

Abstract
Mitochondrial function is essential for the viability of aerobic eukaryotic cells, as mitochondria provide energy through the generation of adenosine triphosphate (ATP), regulate cellular metabolism, provide redox balancing, participate in immune signaling, and can initiate apoptosis. Mitochondria are dynamic organelles that participate in a cyclical and ongoing process of regeneration and autophagy (clearance), termed mitophagy specifically for mitochondrial (macro)autophagy. An imbalance in mitochondrial function toward mitochondrial dysfunction can be catastrophic for cells and has been characterized in several common ophthalmic diseases. In this article, we review mitochondrial homeostasis in detail, focusing on the balance of mitochondrial dynamics including the processes of fission and fusion, and provide a description of the mechanisms involved in mitophagy. Furthermore, this article reviews investigations of ocular diseases with impaired mitophagy, including Fuchs endothelial corneal dystrophy, primary open-angle glaucoma, diabetic retinopathy, and age-related macular degeneration, as well as several primary mitochondrial diseases with ocular phenotypes that display impaired mitophagy, including mitochondrial encephalopathy lactic acidosis stroke, Leber hereditary optic neuropathy, and chronic progressive external ophthalmoplegia. The results of various studies using cell culture, animal, and human tissue models are presented and reflect a growing awareness of mitophagy impairment as an important feature of ophthalmic disease pathology. As this review indicates, it is imperative that mitophagy be investigated as a targetable mechanism in developing therapies for ocular diseases characterized by oxidative stress and mitochondrial dysfunction.
Keywords: disease, mitochondria, mitophagy, dynamics
Mitochondria are the critically important powerhouses of the cell that are essential to cell metabolism and energy production.1,2 Cell viability depends on proper mitochondrial function to execute oxidative phosphorylation, adenosine triphosphate (ATP) synthesis, and apoptosis signaling.3–5 Proper cellular function is maintained in part through macroautophagy. In this recycling process, damaged or dysfunctional organelles and macromolecules are eliminated from the cell through autophagosome formation, lysosome–autophagosome fusion, and enzymatic digestion; the resultant molecules then become available for subsequent use, including organelle regeneration.6–8 Mitochondrial specific macroautophagy, also known as mitophagy,8 occurs in order to clear dysfunctional mitochondria from the cytosol, thus preserving a functional mitochondrial population. Mitophagy also facilitates the maturation and differentiation of several cell types, including erythrocytes from reticulocytes,9,10 lens fiber cells,11 and retinal ganglion cells.12 Conversely, failure of mitophagy to occur results in the accumulation of mitochondria with altered cristae structure, bioenergetics dysfunction, cell death, and, ultimately, organ and tissue damage that can be fatal.13–15
Across a variety of systemic and ocular diseases, there is increasing awareness of the importance of mitochondrial dysfunction and impaired clearance of damaged or dysfunctional mitochondria. Systemic neuromuscular and metabolic diseases that have been correlated with impaired mitochondrial function include Parkinson's disease, Alzheimer's disease, atherosclerosis, hypertension, cardiac hypertrophy, heart failure, and diabetes mellitus.2,16–21 Primary mitochondrial disorders can result in ocular manifestations, including several with gene mutations in mitochondrial DNA (mtDNA) that are well known to ophthalmologists, such as Leber hereditary optic neuropathy (LHON) and chronic progressive external ophthalmoplegia (CPEO).22 Additionally, there are various ophthalmic conditions in which poor mitochondrial function and quality are of emerging importance to the understanding of disease progression, including Fuchs endothelial corneal dystrophy, diabetic corneal endothelial cellular dysfunction, primary open-angle glaucoma, pseudoexfoliation glaucoma, diabetic retinopathy, and age-related macular degeneration.23–29 Given the importance of mitochondrial function in a wide variety of ophthalmic diseases, it is important to understand the critical role of cellular mechanisms and functions involved in maintaining mitochondrial homeostasis in the eye. It is equally important to identify common features of mitochondrial diseases with ocular phenotypes, as these may also help guide future therapy. Current understandings regarding the causes of mitochondrial dysfunction, mechanisms of mitophagy, and alterations to mitophagy in ocular tissues will be reviewed in this article.
Mitochondrial Dysfunction
Mitochondrial dysfunction describes a state in which mitochondria are unable to perform their normal functions (ATP synthesis, signaling between cells, and triggering apoptosis) due to gene mutations, disease, or another contributing condition (such as exposure to toxins).30 Mitochondrial dysfunction is commonly associated with aging.31 Mitochondrial dysfunction can physically be described by the presence of increased local reactive oxygen species (ROS), inflammation, membrane depolarization, mtDNA damage, cristae decomposition, and reduced ATP synthesis.32–36 These hallmarks of mitochondrial dysfunction often act as mediating factors in complex pathophysiologic mechanisms of ocular diseases. Understanding the relationships among these factors expands our knowledge of both normal mitochondrial dynamics and mitochondrial dynamics in disease states.
ROS Imbalance
Oxidative stress causes mitochondrial dysfunction if not retained within normal balance.37 Under normal conditions, free radicals (known as mtROS) are produced as a byproduct of oxidative phosphorylation.38 These free radicals are capable of causing oxidative damage by permanently modifying proteins, lipids, and mtDNA.37 Enough permanent modifications can reduce the function of these molecules. Eventually, the reduced function of organelle components will cause the organelle to partially or fully fail. In the case of mitochondrial damage, the damage is gathered, or “organized,” such that this portion of the mitochondrion can be removed via fission (division) and mitophagy processes.39 An increase in ROS is often associated with disease and cancer phenotypes.40 If not mitigated, the accumulation of free radicals can cause permanent damage.
Inflammatory Response
Permanent damage to the macromolecules that comprise mitochondria (mtDNA damage, protein modifications, lipid alterations) is recognized by the cell as danger-associated molecular patterns (DAMPs) by nucleotide-binding and oligomerization domain (NOD)-like receptors and thioredoxin interacting protein (TXNIP)-activated NLRP3 inflammasomes.41 Pro-inflammatory proteins are converted subsequently to active inflammatory proteins (e.g., pro-IL-1β to IL-1β), leading to cellular death, as demonstrated by Singh et al.41 in diabetic retinopathy.
Membrane Depolarization
Membrane depolarization is described as the long-term change from the normal, steady-state mitochondrial membrane potential (ΔΨm). Under normal physiologic conditions, the proton gradient (ΔpH) and ΔΨm create the transmembrane potential, which is required for H+ transport across the inner mitochondrial membrane (IMM) during oxidative phosphorylation, resulting in the formation of ATP.42 The intermembrane space normally has a positive charge, and the matrix normally has a negative charge. This charge differential represents the proton motive force, which is the driving potential for protons to flow toward the matrix, which results in the production of ATP. If the IMM barrier that maintains this charge polarity becomes compromised, the charge gradient becomes altered, and the mitochondria become depolarized. Mitochondrial depolarization can result in the loss of ATP production and cell stress and ultimately can result in cell death.
mtDNA Damage
Mitochondrial DNA has a higher rate of mutations than nuclear DNA while also having less efficient repair mechanisms. The rate of mtDNA mutations is roughly 15 times that of nuclear DNA and is thought to occur mostly due to the formation of ROS within the mitochondria themselves.43 ROS-mediated DNA damage is measured by quantifying 8-hydroxydeoxyguanosine, the oxidized version of deoxyguanosine. Using this technique, it has been demonstrated in rat hepatocytes that the relative level of 8-hydroxydeoxyguanosine is 16 times higher in mtDNA than nuclear DNA from the same cells.44 ROS-mediated DNA damage can also be quantified using quantitative PCR. Although mtDNA only encodes about 1% of the proteins found in mitochondria, it includes subunits of oxidative phosphorylation proteins. When mtDNA mutations occur in sequences for these proteins, a decrease in ATP production and increased levels of mitochondrial dysfunction and ROS production often occur.43
Cristae Decomposition
Mitochondrial cristae are formed by folding of the IMM and are home to the oxidative phosphorylation proteins. The relationship between biological form and function could not be more evident than when describing mitochondrial cristae. It has been shown that the cristae are dynamic in organization, depending on the cellular energy demands, and can serve as an indicator of overall cell viability. For example, in diabetic donor corneal endothelial cells, mitochondrial cristae are more commonly seen as abnormal or unfolded,25 making oxidative phosphorylation less efficient due to the participating membrane proteins not being close enough together.25,45 In diabetic donor corneal endothelial cells, cristae absence (dropout) is also commonly observed, as well as mitochondrial inclusion bodies. An example of cristae decomposition is shown in Figure 1.25
Figure 1.
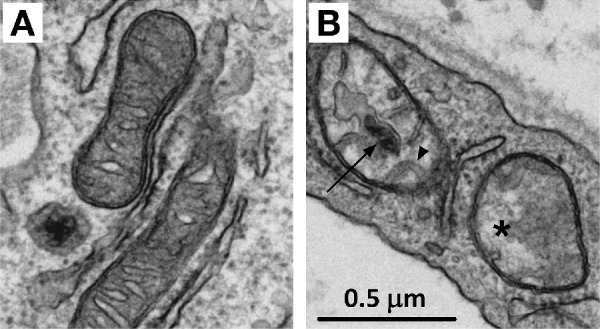
Transmission electron microscopy (TEM) of human mitochondria. (A) Normal, functional, mitochondria display densely packed cristae and no inclusion bodies and often have an average size (0.5–1.0 µm in length) with a discoid shape. (B) Dysfunctional mitochondria display abnormal or unfolded cristae (arrowhead), cristae dropout (asterisk), and inclusion bodies (arrow) and may have smaller or larger than average sizes with a rounded shape.
Reduced ATP Synthesis
Reduced ATP synthesis is the byproduct of the aforementioned forms of mitochondrial damage.46 Reduced oxidative phosphorylation complex protein function may be due to mtROS accumulation, mtDNA mutations that cause protein structure or function defects, cristae dropout yielding decreased numbers of functioning proteins within the mitochondrion, or even depolarization of the membrane, which causes a loss of the H+ gradient. ATP synthesis is often measured to determine mitochondrial output; however, several other metrics of mitochondrial bioenergetics may be used to determine the function of mitochondria within a system, such as mitochondrial respiration and glycolysis as measured by oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), respectively. Determining the predominant cause of functional change requires analysis of mtDNA, mtROS, ΔΨm, and inflammatory markers.
Although mitophagy is typically capable of containing or correcting mitochondrial damage, the dysfunctional dynamics described above can often signal that mitophagy itself has become compromised. The ability to simulate different disease states by inducing changes in mitophagy suggests that the dysregulation of mitophagy plays an important role in the pathophysiology of many ocular diseases, whether or not it is the primary pathogenesis. Studies have also shown that manipulating mitophagy can reverse markers of dysfunctional mitochondrial dynamics, offering promising avenues of exploration for the future development of therapeutics.
Mitophagy in Detail
Mitochondria are dynamic organelles in a coordinated balance of fission (i.e., division) and fusion (i.e., integration/combination), depending on the particular needs of a cell at any given time.47 The integration of mitochondria into networks varies dramatically from one cell type to another. Typically, optimal mitochondrial function may be found in between fully fused/networked and fully divided/fragmented mitochondrial populations48; however, varying densities and intracellular locations of networks within different cell types are found for the enhanced function of that particular cell. For example, skeletal muscle cells have very stable and organized networks, whereas neurons have heterogeneous networks to meet the changing demands of the cell body and synapse.49 Differences in mitochondrial networking also play a role in the efficiency of fission, fusion, and mitophagy within the cell.
