

Abstract
Bone marrow (BM) is the central immunological organ and the origin of hematological diseases. Efficient and specific drug delivery to the BM is an unmet need. We tested delivery of fluorescent indocarbocyanine lipids (ICLs, DiR, DiD, DiI) as a model lipophilic cargo. Systemically injected T-lymphocyte cell line Jurkat delivered ICLs to the BM more efficiently than erythrocytes, and more selectively than PEGylated liposomes. Near infrared imaging showed that the delivery was restricted to the BM, lungs, liver and spleen, with no accumulation in the kidneys, brain, heart, intestines, fat tissue and pancreas. Following systemic injection of ICL-labeled cells in immunodeficient or immunocompetent mice, few cells arrived in the BM intact. However, between 5–10% of BM cells were ICL-positive. Confocal microscopy of intact BM confirmed that ICLs are delivered independently of the injected cells. Flow cytometry analysis showed that the lipid accumulated in both CD11b+ and CD11b- cells, and in hematopoietic progenitors. In a xenograft model of acute myeloid leukemia, a single injection of 10 million Jurkat cells delivered DiD to ~15% of the tumor cells. ICL-labeled cells disappeared from blood almost immediately post-intravenous injection, but numerous cell-derived microparticles continued to circulate in blood. The microparticle particle formation was not due to the ICL labeling or complement attack and was observed after injection of both syngeneic and xenogeneic cells. Injection of microparticles produced in vitro from Jurkat cells resulted in a similar ICL delivery as the injection of intact Jurkat cells. Our results demonstrate a novel delivery paradigm wherein systemically injected cells release microparticles that accumulate in the BM. In addition, the results have important implications for studies involving systemically administered cell therapies.
Keywords: indocyanine lipid, liposome, bone marrow, drug delivery, erythrocyte, Jurkat, leukemia
INTRODUCTION
Hematological malignancies, anemias, and autoimmune diseases originate in the bone marrow (BM). In addition, the microenvironmental niche and availability of growth factors [1] make the BM an ideal site of metastasis for cancers, including prostate, bladder, and breast [2]. Due to the fenestrated nature of the supplying blood vessels (sinusoids), BM is considered a semipermeable organ [3]. Sinusoidal openings allow egress of mature erythrocytes, platelets, and leukocytes into the blood [4]. At the same time, the movement into the BM is more restricted, although the permeability increases due to radiation damage or inflammation [3]. Recent papers demonstrated the accumulation of nano-sized drug delivery systems in the BM; for example, PLGA nanoparticles [5] and quantum dots [6] in mice, and PEGylated liposomes in patients [7]. Negatively charged liposomes that serve as a basis for the anti-leukemic drug CPX-351 (Vyxeos)[8] have been shown to accumulate in the BM and deliver cytarabine and daunorubicin. While the delivery of water-soluble drugs does not pose a problem, there is a wide range of lipophilic drugs, prodrugs and bioactive lipids for which efficient drug delivery systems are needed [9].
Notwithstanding the evidence that conventional drug delivery systems accumulate in the BM, we questioned whether cells are also able to deliver a payload to the BM. Cell-based therapies are some of the oldest types of treatments (e.g., blood transfusion, bone marrow transplantation), which are becoming an emerging frontier in treatment of diseases.[10–12]. There has been substantial research on mechanisms of BM homing and engraftment of hematopoietic stem cells (HSCs) and leukemic cells [13–16]. Homing occurs within hours post the infusion of donor cells, as opposed to engraftment, which takes days [17]. The release of CXCL12 (SDF-1) from BM stroma and recognition via CXCR4 leads to the expression of adhesion molecules, extravasation and retention of the cells in the BM extracellular matrix [1]. Additional emerging factors in the cell homing are selectins [18], integrins, and tetraspanins in the polarized membrane domains [16, 19, 20].
Here, we evaluated cell-based delivery to murine BM using a model fluorescent membrane payload. Indo(carbo)cyanine lipids (ICLs) DiO, DiI, DiD, and DiR are extremely bright fluorescent dyes for labeling of membranes, e.g., for tracking of cells [21, 22] [14, 23]. Our group and others reported that ICLs are extremely stable in membranes with minimal transfer and exchange in plasma [24–26]. Some of the stability can be attributed to the highly lipophilic nature of the dyes and mild cationic charge, allowing the lipids to be stably embedded in the negatively charged cell bilayer.[27] ICLs are highly convenient as they allow multimodal tracking of the accumulation process, as well as the cellular and histological analysis. We used off-shelf cell lines with a documented ability of engraftment in the BM [31, 32]. There is great interest in the use of off-shelf cell lines for therapies, including CAR-T and NK cells [28, 29]. Antigenic determinants of cells can be efficiently deleted with CRISPR-Cas9 or other gene-editing technologies [30], and various kill switches can be engineered. Serendipitously, we observed that cellular microparticles released from cells in vivo, rather than the intact cells, efficiently delivered the lipid payload to the BM. These findings have important implications for drug delivery using off-shelf cell lines, as well as for mechanistic and biodistribution studies involving cell therapies.
RESULTS
1). Cells deliver ICLs to BM more efficiently than erythrocytes and more selectively than liposomes
To compare BM delivery of a lipophilic cargo using different carriers, we labeled T-lymphocyte cells Jurkat 29 and mouse red blood cells (RBCs) with 25 µM DiD (Fig. 1A). The protocol results in a highly efficient incorporation of ICLs, with up to 107 lipid molecules/cell [33] and over 95% of labeled cells and RBCs (Fig. 1B), while assuring high viability. After the labeling, over 95% of Jurkat cells were viable by Trypan blue staining and proliferated in culture similarly to the non-labeled cells (Supplemental Fig. S1). To test the delivery of ICLs via a nanocarrier we prepared DiD-labeled PEGylated liposomes (half-life ~20 hours [26]). Equal amounts of fluorescence of Jurkat, RBCs and liposomes were injected in immunocompetent BALB/c mice. One day post injection, mice were perfused via left ventricle and the BM was analyzed with flow cytometry. According to Fig. 1C–D, there were ~1% DiD+ events in RBC-injected mice, vs. 4% and 8.8% DiD+ events in Jurkat- and liposome-injected mice, respectively. This results suggests that RBCs show lower efficiency of BM delivery than liposomes or cells, and therefore RBCs were not included in subsequent studies.
Fig. 1. Accumulation efficiency of ICLs in murine bone marrow following systemic injection of different lipid carriers:
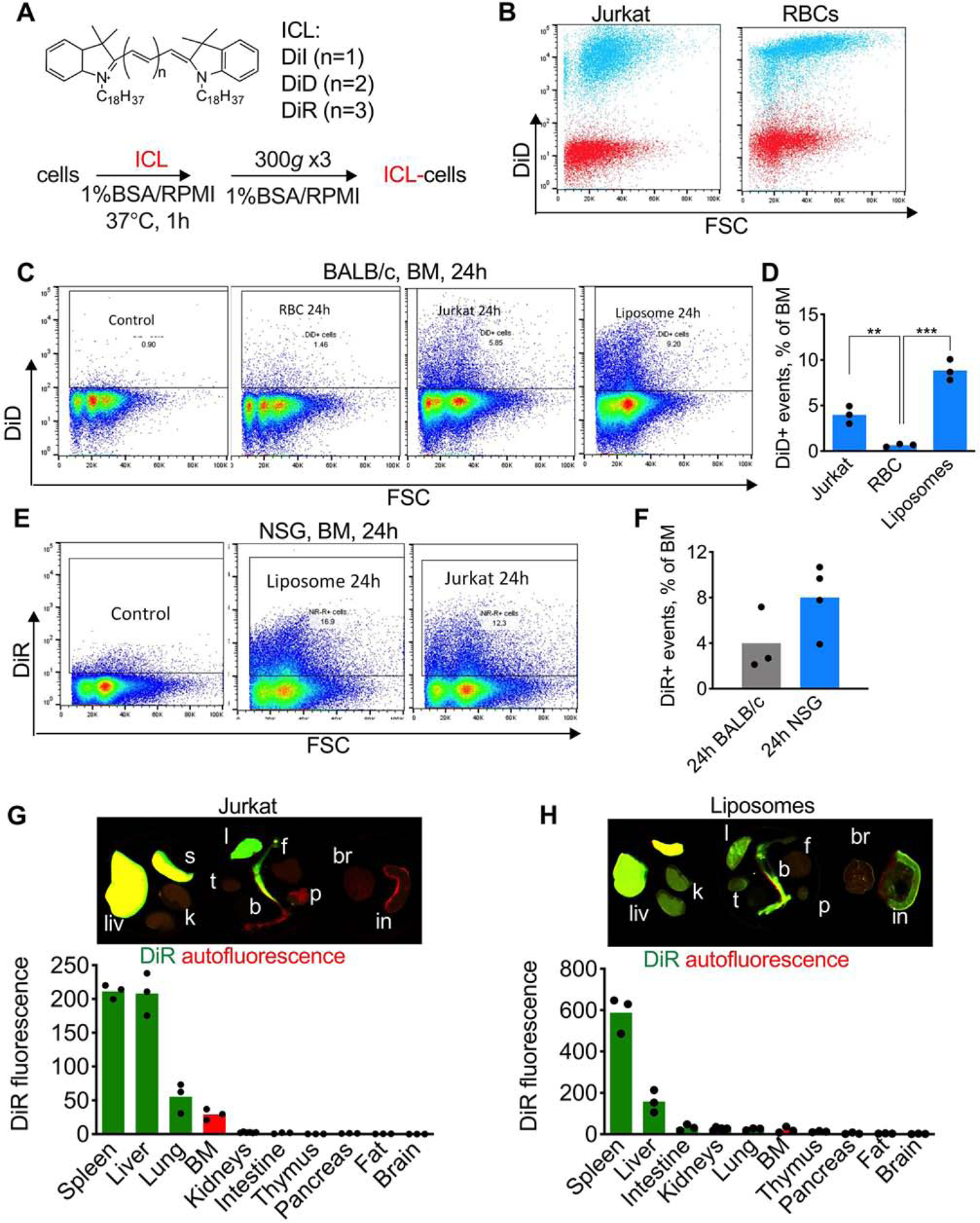
A) Labeling of cells with ICLs; B) DiD labeling efficiency of mouse RBCs and Jurkat cells. Red, before labeling; cyan, after labeling; C-D) DiD-labeled cells, RBCs, or liposomes (long circulating PEGylated liposomes (Doxil® formula)) were injected in BALB/c mice (same dose of fluorescence). Flow cytometry of BM shows much lower delivery via RBCs (n=3, 2-way ANOVA with multiple comparisons); E) similar level of accumulation of DiR after injection of DiR liposomes or DiR Jurkat cells; F) DiR Jurkat accumulation in BALB/c and in NSG mice (difference non-significant, n=3–4; 2-way ANOVA with multiple comparisons); G-H) ex vivo imaging of organs after injection of DiR-labeled Jurkat or liposomes in NSG mice. Organs were scanned with Li-COR Odyssey at 700 nm (autofluorescence, red) and 800 nm (DiR, green). Liv=liver, s=spleen, k=kidney, t=thymus, l=lung, b=bone (femur and tibia), f=fat, p-pancreas, br=brain, in=intestine). Contrast was enhanced in all groups to the same extent. Representative images are shown (3 mice per group). Liposomes show more widespread organ distribution that Jurkat.
