within a given primary tumour (intra-tumoural heterogeneity) as well as its distinct tumour foci and different metastatic sites (inter-tumoural heterogeneity)4,23–27. In addition to the complex presentation of primary tumours, metastatic disease shows a similar level of genomic and phenotypic heterogeneity28–31, which complicates the assessment of molecular alterations and poses potential barriers to the implementation of precision medicine32.
In this Review, we summarize the manifestations of inter-tumoural and intra-tumoural heterogeneity in primary and metastatic prostate cancer. We emphasize the significant contribution of genomics studies in this field and discuss the importance of phenotypic changes. Finally, we provide a framework for assessing heterogeneity and critically discuss the implications for clinical management and research.
Understanding heterogeneity
Within a given tumour, cancer cells and the tumour-associated microenvironment can show alterations in molecular characteristics24. All of these changes show a variable degree of difference between individual tumour cells, cell clusters or topographically or anatomically separated tumour lesions. Such heterogeneities can result in distinct cellular phenotypes that contribute to the overall biological behaviour of a tumour. Owing to advancements in DNA sequencing technologies over the past two decades, heterogeneity at the genetic level has been the focus of a large number of studies, which have revealed important insights into the genomic composition of many solid tumours (including prostate cancer) and allowed a delineation of clonal and subclonal relationships of different tumour cell populations7,25,29,33,34. These studies have shed light on the complex evolution and competition of genomically defined tumour cell subpopulations during tumour progression and in response to therapy2,7,8,25,29,34–36. However, the term heterogeneity can be more broadly defined and applied to other measurable differences, not just at the genomic level2,24. Epigenetic, expression, post-translational, morphological and phenotypic heterogeneities, which all probably contribute to disease progression and clinical manifestations, exist within solid tumours. These non-genomic changes can be used to define heterogeneity, but they only allow an indirect assessment of the underlying clonal and subclonal changes.
Clonal and subclonal alterations.
All human life starts from a single cell that subsequently undergoes clonal expansions, ultimately giving rise to all organ systems. Therefore, all cells in the human body are monoclonal in origin37. On a cell-by-cell level, however, all tissues show evidence of genomic heterogeneity, which arises from insults that occur owing to the infidelity of DNA replication, imperfect DNA repair and other cellular processes36,38. In addition, and probably particularly relevant for prostate cancer, DNA damage occurring during transcription can further contribute to genomic diversity39,40. Therefore, every cell has a slightly different genetic make-up and can harbour numerous unique mutations38,41,42. However, most of these alterations do not affect cell growth and are therefore inconsequential. Neoplastic growth occurs when driver genes are altered such that cell fitness is increased. Most solid tumours are thought to arise from a single cell that gains neoplastic growth potential through accumulation of genomic and heritable epigenetic alterations that change the cellular phenotype43. Driver gene alterations are shared between all cells arising from the initiating cell. Therefore, daughter cells of this tumour-initiating cell, although they share the vast majority of genomic information with all other cells in the body, show distinct driver gene changes that are selected for, giving rise to neoplastic clonal expansion.
Within this clonal cell population, individual cells continue to accumulate additional gene alterations, resulting in subclonal expansions within the tumour mass that exhibit distinct genetic and phenotypic diversity44. A given mutation is considered subclonal if it appears in only a fraction of the tumour cells that otherwise share a set of founding mutations common to all of the cells in the tumour. The detection of a subclonal population is therefore dependent on an assay-specific technical detection threshold and a statistical threshold. More than half of the somatic alterations in an initiating cell are estimated to originate before neoplastic transformation and at least a subset of these changes are shared with adjacent benign cells45. Tracing experiments demonstrate that early non-driver alterations can occur in tissue-resident stem cells from which the glandular network of the prostate is populated46. These changes are distinct from the germline sequence found in other tissue types and can be detected in both benign and cancer tissues25,45,46 (FIG. 1. This finding probably explains why cancer and adjacent benign tissue can often share genomic alterations and also provides an explanation for the high level of intra-tumoural heterogeneity seen in prostate cancer25,47.
Fig. 1 |. Model of clonal progression of prostate cancer.
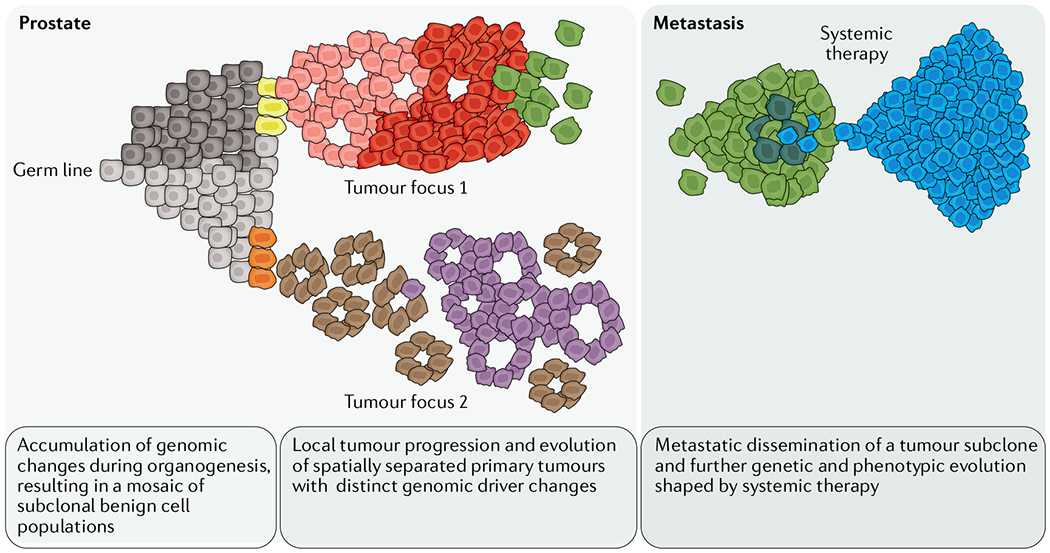
In the context of benign epithelial cells, which can harbour unique genomic alterations, precursor lesions can arise and progress into clonally and spatially distinct foci of invasive carcinoma. Within a given tumour focus, individual cells can acquire additional genomic driver changes, resulting in subclonal tumour cell populations. Subclones can further evolve, replace other tumour cell populations (clonal sweep) and disseminate to distant sites. Disseminated tumour cells can clonally expand, but systemic therapies can induce a major re-sculpting of the subclonal composition selecting for cells with intrinsic or adapted resistance mechanisms. Resistant subclones that pass through therapy-induced clonal bottlenecks contribute to disease recurrence and therapy failure.
Microheterogeneity and macroheterogeneity.
The expansion of a tumour into a clinically detectable mass generates a vast range of genotypes with variable impacts on cell fitness. Given this high level of genetic diversity in tissues, the term heterogeneity needs to be used carefully and in the appropriate context. In particular, we need to consider which level of genetic heterogeneity is clinically relevant36. Microheterogeneity at the single-cell level is subjected to constant selection, which shapes the subclonal cell populations43,48. With the currently available technologies, microheterogeneity is challenging to study and is therefore of uncertain clinical significance at this point. The expansion of a single cell with unique favourable traits into a larger cell mass results in macroheterogeneity, which is more accessible using current methods (see below). Propagation of subclones can lead to complete overgrowth of other less well-adapted cell populations (clonal sweep) and results in further genetic diversification (FIG. 1. Studies applying multiregional sampling approaches, in which several regions of a single tumour mass are analysed, have documented that the vast majority of solid tumours comprise subclones with varying degrees of driver gene alterations7,49–52. On the basis of these studies, most solid tumours represent a mosaic of multiple tumour cell populations that have shared (truncal) and unique (branch) genomic and phenotypic features (FIG. 1). The level of subclonal diversity can vary between different tumours. Importantly, although in some tumour types increased intra-tumoural heterogeneity has been associated with worse clinical outcome, in other settings, tumours with very high subclonal diversity seem to be less aggressive, suggesting a complex relationship between the level of heterogeneity and clinical outcome35,43,53,54.
The unique clonal constellation of prostate cancer.
Like most other tumour types, subclonal heterogeneity is common in prostate cancer25,27,33,55. Studies demonstrate that around 60% of tumour foci show evidence of multiple subclones25,33,47,55. Interestingly, these studies suggest that the subclonal diversity in a given index tumour focus is higher in primary samples than in metastatic samples. In addition, tumours with a more complex sub-clonal architecture have a higher rate of recurrence than monoclonal tumours33,56. Collectively, these data suggest that a high level of subclonal diversity in the primary tumour might increase the chance of metastatic dissemination. Given that the number of subclones capable of metastasis formation is probably very small, the clonal constellation in distant metastases of treatment-naive patients seems more homogeneous.
What sets prostate cancer apart from most other solid tumours is that the vast majority of primary prostate cancers occur multifocally4,26,27. This means that a diseased prostate gland harbours multiple topographically separate tumour foci. A large body of literature demonstrates that these distinct tumour foci show unique non-overlapping mutation profiles4,25,27,29,40,57,58 (FIG. 1), suggesting that these tumours arise independently and follow separate evolutionary trajectories. Therefore, a given patient can harbour more than one genomically and phenotypically distinct prostate cancer (FIG. 2). These clonally independent tumours can show major biological differences and can contribute differently to disease progression and clinical outcome. The multiclonal nature of prostate cancer has important implications for clinical management and research. Therefore, improved understanding of the driving factors involved in the multifocal nature should result in improved understanding of prostate cancer biology. For instance, it remains to be determined which endogenous or exogenous, epithelial cell intrinsic or microenvironmental factors contribute to this field cancerization and enable numerous independent tumour clones to arise in the prostate59–61. In addition, we need to determine in which clinical scenarios multiclonality is most relevant. To this end, defining methods that allow us to capture the heterogeneous nature of prostate cancer is important.
