

Abstract
Amplification of the MYCN oncogene occurs in approximately 25% of primary neuroblastomas and is the single most powerful biological marker of poor prognosis in this disease. MYCN transcriptionally regulates a range of biological processes important for cancer, including cell metabolism. The MYCN-regulated metabolic gene SLC16A1, encoding the lactate transporter monocarboxylate transporter 1 (MCT1), is a potential therapeutic target. Treatment of neuroblastoma cells with the MCT1 inhibitor SR13800 increased intracellular lactate levels, disrupted the nicotinamide adenine dinucleotide (NADH/NAD+) ratio and decreased intracellular glutathione levels. Metabolite tracing with 13C-glucose and 13C-glutamine following MCT1 inhibitor treatment revealed increased quantities of tricarboxylic acid (TCA) cycle intermediates and increased oxygen consumption rate. MCT1 inhibition was highly synergistic with vincristine under cell culture conditions, but this combination was ineffective against neuroblastoma xenografts in mice. Post-treatment xenograft tumors had increased expression of the MCT1 homolog MCT4/SLC16A, a known resistance factor to MCT1 inhibition. We found that MCT4 was negatively regulated by MYCN in luciferase reporter assays and its expression in neuroblastoma cells was increased under hypoxic conditions and following hypoxia-inducible factor (HIF1) induction, suggesting that MCT4 may contribute to resistance to MCT1 inhibitor treatment in hypoxic neuroblastoma tumors. Co-treatment of neuroblastoma cells with inhibitors of MCT1 and LDHA, the enzyme responsible for lactate production, resulted in a large increase in intracellular pyruvate and was highly synergistic in decreasing neuroblastoma cell viability. These results highlight the potential of targeting MCT1 in neuroblastoma in conjunction with strategies that involve disruption of pyruvate homeostasis and indicate possible resistance mechanisms.
Keywords: neuroblastoma, MCT1, lactate, pyruvate, glycolysis, tricarboxylic acid cycle
Introduction
Neuroblastoma is a pediatric solid tumor that originates from persistent neural crest progenitors and is responsible for 15% of cancer-related deaths in children worldwide (1). The survival rate for patients classified as high-risk unfortunately remains at 50% (2, 3), creating a significant need for better therapeutic approaches to improve outcomes for these patients. The master transcription factor MYCN, a member of the MYC family of oncogenes, is an established driver of neuroblastoma progression, but is a challenging therapeutic target (4). Targeting downstream alterations arising from MYCN dysregulation has therefore been a strategic approach in developing novel treatments for high-risk neuroblastoma.
Dysregulated MYC family oncoproteins have been demonstrated to remodel cancer metabolic pathways via direct transcriptional upregulation of metabolic genes, including those involved in glycolysis and glutaminolysis (5–7). Accelerated glycolysis in cancer cells results in the mass conversion of glucose to pyruvate, followed by conversion of pyruvate to lactate by lactate dehydrogenase A (LDHA), in preference to conversion of pyruvate to acetyl-CoA for oxidative phosphorylation via the tricarboxylic acid (TCA) cycle in mitochondria (8). The preferential conversion of pyruvate to lactate is essential in part due to the subsequent production of oxidized nicotinamide adenine dinucleotide (NAD+) which is recycled for continuous glycolysis. Accumulated lactate is exported out of cells by members of the monocarboxylate transporter (MCT) family (9), particularly MCT1 and MCT4, and may contribute to tumorigenesis by serving as an alternative fuel source for aerobic cancer cells, promoting angiogenesis and tumor invasion, and inducing further metabolic reprogramming (10–13). The catabolism of glutamine to glutamate by glutaminases, or glutaminolysis, replenishes TCA cycle intermediates and supports the synthesis of lipids and amino acids (14). In neuroblastoma, MYCN has been demonstrated to promote glutaminolysis by directly activating glutaminase 2 transcription (GLS2) (15).
The therapeutic potential of blocking lactate transport by inhibition of the monocarboxylate transporter 1 (MCT1, encoded by the SLC16A1 gene) has been investigated in preclinical studies of adult cancers and shown to delay tumor growth in lung and breast cancer and Burkitt’s lymphoma xenografts (16–19), while one MCT1 inhibitor has entered early phase clinical trial (ClinicalTrials.gov Identifier NCT01791595). Since MCT1 is a transcriptional target of MYCN (20) with a vital role in lactate homeostasis in cancer cells, this study investigated the biological and metabolic effects of MCT1 inhibitor treatment in preclinical models of high-risk neuroblastoma. The results provide evidence for MCT1 as a potential therapeutic target for this childhood disease.
Results
High MCT1/SLC16A1 expression is associated with poor clinical outcome in primary neuroblastoma and is a feature of MYCN-amplified tumor cells
To determine whether lactate transporter expression is associated with clinical outcome in neuroblastoma, we analysed mRNA expression of SLC16A1 (encoding for MCT1) and its homolog SLC16A3 (encoding for MCT4) in an expression array dataset from a prospectively accrued primary neuroblastoma cohort of 649 patients (21). SLC16A1 expression, but not SLC16A3 expression, was strongly associated with both poor event-free survival (EFS) and overall survival (OS) when mRNA expression was dichotomized at the median (Figure 1A–D). Multivariate Cox regression analyses with SLC16A1 expression and established prognostic indicators for neuroblastoma (age at diagnosis, INSS stage and MYCN amplification status) as variables, revealed that SLC16A1 is an independent prognostic marker for EFS and OS in neuroblastoma (Table 1). SLC16A1 gene expression levels were significantly higher in MYCN-amplified tumors than in MYCN non-amplified tumors (Figure 1E), while SLC16A3 levels were lower in MYCN-amplified tumors (Figure 1F). High expression of lactate dehydrogenase A (LDHA), which generates the MCT1/4 substrate lactate from pyruvate, was also strongly associated with both worse EFS and OS and was higher in MYCN-amplified tumors (Supplementary Figure S1). Multivariate Cox regression analyses revealed that LDHA is also strongly prognostic of EFS and OS, independent of established prognostic indicators for neuroblastoma (Supplementary Table S1). Consistent with tumor mRNA expression data, MYCN-amplified neuroblastoma cell lines typically expressed MCT1 protein at a substantially higher level than MYCN non-amplified cell lines, while MCT4 was expressed at very low levels in MYCN-amplified lines (Supplementary Figure S2). Similarly, LDHA protein levels were substantially higher in MYCN-amplified cell lines, with the exception of SK-N-BE(2) and its sub-clone BE(2)-C, where LDHA protein was absent. These associations suggest the potential importance of lactate homeostasis genes in the biology of MYCN-amplified, high-risk neuroblastoma.
Figure 1. SLC16A1 (MCT1) expression is prognostic for poor outcome in primary neuroblastoma.
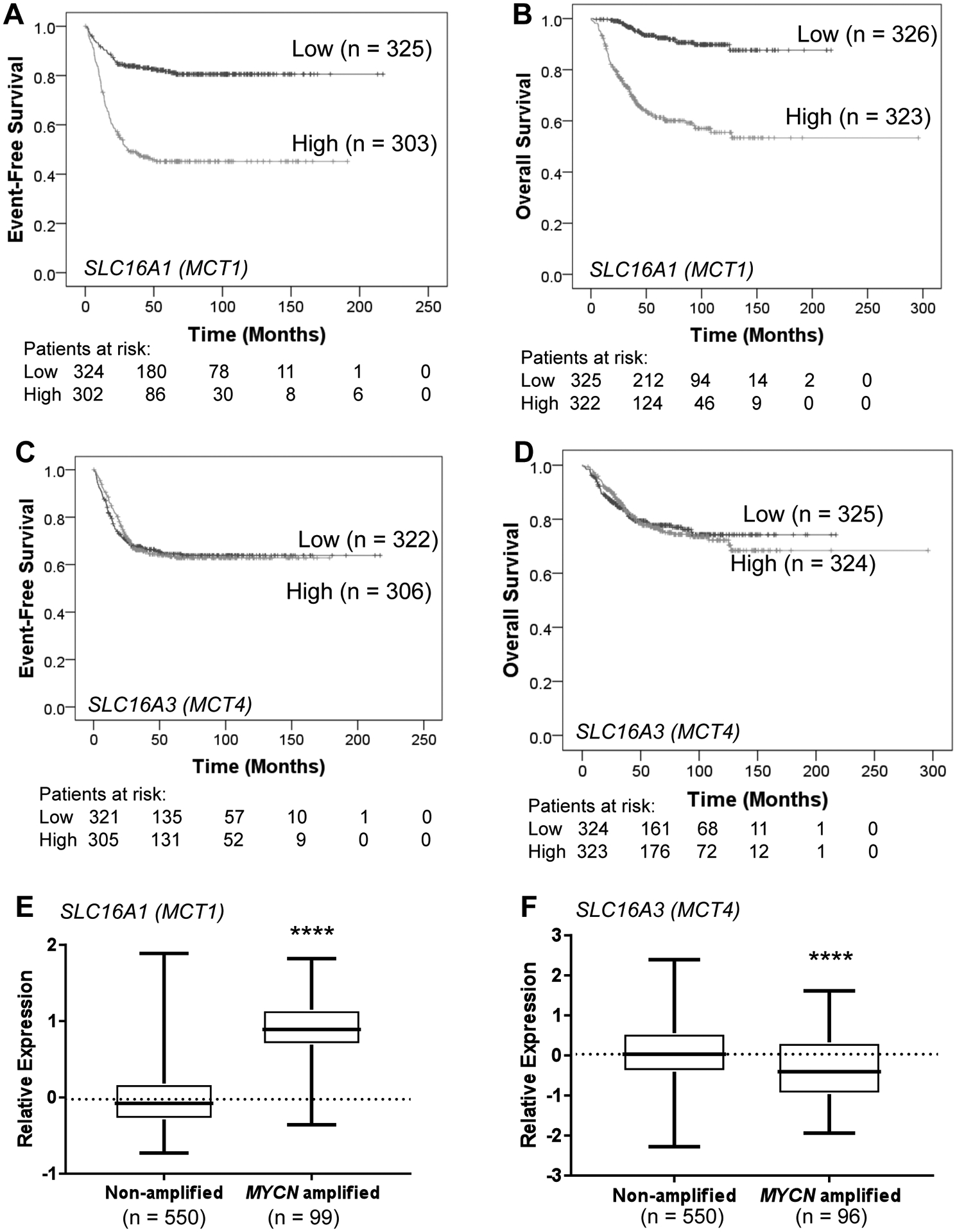
(A–D) Kaplan-Meier curves for event-free survival (EFS) and overall survival (OS) based on SLC16A1 or SLC16A3 mRNA expression in an expression array dataset from a prospectively accrued primary neuroblastoma cohort of 649 patients (21). Gene expression was dichotomized at the median. SLC16A1 EFS: HR = 3.33, 95% CI = 2.48–4.47, P < 0.001. SLC16A1 OS, HR = 6.23, 95% CI = 4.04–9.60, P < 0.001. SLC16A3 EFS: HR = 0.98, 95% CI = 0.75 SLC16A3–1.28, P < 0.001. SLC16A3 OS: HR = 1.04, 95% CI = 0.744–1.44, P < 0.001. (E–F) SLC16A1 mRNA expression was higher in MYCN-amplified compared to MYCN non-amplified tumors, while SLC16A3 expression was lower. ****P<0.0001.