To evaluate the efficiency of BM delivery via cells and liposomes as a function of the immune system, and also to enable ex vivo organ imaging, we labeled Jurkat cells and liposomes with a near infrared ICL DiR and injected them into immunodeficient NOD/LtSz-SCID IL2Rγc null (NSG) mice, which are commonly used for engrafting leukemias in the BM [34]. Liposomes and Jurkat showed similar delivery efficiency to the BM in NSG mice (Fig. 1E). Following DiR Jurkat injection, there were ~2-fold more ICL+ events in NSG mice than in BALB/c mice, but the difference was not statistically significant (Fig. 1F). Organs of NSG mice injected with DiR Jurkat showed accumulation in the liver spleen, lung and BM, with no accumulation in kidneys, intestine, thymus, pancreas, fat and brain (Fig.1G). Lung clearance of intravenously injected cells is a wellknown phenomenon [35, 36]. At the same time, liposomal DiR showed more widespread accumulation in the liver, spleen, lung, kidneys, thymus, pancreas, and small intestine (Fig. 1H). These results suggest that Jurkat cells show somewhat lower delivery efficiency but better selectivity for BM than liposomes.
2). ICLs are delivered to BM independently of the injected cells and accumulate in both immune and non-immune cells
To investigate further the mechanism of BM delivery of ICL via cells, we doubly labeled Jurkat cells with DiR and carboxyfluorescein succinimidyl ester (CFSE), which covalently labels intracellular proteins and enables in vivo tracking of cells [37]. Both dyes efficiently labeled the cells, but their distribution was different: CFSE accumulated mostly in the cytoplasm and nucleus, whereas DiR was localized both in the membrane and inside the cells (Fig. 2A–B). Flow cytometry analysis of BM post injection of DiR/CSFE Jurkat in NSG mice showed ~8% DiR+ events at 2h and 24h, but only 2.5% and 0.5% DiR+/CFSE+ events at 2h and 24h, respectively (Fig. 2C–D). These data suggest that the homing ability of Jurkat is low, and most of the lipid is delivered independently of the cells.
Fig. 2. ICLs accumulate in the BM independently of the injected cells:
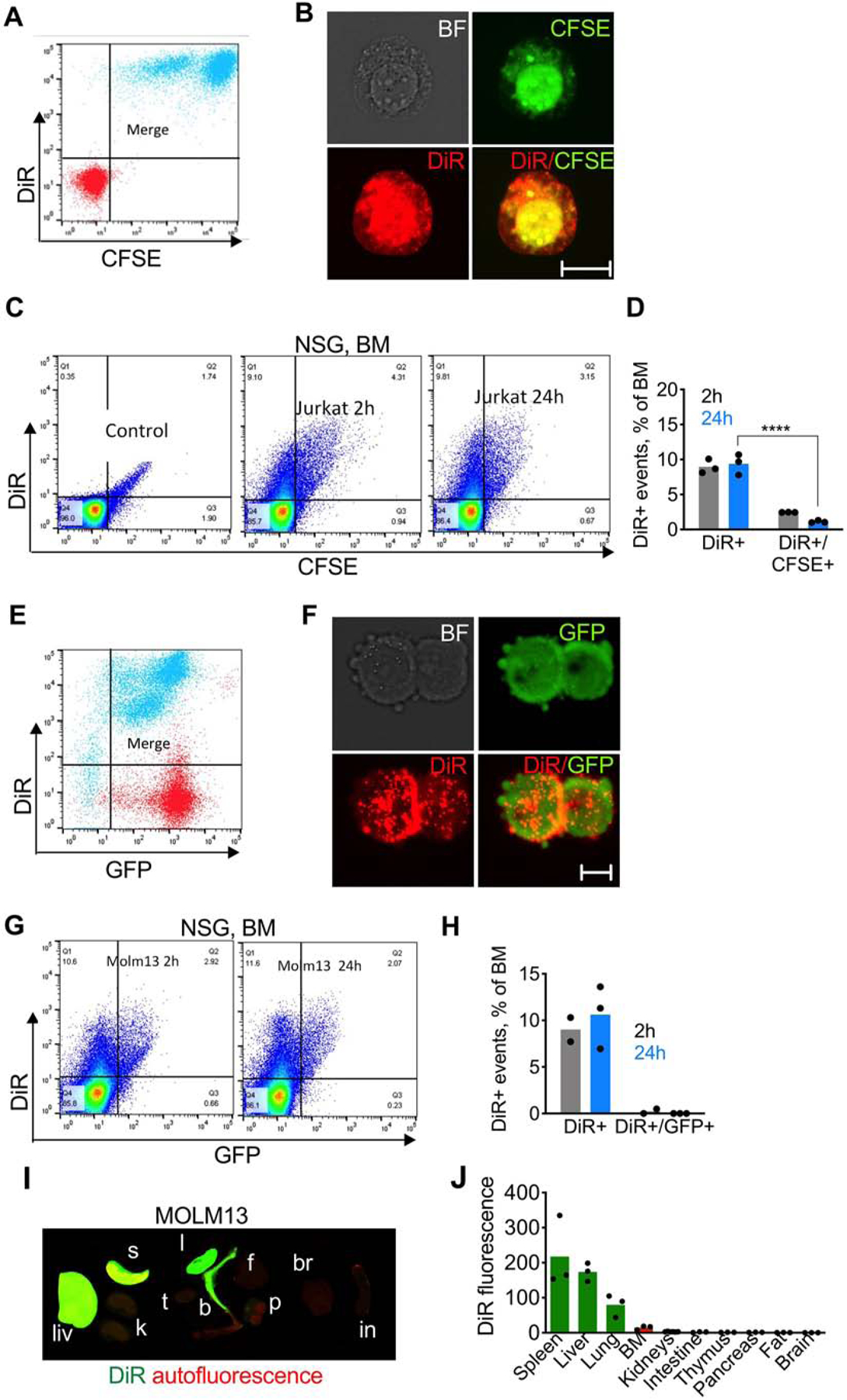
A-B) DiR and CFSE dual labeling of Jurkat cells. Red, before labeling; cyan, after labeling; C-D) flow cytometry of BM shows high percentage of DiR+ events but low percentage of DiR+/GFP+ events (n=3; 2-way ANOVA with multiple comparisons). Control is the BM of non-injected mice; E-F) DiR labeling of GFP-MOLM13 cells. Red, before labeling; cyan, after labeling; G-H) flow cytometry of BM after injection of DiR/GFP-MOLM13 shows high percentage of DiR+ events but almost no DiR+/GFP+ events; I) ex vivo NIR imaging of organs of NSG mice injected with DiR/GFP-MOLM13; J) image quantification.