Fig. 2 |. Visualizing clonal and subclonal heterogeneity in tumour tissues.
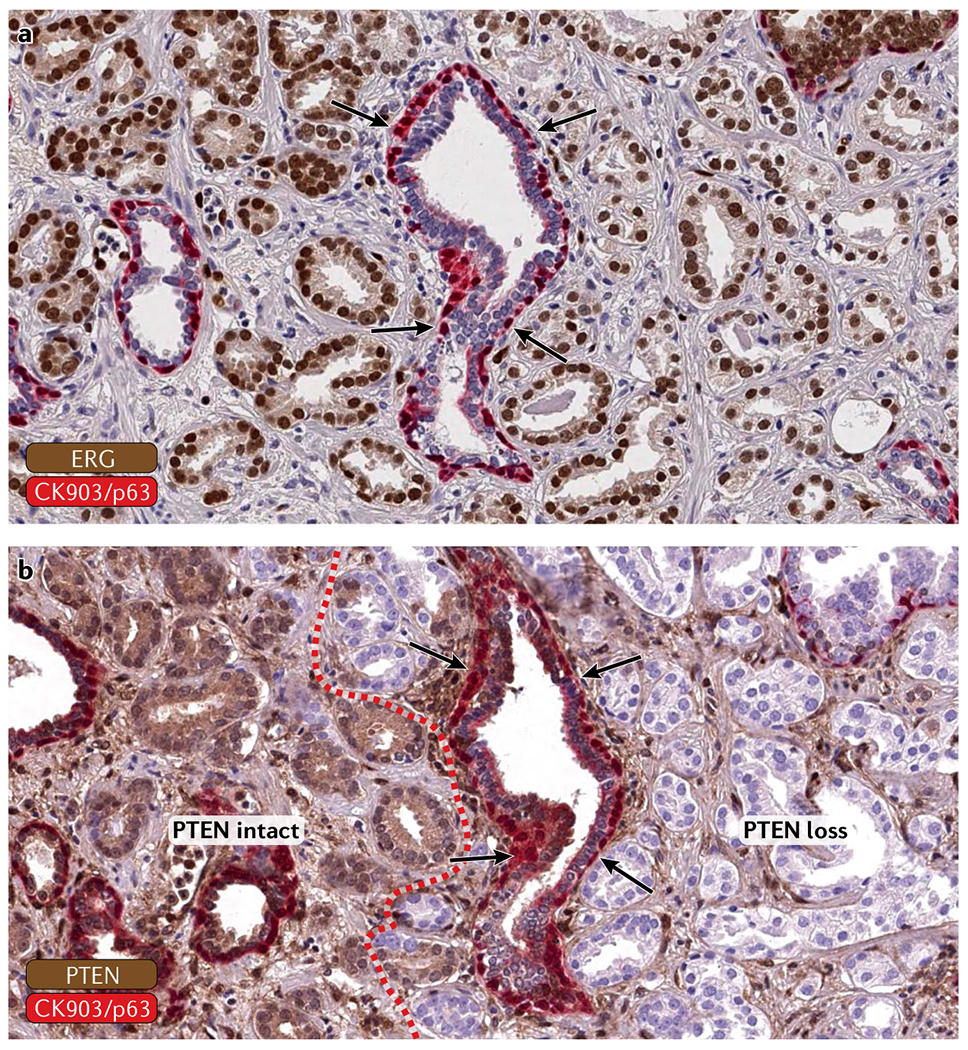
Genetically validated antibodies for ERG and PTEN can be used to highlight clonal and subclonal cancer ceLL populations in tissue sections. a | An ERG+ (brown nuclear stain) tumour that infiltrates between benign glands (highlighted by an intact basal ceLL Layer, red stain). Arrows show a benign gland. b | The tumour shows subclonal loss of PTEN (loss of cytoplasmic staining) in a subset of cancer glands (separated by the red dotted line). Intact basal cells are shown in red. Arrows show a benign gland.
Studying genomic heterogeneity
In situ methods.
Genomic and phenotypic heterogeneity of complex tumour tissues can be assessed using two different methods: in situ approaches, which preserve the tissue architecture and enable cell-by-cell analyses and correlation with histomorphology features; and ex situ assays, in which the tissue is disrupted and extracted analytes can be measured using a variety of assay platforms. The most commonly used in situ methods in molecular pathology — immunohistochemistry and fluorescence in situ hybridization (FISH) — have been used extensively to study prostate cancer. For instance, the detection of TMPRSS2-ERG rearrangements by FISH, which occur in ~50% of prostate cancers, was applied to determine clonal relationships in the setting of multifocal primary tumours and metastatic disease62,63. In addition, FISH-based assessment of the tumour suppressor gene PTEN, which frequently shows subclonal copy-number loss, has contributed to our understanding of the order of genomic events occurring in prostate cancer64. However, FISH can be cumbersome to interpret and requires the use of fluorescence microscopy, thus limiting throughput and potential widespread clinical implementation.
The use of antibodies that can detect oncogenes or tumour suppressors by immunohistochemistry has greatly improved our ability to query genomic alterations directly in tissues. For instance, immunolabelling with antibodies to ERG has been shown to correlate closely with ERG rearrangement status and can therefore be used as a read-out to screen for ERG+ lesions26,65,66. Combination immunostains of ERG and SPINK1, which is expressed in a subset of ERG− tumours, can highlight clonally distinct tumour cell populations directly on standard pathology slides67. Similarly, antibodies to PTEN, p53 and RB1 can be used to highlight clonal and subclonal changes to these important tumour suppressor genes68–70. Therefore, genomic alterations can be visualized in the tumour tissue using surrogate immunohis-tochemistry markers that are strongly associated with certain genetic alterations (FIG. 2). Genetically validated clinical grade immunohistochemical assays, in particular for targets that have potential as clinical prognostic and predictive biomarkers such as PTEN and TP53 alterations, represent extremely useful ancillary diagnostic tools that can easily be implemented in clinical practice68. The major advantages of such in situ methods is that they require a minimal amount of tumour tissue, allow correlation of molecular changes with tumour morphology and can be embedded in already existing workflows in clinical laboratories, ensuring rapid and robust results.
Sequencing approaches.
Although early evidence documenting the clonal heterogeneity of prostate cancer was drawn from the analysis of polymorphic microsatellite markers and comparative genomic hybridization of microdissected tumour foci19,71–73, these technologies have been almost completely replaced by massively parallel sequencing approaches. With increasing accessibility to next-generation sequencing, numerous studies have investigated clonal and subclonal heterogeneity in prostate cancer by using multiregional sampling followed by whole-exome or whole-genome sequencing. All of these spatial genomics studies have in common that multiple tumour foci are macro- or micro-dissected and isolated DNA is subjected to sequencing. Extensive efforts in this space have provided definitive evidence that, in a subset of patients, individual tumour foci do not share any driver gene alterations and likely arose as independent clones, confirming the multiclonal nature of primary prostate cancers27,47,57,74,75. In addition, by using deep sequencing and clustering of variants by their mutant allele fraction, the presence of subclonal populations of cells can be inferred, providing insight into the temporal subclonal evolutionary relationships between different tumour regions28,33,34,55,56,76. Phylogenetic trees can be reconstructed that depict the clonal evolution of truncal and unique alterations34,53,77. Importantly, such analyses provide insights into the origin and timing of cancer evolution and have suggested that early driver gene alterations can occur decades before clinical manifestation and diagnosis34,78,79. Although these approaches allow inferential assessment of clonal evolutionary relationships and macroheterogeneity from bulk tumour sequencing data, they are limited by sequencing depth and the sensitivity of calling algorithms, which are prone to biases48,56.
Single-cell sequencing and other emerging technologies.
Several single-cell analysis platforms have been developed to study microheterogeneity at the level of individual cells, and these tools have been applied in small-scale studies of prostate cancer80–82. Although technically challenging, these extremely powerful approaches have the potential to provide a more comprehensive picture of the diversity of tumour cell populations and enable precise delineation of the clonal dynamics in therapeutic response and emergence of resistance83,84. Such single-cell technologies enable detailed analysis of the cellular tumour composition, genomic alterations and epigenetic states85,86. However, these ex situ approaches rely on dissociation of tumours to single cells, which disrupts tissue architecture. Novel in situ approaches such as digital spatial profiling technologies permit the assessment of mRNA and protein expression by highly scalable next-generation sequencing read-outs directly from tumour tissue sections87,88. The great power of these in situ approaches lies in the preservation of tissue architecture, which allows the cross-referencing of molecular findings with detailed topographical and histomorphological information. Therefore, these tools will be perfectly complementary to single-cell sequencing platforms to understand intra-tumoural heterogeneity.
A major limitation of all of these sequencing-based technologies is that they mostly rely on the assessment of alterations at the RNA or DNA level. However, evidence is growing that transcriptomic, genomic and epigenetic studies are often not adequate for accurate prediction of protein expression89,90. Therefore, efforts have been focused on developing high-throughput proteomics approaches that permit the robust detection of protein expression changes in clinical specimens, including formalin-fixed, paraffin-embedded samples89–91. Application of these approaches has already yielded important new insights into the proteogenomic heterogeneity of prostate cancer and will further improve our ability to determine biologically relevant alterations92.
Primary prostate cancer
Anatomical considerations.
The prostate can be divided into distinct zones93. The peripheral zone, which constitutes the majority of the glandular tissue of the prostate, harbours most cancers, whereas the central and transition zones have a much lower rate of cancer incidence59,94. These anatomically defined zones show distinct gene expression profiles, and lesions arising in different zones show different propensities for certain genomic alterations95. For instance, ERG translocations are common in peripheral zone tumours but rarely seen in lesions arising from the transition zone96. Notably, racial differences have also been associated with distinct zonal distributions of tumours97. These findings suggest that the anatomical site and zone in which tumours arise might influence the spectrum of genomic alterations and potentially the tumour phenotype, providing evidence that tumour location can contribute to tumour heterogeneity.
