

Abstract
Quantification of iron is an important step to assess the iron burden in patients suffering from iron overload diseases, as well as tremendous value in understanding the underlying role of iron in the pathophysiology of these diseases. Current iron determination of total or labile iron, requires extensive sample handling and specialized instruments, whilst being time consuming and laborious. Moreover, there is minimal to no overlap between total iron and labile iron quantification methodologies—i.e. requiring entirely separate protocols, techniques and instruments. Herein, we report a unified-ferene (u-ferene) assay that enables a 2-in-1 quantification of both labile and total iron from the same preparation of a biological specimen. We demonstrate that labile iron concentrations determined from the u-ferene assay is in agreement with confocal laser scanning microscopy techniques employed within the literature. Further, this assay offers the same sensitivity as the current gold standard, inductively coupled plasma mass spectrometry (ICP-MS), for total iron measurements. The new u-ferene assay will have tremendous value for the wider scientific community as it offers an economic and readily accessible method for convenient 2-in-1 measurement of total and labile iron from biological samples, whilst maintaining the precision and sensitivity, as compared to ICP-MS.
Subject terms: Iron, Analytical biochemistry, Chemical biology
Introduction
Iron is an essential element for almost all living organisms; it serves as a prosthetic group for a variety of proteins, due to its high redox potential, involved in vital cellular processes, including energy metabolism, oxygen transport, detoxification of reactive oxygen species, and DNA synthesis1–3. Due to this, not only iron homeostasis but also iron redox-state homeostasis is strictly regulated4,5. Iron homeostasis is tightly regulated by dietary absorption, and undergoes a dynamic cycle of recycling and utilization6. Any changes in iron homeostasis can lead to conditions including iron overload diseases (e.g. thalassemia, hemochromatosis) or iron deficiency causing cognitive defects in children7. Excess cellular iron is shown to induce liver cirrhosis, cardiomyopathy and diabetes mellitus8,9. Localized iron accumulation is often noted in neurodegenerative diseases, including Friedreich’s Ataxia, Parkinson’s, Alzheimer’s disease, and Huntington disease8–10.
Our current understanding points to the fact that the pathogenesis of iron-induced diseases is attributed to the combination of increased iron along with prolonged exposure to the redox active labile iron pool (LIP)3,11,12. Consequently, the accurate, convenient and economic quantification of biological iron—both as total iron and labile iron—is of tremendous value to assess the iron burden in clinics as well as to study the biology of these disease conditions in academia.
Currently used approaches to quantify total iron in biological samples are atomic absorption spectroscopy (AAS)13–17, inductively coupled plasma mass spectrometry (ICP-MS)18–21, electron paramagnetic resonance spectroscopy22,23, and colorimetric methods17,18,21,24–29. Both ICP-MS and AAS are highly sensitive, with ICP-MS enabling iron detection as low as parts per trillion. With the exception of colorimetric methods, all other techniques require special instrumentations, extensive sample manipulation and laborious methodologies. The quantification of labile iron pool has been reviewed elsewhere30 and relies on fluorescent sensors, with an iron binding moiety, that can be investigated via confocal scanning laser microscopy or flow cytometry11,12,31–44. The fluorescent-based approaches offer a methodically defined quantification of labile iron which is often difficult to emulate under different circumstances31,45–47. Further, these redox active iron (Fe2+) probes suffer from lack of metal ion selectivity48,49, need of additional chelators and specific reaction conditions such as acidic solution, indirect measurements of iron based on the free radical formation50, irreversibility of sensing mechanism, and unwanted quenching of fluorescence in the presence of other metal ions51.
As such, robust and easily accessible methods are required to measure iron levels, both labile and total iron, in different biological specimens including both in vitro and in vivo contexts. Colorimetric methods are great alternatives and have been used since the 1970s, however, this approach is still underdeveloped. Several groups have made progress towards more accurate determination of iron17,21,29. However, most of these methods are handicapped by their application only to total iron, typically in vitro conditions. Thus, there is a need to develop a convenient and economical benchtop assay that can quantitatively measure labile and total iron content from a wide variety of biological specimens. Further, an assay to differentiate and quantify redox active iron would profoundly impact the field.
Herein, we report of a unified-ferene assay (u-ferene assay) that enables a 2-in-1 quantification of both labile iron and total iron in biological samples ranging from cultured cells to more complex specimens, including organs, feces and urine, at the same level of precision ICP-MS.
Materials and methods
Ethics approval was obtained from the University of British Columbia (UBC) for experiments conducted at the Center for Blood Research. All ethics approval for animal studies were obtained from UBC and complied with the Canadian Council of Animal Care guidelines and animal care protocol (A18-0276). All animal studies were carried out at the Experimental Therapeutics laboratory, B.C. Cancer Research Centre, Vancouver, BC, Canada.
Materials
Eagle’s Minimum Essential Media (EMEM), Dulbecco’s Phosphate Buffered Saline (PBS), Pierce RIPA Lysis Buffer, bovine serum albumin (BSA) standards and Pierce Coomassie (also referred to as Bradford) protein assay kit was purchased from Thermo Fisher Scientific. Deferoxamine mesylate, fetal bovine serum, ferric ammonium citrate (FAC), ammonium acetate, sodium L-ascorbate, ferene (3-(2-Pyridyl)-5,6-di(2-furyl)-1,2,4-triazine-5′,5′′-disulfonic acid disodium salt), ICP-MS grade iron standards, and concentrated nitric acid were obtained from Sigma-Aldrich unless otherwise mentioned Fast-prep tubes (2 mL) were purchased from MP Biomedicals, LLC-Fisher. ICP-MS grade concentrated nitric acid was acquired from VWR.
Buffers and standards
Preparation of ammonium acetate buffer
Ammonium acetate buffer (pH 4.5, 2.5 M) was prepared using ammonium acetate and glacial acetic acid.
Preparation of iron standards
Iron standards were prepared from ferric chloride in 2% nitric acid, ranging from 0 to 1000 μM. These standards are prepared by mass; mass of ferric chloride and mass of 2% nitric acid are taken into account to determine the concentrations.
Cell culture and treatments
Iron loading cells
Hepatocarcinoma cell line, HepG2 (ATCC HB-8065) cells with passage numbers between 4 to 9, were seeded at 500,000 cells per well in a 12 well-plate and grown for 2 days. Cells were then loaded with iron; cells were treated with media containing 200 μM iron from ferric ammonium citrate (18 mol% iron) each day for 2 days. After 2 days of iron loading, cells were maintained for an additional day in media without iron. Prior to any analysis, cells were washed with 2–3 times PBS.
Cell lysate preparation
HepG2 cells were scraped, pelleted and washed with PBS thrice at 500 g for 5 min. The supernatant was aspirated and cells were lysed in at least 300 μL Pierce RIPA buffer (Thermo Fisher Scientific) with intermittent sonication for 20 min. After lysis, cell debris was pelleted by centrifuging at 21,000 g for 10 min. The supernatant was quantitatively transferred into a clean Eppendorf tube for further analysis.
Protein measurement
Protein content in cell lysates were measured using the Bradford assay, following manufacturer’s protocol. In brief, working solution was prepared using reagent A and B at a 50:1 ratio, respectively. BSA standards (10 μL) and cell lysates (10 μL) were added into a 96 well plate. Then, 200 μL of Bradford working solution were added to each well. This was kept at 37 °C for 30 min, then cooled to room temperature for 5 min. Absorbance was read at 562 nm. Protein concentrations in cell lysates were interpolated using the standard curve generated.
Iron overload mouse model
All the animal studies were carried out at the Experimental Therapeutics laboratory, B.C. Cancer Research Centre, Vancouver, BC, Canada. All the protocols were approved by the Institutional Animal Care Committee (IACC), UBC.