Next, we used a myeloid leukemia cell line that was labeled via stable cytoplasmic expression of green fluorescent protein (GFP-MOLM13), that engrafts in NSG mice [31]. As with Jurkat cells, DiR labeling up to 25µM did not affect proliferation of GFP-MOLM13 (Supplemental Fig. S1). GFP-MOLM13 cells were efficiently labeled with DiR, and some of DiR was localized on the membrane (Fig. 2E–F). Twenty-four hours following systemic injection of DiR/GFP-MOLM13, there ~10% of DiR+ events, but less than 0.5% of DiR+/GFP+ events in the BM (Fig. 2G–H). DiR biodistribution was similar to that of Jurkat cells (Fig. 2I–J). The low level of cell homing was not due to the ICL labeling: injection of non-labeled Jurkat or MOLM13 cells resulted in less than 0.1% of human CD45+ events in the BM of NSG and BALB/c mice (Supplemental Fig. S2). To understand if DiR can be delivered via syngeneic mouse cells, we obtained bone marrow-derived macrophages (BMDM) from BALB/c mice. Following injection of 10 million DiR/CFSE BMDMs (Supplemental Fig. S3) in BALB/c mice, around 2.5% of BM events were DiR+, and less than 0.5% were DiR+/CFSE+ at 24h (Supplemental Fig. S3), suggesting that syngeneic mouse cells also deliver ICL, albeit with lower efficiency than human Jurkat and MOLM13 cells.
The data clearly demonstrate that only a small fraction of injected cells homes to the BM, and ICLs are mostly delivered independently of the cells. To study the distribution of ICLs in non-fixed, intact BM, we developed an ex vivo confocal imaging setup via bone window in femurs and tibia. Jurkat cells were labeled with DiI (ICL analog of Cy3) and CFSE, while blood vessels and BM cells were labeled pre-mortem by injection with DyLight 649 lectin [38, 39]. Two hours post-injection of cells in NSG mice, only a few DiI+/CFSE+ cells were detected per field (Fig. 3, arrowheads), but many sinusoids and BM cells were DiI+ (Fig. 3, asterisks). Upon higher magnification, many DiI+/CFSE+ and DiI+/CFSE- microparticles could be observed both inside and outside the cells (Fig. 3, middle and bottom panels).
Fig. 3. Confocal imaging of ICL distribution via bone window:
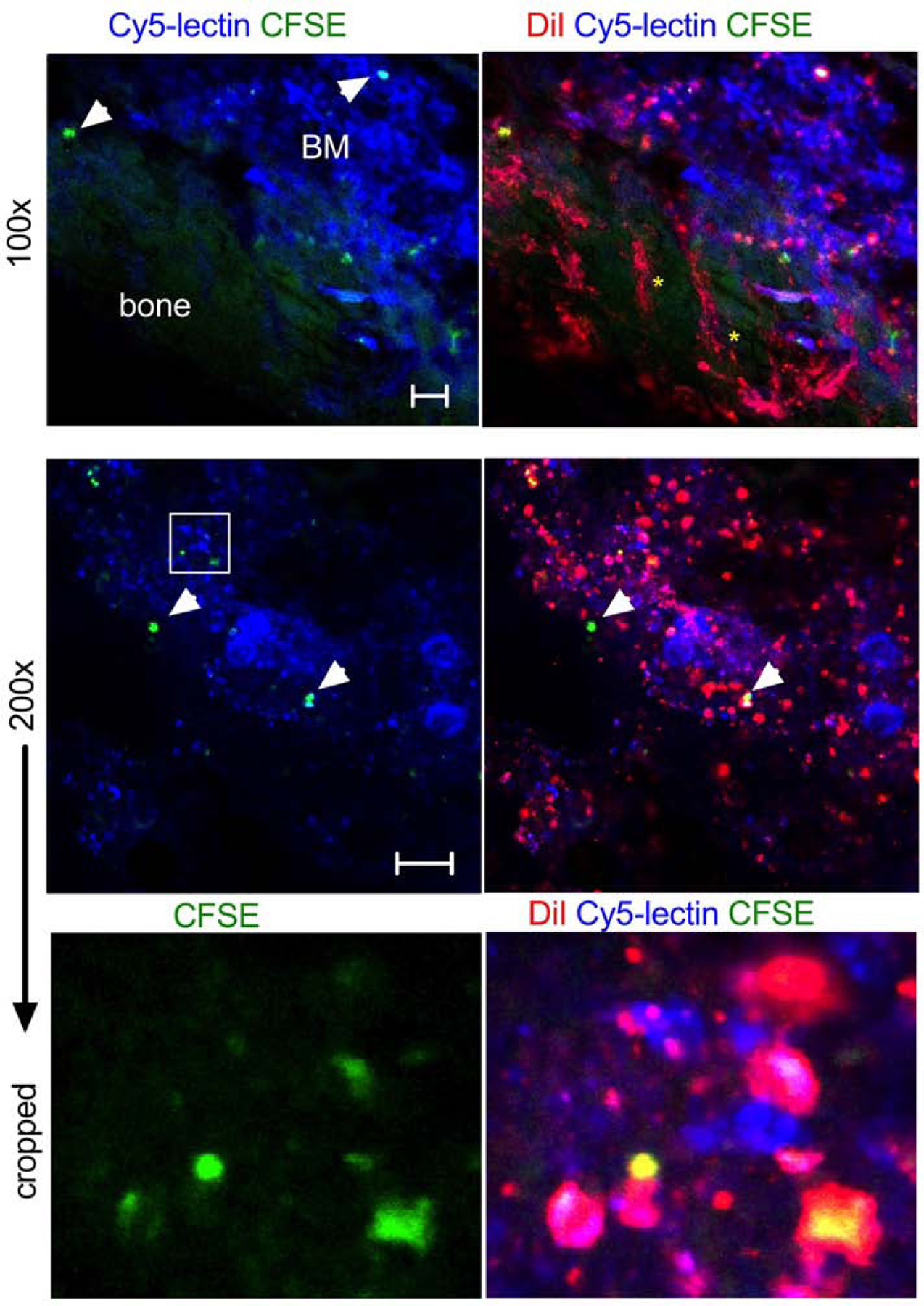
NSG mice were injected with DiI/CFSE Jurkat, followed by DyLight 649-lectin (to label blood vessels and BM cells) and imaged at 2 h (femur or tibia). Low magnification image (top panel) shows the deposition of DiI in cells and BM sinusoids (yellow asterisks). Bone that flanks the window has green autofluorescence. Only a few DiI/CFSE positive cells are visible (arrowheads). Size bar 50µm. High magnification and cropped images (middle and lower panels) show accumulation of DiI-positive and DiI/CFSE-positive particles but few DiI/CFSE-labeled cells (arrowheads). Large megakaryocytes are visible. Some particles appear to be outside the cells. Representative images of 3 mice are shown. Size bar 100 µm;
To identify cell types that take up ICLs in the BM of BALB/c mice, we prepared DiD-labeled Jurkat cells and PEGylated liposomes. According to Table 1 and Supplemental Figs. S4–12, at 24h post injection ~8% of CD11b+ cells were DiD+, and ~3% of CD11b-cells were DiD+. Small percentages of neutrophils, monocytes, eosinophils, T cells and B-cells in the BM were DiD+. A significant proportion of committed hematopoietic progenitors (CD48+/CD150+ and CD48+/CD150-) were also DiD+. Liposomes showed higher uptake of DiD by monocytes and lower uptake by neutrophils and T-lymphocytes. We next questioned whether Jurkat cells could deliver ICLs in a GFP-MOLM13 xenograft model of acute myeloid leukemia. DiD-labeled Jurkat cells (10 million) were injected in mice with advanced disease (~10% of GFP-MOLM13 cells in the BM, Fig. 4A–B). According to Fig. 4A–B, at 24 h post-injection between 2.5% and 16.5% of GFP-MOLM13 cells were DiD+. The disease did not significantly affect overall accumulation efficiency, with ~7% of BM events being DiD+ (Fig. 4B), versus ~10% in healthy NSG mice (Fig. 1F). Confocal microscopy of intact BM via bone window showed that GFP-MOLM13 cells were concentrated in the endosteal region, and some of the tumor cells colocalized with DiD (Fig. 4C).
Table 1. Characterization of DiD+ BM subpopulations:
The values were obtained after subtracting percentages in control group (non-injected mice). HPC1 and HPC2 are hematopoietic progenitor cells at different stages of differentiation. MPP is multipotent progenitor population. The gating strategy is shown in Supplemental data.
| Jurkat-DiD | Liposome-DiD | ||
|---|---|---|---|
| Population | Murine marker | ||
| Phagocytes | CD11b+ | 7.00% | 8.10% |
| Non-phagocytes | CD11b− | 2.90% | 2.20% |
| HPC1 | CD150− CD48+ | 1.00% | 1.70% |
| HPC2 | CD150+ CD48+ | 11.60% | 8.80% |
| MPP | CD150− CD48− | 0.70% | 0.10% |
| Eosinophils | Siglec F+ | 18.00% | 10.80% |
| T cells | CD3+ | 6.60% | 0.40% |
| B cells | CD19+ | 3.40% | 5.30% |
| Neutrophils | Ly6G+ | 2.10% | 0.30% |
| Monocytes | CD11b+ Ly6C high | 17.40% | 20.90% |
Fig. 4. ICL accumulation in leukemic cells:
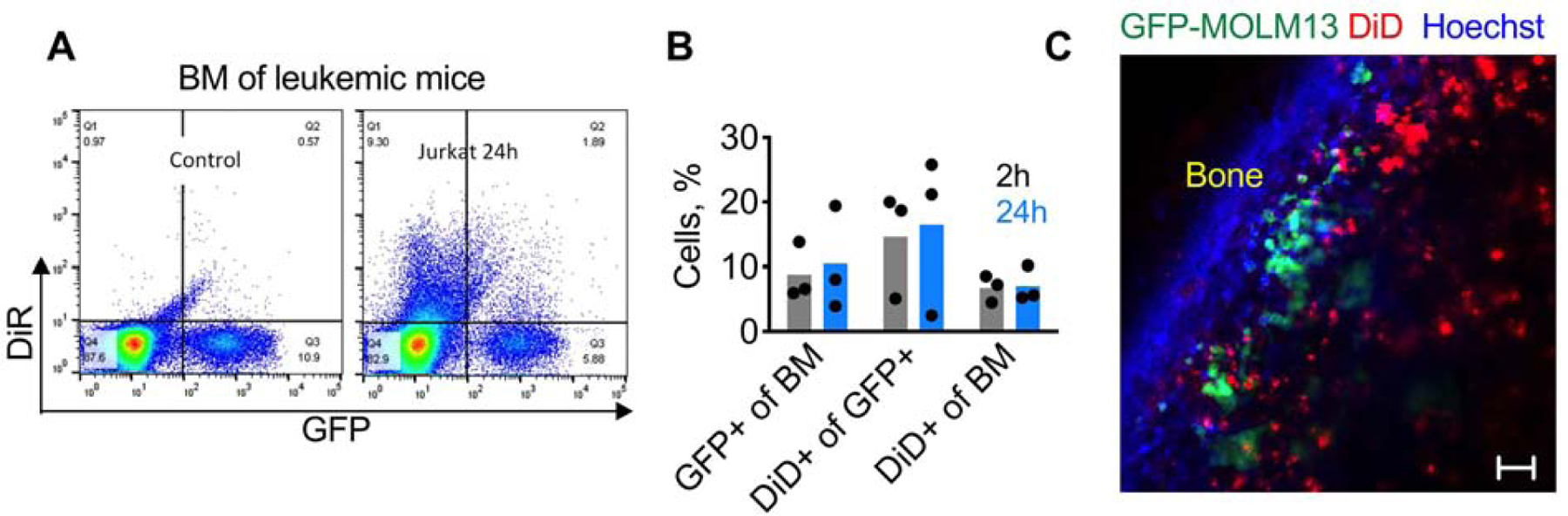
NSG mice were injected i.v with 0.5×106 GFP-MOLM13 cells to produce leukemia, and 7 days later with DiR Jurkat. A) Representative dot-plots of BM of non-injected and Jurkat-injected mice; B) percentage of different cell subpopulations in BM (n=3). There is a variability of disease burden (% tumor cells in BM), as well as the variability of delivery efficiency to the tumor cells; C) confocal microscopy via bone window 24h after injection of DiD Jurkat shows a cluster of GFP+ tumor cells in the endosteal area, some of them containing DiD. Size bar 50 µm.
3). ICL delivery to the BM is mediated via in vivo-released microparticles
Despite the fact that the number of injected DiR/CFSE Jurkat cells (10 million) was comparable to that of total blood leukocytes in an adult mouse, only few DiR+/CFSE+ events were detectable in blood at 2h and 24h post injection in NSG mice (Fig. 5A). Microscopy of a blood smear collected 1 min post-injection of DiR/CFSE Jurkat showed rare DiR+/CFSE+ cells, but numerous DiR+/CFSE+, DiR+/CFSE- and DiR-/CFSE+ microparticles, most of them smaller than RBCs (Fig. 5B). Microparticles were also observed 1 min post-injection of DiR-labeled GFP-MOLM13 cells (Fig. 5B), non-labeled GFP-MOLM13 cells (Supplemental Fig. S13) and autologous DiR/CFSE labeled mouse splenocytes injected in BALB/c mice (Supplemental Fig. S14). The particle number and size were similar for DiR-labeled CFSE Jurkat and non-DiR-labeled CFSE Jurkat (Fig. 5C), suggesting that microparticle formation is not due to the ICL labeling. Incubation of CFSE Jurkat or GFP-MOLM13 cells in vitro in fresh BALB/c serum in presence or absence of 10mM EDTA (inhibitors of complement) did not affect the microparticle release and did not reduce the cell count (Supplemental Fig. S15), which rules out complement as the main mechanism of the release of cellular microparticles. These results suggest that in vivo release of cellular particles is a general phenomenon and is not the artifact of the ICL labeling or complement-mediated destruction of xenogeneic cells in mice.
Fig. 5. Cells release microparticles in vivo:
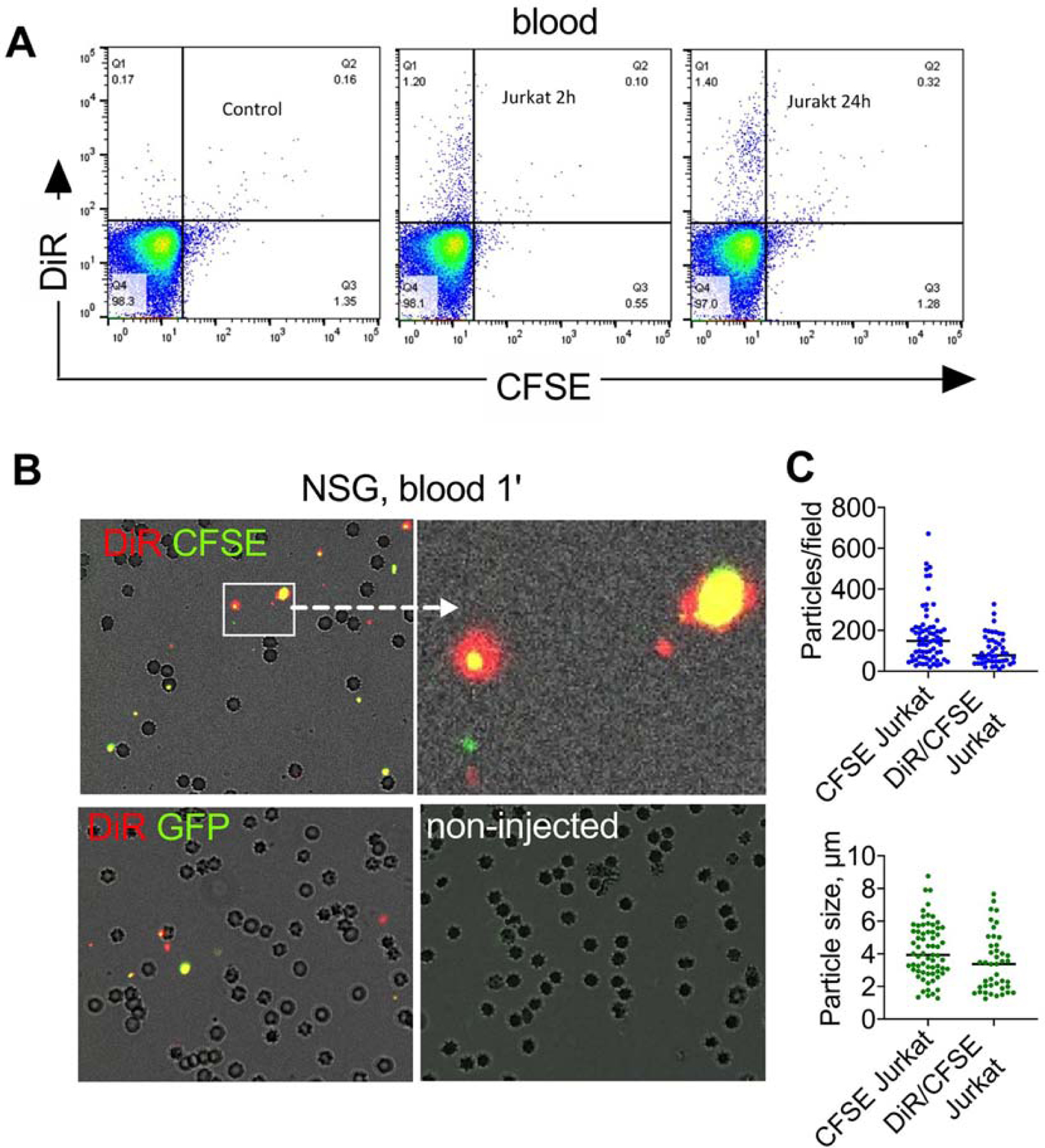
A) Flow cytometry of peripheral blood (RBCs lysed) after injection DiR/CFSE Jurkat cells in NSG mice shows no cells but some DiR+ events; B) microparticles in blood 1 min post-injection of DiR/CFSE Jurkat (top left and center) and DiR/GFP-MOLM13 (top right). Many RBCs are visible. Some of the particles are as large as RBCs, but smaller than intact cells. Fluorescence was enhanced to demonstrate colocalization; graphs show quantification of particles in blood 1 min after injection of DiR/CFSE Jurkat or CFSE Jurkat (n=2 mice, at least 40 microscopic fields counted). Particles were also observed after injection of non-labeled GFP-MOLM13 and DiR/CFSE splenocytes (supplemental data); C) low magnification images of freshly excised organs of NSG mice injected with DiR/CFSE Jurkat (2h post injection) show accumulation of CFSE and DiR in the lungs, but preferential accumulation of DiR in the liver and spleen. Size bar, 100 µm.
Cells are known to release extracellular vesicles, from nano-sized exosomes to microparticles, to larger apoptotic bodies, [40] and cell derived particles can be efficiently labeled with ICLs [41]. Some microparticles could be observed among DiR-labeled cells before injection (not shown). Staining of Jurkat cells painted with DiR with Annexin V showed about 19% of true apoptotic cells (Supplemental Fig. S16). Injection of cell supernatant harvested from Jurkat cells labeled with high concentrations of DiR (50µM and 100µM) did not lead to accumulation of DiR in the BM (Supplemental Fig. S17), suggesting that any microparticles and free DiR released prior to the injection do not contribute to the observed phenomenon. We prepared cellular microparticles from DiR/CFSE Jurkat cells by brief bath sonication. The sonication resulted in microparticles with mean size of ~1µm (Fig. 6A and Supplemental Fig. S18). Flow cytometry and ex vivo NIR imaging after injection of intact cells and microparticles (same injected dose of DiR) showed similar efficiency of BM accumulation and similar organ distribution (Fig. 6B–D). Confocal microscopy via bone window confirmed a similar efficiency of delivery for intact DiI/CFSE Jurkat cells and cell-derived microparticles (Fig. 6E).
Fig. 6. Systemically injected microparticles accumulate in the BM:
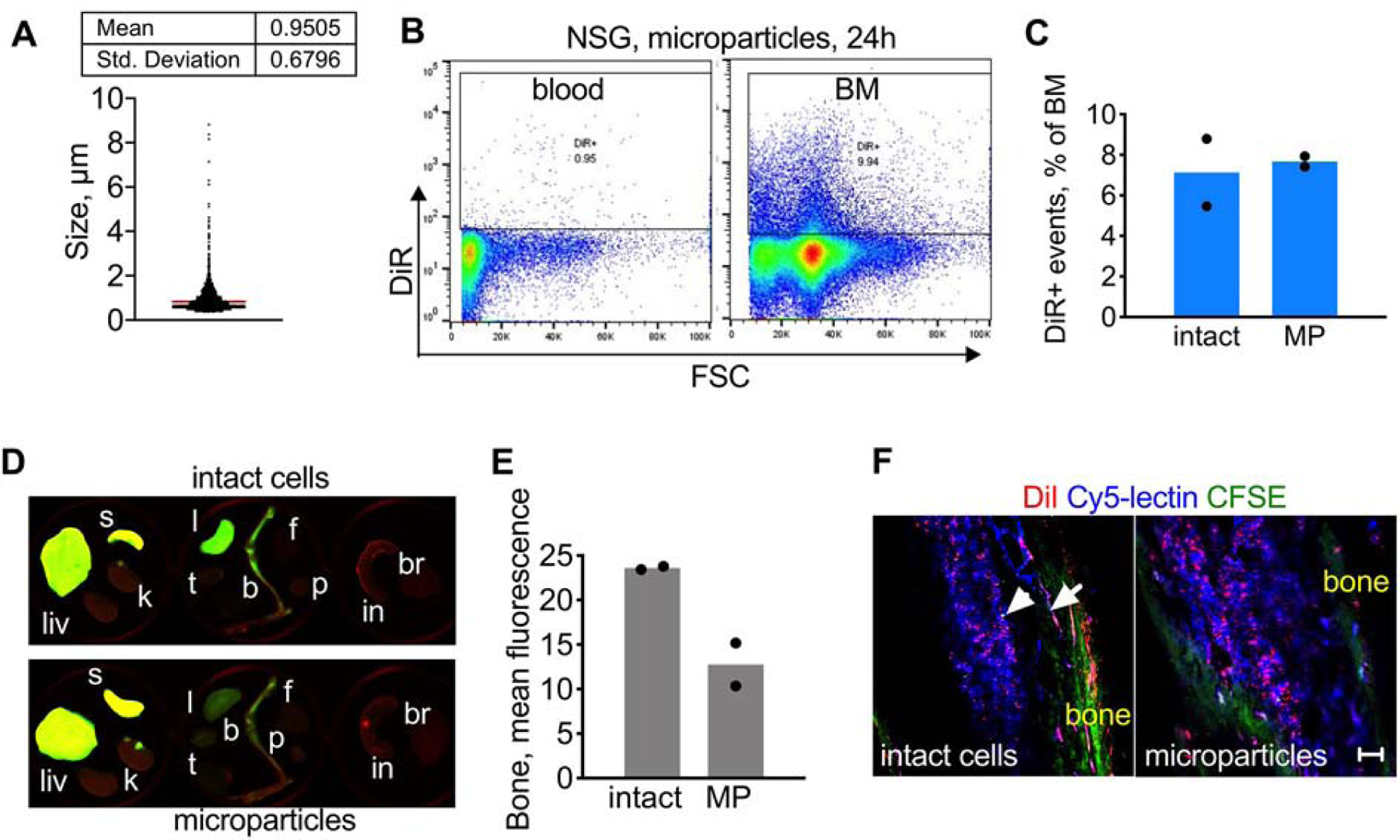
A) DiR/CFSE Jurkat cells were bath sonicated for 5 s to generate microparticles of ~1µm mean size. Quantification of particles was done using microscopy as described in Methods; B) Cell microparticles were injected in NSG mice. The flow cytometry gate was determined based on non-injected control mice (not shown). Microparticles were almost absent in blood 24h post-injection but show BM accumulation; C) same injected dose (DiR) of intact cells and microparticles (MP) produces similar accumulation efficiency; D-E) Organ imaging shows similar bone accumulation for microparticles and intact cells. Organ labels as in Fig. 1. The contrast was enhanced to the same extent; F) DiI/CFSE Jurkat (intact and microparticles) were injected in NSG mice. BM confocal microscopy 2h post-injection shows similar accumulation of DiI for intact cells and microparticles. Arrowhead points to a single DiI/CFSE-labeled cell; arrow shows DiI accumulation in supplying blood vessels. Representative images (2 mice per group) are shown. Size bar, 100µm.
DISCUSSION
We report here a novel phenomenon where systemically injected cells release microparticles in vivo that deliver the payload to the BM. This conclusion is supported by several lines of evidence: 1) the number of ICL-labeled cells in the BM was significantly higher than the number of CFSE- or GFP-labeled cells; 2) as soon as 1 min post-injection, multiple cellular microparticles were detectable in blood; 3) microparticles generated by sonication of cells accumulated in the BM as efficiently as the after injection of the same dose of intact cells; 4) the supernatant purified from ICL-labeled cells showed minimal accumulation in the BM, suggesting that preexisting free particles and dye are not responsible for this phenomenon.
The mechanism of in vivo release of microparticles, as well as the mechanism of preferential accumulation in the BM, needs to be further investigated. Previous studies suggested fragmentation and apoptosis of adoptively transferred stem cells injected in vivo[36, 42]. While we found that a fraction of DiR painted Jurkat cells is apoptotic in vitro, the release of microparticles was observed in blood even after injection of GFP-MOLM13 cells that did not undergo painting and/or incubation in vitro. The release of particles was observed for both human and mouse cells, and was independent of ICL labeling. The very fast release is unlikely to be due to the complement, because the particle release was observed in NSG mice that lack functional hemolytic complement system (C5 and Membrane Attack Complex [43]), and we did not find an increased fragmentation of cells after incubation in complement-sufficient mouse serum. Our current hypothesis that shear forces and interaction with vasculature trigger the fragmentation of cells and release of microparticles. Interestingly, metastatic cells released from tumor site are subjected to shear forces and very few of them survive in the systemic circulation [44] Another open question is why the majority of the particles in the BM was ICL-positive but CFSE-negative. ICLs are very stable in membranes in serum [24–26], ruling out the transfer to serum lipoproteins as the main mechanism of in vivo formation of the particles. It is possible that ICLs are pinched off as part of membrane microdomains in vivo. It is also possible that: a) some microparticles have very low cytoplasmic content; b) CFSE was lost/diluted during in vivo passage of microparticles; While the purpose of this study was to use a model florescent payload rather than actual lipid drug, the results suggest an interesting direction for drug delivery. We previously demonstrated that ICLs can be derivatized with biomolecules and small molecules [45, 46]. In addition, other lipid prodrugs have been developed [47, 48] and those can be incorporated in the cell membrane. The biodistribution of ICLs delivered via particles was limited to the liver, spleen, and BM, which was more selective than liposomes and more efficient than RBCs. RBCs appear to be more suitable for the delivery of membrane payloads to the intravascular compartment as well as long circulating carrier [49] [50]. The studies utilizing cellular microparticles for drug delivery are ongoing in our laboratory.
Our findings explain certain previously described phenomena in experimental hematology/oncology. Thus, intravital imaging studies demonstrated that BM homing of GFP-labeled HSCs is a rare event[51]. Furthermore, some studies have shown that culturing of leukemic cells and HSCs leads to decreased homing [16, 17], likely due to a loss of membrane polarity and receptors that mediate cell adhesion [16]. Shedding of cellular microparticles could lead to a loss of critical receptors and membrane domains, thereby decreasing homing efficiency. Previous reports suggested a lateral transfer of ICLs from HSCs to the surrounding cells in the BM [52, 53], which was ascribed to “microenvironmental contaminants” or trogocytosis by adjacent cells. Based on our data, the release the ICL-labeled particles happens immediately after the injection, well before the cells arrive to the BM. While we used higher ICL concentrations than normally used for cell labeling and tracking (3–10 µM [23]), the labeling did not decrease the cell viability, and the shedding of particles was also observed for cells without the dye. Interestingly, previous report suggested that formalin fixation of leukemic cells prevented the accumulation of DiR in the BM [23], which could be due to the decreased ability of fixed cells to shed particles.
Finally, our findings underscore the pitfalls in the studies that employ membrane dyes for tracking stem cells and cancer cells [23, 54] and suggest a possibility that in vivo generated particles rather than intact cells accumulate in organs and tissues. Given the significant interest in the use of extracellular vesicles for drug delivery [55], our findings represent a new angle of research on the role of cellular particles in organ-specific homing for drug delivery.
Materials and Methods
Materials:
DiD (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine, 4-chlorobenzenesulfonate salt), DiI (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Perchlorate) and DiR (1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide) were from Biotium (Hayward, CA, USA) and were stored as 1 mM sock in ethanol. Whatman Nucleopore Track-Etch Membranes (0.2 µm and 0.1 µm pore size), bovine serum albumin, hydrogen peroxide (H2O2) and ethyl alcohol were from Sigma-Aldrich (St. Louis, MO, USA). Nitrocellulose membrane (0.45 µm) was from Bio-Rad (Hercules, CA, USA). CFSE was from eBioscience (Carlsbad, CA, USA). Hydrogenated soy phosphatidylcholine (HSPC), cholesterol, distearyl phosphatidylethanolamine (DSPE)-PEG-2000 were from Avanti Polar Lipids (Alabaster, AL, USA). RBC Lysis/Fixation Solution was from BD Biosciences (San Diego, CA). FITC Annexin V, Annexin V Binding Buffer, Fc blocker anti-CD16/32 (#101302), PE anti-human CD184 (CXCR4, #306505), PE mouse IgG2a k isotype (#400211), PE/Cy5 anti-mouse CD3 (#100309), PE/Cy7 anti-mouse CD19 (#115520), FITC anti-mouse CD170 (Siglec-F, #155504), APC/Cy7 anti-mouse Ly-6G (#127624), PE/Cy5 anti-mouse CD48 (#103420), Alexa Fluor® 488 anti-mouse CD150 (SLAM, #115916), APC/Fire™ 750 anti-mouse CD115 (CSF-1R, 135535), and FITC anti-mouse/human CD11b (#101206) were from BioLegend (San Diego, CA, USA), anti-CD63 antibody (#SC-5275; RRID: AB_627877) was Santa Cruz Biotechnology, anti-alpha-tubulin antibody (#14–4502-82; RRID: AB_1210456) was from eBioscience. Nuclear staining reagent Hoechst 33342 trihydrochloride tirihydrate was purchased from Life Technologies (Carlsbad, CA, USA). Fetal bovine calf serum, RPMI 1640 and DMEM growth medium supplemented with L-glutamine were from Corning Inc. (New York, NY, USA). DyLight® 649 was from Vector Laboratories (Burlingame, CA, USA). N-ethylmaleimide (NEM) was from Alfa Aesar (Ward Hill, USA).
Cell culture:
Jurkat T-cell leukemia and MOLM13 acute myeloid leukemia cell lines were obtained from Dr. Craig Jordan, University of Colorado. GFP/Luc-MOLM13 cells obtained from Dr. Andreeff, MD Anderson Cancer Center. Cell lines were verified using University of Colorado sequencing core. Cells were grown at 37°C in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 10 mM HEPES, 100 U/ml penicillin and 100 ng/ml streptomycin (all from Corning Inc. New York, NY, USA). Bone marrow derived macrophages (BMDMs) were isolated from femurs of BALB/c female mice (6–10 week old) and differentiated in DMEM medium supplemented with 10% L929-conditioned medium, 10% heat-inactivated FBS and 1% Pen-Strep under 10% CO2 at 37°C as described by us before [56]. Splenocytes were isolated from BALB/c mice by pushing freshly isolated spleens through 40 µm nylon cell strainer (BD Biosciences), washing the cells with 1%BSA/PBS 3 times, and lysis of RBCs according to the manufacturer’s instructions
Cell and RBC labeling:
For CFSE labeling, Jurkat cells in a logarithmic growth phase were collected and washed twice with PBS to remove serum. Cells were resuspended at 1×107/mL in PBS (pre-warmed to room temperature). CFSE was added at 10 µM final concentration, mixed with cells immediately, and cells were incubated for 10 minutes at 37°C in the dark. Labeling was stopped by addition of 200 μl FBS. Cells were incubated on ice for 5 minutes, washed 3 times with RPMI 1640 containing 1% BSA at 330g for 5 min. To label cells with ICLs, CFSE-labeled, or non-labeled cells (2×107 cells per tube) were resuspend in 400 μl of 1% BSA/RPMI and incubated with DiR, DiD or DiI for 1 hour at 37°C in the dark with occasional mixing. Cells were washed three times with 1% BSA/RPMI at 300g for 5 min. Finally, the cells were resuspended in 300 μl of 1% BSA/RPMI. To label RBCs, 50–100 μl of whole blood was collected from the orbital venous plexus of BALB/c mice using a heparinated capillary glass tube, washed three times in 1% BSA/PBS at 1500g, being careful to remove the buffy coat. RBCs were labeled with 25 µM DiR or DiD as described above.
In order to normalize the injected amount of fluorescence for different carriers, labeled RBCs, Jurkat and liposomes were spotted at different dilutions on a nitrocellulose membrane and scanned with Bio-Rad gel scanner at DiD (Cy5) wavelength, or with Li-COR Odyssey at DiR (800 nm) wavelength. The fluorescence signal (mean fluorescence) in 8-bit TIFF images for each formulation was determined with ImageJ software. The formulations of labeled carriers were diluted accordingly in order to inject the equal amount of fluorescence.
Liposome preparation:
Lipids (molar ratios: HSPC/Chol/DSPE-PEG2000/ICL 56.32/38/5.21/0.37) in chloroform were mixed and dried under nitrogen stream. The dry lipid cake was resuspended in PBS for a total lipid composition of 20 mM, then incubated at 60°C for 30 minutes. The solution was then vortexed for 2 minutes, sonicated and vortexed for 30 seconds, repeated three times. The solution was extruded at 60°C using an Avestin manual extruder (Avestin, Ottawa, Canada) through Whatman Nucleopore Track-Etch Membranes (through 200nm pore size 15 times, then through 100nm pore size 15 times). A Zetasizer Nano (Malvern, UK) was used to measure the size of the liposomes in PBS. Liposomes were stored at 4 °C at a final concentration of 20 mM lipid for a maximum period of 2 weeks before use.
Animal experiments:
The University of Colorado Institutional Animal Care and Use Committee (IACUC) approved animal experiments (protocol 103913(11)1D). Mice were treated according to regulations provided by the Office of Laboratory Animal Resources at the University of Colorado. BALB/c and NOD/LtSz-SCID IL2Rγc null (NSG) mice were bred in-house. Mice of 8–10 weeks age (males and females) were used for experiments. In order to determine the distribution of ICLs after injection, mice were injected intravenously with cells, cellular particles obtained from cells, or with fluorescence dose-matched RBCs and liposomes. Mice were sacrificed with carbon dioxide (CO2) followed by cardiac perfusion with PBS through left ventricle. The heart, liver, spleen, kidney, thymus, pancreas, fat, intestine, brain, and limb bones were arranged in a 6-well plate and scanned with a Li-COR Odyssey at 700 nm for autofluorescence and 800 nm for DiR. Mean fluorescence was determined from 8-bit TIFF images using ImageJ software by subtracting the background, drawing a region of interest around the organs, and using “measure” function to determine mean gray values. Such measurement is independent of the organ “area” and is proportional to the dye concentration.
For analysis of ICL accumulation following injection in vivo, 30–50 μl of heparinated blood was obtained. Erythrocytes were lysed with RBC lysis buffer per manufacturer’s instructions. For experiments involving injection of labeled RBCs, RBCs were not lysed. Cells were centrifuged, washed in 1% BSA/PBS and resuspended at 1×106/ml. To collect bone marrow cells, humerus and femur were dissected, the bone marrow cavity was flushed with 8 ml of 1% BSA/PBS, cells were centrifuged at 400g for 5 min, and the supernatant was discarded. Erythrocytes were lysed, the cells were washed three times and resuspended in 1% BSA/PBS.
Flow cytometry analysis:
For bone marrow staining for surface markers, cells were harvested from BM as described above. After lysis of erythrocytes and washing the cell pellet with 1% BSA/PBS, Fc receptors were blocked with an Fc blocker at 4°C for 10 minutes, followed by incubation with antibodies against main surface markers for 30 minutes at 4°C. Cells were analyzed for fluorescence with Guava EasyCyte HT flow cytometer (Luminex Corp., Seattle, WA). Cells were resuspended at ~1 million/ml and at least 10,000 events were counted. Forward scattering threshold was set to >2200 in order to exclude debris. The data were analyzed using FlowJo software v.10 (BD Life Sciences, Ashland, OR). Percentages of BM cells positive for ICLs were determined for injected and control non-injected mice (at least 3 mice per group). The percentages of ICL+ BM cells were calculated after subtraction of control values.
Microscopy:
Zeiss Axio Observer 5 epifluorescent microscope equipped with X-Cite 200DC light source and Axiocam 506 monochromatic camera using Cy7 filter set, catalog number 49007, Chroma Corporation (McHenry, IL, USA) was used. For blood particle imaging, 5 µl of fresh diluted or non-diluted blood was placed on a slide, covered with a cover slip, and at least 20 images per slide were acquired under 200x magnification. Number of particles per image and mean particle size were determined with ImageJ particle count function (after thresholding of background fluorescence). To further study the distribution of fluorescence in the intact bone marrow, a bone window model was generated. Two hours post injection, mice were sacrificed and tibial and femoral bones were dissected and cleaned from muscle and connective tissues. Cortical bone was scraped away with the lateral edge of a 19G needle as close to the endosteum as possible while keeping a thin layer of residual bone. For delineating the vasculature and bone marrow cells, mice were preinjected 30 min prior to the experiment with 50 μl of DyLight 649 tomato lectin (1 mg/ml). Alternatively, mice were preinjected with 40μl Hochest33324 (10 mg/mL) to label nuclei. Bone was placed on a cover slip with the window facing the glass and imaged with Nikon Eclipse AR1HD confocal microscope using 405 nm (Hoechst), 488 nm (CFSE or GFP), 561 nm (DiI) and 640 nm (DyLight 649 or DiD) excitation lasers and corresponding emission filters.
Supplementary Material
Highlights.
Cells delivered a model lipid payload (indocarbocyanine lipid DiR) to murine bone marrow more efficiently than erythrocytes and more selectively than liposomes
The lipid payload was delivered to the bone marrow independently of the injected cells
The lipid payload was delivered to the bone marrow via microparticles released in vivo
The phenomenon was independent of the immune system of the host and was observed for syngeneic and xenogeneic cells
In vitro preformed microparticles efficiently delivered the lipid payload to the bone marrow
Acknowledgments and disclaimers
The study was supported by the NIH grants CA194058 to DS, GM126960 and AG061829 to JS, and the National Natural Science Foundation of China (Grant No. 31771093) and the Fundamental Research Funds for the Central Universities that supported postdoctoral studies of FC (2017TD-27, 2019TD-36). All the work was performed at the University of Colorado Anschutz Medical Campus.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Morrison SJ, Scadden DT, The bone marrow niche for haematopoietic stem cells, Nature, 505 (2014) 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chambers AF, Groom AC, MacDonald IC, Dissemination and growth of cancer cells in metastatic sites, Nat Rev Cancer, 2 (2002) 563–572. [DOI] [PubMed] [Google Scholar]
- [3].Hassanshahi M, Hassanshahi A, Khabbazi S, Su YW, Xian CJ, Bone marrow sinusoidal endothelium: damage and potential regeneration following cancer radiotherapy or chemotherapy, Angiogenesis, 20 (2017) 427–442. [DOI] [PubMed] [Google Scholar]
- [4].Lapid K, Glait-Santar C, Gu-Cohen S, Canaani J, Kollet O, Lapidot T, Egress and Mobilization of Hematopoietic Stem and Progenitor Cells: A Dynamic Multi-facet Process, in: StemBook, Harvard Stem Cell Institute, Cambridge (MA), 2012. [PubMed] [Google Scholar]
- [5].Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, Zhang S, Moschetta M, Seevaratnam D, Zhang Y, Liu J, Memarzadeh M, Wu J, Manier S, Shi J, Bertrand N, Lu ZN, Nagano K, Baron R, Sacco A, Roccaro AM, Farokhzad OC, Ghobrial IM, Engineered nanomedicine for myeloma and bone microenvironment targeting, Proc Natl Acad Sci U S A, 111 (2014) 10287–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Han HS, Niemeyer E, Huang Y, Kamoun WS, Martin JD, Bhaumik J, Chen Y, Roberge S, Cui J, Martin MR, Fukumura D, Jain RK, Bawendi MG, Duda DG, Quantum dot/antibody conjugates for in vivo cytometric imaging in mice, Proc Natl Acad Sci U S A, 112 (2015) 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX, LoRusso PM, Munster PN, Campbell K, Gaddy DF, Leonard SC, Geretti E, Blocker SJ, Kirpotin DB, Moyo V, Wickham TJ, Hendriks BS, (64)Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer, Clin Cancer Res, 23 (2017) 4190–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tardi P, Wan CP, Mayer L, Passive and semi-active targeting of bone marrow and leukemia cells using anionic low cholesterol liposomes, J Drug Target, 24 (2016) 797–804. [DOI] [PubMed] [Google Scholar]
- [9].Mandal A, Patel M, Sheng Y, Mitra AK, Design of Lipophilic Prodrugs to Improve Drug Delivery and Efficacy, Curr Drug Targets, 17 (2016) 1773–1798. [DOI] [PubMed] [Google Scholar]
- [10].Buzhor E, Leshansky L, Blumenthal J, Barash H, Warshawsky D, Mazor Y, Shtrichman R, Cell-based therapy approaches: the hope for incurable diseases, Regen Med, 9 (2014) 649–672. [DOI] [PubMed] [Google Scholar]
- [11].Hu X, Leak RK, Thomson AW, Yu F, Xia Y, Wechsler LR, Chen J, Promises and limitations of immune cell-based therapies in neurological disorders, Nat Rev Neurol, 14 (2018) 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang J, Hubbard-Lucey VM, Pearce L, O’Donnell-Tormey J, Shalabi A, The global landscape of cancer cell therapy, Nature reviews. Drug discovery, 17 (2018) 465–466. [DOI] [PubMed] [Google Scholar]
- [13].Szilvassy SJ, Bass MJ, Van Zant G, Grimes B, Organ-selective homing defines engraftment kinetics of murine hematopoietic stem cells and is compromised by Ex vivo expansion, Blood, 93 (1999) 1557–1566. [PubMed] [Google Scholar]
- [14].Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Cote D, Rowe DW, Lin CP, Scadden DT, Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche, Nature, 457 (2009) 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kerre TCC, De Smet G, De Smedt M, Offner F, De Bosscher J, Plum J, Vandekerckhove B, Both CD34+38+ and CD34+38− Cells Home Specifically to the Bone Marrow of NOD/LtSZ scid/scid Mice but Show Different Kinetics in Expansion, The Journal of Immunology, 167 (2001) 3692. [DOI] [PubMed] [Google Scholar]
- [16].Larochelle A, Gillette JM, Desmond R, Ichwan B, Cantilena A, Cerf A, Barrett AJ, Wayne AS, Lippincott-Schwartz J, Dunbar CE, Bone marrow homing and engraftment of human hematopoietic stem and progenitor cells is mediated by a polarized membrane domain, Blood, 119 (2012) 1848–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Szilvassy SJ, Meyerrose TE, Ragland PL, Grimes B, Differential homing and engraftment properties of hematopoietic progenitor cells from murine bone marrow, mobilized peripheral blood, and fetal liver, Blood, 98 (2001) 2108–2115. [DOI] [PubMed] [Google Scholar]
- [18].Krause DS, Lazarides K, Lewis JB, von Andrian UH, Van Etten RA, Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche, Blood, 123 (2014) 1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Termini CM, Cotter ML, Marjon KD, Buranda T, Lidke KA, Gillette JM, The membrane scaffold CD82 regulates cell adhesion by altering alpha4 integrin stability and molecular density, Mol Biol Cell, 25 (2014) 1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA, Gillette JM, Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin, Oncogene, 35 (2016) 4132–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Progatzky F, Dallman MJ, Lo Celso C, From seeing to believing: labelling strategies for in vivo cell-tracking experiments, Interface Focus, 3 (2013) 20130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zagorodnyuk VP, Kyloh M, Nicholas S, Peiris H, Brookes SJ, Chen BN, Spencer NJ, Loss of visceral pain following colorectal distension in an endothelin-3 deficient mouse model of Hirschsprung’s disease, J Physiol, 589 (2011) 1691–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kalchenko V, Shivtiel S, Malina V, Lapid K, Haramati S, Lapidot T, Brill A, Harmelin A, Use of lipophilic near-infrared dye in whole-body optical imaging of hematopoietic cell homing, J Biomed Opt, 11 (2006) 050507. [DOI] [PubMed] [Google Scholar]
- [24].Bastiat G, Pritz CO, Roider C, Fouchet F, Lignieres E, Jesacher A, Glueckert R, Ritsch-Marte M, Schrott-Fischer A, Saulnier P, Benoit JP, A new tool to ensure the fluorescent dye labeling stability of nanocarriers: a real challenge for fluorescence imaging, J Control Release, 170 (2013) 334–342. [DOI] [PubMed] [Google Scholar]
- [25].Shi G, Mukthavaram R, Kesari S, Simberg D, Distearoyl anchor-painted erythrocytes with prolonged ligand retention and circulation properties in vivo, Advanced healthcare materials, 3 (2014) 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Griffin JI, Wang G, Smith WJ, Vu VP, Scheinman R, Stitch D, Moldovan R, Moghimi SM, Simberg D, Revealing Dynamics of Accumulation of Systemically Injected Liposomes in the Skin by Intravital Microscopy, ACS Nano, 11 (2017) 11584–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gullapalli RR, Demirel MC, Butler PJ, Molecular dynamics simulations of DiI-C18(3) in a DPPC lipid bilayer, Physical chemistry chemical physics : PCCP, 10 (2008) 3548–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benjamin R, Advances in off-the-shelf CAR T-cell therapy, Clin Adv Hematol Oncol, 17 (2019) 155–157. [PubMed] [Google Scholar]
- [29].Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS, Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity, Front Immunol, 8 (2017) 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, Kitaoka F, Takahashi T, Okita K, Yoshida Y, Kaneko S, Hotta A, Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility, Cell stem cell, 24 (2019) 566–578 e567. [DOI] [PubMed] [Google Scholar]
- [31].Carter BZ, Mak PY, Mak DH, Shi Y, Qiu Y, Bogenberger JM, Mu H, Tibes R, Yao H, Coombes KR, Jacamo RO, McQueen T, Kornblau SM, Andreeff M, Synergistic targeting of AML stem/progenitor cells with IAP antagonist birinapant and demethylating agents, Journal of the National Cancer Institute, 106 (2014) djt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jost TR, Borga C, Radaelli E, Romagnani A, Perruzza L, Omodho L, Cazzaniga G, Biondi A, Indraccolo S, Thelen M, Te Kronnie G, Grassi F, Role of CXCR4-mediated bone marrow colonization in CNS infiltration by T cell acute lymphoblastic leukemia, J Leukoc Biol, 99 (2016) 1077–1087. [DOI] [PubMed] [Google Scholar]
- [33].Gaikwad H, Wang G, Smith WJ, Alexander KL, D’Alessandro A, Zhang W, Purev E, Simberg D, Clickable Methyltetrazine-Indocarbocyanine Lipids: A Multicolor Tool Kit for Efficient Modifications of Cell Membranes, Bioconjugate chemistry, 30 (2019) 2106–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sanchez PV, Perry RL, Sarry JE, Perl AE, Murphy K, Swider CR, Bagg A, Choi JK, Biegel JA, Danet-Desnoyers G, Carroll M, A robust xenotransplantation model for acute myeloid leukemia, Leukemia, 23 (2009) 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Visioni A, Kim M, Wilfong C, Blum A, Powers C, Fisher D, Gabriel E, Skitzki J, Intra-arterial Versus Intravenous Adoptive Cell Therapy in a Mouse Tumor Model, Journal of immunotherapy, 41 (2018) 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Leibacher J, Henschler R, Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells, Stem Cell Res Ther, 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Inturi S, Wang G, Chen F, Banda NK, Holers VM, Wu L, Moghimi SM, Simberg D, Modulatory Role of Surface Coating of Superparamagnetic Iron Oxide Nanoworms in Complement Opsonization and Leukocyte Uptake, ACS nano, 9 (2015) 10758–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kuemmel TA, Thiele J, Hafenrichter EG, Varus E, Fischer R, Distribution of lectin binding sites in human bone marrow. Identification by use of an ultrastructural postembedding technique, J Submicr Cytol Path, 28 (1996) 537–551. [PubMed] [Google Scholar]
- [39].Schumacher U, Horny HP, Welsch U, Kaiserling E, Lectin histochemistry of human bone marrow: investigation of trephine biopsy specimens in normal and reactive states and neoplastic disorders, Histochem J, 23 (1991) 215–220. [DOI] [PubMed] [Google Scholar]
- [40].Beer KB, Wehman AM, Mechanisms and functions of extracellular vesicle release in vivo-What we can learn from flies and worms, Cell Adh Migr, 11 (2017) 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Grange C, Tapparo M, Bruno S, Chatterjee D, Quesenberry PJ, Tetta C, Camussi G, Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging, Int J Mol Med, 33 (2014) 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Leibacher J, Dauber K, Ehser S, Brixner V, Kollar K, Vogel A, Spohn G, Schafer R, Seifried E, Henschler R, Human mesenchymal stromal cells undergo apoptosis and fragmentation after intravenous application in immune-competent mice, Cytotherapy, 19 (2017) 61–74. [DOI] [PubMed] [Google Scholar]
- [43].Baxter AG, Cooke A, Complement Lytic Activity Has No Role in the Pathogenesis of Autoimmune Diabetes in Nod Mice, Diabetes, 42 (1993) 1574–1578. [DOI] [PubMed] [Google Scholar]
- [44].Strilic B, Offermanns S, Intravascular Survival and Extravasation of Tumor Cells, Cancer Cell, 32 (2017) 282–293. [DOI] [PubMed] [Google Scholar]
- [45].Smith WJ, Tran H, Griffin JI, Jones J, Vu VP, Nilewski L, Gianneschi N, Simberg D, Lipophilic indocarbocyanine conjugates for efficient incorporation of enzymes, antibodies and small molecules into biological membranes, Biomaterials, 161 (2018) 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gaikwad H, Wang G, Smith WJ, Alexander KL, D’Alessandro A, Zhang W, Purev E, Simberg D, Clickable Methyltetrazine-Indocarbocyanine Lipids: A Multicolor Tool Kit for Efficient Modifications of Cell Membranes, Bioconjugate chemistry, (2019). [DOI] [PMC free article] [PubMed]
- [47].Gabizon AA, Tzemach D, Horowitz AT, Shmeeda H, Yeh J, Zalipsky S, Reduced toxicity and superior therapeutic activity of a mitomycin C lipid-based prodrug incorporated in pegylated liposomes, Clinical Cancer Research, 12 (2006) 1913–1920. [DOI] [PubMed] [Google Scholar]
- [48].Irby D, Du C, Li F, Lipid-Drug Conjugate for Enhancing Drug Delivery, Mol Pharm, 14 (2017) 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Muzykantov VR, Drug delivery by red blood cells: vascular carriers designed by mother nature, Expert opinion on drug delivery, 7 (2010) 403–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brenner JS, Pan DC, Myerson JW, Marcos-Contreras OA, Villa CH, Patel P, Hekierski H, Chatterjee S, Tao JQ, Parhiz H, Bhamidipati K, Uhler TG, Hood ED, Kiseleva RY, Shuvaev VS, Shuvaeva T, Khoshnejad M, Johnston I, Gregory JV, Lahann J, Wang T, Cantu E, Armstead WM, Mitragotri S, Muzykantov V, Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude, Nat Commun, 9 (2018) 2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kim S, Lin L, Brown GAJ, Hosaka K, Scott EW, Extended time-lapse in vivo imaging of tibia bone marrow to visualize dynamic hematopoietic stem cell engraftment, Leukemia, 31 (2017) 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lassailly F, Griessinger E, Bonnet D, “Microenvironmental contaminations” induced by fluorescent lipophilic dyes used for noninvasive in vitro and in vivo cell tracking, Blood, 115 (2010) 5347–5354. [DOI] [PubMed] [Google Scholar]
- [53].Li P, Zhang R, Sun H, Chen L, Liu F, Yao C, Du M, Jiang X, PKH26 can transfer to host cells in vitro and vivo, Stem Cells Dev, 22 (2013) 340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ruan J, Song H, Li C, Bao CC, Fu HL, Wang K, Ni J, Cui DX, DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells, Theranostics, 2 (2012) 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Murphy DE, de Jong OG, Brouwer M, Wood MJ, Lavieu G, Schiffelers RM, Vader P, Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking, Exp Mol Med, 51 (2019) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang G, Serkova NJ, Groman EV, Scheinman RI, Simberg D, Feraheme (Ferumoxytol) Is Recognized by Proinflammatory and Anti-inflammatory Macrophages via Scavenger Receptor Type AI/II, Molecular pharmaceutics, 16 (2019) 4274–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.