The morphological spectrum of primary prostate cancer.
Primary prostate cancers can display a broad range of morphological features and growth patterns, and the appreciation that certain morphologies are associated with distinct patterns of molecular alterations and biological behaviour is increasing98–101. Although the grading system for prostate cancer has been revised several times over the past few decades, the Gleason score remains the most important risk assessment tool for clinical decision-making in localized prostate cancer. Its ingenuity lies in the integration of architectural heterogeneity into a composite score that captures the morphological heterogeneity of prostate cancer98. Importantly, several studies have shown that the number of genomic alterations and the frequency of driver gene changes increases in higher grade tumours69,102,103. Given the heterogeneity of Gleason patterns, the question arises whether morphologically distinct growth patterns in a given tumour focus (for example, Gleason pattern 3 versus Gleason pattern 4) are clonally related. Analyses demonstrate that at least a subset of synchronous adjacent Gleason pattern 3 and Gleason pattern 4 lesions are clonally linked and share initiating driver alterations but show subclonal branching evolution104–108. These findings indicate that Gleason pattern 3 and Gleason pattern 4 lesions can arise from a common ancestral clone but follow divergent genomic and morphological trajectories.
The multifocal nature of prostate cancer.
More than 80% of prostate glands harbouring cancer have two or more topographically and morphologically distinct tumour foci19,109 (FIG. 3). The reported numbers of individual tumour foci in a given prostate are not very consistent, probably due to different definitions of a focus and the challenges in comprehensively sampling the entire prostate in an attempt to detect every tumour focus110. Further complicating the assessment of individual tumour foci is the complex growth pattern of primary prostate cancer, which lacks a well-defined invasion front and shows diffuse infiltrative growth98,110,111. Whereas some tumour foci seem to collide and form large, morphologically indistinguishable tumour masses111, other patients (as shown in FIG. 3) show completely non-contiguous tumour foci with distinct morphological features and histological grades19,20.
Fig. 3 |. Multifocal prostate cancer.
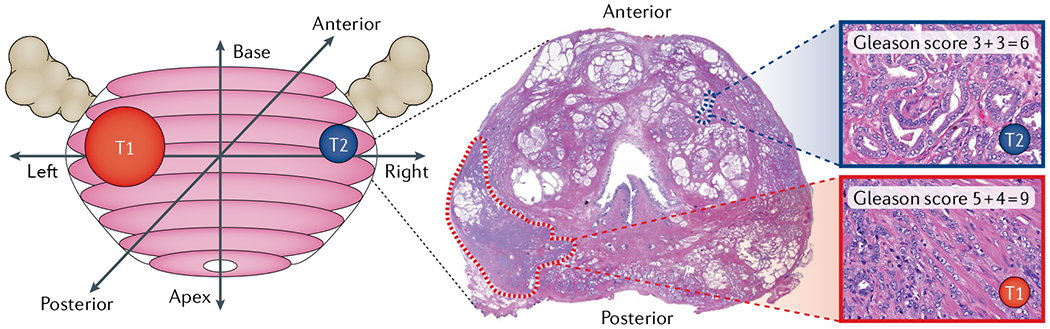
Reconstruction and whole-mount cross-section of a radical prostatectomy specimen with two distinct tumour foci. Note that the larger tumour focus located in the left posterior prostate shows high-grade morphology and extraprostatic extension (Gleason score 5 + 4 = 9), whereas a smaller anterior tumour appears well differentiated (Gleason score 3 + 3 = 6).
Genomic and epigenetic evidence of multiclonal prostate cancer.
Although the multifocal nature of primary prostate cancer suggests a potential polyclonal origin, detailed genomic studies have only recently provided convincing evidence that the majority of topographically distinct tumour foci show non-overlapping mutation profiles, suggestive of an independent clonal origin27,57,62,71,112,113. Multiregional sampling and whole-exome sequencing demonstrated that tumour foci from 3 out of 4 patients with primary prostate cancer showed no overlapping genomic changes57. This finding was further corroborated by whole-genome sequencing of 23 distinct tumour regions in 5 patients, which showed that a given patient can harbour two separate prostate cancers that share no relevant genetic alterations25. In the largest study to date, 2–3 distinct tumour foci were analysed in 41 patients using whole-exome sequencing. Strikingly, 76% of multifocal tumours showed nonoverlapping mutations, including known driver gene alterations, in topographically distinct tumour foci27. Taken together, a large body of literature strongly supports the notion that multiple clonally independent lesions can be present in a diseased prostate gland.
In addition to genomic changes, DNA methylation alterations have been shown to co-segregate with copy-number changes, suggesting that genomic and epigenomic events can evolve on a shared clonal trajectory114. Studies analysing the pattern of DNA methylation alteration in multifocal prostate cancer revealed striking differences in methylation profiles, demonstrating that lesions with distinct genomic alterations also show unique methylation patterns115–117. These studies confirm that DNA methylation alterations can be used to infer clonal relationships and dynamics114–116. These observations pave the way for future studies, which should aim to understand the diversity of other epigenome marks in the context of multiclonal prostate cancer in greater detail117.
Challenges of multiclonality
Challenges of assessing multifocality using biopsies.
Assessing the full clonal and subclonal heterogeneity of prostate cancer is theoretically possible when evaluating a radical prostatectomy specimen. However, multifocality and multiclonality represents a major challenge for prostate cancer diagnosis on needle biopsy samples. Using a dual immunohistochemistry approach for ERG and SPINK1, it was shown that ~25% of biopsies with noncontiguous core involvement sample at least two clonally independent tumours, with the remaining 75% sampling the same tumour going in and out of the plane of section of the biopsy118. This finding highlights the complexity of assessing multifocal or multiclonal disease on core biopsies but also shows that simple immunohistochemical tools provide robust and cost-efficient ways of assessing clonality on standard diagnostic biopsies. Although very powerful for research studies, comprehensive immunohistochemical evaluation of all tumour-containing biopsy samples in an attempt to catalogue clonal heterogeneity is not feasible for routine clinical use. Using a targeted sequencing approach, the heterogeneity of genomic alterations in biopsy samples and matched radical prostatectomy samples has been assessed. On average, only 19% of the mutations detected in the prostatectomy specimen were represented in matched preoperative biopsies119. Although technical improvements can potentially refine biopsy sampling, this finding implies that systematic needle biopsies are probably inadequate for detecting all relevant spatially and molecularly distinct tumour areas. This observation has important implications for clinical practice in which primary tumour samples are often used to make decisions about actionable alterations in distant metastases120. Given the high level of clonal heterogeneity, primary tumour samples need to be selected carefully for genomic studies and, in some patients (where possible), multiregional sampling or direct biopsy of the metastatic lesion should be performed.
Undersampling of high-grade lesions with aggressive molecular features is of particular concern in the setting of active surveillance. For instance, in patients in whom clonally and molecularly distinct tumours with different biological potentials to metastasize can coexist, undersampling of a more aggressive lesion might cause treatment delays and loss of curative opportunities121. Multiparametric MRI, which integrates perfusion, diffusion and anatomical imaging, has greatly improved the accuracy of image-guided tumour mapping122. The widespread use of MRI-guided biopsies has improved the detection rate of high-grade, clinically significant cancers and improvements in imaging approaches are expected to further augment the accuracy of diagnosis123,124. One of the greatest opportunities for MRI and other functional imaging approaches lies in the ability to cross reference in vivo imaging data with molecular tumour features. The identification of imaging characteristics associated with a high risk of clinically significant disease that are functionally linked to defined molecular alterations will eventually enable the detection of molecular intra-tumour heterogeneity and provide an exciting new intersection between radiology and pathology125. Paired with molecular imaging, these approaches will greatly improve tissue sampling and augment the accuracy of precision medicine in localized prostate cancer123,126.
Multifocality as a barrier for biomarker development and implementation.
The multiclonal nature of prostate cancer represents a leading factor contributing to the underperformance of current diagnostic paradigms and molecular biomarkers. For instance, in patients with comprehensive profiling of all tumour-containing material, high degrees of heterogeneity of DNA ploidy and PTEN status were noted in up to 75% of patients18. Along these lines, the high level of molecular heterogeneity poses a major challenge for the implementation of commercial gene expression assays that are aimed at guiding clinical decision-making127,128. Although these expression signatures provide prognostic information and correlate well with tumour grade within a sample, patients with multifocal prostate cancer harbouring both high-grade and low-grade lesions represent a challenge. Expression signatures are usually determined from a single biopsy core that shows the highest Gleason grade. However, risk scores can vary greatly between different tumour foci, and the assessment of a single focus might not capture the complex heterogeneity present in primary tumours129. In addition, in patients with multifocal prostate cancers, analysis of a lower grade component cannot be extrapolated to adjacent high-grade lesions130. Therefore, regardless of the downstream analysis, sampling of different tumour lesions in the setting of multifocal prostate cancer remains a major challenge. The increased use of neo-adjuvant therapies for high-risk patients has provided a unique opportunity to delineate resistance mechanisms and analyse the clonal and subclonal dynamics in primary tumours following androgen deprivation therapies131. For instance, a 2020 study revealed significant differential responses to systemic therapies in clonally and morphologically distinct tumour foci in the primary tumour and therefore highlights the relevance of inter-focal heterogeneity for clinical care132.
Molecular definition of the index lesion.