C57BL/6 mice were iron overloaded with five repeat doses of Fe-Dextran at 300 mg/kg52,53. Mice were administered with Fe-dextran for every 2 days intravenously (Day 1, 3, 5, 8, and 10). Feces and urine were collected in the last 2 days of the study. Mice were euthanized via CO2 asphyxiation (on Day 17) and 200 μL whole blood in EDTA & 150 μL plasma was collected. In addition, entire organs were collected following standard procedure; once removed, they were rinsed with PBS, weighed and frozen in liquid nitrogen and stored at –80 °C. Mouse samples are collectively referred to as biological samples.
Acid digestion of cell lysates and biological samples
Labile iron measurements do not require nitric acid digestion of biological samples, whereas total iron measurements require this acid digestion to release iron from all stores and proteins.
For iron analysis, mice organs were weighed in Fast-Prep tubes and homogenized in 0.5 mL water (BioSpec Product 96+, Bartlesville, OK). Cell lysates (typically 200 μL, unless specified otherwise) and biological samples were transferred into acid-washed glass vials and dried at 100 °C to 120 °C. The evaporation of water hastens the digestion of organic samples with nitric acid. Then, all biological samples were digested with concentrated nitric acid (HNO3)—samples were maintained at 100 °C to 120 °C and small volumes (0.5 to 1.0 mL) of concentrated nitric acid were repeatedly added over the course of 5 days. All digested samples were dried at 120 °C, then cooled to room temperature and weighed.
All the required dilutions were performed by mass. Acid-digested biological samples were resuspended in 4% HNO3 and weighed again; cell lysates and plasma samples were resuspended in 200 μL, and biological samples (organ homogenates, blood, urine and feces) were resuspended in 1.0 mL. All biological samples were further diluted in 2% HNO3 for the quantification of iron, as outlined in the Supplementary Table 1. HNO3 (2%) is used for final dilutions for the longevity of vacuum pumps required by ICP-MS.
Absorbance measurements
For ferene-based iron detection, all mixtures of samples and standards with ferene-containing working solution were spun at 15,000 g for 5 min. Then, 200 μL were transferred into a 96 well plate for absorbance measurements. Absorbance was recorded at 595 nm using a SpectraMax 190 microplate reader from Molecular Devices. Iron concentrations were interpolated from the standard curve generated from the iron standards. These concentrations were normalized to the amount of protein analyzed (i.e., nmole of iron per mg of protein).
The development of the ferene-based iron assay
A modified ferene assay was developed to analyze the labile and total iron content by manipulating ascorbic acid concentration. This section outlines three subsequent methods for the development of this assay, (i) determining iron concentrations in buffer conditions, (ii) distinguishing total and labile iron in cell lysates, and (iii) distinguishing labile and chelatable iron in cell lysates.
Determining iron concentrations in buffer conditions
Preparation of analytes and working solution
Three analytes were prepared with desired concentrations in distilled water;
(Fe) 100 μM of free iron.
(DFO-Fe) 100 μM of iron pre-chelated with 2 mM deferoxamine. 100 μM of iron solution in distilled water was incubated with 2 mM DFO for 48 h.
(DFO) 2 mM free deferoxamine.
Nine working solutions were prepared, each with 5 mM ferene in ammonium acetate buffer (pH 4.5, 2.5 M) with varying concentration of ascorbic acid (0, 1, 5, 10, 25, 50 100, 250, and 1000 mM). All the solutions were filtered (0.2 μm PVDF syringe filter) before use.
Determination of iron concentration
Iron content of the three analytes was measured in nine different sets of working solutions. A fresh calibration curve was generated using iron standards ranging from 0 to 400 μM (as outlined in 2.2.) for each set of working solution. This assay has been conveniently outlined in Supplementary Table 2.
Each set has 100 μL of analyte (either Fe, DFO-Fe, or DFO) and iron standards (eight samples) in separate Eppendorf tubes. To each Eppendorf tube, 100 μL of ammonium acetate buffer (pH 4.5, 2.5 M) and 120 μL working solution were added. The resultant solution was vortexed and left overnight at room temperature. Absorbance was measured as described earlier in “Absorbance measurements”.
Iron concentrations in different analytes in the presence of varying ascorbic acid concentrations were determined by interpolating from the standard curve generated using iron standards (Supplementary Fig. S1). Calibration curves were generated for nine different ascorbic acid concentrations.
Distinguishing total and labile iron in iron overloaded HepG2 cell lysates
Iron overload HepG2 cell lysates were prepared as outlined previously. We define labile iron as iron chelated and detected by ferene using a working solution with a specific low ascorbic concentration (10 mM for final assay-see “Labile iron measurements using the u-ferene assay”) from undigested samples. Similarly, total iron is defined as iron chelated and detected by ferene using a working solution with a particular high ascorbic concentration (1 M for final assay-see “Total iron measurements using the u-ferene assay”) from nitric acid digested samples. In order to distinguish labile and total iron, different working solutions with varying ascorbic acid concentrations were investigated.
Measurement of ferene-bound iron
A set of four working solutions were prepared, each with 5 mM ferene in ammonium acetate buffer (pH 4.5, 2.5 M) with four varying ascorbic acid concentrations (10, 50, 250, and 1000 mM). For labile iron measurements, 100 μL of as made cell lysates were transferred into separate Eppendorf tubes. For total iron measurements, 100 μL of nitric acid digested cell lysates (as outlined in 2.5) were aliquoted into separate Eppendorf tubes. Iron standards (100 μL) were also aliquoted into separate Eppendorf tubes.
Ammonium acetate buffer (pH 4.5, 2.5 M) (100 μL) and 120 μL of working solution were added to all tubes. The samples were vortexed and left overnight at room temperature. The absorbance was measured in 200 μL of the resultant solution, as outlined in “Absorbance measurements”.
Distinguishing labile and chelatable iron in cell lysates
Cell lysates for both non-iron treated and iron overloaded HepG2 cells were prepared as outlined in “Cell culture and treatments”. For the purpose of this investigation, chelatable iron is defined as the portion of labile iron that is chelated by iron chelators and subsequently prevents the ferene-based detection of iron using a working solution at a particular low concentration of ascorbic acid.
Chelator treatment of iron overloaded cell lysates
Deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX) (50 μM each) were prepared in PBS. Iron overloaded cell lysates were treated with chelators; 250 μL of iron loaded HepG2 cell lysates were transferred into a clean Eppendorf tube followed by 50 μL of 50 μM chelator—either DFO, DFP or DFX. As negative controls, 250 μL of non-iron loaded and 250 μL iron loaded HepG2 cell lysates were also transferred into clean Eppendorf tubes followed by 50 μL of PBS only. These samples were left overnight at room temperature.
Measurement of ferene-based iron
For labile iron: Working solution was prepared with 5 mM ferene in ammonium acetate buffer (pH 4.5, 2.5 M) with 10 mM of ascorbic acid. Cell lysates and iron standards (100 μL each) were transferred into separate Eppendorf tubes. 100 μL of ammonium acetate buffer (pH 4.5, 2.5 M) and 120 μL of working solution were added to each tube.
For total iron: Working solution was prepared with 5 mM ferene in ammonium acetate buffer (pH 4.5, 2.5 M) with 1 M of ascorbic acid. Acid digested cell lysates (100 μL) (as outlined in 2.5) and iron standards were transferred into separate Eppendorf tubes. Ammonium acetate buffer (100 μL) (pH 4.5, 2.5 M) and 120 μL working solution were added to each tube.
For iron quantification: These tubes were vortexed and left overnight. Absorbance was measured as described earlier in “Absorbance measurements”.
The unified-ferene assay (u-ferene assay): the finalized protocol
A unified-ferene (u-ferene) assay was developed to enable a 2-in-1 quantification of both labile iron and total iron in a wide variety of biological samples. In this assay, labile and total iron were defined by their experimental parameters; labile iron was defined as iron detected in undigested biological samples using 10 mM ascorbic acid in the working solution, and total iron was defined as iron detected in nitric acid digested biological samples using 1 M ascorbic acid in the working solution.
