

Summary
Since their discovery decades ago, the primary physiological and pathological impact of potassium channels has been attributed to their ion conductance, which sets membrane potential and repolarizes action potentials. For example, Kv3 family channels regulate neurotransmitter release by repolarizing action potentials. Here we report a surprising yet crucial function independent of potassium conductance – by organizing F-actin cytoskeleton in mouse nerve terminals, the Kv3.3 protein facilitates slow endocytosis, rapid endocytosis, vesicle mobilization to the readily releasable pool, and recovery of synaptic depression during repetitive firing. A channel mutation that causes spinocerebellar ataxia inhibits endocytosis, vesicle mobilization, and synaptic transmission during repetitive firing by disrupting the ability of the channel to nucleate F-actin. These results unmask novel functions of potassium channels in endocytosis and vesicle mobilization crucial for sustaining synaptic transmission during repetitive firing. Potassium channel mutations that impair these “non-conducting” functions may thus contribute to the generation of diverse neurological disorders.
Keywords: endocytosis, potassium channels, vesicle mobilization, F-actin, mouse, exocytosis, Kv3 channels, neurological disorders, Spinocerebellar Ataxia Type 13
eTOC blurb
Wu et al. found that Kv3.3 potassium channels facilitate endocytosis, vesicle mobilization, and recovery of short-term synaptic depression by organizing the presynaptic F-actin cytoskeleton. These ‘non-conductive’ functions are impaired by Kv3.3 mutation that causes spinocerebellar ataxia. Thus, by nucleating F-actin, Kv3.3 is crucial for endocytosis, synaptic transmission and neurological disorders.
Introduction
The established role of more than seventy pore-forming α subunits of potassium channels is to alter potassium flux to shape action potentials and generate different types of intrinsic neuronal firing patterns. The fact that the membrane-spanning domains of these channels are highly conserved while their cytoplasmic domains are diverse suggests, however, that they may have additional functions. For example, the main function of Kv3.3 channels, expressed widely in cerebellum, auditory brainstem nuclei, hippocampus and thalamus, is to allow high frequency firing by rapidly repolarizing action potentials (Kaczmarek and Zhang, 2017). When expressed in cell lines, the extended cytoplasmic C-terminal domain of Kv3.3 recruits the Arp2/3 actin nucleating complex to assemble a stable subcortical actin network, implying that Kv3.3 may regulate actin and thus exert “non-conducting” functions (Zhang et al., 2016).
Mutations in the Kv3.3 gene (KCNC3) result in Spinocerebellar Ataxia Type 13 (SCA13), a disease associated with cerebellar degeneration that impairs motor behaviors (Zhang and Kaczmarek, 2016). This disease, when occurring at a late-onset, is also associated with auditory impairment in detecting interaural timing differences (Middlebrooks et al., 2013). One SCA13 mutation, G592R Kv3.3 (Kv3.3G592R), located in the cytoplasmic C-terminus and associated with late onset motor deficits, results in fully functional channels with voltage-dependence identical to those of wild type (WT) channels. Kv3.3G592R channels, however, fail to bind the Arp2/3 complex and fail to generate a subcortical actin network in cell lines (Zhang et al., 2016), implying that the “non-conducting” function of Kv3.3 in actin nucleation could be central to neuronal functions.
Here we tested the role of Kv3.3 channels in calyx of Held nerve terminals, which are required for sound localization by providing a strong excitatory input from the cochlear nucleus to principal neurons of the medial nucleus of the trapezoid body (MNTB) (Borst and Soria van Hoeve, 2012). F-actin in nerve terminals, including calyces (Saitoh et al., 2001; Wen et al., 2016; Wu et al., 2016), is required for vesicle endocytosis following neurotransmitter release, likely by providing mechanical forces required to generate membrane pits (Soykan et al., 2017; Watanabe et al., 2013; Wu et al., 2016). We found that Kv3.3 channels are required for presynaptic actin structure formation, fast and slow endocytosis, vesicle mobilization, and synaptic transmission during repetitive firing in calyces, as well as in cultured hippocampal synapses. Each of these steps is disrupted in nerve terminals lacking Kv3.3 channels or those expressing functional Kv3.3G592R channels that fail to trigger actin nucleation. These results suggest that Kv3.3 channels organize the presynaptic F-actin cytoskeleton to facilitate endocytosis and vesicle mobilization, which maintain synaptic transmission during repetitive firing. Errors in such non-conducting functions of potassium channels may contribute to SCA13 and other diseases.
Results
Kv3.3 knockout or Kv3.3G592R knock-in inhibits slow endocytosis
Whole-cell voltage-clamped recordings were made at 22–24°C in P7–10 mouse calyces of Held, if not mentioned otherwise. A 20-ms depolarization from −80 mV to +10 mV (Depol20ms) was applied to induce slow endocytosis (τ > ~10 s). In WT mice, Depol20ms induced a calcium current (ICa) of 1.7 ± 0.1 nA and a membrane capacitance (Cm) jump (ΔCm) of 343 ± 21 fF (10 calyces, 10 mice, Figures 1A and 1B). The capacitance jump was followed by a mono-exponential decay with a time constant (τ) of 16.6 ± 1.8 s and an initial decay rate (Ratedecay) of 26 ± 3 fF/s (n = 10, Figures 1A and 1B). Although both τ and Ratedecay reflected endocytosis rate, we used Ratedecay for statistics, because τ was often too slow to estimate when endocytosis was blocked (Sun et al., 2010; Wu et al., 2016; Wu et al., 2009).
Figure 1.
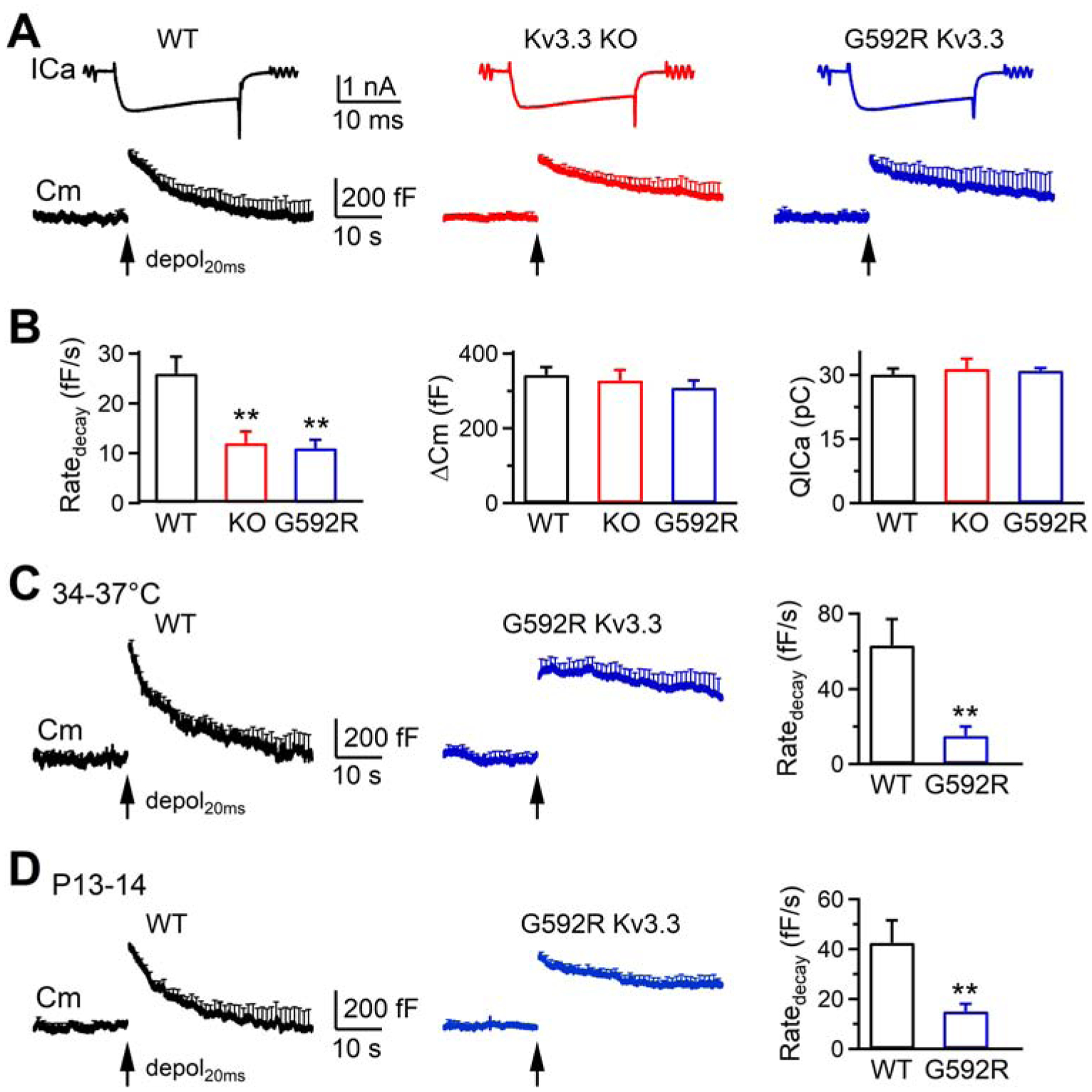
Kv3.3 KO or Kv3.3G592R KI inhibits slow endocytosis at calyces
(A) Sampled ICa and mean Cm trace (mean + s.e.m) induced by depol20ms (arrow) from WT (10 calyces, 10 mice), Kv.3.3−/− (10 calyces, 10 mice) and Kv3.3G592R (10 calyces, 10 mice) calyces from P7–10 mice at 22–24°C. s.e.m. is plotted every 1 s.