Molecular studies have suggested that even small well-differentiated lesions can seed distant metastases29,133. For instance, our group tracked the origin of the lethal metastatic cell clone to a 2.2-mm well-differentiated (Gleason pattern 3) component in the primary tumour in a patient who died of prostate cancer. This lesion harboured a large number of the driver gene changes present in the distant metastasis including mutations in TP53, PTEN and SPOP, which were distinct from the surrounding primary tumour mass29. Although further studies are needed, this observation calls into question the concept of the index lesion as the tumour nodule defined by size and grade and suggests that molecular characterization of such lesions could provide additional insights into what should be considered the dominant clone29,134. This could be particularly relevant for focal therapy approaches that intend to ablate index lesions in a targeted manner to reduce the morbidities ensuing from other forms of treatment for localized disease135. However, it is important to note that in our study this well-differentiated component was present within a larger lesion that was a Gleason score 4 + 3 = 7 tumour29. Although metastases from an isolated Gleason score 3 + 3 = 6 tumour are exceedingly unlikely136, evidence suggests that a Gleason pattern 3 component that is present contiguously with a Gleason pattern 4 tumour (3 + 4 = 7 or 4 + 3 = 7) is molecularly different from a Gleason pattern 3 that is only part of a Gleason score 3 + 3 = 6 tumour137,138.
Regional lymph node metastases
Metastases to the lymph nodes (which can be detected in a subset of men at the time of radical prostatectomy) have been associated with adverse prognosis139. The prevailing hypothesis to explain this association suggests that distant metastases are seeded by lymph node metastases. However, clinical data show that ~30% of men with prostate cancer metastatic to regional lymph nodes are free from recurrence at 10-year follow-up even if left untreated after surgery140. In addition, mounting molecular evidence suggests that in different tumour types, draining lymph nodes represent stopping points rather than spring boards from which more distant metastases are launched141. For instance, in colorectal cancer, phylogenetic reconstruction of clonal relationships between primary tumours, lymph node metastases and distant metastases showed that in 65% of cases, lymphatic and distant metastases arose from independent subclones in the primary tumour142. In prostate cancer, the genomic relationship of matched primary tumour, lymph node and distant metastases has not been systematically studied and only limited data exist on the clonal relationship between primary and metastatic lesions. In a case report of a patient who died of metastatic prostate cancer, we showed that a lymph node metastasis that was present at the time of initial presentation showed no overlap in driver gene alterations with the lethal metastases sampled at the time of death29. In addition, a study comparing copy-number alterations of primary prostate tumours and pelvic lymph node metastases showed that in a substantial fraction of cases (23.3%) the index primary tumour was not clonally related to the locoregional lymph node metastases133. Furthermore, clonal analyses suggest that lymph node metastases often originate from evolutionarily advanced subclones located at the periphery of the prostate gland, in a process that seems to be distinct from clones seeding osseous metastases143. Collectively, these studies suggest that regional lymph node metastases can arise from clonally distinct tumour cell populations and do not necessarily contribute to progression to metastatic disease.
Distant metastatic disease
Processes shaping the clonal constellation in metastases.
The fitness of a clone to form metastases is not only determined by its ability to shed cells into circulation but also by its capability to colonize, expand and eventually develop resistance to systemic therapies144–146 (FIG. 1). In prostate cancer, metastases can take decades to emerge after initial diagnosis of localized disease12,15,147. However, once metastases are established, disease progression is greatly accelerated, suggesting that the early phases of metastasis formation represent a major bottleneck6. Large-scale analyses across solid tumour metastases suggest that the vast majority of metastatic lesions show subclonal homogeneity148. In particular, analyses of treatment-naive metastases have shown that the majority of driver gene alterations are shared between different metastatic sites36,149. By contrast, however, other reports provide evidence of early branching evolution during metastatic dissemination and extensive subclonal heterogeneity in pan-cancer analyses. These findings suggest the presence of widespread and potentially clinically relevant phenotypic and genomic differences between different metastatic sites28,30,77,150. This strikingly divergent assessment of the clonal composition of distant metastases highlights the need for additional studies in this field and probably reflects major differences in the analytical approaches and study cohort characteristics. For instance, the vast majority of studies demonstrating clonal homogeneity have used bulk tumour sequencing of a single metastatic site, whereas studies showing heterogeneity used deep-sequencing approaches and sampled multiple metastatic deposits28,30.
Treatment history might also influence the genomic composition of a tumour. Systemic cancer therapies exert a strong selection pressure that can shape the clonal constellation of a tumour151 (FIG. 1). After therapy, residual tumours might exhibit an altered clonal composition with newly emerging subclones that harbour alterations conferring resistance51,152. Particularly relevant for prostate cancer therapy is the crucial dependence of prostate cancer cells on the androgen receptor (AR) as a core lineage oncogene. Therapies that block AR signalling, which were arguably among the first targeted therapies in oncology, remain the mainstay of therapy for metastatic disease153. However, resistance to AR blockade and progression to metastatic castration-resistant prostate cancer (mCRPC) emerges in almost all patients153. Resistance to androgen deprivation is often associated with genomic alterations of the AR locus, including copy-number gains and gain-of-function mutations. Although very common in mCRPC, these changes are almost never detected in hormone-naive patients, highlighting the strong association between certain genomic alterations and the patient’s treatment history56,154. Second-generation therapies that further suppress either ligand synthesis (for example, abiraterone acetate) or binding to AR (such as enzalutamide) have been shown to be effective in suppressing AR signalling further but can select for clones that show resistance mechanisms that bypass the need for AR signalling for cancer cell growth155. Notably, AR signalling has been shown to regulate a number of key DNA repair genes and AR inhibition is associated with decreased DNA repair activity156,157. Therefore, blockade of the AR signalling axis could potentially result in decreased genomic integrity and contribute to intra-tumoural heterogeneity. More broadly, the contribution of AR blockade to the emergence and selection of subclones with AR alterations, and the potential interference with DNA repair activity, provides a rationale for a combination of AR-targeted therapies and other cytotoxic treatments early in the progression of high-risk patients158.
Morphological and phenotypic heterogeneity in mCRPC.
Metastatic prostate cancer has a wide spectrum of histomorphologies, including features commonly seen in adenocarcinomas such as acinar, cribriform, single-cell and solid growth patterns, but also morphologies consistent with poorly differentiated or anaplastic carcinomas and neuroendocrine differentiation (carcinoid-like pattern and small-cell carcinoma)159–162 (FIG. 4). Morphological patterns can vary greatly between different patients and also show a high level of inter-tumoural heterogeneity within an individual patient. Different morphologies can be present even at a single metastatic site159–162 (FIG. 4g). These diverse histomorphological features suggest profound differences in cellular architecture, metabolism and epigenetic programmes. Although evidence exists that some tumour morphologies are associated with distinct expression profiles, additional studies are needed to investigate the relationship between histomorphological features and the underlying molecular alterations31,163.
Fig. 4 |. Morphological heterogeneity in mCRPC.
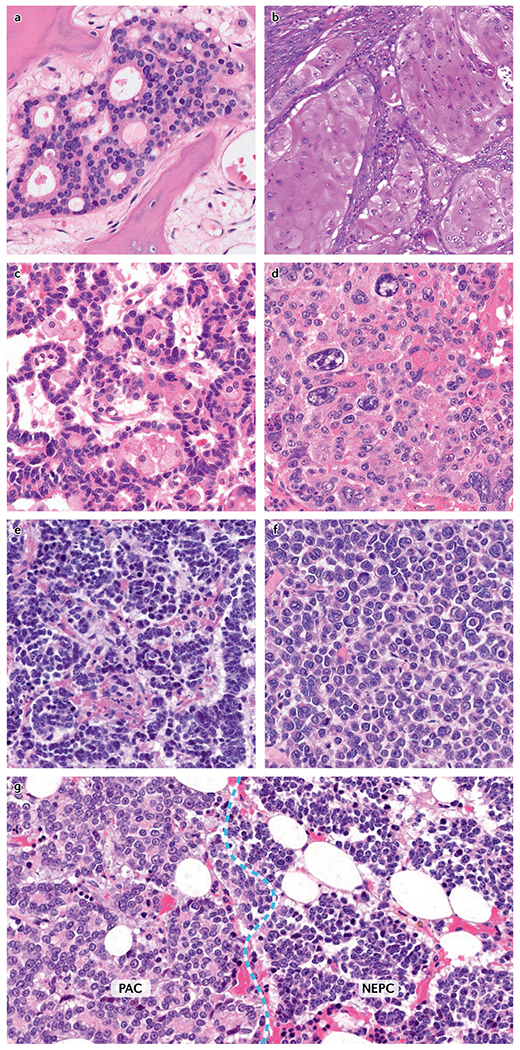
a | Adenocarcinoma with cribriform architecture. b | Adenocarcinoma with squamous differentiation. c | Carcinoid-like differentiation in a neuroendocrine-marker-positive carcinoma. d | Poorly differentiated carcinoma with pleomorphic giant cells. e | Small-cell carcinoma. f | High-grade carcinoma with discohesive cells. g | Hybrid lesion composed of prostatic adenocarcinoma (PAC) with cribriform morphology (left) and small-cell neuroendocrine prostate cancer (NEPC) (right). mCRPC, metastatic castration-resistant prostate cancer.
In contemporary mCRPC cohorts, the majority of metastatic lesions retain some level of prostatic lineage differentiation characterized by the expression of prostate epithelial markers such as AR, NKX3.1 and PSA154,155,159,160,163. However, treatment-associated changes in cellular differentiation have emerged as an important resistance mechanism to AR-directed therapies31,155. Loss of prostate epithelial marker expression (in particular AR) and in some cases gain of neuroendocrine differentiation have been observed in mCRPC155,163. These observations suggest that in response to therapy a subset of tumours can show alterations in prostatic lineage commitment and an increased phenotypic plasticity155,164. On the basis of these findings, metastatic prostate cancers can be classified into subgroups, defined by the expression of AR, AR target genes and neuroendocrine differentiation markers. In addition to tumours with high AR activity, other lesions show low or no AR activity in the absence of neuroendocrine marker expression (double-negative prostate cancers), co-expression of AR and neuroendocrine markers (amphicrine or adenoneuroendocrine prostate cancer) or show loss of AR expression and gain of neuroendocrine differentiation (neuroendocrine prostate cancer, NEPC), with the added complexity of tumours transitioning between these defined phenotypes155,163. As variation exists even within these molecularly defined groups, further studies will probably result in additional refinement and subclassification.