This assay has five components, (1) working solution, (2) ammonium acetate buffer (pH 4.5, 2.5 M), (3) iron standards, (4) the sample of interest (either cell lysates or biological mice samples), and (5) absorbance measurements. “Labile iron measurements using the u-ferene assay” outlines the measurement of labile iron and “Total iron measurements using the u-ferene assay”. outlines the measurement of total iron.
The working solutions:
The working solution is composed of ferene (5 mM) and ascorbic acid (either 10 mM for labile iron measurements, or 1 M for total iron measurements) in an ammonium acetate buffer. This is summarized in the Supplementary Table 3. The volume of working solution is determined by a 1:6 molar ratio between iron in the highest standard to ferene at 5 mM in working solution—i.e. when using 100 μL samples, working solution is calculated such that there is 6 times more moles of ferene than the moles of iron present in 100 μL of 1000 μM iron standard. Hence, this assay is applicable for investigations requiring larger sample volumes.
Ammonium acetate buffer (see also “Buffers and standards”)
Ammonium acetate buffer was added at a 1:1 volume equivalence to all biological samples and iron standards, i.e. when using 100 μL samples, 100 μL of buffer was added.
Iron standards (see also “Buffers and standards”)
When using iron standards for u-ferene assay, 1000 μM was used at the highest iron standard, however, this was not considered due to SpectraMax 190 microplate reader’s limit of detection. Iron standards were prepared using FeCl3 because this is a primary standard used in ICP-MS. The iron standard curve was compared to another primary source, FeSO4, as well to further validate the labile iron measurements and total iron measurements using the u-ferene assay (Supplementary Fig. S5).
Sample of interest (cell lysates and/or biological samples) (see also “Cell culture and treatments” to “Acid digestion of cell lysates and biological samples”)
Different samples are used. Samples of interest can vary in specimen and complexity, including cell culture lysates to homogenates from mouse tissue samples. It is important to highlight that for labile iron measurements, samples do not require nitric acid digestion, whereas total iron measurements require this digestion to release iron from all stores and proteins.
Absorbance measurements
Details are given in “Absorbance measurements”.
Labile iron measurements using the u-ferene assay
Labile iron concentrations were determined in cell lysates and plasma samples. Samples (either cell lysates or plasma samples) (100 μL) and iron standards (100 μL) with concentrations ranging from 0 to 1000 μM were transferred into different clean Eppendorf tubes. Ammonium acetate buffer (pH 4.5, 2.5 M) (100 μL) and labile iron working solution (5 mM ferene and 10 mM ascorbic acid prepared in ammonium acetate buffer pH 4.5, 2.5 M) (120 μL) were added to all Eppendorf tubes. This mixture was vortexed and left overnight at room temperature. Absorbance was measured as described earlier in “Absorbance measurements”.
Total iron measurements using the u-ferene assay
Total iron concentrations were determined in cell lysates, plasma samples, organ homogenates, blood, urine, and feces. The nitric acid digested samples after their final dilution (200 μL) and iron standards (200 μL) ranging from 0 to 1000 μM were transferred into different clean Eppendorf tubes. 200 μL of ammonium acetate buffer (pH 4.5, 2.5 M) and 240 μL of total iron working solution (5 mM ferene and 1 M ascorbic acid prepared in ammonium acetate buffer pH 4.5, 2.5 M) were added to all Eppendorf tubes. This mixture was vortexed and left overnight at room temperature. Absorbance was measured as described in “Absorbance measurements”.
Validation of total iron concentrations using inductively coupled plasma mass spectrometry
Biological samples were nitric acid-digested and resuspended in 4% HNO3, as described previously. These samples were then appropriately diluted in 2% HNO3 with 10 ppb indium (Sigma Aldrich) as the internal standard, as outlined in Supplementary Table 1. Iron standards were also prepared in 2% HNO3 with 10 ppb indium, ranging from 0 to 400 ppb, to generate a standard curve. Iron counts were measured on Agilent 7700 series ICP-MS and total iron concentrations were determined from the standard curve.
Calcein-based labile iron
Intracellular labile iron was measured using Calcein acetoxymethyl ester (Cal-AM), described elsewhere54. This is a non-fluorescent dye that becomes fluorescent after enzymatic modification once it permeates the cell membrane55,56. This fluorophore binds iron stoichiometrically, which quenches its green fluorescence55,56. In short, cells were washed, trypsinized and pelleted at 500 g for 5 min. Then, the cells were resuspended in PBS with 0.2 μM of Cal-AM for 20 min at room temperature. Cellular calcein fluorescence was measured using Beckman Coulter’s flow cytometry. At least 10,000 cells were analyzed and fluorescence was measured by the 488 nm laser and the FITC emission filter (530/20 nm).
Statistical analysis
All data were performed in at least independent triplicates and presented with error bars that correspond to standard deviations. Technical replicates were also performed, but only independent triplicates were used for statistical analysis. All statistical analyses were performed using GraphPad Prism. The statistical tests and appropriate corrections were outlined in the figure legends.
Results
The role of ascorbic acid in iron content measurement by the ferene assay
In order to measure iron using the ferene assay, Fe(III) should be reduced to Fe(II), typically by ascorbic acid, followed by the bidentate chelation of Fe(II) by ferene to form a stable blue complex between pH 3 to 6, with a molar absorptivity of 34,500 L cm−1 mol−157. In addition to ascorbic acid being a potent reducing agent, our working hypothesis was that the concentration of ascorbic acid has strong influence on the decomplexation iron complexes in the analyte (e.g. DFO-Fe). For this purpose, we used DFO, a hexadentate iron (III) chelator, with a pFe3+ of 26 to illustrate the significance of ascorbic acid in iron content measurement by the ferene assay58–60. We first optimized the ascorbic acid concentration in both buffer and in vitro conditions. Iron was measured in three analytes—100 µM of free iron (Fe), 2000 µM DFO only (DFO), and 100 µM iron pre-chelated with 2000 µM DFO (DFO-Fe)—using a working solution composed of 5 mM ferene in ammonium acetate buffer and varying concentrations (0–250 mM) of ascorbic acid (Fig. 1A).
Figure 1.

The effect of changing the working solution’s ascorbic acid concentration on the measurement of ferene-chelatable iron. (A) Changes in OD 595 nm were measured for three analytes, 100 μM of iron, either free or pre-chelated with 2000 μM deferoxamine (DFO), when changing ascorbic acid. A concentration dependent increase in iron concentration was observed for iron pre-chelated with DFO. A two-way ANOVA was performed with Dunnett’s correction, using GraphPad Prism. **** represents p < 0.0001 (B) Labile iron pool, “LIP”, was quantified in iron overload HepG2 cell lysates. Ascorbic acid concentrations in the working solution were varied from 10 mM to 1 M. In LIP measurements, there was a significant increase in the detection of labile iron when using 250 mM (p < 0.0001) and 1 M (p = 0.0080). (C) Total iron was quantified in the same cell lysates after digestion with nitric acid. Ascorbic acid concentrations also varied from 10 mM to 1 M. In total iron measurements, there was a significant increase in iron concentration when using 250 mM (p = 0.0421) and 1 M (p < 0.0001) ascorbic acid. One-way ANOVA was performed with Dunnett’s correction using GraphPad Prism. (D) Total iron was quantified using both ICP-MS and the u-ferene (5 mM ferene and 1 M ascorbic acid) assay in unloaded and iron overloaded HepG2 cells. Similar results were obtained for both ICP-MS and the u-ferene assay; no significant difference is observed between either the control or the iron overload cells. All experiments were done in at least triplicates. Error bars show standard deviations. Statistical analyses were performed using GraphPad Prism.