(B) Ratedecay, ΔCm and QICa (mean + s.e.m.) induced by depol20ms at WT (10 calyces, 10 mice, black), Kv.3.3−/− (10 calyces, 10 mice, red) and Kv3.3G592R (10 calyces, 10 mice, blue) calyces from P7–10 mice at 22–24°C. **: p < 0.01 (t test).
(C) The Cm change (mean + s.e.m.) and Ratedecay (mean + s.e.m.) induced by depol20ms (arrow) at WT (7 calyces, 7 mice) and Kv3.3G592R (7 calyces, 7 mice) calyces from P7–10 mice at 34–37°C. **: p < 0.01 (t test).
(D) Similar to C, except from P13–14 mice at 22–24°C. WT: 8 calyces, 8 mice; Kv3.3G592R: 7 calyces, 7 mice.
Compared to WT calyces, the Ratedecay, but not ΔCm or ICa charge (QICa), was significantly reduced in Kv3.3−/− calyces, and the Cm decay was much prolonged (Figures 1A and 1B, 10 calyces, 10 mice). Similar results were observed in homozygous Kv3.3G592R knock-in (KI) mice (called Kv3.3G592R mice) (10 calyces, 10 mice, Figures 1A and 1B), which we generated with the CRISPR/Cas9 system by replacing the WT channel with a point mutant that generates normal Kv3.3 currents but fails to trigger actin nucleation (Zhang et al., 2016). Similar reduction of Ratedecay was also observed in Kv3.3G592R calyces at physiological 34–37°C (Figure 1C) or in more matured P13–14 calyces (22–24 °C, Figure 1D). Thus, inhibition of slow endocytosis in Kv3.3−/− or Kv3.3G592R calyces is independent of temperature or developmental stage. Given that Kv3.3G592R channels are fully functional channels, but do not trigger Arp2/3-dependent actin nucleation (Zhang et al., 2016), these results suggest that Kv3.3 channels are involved in slow endocytosis by interacting with the Arp2/3 complex.
Kv3.3 knockout or Kv3.3G592R KI inhibits rapid endocytosis at calyces
10 Depol20ms at 10 Hz (Depol20msX10) in WT calyces (P7–10, 22–24°C) induced a ΔCm of 1087 ± 92 fF, followed by a bi-exponential decay with τ of 1.5 ± 0.2 s (20 ± 2.5%) and 23.2 ± 2.7 s that reflected rapid and slow endocytosis, respectively (n = 10, Figure 2A) (Wu et al., 2009). The Ratedecay was 173 ± 13 fF/s (n = 10, Figures 2A and 2B), which reflected mostly (~89%) the rapid component of endocytosis (Sun et al., 2010; Wu et al., 2009). Compared to WT, the Ratedecay, but not QICa, in Kv3.3−/− (n = 10 calyces, 10 mice) or Kv3.3G592R calyces (n = 10 calyces, 10 mice) decreased significantly; the Cm decay was much prolonged in Kv3.3−/− or Kv3.3G592R calyces (Figures 2A and 2B). Similar results were observed in Kv3.3G592R calyces at 34–37°C (Figure 2C), or at more matured P13–14 calyces (22–24°C, Figure 2D). These results suggest that Kv3.3 channel is involved in rapid endocytosis.
Figure 2.
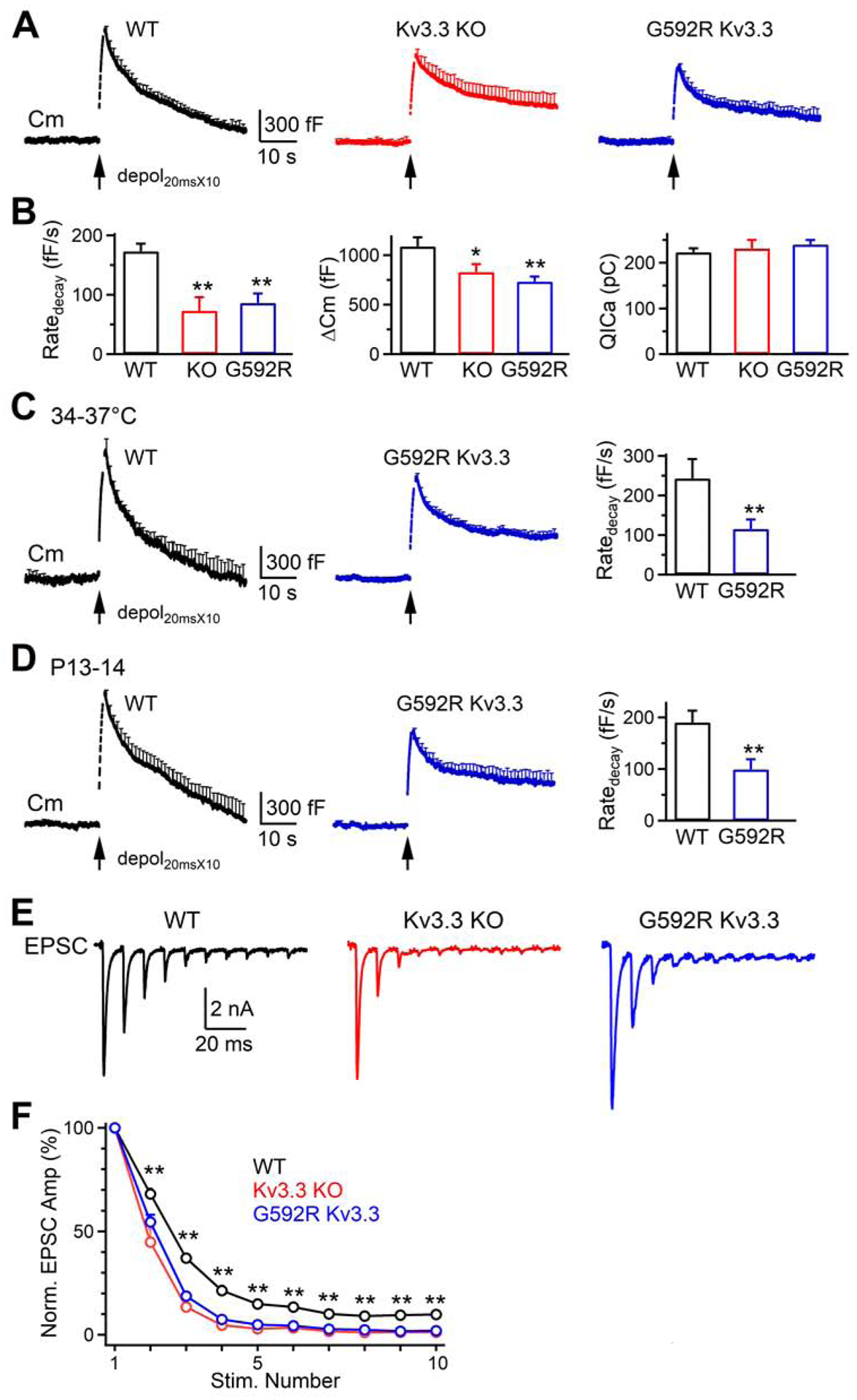
Kv3.3 KO or Kv3.3G592R KI inhibits rapid endocytosis at calyces
(A) Cm traces (mean + s.e.m, every 1 s) induced by depol20msX10 (arrow) in WT (10 calyces, 10 mice), Kv.3.3−/− (10 calyces, 10 mice) and Kv3.3G592R (10 calyces, 10 mice) calyces of P7–10 mice at 22–24°C.
(B) Ratedecay, ΔCm and QICa (mean + s.e.m.) induced by depol20msX10 (arrow) at WT (10 calyces, 10 mice), Kv.3.3−/− (10 calyces, 10 mice) and Kv3.3G592R (10 calyces, 10 mice) calyces from P7–10 mice at 22–24°C. *: p < 0.05; **: p < 0.01 (t test).
(C) The Cm change (mean + s.e.m.) and Ratedecay (mean + s.e.m.) induced by depol20msX10 (arrow) at WT (7 calyces, 7 mice) or Kv3.3G592R (6 calyces, 6 mice, blue) calyces of P7–10 mice at 34–37°C. **: p < 0.01 (t test).
(D) Similar to C, except from P13–14 mice at 22–24°C. WT: 8 calyces, 8 mice; Kv3.3G592R: 7 calyces, 7 mice.
(E-F) Sampled EPSCs (E) and the amplitude of EPSCs (F, mean + s.e.m.) induced by 10 stimuli at 100 Hz at WT (6 neurons, 6 mice), Kv.3.3−/− (5 neurons, 5 mice), and Kv3.3G592R (5 neurons, 5 mice) calyceal synapses of P7–10 mice at 22–24°C. F, amplitudes normalized to the first EPSC (Norm. EPSC Amp) plotted versus stimulation (stim.) number; **: p < 0.01, 2–10th EPSCs in WT are larger than that in Kv.3.3−/− or Kv3.3G592R synapses (t test).
Kv3.3 KO or Kv3.3G592R KI inhibits vesicle replenishment at calyces
Since Depol20ms depletes the readily releasable vesicle pool (RRP), Depol20msX10 repeatedly depletes the RRP that is partially replenished between each Depol20ms (Sakaba and Neher, 2001; Sun and Wu, 2001; Wu and Borst, 1999). Thus, ΔCm induced by Depol20msX10 reflects the RRP replenishment rate. Compared to WT, ΔCm after Depol20msX10, but not Depol20ms (Figure 1B), was reduced in Kv3.3−/− and Kv3.3G592R mice (Figure 2A–D), suggesting that Kv3.3 KO and Kv3.3G592R KI inhibit the RRP replenishment.
