

Abstract
NMDA receptors mediate a slow Ca2+-permeable component of excitatory synaptic transmission, and are involved in numerous normal brain functions including learning and memory. NMDA receptor over-activation can lead to cell death and abnormal excitation in ischemia associated with stroke, traumatic brain injury, and epilepsy. We have explored a series of novel noncompetitive allosteric modulators of NMDA receptor function characterized by an iminothiazolidinone ring. Saturating concentrations of these compounds inhibit NMDA receptors to varying maximal extents, raising the possibility that they may attenuate over-activation in pathological situations while preserving some minimal receptor function, which may limit side-effects. The best in class compounds have sub-micromolar IC50 values and show modest preference for GluN2C- and GluN2D-containing receptors.
Keywords: GluN2, Glutamate, Noncompetitive antagonist, N-Methyl-d-aspartate, Neuroprotection
Ionotropic glutamate receptors (iGluRs) are a family of ligandgated ion channels that mediate the majority of fast excitatory neurotransmission in the mammalian brain. The iGluRs are subdivided into three subtypes based on sequence and structural homology, function, and pharmacology. The N-methyl-d-aspartate (NMDA) subtype of glutamate receptors have slower response time course and are more permeable to calcium ions than other iGluRs.1 Aberrant NMDA receptor activation is thought to play a role in the progression of Parkinson’s disease and the biochemical neuronal damage caused by traumatic brain injury and stroke.2,3 NMDA receptor antagonists have been shown to be neuroprotective in many animal studies of these disorders.2,4 However, clinical trials with multiple classes of NMDA receptor antagonists have been unsuccessful, due in part to complications arising from on-target side-effects.5 It has been hypothesized that next generation therapeutics possessing subunit selectivity and/or submaximal inhibition might avoid these side-effects.5–7 That is, unwanted on-target side-effects could be due to drug actions on a subset of GluN2-containing NMDA receptors and could be avoided by using compounds with GluN2 subunit selectivity.2,6,7 Additionally, side-effects might arise from complete blockade of NMDA receptors, and therefore might be avoided by preserving a low level of activity through use of compounds that produce submaximal inhibition at saturating concentrations.6,7 This latter possibility has not been evaluated due to the lack of candidate compounds that show more than 10% submaximal inhibition at maximally effective concentrations. Recently, some NMDA receptor antagonists have been investigated which may have submaximal inhibition,8 although potency and drug-like properties of these compounds would likely need to be improved before use in pre-clinical testing.
In an attempt to new identify subunit-selective inhibitors, a medium-throughput screen was implemented to identify modulators of GluN2C- or GluN2D-containing NMDA receptors.9 Among the NMDA receptor modulators identified as hits,10–15 compound (1) (Fig. 1) was selected for further investigation due to its distinct structural and pharmacological properties. Compound 1 contains an iminothiazolidinone ring connected to the thiophene moiety through an acetamide linkage. Within the present study, we varied both heterocyclic rings of the parent compound (1) in order to understand the interaction of these compounds with NMDA receptors and the resulting pharmacological effect.
Figure 1.

Generic series chemical structure illustrating modifications to the screening hit explored in this work (1).
Here we report (a) the structure activity relationship (SAR) of a novel class of NMDA receptor antagonists; (b) data supporting the noncompetitive mechanism of action of a compound in this class on NMDA receptors, and (c) the effect of the prototypical compound on neuronal NMDA receptors, and (d) data supporting its therapeutic potential.
The potency and extent of NMDA receptor inhibition by analogues were determined by activating receptors with maximally effective concentrations of glutamate and glycine and evaluating the concentration–effect relationship each of the four diheteromeric NMDA receptors expressed in Xenopus laevis oocytes. Each subtype was expressed in oocytes by co-injecting the cRNA for the rat GluN1–1a (hereafter GluN1) and one of the GluN2 subunits (GluN2A–D). After a sufficient time for robust surface expression of functional NMDA receptors (1–3 days at 16–19 °C), two electrode voltage clamp recordings were performed as described.16 Briefly, NMDA receptor currents were activated by 100 µM glutamate and 30 µM glycine in the absence and then presence of increasing concentrations of the test compounds. The mean steady state current for each concentration of inhibitor tested was used to establish the concentration–response relationships. To determine the maximal pharmacological effect and the concentration required to induce half maximal pharmacological response (IC50), the responses were fitted by the Hill equation, given by
where I is the current amplitude, Inhmax is the percent response remaining in saturating ligand, h is the Hill coefficient and [A] the concentration of ligand.