Table 1.
Multivariate Cox regression analysis of SLC16A1 expression and outcome in neuroblastoma.
| Variable | Event-free survrival (n=628) | Overall survival (n=649) | ||
|---|---|---|---|---|
| HR (95% CI)b | P value | HR (95% CI) | P value | |
| MYCN statusa | 1.39 (0.99–1.94) | 0.055 | 2.43 (1.67–3.54) | <0.001*** |
| INSS tumor stage | 1.62 (1.18–2.24) | 0.003** | 2.30 (1.48–3.59) | <0.001*** |
| Age at diagnosis | 2.22 (1.62–3.03) | <0.001*** | 4.96 (3.16–7.79) | <0.001*** |
| SLC16A1 expression | 2.14 (1.52–3.00) | <0.001*** | 2.35 (1.45–3.81) | 0.001** |
Variables adjusted for in the multivariable analysis included MYCN status (amplified vs non-amplified), INSS tumor stage (1, 2, and 4S vs 3, 4), age at diagnosis (<18 months vs ≥ 18 months) and SLC16A1 expression (dichotomised at the median, low vs high).
HR, hazard ratio; CI, confidence interval.
P<0.01,
P<0.001
The MCT1 inhibitor SR13800 disrupts lactate homeostasis and NAD+/NADH ratio in neuroblastoma cells and decreases cell growth
Next, we directly assessed the importance of MCT1 in the MYCN-amplified cell lines Kelly and LA-N-1, both of which express high levels of MCT1 protein and low levels of MCT4 protein (Supplementary Figure S2). Treatment with the MCT1 inhibitor SR13800 resulted in a large increase in intracellular lactate levels in both cell lines (Figure 2A–B; P<0.05) with a corresponding increase in NADH/NAD+ ratio (Figure 2C–D; P<0.05) and depletion of intracellular GSH (Figure 2E–F). To determine the effects on cell growth, we treated a panel of the neuroblastoma cell lines, as well as the human breast cancer line MCF7 and the human skin fibroblast cell line WI-38, with SR13800 in short-term dose-response assays. The IC50 of SR13800 in the neuroblastoma and MCF7 lines ranged from 14.6μM to 31.3μM (P=0.1379, Table 2). The IC50 for SR13800 in the WI-38 fibroblast cell line was substantially higher at 167.4μM (Table 2). Taken together, these results indicate that MCT1 inhibition effectively disrupts lactate homeostasis and reduces growth in neuroblastoma cells.
Figure 2. Cell biology and metabolic effects of SR13800 on neuroblastoma cells.
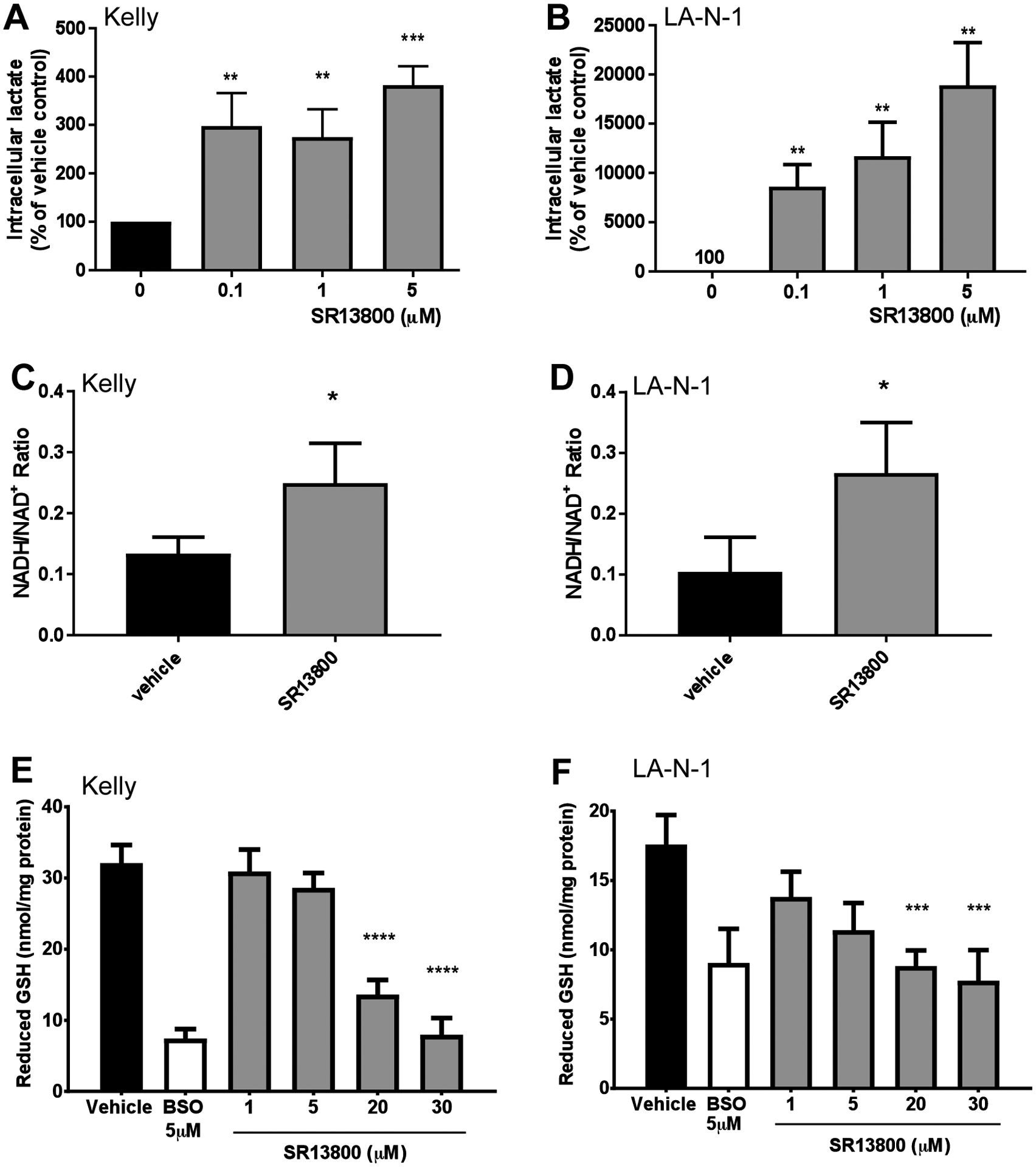
Response of neuroblastoma cell lines Kelly and LA-N-1 to SR13800 treatment. In lactate assays, treatment with the MCT1 inhibitor SR13800 for 4h increased intracellular lactate in Kelly (A) and LA-N-1 (B). (C–D) NADH/NAD+ ratio in Kelly (C) and LA-N-1 (D) cells treated with 0.1μM SR13800 for 6h. (E–F) In GSH assays, Kelly and LA-N-1 were treated with vehicle, 5μM BSO control or SR13800 for 24h and intracellular levels of GSH were determined. Data is presented as means ± SD (n=3). *P < 0.05, ***P<0.001, ****P<0.0001 compared to vehicle control.