Owing to its more aggressive biological behaviour, NEPC has been the focus of numerous studies over the past few years164. NEPC is characterized by loss of AR activity and the expression of neuroendocrine markers such as synaptophysin, chromogranin A, CD56 and INSM1, along with morphological features of small-cell carcinoma or high-grade neuroendocrine carcinoma162,165,166. The study of NEPC is complicated by the focal presence of neuroendocrine differentiation immunohistochemically in ‘usual’ acinar prostate cancer without any clinical relevance162. Recognition is also growing of hybrid tumours that have features of transdifferentiation from usual to neuroendocrine phenotype. At the molecular level, NEPC is enriched in genomic alterations in RB1 and TP53 but also displays striking epigenetic changes, suggesting that NEPC might evolve through subclonal epigenome evolution31,164,167. In cohorts of patients with advanced mCRPC, evidence for NEPC can be found in up to 15–20% of cases155,161,167. However, owing to differences in the clinical and treatment history, and the criteria applied to define neuroendocrine transdifferentiation, some variability exists in the frequency of NEPC reported in different mCRPC cohorts154–161. Therefore, although the subclassification of mCRPC will probably be of great relevance for clinical trial design and ultimately clinical practice, a set of consensus molecular markers and morphological features that enable the distinction of these phenotypic subgroups needs to be defined. To this end, a carefully selected set of biomarkers will need to be validated across different institutions.
Monoclonal origin of lethal distant metastases.
Given the multifocal and multiclonal nature of primary prostate cancer and the often heterogeneous phenotypes observed in anatomically distinct metastases, the question arises whether multiple clones of the primary tumour contribute to the distant metastatic disease burden upon metastatic dissemination (polyclonal seeding) or whether only one primary clone seeds all distant metastases (monoclonal seeding)168. Early studies focusing on the analysis of single gene alterations provided evidence for the monoclonal origin of distant metastases63,169,170. More recently, several whole-exome and whole-genome sequencing studies have demonstrated that anatomically distinct distant metastases share a large number of genomic alterations, confirming that most likely a single clone in the primary tumour gives rise to all distant metastases28–30,114,171–173 (FIGS 1,5).
Fig. 5 |. Schematic of scenarios of clonal evolution of metastatic prostate cancer.
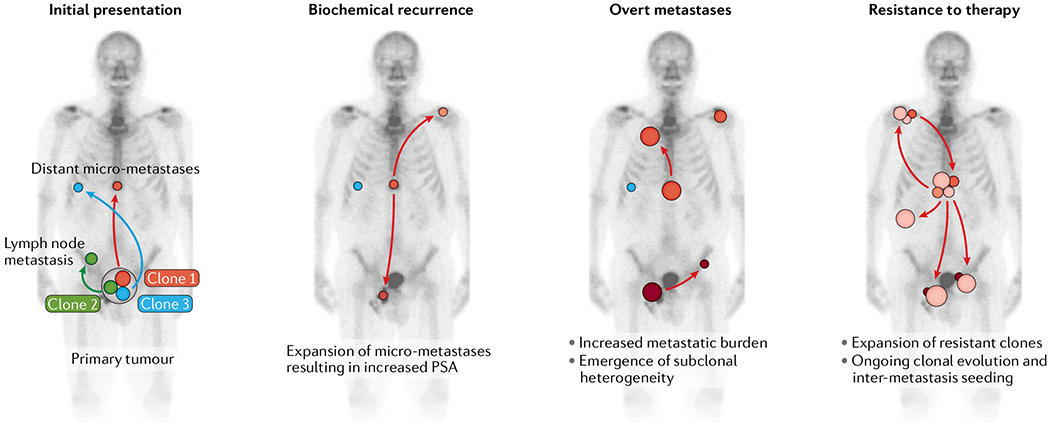
Initial presentation: primary prostate cancers often harbour more than one tumour clone. Distinct clones can have different metastatic potential. Although clone 1 and clone 3 can seed distant micro-metastases, clone 2 is restricted and cannot progress beyond the formation of nodal metastases. Biochemical recurrence: after surgical resection of the prostate, the expansion of micro-metastatic tumour deposits results in increased PSA levels. Note that at this stage, it is possible that more than one clone from the primary tumour can contribute to the pool of the distant micro-metastases. Although some clones expand and seed additional metastases (clone 1), other clones stay dormant or show minimal expansion (clone 3). Overt metastases: as the size of individual metastases increases, the subclonal compositions of the tumours broaden and additional metastases are seeded. Resistance to therapy: systemic therapy results in a re-sculpting of the clonal composition of distant metastases. Clone 3 and several subclonal lesions of clone 1 are effectively eliminated or greatly reduced in their size. A new subclone that arose from clone 1 is therapy resistant, expands and further disseminates. Individual subclones that arise in distinct metastases seed to other sites (inter-metastasis seeding), thereby contributing further to the inter-tumour and intra-tumour heterogeneity.
Homogeneity versus heterogeneity.
Analyses of multiple metastatic deposits at the genomic, epigenetic and transcriptomic levels from men who died of prostate cancer have revealed a high concordance in driver genomic and epigenetic alterations between different metastatic sites in a given patient114,172,173. These findings, which are all based on bulk sequencing and array-based assays, demonstrate limited inter-tumoural differences, which probably do not greatly affect clinically relevant tumour phenotypes114,172. However, deep sequencing combined with bioinformatic reconstruction of subclonal architectures has revealed more complex patterns of subclonal diversity between different metastases from distinct anatomical sites28,30. These studies have demonstrated that individual subclones can seed from one metastasis to another in a polyclonal fashion. Therefore, every metastatic site can be considered a conglomerate of different subclones (FIG. 5). For instance, AR alterations are rarely truncal and different subclones show distinct AR alterations, suggesting convergent evolutionary processes that activate AR signalling28. Detailed phylogenetic reconstructions demonstrate that independent subclones in the primary tumour or metastases can seed to the same metastatic site in distinct waves, indicating that early metastatic dissemination can potentially pave the way for future seeding events28,30. This finding is clinically relevant, as it supports the notion that surgical removal of the primary tumour or ablation of oligometastatic tumour deposits can potentially eliminate sources for additional seeding events and therefore result in improved oncological outcomes in patients with metastatic disease174,175.
Although the number of patients included in these detailed sequencing studies is currently low, it is of interest that the extent of shared truncal genomic alterations between different subclones present in a given patient can vary greatly (range 40–90%)28. An intriguing hypothesis potentially explaining this observation is that truncal mutations in some patients might confer a maximum level of fitness such that additional mutations do not result in further improvements. Other tumours, however, might start out with a founder clone with limited fitness, necessitating the additional accumulation of driver alterations, thus resulting in increased subclonal diversity48. In support of this notion, a study demonstrated that tumours have less genomic subclonal heterogeneity as patients progress towards NEPC in later stages of the disease167. Therefore, clinically more aggressive disease can potentially appear clonally more homogeneous with a high level of shared genomic and epigenetic alterations167. This finding emphasizes the importance of the timing of sampling and suggests that as subclones evolve, the fittest subclonal population can take over the majority of the tumour burden. Such clonal sweeps will result in more homogeneous cell populations (FIGS 1,5). In earlier stages of tumour progression, however, a higher level of subclonal heterogeneity might prevail. Through the passage of clonal bottlenecks, this heterogeneity will become more homogeneous. Therefore, longitudinal sampling efforts are necessary to capture the dynamically changing genomic and epigenomic features of mCRPC167,176.
Assessing inter-tumoural heterogeneity in mCRPC.
Historically, the complex heterogeneity of metastatic disease has been difficult to study, mostly owing to major challenges in obtaining samples from multiple metastatic sites. In addition, comprehensively sampling bone lesions remains a major challenge. A growing number of studies have assessed the molecular alterations in mCRPC from biopsy specimens154,177–179. However, many of the current insights into the biology of mCRPC were derived from rapid autopsy studies, in which multiple metastases are sampled at the time of death28,155,159,160,172,173. One limitation of these studies is that they only provide end-point assessments of heavily pretreated patients and might therefore not be representative of earlier stages of the disease. In addition, most genomic assessments of mCRPC have been restricted to bulk sequencing of tissue samples and data are currently limited on microheterogeneity in mCRPC tissue. However, single-cell sequencing efforts of circulating tumour cells from patients with mCRPC have revealed diverse copy-number profiles, suggesting a high level of genomic heterogeneity at the single-cell level that is not adequately captured by analyses of matched biopsy material180. Given that the level of microheterogeneity is proportional to the size of the metastatic lesion, bulky metastases have an increased chance of harbouring intrinsically resistant cells. This notion provides a rationale for the use of cytoreductive therapies earlier in disease progression36. Indeed, several trials have reported improved outcomes in patients with prostate cancer when hormonal or chemotherapeutic agents were introduced at earlier stages of the disease158,181.
Most studies assessing inter- and intra-tumoural heterogeneity in mCRPC have focused on genomic alterations28–30,171–173. However, interest is growing in profiling epigenome changes and genome-wide chromatin interaction profiles of AR and other key transcription factors in advanced prostate cancer182,183. Epigenetic modifications have been implicated as key drivers of phenotypic plasticity, and the evolution of tumour subclones is probably defined by interactions between genetic and epigenetic factors184. Studies assessing epigenetic intra-tumoural heterogeneity have thus far primarily focused on DNA methylation alterations and have demonstrated that despite an overall high level of clonal conservation of DNA methylation alterations between different metastases, evidence exists for divergent epigenome alterations during tumour progression31,114,167. The potential reversibility of epigenetic changes and the expanding portfolio of drugs that target epigenetic pathways should encourage future research in this field183.