Compounds tested were either obtained from commercial vendors at a purity greater than 90% (assessed by 1H NMR and/or LC/ MS) or synthesized according to the procedures described below. A series of structurally-similar molecules has been shown to have activity against secreted frizzled-related protein 117 (Scheme 1).
Scheme 1.

Synthetic route toward analogues of 1. (a) S8, morpholine, 50 °C. (b) Maleic anhydride, Et2O, 25–35 °C. (c) HOBt, EDCI, DMF, 1 h, then thiourea, 60 °C, 24 h. (d) Acetic anhydride, sodium acetate, 75 °C, then rt, 12–18 h. (e) EtOH, rt, 24–48 h.
To expand the SAR, we also investigated alkyl substitutions at three positions on the thiophene ring (Table 1). Replacement of the methyl ester of 1 with an ethyl ester in compound 14 retained potency. However, exchange of the ethyl group for a methyl group at the position proximal to the sulfur atom in the thiophene ring in analogue 15 resulted in >4-fold decrease in potency. Addition of alkyl substituents at the R2 position of the thiophene ring in compounds 16, 17, and 18 all reduced the potency. The replacement of the alkyl substituents with hydrogen atoms at both the R2 and R3 positions (19) strongly reduced potency over 10-fold. While none of these compounds offered improvements in potency, the data indicates that one or more alkyl substituents on the thiophene ring are necessary for activity, potentially by stabilizing the compound into a hydrophobic binding pocket.
Table 1.
Effect of alkyl substitutions to the thiophene ring on the potency of analogues of 1
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound ID | R1 | R2 | R3 | IC50 (µM)a (% maximal inhibition)b |
|||
| GluN2A | GluN2B | GluN2C | GluN2D | ||||
| 1 | Me | H | Et | 4.8 [4.4, 5.3] (98 ± 0.8) | 9.3 [8.2, 10.5] (97 ± 1.4) | 2.5 [2.1, 3.0] (97 ± 1.0) | 2.6 [2.2, 3.1] (98 ± 0.8) |
| 14 | Et | H | Et | 6.2 [4.3, 9.0] (96 ± 1.3) | 9.7 [8.2, 10.5] (96 ± 1.4) | 3.7 [2.6, 5.1]* (97 ± 0.8) | 2.6 [1.9, 3.7] (94 ± 2.4) |
| 15 | Et | H | Me | 42.8 [38.2, 48.0]* (93 ± 1.8) | 44.9 [39.5, 50.8]* (91 ± 2.3) | 11.2 [9.2, 13.5]* (98 ± 1.0) | 12.3 [11.4, 13.2]* (99 ± 0.6) |
| 16 | Me | Me | Me | 22.3 [19.5, 25.6]* (88 ± 3.5#) | 28.2 [21.9, 36.1]* (63 ± 3.9#) | 9.7 [8.6, 11.0]* (88 ± 1.8#) | 10.4 [9.6, 11.2]* (94 ± 0.9) |
| 17 | Et | Me | Me | 30.2 [16.6, 55.0]* (80 ± 3.5#) | – | 8.1 [6.3, 10.3]* (77 ± 2.7#) | 13.0 [7.9, 21.5]* (81 ± 3.2#) |
| 18 | Me | Et | Me | 26.8 [22.9, 31.3]* (79 ± 2.7#) | 43.8 [37.2, 51.4]* (77 ± 3.7#) | 6.7 [5.3, 8.6]* (85 ± 1.4#) | 9.1 [8.6, 9.6]* (82 ± 0.9#) |
| 19 | Me | H | H | – | – | 41.0 [34.1, 49.4]* (100 ± 0.4) | 43.3 [35.8, 52.4]* (99 ± 0.6) |
IC50 values were obtained by least squares fitting the Hill equation to individual experiments, the Hill slope was allowed to vary between 0 and 1.5.