Table 2.
SR13800 cytotoxicity assays on a panel of neuroblastoma cell lines
| Cell line | SR13800 IC50 (μM) |
|---|---|
| LA-N-1 | 31.3 ± 3.0a |
| Kelly | 31.4 ± 2.1 |
| IMR-32 | 14.6 ± 3.1 |
| BE(2)-C | 18.4 ± 1.8 |
| CHP-134 | 28.9 ± 2.6 |
| SH-SY5Y | 22.3 ± 2.6 |
| MCF7 | 22.3 ± 2.6 |
| WI-38 | 167.4 ± 11 |
Mean ± SD of three independent experiments (n= 3)
SR13800 treatment in Kelly neuroblastoma cells causes an increase in tricarboxylic acid (TCA) cycle activity derived from glucose
The metabolic effects of SR13800 on MYCN-amplified Kelly cells were studied using GC-MS profiling of polar metabolites derived from heavy isotope labeling with 13C-glucose and 13C-glutamine. Examination of overall metabolic quantities (labeled and unlabeled) confirmed a large increase in intracellular lactate upon SR13800 treatment (Figure 3A). Examination of TCA metabolites revealed increases in the amounts of α-ketoglutarate, malate and citrate (Figure 3A), consistent with increased TCA cycle activity (Supplementary Figure S3). Overall metabolic quantification also revealed a large decrease in intracellular aspartate, a metabolite derived from the TCA cycle (Figure 3A). These observations corresponded with decreased lactate secretion and glucose uptake in media samples (Figure 3B–C).
Figure 3. MCT1 inhibition causes increased TCA activity in Kelly neuroblastoma cells.
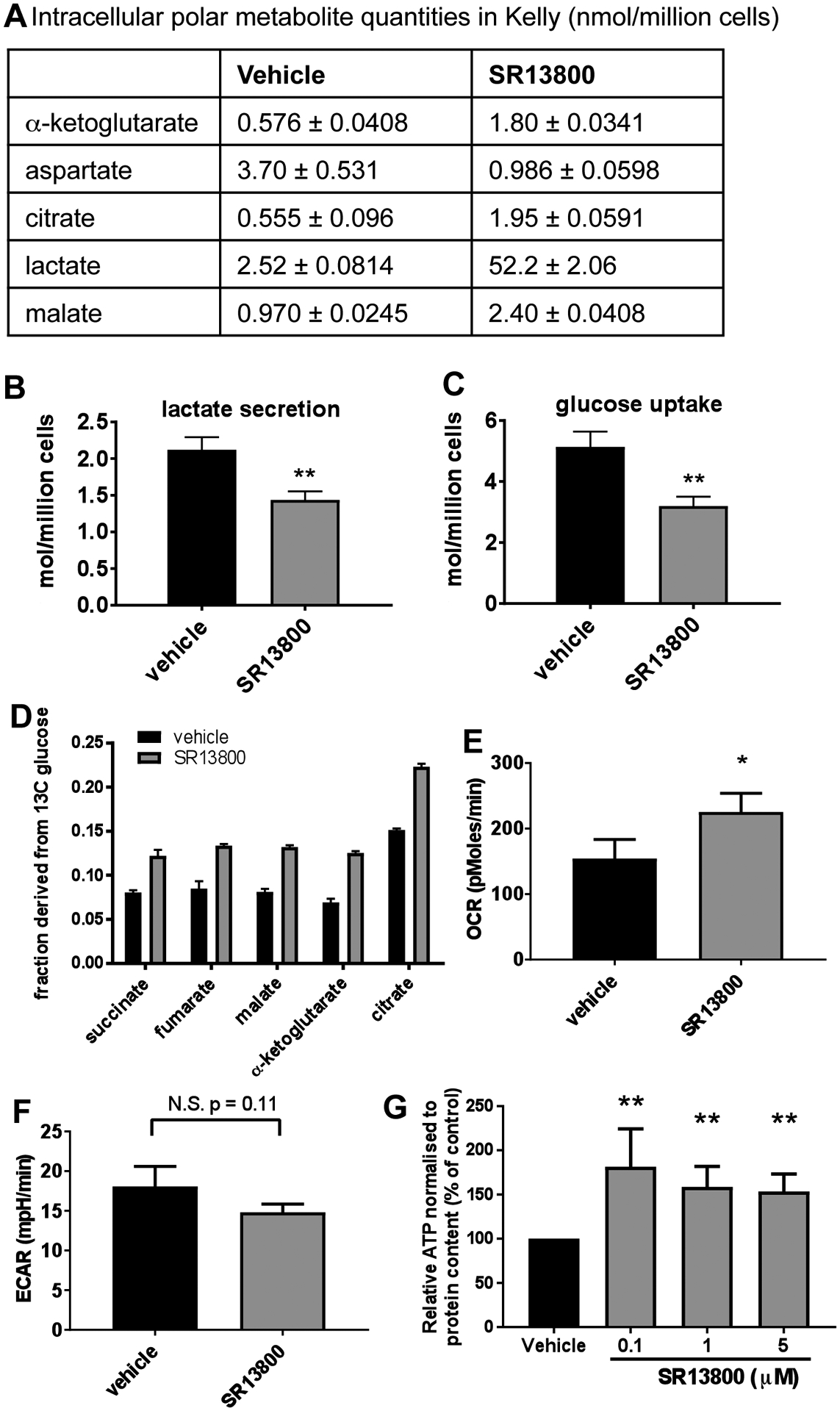
(A) GC-MS metabolic profiling of intracellular polar metabolite quantities in Kelly treated with 0.1μM SR13800 and vehicle treated controls for lactate, TCA intermediates α-ketoglutarate, malate and citrate, and aspartate. YSI measurements on media samples from SR13800-treated Kelly demonstrated decreased lactate secretion (B, 6h) and decreased glucose uptake (C, 24h). (D) Fractional labelling derived from 13C-glucose in TCA metabolites, succinate, fumarate, malate, α-ketoglutarate and citrate fragments in Kelly treated with 0.1μM SR13800. OCR (E) and ECAR (F) 2h post treatment with 1μM SR13800 compared to vehicle treated Kelly (n=3). (G) Relative intracellular ATP levels were determined in Kelly treated with SR13800 (n=3). *P < 0.05, **P<0.01 compared to vehicle control.
Examination of 13C fractional labelling revealed that SR13800 treatment resulted in an increase in the proportion of the TCA intermediates succinate, fumarate, malate, α-ketoglutarate and citrate that were derived from 13C glucose (Figure 3D). While MYCN-amplified neuroblastoma cells can utilise glutamine as an alternative fuel source to glucose (15, 22), examination of the 13C fractional label from 13C glutamine revealed no significant changes in the fraction of any metabolites in Kelly cells in response to SR13800 treatment (Supplementary Table S2). In support of this, cell viability assays conducted on Kelly and LA-N-1 cells treated with SR13800 in the presence or absence of glutamine revealed little change in IC50 values, whether under hypoxic or normoxic conditions (Table 3).
Table 3.
SR13800 cytotoxicity assays in the presence or absence of glutamine.
| SR13800 IC50 (μM) | |||
|---|---|---|---|
| Cell line | Glutamine | Glutamine-free | P value |
| Kelly | 25.4 ± 2.48a | 21.9 ± 1.49 | 0.300 |
| LA-N-1 | 24.8 ± 0.684 | 23.3 ± 1.57 | 0.360 |
Mean ± SD of three independent experiments (n= 3)
These observations suggest that MYCN-amplified neuroblastoma cells redirect pyruvate into the TCA cycle in response to SR13800 treatment, resulting in increased oxidative phosphorylation levels. To further test this possibility, real-time metabolic analyses were performed on live Kelly cells following SR13800 treatment. An increase in oxygen consumption rate (OCR) (Figure 3E, P<0.05 at 2h; Supplementary Figure S4A), a decrease in extracellular acidification rate (ECAR) levels (Figure 3F, P=0.11 at 2h; Supplementary Figure S4B) and an increase in intracellular ATP levels (Figure 3G; P<0.01) were all observed, consistent with increased oxidative phosphorylation.
SR13800 augments the potency of vincristine in neuroblastoma cells
To determine whether SR13800 potentiates the activity of chemotherapeutic agents used in the treatment of high-risk neuroblastoma, LA-N-1 and Kelly cells were treated with SR13800 in combination with vincristine and doxorubicin and evaluated in cell viability assays. Both vincristine and doxorubicin were found to be synergistic with SR13800 in LA-N-1 cells (Figure 4A–B). In Kelly cells, doxorubicin and SR13800 were not synergistic, however vincristine and SR13800 were strongly synergistic with 100% loss of cell viability at the lowest dose combination (Figure 4C–D). This striking synergy was also observed in clonogenic assays (Figure 4E). As vincristine functions primarily by inhibition of mitosis at metaphase, cell cycle analysis was performed following treatment of Kelly cells with SR13800 in combination with vincristine. Low dose treatment with SR13800 or vincristine alone did not significantly affect the percentage of cells in G2/M state, however the combination of SR13800 and vincristine significantly increased G2/M arrest in Kelly cells (Figure 4F), consistent with potentiation of vincristine function.