At concentrations of ascorbic acid lower than 10 mM, there is a significant difference between iron concentrations measured in Fe analyte and DFO-Fe analyte, p < 0.0001 at all concentrations. In other words, there is a significant difference between the iron measured in the two analytes. Interestingly, at ascorbic acid concentrations of 25 mM and above, the iron concentrations measured by ferene in free iron (Fe) and DFO-Fe are not significantly different. DFO only samples show no detection of iron, as expected. This data illustrates that concentration of ascorbic acid has a strong role on the removal of iron from Fe-DFO complex. This observation is of particular interest because it warrants further investigation into the utility of ascorbic acid concentration to discern between labile and total iron with the biological context—labile iron is redox active iron within the cells and total iron is all iron, both redox active and inactive.
The role of ascorbic acid in labile and total iron content measurement by the ferene assay
Next, the role of ascorbic acid was investigated to distinguish the measurement of labile and total iron from cell lysates. HepG2 cells were iron loaded with iron and lysates were prepared. Labile and total iron was measured in cell lysates before (Fig. 1B) and after (Fig. 1C) nitric acid digestion. Four different working solutions were used to measure iron content, consisting of 5 mM ferene and varying ascorbic acid concentrations (10, 50, 250 and 1000 mM). It is important to highlight two aspects. First, only iron that is chelated by ferene enables iron measurements and that ferene chelatable iron is dependent on the ascorbic acid concentrations present in the working solution. Second, cellular iron concentrations are reported as a ratio of iron concentration to protein concentration rather than iron concentration alone61–63 or relative to the number of cells21,64–67. We anticipate that protein concentrations better account for fluctuations associated with handling of cells and reflect changes due to cell death.
In iron overload cell lysates without nitric acid digestion, no difference is observed in the ferene-based iron detection from working solutions with 10 mM or 50 mM ascorbic acid. However, significantly more iron was quantified as ascorbic acid concentration increased from 50 to 250 mM and then from 250 to 1000 mM (p < 0.0001 and p = 0.0080, respectively). This increase is suggestive of iron being released from iron complexes in the undigested cell lysates.
Accounting for the observations made from Fig. 1A, 10 mM of ascorbic acid yields a significant difference in iron detection when comparing free iron and pre-chelated DFO iron, while not being significantly different in iron overloaded cell lysates. Therefore, we can define labile iron as iron measured in undigested samples by 5 mM ferene using a working solution with 10 mM ascorbic acid. The labile iron concentration determined represents the labile iron from various intracellular compartments.
Figure 1C shows the iron determined in iron overload cell lysates that underwent nitric acid digestion. The total iron concentrations determined were significantly increased when comparing working solutions with 50 mM and 250 mM ascorbic acid, and again when comparing 250 mM and 1000 mM ascorbic acid (p = 0.0421 and p < 0.0001). Based on this data, we defined the total iron as iron measured in acid-digested samples by 5 mM ferene using a working solution with 1 M ascorbic acid. The requirement for high ascorbic acid concentrations is likely due to two reason; first, more iron is available after acid-digestion, and second, more of this iron is present in the ferric form which needs to be reduced. This is further corroborated by ICP-MS analysis of these samples, which shows no significant differences in the total iron measurements (Fig. 1D).
Distinguishing “labile” and “chelatable” iron using a 2-in-1 u-ferene assay
Building on the previous observation, we next sought to validate iron measurements using our developed 2-in-1 u-ferene assay. To determine the labile iron pool (LIP) and chelatable labile iron, undigested iron overload HepG2 cells lysates were prepared with and without treatment of clinically approved iron chelators—deferoxamine (DFO), deferiprone (DFP) and deferasirox (DFX)—at 50 µM. Iron concentrations were measured in both sets of samples and are shown in Fig. 2. Chelatable labile iron is defined as the portion of the labile iron that is chelated by the iron chelators. DFO and DFP treatment displayed a significant decrease in labile iron when compared to the untreated iron overloaded cell lysates (p < 0.0001 and p = 0.0431, respectively) (Fig. 2A). Total iron was measured using the total iron protocol and, expectedly, no significant changes were observed (Fig. 2B). Further, there is reproducibility of the total iron content with previous experiments, as shown in Fig. 1C, and ICP-MS, Fig. 1D. Of great importance, the data speaks to the importance of ascorbic acid concentration to differentiate between total and labile iron, as well as chelatable iron with respect to different chelators (Supplementary Fig. S3).
Figure 2.
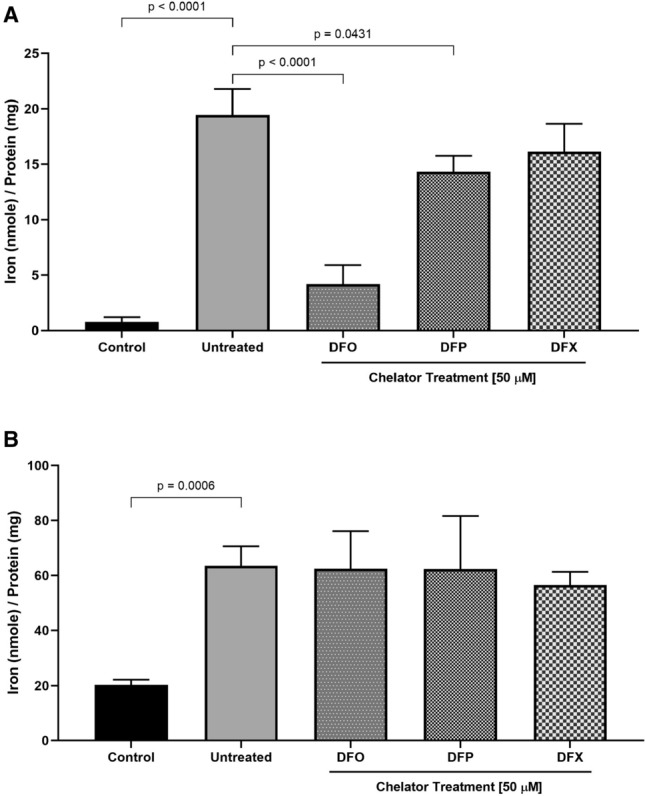
The confirmation of measuring chelatable labile iron in vitro. HepG2 cells were loaded with 200 μM of iron using FAC. Cells were lysed and these lysates were treated with either deferoxamine (DFO), deferiprone (DFP) and deferasirox (DFX) at 50 μM overnight. (A) LIP—measured using 5 mM ferene and 10 mM ascorbic acid in lysates. Chelatable labile iron were measured in cell lysates with iron chelator treatments. Labile iron was measured in cell lysates without iron chelator treatments. (B) Total iron—measured in nitric acid digested cell lysates followed by the ferene-based assay for total iron (5 mM ferene and 1 M ascorbic acid). One-way ANOVA was performed with Tukey’s correction using GraphPad Prism.
Quantification of total iron in various tissue specimens using the u-ferene assay
Total iron content from tissue specimens are shown in Fig. 3. The tissue samples were from iron overloaded mice developed by injecting iron-dextran52,53. Iron concentrations determined either via ICP-MS and the u-ferene assay from different tissue samples are almost identical (Fig. 3A), irrespective of the complexities associated with tissue specimens, which confirms the accuracy and utility of this benchtop technique. In the contest of this experiment, accuracy of the u-ferene assay refers to the comparison of the iron concentrations determined from ICP-MS since ICP-MS is the current gold standard for elemental analysis18. In addition, like ICP-MS, the u-ferene assay is able to measure a wide range of iron concentrations—from 10 μg iron in the whole heart to 10.300 mg iron in the whole liver (1030-fold range of concentration).
Figure 3.