Transcriptional profiling of autopsy specimens has revealed a strong overlap in transcriptional programmes in anatomically distinct metastases114,172. Similarly, phosphoproteomic analyses have demonstrated high inter-individual, but limited intra-individual, heterogeneity at the level of kinase pathway activation185. These studies suggest the conservation of transcriptional and post-translational protein modification patterns across different metastases in most patients with end-stage mCRPC172,185. However, a study investigating the transcriptomic profiles of multiple metastatic sites in 34 patients revealed 5 cases (14.7%) of substantial inter-tumoural heterogeneity at the transcriptomic level163. Inter-tumoural differences have also been observed at the protein expression level for AR and RB1 in a subset of patients186,187. Collectively, these results suggest that although the vast majority of mCRPC lesions in a given patient might show overlapping molecular characteristics, a subset of patients exhibit inter-tumoural heterogeneity in pathways that are relevant for prostate cancer therapy. Notably, however, mRNA and protein-based assays, in particular, are susceptible to pre-analytic changes, which can potentially contribute to major differences in analyte abundance between samples for different anatomical sites. Therefore, RNA sequencing and immunohistochemical data need to be interpreted with this potential bias in mind.
Clinical challenges of heterogeneity in mCRPC.
During the clinical management of mCRPC, different subclones can emerge and disappear, depending on the effectiveness of therapies167,188,189. As described for other tumour types, subclonal evolution can be greatly influenced by therapy24,36,43,51. Every effective therapeutic intervention will restrict and alter the clonal composition of a tumour, eventually resulting in the emergence of resistant clones that can withstand the selection pressure imposed by therapy. Small populations of pre-existing therapy-resistant subclones can re-populate the entire tumour burden, even if the vast majority of tumour cells are sensitive to the therapy24,190. In colorectal cancers, for instance, pre-existing low-frequency mutations in KRAS are strongly selected for and contribute to resistance to EGFR blockade190. In addition, adaptive epigenome changes are thought to contribute to the resistance phenotype31,191. Therefore, therapy resistance is probably driven by an interplay of the selection of pre-existing cell populations and de novo adaptation processes24.
A body of literature supports the notion that truncal alterations predict immediate response to targeted therapies, whereas subclonal alterations can be a major driver of resistance that emerges in response to therapy24,36,190. Although the assessment of a single tumour tissue biopsy sample might miss a subset of alterations, the implications for initial treatment decisions are probably limited. Importantly, genomic alterations that are associated with initial clinical responses to targeted therapies in prostate cancer, such as mutations in DNA repair genes (including BRCA2 and ATM) and mismatch repair genes, have been shown to be truncal and shared between different metastatic sites56,154,172,192. Therefore, a single biopsy sample should be adequate for determining a given patient’s mutation status for such truncal events. On the other hand, a single biopsy sample will probably be insufficient to monitor subsequent resistance mechanisms that emerge in response to targeted therapies193. Particularly relevant for prostate cancer are alterations that change the response to AR-targeted therapies, including genomic changes of the AR locus, which have been shown to occur in a subclonal manner188.
Apart from genomic biomarkers, interest in assessing morphological and other phenotypic changes in mCRPC biopsy tissues is increasing. Given the inter-tumoural heterogeneity in histomorphological features and transcriptomic findings outlined above, a single biopsy might not capture these features adequately. This problem is highlighted by a study that showed discrete regions of NEPC and adenocarcinoma even within the same core needle biopsy in 7 out of 27 patients with NEPC (26%)161. As the presence of neuroendocrine phenotypes has been shown to be associated with a lack of response to AR-targeted therapies, identifying such heterogeneous cell populations early during disease progression is important161,167.
Delineating longitudinal changes.
mCRPC is a dynamic disease necessitating the development of tools for longitudinally monitoring the evolving tumour burden. Serial metastatic tumour biopsies are invasive for patients, technically challenging and do not allow (as described above) a comprehensive sampling of all metastatic sites. Therefore, alternative methods that capture tumour heterogeneity in a less invasive and cost-effective manner are needed. Liquid biopsy strategies, which involve the assessment of circulating plasma cell-free DNA (cfDNA), circulating tumour cells and other analytes, have been developed over the past few years and applied to mCRPC176,194,195. Genomic analyses of cfDNA have been used to investigate the dynamic evolution of AR alterations and provided evidence that resistant subclones can be detected in circulation even before clinical evidence of disease progression under therapy188,189,196. cfDNA-based biomarkers can therefore potentially expand the critical window for therapeutic intervention and provide valuable information on subclonal dynamics167,176,188,190.
Complementary to blood-based assays, functional imaging applications can capture inter-tumoural heterogeneity in mCRPC at the macroscopic level197,198. For instance, a retrospective series showed heterogeneous imaging findings in response to systemic therapies in up to 40% of patients and the study documents the feasibility of using molecular imaging to decipher functional inter-tumoural heterogeneity197. Integration of these new technologies will be increasingly important in clinical practice and will ultimately improve patient care.
Conclusions
Over the past decade, our understanding of the molecular and phenotypic heterogeneity in prostate cancer has matured. Through extensive profiling studies we have learned about the complex constellation of genomic diversity in primary tumours and metastases. In parallel, methods for delineating the heterogeneity of molecular alterations have greatly improved and become more accessible. The clinical challenges posed by the complex molecular heterogeneity of prostate cancer will require a multidisciplinary approach. Insights from imaging and molecular pathology studies will need to be paired with careful clinical annotations to establish features of clinically relevant heterogeneity. In addition, novel computational approaches and deep learning algorithms will be necessary to analyse the resulting multidimensional data. Ultimately, the integration of different levels of information that capture disease heterogeneity will provide an in-depth understanding of tumour biology and will allow us to overcome obstacles to precision medicine posed by intra-tumoural heterogeneity.
Key points.
Primary prostate cancers are often multifocal with spatial and morphologically distinct tumour foci.
Individual tumour foci can show non-overlapping truncal genomic alterations, suggesting that multiple clonally distinct cancers can arise in a given patient.
Intra-tumoural and inter-tumoural heterogeneity present within the prostate gland poses diagnostic challenges.
Despite the multiclonality of primary cancer, clonal bottlenecks imposed by the metastatic process and further by therapeutic interventions seem to select for a single dominant clone in lethal metastatic prostate cancer.
Acknowledgements
The authors thank C. Morrissey (University of Washington), T. Lotan (Johns Hopkins School of Medicine) and W. B. Isaacs (Johns Hopkins School of Medicine), as well as members of the Haffner, Yegnasubramanian and Nelson laboratories, for valuable discussions and suggestions on the manuscript. This work of the authors is supported by the NIH/NCI (P50CA097186, P50CA58236, U01 CA196390, P30 CA006973, R01CA183965), the US Department of Defense Prostate Cancer Research Program (W81XWH-20-1-0111, W81XWH-18-1-0406, W81XWH-18-2-0015), the Prostate Cancer Foundation, the Safeway Foundation, the Commonwealth Foundation and the Irving Hansen Memorial Foundation.
Competing interests
S.Y., W.G.N. and A.M.D.M. are paid consultants to and received sponsored research funding from Cepheid. S.Y. and W.G.N. are co-inventors of intellectual property describing the use of DNA methylation changes as prostate cancer biomarkers and are eligible to earn royalties related to the future sale of any products using those technologies. S.Y. and A.M.D.M. receive sponsored research funding from Janssen. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. The other authors declare no competing interests.
Glossary
- Inter-patient heterogeneity
Differences in tumour genotypes and phenotypes between individual patients.
- Intra-tumoural heterogeneity
Genomic, epigenetic, transcriptomic and phenotypic differences within a tumour mass.
- Inter-tumoural heterogeneity
Differences between anatomically distinct tumour sites within a given patient.
- Multifocality
Spatially distinct and often histomorphologically different tumour lesions within one affected organ.
Footnotes
Peer review information
Nature Reviews Urology thanks Mark Rubin, Kent Mouw and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Alizadeh AA et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med 21, 846–853 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marusyk A Almendro V & Polyak K Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Gerlinger M et al. Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. Eur. Urol 67 729–737 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Mitchell T & Neal DE The genomic evolution of human prostate cancer. Br. J. Cancer 113, 193–198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bruin EC et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yachida S et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlinger M et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med 366, 883–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yap TA Gerlinger M Futreal PA Pusztai L & Swanton C Intratumor heterogeneity: seeing the wood for the trees. Sci. Transl Med 4, 127ps10–127ps10 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Maley CC et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 17, 605–619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel RL Miller KD & Jemal A Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Sartor O & de Bono JS Metastatic prostate cancer. N. Engl. J. Med 378, 645–657 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Nelson WG De Marzo AM & Isaacs WB Prostate cancer. N. Engl. J. Med 349, 366–381 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Attard G et al. Prostate cancer. Lancet 387, 70–82 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Eschenbach von AC The biologic dilemma of early carcinoma of the prostate. Cancer 78, 326–329 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Pound CR et al. Natural history of progression after PSA elevation following radical prostatectomy. JAMA 281, 1591–1597 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Litwin MS & Tan H-J The diagnosis and treatment of prostate cancer: a review. JAMA 317, 2532–2542 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Aihara M Wheeler TM Ohori M & Scardino PT Heterogeneity of prostate cancer in radical prostatectomy specimens. Urology 43, 66–67 (1994). [DOI] [PubMed] [Google Scholar]
- 18.Cyll K et al. Tumour heterogeneity poses a significant challenge to cancer biomarker research. Br. J. Cancer 117, 367–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreoiu M & Cheng L Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum. Pathol 41, 781–793 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Arora R et al. Heterogeneity of Gleason grade in multifocal adenocarcinoma of the prostate. Cancer 100, 2362–2366 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Cheng L et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J. Natl. Cancer Inst 90, 233–237 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Miller GJ & Cygan JM Morphology of prostate cancer: the effects of multifocality on histological grade, tumor volume and capsule penetration. J. Urol 152, 1709–1713 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Spratt DE Zumsteg ZS Feng FY & Tomlins SA Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol 13, 597–610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marusyk A Janiszewska M & Polyak K Intratumor heterogeneity: the rosetta stone of therapy resistance. Cancer Cell 37, 471–484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boutros PC et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet 47, 736–745 (2015). [DOI] [PubMed] [Google Scholar]; Whole-genome sequencing of multiple tumour foci of five primary prostate cancer cases reveals independent tumour cell clones.