Figure 4. Vincristine augments the potency of SR13800 in neuroblastoma cells.
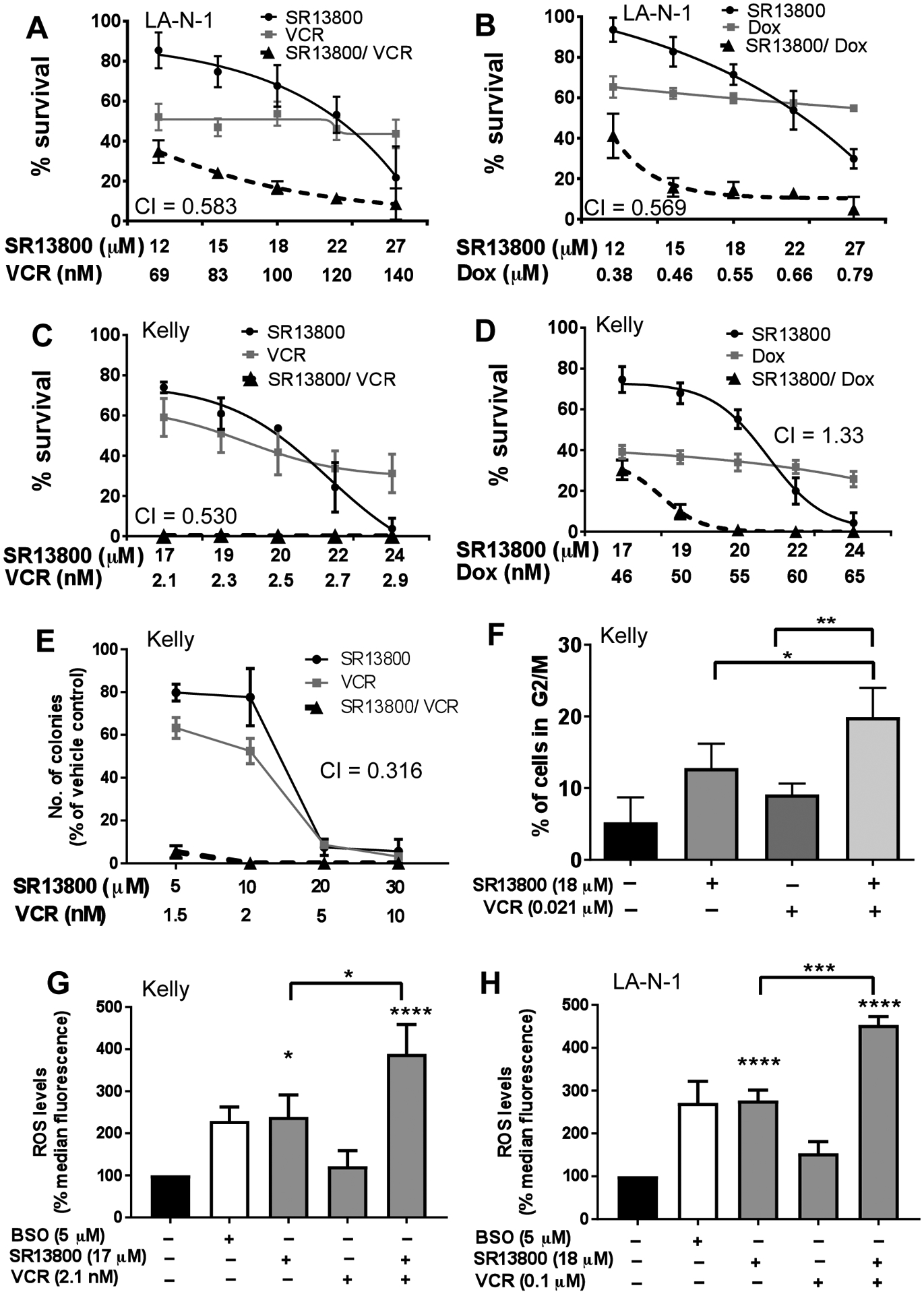
(A–D) In cell viability assays, treatment with SR13800 was synergistic with vincristine (VCR) in both LA-N-1 (A) and Kelly cells (C). SR13800 treatment was also synergistic with doxorubicin (Dox) in LA-N-1 (B) but not Kelly (D) in cell viability assays. CI value of 0.3–0.7 denotes synergism. (E) In colony assays, the combination of vincristine and SR13800 in Kelly was highly synergistic (CI of 0.316). (F) Cell cycle analysis in propidium-iodide stained Kelly cells showed a significant increase of percentage of cells in G2 arrest with the SR13800 (18μM) and vincristine (0.021μM) combination compared to single drug treatment. Analysis at 18h post-treatment. (G–H) ROS analysis in neuroblastoma cells. Kelly (G) were treated with vehicle, BSO control (5μM), SR13800 (17μM) and/or vincristine (2.1×10−3μM). LA-N-1 (H) were treated with vehicle, control BSO (5μM), SR13800 (18μM) and/or vincristine (1μM) for 18h. All data is presented as means ± SD (n=3). *P<0.05, ***P<0.001, ****P<0.0001 compared to vehicle control unless indicated.
Both SR13800 and vincristine are reported to be associated with increased intracellular ROS in cancer cells (17, 23). To investigate whether increased ROS could account for the synergistic decrease in cell viability, LA-N-1 and Kelly cells were treated with SR13800 and vincristine and assayed for ROS levels after 18h treatment. While vincristine treatment alone did not produce a significant increase in intracellular ROS in either cell line at the concentration at which maximal synergy was observed, SR13800 increased ROS to a similar extent as the BSO positive controls (Figure 4G–H). The combination of the two drugs further increased intracellular ROS levels, suggesting that the synergistic effect observed in the viability assays may in part be due to a large increase in intracellular ROS, without further depleting GSH levels (Supplementary Figure S4C).
MCT4 expression is inversely correlated with MYCN in neuroblastoma cells
MYCN has been shown to positively regulate SLC16A1 expression at the transcriptional level (20). Using two MYCN inducible systems, SHEP-TET21/N (Tet-off) and SHEP-MYCN3 (Tet-on), where MYCN is repressed or induced respectively by the addition of doxycycline, we confirmed that MCT1 protein expression paralleled MYCN expression (Figure 5A–B). We also assessed expression of MCT4, which is a resistance factor for MCT1 inhibition in other cancers (16, 24). MCT4 expression was found to be inversely related to MYCN expression in both systems (Figure 5A–B). To confirm these findings in an independent cell line, MYCN was suppressed in Kelly cells using two independent siRNA duplexes (dx1, dx2). MCT4 protein expression was upregulated following MYCN knockdown (Figure 5C), while MCT1 expression was reduced. To exclude MCT4 upregulation as a compensatory response to MCT1 downregulation rather than a direct effect lower MYCN levels, siRNA-mediated MCT1 knockdown was performed on Kelly cells. No significant alteration of MCT4 expression was detected upon MCT1 knockdown (Figure 5D).
Figure 5. MCT4 expression is suppressed by MYCN.