Values in parentheses represent the percentage of inhibition at the saturating concentration of inhibitor. Data are represented as the mean and 95% confidence interval (IC50) or ±SEM (maximal inhibition) from 8 to 13 oocytes from at least 2 frogs. Compounds producing less than 25% inhibition at 30 µM were considered to have no effect (–).
Indicates that the IC50 was significantly different than the IC50 of 1 for the same GluN2 subunit (1 way ANOVA performed on the log transformed IC50, Dunnett’s post hoc test, p <0.05).
Indicates that the maximal inhibition was significantly different than the maximal inhibition of 1 for the same GluN2 subunit (1 way ANOVA, Dunnett’s post hoc test, p <0.05).
The preference for lipophilic functionality on the thiophene ring was further demonstrated through the improved potency observed with several of the compounds summarized in Table 2. Specifically, we evaluated compounds containing a bicyclic thiophene as a replacement for the thiophene ring with short alkyl appendages present in compounds like 1. We hypothesized that the increase in hydrophobic character of these compounds would help reduce the total polar surface area toward a more drug-like value common to CNS drugs. Interestingly, introduction of the tetrahydrobenzothiophene moiety provided several potent NMDA receptor inhibitors.
Table 2.
Effect of substituted tetrahydrobenzothiophenes on the potency of analogues of 1
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound ID | R1 | X | IC50 (μM)a (% maximal inhibition)b |
Solubility (μM) | |||
| GluN2A | GluN2B | GluN2C | GluN2D | ||||
| 20 | CO2Me | CH2 | 3.4 [2.9, 3.9] (81 ± 4.0) | 4.2 [3.6, 4.9] (86 ± 2.0) | 1.0 [0.8, 1.2] (88 ± 1.3) | 1.1 [0.9, 1.2] (86 ± 1.3) | 57 |
| 5 | CO2Et | CH2 | 2.3 [1.5, 3.6] (61 ± 3.0#) | 4.2 [3.0, 6.0] (35 ± 1.5#) | 1.2 [0.7, 1.9] (53 ± 2.4#) | 0.6 [0.4, 0.9] (41 ± 1.6#) | 54 |
| 13 | CO2H | CH2 | – | – | – | – | 19 |
| 21 | CN | CH2 | N.D. | N.D. | 2.9 [2.4, 3.5]†,* (59 ± 2.4#) | 4.3 [2.5, 7.5]†,* (63 ± 4.2#) | 36 |
| 4 | CO2Et | CHMe | 0.9 [0.7, 1.2]* (88 ± 3.1) | 2.2 [1.0, 4.5] (70 ± 7.1) | 0.7 [0.4, 1.2] (80 ± 1.5) | 0.4 [0.3, 0.5]* (76 ± 2.9) | 43 |
| 22 | CO2Me | CHMe | 0.7 [0.6, 0.8]* (80 ± 3.4) | 0.6 [0.4, 0.8]* (74 ± 5.0) | 0.3 [0.3, 0.4]* (95 ± 1.5) | 0.3 [0.2, 0.4]* (94 ± 1.6) | 20 |
| 10 | CO2Et | CHEt | 3.4 [2.3, 5.1]† (58 ± 6.5#) | 2.2 [1.8, 2.7]† (59 ± 3.8#) | 0.9 [0.6, 1.3]† (80 ± 3.2) | 1.5 [1.0, 2.3]† (73 ± 3.7#) | 73 |
| 8 | CO2Et | CMe2 | 16.1 [5.6, 46.3]†,* (72 ± 7.5) | 9.2 [2.9, 29.0]† (44 ± 8.0#) | 9.3 [5.4, 16.1]* (89 ± 4.0) | 8.8 [6.0, 12.9]* (90 ± 4.2) | 37 |
| 23 | CO2Me | Ct-Bu | – | – | – | – | 48 |
| 11 | CO2Et | CF2 | 5.2 [3.2, 8.2] (84 ± 2.0) | 8.0 [5.4, 11.9] (74 ± 3.8) | 1.4 [0.8, 2.4] (75 ± 1.8#) | 1.5 [0.9, 2.8] (65 ± 3.8#) | 35 |
| 6 | CO2Et | S | 6.2 [4.9, 7.8] (82 ± 1.7) | 12.2 [9.9, 15.2]* (78 ± 3.4) | 0.8 [0.7, 1.1] (88 ± 1.2) | 0.8 [0.6, 1.1] (84 ± 1.2) | |
| 7 | CO2Et | O | 3.0 [1.8, 5.0] (74 ± 2.0) | 5.6 [3.5, 8.9] (69 ± 3.7) | 2.9 [2.1, 4.0]* (59 ± 2.6#) | 2.1 [1.1, 3.9] (41 ± 3.9#) | |
IC50 values were obtained by least squares fitting the Hill equation to individual experiments, the Hill slope was allowed to vary between 0 and 1.5.