Inhibition of endocytosis by many other approaches, such as block of dynamin, actin, calmodulin, calcium influx, or SNARE proteins, also slows down RRP replenishment, suggesting that endocytosis may facilitate RRP replenishment by clearing the active zone perturbed by exocytosis (Hosoi et al., 2009; Hua et al., 2013; Neher, 2010; Wu et al., 2014; Wu et al., 2016; Wu et al., 2009; Wu and Wu, 2009). Accordingly, inhibition of endocytosis and RRP replenishment by Kv3.3 KO and Kv3.3G592R KI suggests that Kv3.3 may facilitate RRP replenishment by promoting active zone clearance.
Kv3.3 KO or Kv3.3G592R KI inhibits EPSCs during repetitive stimuli
To determine whether Kv3.3 regulates synaptic transmission, we recorded EPSCs induced by 10 action potentials (AP) at 100 Hz at calyx of Held synapses. The amplitude of the EPSC induced by each AP decreased during the train, reflecting short-term depression (Figure 2E) (Wu and Borst, 1999; Xu and Wu, 2005). Such a decrease was significantly larger in Kv3.3−/− or Kv3.3G592R synapses (Figures 2E and 2F), suggesting that Kv3.3 KO and Kv3.3G592R KI caused more short-term synaptic depression. Thus, both presynaptic Cm and postsynaptic EPSC recordings suggest that Kv3.3 may slow down short-term depression and thus help maintain synaptic transmission during repetitive firing.
Kv3.3 KO or Kv3.3G592R KI inhibits actin network at calyces
In heterologous cells, the Kv3.3 channel nucleates a substantial actin network via its cytoplasmic C-terminal domain which couples to the Arp2/3 complex, whereas the Kv3.3G592R channel, which does not bind to the Arp2/3 complex, fails to nucleate the subcortical cytoskeleton (Zhang et al., 2016). This finding and the observed inhibition of slow and rapid endocytosis by Kv3.3 KO or Kv3.3G592R KI (Figures 1 and 2), suggest that the Kv3.3 channel is involved in mediating endocytosis by nucleating an actin network at nerve terminals. Two sets of evidence further support this suggestion.
First, we carried out electron immunomicroscopy using horseradish peroxidase-diaminobenzidine staining to examine the subcellular distribution of Kv3.3 immonoreactivity within the terminals. In WT mice, dense immunostaining was detected in patches at the calyx plasma membrane, particularly on membranes facing the postsynaptic cell adjacent to clusters of synaptic vesicles (Figure 3A). A similar distribution at the presynaptic plasma membrane was found in Kv3.3G592R calyces (Figure 3A). In contrast, no Kv3.3 diaminobenzidine staining was detected in Kv3.3−/− calyces (Figure 3A). Thus, Kv3.3 channels are localized at nerve terminal membranes facing the postsynaptic neuron.
Figure 3.
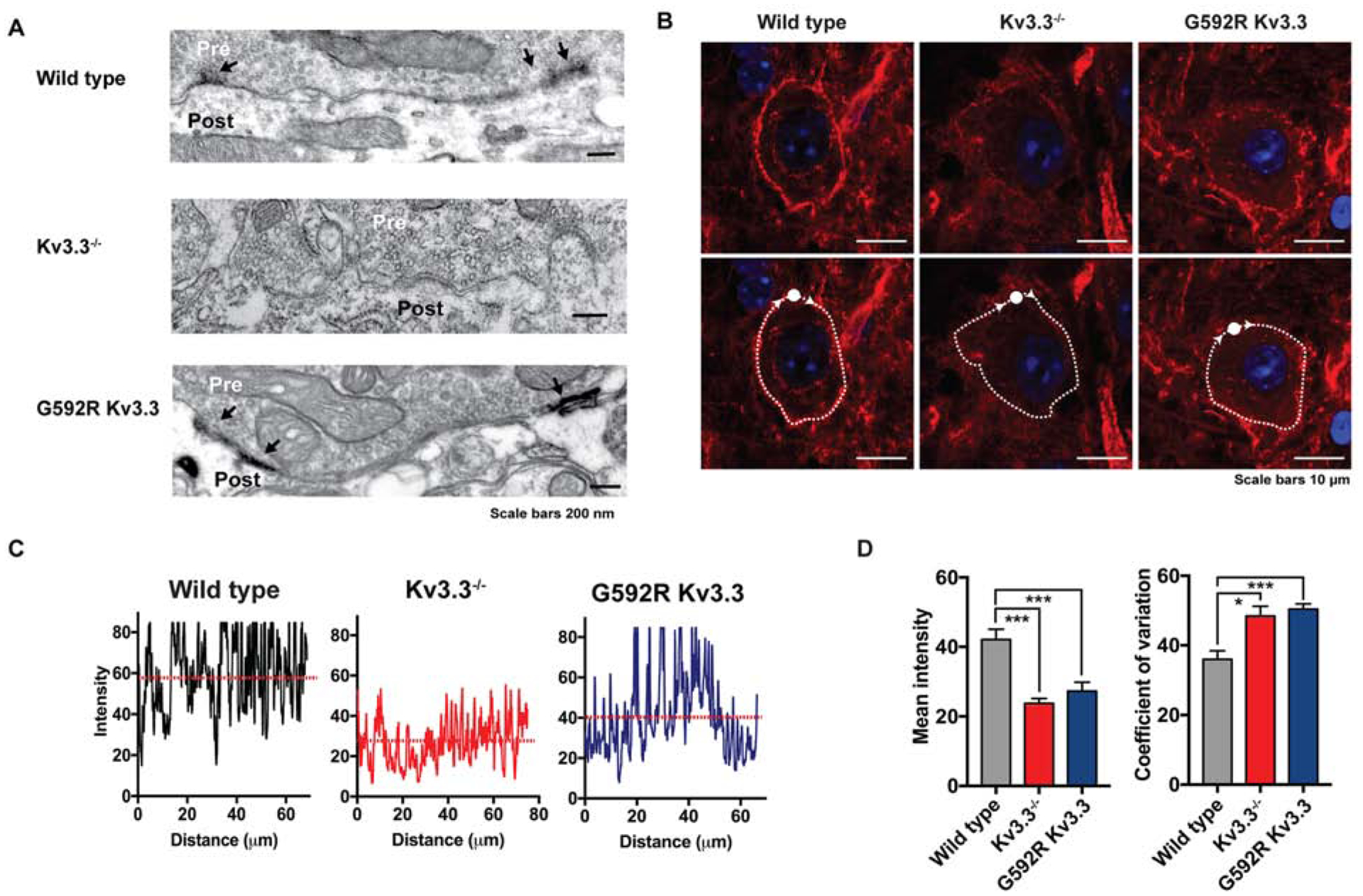
Kv3.3 KO or Kv3.3G592R KI reduces F-actin at calyces
(A) Electron microscopic images showing Kv3.3 immunoreactivity (arrows) at the calyces of WT, Kv3.3−/− or Kv3.3G592R mouse. Results are representative of 8–36 immunostained sections from 1, 2 or 1 animal, respectively.
(B) Phalloidin labelling of F-actin at WT, Kv3.3−/−, and Kv3.3G592R calyces visualized using Airy-scan confocal microscopy. Upper panels: a dense ring of presynaptic F-actin staining surrounds MNTB neurons in WT; the intensity of actin staining is greatly reduced in Kv3.3−/− and Kv3.3G592R mice. Lower panels: the same sections with superimposed traces of lines following the circumference of the calyx. Such traces were used in panels C and D to quantify coefficient of variability for the intensity of pixels in the ring.
(C) Representative plots of pixel intensity across the circumference of WT, Kv3.3−/− and Kv3.3G592R cells.
(D) Bar graphs depicting mean pixel intensity and coefficient of variability (+ s.e.m.) for the three conditions. (*: p < 0.02; ***: p < 0.005, Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test).
Second, we stained sections of the MNTB using phalloidin to examine the distribution of F-actin using Airy-scan confocal microscopy. As documented previously (Saitoh et al., 2001; Wu et al., 2016), a dense ring of F-actin staining was found to surround the postsynaptic MNTB neurons in sections from WT mice (Figure 3B). Previous work has demonstrated that this actin staining is located in nerve terminals, where it colocalizes with the vesicular glutamate transporter 1 that labels the calyx (Wu et al., 2016). In contrast, the pattern of actin staining around MNTB neurons from Kv3.3−/− and Kv3.3G592R mice was strikingly different (Figure 3B; calyces imaged: WT, 92, 3 mice; Kv3.3−/−, 127, 3 mice; Kv3.3G592R, 113, 3 mice). From visual inspection, it was apparent that the density of calyceal actin staining was greatly reduced in Kv3.3−/− and Kv3.3G592R neurons compared to that from WT. In contrast, the level of phalloidin staining in blood vessels was not different among WT, Kv3.3−/− and Kv3.3G592R slices. This observation was used to quantify the differences in neuronal staining, by normalizing the intensity of all pixels in a ring surrounding each MNTB neuron to the maximal intensity found for blood vessels that were found on all sections (Figure 3B). We calculated the mean intensity of pixels surrounding neurons (10 calyces, 3 mice, staining was visualized across the circumference of the cell). Consistent with the visual observations, the mean of the intensity values was significantly lower in Kv3.3−/− and Kv3.3G592R mice compared to WT (Figure 3D; Kv.3.3−/−, p = 0.0002; Kv3.3G592R, p = 0.0038, Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test).