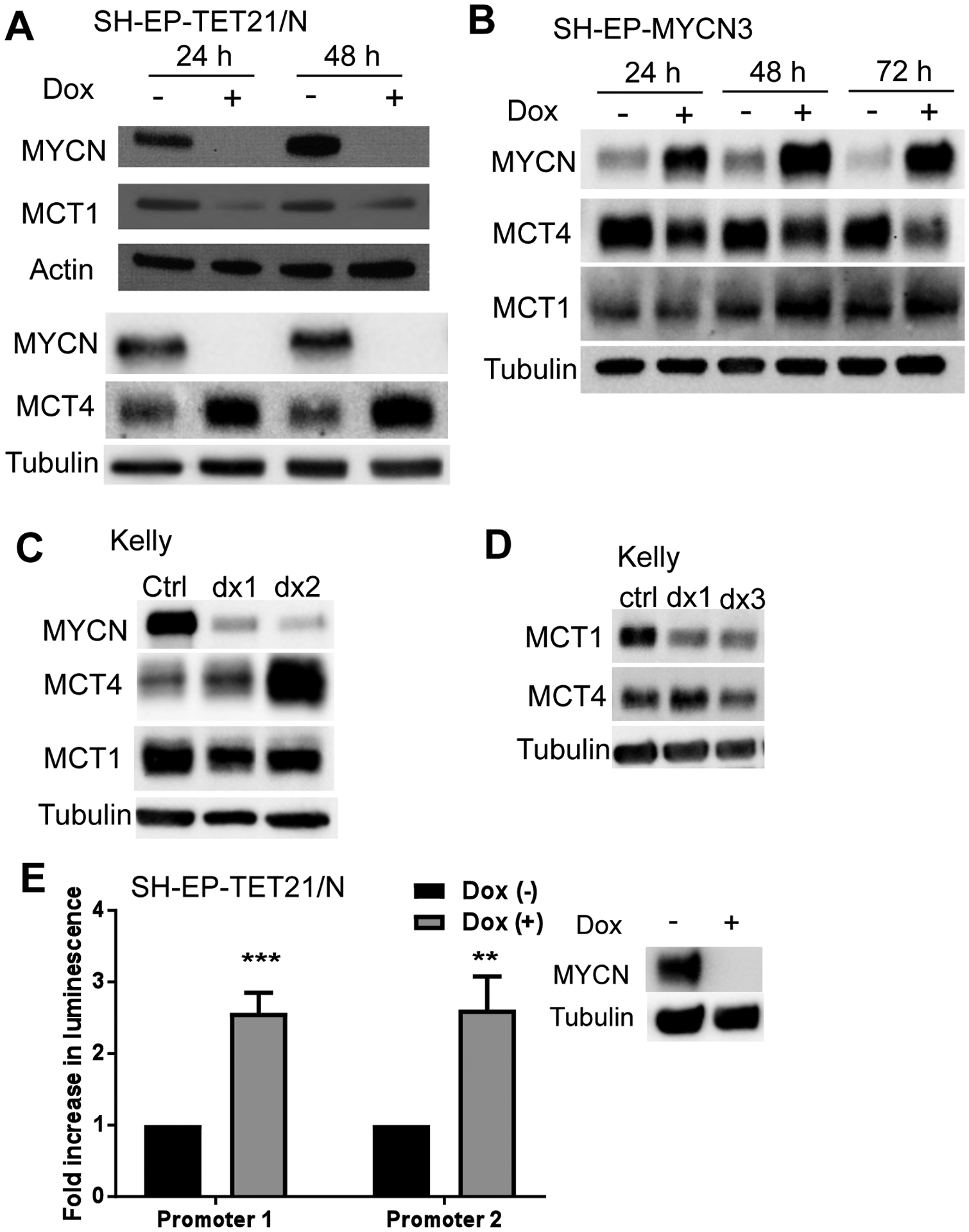
MCT1 expression is associated with MYCN in neuroblastoma cells (A–D). In both SH-EP-Tet21/N cells (MYCN Tet-off system) (A) and SH-EP-MYCN3 (MYCN Tet-On system) (B) neuroblastoma cells at 24h and 48h post-treatment with doxycycline, MCT1 expression was positively associated with MYCN expression while MCT4 was negatively associated with MYCN expression. (C) MCT4 expression was upregulated in Kelly cells when endogenous MYCN expression was suppressed with two independent MYCN siRNAs. However, MCT4 expression is unaffected by MCT1 knockdown using two different siRNAs in Kelly neuroblastoma cells (D). Actin or tubulin served as a loading control. Representative blots from three independent experiments. (E) In luciferase reporter assays, increased luciferase activity was detected in SH-EP-TET21/N cells transfected with two SLC16A3/MCT4 promoter/luciferase gene reporter constructs when MYCN expression was suppressed with doxycycline (Dox). Data presented as means ± SD (n=3). ***P<0.001 compared to no doxycycline treatment. **P<0.01, ***P<0.001 compared to vehicle control.
To examine for possible transcriptional repression of SLC16A3 by MYCN, two overlapping promoter regions of SLC16A3 were cloned into the luciferase reporter plasmid, pGL3-Basic, which was subsequently transfected into SHEP-Tet21/N cells and tested for luciferase activity. Luciferase activity of both SLC16A3 promoter/pGL3-Basic constructs was increased by over two-fold following doxycycline-induced MYCN downregulation (Figure 5E; P<0.01), suggesting that MYCN negatively regulates SLC16A3 transcription.
In vivo resistance to MCT1 inhibitor treatment is associated with increased levels of MCT4 protein
To determine whether the combination of SR13800 and vincristine is effective in vivo, Balb/c nude mice were xenografted with Kelly cells and established tumors treated with SR13800 alone or in combination with vincristine (Figure 6A). While vincristine modestly delayed tumor growth compared to the vehicle-treated group (P=0.01), SR13800 alone or combined with vincristine were both ineffective at delaying tumor growth (P=0.77 for SR13800, P=0.11 for SR13800/VCR). Tumors harvested at endpoint from mice treated with SR13800 or SR13800 and vincristine in combination demonstrated increased MCT4 protein expression (Figure 6B), potentially contributing to MCT1 inhibitor resistance.
Figure 6. MCT4 expression is elevated in neuroblastoma under conditions of hypoxia and HIF-1α induction.
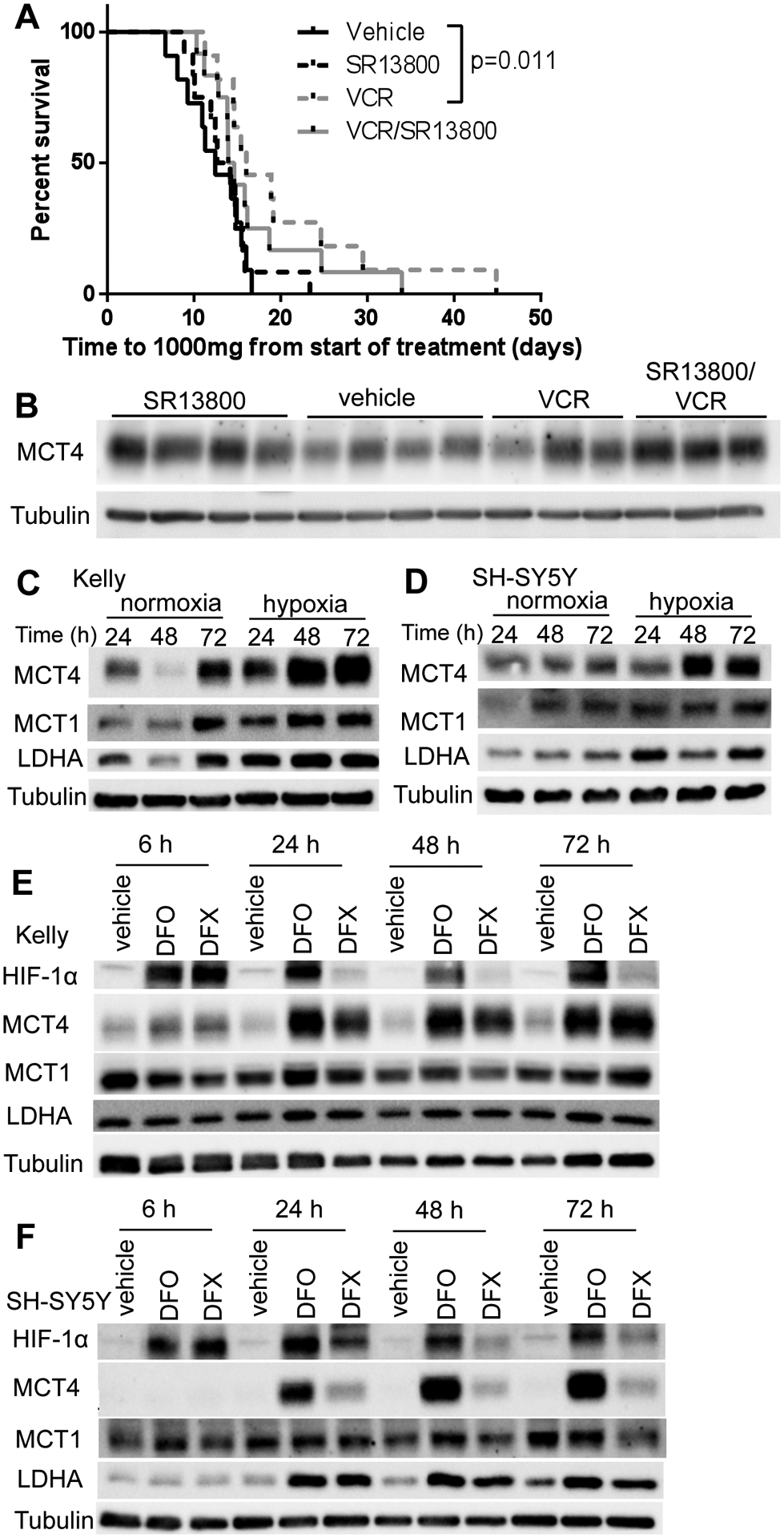
(A) Response of Kelly xenografts in BALB/c nude mice to vehicle, SR13800 (30 mg/kg), vincristine (VCR, 0.2 mg/kg) and VCR/SR13800. (B) Tumor protein levels of MCT4 harvested at end point. (C–D) MYCN-amplified Kelly (C) and MYCN non-amplified SH- SY5Y (D) cells were incubated in normoxic or hypoxic conditions for 24, 48 and 72h and samples were analysed by Western blot for MCT1, MCT4 and LDHA. Kelly (E) and SH-SY5Y (F) were treated with HIF inducers desferrioxamine (DFO, 20mM) or deferasirox (DFX, 100mM) over a timecourse (6h–72h) and levels of HIF1α, MCT1, MCT4 and LDHA were analysed by Western blot. Tubulin served as a loading control. Representative blots from three independent experiments.