Values in parentheses represent the percentage of inhibition at the saturating concentration of inhibitor. Data are represented as the mean and 95% confidence interval (IC50) or ±SEM (maximal inhibition) from 4 to 17 oocytes at least 2 frogs. Compounds producing less than 25% inhibition at 30 µM were considered to have no effect (–). N.D. indicates where the calculated IC50 value was greater than the measured solubility. Curves were fit with data at concentrations at and below the solubility for each compound. Solubility was determined by using the intersection point from a linear segmental regression on nephelometry data from range of solution concentrations (3.25–100 µM) of each analogue in TEVC electrophysiological solution (mean, n = 2 in quadruplicate).
Indicates that the Hill coefficient held constant (h = 1) to obtain estimates of lower potency or less soluble compounds.
Indicates that the IC50 was significantly different than the IC50 of 20 for the same GluN2 subunit (1 way ANOVA performed on the log transformed IC50, Dunnett’s post hoc test, p <0.05).
Indicates that the maximal inhibition was significantly different than the maximal inhibition of 20 for the same GluN2 subunit (1 way ANOVA, Dunnett’s post hoc test, p <0.05).
Comparison of both 20 and 5 showed that changing from the methyl ester to the ethyl ester did not significantly alter potency. When compared to the original screening hit (1, Table 1), analogues with the fused cyclohexyl ring demonstrated either a 2- (20) or 3-fold increase (5) in potency at GluN2D-containing receptors. To our surprise, they also exhibited varying degrees of submaximal inhibition at all receptor combinations tested. The specificity of the R1 position is highlighted by the loss of activity of the carboxylic acid 13. Interestingly, the nitrile substituted analogue (21) appears to be less potent than 1 and has reduced activity (60% maximal inhibition). This result supported the hypothesis that this portion of the molecule interacts with a pocket which prefers hydrophobicity.
Additional analogues were generated containing the bicyclic thiophene moiety in attempt to expand the SAR surrounding this favorable constituent and improve potency, selectivity, or both. Various substitutions were made exclusively at the 6-position of the tetrahydrobenzothiophene ring system so as to avoid regioisomers that could form in the condensation reaction toward the aminothiophene building blocks (Scheme 1). Introduction of a single methyl unit onto the cyclohexyl ring (4) resulted in slightly improved activity compared to its unsubstituted counterpart (~2 fold, 5). The potency of 22, the methyl ester analogue of 4, was further improved, and showed submicromolar IC50 values for all NMDA receptor subunits. An increase in steric bulk at the position denoted X (compounds 10, 8, and 23) may have decreased inhibitor potency. This suggests that the antagonist binding site has a finite size and is able to accommodate ligands up to a specific molecular volume in this portion of the pocket. While the gemdifluoro isostere (11) was well tolerated, it possessed lower potency at GluN2D-containing receptors (IC50 = 2.4 µM) than 5 and 4 (IC50 values <1 µM). The incorporation of heteroatoms (X = S or O, 6 and 7, respectively) resulted in a low micromolar inhibitor (6, X = S), with some improvement of selectivity for GluN2D-containing receptors over GluN2A (~7-fold selectivity).