Mitochondrial fission ensures mitochondrial integrity by isolating damaged or dysfunctional mitochondria for subsequent removal from the functional mitochondrial population via mitophagy.39 Fission helps facilitate mitophagy in two ways. First, data suggest that small, fragmented mitochondria are more readily incorporated into autophagosomes compared to larger mitochondria.50 Second, mitochondrial fission events result in two distinct mitochondria with higher or lower membrane potentials, where the mitochondria with lower membrane potentials are subsequently taken up by autophagosomes and degraded via mitophagy.51,52 Mitochondrial fission in humans is mediated primarily by the GTPase dynamin-related protein 1 (DRP1), but additional proteins are also required, including fission protein 1 (FIS1), the mitochondrial dynamics proteins Mid49 and Mid51, and mitochondrial fission factor (MFF).53–55
Mitochondrial fusion helps maintain mitochondrial integrity by facilitating mitochondrial biogenesis and networking.56 Mitochondrial biogenesis is a general term describing the ability of the cell to generate the proteins required to maintain or synthesize new mitochondria and is stimulated by several endogenous signals, such as ROS, nitrogen oxide, ratios of nicotinamide adenine dinucleotide (NAD+/NADH), and adenosine monophosphate (AMP)-activated protein, as well as exogenous signals such as estrogen.57 Mitochondria first attempt to maintain their structure and integrity at the protein level through the activity of a number of antioxidants, DNA repair proteins, and protein repair proteins.58 Mitochondrial fusion is instrumental in this process by helping to distribute mitochondrial proteins and facilitating the replacement of damaged mitochondrial DNA.51,59 In humans, mitochondrial fusion is mediated by the dynamin-related GTPases mitofusin 1 and 2 (MFN1 and MFN2) and optic atrophy 1 (OPA1), which mediate outer mitochondrial membrane (OMM) and IMM fusion, respectively. Mutations in OPA1 (also known as dynamin-like 120-kDa mitochondrial protein) are associated with dominant optic atrophy, which can lead to blindness.60–62
The balance between mitochondrial fission and fusion appears to be closely linked to mitophagy, as manipulating either fission or fusion within a cell has been shown to alter mitophagy pathways.51,63 For example, inducing fission with silibinin increases mitophagy in breast cancer cells,64 whereas inhibiting TXNIP-mediated fission with short hairpin RNA (shRNA) decreases mitochondrial fragmentation and does not induce mitophagy in a human retinal pigment epithelial (HRPE) cell model of diabetic retinopathy.65
Mitophagy plays a key role in protecting mitochondrial dynamics from becoming dysfunctional. Our current understanding of mitophagy in normal states is incomplete, and there are many questions left to be answered before dysregulation of mitophagy in the context of ocular disease is well understood. There are currently several documented mitophagy pathways. Many pathways share similar components, including signaling proteins and recruitment of autophagosomes; however, many details have yet to be discovered. Even in diseases where mitophagy does not appear to be the primary pathogenesis, our growing understanding of the functions, mechanisms, and regulation of mitophagy offers possibilities for corrective intervention. Below, we describe two of the most established pathways to date, as well as one possible pathway that has been investigated recently.
PINK1/Parkin-Mediated Mitophagy
The PTEN-induced kinase 1 (PINK1)/parkin mitophagy pathway is the most characterized mitophagy pathway, mediated by proteins PINK1 and parkin, both of which have known mutations associated with Parkinson's disease.66 Under normal conditions, newly synthesized full-length (64-kDa) PINK1 associates with translocase of the outer membrane (TOM) complex and is transported through the OMM. Then, full-length PINK1 is transported to the IMM through the translocase inner membrane (TIM) complex and processed to the cleaved version (52-kDa) of PINK1 by two proteins, mitochondrial processing peptidase (MPP) and mitochondrial presenilins-associated rhomboid-like protein (PARL).67 The cleaved version of PINK1 is degraded subsequently by MG132-sensitive protease.
However, in states of mitochondrial dysfunction, the PINK1/parkin pathway can be activated to induce mitophagy as a corrective mechanism. When the mitochondrial membrane is depolarized, full-length PINK1 becomes tethered to the OMM by TOM. It has been shown that BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) stabilizes full-length PINK1 protein (64-kDa) at the OMM, which in turn recruits parkin to the mitochondrial surface.68 Several PINK1 proteins will accumulate at the OMM, which in turn recruits parkin to the OMM. PINK1 phosphorylates parkin, which ubiquitinates OMM proteins, including MFN, TOM, voltage-dependent anion-selective channel protein (VDAC), MFF, and FIS1. This ubiquitination recruits downstream mitophagy factors including microtubule-associated proteins 1A/1B light chain 3 (LC3), histone deacetylase 6 (HDAC6), p97, sequestosome-1 (p62/SQSTM1), and contactin.69 All of these proteins are direct regulators of autophagosome component recruitment. Autophagosome recruitment signaling is summarized in Figure 2A. Finally, the membrane of the autophagosome membrane fuses with a lysosome membrane positive for lysosome-associated membrane glycoprotein-1 (LAMP1), resulting in a mature autophagolysosome.70 Herein, the mitochondrion is digested. This process can be quantified in cells using MitoTracker + LC3II + LAMP1 colocalization71 or measurement of LC3II, p62, and contactin protein expression with western blotting or ELISA. LC3II is the autophagy “active” form of cytosolic LC3, conjugated to phosphatidylethanolamine and embedded within autophagosomes.72 When assessing for active mitophagy, the colocalization of LC3II with antibodies for mitochondrial-specific proteins such as TOM20, VDAC, or electron transport chain complexes can be compared among samples of interest.73 Furthermore, mitophagy flux, or the net movement of mitochondria out of the cell, can be quantified as the ratio of mitophagy in the presence of lysosomal inhibitors to mitophagy in the absence of the same inhibitors, or by dual assessment of mitophagy and mitochondrial quantity.74
Figure 2.

Mitophagy protein signaling mechanisms. (A) PINK1/parkin-mediated mitophagy. Following the stabilization of PINK1 to the OMM, possibly by BNIP3, parkin is recruited to the OMM, resulting in the phosphorylation of parkin by PINK1. This event then triggers parkin to polyubiquitinate an OMM protein (VDAC), which then recruits a phagophore through proteins p62 and LC3. Interactions between the polyubiquitinated OMM protein and LC3/p62 initiate the mitophagy process. (B) Non-parkin-mediated mitophagy is initiated through proteins on the OMM (FUNDC1, NIX, or BNIP3) that bind directly to LC3 on the phagophore membrane, initiating the mitophagy process. (C) Non-parkin, syntaxin 17 (STX17)-mediated mitophagy occurs when the expression of FIS1 is downregulated, allowing for the accumulation of STX17 proteins on the OMM. The accumulation of STX17 proteins on the OMM recruits ATG14, which interacts with LC3 on the phagophore membrane.
Non-Parkin-Mediated Mitophagy
There are several mitophagy mechanisms initiated through signals not involving parkin protein. In general, these pathways are initiated by mitochondrial membrane depolarization, hypoxia, or post-translational modifications. They are regulated by gamma-aminobutyric acid receptor-associated protein (GABARAP) and LC3 proteins on the autophagosome membrane. These autophagosome proteins interact with proteins on the OMM, including BNIP3, FUN14 domain-containing protein 1 (FUNDC1), peptidyl-prolyl cis-trans isomerase FKBP8, B-cell lymphoma 2 (BCL-2), and/or BNIP3-like protein (BNIP3L/NIX), which are known mitophagy receptors.75,76 This interaction initiates the mitophagy process, resulting in mitochondrial clearance (Fig. 2B). Syntaxin 17 (STX17) also initiates mitophagy through a non-parkin-mediated pathway. STX17 trafficking from the endoplasmic reticulum (ER) to mitochondria is controlled through interactions with FIS1 under normal conditions. Loss of FIS1 at the mitochondrial membrane causes a buildup of STX17 trafficking to mitochondria, which in turn recruits an autophagy-related 14 (ATG14)-containing PI3K complex.77 STX17 interacts with ATG14, which then drives mitophagy (Fig. 2C).77,78
Primary Ocular Disease and Mitophagy
Many ocular conditions are associated with changes in mitophagy and mitochondrial dysfunction. Studying these associations gives insight into the pathophysiology of these diseases. Evidence of mitophagy impairment by different regulatory mechanisms in the cycle of mitochondrial dynamics can present as mitochondrial elongation or fragmentation. Buildup of fragmented mitochondria generally indicates the presence of fission but also a dysfunction related to clearance (mitophagy). Buildup of swollen mitochondria in conjunction with a decrease in mitochondrial function indicates a reduction in fission as well as mitophagy. Fully swollen mitochondria should be broken down into functional and dysfunctional counterparts through the process of fission, and the dysfunctional counterparts (if not the entire mitochondrion) should be eliminated through mitophagy. A decrease in mitochondrial bioenergetics despite an increase in mitochondrial counts and/or surface area is a key indication of mitophagy signaling failure. Many primary ocular diseases demonstrate evidence of abnormal mitochondrial morphology, mitochondrial counts, or mitochondrial protein expression that points to impaired or dysregulated mitophagy. In general, the primary ocular diseases covered in this review demonstrate mitophagy imbalance and mitochondrial dysfunction that contribute to the disease state but are secondary to the primary pathogenesis. Details of these changes with respect to specific ocular diseases are outlined below. A summary of specific mitophagy protein changes associated with the following diseases and their observed effects is provided in the Table.
Table.
Mitophagy Protein Changes Associated With Primary Ocular Diseases
| Disease | Observed Effect | Mitophagy-Related Proteins | Relevant Study |
|---|---|---|---|
| Fuchs endothelial corneal dystrophy | Reduced oxidative phosphorylation proteins, increased mitochondrial fragmentation | LC3II, LAMP1 (increased); MFN2 (reduced) | Benischke et al.23 |
| ROS-mediated mitophagy signaling independent of PINK1 and DRP1 proteins | PINK1, parkin, DRP1 | Miyai et al.24 | |
| Primary open-angle glaucoma | ATP deficiency, increased number of mitochondria, disrupted mitochondrial cristae, smaller mitochondrial surface area | LAMP1 (reduced) | Coughlin et al.26 |
| Reduced oxidative damage, reduced RGC death | UCP2 (Ucp2 knockout) | Hass and Barnstable111 | |
| Increased LC3-II/LC3-I, increased LAMP1, increased mitophagosomes | Parkin (overexpressed) | Dai et al.112 | |
| Increased mitochondrial fusion, higher mitochondrial surface area, increased parkin expression | OPA1 (overexpressed) | Hu et al.113 | |
| Buildup of dysfunctional, fractionated mitochondria; RGC death | Optineurin (mutation) | Ito et al.,115 Rezaie et al.,116 Wong et al.117 | |
| Pseudoexfoliation glaucoma | Reduced mitophagy, dysfunctional mitochondria, cellular aggregopathy | LC3II (accumulation) | Want et al.29 |
| Diabetic retinopathy | Decreased mitochondrial fragmentation, reduced mitophagy, decreased lysosomal enlargement | TXNIP (knockdown) | Devi et al.65 |
| Increased mitochondrial dysfunction, increased mitochondrial membrane depolarization, increased mitophagic flux, lysosomal enlargement | TXNIP (upregulation) | Devi et al.65,133 | |
| Increased NLRP3-mediated inflammation and cell death | TXNIP (upregulation) | Singh et al.,41 Devi et al.133 | |
| Reduced mitophagy and increased apoptosis | PINK1, parkin (downregulation) | Zhang et al.137 | |
| Increased mitophagy, decreased apoptosis, decreased vascular endothelial growth factor expression, increased pigment epithelium-derived factor expression | PINK1, parkin (increased) | Zhou et al.138 | |
| Age-related macular degeneration | Increased mitophagy, decreased ferroptosis | Iron (chelation) | Stenirri et al.,151 Allen et al.,152 Wang et al.153 |
| Increased mitochondrial fragmentation | pDRP1 (S616; increased) | Chan et al.155 | |
| Decreased mitochondrial counts, decreased antimycin A-induced cell death | LC3B, p62 (increased) | Hytti et al.156 | |
| Protein aggregation, mitochondrial dysfunction, increased oxidative stress | PINK1, parkin, LC3B/ATP synthase beta colocalization (increased); LAMP1/ATP synthase beta colocalization (decreased) | Sridevi Gurubaran et al.27 |
Corneal Endotheliopathy
The cornea is at risk for increased free radical production and oxygen-mediated damage due to its direct exposure to environmental oxygen and ultraviolet light. Corneal endothelial cells (CECs) are especially susceptible to oxidative damage because they are at homeostasis under tightly regulated low-oxygen conditions in the anterior chamber,79 have high rates of mitochondrial activity as they utilize active ion pumping mechanisms to counteract the passive leak of aqueous humor into the stroma, and do not proliferate in response to injury. With the emergence of single-layer endothelial cell transplant techniques (Descemet membrane endothelial keratoplasty and Descemet stripping automated endothelial keratoplasty [DSAEK]), it has become imperative to understand these cells in more detail in order to maximize the success of surgical outcomes. Two common diseases that feature impaired CEC function, Fuchs endothelial corneal dystrophy (FECD) and diabetes mellitus, demonstrate the effects of altered mitophagy on disease progression, as discussed below.
Fuchs Endothelial Corneal Dystrophy. FECD, the most prevalent indication for endothelial keratoplasty in the United States,80 manifests as a result of genetic mutations (spontaneous and familial), as well as non-heritable risk factors including gender (female predominance) and smoking. The clinical hallmarks of FECD include cell death and extracellular matrix deposition, resulting in corneal edema and characteristic excrescences observable on the posterior cornea (guttae).81 Cellular and ultracellular characteristics of FECD include channel protein dysfunction, mitochondrial dysfunction, ROS accumulation, ER stress, DNA alterations, unfolded protein response, and endothelial cell apoptosis/dropout.81,82 Although FECD is a complex disease with several different primary mechanisms involved, secondary mitochondrial dysfunction and mitophagy play a central role in the decline of endothelial cell viability during the progression of this disease.