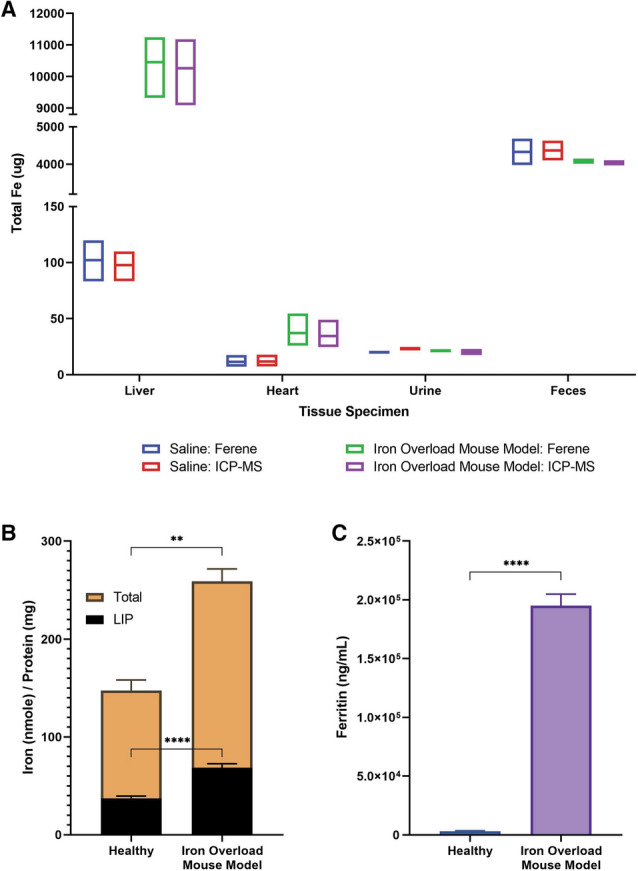
Quantification of total iron in various tissue specimens; organs (liver and heart), fluids (urine and plasma), and solid excrements (feces). (A) Total iron was measured in both saline (red and blue) and iron-dextran iron overload mouse model groups (green and purple) comparing the ferene assay (blue and green) to ICP-MS (red and purple). The total iron measurements used 5 mM ferene and 1 M ascorbic acid as the working solution in the u-ferene assay. (B) Plasma labile iron (5 mM ferene and 10 mM ascorbic acid) and total iron (5 mM ferene and 1 M ascorbic acid) were measured from healthy and iron overloaded mice using the ferene assay. (C) Plasma ferritin concentrations were measured using an ELISA assay from health and iron overload mice. Three biological replicates were measured per group. Unpaired t-test were performed using GraphPad Prism. **** represents p < 0.0001 and ** represents p < 0.01.
Furthermore, we applied the u-ferene assay to plasma specimens as well; significantly higher concentrations of both labile and total plasma iron (p < 0.0001 and p = 0.0055, respectively) than healthy mice controls were observed (Fig. 3B). The increase in total plasma iron was further corroborated by the significant elevation in ferritin concentrations (p < 0.0001) (Fig. 3C). This demonstrates the potential of using this convenient assay to investigate differences in labile plasma iron (and non-transferrin bound iron (NTBI)) in various disease states.
Discussion
Robust and accessible methods are required to measure iron levels, both labile and total iron, in different tissue specimens. Current approaches to quantitate iron exist, however they require specialised methodologies, extensive sample preparation and costly equipment13–23. Colorimetric methods provide an accessible, economic and are routinely refined for better sensitivities, convenient sample preparation and robust applications. Several chromogenic substrates, specific for chelatable ferrous iron, absorb strongly in the UV/Vis region, including bathophenanthroline sulfonate, ferrozine or ferene17,21,25,26,68–70. Reimer et al. developed a common assay for quantification of iron in astrocytes, using permanganate-mediated digestion and ferrozine-based colorimetric iron determination17. Hedayati et al. then improved Reimer et al. assay’s sensitivity by using nitric acid-mediated digestion and ferene-based colorimetric iron determination21. Ferene binds iron optimally between pH 3 to 6, with pFe2+ of 1557. However, the quantification of both labile and total iron from the same biological samples is yet to be reported using a ferene assay. It is worthwhile to mention that the quantification of labile iron pools are often tied to the specific fluorescent sensor, environmental variations within cells, and solvent systems used for calibrations47. As such, it is difficult to directly translate these concentrations to other modes of analysis.
The labile iron pool is critical for cellular iron metabolism and exists in dynamic equilibrium between different organelles3,11,71. Current methods to trace and quantitate labile iron concentration measure changes in fluorescence from iron sensors, which either turn-on (fluorescence enhances) and turn-off (fluorescence quenches) in the presence of iron11,31. These fluorescence techniques to measure metals is a rapidly growing approach offering spatial and temporal resolution. However, the use of fluorescence sensors are greatly hindered by limited commercially available probes, poor metal selectivity, mediocre signal-to-noise ratio, and requires specialized instruments (fluorescence microscopy or flow cytometry)11,31. Taken together, this undermines the absolute quantification of the labile iron pool.
In this investigation, we report an easy-to-use ferene-based protocol, u-ferene assay, that enables a 2-in-1 detection of both total and labile iron in biological samples, by varying ascorbic acid concentrations. Ascorbic acid is one of the low-molecular ligands that reduces ferric iron to ferrous iron, which then enables ferene, a bidentate chelator with a pFe2+ of 15, to chelate ferrous iron6,57,72. Moreover, this reducing agent also has the potential to interrupt stably chelated iron. To assess the ferene-based detection of iron, iron pre-chelated with DFO, was used. We used DFO as our model iron chelator because it has been extensively investigated and offers a particular advantage due to its hexacoordinate sphere with iron and high affinity (pFe3+ of 26)58–60. Based on our preliminary screening experiments, it is confirmed that ascorbic acid concentration has a central role in these measurements; significant release of iron from DFO-Fe coordination sphere was observed with the concentration of ascorbic acid above 10 mM (Fig. 1A). In other words, ascorbic acid above 10 mM is sufficient to reduce all iron pre-chelated with DFO. Therefore, we chose 10 mM ascorbic concentration for the determination of labile iron. This is because the iron measured at 10 mM ascorbic concentration sufficiently discerns between the iron only analyte and the iron pre-chelated with DFO analyte (p < 0.0001). It is important to note that these working concentrations may change when using other iron chelators. Within a biological context, DFO bound iron serves as an indicator for stably bound iron, and at 10 mM ascorbic acid, we can conservatively discern between chelatable and unchelatable iron.
In this report, we defined labile iron as redox active iron quantified using 5 mM ferene and 10 mM ascorbic acid from a biological sample. The labile iron concentrations in non-iron loaded HepG2 cells were found to be 0.8 nmole ± 0.2 nmole/mg of protein (4 μM) (Fig. 2A) by the u-ferene assay. This is consistent with the reported values using fluorescent based labile iron determination methods. It has been reported that there is an average of 2 to 5 μM (equivalent to 0.3 to 0.9 nmole) of intracellular chelatable iron within normal rat hepatocytes46,47,73. Moreover, the increase in labile iron after iron loading HepG2 cells was also shown using calcein as a metallosensor for intracellular labile iron (Supplementary Fig. S2). While there is agreement in the labile iron concentrations determined by two different techniques in two different hepatocyte preparations, it is important to note that these diversities pose a challenge to validate labile iron concentrations thoroughly.
On the other hand, total iron is defined as all iron, redox active and unchelatable stored iron, quantified using 5 mM ferene and 1 M ascorbic acid after nitric acid digestion in a biological sample. We noticed 10 mM ascorbic acid concentration is not enough to completely reduce the iron after acid digestion of cell lysates (Fig. 1C) and ferene-based iron detection increased with ascorbic acid concentration. We hypothesize that this is due to the equilibrium between Fe3+ and Fe2+ under acidic conditions requiring more ascorbic acid to reduce the iron, since all the stable and otherwise unchelated iron have been released via nitric acid digestion. At 1000 mM ascorbic acid concentration, the total iron measurements agreed with ICP-MS analysis (Fig. 1D). Thus, by optimizing for ascorbic acid concentration needed to sufficiently reduce iron for the detection by ferene, we were able to quantitate both labile iron and total iron (Fig. 1).