Lastly we investigated the aminothiazolidinone ring to identify the molecular determinants of the pharmacological interaction (Table 3). None of the changes tested to this group were well-tolerated. Specifically, replacement of the imine in 5 with a carbonyl group in 24 and 25 abolished activity at all receptor subtypes. Similar results were observed for 26 where both nitrogen atoms were alkylated. In addition, replacement of the iminothiazolidinone moiety for the imidazolidinedione ring in compound 27 abrogated activity. Two additional compounds where the A-ring was replaced with a different heterocycle (thiophene, thio-pyridine) also resulted in a complete loss of activity (data not shown). Taken together, the data suggest that the presence and position of the hydrogen bond donating nitrogen atoms in this ring are important for activity.
Table 3.
Effect of A-ring substitutions on the potency of analogues of 1
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound ID | X | Y | Z | R1 | R2 | IC50 (µM)a (% maximal inhibition)b |
|||
| GluN2A | GluN2B | GluN2C | GluN2D | ||||||
| 5 | NH | NH | S | CH2–(CH2)2–CH2 | 2.3 [1.5, 3.6] (61 ± 3.0) |
4.2 [3.0, 6.0] (35 ± 1.5) |
1.2 [0.7, 1.9] (53 ± 2.4) |
0.6 [0.4, 0.9] (41 ± 1.6) |
|
| 24 | NH | O | S | CH2–CH2–CH2 | – | – | – | – | |
| 25 | NH | O | S | CH2–(CH2)2–CH2 | – | – | – | – | |
| 26 | NMe | NMe | S | CH2–(CH2)2–CH2 | – | – | – | – | |
| 27 | NH | O | NH | CH2–(CH2)2–CH2 | – | – | – | – | |
IC50 values were obtained by least squares fitting the Hill equation to individual experiments, the Hill slope was allowed to vary between 0 and 1.5.
Values in parentheses represent the percentage of inhibition at the saturating concentration of inhibitor. Data for compound 5 are shown again for comparison. Data are represented as the mean and 95% confidence interval (IC50) or ±SEM (maximal inhibition) from 6 to 13 oocytes at least 2 frogs. Compounds producing less than 25% inhibition at 30 µM were considered to have no effect (–).
Since all compounds were tested as the racemic mixture, we sought to determine whether the enantiomers had different degrees of potency. While the separation of enantiomers for compound 11 was successfully accomplished using chiral semi-preparative HPLC, we found that the molecules were prone to rapid racemization. This observation is consistent with reports of racemization with the structurally-related glitazone class of peroxisome proliferator-activated receptor (PPAR) activators.18 We suspect that this facile process occurs via enolization at the stereogenic center. Consequently, this rapid racemization prohibited biological evaluation of the enantiomers.
A key feature of this class of inhibitors is their ability to exert submaximal inhibition at saturating concentrations. This could reflect low solubility, which can lead to precipitation of compound at higher concentrations, setting an effective ceiling on the maximal concentration observed for a given compound in aqueous solution. We therefore determined the aqueous solubility of the most active compounds using nephelometry. Briefly, solubility was determined by measuring the forward light scattering of compounds in electrophysiological solutions (3.25–100 µM) and fitting the data with a linear segmental regression fit, where the intersection point was deemed to be the maximum solubility (NEPHELOstar, BMG Labtech, Cary, NC). The estimated maximal solubility for all active compounds was greater than 20 µM (except for 10 and 21), and was 10 times higher than the IC50 (except for 10, 8, 7 and 21), suggesting that the submaximal inhibition observed by saturating concentration of several analogues in this series does not reflect loss of material from solution (Table 2).