MCT4 is upregulated by hypoxia
As MCT4 is regulated by hypoxia inducible factor HIF-1α (25), we investigated the impact of hypoxia and HIF-1α induction on MCT4 protein expression in neuroblastoma cells. Kelly, LA-N-1, and MYCN non-amplified SH-SY5Y cells were maintained under both normoxic and hypoxic (1% oxygen) conditions over a period of 24h–72h and analyzed by Western blot. Under hypoxic conditions, MCT4 expression was upregulated in each cell line, particularly at the later time points, while no consistent changes were observed with MCT1 expression between normoxic and hypoxic conditions (Figure 6C–D, Supplementary Figure S5A). Cells were also treated with three different HIF-1α inducers, including the iron chelators desferrioxamine (DFO) and deferasirox (DFX) (26) and the prolyl hydroxylase inhibitor dimethyloxalylglycine (DMOG) (27), each of which increased MCT4 expression (Figure 6E–F, Supplementary Figure S5B, C). We also observed that 5μM SR13800 treatment of Kelly cells in vitro caused small increases in MCT4 protein expression after 72h treatment (Supplementary Figure S5D), suggesting that SR13800 treatment may partially contribute to the increased tumor expression of MCT4.
SR13800 modulates pyruvate levels in neuroblastoma cells and is highly synergistic with the LDHA inhibitor FX11 in reducing cell viability
Although MCT1 is widely recognized to regulate lactate transport, the inhibition of MCT1-mediated pyruvate export may also lead to cancer cell apoptosis (18). Accordingly, we investigated the effect of SR13800 treatment on pyruvate levels in neuroblastoma cells. SR13800 treated Kelly cells demonstrated a decrease in extracellular pyruvate as detected by GC-MS (Supplementary Figure S6A). To further modulate pyruvate levels, we treated with the LDHA inhibitor FX11, which inhibits the conversion of pyruvate to lactate. Although FX11 decreased intracellular lactate and ATP in Kelly and LA-N-1 cells (Supplementary Figure S6B–E), FX11 treatment had markedly few effects on the polar metabolic profile of Kelly cells (Supplementary Table S3). One notable exception was that FX11 treatment caused a significant increase in levels of extracellular pyruvate in Kelly (Supplementary Figure S6F). To follow up on this observation, we performed extracellular and intracellular pyruvate assays with Kelly and LA-N-1 treated with either SR13800 or FX11, or both drugs in combination. SR13800 treatment decreased extracellular pyruvate in both cell lines (Figure 7A–B) and increased intracellular pyruvate in LA-N-1, however in Kelly intracellular pyruvate levels remained unchanged (Figure 7C–D). While LA-N-1 showed a significant increase in intracellular pyruvate upon FX11 treatment, which would be expected with LDHA inhibition, Kelly instead showed a significant increase in extracellular pyruvate (Figure 7A–D). This suggested that excess accumulation of pyruvate caused by FX11 treatment is effluxed from Kelly cells, and this efflux is likely to be mediated by MCT1.
Figure 7. SR13800 and FX11 treatment altered pyruvate levels in neuroblastoma cells and are synergistic with each other in cell viability assays.
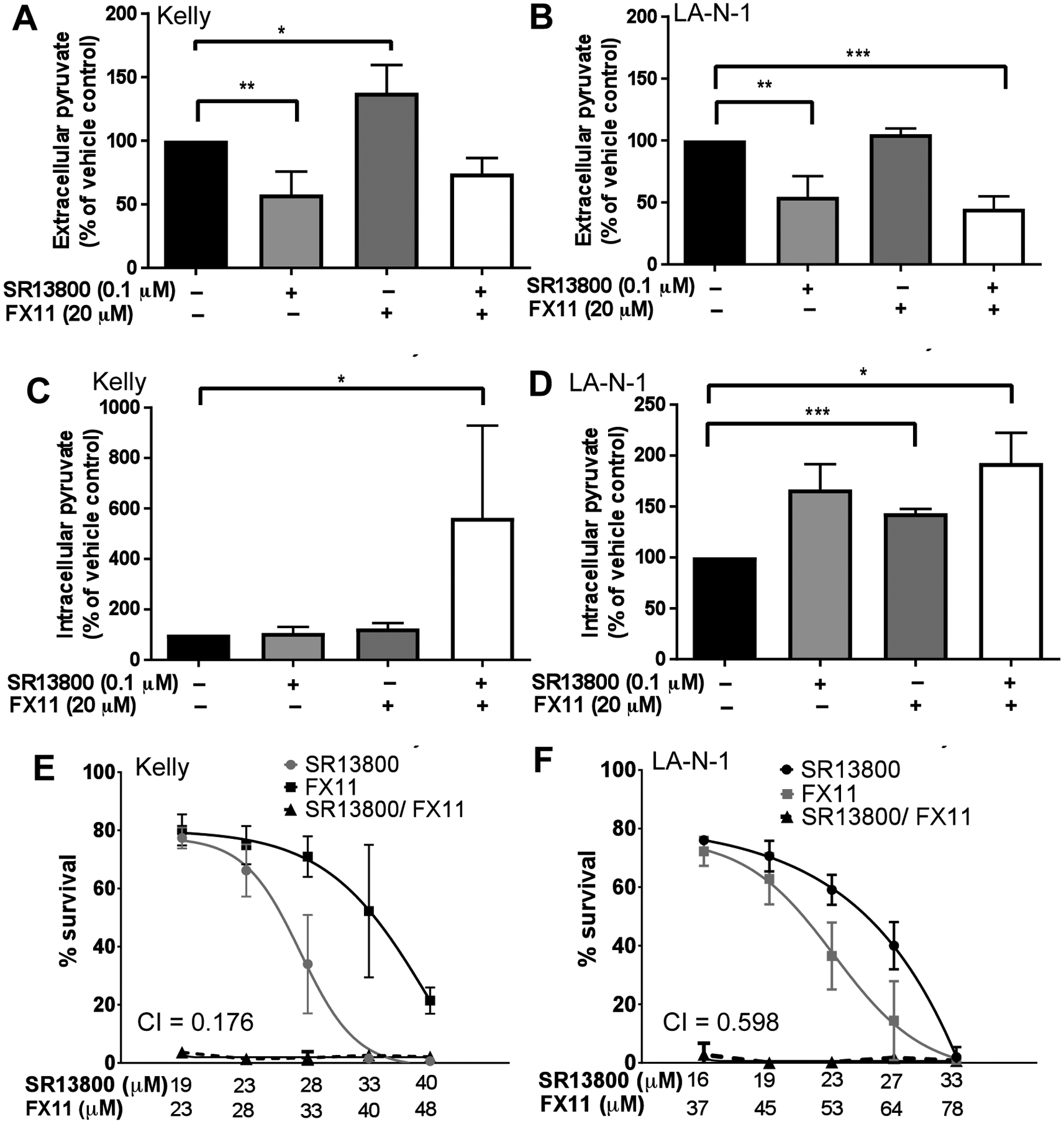
Pyruvate assays on Kelly (A, C) and LA-N-1 (B, D) treated for 6h (intracellular) or 24h (extracellular) with SR13800 (0.1μM), FX11 (20μM) or both drugs in combination. (E–F) In cell viability assays, the combination of SR13800 and FX11 was highly synergistic in reducing cell viability in both (E) Kelly (CI = 0.176) and (F) LA-N-1 (CI = 0.598). Data presented as means ± SD (n=3). *P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001 compared to vehicle control.
Taken together, these observations led to the hypothesis that the combination of SR13800 and FX11 leads to an accumulation of intracellular pyruvate in both cell lines, regardless of the differential response between the two cell lines to SR13800 and FX11 as individual agents. Indeed, combined treatment of SR13800 and FX11 on Kelly resulted in a large increase in intracellular pyruvate (Figure 7C). A small increase in intracellular pyruvate was also detected in LA-N-1 for the combination versus single treatments (Figure 7D). To investigate if accumulation of intracellular pyruvate by the combined treatment of SR13800 and FX11 is associated with inhibition of cell growth to neuroblastoma cells, cell viability assays combining SR13800 and FX11 were performed on Kelly and LA-N-1 cells. Strikingly, this combination was strongly synergistic in both Kelly and LA-N-1 cells, resulting in almost 100% loss of cell viability (Figure 7E–F).
Discussion
The clinical application of MCT1 inhibitors to block export of excess intracellular lactate, a critical step in maintenance of high glycolytic rates in cancer cells, is under investigation for adult cancer therapy. This study investigated whether MCT1 inhibition has antitumor activity in neuroblastoma, a pediatric tumor characterized by MYCN amplification. Our finding that high expression of SLC16A1/MCT1 is strongly and independently prognostic for poor survival in patients and is associated with MYCN amplification, supports MCT1 as a potential therapeutic target in this tumor. MCT1 expression was increased with high levels of MYCN, and, interestingly, we showed for the first time that high MYCN levels repress MCT4 expression. Strong synergy was observed between the MCT1 inhibitor SR13800 and vincristine in vitro. Despite this finding, this combination was ineffective in vivo in Kelly neuroblastoma xenografts. Our observations suggest that resistance mechanisms could include an increase in MCT4 levels in the hypoxic neuroblastoma tumor environment, and SR13800 treatment itself leading to an increase in MCT4 levels as well as increased OCR in Kelly cells. SR13800 was also highly synergistic with the LDHA inhibitor FX11 in vitro, and this synergy was associated with an accumulation of intracellular pyruvate. Because MCT4 has a very low affinity for pyruvate transport (9), combining an MCT1 inhibitor with an LDHA inhibitor could circumvent induced MCT4 resistance. Therefore, the therapeutic potential of this combination should be tested when suitable LDHA inhibitors become available.