- 26.Fraser M Berlin A Bristow RG & van der Kwast T Genomic, pathological, and clinical heterogeneity as drivers of personalized medicine in prostate cancer. Urol. Oncol 33, 85–94 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Løvf M et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur. Urol 75, 498–505 (2019). [DOI] [PubMed] [Google Scholar]; Detailed assessment of 41 cases shows that 76% of multifocal primary tumours are genomically distinct, providing strong evidence of the multiclonality of prostate cancer.
- 28.Gundem G et al. The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Seminal study demonstrating the complex clonal architecture of lethal metastatic prostate cancer.
- 29.Haffner MC et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Invest 123, 4918–4922 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong MKH et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun 6, 6605–6612 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates the clonal dynamics and complex seeding pattern of advanced prostate cancer.
- 31.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive assessment of the clonal relationship of NEPC and the role of DNA methylation changes in lineage plasticity.
- 32.Lipinski KA et al. Cancer evolution and the limits of predictability in precision cancer medicine. Trends Cancer 2, 49–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espiritu SMG et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 173, 1003–1013.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Turajlic S & Swanton C Metastasis as an evolutionary process. Science 352, 169–175 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Turajlic S et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell 173, 595–610.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiter JG et al. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 19, 639–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomasetti C Li L & Vogelstein B Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martincorena I & Campbell PJ Somatic mutation in cancer and normal cells. Science 349, 1483–1489 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Haffner MC et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet 42, 668–675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haffner MC De Marzo AM Meeker AK Nelson WG & Yegnasubramanian S Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin. Cancer Res 17, 3858–3864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Blokzijl F et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greaves M & Maley CC Clonal evolution in cancer. Nature 481306–313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGranahan N et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl Med 7, 283ra54–283ra54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomasetti C Vogelstein B & Parmigiani G Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl Acad. Sci. USA 110, 1999–2004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moad M et al. Multipotent basal stem cells, maintained in localized proximal niches, support directed long-ranging epithelial flows in human prostates. CellReports 20, 1609–1622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooper CS et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet 47, 367–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barber LJ Davies MN & Gerlinger M Dissecting cancer evolution at the macro-heterogeneity and micro-heterogeneity scale. Curr. Opin. Genet. Dev 30, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nik-Zainal S et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McPherson A et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet 48, 758–767 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Ding L et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andor N et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med 22, 105–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mroz EA et al. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 119, 3034–3042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baca SC et al. Punctuated evolutionof prostate cancer genomes. Cell 153, 666–677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wedge DC et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet 50, 682–692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindberg J et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur. Urol 63, 347–353 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Van Etten JL & Dehm SM Clonal origin and spread of metastatic prostate cancer. Endocr. Relat. Cancer 23, R207–R217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Marzo AM et al. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 7, 256–269 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tyekucheva S et al. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun 8, 420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nonn L Ananthanarayanan V & Gann PH Evidence for field cancerization of the prostate. Prostate 69, 1470–1479 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehra R et al. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res 67, 7991–7995 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Mehra R et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res 68, 3584–3590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han B et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod. Pathol 22, 1083–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park K et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia 12, 590–598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furusato B et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis 13, 228–237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu Z et al. Clonal evaluation of early onset prostate cancer by expression profiling of ERG, SPINK1, ETV1, and ETV4 on whole-mount radical prostatectomy tissue. Prostate 80, 38–50 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Jamaspishvili T et al. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol 15, 222–234 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guedes LB et al. Analytic, preanalytic, and clinical validation of p53 IHC for detection of TP53 missense mutation in prostate cancer. Clin. Cancer Res 23, 4693–4703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan H-L et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res 20, 890–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi M et al. Molecular analysis of multifocal prostate cancer by comparative genomic hybridization. Prostate 68, 1715–1724 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Bostwick DG et al. Independent origin of multiple foci of prostatic intraepithelial neoplasia: comparison with matched foci of prostate carcinoma. Cancer 83, 1995–2002 (1998). [DOI] [PubMed] [Google Scholar]
- 73.Cheng L et al. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer 85, 2017–2022 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Wu B et al. Intratumoral heterogeneity and genetic characteristics of prostate cancer. Int. J. Cancer 146, 3369–3378 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Boutros PC Fraser M van der Kwast T & Bristow RG Clonality of localized and metastatic prostate cancer. Curr. Opin. Urol 26, 219–224 (2016). [DOI] [PubMed] [Google Scholar]
- 76.VanderWeele DJ et al. Genomic heterogeneity within individual prostate cancer foci impacts predictive biomarkers of targeted therapy. Eur. Urol. Focus 5, 416–424 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Macintyre G et al. How subclonal modeling is changing the metastatic paradigm. Clin. Cancer Res 23, 630–635 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Mitchell TJ et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 173, 611–623.e172018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerstung M et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baslan T & Hicks J Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer 17, 557–569 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Alexander J et al. Utility of single-cell genomics in diagnostic evaluation of prostate cancer. Cancer Res 78, 348–358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Su F et al. Spatial intratumor genomic heterogeneity within localized prostate cancer revealed by single-nucleus sequencing. Eur. Urol 74, 551–559 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Wang Y et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim C et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 173, 879–893.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grosselin K et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Li G et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 16, 991–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berglund E et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun 9, 2419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Decalf J Albert ML & Ziai J New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J. Pathol 247, 650–661 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Latonen L et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun 9, 1176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sinha A et al. The proteogenomic landscape of curable prostate cancer. Cancer Cell 35, 414–427.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu Y et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol 13, 2305–2328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Charmpi K et al. Proteogenomic heterogeneity of localized human prostate cancer progression. Preprint at bioRxiv 10.1101/2020.02.16.950378 (2020). [DOI] [Google Scholar]
- 93.McNeal JE The zonal anatomy of the prostate. Prostate 2, 35–49 (1981). [DOI] [PubMed] [Google Scholar]
- 94.McNeal JE Redwine EA Freiha FS & Stamey TA Zonal distribution of prostatic adenocarcinoma. Correlation with histologic pattern and direction of spread. Am. J. Surg. Pathol 12, 897–906 (1988). [DOI] [PubMed] [Google Scholar]
- 95.Shaikhibrahim Z et al. Genes differentially expressed in the peripheral zone compared to the transitional zone of the normal human prostate and their potential regulation by ETS factors. Mol. Med. Rep 5, 32–36 (2012). [DOI] [PubMed] [Google Scholar]
- 96.Guo CC Zuo G Cao D Troncoso P & Czerniak BA Prostate cancer of transition zone origin lacks TMPRSS2-ERG gene fusion. Mod. Pathol 22, 866–871 (2009). [DOI] [PubMed] [Google Scholar]
- 97.Sundi D et al. Pathological examination of radical prostatectomy specimens in men with very low risk disease at biopsy reveals distinct zonal distribution of cancer in black American men. J. Urol 191, 60–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Epstein JI Prostate cancer grading: a decade after the 2005 modified system. Mod. Pathol 31, S47–S63 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Elfandy H et al. Genetic and epigenetic determinants of aggressiveness in cribriform carcinoma of the prostate. Mol. Cancer Res 17, 446–456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hollemans E et al. Large cribriform growth pattern identifies ISUP grade 2 prostate cancer at high risk for recurrence and metastasis. Mod. Pathol 32, 139–146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schweizer MT et al. Genomic characterization of prostatic ductal adenocarcinoma identifies a high prevalence of DNA repair gene mutations. JCO Precis. Oncol 3, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rubin MA Girelli G & Demichelis F Genomic correlates to the newly proposed grading prognostic groups for prostate cancer. Eur. Urol 69, 557–560 (2016). [DOI] [PubMed] [Google Scholar]
- 103.Lotan TL et al. PTEN loss as determined by clinical-grade immunohistochemistry assay is associated with worse recurrence-free survival in prostate cancer. Eur. Urol. Focus 2, 180–188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kovtun IV et al. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res 73, 3275–3284 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Sowalsky AG Ye H Bubley GJ & Balk S P Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer Res 73, 1050–1055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sowalsky AG et al. Gleason score 7 prostate cancers emerge through branched evolution of clonal Gleason pattern 3 and 4. Clin. Cancer Res 23, 3823–3833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ye H & Sowalsky AG Molecular correlates of intermediate- and high-risk localized prostate cancer. Urol. Oncol 36, 368–374 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.VanderWeele DJ et al. Low-grade prostate cancer diverges early from high grade and metastatic disease. Cancer Sci 105, 1079–1085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karavitakis M Ahmed HU Abel P D., Hazell, S. & Winkler, M. H. Tumor focality in prostate cancer: implications for focal therapy. Nat. Rev. Clin. Oncol 8, 48–55 (2011). [DOI] [PubMed] [Google Scholar]
- 110.Humphrey PA Complete histologic serial sectioning of a prostate gland with adenocarcinoma. Am. J. Surg. Pathol 17, 468–472 (1993). [DOI] [PubMed] [Google Scholar]
- 111.van Royen ME et al. Three-dimensional microscopic analysis of clinical prostate specimens. Histopathology 69, 985–992 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Barry M Perner S Demichelis F & Rubin MA TMPRSS2-ERG fusion heterogeneity in multifocal prostate cancer: clinical and biologic implications. Urology 70, 630–633 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ruijter ET et al. Molecular analysis of multifocal prostate cancer lesions. J. Pathol 188, 271–277 (1999). [DOI] [PubMed] [Google Scholar]