Another difference evident on visual inspection was that the integrity of the actin “ring” was disrupted. In contrast to the appearance of a more-or-less contiguous pattern of staining around the MNTB neurons of WT mice, the actin distribution around the circumference of neurons from Kv3.3−/− and Kv3.3G592R mice was more punctate (Figure 3C). To quantify this observation, we determined the coefficient of variability for the intensity of pixels surrounding neurons. Consistent with visual observations, the coefficient of variability for Kv3.3−/− and Kv3.3G592R was greater than that for WT neurons (Figure 3D, Kv.3.3−/−, p = 0.011; Kv3.3G592R, p = 0.0004, Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test).
Kv3.3 KO or Kv3.3G592R KI inhibits endocytosis at hippocampal synapses
To determine whether results obtained at calyces apply to conventional small nerve terminals, we examined endocytosis at hippocampal cultures transfected with a pH-sensitive synaptophysin-pHluorin2X (SypH) (Sankaranarayanan and Ryan, 2000; Sun et al., 2010). At 34–37°C, application of 1, 40 or 200 APs at 20 Hz induced SypH fluorescence (FSypH) increase (ΔF) and decrease, reflecting exo- and endocytosis, respectively (Figure 4A–F). Compared to WT cultures, decay of FSypH and the initial decay rate (Ratedecay) were significantly slower in Kv3.3−/− or Kv3.3G592R cultures after 1, 40 or 200 APs (Figure 4A–F). For example, in WT after 200 APs, the mono-exponential decay τ was 15.4 ± 1.6 s, reflecting slow endocytosis; the Ratedecay was 9 ± 1%/s (17 experiments, 6 cultures, each culture from 3–5 mice, Figures 4E and 4F).
Figure 4.
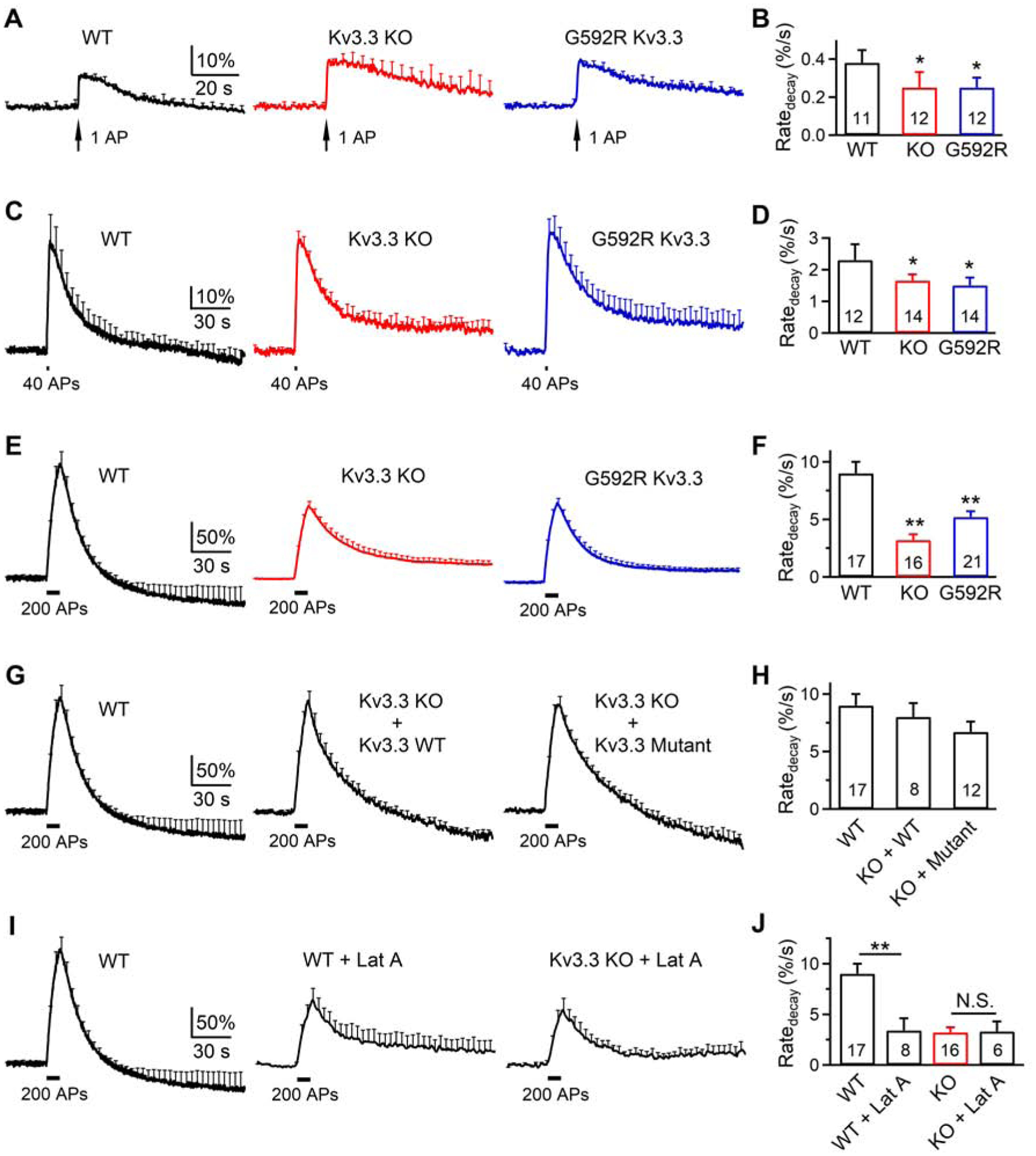
Kv3.3 KO or Kv3.3G592R KI inhibits endocytosis at hippocampal synapses
(A-B) FSypH (mean + s.e.m., A) and Ratedecay (mean + s.e.m., B) induced by 1 AP in WT, Kv.3.3−/− or Kv3.3G592R boutons at 34–37°C. FSypH is normalized to baseline; s.e.m. (A) is plotted every 4 s. The experimental number is written inside the bar (B, applies to panel A-B); each experiment contained 20–30 boutons; 1–3 experiments were taken from 1 culture; each culture was from 3–5 mice. Ratedecay was calculated from FSypH normalized to the baseline. *: p < 0.05; **: p < 0.01; t test (compared to WT).
(C-F) Similar to A-B, respectively, except that the stimulus was 40 APs (C-D) or 200 APs (E-F) at 20 Hz.
(G-H) Similar to E-F (200 APs), respectively, except from WT, or Kv.3.3−/− cultures overexpressed with WT Kv3.3 (KO + WT) or a mutant (W496F) Kv3.3 with no ionic conductance (KO + Mutant).
(I-J) Similar to E-F, respectively, except from WT cultures, WT cultures with 20 μM latrunculin A in the bath (WT + Lat A), or Kv.3.3−/− cultures with Lat A in the bath (KO + Lat A). N.S.: not significant (p > 0.05, t test).
See also Figures S1–S4.
Interestingly, FSypH increase after 200 APs was followed by a decay below baseline by a value of 18 ± 8% of ΔF at 120 s later (17 experiments, Figure 4E). Thus, endocytosis overshoot is not limited to large terminals where it was first reported (Renden and Von Gersdorff, 2007; Wu et al., 2009), but may also be used by small conventional boutons.
In Kv3.3−/− or Kv3.3G592R cultures, FSypH decay was slower with a significantly lower Ratedecay as compared to that in WT (Figures 4E and 4F), reflecting inhibition of endocytosis. Such a slower Ratedecay was observed regardless of whether FSypH was normalized to its baseline (Figure 4F) or ΔF (Figure S1A). The endocytosis overshoot was not observed in Kv3.3−/− or Kv3.3G592R cultures. These results (Figure 4A–F) suggest that Kv3.3 KO or Kv3.3G592R KI inhibits endocytosis and endocytosis overshoot at hippocampal synapses. Consistent with results obtained at calyces (Figures 1 and 2), ΔF after 200 APs at 20 Hz was reduced by Kv3.3 KO or Kv3.3G592R KI (Figures 4E, 4F and S2A), suggesting that reduction of exocytosis during repetitive firing may be caused by impairment of the RRP replenishment.
Kv3.3 regulates endo- and exocytosis by regulating actin, but not K+ permeability
Three sets of evidence suggest that the observed regulation of exo- and endocytosis by Kv3.3 is independent of K+ channel conductance. First, Ratedecay and ΔCm at calyces were measured at the voltage-clamp mode with K+ currents abolished by 3,4-diaminopyridine, tetraethylammonium chloride, and cesium (see Methods). Reduction of Ratedecay and ΔCm after depol20msx10 in Kv3.3−/− or Kv3.3G592R calyces (Figures 1 and 2) is thus independent of K+ currents.
Second, at hippocampal synapses, Ratedecay and ΔF after 200 APs at 20 Hz were reduced by either Kv3.3 KO or Kv3.3G592R KI (Figures 4E and 4F). Because Kv3.3G592R has a nearly normal Kv3.3 channel conductance, this suggests that Kv3.3 regulates endo- and exocytosis independently of channel conductivity.
Third, to probe further the role of K+ conductance, we generated a non-conducting Kv3.3 channel by inserting a point mutation W496F at the pore region (Minassian et al., 2012; Perozo et al., 1993). This mutation, W496F Kv3.3, abolished ionic conductance when expressed in CHO cells (Figure S3). At Kv3.3−/− boutons, the reduction of Ratedecay and ΔF and the block of endocytosis overshoot after 200 APs at 20 Hz were rescued to the WT level by overexpression of either WT Kv3.3 or the W496F Kv3.3 pore mutant (Figures 4G and 4H), confirming that Kv3.3 regulates endo- and exocytosis independently of its conductance.