We next examined the mechanism of action for the prototypical compound 4, which showed in separate experiments 73–81% inhibition at saturating concentrations against all GluN2 subunits (n = 4–6, p <0.05 vs complete inhibition, extra sum-of-squares F test). We first evaluated whether the inhibition was derived from interference with the binding of either co-agonist. We tested whether the extent of 4 inhibition was reduced in the presence of increased concentrations of glutamate (1000 µM) or glycine (300 µM), which would be predicted if 4 acted as a competitive antagonist at the glutamate or glycine binding sites. Submaximal inhibition of the diheteromeric NMDA receptor responses by 5 µM 4 could not be surmounted by a 10-fold increase in glutamate or glycine concentration, indicating that the compound acts by a noncompetitive mechanism (Fig. 2).
Figure 2.

Noncompetitive inhibition of recombinant diheteromeric NMDA receptor by 4. The mean inhibition by 5 µM 4 of NMDA receptor current responses to saturating concentrations of glutamate and glycine (100 µM and 30 µM, respectively) is compared to inhibition in elevated concentrations of either (A) glutamate (1000 µM) or (B) glycine (300 µM). Data shown are the mean inhibition (±SEM) from 4 to 6 oocytes, there are no significant differences at different co-agonist concentrations.
We also examined whether the inhibition observed with 4 was voltage-dependent. Two electrode voltage clamp recordings of NMDA receptor current responses to a maximally effective concentration of glutamate and glycine with or without 4 were made at different membrane potentials between ‒60 and +60 mV and in the absence of Mg2+. The degree of inhibition produced by 3 µM 4 was the same at all voltages tested (n = 5, ANOVA p = 0.99). In addition, the mean reversal potential (VREV) was not significantly different between control response to glutamate and glycine and the response to glutamate and glycine plus 3 µM 4 (VREV-Control = ‒13.0 ± 0.6, VREV-1794-2 = ‒11.9 ± 0.5, n = 5, t-test p = 0.22, Fig. 3). These data suggest that 4 is a negative allosteric modulator of NMDA receptors, since its effects are noncompetitive and voltage-independent.
Figure 3.

Voltage-independent mechanism of GluN1/GluN2C receptor inhibition by 4. The mean current–voltage relationship is shown for activated GluN1/GluN2C receptor responses activated by maximally effective concentration of glutamate and glycine. Current responses were recorded from oocytes in the absence of extracellular Mg2+, and in the absence and presence of 3 µM 4. Data are averaged from 5 oocytes and are shown ±SEM. The smooth curve shows a 3rd order polynomial fit to the data.
To examine if this compound class can inhibit native NMDA receptors, we prepared primary cultures of p6–8 rat cerebellar granule cells as previously described,19 and performed patch clamp recordings of NMDA receptor responses as previously described.20 NMDA receptors in granule cells were activated with 100 µM NMDA and 30 µM glycine and then inhibited by co-applying NMDA and glycine with different concentrations of 4. The mean inhibitory responses of the varying concentrations of 4 were fit by the Hill equation (Fig. 4). The IC50 of 4 in this preparation was similar to the IC50 of the compound on GluN2B-containing diheteromeric receptors expressed in oocytes. This result confirms that this series of negative allosteric modulator is active at native receptors.
Figure 4.

Negative allosteric modulation by 4 of NMDA receptors expressed on cultured primary granule cells from rat cerebellum. (A) Representative patch clamp recording of the current response under voltage clamp from a primary cerebellar granule cell (VHOLD −70 mV). The steady-state current response was attenuated by 30 µM 12 during the co-application of 100 µM NMDA plus 50 µM glycine. (B) Concentration–effect relationship of 4 on NMDA-induced currents in primary cerebellar granule cell cultures with an IC50 = 3.4 µM. Date are mean ± SEM (n = 5), data were fitted by the Hill equation.