- 114.Aryee MJ et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci. Transl Med 5, 169ra10–169ra10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mundbjerg K et al. Identifying aggressive prostate cancer foci using a DNA methylation classifier. Genome Biol 18, 3–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brocks D et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. CellReports 8, 798–806 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Stelloo S et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun 9, 4900–4912 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fontugne J et al. Clonal evaluation of prostate cancer foci in biopsies with discontinuous tumor involvement by dual ERG/SPINK1 immunohistochemistry. Mod. Pathol 29, 157–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kristiansen A et al. Somatic alterations detected in diagnostic prostate biopsies provide an inadequate representation of multifocal prostate cancer. Prostate 79, 920–928 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Brastianos PK et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov 5, 1164–1177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Haffner MC De Marzo AM Yegnasubramanian S Epstein JI & Carter HB Diagnostic challenges of clonal heterogeneity in prostate cancer. J. Clin. Oncol 33, e38–e40 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Stabile A et al. Multiparametric MRI for prostate cancer diagnosis: current status and future directions. Nat. Rev. Urol 17, 41–61 (2020). [DOI] [PubMed] [Google Scholar]
- 123.Valerio M et al. Detection of clinically significant prostate cancer using magnetic resonance imaging-ultrasound fusion targeted biopsy: a systematic review. Eur. Urol 68, 8–19 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Sathianathen NJ et al. Accuracy of the magnetic resonance imaging pathway in the detection of prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis 22, 39–48 (2019). [DOI] [PubMed] [Google Scholar]
- 125.Harmon SA Tuncer S Sanford T Choyke PL & Türkbey B Artificial intelligence at the intersection of pathology and radiology in prostate cancer. Diagn. Interv. Radiol 25, 183–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Houlahan KE et al. Molecular hallmarks of multiparametric magnetic resonance imaging visibility in prostate cancer. Eur. Urol 76, 18–23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cucchiara V et al. Genomic markers in prostate cancer decision making. Eur. Urol 73, 572–582 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Loeb S & Ross AE Genomic testing for localized prostate cancer: where do we go from here? Curr. Opin. Urol 27, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wei L et al. Intratumoral and intertumoral genomic heterogeneity of multifocal localized prostate cancer impacts molecular classifications and genomic prognosticators. Eur. Urol 71, 183–192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Salami SS et al. Transcriptomic heterogeneity in multifocal prostate cancer. JCI Insight 3, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sowalsky AG et al. Neoadjuvant-intensive androgen deprivation therapy selects for prostate tumor foci with diverse subclonal oncogenic alterations. Cancer Res 78, 4716–4730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wilkinson S et al. A case report of multiple primary prostate tumors with differential drug sensitivity. Nat. Commun 11, 837–838 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kneppers J et al. Frequent clonal relations between metastases and non-index prostate cancer lesions. JCI Insight 4, e124756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Epstein JI Amin MB Reuter VE & Humphrey PA Contemporary Gleason grading of prostatic carcinoma: an update with discussion on practical issues to implement the 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol 41, e1–e7 (2017). [DOI] [PubMed] [Google Scholar]
- 135.Valerio M et al. New and established technology in focal ablation of the prostate: a systematic review. Eur. Urol 71, 17–34 (2017). [DOI] [PubMed] [Google Scholar]
- 136.Ross HM et al. Do adenocarcinomas of the prostate with Gleason score (GS) ≤6 have the potential to metastasize to lymph nodes? Am. J. Surg. Pathol 36, 1346–1352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Trock BJ et al. PTEN loss and chromosome 8 alterations in Gleason grade 3 prostate cancer scores predicts the presence of un-sampled grade 4 tumor: implications for active surveillance. Mod. Pathol 29, 764–771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lotan TL et al. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol 28, 128–137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moschini M et al. Natural history of clinical recurrence patterns of lymph node-positive prostate cancer after radical prostatectomy. Eur. Urol 69, 135–142 (2016). [DOI] [PubMed] [Google Scholar]
- 140.Touijer KA Mazzola CR Sjoberg DD Scardino PT & Eastham JA Long-term outcomes of patients with lymph node metastasis treated with radical prostatectomy without adjuvant androgen-deprivation therapy. Eur. Urol 65, 20–25 (2014). [DOI] [PubMed] [Google Scholar]
- 141.Chaffer CL & Weinberg RA A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Naxerova K et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 357, 55–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mangiola S et al. Comparing nodal versus bony metastatic spread using tumour phylogenies. Sci. Rep 6, 33918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pienta KJ Robertson BA Coffey DS & Taichman RS The cancer diaspora: metastasis beyond the seed and soil hypothesis. Clin. Cancer Res 19, 5849–5855 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van der Toom EE Verdone JE & Pienta KJ Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr. Opin. Biotechnol 40, 9–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Massagué J & Obenauf AC Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Antonarakis ES et al. The natural history of metastatic progression in men with prostate-specific antigen recurrence after radical prostatectomy: long-term follow-up. BJU Int 109, 32–39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Reiter JG et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Marusyk A et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Merlo LMF Pepper JW Reid BJ & Maley CC Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 6, 924–935 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Landau DA et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Watson PA, Arora VK & Sawyers CL Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bluemn EG et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 32, 474–489.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Polkinghorn WR et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 3, 1245–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goodwin JF et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov 3, 1254–1271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sweeney CJ et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N. Engl. J. Med 373, 737–746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roudier MP et al. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum. Pathol 34, 646–653 (2003). [DOI] [PubMed] [Google Scholar]
- 160.Shah RB et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res 64, 9209–9216 (2004). [DOI] [PubMed] [Google Scholar]
- 161.Aggarwal R et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol 36, 2492–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Epstein JI et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol 38, 756–767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Labrecque MP et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Invest 130, 4492–4505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Definition of molecular subclasses of metastatic prostate cancers by comprehensive expression analyses.
- 164.Beltran H et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res 25, 6916–6924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nadal R Schweizer M Kryvenko ON Epstein JI & Eisenberger MA Small cell carcinoma of the prostate. Nat. Rev. Urol 11, 213–219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Quintanal-Villalonga Á et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol 20, 2429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Beltran H et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Invest 130, 1653–1668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Beltran H & Demichelis F Prostate cancer: intrapatient heterogeneity in prostate cancer. Nat. Rev. Urol 12, 430–431 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Stapleton AM et al. Primary human prostate cancer cells harboring p53 mutations are clonally expanded in metastases. Clin. Cancer Res 3, 1389–1397 (1997). [PubMed] [Google Scholar]
- 170.Chesire DR Ewing CM Sauvageot J Bova GS & Isaacs WB Detection and analysis of beta-catenin mutations in prostate cancer. Prostate 45, 323–334 (2000). [DOI] [PubMed] [Google Scholar]
- 171.Grasso CS et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kumar A et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med 22, 369–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Assessment of genomic and transcriptomic heterogeneity in mCRPC reveals shared driver alterations and highly similar expression pattern in anatomically distinct metastases.
- 173.Liu W et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med 15, 559–565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Heidenreich A et al. Cytoreductive radical prostatectomy in men with prostate cancer and skeletal metastases. Eur. Urol. Oncol 1, 46–53 (2018). [DOI] [PubMed] [Google Scholar]
- 175.Phillips R et al. Outcomes of observation vs stereotactic ablative radiation for oligometastatic prostate cancer: the ORIOLE phase 2 randomized clinical trial. JAMA Oncol 6, 650–659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wu A et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J. Clin. Invest 130, 1991–2000 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mateo J et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Invest 130, 1743–1751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Quigley DA et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174, 758–769.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.van Dessel LF et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun 10, 5251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lambros MB et al. Single-cell analyses of prostate cancer liquid biopsies acquired by apheresis. Clin. Cancer Res 24, 5635–5644 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Hussain M et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med 378, 2465–2474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stelloo S Bergman AM & Zwart W Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr. Relat. Cancer 26, R267–R285 (2019). [DOI] [PubMed] [Google Scholar]
- 183.Yegnasubramanian S De Marzo AM & Nelson WG Prostate cancer epigenetics: from basic mechanisms to clinical implications. Cold Spring Harb. Perspect. Med 9, a030445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Feinberg AP Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440 (2007). [DOI] [PubMed] [Google Scholar]
- 185.Drake JM et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl Acad. Sci. USA 110, E4762–E4769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nava Rodrigues D et al. RB1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin. Cancer Res 25, 687–697 (2019). [DOI] [PubMed] [Google Scholar]
- 187.Li Q et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun 9, 3600–3617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Carreira S et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med 6, 254ra125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Romanel A et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl Med 7, 312re10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Diaz LA et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sharma SV et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Abida W et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl Acad. Sci. USA 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Quigley D et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov 7, 999–1005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wu A & Attard G Plasma DNA analysis in prostate cancer: opportunities for improving clinical management. Clin. Chem 65, 100–107 (2019). [DOI] [PubMed] [Google Scholar]
- 195.Mahon KL et al. Methylated glutathione S-transferase 1 (mGSTP1) is a potential plasma free DNA epigenetic marker of prognosis and response to chemotherapy in castrate-resistant prostate cancer. Br. J. Cancer 111, 1802–1809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Antonarakis ES et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med 371, 1028–1038 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Morin F et al. Metabolic imaging of prostate cancer reveals intrapatient intermetastasis response heterogeneity to systemic therapy. Eur. Urol. Focus 3, 639–642 (2017). [DOI] [PubMed] [Google Scholar]
- 198.Fox JJ et al. Positron emission tomography/computed tomography-based assessments of androgen receptor expression and glycolytic activity as a prognostic biomarker for metastatic castration-resistant prostate cancer. JAMA Oncol 4, 217–224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]