To further validate this assay’s capacity to quantify chelatable iron in cell lysates, the labile iron pool was perturbed by the introduction of high-affinity iron (III) chelators with stability constants above log(20) and the iron content was determined by the u-ferene assay (Fig. 2A). The monitoring of different cellular iron pools is of utmost importance, especially within the field of hereditary and transfusion-dependent iron overload, since the production of reactive oxygen species has been correlated to changes in the labile iron pool74,75. Cell lysates were used for two reasons; first, it ensured changes in the labile iron pool without affecting the total iron and second, it demonstrates the use of the same lysate for both labile and total iron determination. Upon treatment with DFO and DFP, the labile iron pool is significantly lowered because these chelators strongly bind to the intracellular ferric iron hence reducing the measurable labile iron pool (Fig. 2A). DFX treatment shows only a slight decrease in the labile iron pool. This may speak to the tunability of this assay, with respect to ascorbic acid concentration, for other chelators (Supplementary Fig. S3). Of note, total iron concentration is not different across treatments since only the lysates were manipulated.
Next, total iron was quantified using the same u-ferene assay with two modifications; first, biological samples underwent nitric acid digestion to release all iron21 and second, 1 M ascorbic acid concentration in the working solution was used. Total iron concentrations loaded within HepG2 cells were in the range of 3.5 to 5.0 μg iron per mg protein (70 to 90 nmole iron per mg protein across the different iron loading concentrations), which has been validated by ICP-MS (Fig. 1D) and is in agreement with earlier reports further confirming the utility of our assay76–78. Moreover, the total iron content did not change despite increasing iron loading concentrations. This pattern of iron accumulation has been observed to be unique in HepG2 cells (Fig. 2B)78.
To further explore the robustness of the u-ferene assay, we determined iron content from various biological specimens collected from mice (Fig. 3 & Supplementary Fig. S4) and compared the data with ICP-MS. For the sake of clarity, we have loosely categorised the different biological specimens in three groups; fluids (plasma and urine), organs (liver, spleen, kidneys, and heart) and solid excrement (feces). The total iron determined from the u-ferene assay is almost identical to that determined from ICP-MS in all biological specimens (Fig. 3)—the precision (i.e. standard deviations) of the u-ferene assay parallels that of ICP-MS. This also highlights the sensitivity of the u-ferene assay by offering a wide range of concentrations of iron detection (0 to 1000 μM), principally through acid-cleaned vials, sample manipulation and appropriate dilutions.
In addition, plasma iron from both groups were quantified with respect to labile and total iron. Both labile and total iron measurements were significantly elevated in the iron overload group when compared to the saline group (p < 0.0001 and p = 0.0055, respectively). This increase in total iron was further supported by the increase in ferritin concentration, as measured by the ferritin ELISA kit. Plasma, serum and blood specimens are readily tested clinical samples, and speak to potential adaptability of this assay. To further explore that source of the labile iron measured by the u-ferene assay and to determine whether the experimental conditions used in labile iron pool measurements could remove iron from proteins, in vitro experiments were performed using holo-transferrin (holo-Tf) (Supplementary Fig. S6). This suggests that some iron can be stripped and measured from proteins, like transferrin, using a working solution for labile iron measurements (5 mM ferene and 10 mM ascorbic acid). This might explain the elevated labile iron measured (a combination of potentially chelatable free iron (non-transferrin bound iron (NTBI)) and some additional iron released from proteins) in the healthy control (Fig. 3A). On the other hand, the working solution for total iron measurements (5 mM ferene and 1 M ascorbic acid) in concert with nitric acid digestion is sufficient to measure all iron since the u-ferene assay measures the same concentration of iron as provided by Sigma Aldrich's certificate of analysis.
Taken all together, using our u-ferene colorimetric benchtop assays enables the quantification of iron—both labile and total iron—in a variety of biological specimens. This assay offers the precision and sensitivity similar to that of ICP-MS and fluorescent based methods with the added cost-saving and readily accessible methodology.
Conclusions
We reported the development of an economic and readily accessible alternative assay for the measurement of both labile and total iron using the unified-ferene (u-ferene) assay, whilst maintaining the accuracy and sensitivity as compared to the current gold standard. This assay enables a 2-in-1 quantification of iron from the same preparation of a biological specimen, circumventing tedious, expensive and laborious methodologies. We demonstrated that labile iron can be distinguished from total iron using different ascorbic acid concentrations in the working solutions as well as performing acid-digestion on samples. This method provides adequate quantification of both chelatable and labile iron as well. Iron quantification, both labile and total iron, agree with literature as well as comparison with other currently available techniques. Quantification of iron is a necessity to assess the iron burden in patients suffering from iron overload diseases, as well as tremendous value in understanding the underlying role of iron in the pathophysiology of these diseases. Thus, the u-ferene assay brings considerable value for the wider scientific community.
Supplementary information
Acknowledgements
The authors thank the Macromolecular Hub (CBR) for the use of their research facilities.
Author contributions
Data were generated by U.A., S.A., A.G. and V.B. Data analysis was performed by U.A., S.A. and J.K. Manuscript was prepared by U.A., S.A. and J.K. with inputs from other authors. J.K. provided the grant support and supervision of the project.
Funding
The authors acknowledge the funding by Canadian Institutes of Health Research (CIHR), Natural Sciences and Engineering Council of Canada (NSERC), Canada Foundation for Innovation (CFI), and British Columbia Knowledge Development Fund. JNK holds a Career Investigator Scholar award from the Michael Smith Foundation for Health Research (MSFHR). SA acknowledges a MSFHR postdoctoral fellowship.
Data availability
All data is available in the main text or the supplementary materials.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Usama Abbasi and Srinivas Abbina.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-85387-z.