Halilovic et al.83 showed that corneas from FECD patients have increased mtDNA damage, and expanded ex vivo FECD cells have decreased ATP production as a result of oxidative stress. These findings indicate that reduced mitochondrial respiratory capacity and reduced mitochondrial protein expression are central features of the FECD cellular disease phenotype. Importantly, ATP production deficits were rescued with the antioxidant N-acetylcysteine (NAC), indicating the importance of maintaining proper redox balance in CEC function. Furthermore, Benischke et al.23 showed that the autophagy-related proteins LC3-II and LAMP1 accumulate while also showing a reduced amount of the fusion-related protein MFN2 in corneas from FECD patients compared to controls. This protein expression imbalance favors both more mitochondrial fission and mitophagy and less fusion and mitochondrial networking in FECD. In addition, this study indicates that carbonyl cyanide m-chlorophenylhydrazine (CCCP)-induced mitochondrial depolarization in FECD immortalized CECs led to decreased MFN2 protein expression, which further reduced mitochondrial fusion and networking. Of note, blocking mitophagy using bafilomycin—a chemical compound that inhibits the fusion of autophagosomes and lysosomes, inhibits vacuolar ATPase (V-ATPase), and causes lysosomal basification, thereby preventing lysosomal degradation of autophagosome contents—has been shown to rescue some of these FECD phenotypes. Specifically, using bafilomycin increased MFN2 expression and MFN2/LC3 colocalization. Because MFN2 plays a role in both fusion and apoptosis prevention, its decreased expression results in decreased fusion, increased fission, and increased risk of cell death. Overall, this study indicates that decreased expression of MFN2 in FECD is due to imbalanced mitophagy.
Miyai et al.24 showed that there are two different mechanisms responsible for the aberrant activation of mitophagy in FECD, using two activators of mitophagy, CCCP and menadione. CCCP induces mitochondrial membrane depolarization, and menadione generates mitochondrial ROS. First, the authors showed that tissues from patients with FECD have increased accumulations of PINK1, phosphorylated DRP1, and phosphorylated parkin and decreased levels of DRP1 and parkin. These findings indicate mitophagy activation in FECD via the PINK1/parkin mechanism. Then, in immortalized cell cultures of human CECs with and without FECD, administration of CCCP caused an increase in parkin-mediated mitophagy without significant changes in PINK1 and DRP1 protein levels. In the same cells, they showed that when menadione-induced ROS is the activator of mitophagy (corresponding to the increased ROS shown in FECD),84,85 there is increased proteasome degradation of parkin and downregulated expression of both PINK1 and DRP1 that can be prevented by blocking autophagolysosome formation with bafilomycin.24 This study offers the possibility that there are two unique mitophagy pathways at play in FECD pathobiology, one initiated by mitochondrial depolarization that is mediated by Parkin, PINK1, and DRP1 and another initiated by ROS that is mediated by parkin through a separate mechanism that is independent of PINK1 and DRP1. However, CCCP has been shown to cause both mitochondrial membrane depolarization and increased ROS,86 and menadione-induced ROS causes membrane depolarization downstream.24 Thus, the sole effects of mitochondrial membrane depolarization or ROS may not have been isolated in this study so additional work is needed to clarify the cause and effect of possible unique mitophagy mechanisms at play in FECD pathogenesis. Finding the activation of multiple mitophagic pathways fits in well with previous studies showing that corneas from FECD patients demonstrate decreased mitochondrial density and decreased mass and are consistent with a pathogenesis of increased fission and mitophagy upregulation in FECD.24,85 Patient corneal tissues with FECD also demonstrate decreased expression of antioxidants (such as superoxide dismutase and peroxiredoxins)84 and increased levels of glycation end products (a sign of increased oxidative stress),87 correlating with the model of increased ROS induced by menadione.
In summary, corneas with FECD demonstrate increased activation of mitophagy that is likely pathological. Increased mitochondrial depolarization, increased ROS, and reduced fusion protein levels (MFN2) lead to increased mitochondrial degradation through a PINK1/parkin pathway as well as a distinct parkin-dependent and PINK1- and DRP1-independent pathway. Aberrant mitochondrial degradation ultimately results in cellular death. Furthermore, inhibition of mitophagy has been shown to rescue FECD phenotypes.
Diabetic Corneal Endothelial Pathology. Type II diabetes mellitus (T2DM) has dramatic pathological effects on the lens, retina, choroid, corneal nerves, and corneal epithelial cells. Often, these changes result in decreased visual acuity that can become permanent. More recently, the effects of T2DM on CECs have become better understood, especially with respect to endothelial keratoplasty and surgical outcomes.88,89 Prior to transplant, graft tissues from donors with advanced diabetes have lower than average CEC densities,90 and 3 years following DSAEK surgery diabetic donor graft tissues display more cell loss compared to nondiabetic control tissues, as well as increased graft failure rates.91,92 Advanced diabetic donor tissues also have altered extracellular matrix properties impacting their biomechanical stiffness.93 Beyond these gross morphological and functional changes, CECs affected by T2DM have indications that mitochondrial quality is also reduced.
Aldrich et al.25 investigated the impact of diabetes and disease severity on CEC mitochondrial function using human ex vivo donor corneal tissue. Donor CECs with advanced diabetes (n = 12) had reduced spare respiratory capacity (mean ratio = 0.24; P = 0.001) compared to nondiabetic control donor CECs (n = 102) as measured using a Seahorse XFe24 extracellular flux analyzer (Agilent, Santa Clara, CA, USA). Although mitochondrial function is reduced in advanced T2DM human donor CECs, diabetic mitochondria have the same average count per cell but 60% higher mean surface area compared to controls as determined by transmission electron microscopy (TEM) morphometrics (P = 0.052).25 Mitochondrial cristae in diabetic CECs are absent or disorganized, and the overall appearance of the mitochondria is swollen with dark inclusion bodies present. Taken together, the functional and morphological data indicate that mitochondria in advanced diabetic CECs are damaged but not being eliminated from the cell as they would be under normal conditions, indicative of reduced mitophagy. Retention of dysfunctional mitochondria may lead to increased susceptibility to cellular dysfunction and death, but this subject requires further investigation.
Overall, T2DM CECs appear to acquire mitochondrial dysfunction and extracellular matrix abnormalities (possibly due to local exposures to hyperglycemia) that result ultimately in cell death. The evidence also suggests that the effects of T2DM on CEC mitophagy appear to be the opposite of the effects of FECD on CEC mitophagy. Although reduced mitochondrial function in FECD appears to be the result of increased mitophagy leading to overactive clearance and lower overall mitochondrial counts, reduced mitochondrial function in T2DM may be attributable to a decline in mitophagy that results in intracellular accumulation of dysfunctional mitochondria. There is also evidence that dysfunctional diabetic mitochondria are not undergoing fission when necessary, resulting in larger mitochondrial size, as dysfunctional components are being retained within the mitochondria rather than removed. This is supported by the decline in function despite the increased size. Further investigation is required to determine if fission proteins are affected by diabetes in corneal endothelial cells.
Lens Maturation and Cataract
The mature crystalline lens is a product of cellular maturation and organelle degradation, resulting in an optically clear path for light to traverse and focus on its way to the retinal outer segments.11,94 The mature lens requires an organelle-free zone (OFZ) void of mitochondria, nuclei, endoplasmic reticulum, and Golgi apparatus.95 This OFZ allows for minimally obstructed passage of light through the lens, and it has been shown that uncleared mitochondria, nuclei, nucleic fragments, and DNA in the OFZ causes light scattering in animal models.96 Since the discovery of organelle degradation in the lens, autophagy and mitophagy have been shown to play a vital role in the degradation and clearance of mitochondria specifically in lens maturation.
Costello et al.11 showed that mitophagy is evident in both adult human lens epithelial cells and differentiating lens fiber cells (as determined using TEM analysis) as well as in E12 chick embryo lenses (as determined with TOM20 and LC3B colocalization immunohistochemistry). Furthermore, mitophagy was widespread in chick embryo lenses, indicating a role in cellular differentiation during the development of the lens, similar to that found in the maturation process of erythrocytes from reticulocytes.9 Brennan et al.97 found that the process of mitophagy in differentiating lens fibers begins in parallel with a spike of BNIP3L/NIX protein expression, and the highest concentration of BNIP3L/NIX RNA expression was found in the same geographic area of embryonic chick lenses where organelle elimination was occurring. These findings were confirmed in mouse lenses, where BNIP3L protein expression is consistently higher in regions of lens fiber differentiation; conversely, BNIP3L−/− mice retained organelles (including mitochondria) in the same regions of the lens.97 If dysfunctional mitochondrial components and other autophagy-related vesicles are not eliminated from the lens during fiber cell differentiation, they build up in the OFZ, eventually causing cataract, one of the leading causes of vision loss.96,98
Glaucomatous Optic Neuropathy
Glaucoma is a leading cause of blindness. It is associated most often with the modifiable risk factor of elevated intraocular pressure (IOP) and eventually leads to retinal ganglion cell (RGC) loss and optic nerve damage. RGC death is a hallmark of glaucomatous optic neuropathy, and glaucomatous RGCs are shorter and more fragmented.99 RGCs have very long axons that rely heavily on a dense population of mitochondria for energy and metabolism. These cells, therefore, are very susceptible to oxidative stress and mitochondrial dysfunction.100,101 Several mitochondria protein-coding genes are differentially expressed in glaucoma,102 and an emerging role of mitophagy in glaucomatous optic neuropathy is being elucidated. Below, we review two common types of glaucoma and the role of mitophagy as characterized using different animal models.
Primary Open-Angle Glaucoma. Primary open-angle glaucoma (POAG) accounts for roughly three-quarters of all glaucoma cases103 and is characterized by visual field loss attributed to RGC death and optic nerve head (ONH) excavation.104 Mitochondrial dysfunction plays a critical role in POAG.105 Various animal models have been useful in recapitulating human disease phenotypes and performing targeted investigations of mitochondrial pathophysiology to further explore the role of mitophagy in this disease.
DBA/2J Mouse Model . This commonly used glaucoma mouse model displays RGC damage that is similar to human glaucomatous optic neuropathy. ONH axonal degeneration has been described in human POAG and in this mouse model. However, compared to control mouse (DBA/2wt-gpnmb) axons, DBA/2J axons display increased counts of smaller mitochondria with disrupted cristae (unrelated to increased organelle production) that occur before axonal breakdown is observed,26 indicating a reduced capacity for energy production that is in concordance with decreased mitochondrial respiration observed in human POAG lymphocytes.106 Glaucomatous mitochondria in this model have disrupted cristae and smaller surface areas than controls, suggestive of an increase in fission. Although levels of mitophagy-related proteins PINK1 and parkin were not significantly altered, suggesting no change in the induction of mitophagy, the lysosomal protein LAMP1 was significantly reduced in glaucoma model mice, suggesting that clearance of autophagosomes was dysfunctional, potentially impacting mitophagy. Levels of peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α, a protein involved in mitochondrial biogenesis, were also found to be low, indicating that mitochondrial production had not increased and the increased mitochondrial counts were more likely due to either an increase in fission or a decrease in mitophagic clearance. Because this study did not use lysosomal inhibitors to investigate mitophagic flux, strong conclusions about a decrease in mitophagy are not able to be drawn from this data. This study indicates that destruction and elimination of poorly functioning mitochondria are segregated from other portions of mitochondria through fission, and there is room to investigate the possibility of reduced mitophagy in the DBA/2J mouse ONH axons.
Ucp2 Knockout Mouse Model . Mitochondrial dysfunction can result in increased levels of damaging ROS in the eye, and restoration of mitochondrial homeostasis and antioxidative support to neurons of the optic nerve has become a focus in research to prevent glaucomatous optic neuropathy.101,107–110 Using mice with a genetic background of elevated ROS and increased susceptibility to stress, Hass and Barnstable111 injected microbeads into the anterior chamber to increase IOP and replicated the increased risk for ONH damage in humans. They found that RGC-specific deletion of mitochondrial uncoupling protein 2 (Ucp2) reduced the amount of ROS-mediated protein modifications and RGC death. Deletion of Ucp2 does increase mitochondrial depolarization and free radical species; however, it also increases mitophagy, which eliminates damaged mitochondria before they become toxic to the cells and provides protection for RGCs. Increased mitophagy was quantified by measuring colocalization of LC3B and TOM20, mitochondrial surface area, and counts per cell, as well as expression of BNIP3L/NIX protein and gene expression. Ucp2 may provide a promising target for the increase of mitophagy in glaucoma to prevent RGC death at the ONH.
Chronic Hypertensive Glaucoma Sprague – Dawley Rat Model . Dai et al.112 created glaucoma model rats using translimbal laser photocoagulation that reproduce elevated ocular hypertension and POAG risk similar to those in humans. They demonstrated in this rat model that there is an increase in mitochondrial cristae decomposition as determined by cristae appearance scaling in TEM images as well as an increase in RGC death. Although an abundance of autophagosomes and mitophagosomes was observed in these cells and the ratio of LC3-II/LC3-I was increased, levels of LAMP1 were decreased, suggesting that dysfunction in phagolysosome maturation and mitochondrial clearance might account for the increase of dysfunctional mitochondria.112 When overexpressing parkin in this model using adeno-associated virus 2 (AAV2)-parkin, a decrease in RGC death was observed. The levels of LC3-II/LC3-I and LAMP1 both increased in response to IOP elevation, and, although they are not specific to mitophagy, they add plausibility to the possibility of an increase in mitophagy that would be expected from parkin overexpression in RGCs and that could potentially impact cellular viability. Increasing expression of mitophagy signaling protein parkin in hypertensive rats provides evidence that clearance of dysfunctional mitochondria via mitophagy increases RGC survival.
From the same research group, Hu et al.113 injected the same hypertensive glaucoma model rats with either AAV2–OPA1 or AAV2-null constructs into the vitreous. Rats transfected with OPA1 had increased fusion morphology, greater mitochondrial surface area, and higher mitochondrial quality in optic nerve RGCs. The overexpression of OPA1 also increased parkin expression in the retina. As shown in the previous study, overexpression of parkin increased clearance of dysfunctional mitochondria, which was associated with RGC viability. The increase of both the fusion protein, OPA1, and the mitophagy protein, parkin, led to a simultaneous increase in mitochondrial surface area and quality, as well as a decrease of dysfunctional mitochondria, as shown in the OPA1 overexpression hypertension rats. Overexpression of OPA1 and parkin in this glaucoma rat model led to increased mitophagy and resulted in decreased RGC death, demonstrating the protective effect of efficient mitochondrial clearance in retaining a functional mitochondrial population in these cells.
All three of these different glaucoma animal models have common themes. Without intervention, IOP increases RGC death, mitochondrial dysfunction, and mitochondrial buildup, indicative of dysfunctional mitophagy. Interventions to increase mitophagy in these models of glaucoma restored mitophagy and reduced RGC death. This hypothesis has been further confirmed using mito-QC reporter (mCherry-GFP mitophagy reporter plasmid) mouse-derived RGCs. In these studies, induction of glaucoma was achieved using paraquat, tert-butyl hydroperoxide (tBOOH), staurosporine, or nerve crush surgery in vivo.114 These glaucoma-relevant stressors all induced an increase in mitophagy (quantified using mito-QC) in RGCs. Furthermore, RGC stress was rescued using the mitophagy inducer, iron chelator deferiprone (DFP). Mito-QC reporter allowed for direct quantification of mitophagy in these models.
Other studies have shown that mitophagy dysfunction in POAG may also be attributed in part to optineurin.115 Mutations in optineurin are directly associated with POAG.116 Optineurin is an autophagy adaptor protein that has binding domains for both ubiquitin and LC3; it is responsible for the signaling involved in the engulfment of mitochondria by autophagosomes.117,118 Therefore, it is possible that POAG patients with optineurin mutations have deficient mitophagy, resulting in the buildup of dysfunctional and fractionated mitochondria and potentially contributing to disease pathogenesis. Although the evidence strongly suggests that mitophagy imbalance impairs RGC viability, the cause of the imbalance requires further investigation.
Pseudoexfoliation Glaucoma. Pseudoexfoliation glaucoma (PXG) is a subset of POAG in which a systemic age-related elastinopathy (pseudoexfoliation syndrome) presents with ocular manifestations featuring fibrillar deposition in anterior segment structures, including trabecular meshwork.119–123 Patients with PXG harbor primary tenon fibroblast cells (isolated from trabecular meshwork) with mitochondrial dysfunction, as well as a dysregulated lysosome–autophagosome formation compared to age-matched POAG control cells.124 This results in a reduction of autophagy events and may explain both the greater presence of dysfunctional mitochondria in the cells (reduced mitophagy) and an accumulation of material in the extracellular matrix (cellular aggregopathy) compared to POAG cells.124 Compared to POAG tenon fibroblasts, PXG fibroblasts have a 3.9-fold higher accumulation of protein LC3II but a 53% decrease in autophagic flux out of the cell, indicating that autophagosome markers are upregulated but clearance is dysfunctional, similar to the chronic hypertensive glaucoma Sprague–Dawley rat model.29 Mitochondrial membrane potential was more diminished in PXG cells than POAG cells. These findings together indicate a decrease in efficient mitophagy and begin to explain the buildup of dysfunctional mitochondria in the PXG cells.29
In summary, elevated IOP causes mitochondrial dysfunction and reduced mitophagy in RGCs at the ONH in glaucoma animal models and in cell cultures of patient-derived trabecular meshwork tenon fibroblasts. These deficiencies lead to the buildup of small, dysfunctional mitochondria within these cells and may be a contributing factor to increased cell death. Although increased IOP does increase mitophagy mechanisms at a protein level, overexpression of mitophagy proteins is required to initiate enough mitophagy to clear these dysfunctional mitochondria and rescue cell death.
Retinopathy
Broadly, any retinal degeneration affecting outer-segment rod and cone photoreceptor cells can result in permanent vision loss. Mitochondrial dysfunction is a prominent part of the pathophysiology in diabetic retinopathy and age-related macular degeneration, two common diseases that result in degenerative retinopathy and vision loss. Mitophagy impairment specifically has been linked to retinal degeneration in the Park2−/− mouse. In this model, it has been shown that in animals lacking parkin protein mitophagy is impaired in retinal photoreceptors following light exposure.125 An increased number of damaged mitochondria is observed in these knockout mice, suggesting that damaged mitochondria are not properly cleared and furthermore that damaged mitochondrial buildup may contribute to the retinal degeneration observed in Park2−/− mouse photoreceptors following light exposure.125 Other research indicates that retinal outer segment mitophagy occurs independent of PINK1/parkin signaling. McWilliams et al.126 demonstrated that basal levels of mitophagy occur in the outer nuclear layer of the photoreceptors at similar levels in both wild-type (WT) and Pink1−/− knockout mice. They also demonstrated that PINK1 protein expression was much lower compared to other tissues with high metabolic demand (liver, brain, heart, pancreas) in the WT mouse. Although these two models are contradictory, further research may indicate multiple mitophagy pathways responsible for basal, homeostatic mitophagy (parkin-independent) compared to pathways activated during stress (parkin-dependent). Nonetheless, effective mitophagy is essential for retinal development and viability.127 Diabetic retinopathy and age-related macular degeneration (AMD) are reviewed below in the context of mitophagy impairment.
Diabetic Retinopathy. As a microvascular complication of diabetes, diabetic retinopathy is characterized by breakdown of the blood–retina barrier, a barrier normally maintained by RPE cells,128 with accompanying vascular endothelial cell and/or pericyte dropout, endothelial cell basement membrane thickening, and vascular leakage with extravascular deposition of lipids and proteins.129 Understanding the pathophysiological effects of diabetes on retinal blood vessels may provide early intervention options that could prevent vision loss. Several cell culture and animal model investigations offer insight into the role of mitophagy, specifically as a potential target in diabetic retinopathy.
ARPE-19 Cell Culture Models . TXNIP is an important participant in mitochondrial degradation in the retinal pigment epithelium (RPE) during the pathogenesis of diabetic retinopathy. When TXNIP binds to thioredoxin, it reduces the ability of thioredoxin to scavenge free radicals; therefore, TXNIP is associated with increased ROS.65,130 TXNIP upregulation is associated with mitochondrial membrane depolarization and has been shown to be increased in hyperglycemic conditions, specifically associated with diabetes in RPE cells.41,65 Adult retinal pigment epithelial cell line-19 (ARPE-19) cells are commonly used in diabetic retinopathy and AMD research due to their retained differentiated expression of the RPE markers cellular retinaldehyde-binding protein (CRALBP) and RPE65 in vitro, apical/basal polarity, and formation of tight junctions.131 Using cultures of ARPE-19 cells, Devi et al.65 demonstrated that hyperglycemia causes increased TXNIP expression, increased mitochondrial dysfunction, increased mitochondrial fragmentation, increased mitophagic flux (confirmed with the use of the flux inhibitors NAC and amlexanox), and increased lysosomal enlargement. Lysosomal enlargement occurs when mitophagic flux exceeds the digestive capacity of the lysosomes, which in turn causes poor or inactive lysosomal enzymatic activity. Devi et al.132 further showed that knocking down TXNIP expression using shRNA under hyperglycemic conditions (25-mM glucose compared to 5.5-mM glucose) decreased mitochondrial fragmentation, mitophagy, and lysosomal enlargement. Similar results were also shown in Müller cells. Furthermore, TXNIP upregulation is associated with NLRP3 activation and plays a role in the increased inflammatory-associated cell death in chronic disease conditions including diabetic retinopathy.41,133 It is plausible that targeting TXNIP may be a useful therapy for patients with diabetic complications, including retinopathy.28
Similar results have been shown in studies of cytoplasmic hybrid (cybrid) cell lines that harbor mtDNA variants that have been used to investigate diabetic mitochondriopathy.134,135 Diabetes-susceptible cybrids showed an increase in mitochondrial ROS production and have been able to replicate an association between higher levels of oxidative stress and decreased insulin sensitivity seen in humans.135 Using both diabetes-susceptible and mitochondria-protective haplotypes, Kuo et al.136 demonstrated that diabetes-susceptible cybrids had more fragmented mitochondria, less mitochondrial biogenesis, less expression of mitochondrial fusion-related proteins, ineffective mitophagy, and increased apoptosis. Insulin exposure promotes a pro-fusion phenotype but more so in mitochondria-protective cybrids. Increased mitochondrial networking and ATP production were also found following insulin exposure, but only in cybrids harboring diabetes-protective mtDNA variants. Overall, cybrids with diabetes-susceptible mtDNA variants have been found to be resistant to insulin and have ineffective mitophagy, low ATP production, and increased cell death.136 Ineffective mitophagy is demonstrated by an increased number of fragmented mitochondria present in the cells despite an increased number of lysosomes compared to mitochondria-protective haplotypes. Antioxidant (NAC) supplementation improved these phenotypes in diabetes-susceptible cybrids, indicating a causative role of oxidative stress in the mitochondrial dysfunction and altered mitophagy in these particular cells. mtDNA variants that are diabetes susceptible likely cause an increase in mitochondrial oxidative damage that in turn propagates mitochondrial dysfunction and ineffective mitophagy.
Using a culture of ARPE-19 RPE cells, Zhang et al.137 showed that hyperglycemia (50-mM glucose) induces ROS, reduces mitophagy, and increases cellular apoptosis. In comparison, lower but still elevated glucose conditions (15-mM glucose) also resulted in increased ROS levels but increased mitophagy and did not increase apoptosis. Additional assays indicate that the mitophagy reduction in hyperglycemic conditions is due to ROS inhibition of PINK1 and downregulation of parkin protein expression. Supplementing hyperglycemic cells with ROS scavengers mitigated the toxic effects of high glucose on ARPE-19 cells. This study demonstrates the intimate relationship among elevated ROS, reduced mitophagy, and increased cell death in the hyperglycemic environment of diabetic retinopathy and suggests a potential therapeutic role for ROS mitigation in the treatment of diabetic retinopathy.
Rat Müller Cell Culture and db/db Mouse Model . Zhou et al.138 reported similar findings in both a hyperglycemic rat Müller cell culture model (HG rMC-1) and diabetic mouse (db/db) retinas. Rat Müller cells demonstrate the same oxidative stress, inflammatory response, and excessive production of vascular endothelial growth factor (VEGF) observed in human Müller cells with diabetic retinopathy, and the db/db mouse model replicates the neurodegenerative features of diabetic retinopathy in humans well, including increased apoptosis in ganglion cell, endothelial, and pericyte layers and markers of increased gliosis in the retina.139 The study treated both cells and mice with notoginsenoside R1 (NGR1), a saponin that exhibits antiinflammatory properties and scavenges ROS and is used to treat diabetic encephalopathy and microvascular disorders.138,140 Treatment with NGR1 led to enhancement of PINK1 and parkin expression, increased mitophagy autophagosomes seen on TEM, increased LC3II/LC3I ratio, increased colocalization of GFP-LC3 and MitoTracker labeling, and decreased p62. Although flux was not measured using lysosomal inhibitors, the study demonstrated that in cells treated with NGR1 but where PINK1 expression was silenced by a siRNA, the increased LC3II/LC3I ratio and decreased p62 were reversed, suggesting that the effects of NGR1 were the result of a PINK1-mediated activation of mitophagy. Ultimately, NGR1 treatment prevented the progression of diabetic retinopathy by reducing apoptosis, decreasing VEGF expression, and increasing PEDF expression in both models.138 This study supports a role for antioxidant therapy in the prevention of diabetic retinopathy through the targeting of PINK1/parkin-mediated mitophagy.
These featured cell culture and animal studies modeling diabetic retinopathy commonly show that hyperglycemic conditions associated with diabetes cause insulin resistance, lower ATP production, increase ROS, increase mitochondrial dysfunction, decrease mitophagy in RPE and Müller cells, and ultimately replicate the cell death associated with diabetic retinopathy in humans. Some of these models also highlight additional responses to hyperglycemia, including inflammation, that accompany the mitochondrial responses to hyperglycemia in RPE and Müller cells. Using a dual-pathway therapy such as NGR1, which decreases inflammation and increases mitophagy, may provide a promising approach for preventing diabetic retinopathy disease progression and damage. However, the timing of mitophagy management will require further investigation due to evidence in hyperglycemic mitoQC-Ins2Akita/+mice showing that mitophagy increases in the outer retina during early stages of diabetes and then decreases during advanced stages of the disease.141
Age-Related Macular Degeneration. AMD ultimately results in damage to the retinal outer segments and subretinal inflammation and fibrosis142–144; mitophagy-related studies of AMD have focused primarily on the viability of the RPE, which becomes damaged and atrophic in earlier stages of this disease. In AMD, RPE cells display severe disruptions in IMM and OMM structure, alterations in mitochondrial size, and changes in dynamic cytosolic organization.145 Increased oxidative stress and related damage in the RPE are thought to play a pivotal role in the progression of AMD,146 and the high constitutive energy demands of the retina and accumulation of ROS with age compound oxidative stress on the choroid and RPE in this disease.147 Understanding the pathophysiological effects of oxidative stress on the RPE may provide early intervention options that could prevent AMD disease progression and vision loss.
Redox and mitophagy impairments have been characterized in patients with AMD. In RPE cells isolated from deceased donor eyes, nicotinamide adenine dinucleotide (NAD+) has been shown to be decreased in AMD.148 NAD+ is an essential factor for efficient metabolism, and when NAD+ levels drop during normal aging the amount of mitophagy also slows. Increasing NAD+ levels in human RPE cell cultures reduces ROS, reduces cell death, and increases the amount of mitophagy, resulting in a promising potential therapy for preventing AMD progression.149,150 In another example, Stenirri et al.151 showed that mitochondrial ferritin (FtMt) mutations are associated with AMD in a study of 50 patients with various stages of disease, including exudative, nonexudative, and early. FtMt is an iron-storing antioxidant protein, and it has been shown that iron chelation intended to decrease cytosolic iron induces mitophagy through a PINK1/parkin-independent signaling pathway.152 FtMt also protects against ferroptosis,153 a non-apoptotic form of programmed cell death that is iron dependent and driven by ROS-induced lipid peroxidation.154 FtMt protects against ferroptosis directly by regulating mitochondrial iron storage and possibly indirectly through the control of mitophagy, providing another potential AMD therapeutic strategy.
Several experimental investigations have also explored mitophagy impairment and rescue in RPE cells and AMD, as described below.
NaIO3 ARPE-19 Cell Culture Model. Chan et al.155 showed that sodium iodate (NaIO3)-induced oxidative toxicity in ARPE-19 immortalized human RPE cells recapitulates features of AMD, including decreased resting oxygen consumption rate and ATP turnover, increased cytosolic but not mitochondrial ROS, and increased cell apoptosis. When RPE cells are treated with NaIO3 and bafilomycin A1, LC3II and TOM20 punctate co-labeling identified by immunohistochemistry and confocal microscopy becomes significantly increased compared to bafilomycin treatment alone or no treatment, demonstrating the presence of rapid mitophagic flux active in this model of AMD. The study also demonstrates that NaIO3-treated cells display increased mitochondrial fragmentation, as well as an increase in phospho-DRP1 (DRP1 S616, pro-fission form of DRP1) but no increase in fusion proteins MFN1, MFN2, or OPA1. These findings make it tempting to draw the conclusion that ROS exacerbate the mitochondrial dysfunction present in this cell culture model and should be the target of therapy for this condition. But, surprisingly, treating healthy RPE cells with the antioxidants NAC or Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a cell-permeable, water-soluble analog of vitamin E) led to similar mitochondrial fragmentation and increases in pDRP1 (S616). Treating NaIO3-dosed cells with NAC or Trolox further augmented these mitochondrial phenotypes. These findings indicate that lowering levels of cytosolic ROS increases mitochondrial fragmentation, supporting the idea that ROS signaling is actually beneficial for maintaining mitochondrial dynamic balance under stressful conditions. However, too much cytosolic ROS leads to permanent DNA, lipid, and protein damage. Taken together, this study indicates that the mechanism of RPE cell death in AMD as modeled by NaIO3-induced cellular toxicity may be attenuated by cytosolic ROS-induced mitophagy and restoration of mitochondrial dynamic balance, complicating the attractive possibility of using antioxidants as a therapeutic in this disease.155
Human Primary RPE and ARPE-19 Cell Culture . Hytti et al.156 studied the effects of antimycin A (AMA, an inducer of ROS and inhibitor of oxidative phosphorylation) on both primary human RPE cells and immortalized ARPE-19 cells. The mitochondria in AMA-treated cells had a rounded shape and disrupted cristae in TEM image analyses of both cell types. After over 24 hours of AMA exposure, mitochondrial counts decreased, indicating clearance of damaged mitochondria. AMA also increased ARPE-19 protein expression of both autophagy markers LC3B and p62. As expected, RPE cell death increased with AMA treatment; however, cell death was increased by treatment with bafilomycin A1. This finding indicates that, although RPE cell death increases with AMA-induced mitochondrial dysfunction, intact mitophagy is a compensatory mechanism that attenuates the extent of cell death. Therefore, proper mitophagy function is protective in AMD pathogenesis by upregulating clearance of damaged mitochondria, reducing cell death.156
NFE2L2/PGC-1α−/ − Mouse Model. Sridevi Gurubaran et al.27 investigated a nuclear factor erythroid-2-related factor 2 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha double knockout (NFE2L2/PGC-1α−/−) mouse model of dry AMD that demonstrates protein aggregation, mitochondrial dysfunction, and oxidative stress. These features of the model correlate well with the excessive ROS production, protein accumulation caused by oxidative stress, higher levels of RPE mtDNA damage, reduced ATP production, and reduced mitochondrial counts observed in patients with AMD.146,157 In this model, the investigators performed confocal microscopy of the affected RPE cells and observed that colocalization of mitophagy-related proteins PINK1 and parkin was increased, and colocalization of LC3B and ATP synthase β was also increased.27 On the other hand, they discovered that colocalization of LAMP2 and ATP synthase β was not increased in these double-knockout RPE cells. Because LC3B is a marker of the phagosome and LAMP2 is a marker of the lysosome, the authors speculated that their results indicate that increased mitophagy signaling but decreased lysosome recruitment occurs in AMD. However, because they did not investigate mitophagic flux along with monitoring steady-state levels of mitophagic proteins, it is unclear whether the increase in PINK1, parkin, and the colocalization of LC3B and ATP synthase β was due to increased activation upstream of these steps or impaired progression through late mitophagy downstream of these steps, causing a buildup of earlier intermediates in the pathway. The decreased colocalization of LAMP2 and ATP synthase β is difficult to interpret for the same reason. RAS-related GTP-binding proteins (Rab family proteins) are key proteins essential for mediating the fusion of lysosomes and autophagosomes. In this same investigation, the authors determined that, in the double-knockout RPE cells, Rab5 and Rab7 proteins colocalized with autofluorescent protein aggregates in the perinuclear space. The authors concluded that these autofluorescent aggregates most likely represent lipofuscin, an inhibitor of lysosomal protein degradation, as well as a potential participant in AMD progression.158 In all, this model shows that autofluorescent aggregates consistent with lipofuscin may inhibit mitophagy directly in RPE cells during the progression of AMD by preventing mitochondrial clearance, thereby allowing buildup of dysfunctional mitochondria in the cells.
In summary, AMD progression results in part from increased ROS, mitochondrial dysfunction, and mitophagy dysregulation that result in reduced RPE survival. Conversely, increasing the clearance of damaged mitochondria via mitophagy increases RPE cell viability. Targeting mitophagy-associated proteins such as FtMt or increasing NAD+ may be possible treatments for the prevention of AMD progression.
Ocular Phenotypes of Systemic Mitochondrial Disease Related to Mitophagy
Several mitochondrial diseases have ophthalmic manifestations, including Kearns–Sayre syndrome (KSS); myoclonic epilepsy with ragged red fibers (MERRF); mitochondrial encephalopathy lactic acidosis stroke (MELAS); neuropathy, ataxia, and retinitis pigmentosa (NARP); LHON; CPEO; and autosomal dominant optic atrophy (DOA).159–161 Many of these diseases have phenotypes related to myopathy due to the high energy demand of sarcomeres,162,163 and ocular phenotypes can be the first observed in mitochondrial diseases.164 Mitochondrial diseases give great insight into the effects of altered mitophagy on disease pathology because the mechanism is the result of specific gene mutations.
mtDNA Point Mutations and Deletions Affecting Extraocular Muscles—KSS and MERRF
KSS, often categorized as a subset of CPEO,164 is characterized by progressive external ophthalmoplegia and pigmentation of the retina that may affect visual acuity. Deletions in mtDNA associated with KSS are associated with protein damage, inhibition of ubiquitin-associated proteasome activity, and activation of autophagy.163 The increase in autophagy is indicated by an increase in SNAP receptor proteins (SNAREs), specifically syntaxin 16 (of note, syntaxin 17 is the more commonly implicated SNARE protein associated with mitophagy).77,165 It is possible that increased autophagy in this disease and other primary mitochondriopathies also results in increased mitophagy.
MERRF ocular manifestations can include myoclonus, optic nerve degeneration, and retinal pigmentation. MERRF, most commonly caused by a point mutation in mtDNA tRNA(Lys), affects the synthesis of mtDNA-encoded proteins and subsequently causes impaired mitochondrial respiration due to decreased expression of respiratory chain complexes.166 Fibroblasts derived from patients containing the MERRF mutation m.8344A>G demonstrate rounded, depolarized mitochondria, increased ROS, decreased ATP production, and decreased cell proliferation rates. The MERRF fibroblasts also showed an increase in the ratio of LC3-II to LC3 I, an increase in LysoTracker staining, and increased levels of LC3 and cytochrome c colocalization compared to fibroblasts from control patients.166 These findings were all partially ameliorated by supplementing the cells with antioxidant coenzyme Q10. Because flux was not measured, it was not clear whether the increase in steady-state concentrations of mitophagy markers reflects increased activation in early mitophagy or decreased flux through late mitophagy. However, the difference between MERRF fibroblasts and control fibroblasts in markers relating to mitophagy and mitochondrial homeostasis, taken in conjunction with the ability of coenzyme Q10 to markedly improve mitochondrial function in MERRF fibroblasts, suggests that regulation of mitophagy could play an important role in the pathogenesis of MERRF. Using patient-derived MERRF fibroblasts, Villanueva-Paz et al.167 demonstrated that mitophagy is increased compared to control cells. Flux was assessed using bafilomycin, and the study demonstrated that these cells specifically have increased parkin-mediated mitophagy. Coenzyme Q10 offers a potential therapy for MERRF patients because it helps prevent recruitment of parkin protein to mitochondria.167
Other mtDNA Point Mutations—MELAS
MELAS is characterized by muscle weakness, headaches, and strokes that can be accompanied by vision abnormalities and altered levels of consciousness. Fibroblast culture studies for MELAS showed that mitochondria are associated with higher mutational load, reduced antioxidants superoxide dismutase 2 (SOD2) and catalase, increased mitophagy, and lower mitochondrial density, the latter of which is further associated with lower AMP-activated protein kinase (AMPK) activity and reduced autophagic flux, resulting in the buildup of autophagolysosome complexes in the cytosol.168 These phenotypes are greatly reduced by activating AMPK with supplementation of coenzyme Q10, which was found to increase mitochondrial biogenesis, increase antioxidant expression, and decrease mitophagy (as quantified by cytochrome c and LC3 colabeling). Additional fibroblast cultures containing point mutations in mtDNA tRNAs as found in MELAS and MERRF have been shown to have higher levels of mitochondrial depolarization, higher levels of mitophagy, and larger cell sizes, but similar levels of intact mitochondrial content.169 These findings indicate not only mitochondrial dysfunction but also a decrease in their clearance. The larger cell size is possibly due to an accumulation of dysfunctional mitochondria, but more research is necessary for clarification.
Mutations in ATPase Subunit 6—NARP and LHON
Muscle weakness, tingling, problems with balance, dementia, and vision loss due to retinitis pigmentosa are hallmarks of NARP syndrome. In a human NARP patient fibroblast cybrid cell culture, Walczak et al.170 demonstrated that mitochondria are more fragmented than control cells (human osteosarcoma line 143B) and have higher levels of fission- and mitophagy-related proteins, including FIS1, MFF, LC3II, and beclin-1. p62, an autophagy marker often inversely correlated to amount of autophagy, as it is degraded during the process, was decreased in NARP cells.8 Although p62 has been shown to localize to mitochondria and participate in mitochondrial specific functions, this has not specifically been shown in these NARP cells.171 Lower p62 levels in NARP cells may indicate increased mitophagy; however, this notion requires further investigation. Overall, the findings indicate that the mitochondrial stress associated with NARP mtDNA point mutations in ATPase subunit 6 upregulates mitochondrial fission and autophagy/mitophagy in an attempt to enrich the cellular mitochondrial population.
LHON is characterized by RGC degeneration and subsequent loss of central vision, predominately in young men.172 Dombi et al.173 showed that mtDNA mutation m.13051G>A is associated with increased mitophagy (assessed by LC3 and TOM20 colocalization in addition to mtDNA quantification) in patient-derived fibroblasts with LHON/Leigh phenotypes. The use of idebenone, a synthetic antioxidant similar to coenzyme Q10, reduced mitophagy in the fibroblast cultures and is thought to provide a possible therapeutic effect. Zhang et al.174 demonstrated reduced LC3II and accumulation of p62 in cybrid mtDNA ND5 12338T>C mutant cells, possibly suggesting decreased autophagy/mitophagy in contrast to the Dombi model, although mitophagic flux was not measured. Of note, the Zhang group's findings occurred despite the cells having reduced ATP, reduced membrane potential, and increased production of ROS. The reduced mitophagy phenotype in LHON is supported by Sharma et al.,175 who demonstrated that activation of mitophagy in cells harboring LHON mtDNA mutations rescues mitochondrial function and cell survival. Work by Jankauskaitė et al. (2020)172 and Kodroń et al. (2019)176 also showed higher mitochondrial mass, lower mitophagic receptor BNIP3, and lower autophagic flux present in LHON lymphoblast cultures treated with testosterone, as well as in cybrid LHON mutation cultures, supporting a reduced mitophagy phenotype in LHON.172,176 Genome-wide linkage studies have further shown two PARL SNPs associated with LHON mitochondrial disease.177 PARL protease is responsible for processing the antiapoptotic form of OPA1, which subsequently prevents the release of mitochondrial cytochrome c into the cytosol, normally an intrinsic signal initiating apoptosis.178 Finally, in the Ndufs4–/– mouse model of LHON, knocking out mir181a/b increased mitophagy, as measured by an increase in the expression of parkin and p62 protein expression, and enhanced mitochondrial biogenesis, as measured by mtDNA quantitative PCR and complex I, II, and IV biochemical activity.179 Altogether, most evidence suggests that LHON patients have a reduced amount of mitophagy and could therefore benefit from a therapeutic strategy that increases mitophagy.
Mutations in OPA1—CPEO and DOA
CPEO is characterized by a progressive inability to move the eyelids and eyebrows. Although there are a variety of genetic etiologies accounting for CPEO,164 Carelli et al.180 characterized two families with CPEO in Italy and found they had missense mutations in OPA1, decreased OPA1 protein expression despite normal mRNA expression, fragmented mitochondria, and increased mitophagy.
DOA is an optic neuropathy caused by mutations in the OPA1 gene.181 Dermal fibroblasts from patients with severe DOA (DOA plus) were assayed for changes in mitophagy, measured by LC3II and TOM20 autophagosome–mitochondria colocalization immunohistochemistry, with flux being measured using chloroquine. OPA–/–-, OPA+/–-, and OPA siRNA-silenced cells all demonstrated increased mitophagy compared to control and siRNA scramble cells. These events were reduced by knocking down ATG7, the gene that encodes ATG7, which is essential for the conversion of cytosolic LC3 to LC3II by conjugation to phosphatidylethanolamine. Results in DOA plus cells were compared to fibroblasts from another patient, in this case with a dominant negative MFN2 mutation, which also had increased mitophagy. This phenotype mimics that found in FECD cell lines, which are also deficient in MFN2 protein.23 Mitophagy events in DOA plus fibroblasts were resistant to effects of idebenone, unlike those with the LHON m.13051G>A mutation described above.173,181 In contrast, Moulis et al.182 showed that mitophagy protein BNIP3, autophagy, and mitophagy are decreased in the OPA1-depleted (siOPA1) cortical cell culture model.182 However, they also showed that, in a DOA mouse model, the OPA1enu/+ mouse, BNIP3 protein expression was decreased at an early age and then increased with age. This may indicate an early attempt for disease compensation.
In summary, mtDNA variants cause an imbalance in mitochondrial function and quality of affected cells. The mitochondrial diseases outlined above all have ocular phenotypes; however, the direct connection between mitophagy imbalance and ocular phenotype largely remains unclear. Because these mitochondrial diseases manifest in many tissues, they can generally be studied using patient-derived fibroblasts to help elucidate the links between mitophagy and clinical findings. The insights gained from these primary mitochondrial diseases are easier to understand from a mechanistic viewpoint because causative mutations and affected proteins are known. In DOA, for example, OPA and MFN2 mutations are harbored in patient-derived fibroblast populations, and in both cases mitophagy is increased. This is expected from a mitochondrial dynamics perspective; because both of these proteins are associated with fusion, their absence will shift the dynamic toward fission, and ultimately mitophagy increases. These insights from primary mitochondrial disease can be used to better understand complex genetic diseases with secondary mitochondrial dysfunction such as FECD. Expression of MFN2 (a fusion protein) is reduced in FECD; predictably, CECs harbor smaller mitochondria and display increased mitophagy. Understanding known mitophagy changes and their mechanisms in primary mitochondrial diseases can therefore provide great insight into pathological mechanisms and possible treatment strategies of other diseases with similar phenotypes.
Conclusions
There is abundant evidence supporting a connection between mitochondrial quality and cell viability. Whether mitochondrial counts increase or decrease, their overall quality is essential for survival of the cell. Observing several ocular diseases including FECD, diabetic endotheliopathy, glaucoma, diabetic retinopathy, and age-related macular degeneration, one theme is common: When mitochondrial quality is reduced, the balance of mitochondrial dynamics in affected ocular cells is skewed toward either increased fission or fusion, and retention of dysfunctional mitochondrial mass results. Proper mitochondrial turnover, or mitophagy, is a key process in maintaining functional mitochondria within in the cell.
Many of the aforementioned studies demonstrated that manipulation of mitophagy can lead to improved mitochondrial function and cell survival; however, manipulation of mitophagy can be direct or indirect. For example, Benischke et al.23 showed that blocking mitophagy with bafilomycin directly rescued some FECD phenotypes, indicating that direct manipulation of mitophagy in some ocular diseases may prove to be a promising therapy. On the other hand, in ARPE-19 cells, Zhang et al.137 reported that hyperglycemia induced ROS, reduced mitophagy, and increased cellular apoptosis and that supplementing these cells with ROS scavengers mitigated the toxic effects of hyperglycemia. Their study suggests that elevated ROS was the reason for reduced mitophagy and increased cell death in the hyperglycemic environment of diabetic retinopathy; thus, therapies targeting ROS may help prevent cell death through increasing mitophagy. The question of how best to restore mitochondrial quality in ocular disease with secondary mitochondriopathies may be aided by studying the effects of primary mitochondriopathies with ocular phenotypes.
Regulation of mitophagy, either directly or indirectly, provides a key target for restoring the balance of mitochondrial dynamics, improving mitochondrial quality, and preventing cellular dysfunction and death in several common ocular diseases. Many of the studies reviewed herein showed protective effects of antioxidants related to mitophagy and the expression of key mitophagy proteins, regardless of the disease or cell type being used. Small molecule or protein interventions that target mitophagy may also provide promising targets for the treatment of ocular diseases characterized by impaired mitophagy. This review demonstrates overwhelmingly the connections that exist among oxidative stress, mitochondrial function, mitophagy, and cell viability in ocular diseases.
Acknowledgments
The authors thank Chantal Allamargot, PhD, and the University of Iowa Central Microscopy Research Facility for assistance with electron microscopy.
Supported in part by the M.D. Wagoner & M.A. Greiner Cornea Excellence Fund, the Beulah and Florence Usher Chair in Cornea/External Disease and Refractive Surgery, the UIHC Cornea Research Fund, Lloyd and Betty Schermer, and Robert and Joell Brightfelt.
Disclosure: J.M. Skeie, (N); D.Y. Nishimura, (N); C.L. Wang, (N); G.A. Schmidt, (N); B.T. Aldrich, (N); M.A. Greiner, (N)
References
- 1. Siekevitz P. Powerhouse of the cell. Sci Am . 1957; 197(1): 131–140. [Google Scholar]
- 2. Willis EJ. The powerhouse of the cell. Ultrastruct Pathol . 1992; 16(3): iii–vi. [DOI] [PubMed] [Google Scholar]
- 3. Ahn M, Oh E, McCown EM, Wang X, Veluthakal R, Thurmond DC.. A requirement for PAK1 to support mitochondrial function and maintain cellular redox balance via electron transport chain proteins to prevent β-cell apoptosis. Metabolism . 2020; 115: 154431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Surgucheva I, Shestopalov VI, Surguchov A.. Effect of gamma-synuclein silencing on apoptotic pathways in retinal ganglion cells. J Biol Chem . 2008; 283(52): 36377–36385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshizumi T, Imamura H, Taku T, et al.. RLR-mediated antiviral innate immunity requires oxidative phosphorylation activity. Sci Rep . 2017; 7(1): 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yim WW, Mizushima N.. Lysosome biology in autophagy. Cell Discov . 2020; 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng Y, He D, Yao Z, Klionsky DJ.. The machinery of macroautophagy. Cell Res . 2014; 24(1): 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klionsky DJ, Abdelmohsen K, Abe A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy . 2016; 12(1): 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gronowicz G, Swift H, Steck TL.. Maturation of the reticulocyte in vitro. J Cell Sci . 1984; 71: 177–197. [DOI] [PubMed] [Google Scholar]
- 10. Schweers RL, Zhang J, Randall MS, et al.. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA . 2007; 104(49): 19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costello MJ, Brennan LA, Basu S, et al.. Autophagy and mitophagy participate in ocular lens organelle degradation. Exp Eye Res . 2013; 116: 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Esteban-Martinez L, Sierra-Filardi E, McGreal RS, et al.. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J . 2017; 36(12): 1688–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stallons LJ, Funk JA, Schnellmann RG.. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep . 2013; 1(3): 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suliman HB, Keenan JE, Piantadosi CA.. Mitochondrial quality-control dysregulation in conditional HO-1(-/-) mice. JCI Insight . 2017; 2(3): e89676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durga Devi T, Babu M, Makinen P, et al.. Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am J Pathol . 2017; 187(12): 2659–2673. [DOI] [PubMed] [Google Scholar]
- 16. Disatnik MH, Hwang S, Ferreira JC, Mochly-Rosen D.. New therapeutics to modulate mitochondrial dynamics and mitophagy in cardiac diseases. J Mol Med (Berl) . 2015; 93(3): 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siasos G, Tsigkou V, Kosmopoulos M, et al.. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med . 2018; 6(12): 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Higgins GC, Coughlan MT.. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol . 2014; 171(8): 1917–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ganguly G, Chakrabarti S, Chatterjee U, Saso L.. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Des Devel Ther . 2017; 11: 797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rovira-Llopis S, Banuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM.. Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol . 2017; 11: 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez-Franquesa A, Patti ME.. Insulin resistance and mitochondrial dysfunction. Adv Exp Med Biol . 2017; 982: 465–520. [DOI] [PubMed] [Google Scholar]
- 22. Chinnery PF. Mitochondrial disorders overview. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® . Seattle, WA: University of Washington, Seattle; 2014. [Google Scholar]
- 23. Benischke AS, Vasanth S, Miyai T, et al.. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci Rep . 2017; 7(1): 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyai T, Vasanth S, Melangath G, et al.. Activation of PINK1-Parkin-mediated mitophagy degrades mitochondrial quality control proteins in Fuchs endothelial corneal dystrophy. Am J Pathol . 2019; 189(10): 2061–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aldrich BT, Schlotzer-Schrehardt U, Skeie JM, et al.. Mitochondrial and morphologic alterations in native human corneal endothelial cells associated with diabetes mellitus. Invest Ophthalmol Vis Sci . 2017; 58(4): 2130–2138. [DOI] [PubMed] [Google Scholar]
- 26. Coughlin L, Morrison RS, Horner PJ, Inman DM.. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci . 2015; 56(3): 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sridevi Gurubaran I, Viiri J, Koskela A, et al.. Mitophagy in the retinal pigment epithelium of dry age-related macular degeneration investigated in the NFE2L2/PGC-1α–/– mouse model. Int J Mol Sci . 2020; 21(6): 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh LP, Yumnamcha T, Swornalata Devi T. Mitophagic flux deregulation, lysosomal destabilization and NLRP3 inflammasome activation in diabetic retinopathy: potentials of gene therapy targeting TXNIP and the redox system. Ophthalmol Res Rep . 2018; 3(1): ORRT–126. [PMC free article] [PubMed] [Google Scholar]
- 29. Want A, Gillespie SR, Wang Z, et al.. Autophagy and mitochondrial dysfunction in tenon fibroblasts from exfoliation glaucoma patients. PLoS One . 2016; 11(7): e0157404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN.. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med . 2018; 50(2): 121–127. [DOI] [PubMed] [Google Scholar]
- 31. Akbari M, Kirkwood TBL, Bohr VA.. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res Rev . 2019; 54: 100940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Green DR, Galluzzi L, Kroemer G.. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science . 2011; 333(6046): 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Srinivasan S, Guha M, Kashina A, Avadhani NG.. Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim Biophys Acta Bioenerg . 2017; 1858(8): 602–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prakash YS, Pabelick CM, Sieck GC.. Mitochondrial dysfunction in airway disease. Chest . 2017; 152(3): 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brand MD, Nicholls DG.. Assessing mitochondrial dysfunction in cells. Biochem J . 2011; 435(2): 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee HC, Wei YH.. Mitochondria and aging. Adv Exp Med Biol . 2012; 942: 311–327. [DOI] [PubMed] [Google Scholar]
- 37. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res . 2017; 39(1): 73–82. [DOI] [PubMed] [Google Scholar]
- 38. Schofield JH, Schafer ZT.. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal . 2021; 34(7): 517–530. [DOI] [PubMed] [Google Scholar]
- 39. Zorov DB, Vorobjev IA, Popkov VA, et al.. Lessons from the discovery of mitochondrial fragmentation (fission): a review and update. Cells . 2019; 8(2): 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li X, Fang P, Yang WY, et al.. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol . 2017; 95(3): 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh LP, Devi TS, Yumnamcha T.. The role of Txnip in mitophagy dysregulation and inflammasome activation in diabetic retinopathy: a new perspective. JOJ Ophthalmol . 2017; 4(4), doi: 10.19080/jojo.2017.04.555643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zorova LD, Popkov VA, Plotnikov EY, et al.. Mitochondrial membrane potential. Anal Biochem . 2018; 552: 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV.. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int . 2014; 2014: 238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Richter C, Park JW, Ames BN.. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA . 1988; 85(17): 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci . 2016; 41(3): 261–273. [DOI] [PubMed] [Google Scholar]
- 46. Schutt F, Aretz S, Auffarth GU, Kopitz J.. Moderately reduced ATP levels promote oxidative stress and debilitate autophagic and phagocytic capacities in human RPE cells. Invest Ophthalmol Vis Sci . 2012; 53(9): 5354–5361. [DOI] [PubMed] [Google Scholar]
- 47. Jang SY, Kang HT, Hwang ES.. Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J Biol Chem . 2012; 287(23): 19304–19314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zamponi N, Zamponi E, Cannas SA, Billoni OV, Helguera PR, Chialvo DR.. Mitochondrial network complexity emerges from fission/fusion dynamics. Sci Rep . 2018; 8(1): 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Glancy B, Kim Y, Katti P, Willingham TB.. The functional impact of mitochondrial structure across subcellular scales. Front Physiol . 2020; 11: 541040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Twig G, Shirihai OS.. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal . 2011; 14(10): 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Twig G, Elorza A, Molina AJ, et al.. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J . 2008; 27(2): 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ashrafi G, Schwarz TL.. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ . 2013; 20(1): 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mozdy AD, McCaffery JM, Shaw JM.. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol . 2000; 151(2): 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT.. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep . 2011; 12(6): 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gandre-Babbe S, van der Bliek AM.. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell . 2008; 19(6): 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gottlieb RA, Piplani H, Sin J, et al.. At the heart of mitochondrial quality control: many roads to the top [published online ahead of print February 5, 2021]. Cell Mol Life Sci , 10.1007/s00018-021-03772-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Suliman HB, Piantadosi CA.. Mitochondrial quality control as a therapeutic target. Pharmacol Rev . 2016; 68(1): 20–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM 3rd, Bohr VA.. Protecting the mitochondrial powerhouse. Trends Cell Biol . 2015; 25(3): 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Legros F, Lombes A, Frachon P, Rojo M.. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell . 2002; 13(12): 4343–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brown J Jr, Fingert JH, Taylor CM, Lake M, Sheffield VC, Stone EM. Clinical and genetic analysis of a family affected with dominant optic atrophy (OPA1). Arch Ophthalmol . 1997; 115(1): 95–99. [DOI] [PubMed] [Google Scholar]
- 61. Schnieders MJ, Goar W, Griess M, et al.. A novel mutation (LEU396ARG) in OPA1 is associated with a severe phenotype in a large dominant optic atrophy pedigree. Eye (Lond) . 2018; 32(4): 843–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Romagnoli M, La Morgia C, Carbonelli M, et al.. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann Clin Transl Neurol . 2020; 7(4): 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arnoult D, Rismanchi N, Grodet A, et al.. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol . 2005; 15(23): 2112–2118. [DOI] [PubMed] [Google Scholar]
- 64. Si L, Fu J, Liu W, et al.. Silibinin-induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells. Arch Biochem Biophys . 2020; 685: 108284. [DOI] [PubMed] [Google Scholar]
- 65. Devi TS, Yumnamcha T, Yao F, Somayajulu M, Kowluru RA, Singh LP.. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol Open . 2019; 8(4): bio038521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pickrell AM, Youle RJ.. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron . 2015; 85(2): 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jin SM, Youle RJ.. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci . 2012; 125(pt 4): 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang T, Xue L, Li L, et al.. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem . 2016; 291(41): 21616–21629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gao J, Qin S, Jiang C.. Parkin-induced ubiquitination of Mff promotes its association with p62/SQSTM1 during mitochondrial depolarization. Acta Biochim Biophys Sin (Shanghai) . 2015; 47(7): 522–529. [DOI] [PubMed] [Google Scholar]
- 70. Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL.. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol . 2014; 206(5): 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu W, Xu H, Wang Z, et al.. PINK1-Parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One . 2015; 10(7): e0132499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tanida I, Ueno T, Kominami E.. LC3 and autophagy. Methods Mol Biol . 2008; 445: 77–88. [DOI] [PubMed] [Google Scholar]
- 73. Williams JA, Ding WX.. Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biol Chem . 2018; 399(2): 147–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mauro-Lizcano M, Esteban-Martinez L, Seco E, et al.. New method to assess mitophagy flux by flow cytometry. Autophagy . 2015; 11(5): 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Boland ML, Chourasia AH, Macleod KF.. Mitochondrial dysfunction in cancer. Front Oncol . 2013; 3: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. von Stockum S, Marchesan E, Ziviani E.. Mitochondrial quality control beyond PINK1/Parkin. Oncotarget . 2018; 9(16): 12550–12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Xian H, Yang Q, Xiao L, Shen HM, Liou YC.. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun . 2019; 10(1): 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mercer TJ, Gubas A, Tooze SA.. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem . 2018; 293(15): 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Siegfried CJ, Shui YB, Bai F, Beebe DC.. Central corneal thickness correlates with oxygen levels in the human anterior chamber angle. Am J Ophthalmol . 2015; 159(3): 457–462.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sarnicola C, Farooq AV, Colby K.. Fuchs endothelial corneal dystrophy: update on pathogenesis and future directions. Eye Contact Lens . 2019; 45(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 81. Vedana G, Villarreal G Jr, Jun AS. Fuchs endothelial corneal dystrophy: current perspectives. Clin Ophthalmol . 2016; 10: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nanda GG, Alone DP.. REVIEW: Current understanding of the pathogenesis of Fuchs’ endothelial corneal dystrophy. Mol Vis . 2019; 25: 295–310. [PMC free article] [PubMed] [Google Scholar]
- 83. Halilovic A, Schmedt T, Benischke AS, et al.. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs endothelial corneal dystrophy. Antioxid Redox Signal . 2016; 24(18): 1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jurkunas UV, Bitar MS, Funaki T, Azizi B.. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am J Pathol . 2010; 177(5): 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jurkunas UV. Fuchs endothelial corneal dystrophy through the prism of oxidative stress. Cornea . 2018; 37(suppl 1): S50–S54. [DOI] [PubMed] [Google Scholar]
- 86. Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC.. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem . 2017; 292(40): 16697–16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang Z, Handa JT, Green WR, Stark WJ, Weinberg RS, Jun AS.. Advanced glycation end products and receptors in Fuchs’ dystrophy corneas undergoing Descemet's stripping with endothelial keratoplasty. Ophthalmology . 2007; 114(8): 1453–1460. [DOI] [PubMed] [Google Scholar]
- 88. Price MO, Lisek M, Feng MT, Price FW Jr. Effect of donor and recipient diabetes status on Descemet membrane endothelial keratoplasty adherence and survival. Cornea . 2017; 36(10): 1184–1188. [DOI] [PubMed] [Google Scholar]
- 89. Janson BJ, Terveen DC, Benage MJ, et al.. DMEK outcomes using nondiabetic grafts for recipients with diabetes mellitus. Am J Ophthalmol Case Rep . 2019; 15: 100512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liaboe CA, Aldrich BT, Carter PC, et al.. Assessing the impact of diabetes mellitus on donor corneal endothelial cell density. Cornea . 2017; 36(5): 561–566. [DOI] [PubMed] [Google Scholar]
- 91. Terry MA, Aldave AJ, Szczotka-Flynn LB, et al.. Donor, recipient, and operative factors associated with graft success in the Cornea Preservation Time Study. Ophthalmology . 2018; 125(11): 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lass JH, Benetz BA, Patel SV, et al.. Donor, recipient, and operative factors associated with increased endothelial cell loss in the Cornea Preservation Time Study. JAMA Ophthalmol . 2019; 137(2): 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schwarz C, Aldrich BT, Burckart KA, et al.. Descemet membrane adhesion strength is greater in diabetics with advanced disease compared to healthy donor corneas. Exp Eye Res . 2016; 153: 152–158. [DOI] [PubMed] [Google Scholar]
- 94. Bassnett S, Shi Y, Vrensen GF.. Biological glass: structural determinants of eye lens transparency. Philos Trans R Soc Lond B Biol Sci . 2011; 366(1568): 1250–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bassnett S. Mitochondrial dynamics in differentiating fiber cells of the mammalian lens. Curr Eye Res . 1992; 11(12): 1227–1232. [DOI] [PubMed] [Google Scholar]
- 96. Pendergrass W, Penn P, Possin D, Wolf N.. Accumulation of DNA, nuclear and mitochondrial debris, and ROS at sites of age-related cortical cataract in mice. Invest Ophthalmol Vis Sci . 2005; 46(12): 4661–4670. [DOI] [PubMed] [Google Scholar]
- 97. Brennan LA, McGreal-Estrada R, Logan CM, Cvekl A, Menko AS, Kantorow M.. BNIP3L/NIX is required for elimination of mitochondria, endoplasmic reticulum and Golgi apparatus during eye lens organelle-free zone formation. Exp Eye Res . 2018; 174: 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gilliland KO, Freel CD, Johnsen S, Craig Fowler W, Costello MJ. Distribution, spherical structure and predicted Mie scattering of multilamellar bodies in human age-related nuclear cataracts. Exp Eye Res . 2004; 79(4): 563–576. [DOI] [PubMed] [Google Scholar]
- 99. Kim KY, Perkins GA, Shim MS, et al.. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis . 2015; 6: e1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lin WJ, Kuang HY.. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy . 2014; 10(10): 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lee S, Van Bergen NJ, Kong GY, et al.. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp Eye Res . 2011; 93(2): 204–212. [DOI] [PubMed] [Google Scholar]
- 102. Khawaja AP, Cooke Bailey JN, Kang JH, et al.. Assessing the association of mitochondrial genetic variation with primary open-angle glaucoma using gene-set analyses. Invest Ophthalmol Vis Sci . 2016; 57(11): 5046–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kapetanakis VV, Chan MP, Foster PJ, Cook DG, Owen CG, Rudnicka AR.. Global variations and time trends in the prevalence of primary open angle glaucoma (POAG): a systematic review and meta-analysis. Br J Ophthalmol . 2016; 100(1): 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu Y, Allingham RR.. Major review: molecular genetics of primary open-angle glaucoma. Exp Eye Res . 2017; 160: 62–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu H, Mercieca K, Prokosch V.. Mitochondrial markers in aging and primary open-angle glaucoma. J Glaucoma . 2020; 29(4): 295–303. [DOI] [PubMed] [Google Scholar]
- 106. Abu-Amero KK, Morales J, Bosley TM.. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci . 2006; 47(6): 2533–2541. [DOI] [PubMed] [Google Scholar]
- 107. Kong GY, Van Bergen NJ, Trounce IA, Crowston JG.. Mitochondrial dysfunction and glaucoma. J Glaucoma . 2009; 18(2): 93–100. [DOI] [PubMed] [Google Scholar]
- 108. Garcia-Medina JJ, Garcia-Medina M, Garrido-Fernandez P, et al.. A two-year follow-up of oral antioxidant supplementation in primary open-angle glaucoma: an open-label, randomized, controlled trial. Acta Ophthalmol . 2015; 93(6): 546–554. [DOI] [PubMed] [Google Scholar]
- 109. Shen C, Chen L, Jiang L, Lai TY.. Neuroprotective effect of epigallocatechin-3-gallate in a mouse model of chronic glaucoma. Neurosci Lett . 2015; 600: 132–136. [DOI] [PubMed] [Google Scholar]
- 110. Kimura A, Namekata K, Guo X, Noro T, Harada C, Harada T.. Targeting oxidative stress for treatment of glaucoma and optic neuritis. Oxid Med Cell Longev . 2017; 2017: 2817252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hass DT, Barnstable CJ.. Mitochondrial uncoupling protein 2 knock-out promotes mitophagy to decrease retinal ganglion cell death in a mouse model of glaucoma. J Neurosci . 2019; 39(18): 3582–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dai Y, Hu X, Sun X.. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis . 2018; 9(2): 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hu X, Dai Y, Zhang R, Shang K, Sun X.. Overexpression of optic atrophy type 1 protects retinal ganglion cells and upregulates Parkin expression in experimental glaucoma. Front Mol Neurosci . 2018; 11: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rosignol I, Villarejo-Zori B, Teresak P, et al.. The mito-QC reporter for quantitative mitophagy assessment in primary retinal ganglion cells and experimental glaucoma models. Int J Mol Sci . 2020; 21(5): 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ito YA, Di Polo A.. Mitochondrial dynamics, transport, and quality control: a bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion . 2017; 36: 186–192. [DOI] [PubMed] [Google Scholar]
- 116. Rezaie T, Child A, Hitchings R, et al.. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science . 2002; 295(5557): 1077–1079. [DOI] [PubMed] [Google Scholar]
- 117. Wong YC, Holzbaur EL.. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA . 2014; 111(42): E4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Weil R, Laplantine E, Curic S, Genin P.. Role of optineurin in the mitochondrial dysfunction: potential implications in neurodegenerative diseases and cancer. Front Immunol . 2018; 9: 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ovodenko B, Rostagno A, Neubert TA, et al.. Proteomic analysis of exfoliation deposits. Invest Ophthalmol Vis Sci . 2007; 48(4): 1447–1457. [DOI] [PubMed] [Google Scholar]
- 120. Zenkel M. Extracellular matrix regulation and dysregulation in exfoliation syndrome. J Glaucoma . 2018; 27(suppl 1): S24–S28. [DOI] [PubMed] [Google Scholar]
- 121. Zenkel M, Schlotzer-Schrehardt U.. Expression and regulation of LOXL1 and elastin-related genes in eyes with exfoliation syndrome. J Glaucoma . 2014; 23(8 Suppl 1): S48–S50. [DOI] [PubMed] [Google Scholar]
- 122. Aboobakar IF, Allingham RR.. Genetics of exfoliation syndrome and glaucoma. Int Ophthalmol Clin . 2014; 54(4): 43–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. John SW, Harder JM, Fingert JH, Anderson MG.. Animal models of exfoliation syndrome, now and future. J Glaucoma . 2014; 23(8, suppl 1): S68–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bernstein AM, Ritch R, Wolosin JM.. Exfoliation syndrome: a disease of autophagy and LOXL1 proteopathy. J Glaucoma . 2018; 27(suppl 1): S44–S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chen Y, Sawada O, Kohno H, et al.. Autophagy protects the retina from light-induced degeneration. J Biol Chem . 2013; 288(11): 7506–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. McWilliams TG, Prescott AR, Montava-Garriga L, et al.. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab . 2018; 27(2): 439–449.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. McWilliams TG, Prescott AR, Villarejo-Zori B, Ball G, Boya P, Ganley IG.. A comparative map of macroautophagy and mitophagy in the vertebrate eye. Autophagy . 2019; 15(7): 1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Xu HZ, Song Z, Fu S, Zhu M, Le YZ.. RPE barrier breakdown in diabetic retinopathy: seeing is believing. J Ocul Biol Dis Infor . 2011; 4(1-2): 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rodriguez ML, Perez S, Mena-Molla S, Desco MC, Ortega AL.. Oxidative stress and microvascular alterations in diabetic retinopathy: future therapies. Oxid Med Cell Longev . 2019; 2019: 4940825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Singh LP. Thioredoxin interacting protein (TXNIP) and pathogenesis of diabetic retinopathy. J Clin Exp Ophthalmol . 2013; 4, doi: 10.4172/2155-9570.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM.. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res . 1996; 62(2): 155–169. [DOI] [PubMed] [Google Scholar]
- 132. Devi TS, Somayajulu M, Kowluru RA, Singh LP.. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis . 2017; 8(5): e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Devi TS, Lee I, Huttemann M, Kumar A, Nantwi KD, Singh LP.. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Müller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res . 2012; 2012: 438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lin TK, Lin HY, Chen SD, et al.. The creation of cybrids harboring mitochondrial haplogroups in the Taiwanese population of ethnic Chinese background: an extensive in vitro tool for the study of mitochondrial genomic variations. Oxid Med Cell Longev . 2012; 2012: 824275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Weng SW, Kuo HM, Chuang JH, et al.. Study of insulin resistance in cybrid cells harboring diabetes-susceptible and diabetes-protective mitochondrial haplogroups. Mitochondrion . 2013; 13(6): 888–897. [DOI] [PubMed] [Google Scholar]
- 136. Kuo HM, Weng SW, Chang AY, et al.. Altered mitochondrial dynamics and response to insulin in cybrid cells harboring a diabetes-susceptible mitochondrial DNA haplogroup. Free Radic Biol Med . 2016; 96: 116–129. [DOI] [PubMed] [Google Scholar]
- 137. Zhang Y, Xi X, Mei Y, et al.. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. Biomed Pharmacother . 2019; 111: 1315–1325. [DOI] [PubMed] [Google Scholar]
- 138. Zhou P, Xie W, Meng X, et al.. Notoginsenoside R1 ameliorates diabetic retinopathy through PINK1-dependent activation of mitophagy. Cells . 2019; 8(3): 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhang SY, Li BY, Li XL, et al.. Effects of phlorizin on diabetic retinopathy according to isobaric tags for relative and absolute quantification-based proteomics in db/db mice. Mol Vis . 2013; 19: 812–821. [PMC free article] [PubMed] [Google Scholar]
- 140. Zhang HS, Wang SQ.. Notoginsenoside R1 inhibits TNF-alpha-induced fibronectin production in smooth muscle cells via the ROS/ERK pathway. Free Radic Biol Med . 2006; 40(9): 1664–1674. [DOI] [PubMed] [Google Scholar]
- 141. Hombrebueno JR, Cairns L, Dutton LR, et al.. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight . 2019; 4(23): e129760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mullins RF, Grassi MA, Skeie JM.. Glycoconjugates of choroidal neovascular membranes in age-related macular degeneration. Mol Vis . 2005; 11: 509–517. [PubMed] [Google Scholar]
- 143. Skeie JM, Mullins RF.. Elastin-mediated choroidal endothelial cell migration: possible role in age-related macular degeneration. Invest Ophthalmol Vis Sci . 2008; 49(12): 5574–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF.. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci . 2010; 51(10): 5336–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Brown EE, Lewin AS, Ash JD.. Mitochondria: potential targets for protection in age-related macular degeneration. Adv Exp Med Biol . 2018; 1074: 11–17. [DOI] [PubMed] [Google Scholar]
- 146. Kaarniranta K, Uusitalo H, Blasiak J, et al.. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog Retin Eye Res . 2020; 79: 100858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hyttinen JMT, Viiri J, Kaarniranta K, Blasiak J.. Mitochondrial quality control in AMD: does mitophagy play a pivotal role? Cell Mol Life Sci . 2018; 75(16): 2991–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang M, Jiang N, Chu Y, et al.. Dysregulated metabolic pathways in age-related macular degeneration. Sci Rep . 2020; 10(1): 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wei Q, Hu W, Lou Q, Yu J.. NAD+ inhibits the metabolic reprogramming of RPE cells in early AMD by upregulating mitophagy. Discov Med . 2019; 27(149): 189–196. [PubMed] [Google Scholar]
- 150. Zhu Y, Zhao KK, Tong Y, et al.. Exogenous NAD(+) decreases oxidative stress and protects H2O2-treated RPE cells against necrotic death through the up-regulation of autophagy. Sci Rep . 2016; 6: 26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Stenirri S, Santambrogio P, Setaccioli M, et al.. Study of FTMT and ABCA4 genes in a patient affected by age-related macular degeneration: identification and analysis of new mutations. Clin Chem Lab Med . 2012; 50(6): 1021–1029. [DOI] [PubMed] [Google Scholar]
- 152. Allen GF, Toth R, James J, Ganley IG.. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep . 2013; 14(12): 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wang YQ, Chang SY, Wu Q, et al.. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci . 2016; 8: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Dixon SJ, Lemberg KM, Lamprecht MR, et al.. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012; 149(5): 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chan CM, Huang DY, Sekar P, Hsu SH, Lin WW.. Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J Biomed Sci . 2019; 26(1): 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Hytti M, Korhonen E, Hyttinen JMT, et al.. Antimycin A-induced mitochondrial damage causes human RPE cell death despite activation of autophagy. Oxid Med Cell Longev . 2019; 2019: 1583656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Felszeghy S, Viiri J, Paterno JJ, et al.. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol . 2019; 20: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sparrow JR, Dowling JE, Bok D.. Understanding RPE lipofuscin. Invest Ophthalmol Vis Sci . 2013; 54(13): 8325–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Al-Enezi M, Al-Saleh H, Nasser M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr J Ophthalmol . 2008; 15(2): 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des . 2014; 20(35): 5507–5509. [DOI] [PubMed] [Google Scholar]
- 161. Graeber MB, Muller U.. Recent developments in the molecular genetics of mitochondrial disorders. J Neurol Sci . 1998; 153(2): 251–263. [DOI] [PubMed] [Google Scholar]
- 162. Tsang SH, Aycinena ARP, Sharma T.. Mitochondrial disorder: Kearns-Sayre syndrome. Adv Exp Med Biol . 2018; 1085: 161–162. [DOI] [PubMed] [Google Scholar]
- 163. Alemi M, Prigione A, Wong A, et al.. Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic Biol Med . 2007; 42(1): 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kisilevsky E, Freund P, Margolin E.. Mitochondrial disorders and the eye. Surv Ophthalmol . 2020; 65(3): 294–311. [DOI] [PubMed] [Google Scholar]
- 165. Hamacher-Brady A, Brady NR.. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci . 2016; 73(4): 775–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. De la Mata M, Garrido-Maraver J, Cotan D, et al.. Recovery of MERRF fibroblasts and cybrids pathophysiology by coenzyme Q10. Neurotherapeutics . 2012; 9(2): 446–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Villanueva-Paz M, Povea-Cabello S, Villalon-Garcia I, et al.. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim Biophys Acta Mol Basis Dis . 2020; 1866(6): 165726. [DOI] [PubMed] [Google Scholar]
- 168. Garrido-Maraver J, Paz MV, Cordero MD, et al.. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim Biophys Acta . 2015; 1852(11): 2535–2553. [DOI] [PubMed] [Google Scholar]
- 169. James AM, Wei YH, Pang CY, Murphy MP.. Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem J . 1996; 318(pt 2): 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Walczak J, Partyka M, Duszynski J, Szczepanowska J.. Implications of mitochondrial network organization in mitochondrial stress signalling in NARP cybrid and Rho0 cells. Sci Rep . 2017; 7(1): 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW.. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta . 2013; 1833(3): 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Jankauskaite E, Ambroziak AM, Hajieva P, et al.. Testosterone increases apoptotic cell death and decreases mitophagy in Leber's hereditary optic neuropathy cells. J Appl Genet . 2020; 61(2): 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Dombi E, Diot A, Morten K, et al.. The m.13051G>A mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology . 2016; 86(20): 1921–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Zhang J, Ji Y, Lu Y, et al.. Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T>C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum Mol Genet . 2018; 27(11): 1999–2011. [DOI] [PubMed] [Google Scholar]
- 175. Sharma LK, Tiwari M, Rai NK, Bai Y.. Mitophagy activation repairs Leber's hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum Mol Genet . 2019; 28(3): 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kodron A, Hajieva P, Kulicka A, Paterczyk B, Jankauskaite E, Bartnik E.. Analysis of BNIP3 and BNIP3L/Nix expression in cybrid cell lines harboring two LHON-associated mutations. Acta Biochim Pol . 2019; 66(4): 427–435. [DOI] [PubMed] [Google Scholar]
- 177. Phasukkijwatana N, Kunhapan B, Stankovich J, et al.. Genome-wide linkage scan and association study of PARL to the expression of LHON families in Thailand. Hum Genet . 2010; 128(1): 39–49. [DOI] [PubMed] [Google Scholar]
- 178. Liu X, Kim CN, Yang J, Jemmerson R, Wang X.. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell . 1996; 86(1): 147–157. [DOI] [PubMed] [Google Scholar]
- 179. Indrieri A, Carrella S, Romano A, et al.. miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol Med . 2019; 11(5): e8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Carelli V, Musumeci O, Caporali L, et al.. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol . 2015; 78(1): 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Liao C, Ashley N, Diot A, et al.. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology . 2017; 88(2): 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Moulis MF, Millet AM, Daloyau M, et al.. OPA1 haploinsufficiency induces a BNIP3-dependent decrease in mitophagy in neurons: relevance to dominant optic atrophy. J Neurochem . 2017; 140(3): 485–494. [DOI] [PubMed] [Google Scholar]