The regulation of metabolites is a complex homeostatic process involving large numbers of regulators, enzymes, transporters, signalling pathways and metabolites, and an accumulation of metabolites can cause cascading effects on the metabolic network (28). Cancer cells have considerable capacity to undergo continuous global metabolic rewiring to adapt to changing environment conditions (29) and are flexibly respond to fluctuations in metabolites and activate or repress pathways via the action of master transcription factors such as MYCN and HIF-1α (6). Relatively little is understood about the metabolic phenotype of neuroblastoma, its metabolic preferences, or its capacity to switch between different metabolic pathways in response to various stimuli, nutrients or pathway inhibition (6, 30).
Recent studies have uncovered potential glycolytic and glutaminolytic targets for neuroblastoma, but few reports to date have studied the metabolic profile of neuroblastoma cells. We showed that SR13800 treatment caused an increase in TCA activity in Kelly cells, suggesting that pyruvate is redirected to the TCA cycle, which also corresponded to an increase in intracellular ATP levels. However, this contrasts with SR13800 administered to Raji lymphoma cells which did not alter levels of TCA intermediates, but instead showed decreased ECAR without changing basal OCR, and decreased intracellular ATP levels, most likely caused by reduction in glycolytic rates (17). The ability of neuroblastoma cells to redirect excess pyruvate into the TCA cycle and generate additional ATP via oxidative phosphorylation is in concordance with a recent report showing that human lung NSCLC tumors can take up lactate and pyruvate to supplement the TCA cycle (10). The decreased aspartate levels in response to SR13800 treatment in Kelly cells may indicate reduced utilisation of the TCA cycle to produce aspartate from oxaloacetate. This may be due to limited NAD+ availability leading to inhibition of malate dehydrogenase activity, or alternatively, aspartate may be rapidly used as a precursor for other metabolites as a result of MCT1 inhibition. Our data also indicate that while these neuroblastoma cells can compensate for excess pyruvate caused by MCT1 inhibition by redirecting pyruvate to the TCA cycle, the addition of an LDHA inhibitor abolishes pyruvate homeostasis. Thus, an in-depth understanding of neuroblastoma metabolic pathways will be helpful in developing and refining relevant therapies for the disease.
An important resistance mechanism for MCT1 inhibitors is compensatory upregulation of the related lactate transporter MCT4 (16, 25). While MYCN is known to directly regulate MCT1 transcription (20), our study made the novel finding that MYCN normally suppresses SLC16A3/MCT4 transcription in neuroblastoma cells. MYCN, a well characterized transcriptional activator, also acts as a gene repressor via binding to Sp1 (31, 32), and this function may contribute to neuroblastoma progression by inhibiting genes that antagonise tumor growth such as angiogenic inhibitors and pro-apoptotic genes (32, 33). Although MYCN is active in remodeling metabolic pathways (6, 34), there is evidence for MYCN acting as a repressor of metabolic genes. The ability of MYCN to activate SLC16A1/MCT1 transcription and simultaneously repress SLC16A3/MCT4 is reflected in the pattern of endogenous expression in the cell lines. However, MCT1 inhibitor treatment was ineffective in vivo, which may suggest that the regulation of MCT4 by HIF1α in neuroblastoma tumors is likely to overcome the repressive ability of MYCN, resulting in MCT4-mediated resistance to MCT1 inhibition. In support of this, a recent study also indicated that some solid tumor cell lines, including neuroblastoma and lung cancer lines, demonstrated only partial sensitivity to MCT1 inhibition and generally had higher levels SLC16A3 mRNA expression compared to hematological tumors (35).
The combinatorial use of metabolic inhibitors with conventional cancer agents has been investigated as a strategy to overcome resistance, generally targeting the importance of metabolite balance for cellular or chemoresistance responses or cell damage repair (36–38). In our in vitro studies, we found that the effect of SR13800 was greatly enhanced in combination with vincristine, resulting in increased intracellular ROS and G2/M arrest. However, this combination administered in vivo was ineffective partially due to MCT4 upregulation as a result of functional inhibition of MCT1 via SR13800. Another possible strategy that may avoid MCT4 upregulation may be to exploit cancer cells with high MCT1 expression by developing a MCT1 targeting substrate agent like 3-BrPA (20, 39). While 3-BrPA is not suitable for clinical use, 3-BrOP, a derivative of 3-BrPA, has been reported to have activity against neuroblastoma cells (40) and it would be interesting to investigate if existing analogs can be further developed as MCT1 substrate targets.
SR13800 has inhibitory activity not only against MCT1 but also the related transporter MCT2, which can also export pyruvate (41) and is expressed by various cancers including neuroblastoma (20, 42). It is possible that the effectiveness of combining MCT1 and LDHA inhibition in dramatically reducing neuroblastoma cell viability is partially attributable to the action of SR13800 on MCT2. Regardless, the very high synergy demonstrated by this combination suggests that neuroblastoma cells are particularly sensitive to large increases in intracellular pyruvate and are unable to adequately redirect it to other pathways. Pyruvate is also required by histone deacetylases (HDACs), and pro-survival proteins which induce apoptosis upon association with pyruvate (43). Alternatively, accumulation of pyruvate may exert added pressure on the TCA cycle, leading to increases in ROS activity, although we could find no evidence for this in our analyses. Unfortunately, FX11 is unlikely to be suitable for clinical use particularly due to poor solubility and insufficient potency (44, 45). Although improved LDHA inhibitors have been developed, these have not been reported as suitable for in vivo use, but once such development occurs these may warrant testing in combination with MCT1 inhibitors.
In conclusion, this study has investigated the potential application of MCT1 inhibition as a therapeutic strategy for MYCN-amplified high risk neuroblastoma. In particular, the combination of MCT1 and LDHA inhibition was found to be highly synergistic in decreasing cell viability of neuroblastoma cells. An interesting novel discovery is the role of MYCN as a repressor of MCT4 while positively regulating MCT1. Although in vivo Kelly xenografts were resistant to MCT1 inhibitor treatment likely via various resistance mechanisms, importantly, our results suggest that there may be unique opportunities to develop MCT1 inhibition strategies for neuroblastoma therapy in combination with strategies that disrupt pyruvate homeostasis.
Materials and methods
General reagents
DMEM and RPMI media, FCS and Lipofectamine 2000 and RNAiMAX transfection reagents were obtained from Life Technologies Australia (Mulgrave, VIC). MCT1 inhibitor SR13800 and LDHA inhibitor FX11 were obtained from Merck Australia (Bayswater, VIC). Vincristine was obtained from Selleck (Houston, Texas). Deferasirox was obtained from Ontario Chemicals (Canada). All other chemicals were obtained from Sigma-Aldrich Australia (Castle Hill, NSW).
Reactive oxygen species (ROS) assay and cell cycle analysis
For ROS assays, after drug treatment for 18h, cells were incubated with 2′,7′-Dichlorofluorescein diacetate (DCF) (Sigma-Aldrich) for 1h at 37°C, harvested and analysed on a FACSCalibur (BD Biosciences) using FlowJo Software (Ashland, Oregon, USA).
Cell cycle analysis was performed as described (46) on cells treated with vehicle, SR13800 and/or vincristine for 18h, harvested and stained with propidium iodide (25μg/mL, Sigma-Aldrich) containing RNAse (2μg/mL)(Roche Diagnostics, Castle Hill, NSW), then fixed with 70% ethanol before flow analysis.
13C metabolic labeling, metabolomic and real-time metabolic analyses
13C-metabolic labeling was performed following published methods (47). Kelly were plated at 5 × 105/well in 6-well plates in RPMI/10% FBS overnight before medium was changed to MEM (Sigma-Aldrich #51416C) containing 10% FBS, NEAA, 2g/L glucose and 2mM 50% 13C-uniformly labeled glutamine. For 13C glucose labeling, the same medium was used except that the glucose component was 50% 13C-uniformly labeled glucose (Sigma-Aldrich) and glutamine was unlabelled (2mM). Inhibitor or vehicle was added to cells for 6h or 24h before harvesting for media and polar metabolite fractions (47). GC-MS metabolomic analyses were performed as published (48) on polar metabolite or media fractions. Media was also analysed for glucose and lactate using a YSI model 7100 enzyme analyzer.
For real-time metabolic analyses, 80,000 Kelly cells were plated in wells of a XF24 plate (Seahorse Bioscience), and next day media was replaced with base media containing 2g/L glucose and 200 mM glutamine and analysed on a Seahorse XF24-3 Bioanalyser. After determination of baseline ECAR and OCR, 1μM of SR13800 or vehicle was injected onto cells. Results were normalized by cell count in five fields of methanol-fixed cells stained with May-Grunwald (Sigma-Aldrich) and Giemsa (Sigma-Aldrich).
Generation of MCT4 promoter constructs and luciferase reporter assay
Two overlapping promoter regions of the human SLC16A3 (MCT4) gene were PCR-amplified and cloned into luciferase reporter vector pGL3- Basic (Promega) using standard procedures. Promoter 1 (−1600 to +500 from the transcriptional start site (TSS) of NM_001206950.1) and promoter 2 (−1000 to +600 from the TSS of NM_004207.3) (25) were PCR-amplified using primers (Supplementary Table S6) on genomic DNA from Kelly cells. MCT4 constructs or empty pGL3-Basic luciferase vector were transiently co-transfected with Renilla construct (Promega) into SHEP-TET/21N and after 4h, media was replaced with or without doxycycline (2μg/mL). At 48h post-transfection, cells were assayed using a Dual Luciferase Kit (Promega).
Animal studies
All animal studies were approved by the University of New South Wales Animal Care and Ethics Committee and conducted according to the Animal Research Act, 1985 (NSW, Australia) and the Australian Code of Practice for Care and Use of Animals for Scientific Purposes (2013). The toxicity of single doses of SR13800 (in 1:1:8 DMSO-Tween80-water) administered i.p. was evaluated in Balb/c mice, and the maximum tolerable dose of 30 mg/kg was evaluated for toxicity in combination with vincristine (0.2 mg/kg). 107 Kelly cells were subcutaneously engrafted into one flank of Balb/c nude mice with Matrigel Matrix (BD Biosciences). When tumor size reached 100 mg as measured by vernier calipers, mice were administered i.p. vehicle, SR13800 (30 mg/kg) for five days per week for the duration of the experiment and/or vincristine (0.2 mg/kg) for five consecutive days total. Mice were sacrificed when tumor size reached 1000 mg.
Statistical Analysis
Data are presented as means ± standard deviation (SD) of three independent experiments unless otherwise stated. All statistical analyses were performed using GraphPad Prism (San Diego, CA, USA). P<0.05 was considered statistically significant. Unless indicated, two-sided Student’s t-test was used to evaluate statistical differences. Combination index (CI) was calculated using the Calcusyn program (Biosoft, Cambridge, UK), where a CI<1 indicates synergy, CI = 1 indicates additive effect and CI>1 indicates antagonistic effect (49). Survival analyses were computed by the Kaplan-Meier method and compared between groups using the log-rank test. Univariate and multivariate analyses for event-free survival (EFS) and overall survival (OS) were performed using SPSS version 24 (IBM, Mainz, Germany) as described (50).
Supplementary Material
Acknowledgements
We thank Donna Lai and Sheng Hua from the Bosch Molecular Biology Facility, University of Sydney for support with Seahorse Analyses, and Alan Truong for technical support. This work was supported by a Balnaves Foundation Young Investigator Grant (DMTY) and a National Health and Medical Research Council Program Grant APP1016699 (MH, MDN).
Footnotes
Competing Interests: The authors declare there are no competing financial interests in this work.
References
- 1.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010; 24: 65–86. [DOI] [PubMed] [Google Scholar]
- 2.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. 2016; 2: 16078. [DOI] [PubMed] [Google Scholar]
- 3.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009; 27: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013; 3: a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008; 105: 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int J Biochem Cell Biol. 2015; 59: 167–181. [DOI] [PubMed] [Google Scholar]
- 7.Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012; 22: 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halestrap AP, Wilson MC. The monocarboxylate transporter family--role and regulation. IUBMB Life. 2012; 64: 109–119. [DOI] [PubMed] [Google Scholar]
- 10.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017; 171: 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 118: 3930–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010; 9: 3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012; 72: 5130–5140. [DOI] [PubMed] [Google Scholar]
- 14.Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. [Review]. 2016; 35: 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao D, Ren P, Su H, Yue M, Xiu R, Hu Y, et al. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget. 2015; 6: 40655–40666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polanski R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014; 20: 926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014; 74: 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong CS, Graham NA, Gu W, Espindola Camacho C, Mah V, Maresh EL, et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep. 2016; 14: 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis NJ, Mooney L, Hopcroft L, Michopoulos F, Whalley N, Zhong H, et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget. 2017; 8: 69219–69236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan L, Xiu R, Ren P, Yue M, Su H, Guo G, et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene. 2016; 35: 3037–3048. [DOI] [PubMed] [Google Scholar]
- 21.Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013; 4: e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. 2015; 235: 90–100. [DOI] [PubMed] [Google Scholar]
- 23.Groninger E, Meeuwsen-De Boer GJ, De Graaf SS, Kamps WA, De Bont ES. Vincristine induced apoptosis in acute lymphoblastic leukaemia cells: a mitochondrial controlled pathway regulated by reactive oxygen species? Int J Oncol. 2002; 21: 1339–1345. [DOI] [PubMed] [Google Scholar]
- 24.Le Floch R, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM, et al. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2011; 108: 16663–16668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006; 281: 9030–9037. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y, Rempe DA. Prophylactic neuroprotection against stroke: low-dose, prolonged treatment with deferoxamine or deferasirox establishes prolonged neuroprotection independent of HIF-1 function. J Cereb Blood Flow Metab. 2011; 31: 1412–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang XL, Yan ZW, Sheng WW, Xiao J, Zhang ZX, Ye ZB. Activation of hypoxia-inducible factor-1 ameliorates postischemic renal injury via inducible nitric oxide synthase. Mol Cell Biochem. 2011; 358: 287–295. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan LB, Gui DY, Heiden MGV. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016; 16: 680–693. [DOI] [PubMed] [Google Scholar]
- 29.Galluzzi L, Kepp O, Heiden MGV, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. [Review]. 2013; 12: 829–846. [DOI] [PubMed] [Google Scholar]
- 30.Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, et al. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014; 20: 280–294. [DOI] [PubMed] [Google Scholar]
- 31.Valli E, Trazzi S, Fuchs C, Erriquez D, Bartesaghi R, Perini G, et al. CDKL5, a novel MYCN-repressed gene, blocks cell cycle and promotes differentiation of neuronal cells. Biochim Biophys Acta. 2012; 1819: 1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gherardi S, Valli E, Erriquez D, Perini G. MYCN-mediated transcriptional repression in neuroblastoma: the other side of the coin. Front Oncol. 2013; 3: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fotsis T, Breit S, Lutz W, Rossler J, Hatzi E, Schwab M, et al. Down-regulation of endothelial cell growth inhibitors by enhanced MYCN oncogene expression in human neuroblastoma cells. Eur J Biochem. 1999; 263: 757–764. [DOI] [PubMed] [Google Scholar]
- 34.Qing G, Skuli N, Mayes PA, Pawel B, Martinez D, Maris JM, et al. Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 2010; 70: 10351–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noble RA, Bell N, Blair H, Sikka A, Thomas H, Phillips N, et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. 2017; 102: 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006; 25: 4633–4646. [DOI] [PubMed] [Google Scholar]
- 37.Wagner W, Ciszewski WM, Kania KD. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun Signal. 2015; 13: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu R-h, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of Glycolysis in Cancer Cells: A Novel Strategy to Overcome Drug Resistance Associated with Mitochondrial Respiratory Defect and Hypoxia. Cancer Res. 2005; 65: 613–621. [PubMed] [Google Scholar]
- 39.Birsoy K, Wang T, Possemato R, Yilmaz OH, Koch CE, Chen WW, et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Genet. [10.1038/ng.2471]. 2013; 45: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy AG, Zage PE, Akers LJ, Ghisoli ML, Chen Z, Fang W, et al. The combination of the novel glycolysis inhibitor 3-BrOP and rapamycin is effective against neuroblastoma. Invest New Drugs. 2012; 30: 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin R-Y, Vera JC, Chaganti RSK, Golde DW. Human Monocarboxylate Transporter 2 (MCT2) Is a High Affinity Pyruvate Transporter. J Biol Chem. 1998; 273: 28959–28965. [DOI] [PubMed] [Google Scholar]
- 42.Pertega-Gomes N, Vizcaino JR, Felisbino S, Warren AY, Shaw G, Kay J, et al. Epigenetic and oncogenic regulation of SLC16A7 (MCT2) results in protein over-expression, impacting on signalling and cellular phenotypes in prostate cancer. Oncotarget. 2015; 6: 21675–21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thangaraju M, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009; 417: 379–389. [DOI] [PubMed] [Google Scholar]
- 44.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010; 107: 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colon M, et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013; 1: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tu Y, Cheng S, Zhang S, Sun H, Xu Z. Vincristine induces cell cycle arrest and apoptosis in SH-SY5Y human neuroblastoma cells. Int J Mol Med. 2013; 31: 113–119. [DOI] [PubMed] [Google Scholar]
- 47.Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011; 286: 42626–42634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ratnikov B, Aza-Blanc P, Ronai ZA, Smith JW, Osterman AL, Scott DA. Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget. 2015; 6: 7379–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004; 10: 7994–8004. [DOI] [PubMed] [Google Scholar]
- 50.Henderson MJ, Haber M, Porro A, Munoz MA, Iraci N, Xue C, et al. ABCC multidrug transporters in childhood neuroblastoma: clinical and biological effects independent of cytotoxic drug efflux. J Natl Cancer Inst. 2011; 103: 1236–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.