References
- 1.Koppenol WH. The Haber-Weiss cycle–70 years later. Redox Rep. Commun. Free Radic. Res. 2001;6:229–234. doi: 10.1179/135100001101536373. [DOI] [PubMed] [Google Scholar]
- 2.Anderson GJ. Mechanisms of iron loading and toxicity. Am. J. Hematol. 2007;82:1128–1131. doi: 10.1002/ajh.21075. [DOI] [PubMed] [Google Scholar]
- 3.Kruszewski M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Winn NC, Volk KM, Hasty AH. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight. 2020;5:2. doi: 10.1172/jci.insight.132964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallace DF. The regulation of iron absorption and homeostasis. Clin. Biochem. Rev. 2016;37:51–62. [PMC free article] [PubMed] [Google Scholar]
- 6.Lane DJR, et al. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta. 2015;1853:1130–1144. doi: 10.1016/j.bbamcr.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 7.Kassebaum NJ. The global burden of anemia. Hematol. Oncol. Clin. North Am. 2016;30:247–308. doi: 10.1016/j.hoc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Fleming RE, Ponka P. Iron overload in human disease. N. Engl. J. Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 9.Dev S, Babitt JL. Overview of iron metabolism in health and disease. Hemodial. Int. Int. Symp. Home Hemodial. 2017;21:S6–S20. doi: 10.1111/hdi.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons DA, et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- 11.Kakhlon O, Cabantchik ZI. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002;33:1037–1046. doi: 10.1016/S0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 12.Chutvanichkul B, Vattanaviboon P, Mas-Oodi S, Pratya Y, Wanachiwanawin W. Labile iron pool as a parameter to monitor iron overload and oxidative stress status in β-thalassemic erythrocytes. Cytometry B Clin. Cytom. 2018;94:631–636. doi: 10.1002/cyto.b.21633. [DOI] [PubMed] [Google Scholar]
- 13.Kimber CL, Mukherjee T, Deller DJ. In vitro iron attachment to the intestinal brush border. Am. J. Dig. Dis. 1973;18:781–791. doi: 10.1007/BF01070848. [DOI] [PubMed] [Google Scholar]
- 14.Slavin W. Methods in Enzymology. Academic Press; 1988. Atomic absorption spectrometry; pp. 117–145. [DOI] [PubMed] [Google Scholar]
- 15.Bosscher D, et al. In vitro availability of calcium, iron, and zinc from first-age infant formulae and human milk. J. Pediatr. Gastroenterol. Nutr. 2001;32:54–58. doi: 10.1097/00005176-200101000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Felice MR, et al. Post-transcriptional regulation of the yeast high affinity iron transport system. J. Biol. Chem. 2005;280:22181–22190. doi: 10.1074/jbc.M414663200. [DOI] [PubMed] [Google Scholar]
- 17.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 18.Jenner GA, Longerich HP, Jackson SE, Fryer BJ. ICP-MS—A powerful tool for high-precision trace-element analysis in Earth sciences: Evidence from analysis of selected USGS reference samples. Chem. Geol. 1990;83:133–148. doi: 10.1016/0009-2541(90)90145-W. [DOI] [Google Scholar]
- 19.Patil US, et al. In vitro/in vivo toxicity evaluation and quantification of iron oxide nanoparticles. Int. J. Mol. Sci. 2015;16:24417–24450. doi: 10.3390/ijms161024417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue Y, et al. Endoplasmic reticulum-mitochondria junction is required for iron homeostasis. J. Biol. Chem. 2017 doi: 10.1074/jbc.M117.784249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedayati M, et al. An optimised spectrophotometric assay for convenient and accurate quantitation of intracellular iron from iron oxide nanoparticles. Int. J. Hyperth. 2018;34:373–381. doi: 10.1080/02656736.2017.1354403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: Evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 2004;24:949–954. doi: 10.1161/01.ATV.0000124892.90999.cb. [DOI] [PubMed] [Google Scholar]
- 23.Woodmansee AN, Imlay JA. Methods in Enzymology. Academic Press; 2002. Quantitation of intracellular free iron by electron paramagnetic resonance spectroscopy; pp. 3–9. [DOI] [PubMed] [Google Scholar]
- 24.Viollier E, Inglett PW, Hunter K, Roychoudhury AN, Van Cappellen P. The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem. 2000;15:785–790. doi: 10.1016/S0883-2927(99)00097-9. [DOI] [Google Scholar]
- 25.Derman DP, et al. A systematic evaluation of bathophenanthroline, ferrozine and ferene in an ICSH-based method for the measurement of serum iron. Ann. Clin. Biochem. 1989;26(Pt 2):144–147. doi: 10.1177/000456328902600209. [DOI] [PubMed] [Google Scholar]
- 26.Artiss JD, Vinogradov S, Zak B. Spectrophotometric study of several sensitive reagents for serum iron. Clin. Biochem. 1981;14:311–315. doi: 10.1016/S0009-9120(81)91065-1. [DOI] [PubMed] [Google Scholar]
- 27.Carter P. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine) Anal. Biochem. 1971;40:450–458. doi: 10.1016/0003-2697(71)90405-2. [DOI] [PubMed] [Google Scholar]
- 28.Eskelinen S, Haikonen M, Räisänen S. Ferene-S as the chromogen for serum iron determinations. Scand. J. Clin. Lab. Invest. 1983;43:453–455. doi: 10.3109/00365518309168286. [DOI] [PubMed] [Google Scholar]
- 29.Jeitner TM. Optimized ferrozine-based assay for dissolved iron. Anal. Biochem. 2014;454:36–37. doi: 10.1016/j.ab.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 30.Hirayama T, Nagasawa H. Chemical tools for detecting Fe ions. J. Clin. Biochem. Nutr. 2017;60:39–48. doi: 10.3164/jcbn.16-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lv H, Shang P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics. 2018;10:899–916. doi: 10.1039/C8MT00048D. [DOI] [PubMed] [Google Scholar]
- 32.Prus E, Fibach E. Flow cytometry measurement of the labile iron pool in human hematopoietic cells. Cytom Part J. Int. Soc. Anal. Cytol. 2008;73:22–27. doi: 10.1002/cyto.a.20491. [DOI] [PubMed] [Google Scholar]
- 33.Espósito BP, Epsztejn S, Breuer W, Cabantchik ZI. A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal. Biochem. 2002;304:1–18. doi: 10.1006/abio.2002.5611. [DOI] [PubMed] [Google Scholar]
- 34.Jacobs A. An intracellular transit iron pool. Ciba Found. Symp. 1976;2:91–106. doi: 10.1002/9780470720325.ch5. [DOI] [PubMed] [Google Scholar]
- 35.Jacobs A. Low molecular weight intracellular iron transport compounds. Blood. 1977;50:433–439. doi: 10.1182/blood.V50.3.433.433. [DOI] [PubMed] [Google Scholar]
- 36.Chen H, et al. Citrate-based fluorophore-modified cellulose nanocrystals as a biocompatible fluorescent probe for detecting ferric ions and intracellular imaging. Carbohydr. Polym. 2019;224:115198. doi: 10.1016/j.carbpol.2019.115198. [DOI] [PubMed] [Google Scholar]
- 37.Gao G, et al. A simple and effective dansyl acid based “turn-on” fluorescent probe for detecting labile ferrous iron in physiological saline and live cells. Talanta. 2020;215:120908. doi: 10.1016/j.talanta.2020.120908. [DOI] [PubMed] [Google Scholar]
- 38.Mahapatra M, et al. Fluorescent terpolymers via in situ allocation of aliphatic fluorophore monomers: Fe(III) sensor, high-performance removals, and bioimaging. Adv. Healthc. Mater. 2019;8:1900980. doi: 10.1002/adhm.201900980. [DOI] [PubMed] [Google Scholar]
- 39.Kumar TU, Pawar S, Nag A, Bhattacharya A. Selective sensing of Iron by pyrrolo[2,3-c]quinolines. J. Fluoresc. 2019;29:271–277. doi: 10.1007/s10895-018-02337-1. [DOI] [PubMed] [Google Scholar]
- 40.Tümay SO, Haddad Irani-nezhad M, Khataee A. Design of novel anthracene-based fluorescence sensor for sensitive and selective determination of iron in real samples. J. Photochem. Photobiol. Chem. 2020;402:112819. doi: 10.1016/j.jphotochem.2020.112819. [DOI] [Google Scholar]
- 41.Arockiam JB, Ayyanar S. Benzothiazole, pyridine functionalized triphenylamine based fluorophore for solid state fluorescence switching, Fe3+ and picric acid sensing. Sens. Actuators B Chem. 2017;242:535–544. doi: 10.1016/j.snb.2016.11.086. [DOI] [Google Scholar]
- 42.Maity M, Bera K, Pal U, Khamaru K, Maiti NC. Sensing of iron(III) ion via modulation of redox potential on biliverdin protected silver nanosurface. ACS Appl. Nano Mater. 2018;1:6099–6111. doi: 10.1021/acsanm.8b01311. [DOI] [Google Scholar]