The regulation of endocytosis by a presynaptic K+ channel is specific to Kv3.3, and is unaffected by Kv3.1, the other major Kv3 family member expressed in many presynaptic terminals, including calyces of Held (Choudhury et al., 2020; Kaczmarek and Zhang, 2017; Song et al., 2005). Specifically, we found that, in contrast to Kv3.3−/− cultures, endocytosis in Kv3.1−/− mouse hippocampal cultures was not significantly different from that in WT (Figure S4). This is consistent with facts that i) of these two channels, only Kv3.3 has the specific sequences in its longer cytoplasmic C-terminus that link it to actin polymerization (Zhang et al., 2016); ii) transfection of Kv3.3, but not Kv3.1, leads to assembly of a subcortical actin cytoskeleton in CHO cells; and iii) Kv3.3, but not Kv3.1, is localized to the release face of calyces, while Kv3.1 is expressed on the “back” face (Elezgarai et al., 2003).
Considering that F-actin is crucial for all kinetically distinguishable forms of endocytosis at calyces and hippocampal synapses (Wu et al., 2016), our finding that Kv3.3 KO or Kv3.3G592R KI reduced the F-actin cytoskeleton (Figure 3) and Ratedecay at these nerve terminals (Figures 1, 2, 4) suggests that Kv3.3 regulates endocytosis by organizing F-actin. As a further test of this hypothesis, we analyzed the effects of latrunculin A, which depolymerizes F-actin, on WT and Kv3.3−/− boutons. Latrunculin A reduced Ratedecay and ΔF, and block endocytosis overshoot after 200 APs at 20 Hz in WT boutons, but did not significantly reduce Ratedecay or ΔF in Kv3.3−/− boutons (Figures 4I, 4J, S1B and S2B). Thus, Kv3.3 KO occluded the effects of latrunculin A, suggesting that inhibition of endocytosis (including overshoot) and exocytosis by Kv3.3 KO is via the F-actin pathway.
Discussion
We found that complete knockout of Kv3.3 or its replacement by Kv3.3G592R, a channel that has normal electrophysiological function but does not bind the Arp2/3 actin-nucleating complex (Zhang et al., 2016), inhibited slow endocytosis, rapid endocytosis, endocytosis overshoot, the RRP replenishment, the EPSC during repetitive firings, and the F-actin network at the large calyx-type synapses (Figures 1–3). Kv3.3 KO or Kv3.3G592R KI inhibited endocytosis and endocytosis overshoot induced by 1–200 APs at 20 Hz, and inhibited exocytosis induced by 200 APs at small conventional hippocampal synapses (Figure 4). These inhibitions were not due to regulation of the Kv3.3 channel conductance, but to the F-actin network, via the binding of the Kv3.3 C-terminus with Arp2/3 (Figures 1–4). Thus, the Kv3.3 channel may facilitate synaptic transmission by organizing the F-actin network at nerve terminals to facilitate endocytosis and RRP replenishment. These findings have widespread implications for four aspects of synapse function, as discussed below.
First, Kv3.3 channels are expressed at high levels in nerve terminals of multiple types of neurons, including auditory brainstem neurons, mossy fibers of hippocampal granule cells and cortical interneurons, and many of these are characterized by high rates of firing and synaptic transmission (Chang et al., 2007; Kaczmarek and Zhang, 2017; Li et al., 2001; Zhang et al., 2016). At the calyx of Held, ultrastructural examination reveals Kv3.3 to be localized in the plasma membrane facing the postsynaptic neuron (Figure 3A) and previous work has documented a dense F-actin network under this membrane that faces the postsynaptic cell (Wen et al., 2016; Wu et al., 2016), suggesting Kv3.3 and F-actin are co-localized at this location. F-actin is involved in mediating not only slow and rapid endocytosis, but also other forms of endocytosis, such as bulk endocytosis, endocytosis overshoot and ultrafast endocytosis at large calyx nerve terminals and small, conventional hippocampal boutons (Soykan et al., 2017; Watanabe et al., 2014; Wu et al., 2016). Thus, regulation of F-actin by the Kv3.3 channel may control not only the most common forms of endocytosis studied here, the slow and fast endocytosis, but also other kinetically distinguishable forms of endocytosis at nerve terminals that fire at high rates and that express this channel.
Second, actin may provide force to invaginate the membrane during endocytosis in yeast (Engqvist-Goldstein and Drubin, 2003) and mammalian non-secretory cells (Boulant et al., 2011; Kaksonen and Roux, 2018; Mettlen et al., 2018). At synapses, actin β or γ isoform KO or pharmacological inhibition of F-actin polymerization inhibits shallow pit formation, suggesting that actin is involved in providing forces for membrane invagination (Watanabe et al., 2014; Wu et al., 2016). Accordingly, Kv3.3 may organize a subcortical actin network to mediate membrane invagination, the initial step of endocytosis. The present work revealed the unexpected link between Kv3.3 and the endocytic machinery that recycles vesicles and sustains synaptic transmission during repetitive firing. We suggest including Kv3.3 channel as an integral component of the endocytic machinery at the fast-firing nerve terminals. As future studies may uncover more seemingly endocytosis-unrelated mechanisms that may regulate endocytosis, the integral machinery regulating endocytosis at nerve terminals may be much larger than the traditional endocytic machinery in non-excitable cells. An interesting question for the future is to determine whether other K+ channels or related proteins substitute for Kv3.3 to organize F-actin at nerve terminals that fire at lower rates and that do not express Kv3.3, although, in principle, any regulation of the F-actin assembly pathway, such as by actin-nucleating formins (Soykan et al., 2017; Wen et al., 2016), may regulate endocytosis.
Third, we found that Kv3.3 facilitates replenishment of the RRP and helps to counteract short-term synaptic depression during repetitive firing (Figures 2 and 4). Because many endocytic players, including actin and dynamin, have been suggested to facilitate RRP replenishment via active zone clearance (Hosoi et al., 2009; Wu et al., 2014; Wu et al., 2016; Wu et al., 2009), the key role of the Kv3.3 channel may also be to facilitate active zone clearance and, as a result, RRP replenishment. Replenishment of the RRP is crucial for sustaining synaptic transmission during repetitive firing at nearly all synapses (Wu et al., 2014; Zucker and Regehr, 2002). Thus, the Kv3.3 channel may help to sustain synaptic transmission during repetitive firing by promoting nucleation of F-actin close to release sites.
Fourth, the present work provides a ready explanation for some of the phenotypes associated with the human disease SCA13. The MNTB is a key component of auditory brainstem circuitry that computes inter-aural timing and level differences, and its function requires sustained synaptic transmission at rates of hundreds of Hertz. A deficit in transmission at this synapse is, therefore, fully consistent with the inability of SCA13 patients to localize sounds in space (Middlebrooks et al., 2013). These deficits in transmission in other Kv3.3-expressing neurons are also likely to contribute eventually to cerebellar atrophy and motor symptoms caused by Kv3.3 channel mutations.
Interestingly, we found that hippocampal synapses use endocytosis overshoot (Figure 4), an endocytic mode previously reported only in giant nerve terminals and endocrine cells (Artalejo et al., 1995; Renden and Von Gersdorff, 2007; Wu et al., 2014; Wu et al., 2009; Xue et al., 2012). Thus, like rapid and slow endocytosis (Wu et al., 2014), endocytosis overshoot may participate in retrieving vesicles widely in small conventional boutons, large nerve terminals and endocrine cells. It has been suggested that endocytosis overshoot reflects a preexisting membrane pool ready to be retrieved via a dynamin- and calcineurin-dependent mechanism upon calcium influx (Xue et al., 2012). Our finding of endocytosis overshoot at hippocampal synapses is consistent with, and may explain, the existence of a readily retrieval pool of synaptic proteins that are retrieved upon depolarization at hippocampal synapses (Fernandez-Alfonso et al., 2006; Hua et al., 2011; Wienisch and Klingauf, 2006). Block of the endocytosis overshoot by inhibition of F-actin, Kv3.3 KO or Kv3.3G592R KI (Figure 4) suggests that F-actin is involved in mediating endocytosis overshoot, and Kv3.3 channels may regulate endocytosis overshoot by promoting nucleation of F-actin at nerve terminals.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ling-Gang Wu (wul@ninds.nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data produced for this manuscript are available from the Lead Contact (wul@ninds.nih.gov) upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animal care and use were carried out according to NIH guidelines and were approved by the NIH Animal Care and Use Committee. Kv3.3−/− mice (Zagha et al., 2008; Zhang et al., 2016), were obtained by homozygous breeding using standard mouse husbandry procedures. To generate the G592R Kv3.3 point mutant mouse strain, we used the CRISPR/Cas9 system as previously described (Henao-Mejia et al., 2016). In brief, a gRNA close to the genomic location of the desired mutation was designed to maximize cutting efficiency and minimize off-target effects using the CRISPOR algorithm. Cas9 mRNA, a single guide RNA (gRNA), and a repair template including the point mutation were in vitro transcribed. Superovulated female C57BL/6J mice were mated to stud C57BL/6J males, and fertilized embryos collected from the oviducts. The Cas9 mRNA, gRNA, and repair template were injected into the cytoplasm of ~175 fertilized eggs and then transferred into uterus of pseudopregnant female mice (gRNA sequence: CACCCCCACCACGGCAGCGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTT; Repair template: GCAGCCTGGCTCACCCAACTACTGCAAGCCTGACCCCCCGCCTCCACCCCCACCACACCCCCACCACGGCAGCCGTGGCATAAGCCCACCGCCGCCCATCACCCCTCCTTCCATGGGGGTGAATGT). F0 founder mice were screened for the insertion of the point mutation and the absence of additional mutations in the nearby regions to the target sequence for the selected gRNA to ensure adequate DNA repair of the specific genomic locus. Out of nine F0 founders, two containing the desired point mutation and perfect repair of the locus were identified using standard PCR and sequencing methodologies. One of these positive F0 founders bearing a heterozygous mutation was bred to C57BL/6J WT mice for three generations to obtain heterozygous mice and breed out potential off target mutations. Heterozygous mice were then bred to each other to obtain littermate mutant and WT mice for experimentation. The Kv3.1−/− mice generated previously (Ho et al., 1997; Macica et al., 2003) were maintained in the lab by breeding with heterozygous mice.
P0 mice of either sex were used for hippocampal culture experiments. P7–14 mice of either sex were used for the preparation of brain slices where the membrane capacitance of the calyces was recorded. 8-week-old mice were used for immunostaining of Kv3.3 channels and F-actin labeling shown in Figure 3.
Cell lines
CHO cells [identifier: CHO/DHFR (−), from Dr. Kaczmarek’s lab] were grown in Iscove’s modified Dulbecco’s medium (Invitrogen) supplemented with 10% fetal bovine serum (heat-inactivated), 100 units/ml penicillin/streptomycin, 5% HT supplement (Invitrogen) in a 5% CO2 incubator at 37 °C.
Primary cell cultures
Hippocampal CA1-CA3 region from P0 mice of either sex was dissected, dissociated, and plated on Poly-D-lysine treated coverslips, which produced mouse hippocampal culture used in this study (Sankaranarayanan and Ryan, 2000; Sun et al., 2010).
METHOD DETAILS
Brain slice preparation, capacitance recordings, and EPSC recordings
Parasagittal or transverse brainstem slices (200 μm thick) containing the MNTB were prepared from 7–14 days old male or female mice using a vibratome (Sun and Wu, 2001; Wu et al., 2009). Whole-cell capacitance measurements of the calyces in parasagittal slices were made with the EPC-9 amplifier with a software lock-in amplifier (1000 Hz sine wave, peak-to-peak voltage ≤ 60 mV, HEKA, Lambrecht, Germany). We pharmacologically isolated presynaptic Ca2+ currents with a bath solution (~22–24°C or 34–37°C when mentioned) containing (in mM): 105 NaCl, 20 TEA-Cl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol, 2 sodium pyruvate, 0.001 tetrodotoxin (TTX), 0.1 3,4-diaminopyridine, pH 7.4 when bubbled with 95% O2 and 5% CO2. The presynaptic pipette contained (in mM): 125 Cs-gluconate, 20 CsCl, 4 MgATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, 0.05 BAPTA, pH 7.2, adjusted with CsOH. If not mentioned otherwise, all reagents were purchased from Sigma (St. Louis, MO).
In temperature (34–37°C) experiments, the continuously flowing solution reached the slice chamber via a tube, which was heated to ~40–42°C right (at ~10–15 cm) before the solution reached the chamber. With a flow rate of ~2.5 ml/min, the chamber temperature was maintained at 34–37°C, as confirmed with a thermometer. Slices were at 34–37°C for ~15–20 min before patching.
For postsynaptic whole-cell voltage-clamp recordings of EPSCs in transverse slices, an Axopatch 200B amplifier (Axon Instruments Inc, Foster City, CA) was used. Postsynaptic pipette (2 – 3 MΩ) solution contained (in mM): 125 K-gluconate, 20 KCl, 4 MgATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, 0.5 EGTA, pH 7.2, adjusted with KOH. The series resistance (< 15 MΩ) was compensated by 95% (lag 10 μs). The bath solution contained (in mM): 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 25 dextrose, 0.4 ascorbic acid, 3 myo-inositol, 2 sodium pyruvate, 0.05 D, L-2-amino-5-phosphonovaleric acid, 0.01 bicuculline and 0.01 strychnine, pH 7.4 when bubbled with 95% O2 and 5% CO2. A bipolar stimulating electrode was positioned at the midline of the trapezoid body, through which the presynaptic axons of MNTB synapses pass. A stimulus (5–20 V, 0.1 ms) was delivered every 20 s to induce a presynaptic action potential and the resulting EPSC was recorded at the postsynaptic cell (Wu et al., 1998). The postsynaptic neurons of the MNTB were whole-cell voltage-clamped at a holding potential of −80 mV, a potential at which the EPSC is mediated by AMPA receptors.
Hippocampal culture
Mouse hippocampal culture was prepared as described previously (Sankaranarayanan and Ryan, 2000; Sun et al., 2010). Hippocampal CA1-CA3 regions from P0 mice were dissected, dissociated, and plated on Poly-D-lysince treated coverslips. Cells were maintained at 37°C in a 5% CO2 humidified incubator with a culture medium consisting of Neurobasal A, 1% GlutaMAX-1, 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2% B-27 (Invitrogen, Carlsbad, CA), and 3 μM cytosine β-D-arabinofuranoside. On 5–7 days after plating, neurons were transfected with plasmids using Lipofectamine LTX (Invitrogen, Carlsbad, CA).
Hippocampal cultures were transfected with a plasmid containing SypH alone. Action potential was evoked by a 1 ms pulse (20 mA) through a platinum electrode. The bath solution contained (in mM): 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES (buffered to pH 7.4), 30 glucose, 0.01 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX), and 0.05 D, L-2-amino-5-phosphonovaleric acid. In temperature experiments, we heated the culture chamber using a temperature controller (TC344B, Warner Instruments, Hamden, CT). Imaging was performed after the culture was at 34–37°C for 15–30 min. The temperature was verified with another small thermometer (BAT-7001H, Physitemp Instruments, Clifton, NJ) in the chamber. SypH images were acquired at 10 Hz using Nikon A1 confocal microscope (60X, 1.4 NA), and analyzed with Nikon software. All boutons showing fluorescence increases were analyzed (region of interest: 2 μm X 2 μm). Each data group was obtained from at least three batches of cultures.
Electron Microscopy
Mice (8 weeks old) were anaesthetized and transcardially perfused with 4% PFA and 0.1% glutaraldehyde. After post-fixation overnight, vibratome sections (50 μm) containing the cerebellum were washed and then then immunostained for Kv3.3 for 48h at 4°C using an anti-Kv3.3 antibody raised in rabbit (Alomone labs, APC-102). The sections were then incubated with biotinylated goat anti-rabbit secondary antibodies (Vector, 1:250) for 1.5h at room temperature followed by incubation in Avidin biotin-peroxidase (Vector, 1:200) for 1.5h at room temperature. Immunoreactivity was then visualized with a diaminobenzidine (DAB)/glucose oxidase reaction (Shu et al., 1988). The sections were then osmicated (15 min in 1% osmium tetroxide) and dehydrated in increasing ethanol concentrations. During the dehydration, 1% uranyl acetate was added to the 70% ethanol to enhance ultrastructural membrane contrast. Flat embedding in Durcupan followed dehydration. Ultrathin sections were cut on a Leica ultramicrotome, collected on Formvar-coated single-slot grids, and analyzed with a Tecnai 12 Biotwin electron microscope (FEI).
Confocal Microscopy
Mice (8-week-old) were anaesthetized and decapitated. Vibratome sections (200 μm) containing the MNTB were prepared. Following fixation overnight in 4% PFA, sections were washed and then permeabilized with 0.1% Triton X-100 (Sigma Aldrich 9002-93-1) for 1h hour at RT, washed and stained with AlexaFluor™ 594-conjugated phalloidin (ThermoFisher A12381) at a concentration of approximately 165 nM per manufacturer’s protocol diluted in 0.5% Triton X-100 for 2h at room temperature. Nuclei were stained with Hoechst 33342 (Thermofisher R37605) at a concentration of 2 drops per mL per manufacturer’s protocol for 30 min at room temperature. Sections were then mounted onto slides and imaged using the LSM880 Airyscan microscope (https://www.zeiss.com/microscopy/us/products/confocal-microscopes/lsm-880-with-airyscan-.html).
Compared to traditional confocal microscopy, the Airyscan feature produces a 4–8x improvement in the signal-to-noise ratio (SNR), with improvements in both sensitivity and resolution. Its high speed of acquisition also bypasses the need to sacrifice acquiring a high-resolution image for fear of bleaching the sample. Images were acquired at 63x magnification and 4096×4096 pixel resolution. Fluorescent excitation was conducted using 405 and 561 nm laser lines. For each image, the 561 nm laser gain was adjusted according to the intensity of blood vessels, which are plentiful in the MNTB and of maximal intensity relative to other F-actin-containing structures. There was no significant difference in the laser gain values used for antitative analysis of images of sections from WT, from Kv3.3−/− and Kv3.3G592R mice (p = 0.5867 in Brown-Forsythe ANOVA test and p = 0.6934 in Welch ANOVA test). This allowed direct comparison of intensity across all images at all z-planes, accounting for both differences in dye permeation between samples and within a sample. Fluorescence intensity was measured pixel-by-pixel (1 pixel = 0.0183 μm) along the ring of F-actin for each calyx using ImageJ software (NIH), producing quantitative data that distinguished between points less than 0.02 μm apart. These values were then used to calculate the mean intensity and coefficient of variability of each calyx.
Generation of W496F Kv3.3 mutation
The W496F mutation was introduced into Kv3.3 by full-length PCR amplification of plasmid DNA with sense and antisense primers containing the mutation using Pfu polymerase (Stratagene). The 5’ primer sequence for the PCR reaction was gaacatccccatcggcttcttctgggctgtggtcaccatgac, and the 3’ primer sequence for PCR reaction was gtcatggtgaccacagcccagaagaagccgatggggatgttc. Parental (wild type) DNA was digested with DpnI (New England Biolabs), and 1 μl of this reaction was used for transformation of competent Escherichia coli XL1Blue strain (Stratagene). Miniprep DNA was used for sequencing to confirm the presence of the desired mutation. The whole Kv3.3 cDNA was sequenced to verify no other undesired mutations were introduced during PCR.
Recordings of K+ currents in CHO cells
To test the effects of the mutation on Kv3.3 currents, CHO cells were grown in Iscove’s modified Dulbecco’s medium (Invitrogen) supplemented with 10% fetal bovine serum (heat-inactivated), 100 units/ml penicillin/streptomycin, 5% HT supplement (Invitrogen) in a 5% CO2 incubator at 37 °C. Cells were seeded 1 day before transient transfection. Lipofectamine (Invitrogen) was used to co-transfect CHO cells with wild type Kv3.3 and GFP or with the Kv3.3 pore mutation W496F Kv3.3 and GFP. To record whole cell currents, patch electrodes were pulled from 1.5-mm OD borosilicate capillary glass (World Precision Instruments, resistances ~2–3 MΩ). The intracellular solution consisted of (in mM) 97.5 K-gluconate, 32.5 KCl, 10 HEPES, 5 EGTA, pH 7.2, with KOH. The bath solution consisted of (in mM) 140 NaCl, 5.4 KCl, 1.3 CaCl2, 25 HEPES, 33 glucose, pH 7.4, with NaOH. Series resistance was 2–4 MΩ and was compensated by 80–85%. The data were acquired at 10 kHz and filtered at 5 kHz. Data were acquired using pClamp8 software (Molecular Devices). All electrophysiological data were analyzed using Clampfit 9 software (Molecular Devices, Sunnyvale, CA), and statistical analysis was carried out using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data collection and measurements of the time constant and Ratedecay
Calyx capacitance was measured within 10 min after break-in to avoid rundown (Wu et al., 2009). The τ was measured from exponential fit. Ratedecay at calyces was measured between 0.5 – 4 s after depol20ms that induced slow endocytosis, but between 0.5 – 1.5 s after depol20msX10 that induced rapid endocytosis. The first 0.5 s trace was not used (and thus not shown in figures) to avoid capacitance artifact contamination (Wu et al., 2005; Yamashita et al., 2005). We used depol20msX10 to induce rapid endocytosis, because the Ratedecay after depol20msX10 reflected mostly (~80%) the rapid component of endocytosis (Sun et al., 2010; Wu et al., 2009).
For SypH signal in hippocampal cultures, the Ratedecay was measured from FSypH in the first 4–10 s after stimulation. For measurements of the Ratedecay in nearly all graphs, FSypH was normalized to the baseline FSypH before stimulation (baseline FSypH was normalized as 100%). Only in Figure S1 where the normalized Ratedecay was measured from FSypH with a different normalization method: ΔF, the FSypH increase induced by stimulation, but not the FSypH baseline, was normalized as 100%. Such a normalization removed the impact of ΔF on the measurement of the Ratedecay.
Fluorescence intensity in Figure 3 was measured pixel-by-pixel (1 pixel = 0.0183 μm) along the ring of F-actin for each calyx using ImageJ software (NIH), producing quantitative data that distinguished between points less than 0.02 μm apart. These values were then used to calculate the mean intensity and coefficient of variability of each calyx.
Statistical tests
Data were expressed as mean ± s.e.m. Replicates are indicated in results and figure legends. The statistical test used is t test or ANOVA. Although the statistics were performed based on the number of cells or experiments, each group of data was collected from at least four mice. For imaging in hippocampal cultures, each group of data was collected from at least 6 experiments; each experiment contained 20–30 boutons; 1–3 experiments were taken from 1 culture; each culture was from 3–5 mice. We determined these sample-sizes based on the experience in the past several decades. Blinding was not used for data collection or analysis. We did not include mice for use on a random basis, but based on their genotypes (described in the main text) and ages. We used P7–14 mice for calyx recordings, P0 mice for hippocampal cultures, and 8 weeks old mice for electron microscopy.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| 3,3-Diaminobenzidine tetrahydrochloride (DAB) | EMS | 13080 |
| 3,4-Diaminopyridine (3.4-DAP) | Sigma | D7148 |
| 6-Cyano-7-nitroquinoxaline-2, 3-dione (CNQX) | Tocris | 0190 |
| Absolute Ethanol | Decon Labs | 2716 |
| B27 supplement, serum free | Gibco | 17504044 |
| Bicuculline | Sigma | 14340 |
| BioMix Red 2X | Thermo Fisher Scientific | BIO25006 |
| Biotinylated goat anti rabbit antibody | Vector Labs | BA-1000 |
| Bovine serum albumin | Sigma | A2153 |
| Bovine serum albumin | American Analytical | AB01088 |
| Collagenase P | Sigma | 11249002001 |
| Cytosine β-D-arabinofuranoside | Sigma | C1768 |
| D(−)-2-Amino-5-phosphonovaleric acid | Tocris | 3693 |
| DMEM medium | Thermo Fisher Scientific | 11885–092 |
| DNase | Worthington | LK003172-D2 |
| Durcupan ACM Epoxy Resin | EMS | 14040 |
| Embed-812 | EMS | 13940 |
| Fetal Bovine Serum | Thermo Fisher Scientific | 10082–147 |
| Formvar | EMS | 15810 |
| German glass coverslips with mouse Laminin coating over PDL layer | Neuvitro | GG-25-Laminin |
| GlutaMAX-1 | Gibco | 35050–061 |
| HBSS (with Ca and Mg) | Thermo Fisher Scientific | 14025–134 |
| HBSS (without Ca and Mg) | Thermo Fisher Scientific | 14175–103 |
| HEPES | Thermo Fisher Scientific | 15630080 |
| HT supplement | Thermo Fisher Scientific | 11067030 |
| IMDM medium | Thermo Fisher Scientific | 12440053 |
| Insulin | Sigma | I5500 |
| Latrunculin A | Tocris | 3973 |
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| Lipofectamine LTX with PLUS | Thermo Fisher Scientific | 15338–100 |
| Neurobasal-A | Thermo Fisher Scientific | 10888–022 |
| Normal goat serum | Vector Laboratories | S-1000–20 |
| NucBlue Live ReadyProbes | Thermo Fisher Scientific | R37605 |
| Opti-MEM | Thermo Fisher Scientific | 31985070 |
| Osmium Tetroxide | EMS | 19140 |
| Ovomucoid inhibitor | Worthington | LK003182 |
| Papain | Worthington | LK003178 |
| Penicillin-Streptomycin solution | Thermo Fisher Scientific | 15140–122 |
| Phosphate buffered saline (PBS) | Gibco | 10010023 |
| Pierce 16% formaldehyde, methanol free | Thermo Fisher Scientific | 28908 |
| Propylene Oxide | EMS | 20401 |
| Rabbit Kv3.3 antibody | Alomone Labs | APC-102 |
| Single slot grids | EMS | M2010-CR |
| Tetraethylammonium chloride (TEA-Cl) | Sigma | T2265 |
| Tetrodotoxin (TTX) | Tocris | 1078 |
| Triton X-100 | Sigma | T8787 |
| Trypsin inhibitor | Sigma | T9253 |
| Uranyl Acetate | EMS | 22400 |
| Basic Primary Neurons Nucleofector Kit | Lonza | VVPI-1003 |
| Elite ABC-HRP kit | Vector Labs | PK-6100 |
| QuickExtract DNA Extraction Solution | Lucigen | QE09050 |
| QuikChange II XL Site-Directed Mutagenesis Kid | Agilent | 200521 |
| CHO cell | Kaczmarek lab | CHO/DHFR (−) |
| G592R Kv3.3 knock-in mice | Kaczmarek lab | N/A |
| Kv3.1 KO mice | Kaczmarek lab | N/A |
| Kv3.3 KO mice | Kaczmarek lab | N/A |
| B6 line mice | Kaczmarek lab | N/A |
| Kv3.3 WT | Kaczmarek lab | N/A |
| pH-sensitive pHluorin 2X (SypH) | Yong-Ling Zhu lab | N/A |
| rKv3.3/pcDNA3 | Kaczmarek lab | N/A |
| W496F Kv3.3 mutant | Kaczmarek lab | N/A |
| Clampfit 9 | Molecular Devices | https://mdc.custhelp.com/app/answers/detail/a_id/18826/~/axon%E2%84%A2-pclamp%E2%AE-9-electrophysiology-data-acquisition-%26-analysis-software-download |
| Huygens Professional | Scientific Volume Imaging | https://svi.nl/Huygens-Professional |
| Igor Pro | WaveMetrics | http://www.wavemetrics.com/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| LAS X | Leica | https://www.leicabiosystems.com/ |
| Matlab | MathWorks | https://www.mathworks.com/products/matlab.html |
| NIS-Elements AR 4.1 (Nikon A1 confocal microscope) | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements/nis-elements-advanced-research |
| Python v3.5.2 | Python Core Team | https://www.python.org |
| Pulse v8.67 (EPC-9 amplifier) | HEKA Elektronik | https://www.heka.com |
Highlights.
Kv3.3 channel facilitates slow and rapid endocytosis by nucleating F-actin at synapse
Kv3.3 channel facilitates vesicle mobilization and recovery of short-term depression
Linking Kv3.3-related spinocerebellar ataxia to endocytosis and vesicle mobilization
Kv3.3 non-conductive functions are crucial for synaptic functions and disorders
Acknowledgements:
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (ZIA-NS003009-13, L.G.W), and NIH grant DC 01919 (L.K.K.). We thank Dr. Yongling Zhu (Northwestern University, Chicago) for providing synaptophysin-pHluorin2X plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
References
- Artalejo CR, Henley JR, McNiven MA, and Palfrey HC (1995). Rapic endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP, and dynamin but not clathrin. Proc Natl Acad Sci USA 92, 8328–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, and Soria van Hoeve J (2012). The calyx of held synapse: from model synapse to auditory relay. Annu Rev Physiol 74, 199–224. [DOI] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, and Kirchhausen T (2011). Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol 13, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SY, Zagha E, Kwon ES, Ozaita A, Bobik M, Martone ME, Ellisman MH, Heintz N, and Rudy B (2007). Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. The Journal of comparative neurology 502, 953–972. [DOI] [PubMed] [Google Scholar]
- Choudhury N, Linley D, Richardson A, Anderson M, Robinson SW, Marra V, Ciampani V, Walter SM, Kopp-Scheinpflug C, Steinert JR, et al. (2020). Kv3.1 and Kv3.3 subunits differentially contribute to Kv3 channels and action potential repolarization in principal neurons of the auditory brainstem. J Physiol 598, 2199–2222. [DOI] [PubMed] [Google Scholar]
- Elezgarai I, Diez J, Puente N, Azkue JJ, Benitez R, Bilbao A, Knopfel T, Donate-Oliver F, and Grandes P (2003). Subcellular localization of the voltage-dependent potassium channel Kv3.1b in postnatal and adult rat medial nucleus of the trapezoid body. Neuroscience 118, 889–898. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Kwan R, and Ryan TA (2006). Synaptic Vesicles Interchange Their Membrane Proteins with a Large Surface Reservoir during Recycling. Neuron 51, 179–186. [DOI] [PubMed] [Google Scholar]
- Henao-Mejia J, Williams A, Rongvaux A, Stein J, Hughes C, and Flavell RA (2016). Generation of Genetically Modified Mice Using the CRISPR-Cas9 Genome-Editing System. Cold Spring Harb Protoc 2016, pdb prot090704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CS, Grange RW, and Joho RH (1997). Pleiotropic effects of a disrupted K+ channel gene: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci U S A 94, 1533–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Holt M, and Sakaba T (2009). Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron 63, 216–229. [DOI] [PubMed] [Google Scholar]
- Hua Y, Sinha R, Thiel CS, Schmidt R, Huve J, Martens H, Hell SW, Egner A, and Klingauf J (2011). A readily retrievable pool of synaptic vesicles. Nat Neurosci 14, 833–839. [DOI] [PubMed] [Google Scholar]
- Hua Y, Woehler A, Kahms M, Haucke V, Neher E, and Klingauf J (2013). Blocking endocytosis enhances short-term synaptic depression under conditions of normal availability of vesicles. Neuron 80, 343–349. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK, and Zhang Y (2017). Kv3 Channels: Enablers of Rapid Firing, Neurotransmitter Release, and Neuronal Endurance. Physiol Rev 97, 1431–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M, and Roux A (2018). Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 19, 313–326. [DOI] [PubMed] [Google Scholar]
- Li W, Kaczmarek LK, and Perney TM (2001). Localization of two high-threshold potassium channel subunits in the rat central auditory system. J Comp Neurol 437, 196–218. [DOI] [PubMed] [Google Scholar]
- Macica CM, von Hehn CAA, Wang L-Y, Ho C-S, Yokoyama S, Joho RH, and Kaczmarek LK (2003). Modulation of the Kv3.1b Potassium Channel Isoform Adjusts the Fidelity of the Firing Pattern of Auditory Neurons. The Journal of Neuroscience 23, 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M, Chen PH, Srinivasan S, Danuser G, and Schmid SL (2018). Regulation of Clathrin-Mediated Endocytosis. Annu Rev Biochem 87, 871–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlebrooks JC, Nick HS, Subramony SH, Advincula J, Rosales RL, Lee LV, Ashizawa T, and Waters MF (2013). Mutation in the kv3.3 voltage-gated potassium channel causing spinocerebellar ataxia 13 disrupts sound-localization mechanisms. PLoS One 8, e76749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian NA, Lin MC, and Papazian DM (2012). Altered Kv3.3 channel gating in early-onset spinocerebellar ataxia type 13. J Physiol 590, 1599–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (2010). What is Rate-Limiting during Sustained Synaptic Activity: Vesicle Supply or the Availability of Release Sites. Front Synaptic Neurosci 2, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, MacKinnon R, Bezanilla F, and Stefani E (1993). Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron 11, 353–358. [DOI] [PubMed] [Google Scholar]
- Renden R, and Von Gersdorff H (2007). Synaptic vesicle endocytosis at a CNS nerve terminal: faster kinetics at physiological temperatures and increased endocytotic capacity during maturation. J Neurophysiol 98, 3349–3359. [DOI] [PubMed] [Google Scholar]
- Saitoh N, Hori T, and Takahashi T (2001). Activation of the epsilon isoform of protein kinase C in the mammalian nerve terminal. Proc Natl Acad Sci U S A 98, 14017–14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, and Neher E (2001). Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, and Ryan TA (2000). Real-time measurements of vesicle-SNARE recycling in synapses of the central nervous system. Nat Cell Biol 2, 197–204. [DOI] [PubMed] [Google Scholar]
- Shu SY, Ju G, and Fan LZ (1988). The glucose oxidase-DAB-nickel method in peroxidase histochemistry of the nervous system. Neuroscience letters 85, 169–171. [DOI] [PubMed] [Google Scholar]
- Song P, Yang Y, Barnes-Davies M, Bhattacharjee A, Hamann M, Forsythe ID, Oliver DL, and Kaczmarek LK (2005). Acoustic environment determines phosphorylation state of the Kv3.1 potassium channel in auditory neurons. Nat Neurosci 8, 1335–1342. [DOI] [PubMed] [Google Scholar]
- Soykan T, Kaempf N, Sakaba T, Vollweiter D, Goerdeler F, Puchkov D, Kononenko NL, and Haucke V (2017). Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 93, 854–866. [DOI] [PubMed] [Google Scholar]
- Sun JY, and Wu LG (2001). Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynatpic currents at a central synapse. Neuron 30, 171–182. [DOI] [PubMed] [Google Scholar]
- Sun T, Wu XS, Xu J, McNeil BD, Pang ZP, Yang W, Bai L, Qadri S, Molkentin JD, Yue DT, et al. (2010). The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J Neurosci 30, 11838–11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Rost BR, Camacho-Perez M, Davis MW, Sohl-Kielczynski B, Rosenmund C, and Jorgensen EM (2013). Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Trimbuch T, Camacho-Perez M, Rost BR, Brokowski B, Sohl-Kielczynski B, Felies A, Davis MW, Rosenmund C, and Jorgensen EM (2014). Clathrin regenerates synaptic vesicles from endosomes. Nature 515, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PJ, Grenklo S, Arpino G, Tan X, Liao HS, Heureaux J, Peng SY, Chiang HC, Hamid E, Zhao WD, et al. (2016). Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat Commun 7, 12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienisch M, and Klingauf J (2006). Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat Neurosci 9, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Wu LG, and Borst JGG (1999). The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron 23, 821–832. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JGG, and Sakmann B (1998). R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci USA 95, 4720–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Hamid E, Shin W, and Chiang HC (2014). Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol 76, 301–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Xu J, Wu XS, and Wu LG (2005). Activity-dependent acceleration of endocytosis at a central synapse. J Neurosci 25, 11676–11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Lee SH, Sheng J, Zhang Z, Zhao WD, Wang D, Jin Y, Charnay P, Ervasti JM, and Wu LG (2016). Actin Is Crucial for All Kinetically Distinguishable Forms of Endocytosis at Synapses. Neuron 92, 1020–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, McNeil BD, Xu J, Fan J, Xue L, Melicoff E, Adachi R, Bai L, and Wu LG (2009). Ca(2+) and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci 12, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, and Wu LG (2009). Rapid endocytosis does not recycle vesicles within the readily releasable pool. J Neurosci 29, 11038–11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, and Wu LG (2005). The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron 46, 633–645. [DOI] [PubMed] [Google Scholar]
- Xue L, McNeil BD, Wu XS, Luo F, He L, and Wu LG (2012). A membrane pool retrieved via endocytosis overshoot at nerve terminals: a study of its retrieval mechanism and role. J Neurosci 32, 3398–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T, and Takahashi T (2005). Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science 307, 124–127. [DOI] [PubMed] [Google Scholar]
- Zagha E, Lang EJ, and Rudy B (2008). Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci 28, 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Kaczmarek LK (2016). Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol 594, 4677–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang XF, Fleming MR, Amiri A, El-Hassar L, Surguchev AA, Hyland C, Jenkins DP, Desai R, Brown MR, et al. (2016). Kv3.3 Channels Bind Hax-1 and Arp2/3 to Assemble a Stable Local Actin Network that Regulates Channel Gating. Cell 165, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data produced for this manuscript are available from the Lead Contact (wul@ninds.nih.gov) upon reasonable request.