We also investigated the potential neuroprotective effects of this class of inhibitors. Cultured primary hippocampal neurons were prepared as previously described,21 maintained for 7–10 days in vitro at 37 °C in 5/95% CO2/O2. Neurons were challenged by replacing the cell’s media with a similar media but lacking Mg2+ and supplemented with 100 µM NMDA and 30 µM glycine for 1 h, then normal media was replaced. The extent of cell death was measured in the absence and presence of 4 by measuring lactate dehydrogenase (LDH) activity spectrophotometrically in the supernatant of the cells 20–24 h after treatment (22). LDH is an intracellular enzyme that is released into the media upon cell rupture. Treatment of cells with NMDA and glycine induces a significant increase in supernatant LDH as compared to control (Fig. 5). This increase in cell death was reduced significantly by the addition of 30 µM 4 (n = 6, ANOVA, Dunnett’s post hoc test, p = 0.001). For comparison, 10 µM (+)MK-801, a potent NMDA channel blocker (Kd = 37.2 ± 2.7 nM),23 was also able to attenuate the increase in the extracellular LDH caused by NMDA receptor over-activation.
Figure 5.
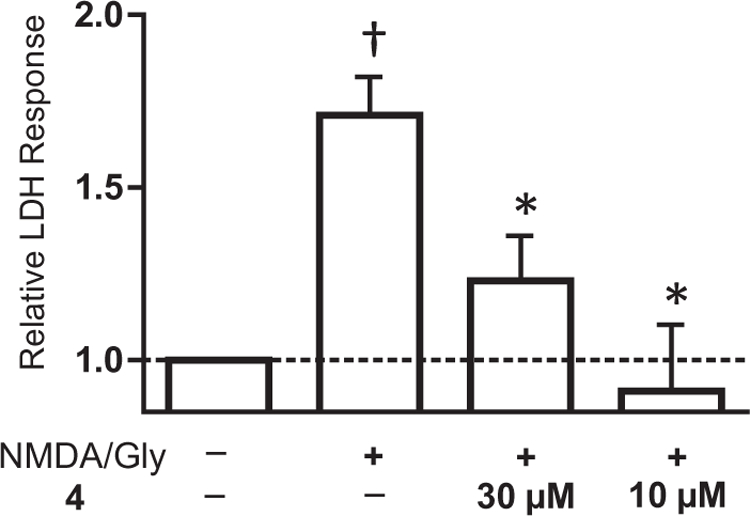
Neuroprotection by 4 against NMDA-induced excitotoxicity of cultured primary hippocampal neurons. Attenuation of excitotoxicity produced by 100 µM NMDA plus 30 µM glycine is shown for 30 µM 4 and the open channel blocker MK- 801 (10 µM). Cell death was assessed by release of the intracellular enzyme lactate dehydrogenase (22), and expressed relative to the concentration of LDH released into culture media upon in vehicle treated cells (*p <0.05 as compared to NMDA/Gly response and †p <0.05 as compared to control). Measurements were made in triplicate from 6 independent experiments, and are shown as the mean ± SEM.
In summary, a novel series of iminothiazolidinone-based compounds were synthesized and evaluated for NMDA receptor inhibition. The SAR data showed that the potency of this series is dependent upon the substitution around the aminothiophene ring. This series demonstrated limited subunit-selectivity, but had the interesting effect of exerting submaximal inhibition at saturating concentrations, which did not reflect solubility limits in aqueous solution. The mechanism of action for 4 was noncompetitive and voltage-independent. The effectiveness of this compound against native neuronal NMDA receptors was investigated in cerebellar granule cells, where 4 showed strong inhibition of responses to the agonist NMDA. The inhibition of NMDA receptors by 4 reduced cell death in cultured primary hippocampal neurons challenged with NMDA, confirming its neuroprotective potential. Thus, the efficacy of this class of compounds coupled with the submaximal receptor block at saturating concentrations may allow development of analogues that are effective neuroprotectants with a reduced side-effect profile.
Acknowledgments
This work was supported by the NIH (NS065371, SFT) and the Michael J. Fox Foundation. Some of the authors are Board members (DCL) or paid consultants (SFT) of NeurOp Inc, a company developing glutamate receptor modulators for therapeutic use.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.10.046.
References and notes
- 1.Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Pharmacol. Rev 2010, 62, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallett PJ; Standaert DG Pharmacol. Ther 2004, 102, 155. [DOI] [PubMed] [Google Scholar]
- 3.Lipton SA; Rosenberg PA N. Engl. J. Med 1994, 330, 613. [DOI] [PubMed] [Google Scholar]
- 4.Palmer GC Curr. Drug Targets 2001, 2, 241. [DOI] [PubMed] [Google Scholar]
- 5.Ikonomidou C; Turski L Lancet Neurol 2002, 1, 383. [DOI] [PubMed] [Google Scholar]
- 6.Koller M; Urwyler S Expert Opin. Ther. Pat 2010, 20, 1683. [DOI] [PubMed] [Google Scholar]
- 7.Santangelo RM; Acker TM; Zimmerman SS; Katzman BM; Strong KL; Traynelis SF; Liotta DC Expert Opin. Ther. Pat 2012, 22, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa BM; Irvine MW; Fang G; Eaves RJ; Mayo-Martin MB; Laube B; Jane DE; Monaghan DT Neuropharmacology 2012, 62, 1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen KB; Mullasseril P; Dawit S; Kurtkaya NL; Yuan H; Vance KM; Orr AG; Kvist T; Ogden KK; Le P; Vellano KM; Lewis I; Kurtkaya S; Du Y; Qui M; Murphy TJ; Snyder JP; Bräuner-Osborne H; Traynelis SF J. Pharmacol. Exp. Ther 2010, 333, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acker TM; Yuan H; Hansen KB; Vance KM; Ogden KK; Jensen HS; Burger PB; Mullasseril P; Snyder JP; Liotta DC; Traynelis SF Mol. Pharmacol 2011, 80, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acker TM; Khatri A; Vance KM; Slabber C; Bacsa J; Snyder JP; Traynelis SF; Liotta DC J. Med. Chem 2013, 56, 6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosley CA; Acker TM; Hansen KB; Mullasseril P; Andersen KT; Le P; Vellano KM; Bräuner-Osborne H; Liotta DC; Traynelis SF J. Med. Chem 2010, 53, 5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullasseril P; Hansen KB; Vance KM; Ogden KK; Yuan H; Kurtkaya NL; Santangelo R; Orr AG; Le P; Vellano KM; Liotta DC; Traynelis SF Nat. Commun 2010, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santangelo Freel R; Ogden K; Strong K; Khatri A; Chepiga K; Jensen H; Traynelis S; Liotta DJ Med. Chem 2013, 56, 5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmerman SS; Khatri A; Garnier-Amblard EC; Mullasseril P; Kurtkaya NL; Gyoneva S; Hansen KB; Traynelis SF; Liotta DC J. Med. Chem 2014, 57, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen KB; Tajima N; Risgaard R; Perszyk RE; Jorgensen L; Vance KM; Ogden KK; Clausen RP; Furukawa H; Traynelis SF Mol. Pharmacol 2013, 84, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi M; Stauffer B; Bhat R; Billiard J; Ponce-de-Leon H; Seestaller-Wehr L; Fukayama S; Mangine A; Moran R; Krishnamurthy G; Bodine P; Gopalsamy A Bioorg. Med. Chem. Lett 2009, 19, 6337. [DOI] [PubMed] [Google Scholar]
- 18.Sohda T; Mizuno K; Kawamatsu Y Chem. Pharm. Bull 1984, 32, 4460. [DOI] [PubMed] [Google Scholar]
- 19.Zheng F; Erreger K; Low CM; Banke T; Lee CJ; Conn PJ; Traynelis SF Nat. Neurosci 2001, 4, 894. [DOI] [PubMed] [Google Scholar]
- 20.Traynelis SF; Cull-Candy SG J. Physiol 1991, 433, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beaudoin GMJ; Lee S-H; Singh D; Yuan Y; Ng Y-G; Reichardt LF; Arikkath J Nat. Protoc 2012, 7, 1741. [DOI] [PubMed] [Google Scholar]
- 22.Koh JY; Choi DW J. Neurosci. Methods 1987, 20, 83. [DOI] [PubMed] [Google Scholar]
- 23.Wong EH; Kemp JA; Priestley T; Knight AR; Woodruff GN; Iversen LL Proc. Natl. Acad. Sci. U.S.A 1986, 83, 7104. [DOI] [PMC free article] [PubMed] [Google Scholar]