- 43.Taher MA, Asadollahzadeh H, Fazelirad H. Determination of trace amounts of iron by a simple fluorescence quenching method. Anal. Methods. 2015;7:6726–6731. doi: 10.1039/C5AY00859J. [DOI] [Google Scholar]
- 44.Başoğlu A, et al. Simple time-saving method for iron determination based on fluorescence quenching of an azaflavanon-3-ol compound. J. Agric. Food Chem. 2015;63:2654–2659. doi: 10.1021/jf505336d. [DOI] [PubMed] [Google Scholar]
- 45.Petrat F, de Groot H, Sustmann R, Rauen U. The chelatable iron pool in living cells: A methodically defined quantity. Biol. Chem. 2002;383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 46.Petrat F, de Groot H, Rauen U. Determination of the chelatable iron pool of single intact cells by laser scanning microscopy. Arch. Biochem. Biophys. 2000;376:74–81. doi: 10.1006/abbi.2000.1711. [DOI] [PubMed] [Google Scholar]
- 47.Petrat F, de Groot H, Rauen U. Subcellular distribution of chelatable iron: A laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 2001;356:61–69. doi: 10.1042/bj3560061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Praveen L, Reddy MLP, Varma RL. Dansyl-styrylquinoline conjugate as divalent iron sensor. Tetrahedron Lett. 2010;51:6626–6629. doi: 10.1016/j.tetlet.2010.10.043. [DOI] [Google Scholar]
- 49.Petrat F, Rauen U, Groot H. Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology. 1999;29:1171–1179. doi: 10.1002/hep.510290435. [DOI] [PubMed] [Google Scholar]
- 50.Chen J-L, et al. High selective determination iron(II) by its enhancement effect on the fluorescence of pyrene-tetramethylpiperidinyl (TEMPO) as a spin fluorescence probe. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2006;63:438–443. doi: 10.1016/j.saa.2005.04.057. [DOI] [PubMed] [Google Scholar]
- 51.Au-Yeung HY, Chan J, Chantarojsiri T, Chang CJ. Molecular imaging of labile iron(II) pools in living cells with a turn-on fluorescent probe. J. Am. Chem. Soc. 2013;135:15165–15173. doi: 10.1021/ja4072964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moon SN, et al. Establishment of secondary iron overloaded mouse model: Evaluation of cardiac function and analysis according to iron concentration. Pediatr. Cardiol. 2011;32:947–952. doi: 10.1007/s00246-011-0019-4. [DOI] [PubMed] [Google Scholar]
- 53.Yatmark P, et al. Iron distribution and histopathological characterization of the liver and heart of β-thalassemic mice with parenteral iron overload: Effects of deferoxamine and deferiprone. Exp. Toxicol. Pathol. 2014;66:333–343. doi: 10.1016/j.etp.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Breuer W, Epsztejn S, Cabantchik ZI. Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II) J. Biol. Chem. 1995;270:24209–24215. doi: 10.1074/jbc.270.41.24209. [DOI] [PubMed] [Google Scholar]
- 55.Tenopoulou M, Kurz T, Doulias P-T, Galaris D, Brunk UT. Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it? Biochem. J. 2007;403:261–266. doi: 10.1042/BJ20061840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas F, et al. Calcein as a fluorescent probe for ferric iron application to iron nutrition in plant cells. J. Biol. Chem. 1999;274:13375–13383. doi: 10.1074/jbc.274.19.13375. [DOI] [PubMed] [Google Scholar]
- 57.Hennessy DJ, Reid GR, Smith FE, Thompson SL. Ferene—a new spectrophotometric reagent for iron. Can. J. Chem. 1984;62:721–724. doi: 10.1139/v84-121. [DOI] [Google Scholar]
- 58.Hider RC, Kong X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 59.Spasojević I, Armstrong SK, Brickman TJ, Crumbliss AL. Electrochemical behavior of the Fe(III) complexes of the cyclic hydroxamate siderophores alcaligin and desferrioxamine E. Inorg. Chem. 1999;38:449–454. doi: 10.1021/ic980635n. [DOI] [PubMed] [Google Scholar]
- 60.Ma Y, Zhou T, Hider RC. pFe3+ determination of multidentate ligands by a fluorescence assay. Analyst. 2015;140:3603–3606. doi: 10.1039/C5AN00123D. [DOI] [PubMed] [Google Scholar]
- 61.Zanninelli G, et al. Chelation and mobilization of cellular iron by different classes of chelators. Mol. Pharmacol. 1997;51:842–852. doi: 10.1124/mol.51.5.842. [DOI] [PubMed] [Google Scholar]
- 62.Briley-Saebo K, et al. Hepatic cellular distribution and degradation of iron oxide nanoparticles following single intravenous injection in rats: Implications for magnetic resonance imaging. Cell Tissue Res. 2004;316:315–323. doi: 10.1007/s00441-004-0884-8. [DOI] [PubMed] [Google Scholar]
- 63.Huang X, et al. Ferrous ion autoxidation and its chelation in iron-loaded human liver HepG2 cells. Free Radic. Biol. Med. 2002;32:84–92. doi: 10.1016/S0891-5849(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 64.Gao J, Zhao N, Knutson MD, Enns CA. The hereditary hemochromatosis protein, HFE, inhibits iron uptake via down-regulation of Zip14 in HepG2 cells. J. Biol. Chem. 2008;283:21462–21468. doi: 10.1074/jbc.M803150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ge Y, et al. Effect of surface charge and agglomerate degree of magnetic iron oxide nanoparticles on KB cellular uptake in vitro. Colloids Surf. B Biointerfaces. 2009;73:294–301. doi: 10.1016/j.colsurfb.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 66.Spira D, et al. Labeling human melanoma cells with SPIO: in vitro observations. Mol. Imaging. 2016;15:1536012115624915. doi: 10.1177/1536012115624915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soenen SJH, Nuytten N, Meyer SFD, Smedt SCD, Cuyper MD. High intracellular iron oxide nanoparticle concentrations affect cellular cytoskeleton and focal adhesion kinase-mediated signaling. Small. 2010;6:832–842. doi: 10.1002/smll.200902084. [DOI] [PubMed] [Google Scholar]
- 68.Pieroni L, et al. Comparison of bathophenanthroline sulfonate and ferene as chromogens in colorimetric measurement of low hepatic iron concentration. Clin. Chem. 2001;47:2059–2061. doi: 10.1093/clinchem/47.11.2059. [DOI] [PubMed] [Google Scholar]
- 69.Ceriotti, F. & Cerlotti, G. Improved direct specific determination of serum iron and total iron-bindingcapacity. 6. [PubMed]
- 70.Iron Panel of the International Committee for Standardization in Haematology. Revised recommendations for the measurements of the serum iron in human blood. Br. J. Haematol.75, 615–616 (1990). [DOI] [PubMed]
- 71.Hider RC, Kong X. Iron speciation in the cytosol: An overview. Dalton Trans. 2013;42:3220–3229. doi: 10.1039/C2DT32149A. [DOI] [PubMed] [Google Scholar]
- 72.Sherman HG, et al. New perspectives on iron uptake in eukaryotes. Front. Mol. Biosci. 2018;5:2. doi: 10.3389/fmolb.2018.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma Y, de Groot H, Liu Z, Hider RC, Petrat F. Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes. Biochem. J. 2006;395:49–55. doi: 10.1042/BJ20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zanninelli G, et al. The labile iron pool of hepatocytes in chronic and acute iron overload and chelator-induced iron deprivation. J. Hepatol. 2002;36:39–46. doi: 10.1016/S0168-8278(01)00222-7. [DOI] [PubMed] [Google Scholar]
- 75.Cabantchik ZI. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014;5:2. doi: 10.3389/fphar.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ternes N, Scheiber-Mojdehkar B, Landgraf G, Goldenberg H, Sturm B. Iron availability and complex stability of iron hydroxyethyl starch and iron dextran—a comparative in vitro study with liver cells and macrophages. Nephrol. Dial. Transplant. 2007;22:2824–2830. doi: 10.1093/ndt/gfm315. [DOI] [PubMed] [Google Scholar]
- 77.Popovic Z, Templeton DM. Iron accumulation and iron-regulatory protein activity in human hepatoma (HepG2) cells. Mol. Cell. Biochem. 2004;265:37–45. doi: 10.1023/B:MCBI.0000044313.19574.c6. [DOI] [PubMed] [Google Scholar]
- 78.Hirsh M, Konijn AM, Iancu TC. Acquisition, storage and release of iron by cultured human hepatoma cells. J. Hepatol. 2002;36:30–38. doi: 10.1016/S0168-8278(01)00221-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials.