

Abstract
A diet high in fat and sucrose (HFHS), the so-called Western diet promotes metabolic syndrome, a significant comorbidity for individuals with spinal cord injury (SCI). Here we demonstrate that the spinal cord of mice consuming HFHS expresses reduced insulin-like growth factor 1 (IGF-1) and its receptor and shows impaired tricarboxylic acid cycle function, reductions in PLP and increases in astrogliosis, all prior to SCI. After SCI, Western diet impaired sensorimotor and bladder recovery, increased microgliosis, exacerbated oligodendrocyte loss and reduced axon sprouting. Direct and indirect neural injury mechanisms are suggested since HFHS culture conditions drove parallel injury responses directly and indirectly after culture with conditioned media from HFHS-treated astrocytes. In each case, injury mechanisms included reductions in IGF-1R, SIRT1 and PGC-1α and were prevented by metformin. Results highlight the potential for a Western diet to evoke signs of neural insulin resistance and injury and metformin as a strategy to improve mechanisms of neural neuroprotection and repair.
Keywords: Astrogliosis, Myelin, Insulin, Spinal cord injury, Metformin, Western diet
1. Introduction
Traumatic spinal cord injury (SCI) is a devastating often paralyzing condition with an annual incidence of 15–40 cases per million in the USA and an estimated annual cost of more than $7 billion (National Spinal Cord Injury Statistical Center, 2017). That these injuries impart a tremendous emotional impact on the individual and their families is an understatement. After SCI, cellular and signaling perturbations cause immediate loss of function and result in a secondary cascade of pathophysiological events that worsen the outcome of the initial injury. Post-trauma events include inflammation, astrogliosis and neuron and axonal degeneration, in addition to apoptosis of myelin producing oligodendrocytes (Alizadeh et al., 2019). Interventions to improve outcomes in experimental models of SCI and in individuals with SCI span delivery of neural stem cells, growth factors and rehabilitation, including neuromodulation to promote plasticity and motor relearning (Katoh et al., 2019; Muir et al., 2019; Terson de Paleville et al., 2019; Venkatesh et al., 2019). Each of these therapeutic strategies would be enhanced by lessening secondary injury and improving the microenvironment available for repair.
There is growing recognition that consumption of a diet high in saturated fat and sucrose, the so-called Western diet, contributes to the development of metabolic syndrome. The presence of metabolic syndrome is characterized as having at least 3 of the following: obesity, insulin resistance, hypertension, elevated triglycerides, low HDL cholesterol and a pro-inflammatory state. Metabolic syndrome is a well-recognized risk factor for the development of cardiovascular disease and type II diabetes and currently affects more than one quarter of the World’s population (Steinberger et al., 2003; Stubbs and Lee, 2004). Systemic metabolic dysfunction is more recently recognized as a risk factor for the development of Alzheimer’s Disease (Emmerzaal et al., 2015; Haan, 2006) and exacerbates functional decline after traumatic brain injury (TBI) (Lee et al., 2016; Mowery et al., 2009). Obesity is also associated with a significant increase in the risk of multiple sclerosis (MS) (Hedstrom et al., 2014) and insulin resistance is linked to increased expanded disability status scale scores (Oliveira et al., 2014).
The development of metabolic syndrome is of particular significance to individuals with SCI (Nash et al., 2016; Yahiro et al., 2019) and to the potential for neural repair. As many as 55% of individuals with SCI also develop systemic metabolic dysfunction due in part to changes in physical activity and muscle/fat mass ratios, as well as dietary considerations (Gater Jr et al., 2019; Gilbert et al., 2014; Groah et al., 2011; Moore et al., 2018). Each of the cardinal features of metabolic syndrome, including insulin resistance; hypertension; and a systemic pro-inflammatory/pro-thrombotic, fibrinolytic state, could negatively impact the response of the central nervous system (CNS) to injury and its capacity for repair. A better understanding of the pathophysiologic effects of systemic metabolic dysfunction on injury and repair mechanisms in the intact and injured CNS is therefore of critical importance to considerations of recovery and to the development of best practices for identification and implementation of treatment strategies.
Experimental models to study the impact of metabolic dysfunction on the physiology and pathophysiology of major organ systems, including the CNS, include both genetic and Western diet-induced obesity models. With regard to the CNS, experimental obesity exacerbates neurodegeneration in rodent models of Alzheimer’s (Graham et al., 2016; Martino Adami et al., 2017; Theriault et al., 2016). Experimental obesity also exacerbates neuroinflammation in the experimental autoimmune encephalomyelitis model of MS (Ji et al., 2019; Timmermans et al., 2014) and after experimental TBI (Hoane et al., 2011; Karelina et al., 2016; Sherman et al., 2016; Tyagi et al., 2013). There is much more limited information regarding the impact of obesity in models of SCI; however, transcriptional analysis points to perturbed spinal cord metabolism (Spann et al., 2017), and recent studies also suggest reductions in functional recovery after injury (Harris et al., 2019). Given the high risk for the development of metabolic syndrome after SCI and its role as a significant co-morbidity, we investigated the impact of Western diet-induced obesity on metabolic function and energy homeostasis in the adult mouse spinal cord and its response to traumatic injury, including the capacity for repair. We complemented these studies with in vitro approaches to identify the potential cellular and molecular mechanisms involved.
Through the application of in vivo and ex vivo approaches, the current studies demonstrate that a Western diet not only results in systemic insulin resistance, but also impairs key intermediates in the insulin signaling pathway across spinal cord neuron and glial compartments. These changes were accompanied by a significant reduction in spinal cord tricarboxylic acid (TCA) cycle function, astrogliosis and microgliosis, and reductions in PLP in the intact spinal cord. After SCI, Western diet consumption not only exacerbated myelin loss and microgliosis, and blunted signs of neural repair, but was also associated with overall reductions in recovery of sensorimotor and bladder function. Neural culture under HFHS conditions suggests pathogenic outcomes may occur through direct mechanisms, in addition to indirect-astrocyte-related events driving reduced IGF-1R signaling and PGC-1α expression. Of potential therapeutic significance, HFHS-mediated neural injury responses in vitro were largely prevented by the insulin-sensitizing agent metformin. Together, these studies highlight previously unrecognized Western diet-associated metabolic mechanisms positioned to exacerbate CNS injury and impair recovery, and additionally point to insulin-sensitizing strategies as a new target for therapeutic innervation.
2. Material and methods
2.1. Animals and diets
The impact of consuming a high-fat high-sucrose (HFHS) diet on metabolic parameters in the adult spinal cord and on recovery after traumatic injury was investigated using 10 wk. old male or female C57BL6J mice obtained from Jackson Laboratories (Catalog #000664, Bar Harbor, ME, USA). All mice were housed under environmentally-controlled conditions (22–24 °C with a 12 h light/dark cycle). For experiments examining the impact of HFHS on the intact spinal cord male mice were randomized and provided unlimited access to either a regular diet (RD) or a HFHS diet for 12 wk. (n = 8 per group). For experiments involving SCI, two groups of 15 female mice were fed either a RD or a HFHS diet ad libitum for 7 wk. Five mice in each group were then randomized to a non-injury group and continued on the same diets for the next 4 wk. The remaining 10 mice in each group received lateral compression SCI after which they continued on the same RD or HFHS diet for an additional 14 or 30 d post injury (dpi) prior to perfusion for histological analysis.
The RD (D12450K) and the HFHS (D15010302) diets were obtained from Research Diets, Inc. (New Brunswick, NJ, USA). The RD was comprised of 20% kcal from protein, 10% kcal from fat, and 70% kcal from carbohydrates and contained 10% cholesterol and 0% sucrose. In comparison, the HFHS diet was comprised of 20% kcal from protein, 60% kcal from fat, and 20% kcal from carbohydrates and contained 60% cholesterol and 19% sucrose (see Table S1 for detailed diet composition). Mice were monitored weekly for body weight and food intake. All molecular and histological outcome measures were assessed without knowledge of the dietary intervention.
2.2. Experimental compression SCI
The impact of HFHS consumption on recovery after SCI was examined using an experimental model of compression SCI (Plemel et al., 2008). Prior to spinal cord compression injury, mice were deeply anesthetized with xylazine (10 mg/kg; Akorn, Inc. Lake Forest, IL, USA) and ketamine (100 mg/kg; Fort Dodge, IA, USA). A laminectomy was performed leaving the dura intact and Dumont #2 forceps with a 0.25 mm spacer was applied to laterally compress the spinal cord at T8–9 for 14 s. Immediately after surgery, 1 ml of saline was administered intraperitoneally to replace lost blood volume. Baytril (10 mg/kg; Bayer Healthcare, LLC; Animal Health Division; Shawnee Mission, Kanas, USA) was delivered once immediately post-surgically. Buprenex (0.05 mg/kg; Reckitt Benckiser Healthcare, England) were delivered every 12 h for 3 d post-operatively to minimize infection risk and pain, respectively. Bladders of injured mice were manually vacated twice daily until recovery of spontaneous bladder release. All experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee and completed in accordance with NIH Guidelines.
2.3. Glucose tolerance test
Systemic insulin resistance was documented in mice consuming a HFHS diet at 7 or 12 wk. using standard glucose tolerance testing (GTT). Briefly, prior to the test, mice were fasted for 6 h and blood collected to determine basal glucose levels (0 min). Mice were subsequently injected with a bolus (125 μl) of glucose (1.0–1.5 g/kg; Sigma-Aldrich, St. Louis, MO, USA) and blood was collected at 30, 60, 90 and 120 min thereafter for glucose level measurements. Mean blood glucose values ± SEM (mg/dl) were determined for each group using a glucose meter (Bayer Breeze2; Bayer Ascensia; NJ, USA) and plotted versus time (Schafer et al., 2016).
2.4. Neurobehavioral outcome measures
2.4.1. Open field test
The open field test was used to assess seven categories of locomotor recovery according to the Basso Mouse Scale (BMS) (Basso et al., 2006). Baseline measurements for each mouse were collected prior to SCI (0 dpi), the day after injury (1 dpi) and weekly thereafter until 30 dpi. Mice were videotaped navigating a Plexiglas open field for 3 min. Two observers evaluated each video for ankle movement, plantar placement, stepping, coordination, paw position, trunk stability and tail position, generating a BMS score (0 to 9) and a BMS subscore of 0 to 11 (Radulovic et al., 2016).
2.4.2. Inclined plane test
The inclined plane test was used to evaluate hind limb strength necessary to maintain a horizontal position on an inclined plane, with larger angles associated with better recovery. The last angle at which each mouse could maintain a stance for 5 s before turning or falling was recorded and averaged across 3 attempts (Joshi and Fehlings, 2002). All mice received training in the inclined plane test prior to surgery, and testing occurred weekly from 7 to 30 dpi.
2.4.3. Monitoring bladder fullness
Following surgery, bladders were manually expressed twice daily via the application of pressure. The amount of pressure needed and amount of urine expressed were determined to rank bladder function on a scale from 0 to 3 (3 = dysfunctional, large amount of urine expressed by medium to high pressure; 2 = partially functional, medium amount of urine expressed after medium pressure; 1 = mostly functional, small amount of urine expressed after slight pressure; and 0 = fully functional, no pressure needed for urine expression). This procedure was carried out until mice recovered full bladder release (Plemel et al., 2008).
2.5. Neuropathological outcomes
2.5.1. Immunohistochemistry
Histopathological outcomes were quantified at 14 or 30 dpi and compared to uninjured controls (n = 5 female mice per time point in each group). Mice were deeply anesthetized (Pentobarbital; 60 mg/ml; Abbott Laboratories; Chicago, IL) and transcardially perfused with 4% paraformaldehyde (pH 7.2). Spinal cords were dissected and cut into 2 mm transverse segments with the injury epicenter and two 2 mm blocks above and two below embedded in a single paraffin block. 6 μm horizontal sections taken from these blocks were deparaffinized and immunostained for IGF-1 receptor (IGF-1R), myelin proteins (MBP, PLP), myelinating cells (Olig2, CC-1), astrocytes (GFAP, ALDH1L1), microglia (Iba1), neurons (NeuN), neurofilament protein (NFH), serotonin (5-HT), synaptic elements (Synapsin 1) and growth cones (GAP43) using the primary antibodies detailed in Table 1.
Table 1.
Antibodies used for Immunohistochemistry.
| Primary antibody | Source | Company (catalog #) | RRID number | Dilution |
|---|---|---|---|---|
| 5-HT | Rabbit | Sigma-Aldrich (S5545) | AB_572263 | 1:4000 |
| ALDH1L1 | Rabbit | Abcam (ab87117) | AB_10712968 | 1:1000 |
| CC-1 | Mouse | Abcam (ab16794) | AB_443473 | 1:150 |
| GAP43 | Rabbit | Abcam (ab75810) | AB_1310252 | 1:200 |
| GFAP | Rabbit | Dako (Z0334) | AB_10013382 | 1:5000 |
| Iba-1 | Rabbit | Wako (019–19,741) | AB_839504 | 1:3000 |
| Insulin receptor-1 (IGF-1R) | Rabbit | Abcam (ab39675) | AB_731541 | 1:100 |
| MBP | Rat | Millipore (MAB386) | AB_94975 | 1:750 |
| NeuN | Mouse | Millipore (MAB337) | AB_2313673 | 1:2000 |
| Neurofilament 200 (NFH) | Rabbit | Sigma-Aldrich (N4142) | AB_477272 | 1:1300 |
| Olig2 | Rabbit | Millipore (AB9610) | AB_10141047 | 1:500 |
| PLP | Rabbit | Abcam (ab28486) | AB_776593 | 1:750 |
Immunostaining for a given antigen across each segmental level was performed simultaneously for all individual animals in a group. Sections were deparaffinized, and processed for antigen retrieval (sodium citrate, 10 mM, pH 6.0) and pre-incubated in 20% normal goat serum (NGS) with 0.25% Triton x-100 in PBS for 20 min prior to immunostaining. Primary antibodies were applied overnight in 10% NGS at 4 °C. For immunoperoxidase staining, subsequent incubation with a species-appropriate biotinylated secondary antibody (donkey anti-mouse (Jackson ImmunoResearch, West Grove, PA, 715–066-151, 1:200), donkey anti-rabbit (Jackson, 711–065-152, 1:200), or goat anti-rat (Jackson, 112–066-072, 1:200)) occurred at RT in 1% NGS followed by immunoperoxidase staining (Vector Laboratories; Burlingame, CA, PK-6100) to yield a brown reaction product after incubation with 3,3′-diamino-benzidine (DAB, Sigma D5637). Alternatively, sections were stained with hematoxylin and eosin (H&E) or by HRP-based avidin-biotin immunochemistry in combination with Eriochrome. In most cases, sections were also counterstained with methyl green (Vector Laboratories; H-3402) to visualize nuclei.
Quantification of antigen staining included 5 sections per animal sampled randomly from across the 2 mm blocks of spinal cord that were collected from the injury epicenter (0) and from the 4 mm of spinal cord immediately above (+4 mm) or below (−4 mm). All images were captured under constant illumination (Olympus BX51 microscope and DP72 camera equipped with CellSens software 1.9; Olympus; Center Valley, PA, USA). Any changes in immunostaining density or cell counts were considered either in the gray or white matter areas of the spinal cord alone or collectively. Depending on the protein being quantified, images were taken at 5× (ALDH1L1, Eriochrome + NFH, GFAP, MBP, NFH, Synapsin-1) to encompass the entire spinal cord, at 20× (CC-1, Iba-1, MBP, NeuN, Olig2, PLP) to encompass the ventral horn gray matter and adjacent lateral white matter funiculus, or 40× (5-HT, GAP43, IGF-1R) to include only the ventral horn gray matter. Images were collected from both the right and left sides of the spinal cord with an average density or count for each individual animal determined. All density measurements were made from digital images using KS-400 image analysis software (Carl Zeiss Vision; Hallbergmoos, Germany) (Scarisbrick et al., 1999). Briefly, a threshold for positive staining was set individually for each stain within the program and this was applied uniformly across all animals. All counts and staining densities were normalized to the area quantified (mm2) with area measurements made using ImageJ (U. S. National Institutes of Health, Bethesda, MD). The number of Olig2+ or CC-1+ oligodendrocytes or NeuN+ ventral horn motoneurons were counted manually from both left and right lateral ventral horn 20× images. Motoneurons were defined by NeuN expression and having a diameter greater than 20 μm. Counts of Olig2+ or CC-1+ positive oligodendrocytes included only those with a methyl green positive nucleus.
For microglial skeletal analysis, images were taken at 100× with oil immersion from the ventral horn gray matter. Images were converted to binary, then skeletonized using ImageJ software. The Analyze Skeleton plugin (http://imagejdocu.tudor.lu/) was applied to all skeletonized images to quantify the microglial number; somal size; number of branches, junctions and end points; and the longest branch length. We summarized the data from the Analyze Skeleton plugin and averaged data from both sides of the ventral horn.
2.5.2. Unbiased stereology for spinal cord volume estimation
Spinal cord sections were stained with Eriochrome cyanine (Sigma-Aldrich) and neurofilament (NF-200) to identify damaged myelin and axons. Stained tissue sections were captured digitally with the Olympus BX51 microscope and DP72 camera equipped with CellSens software 1.9 (Olympus; Center Valley, PA), under constant illumination. Photographs were analyzed by Stereo Investigator software (MBF Bioscience; Williston, VT, USA). The Cavalieri estimator probe was used for stereological analysis for determination of spared white matter volume. The spinal cord sections were viewed with a 5× objective, and contours were drawn and grid spacing was placed at 100 μm with 6 μm section thickness with a resulting coefficient of error (CE) for analysis = 1 ranged from 0.022 to 0.069 for all mice. These values were used to ensure that adequate numbers of sample sites were visited and adequate numbers of markers indicating injured myelin were acquired, consistent with accepted counting rules for stereology (Schmitz and Hof, 2005).
2.6. Cell culture
2.6.1. Primary cortical astrocytes
Primary murine astrocytes were purified from mixed glial cultures obtained from the cortices of postnatal d 1–3 male and female pups. Mixed glial preps were grown in DMEM, containing 2 mM Glutamax, 1 mM sodium pyruvate, 20 mM HEPES, 5 μg/ml insulin, 100 U/ml penicillin and 100 μg/ml streptomycin (P/S), and 10% heat-inactivated fetal bovine serum (Atlanta Biologicals; Lawrenceville, GA, USA). All reagents were purchased from Gibco (Invitrogen, Carlsbad, CA, USA). After 10–12 d in vitro (DIV), mixed glial cultures were shaken overnight (225 RPM) to remove microglia and oligodendrocytes as previously detailed (Burda et al., 2013; Yoon et al., 2017). Astrocytes were tryp-sinized (0.025%) and plated at a density of 450,000 cells/well on poly-L-lysine- (Sigma-Aldrich) coated 6-well plates and grown for 2–3 d in serum-free Neurobasal A media containing 1% N2, 2% B27, 100 U/ml P/S, 1 mM sodium pyruvate, 0.45% glucose, 0.5% Glutamax, 0.1% BSA, and 50 μM β-mercaptoethanol. To model high-fat conditions, cultures were treated with or without palmitate (PA, 100 μM) for 24 h. We next determined if an insulin sensitizing agent – metformin (100 μM) – affected astroglial responses to PA. The potential involvement of an astrocyte-secreted factor in oligodendrocyte and neuron responses to PA was examined by collecting 24 h-treated cell culture supernatants. Supernatants were concentrated with Vivaspin 2, 100 k to 10 kDa molecular weight cutoff sample concentrators (Sigma-Aldrich) and stored frozen as 10× concentrates for downstream use (Liddelow et al., 2017).
2.6.2. Oli-neu cells
Oli-neu cells are a t-neu oncogene transduced primary oligodendrocyte cell line (Huitinga et al., 1995). All morphology, signaling, and myelin gene expression studies using Oli-neu oligodendrocytes were performed in media containing DMEM, 1% N2, 2 mM Glutamax, 1 mM sodium pyruvate, 20 mM HEPES buffer, and 50 μM β-mercaptoethanol (Sigma-Aldrich), and all reagents were purchased from Gibco (Invitrogen). To evaluate the impact of excess saturated fatty acid alone or in conjunction with insulin sensitization on Oli-neu oligodendrocytes, cultures were treated with (or without (control)) 100 μM PA, 100 μM metformin, or a combination of PA and metformin for 24 h prior to RNA isolation.
2.6.3. Primary oligodendrocyte cultures
Primary oligodendrocyte cells were isolated from mixed glia cultures by overnight shaking and panning on plastic as previously described (Burda et al., 2013). OPCs were plated 90,000 cells/well on poly-L-lysine- (Sigma-Aldrich) coated 24-well plates and grown for 24 h in serum-free oligodendrocyte differentiation medium containing Neurobasal A medium, 1% N2, 2% B27, P/S, 1 mM sodium pyruvate, 0.45% glucose, 0.1% BSA, 50 μM β-mercaptoethanol, and 40 ng/ml T3 before treatment with (or without – control) PA (100 μM) and/or metformin (100 μM). After 72 h, cells were harvested for RNA isolation.
2.6.4. Primary cortical neurons
Primary cortical neurons were isolated from embryonic day 15 C57BL6/J mice (Radulovic et al., 2013) and plated at a density of 250,000 cells per well on 6-well plates coated with poly-D-lysine (50 μg/ml) and laminin (10 μg/ml). Cortical neurons were grown in defined media consisting of Neurobasal A media supplemented with 2% B27 supplement, 500 μM L-glutamine, 100 U/ml penicillin and 100 μg/ ml streptomycin from Gibco (Invitrogen), 2/3 of which was changed every 2–3 d. Neurons were allowed to mature for 96 h in vitro and then left untreated or treated with 100 μM PA and/or 100 μM metformin for an additional 72 h before being harvested for RNA isolation.
2.6.5. Primary oligodendrocytes cultured with astrocyte-conditioned media
To determine the potential for indirect (astrocyte-mediated) effects of excess fatty acid (PA) on myelinating cells in vitro, primary murine oligodendrocyte cultures derived from cortical mixed glial preparations were plated at 90,000 cells/well on PLL-coated 24-well plates and cultured for 2 d in serum-free oligodendrocyte differentiation medium (see above) (Yoon et al., 2015). Media was then replaced with astrocyte -conditioned medium (ACM) diluted to 1× concentration in serum-free oligodendrocyte differentiation medium and cells cultured for an additional 24 h prior to RNA isolation. In all cases, RNA was isolated from cultured cells using RNA STAT-60 (Tel-Test; Friendswood, TX, USA), as previously described (Yoon et al., 2017).
2.6.6. Organotypic spinal cord slice culture
To determine the impact of excess fatty acid on spinal cord slices, spinal cords from C57BL/6 P7 pups were dissected at 4 °C media consisting of Advanced DMEM-F12, 1.5% glucose, and 1% P/S, then transferred to HBSS with 1.5% glucose on ice. Spinal cords were embedded in 7% Agar (16520–050, Invitrogen) and cut serially at 350 μm using a tissue chopper (McIlwain tissue chopper; the Mickle Laboratory Engineering CO. LTD.; Goose Green, United Kingdom). Six slices encompassing thoracic to lumbar segments were cultured on a 0.4 μm insert (PIHPO3050, Millipore) in 6-well plates. Spinal cord slices were cultured in Neurobasal A medium, 50% horse serum, 1.5% glucose, 1% glutamax, 2.5% HEPES and 1% P/S and incubated at 36 °C. After 7 DIV, slice culture media was changed to serum-free Neurobasal A media containing 1% N2 supplement, 2% B27 supplement, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% glucose, and 1% glutamax, with (or without (control)) PA (100 μM) and/or 100 μM metformin treatment for 72 h. All reagents were purchased from Gibco (Invitrogen). Organotypic slices were fixed with 4% PFA for 24 h and stained by immunofluorescence for NFH, GFAP, Olig2 and MBP. Species-appropriate fluorescent secondary antibodies included: goat anti-rabbit (Alexa Fluor 488, 111–545-047), goat anti-mouse (Cy3, 115–166-062), and goat anti-rat (Alexa Fluor 647, Cy5, 112–606-143) (Jackson ImmunoResearch Laboratories). See Table 2 for antibody information. Slices were finally incubated with DAPI (D1306; Thermo Fisher Scientific; Waltham, MA, USA) prior to being slide-mounted with Fluoromount-G® mounting (0100–01; SouthernBiotech; Birmingham, AL, USA) and imaged using a confocal microscope (LSM 780; Carl Zeiss; Oberkochen, Germany). ImageJ was used for automated quantification of staining intensity per area.
Table 2.
Antibodies used for Immunofluorescence.
| Primary antibody | Source | Company (catalog #) | RRID number | Dilution |
|---|---|---|---|---|
| DAPI | Thermo Fisher Scientific (D1306) |
AB_2629482 | 1:5000 | |
| GFAP | Rabbit | Dako (Z0334) | AB_10013382 | 1:5000 |
| MBP | Rat | Millipore (MAB386) | AB_94975 | 1:750 |
| NF | Rabbit | Sigma-Aldrich (N4142) | AB_477272 | 1:1300 |
| Olig2 | Mouse | Millipore (MABN50) | AB_10807410 | 1:500 |
2.7. RNA transcription
Real-time quantitative reverse-transcription PCR (qRT-PCR) was used to determine the impact of HFHS consumption on systemic insulin resistance by evaluating expression of insulin signaling partners in the spinal cord (IGF-1, IGF-2, IGF-1R, insulin and insulin receptor (InsR)). qRT-PCR was also used to quantify the impact of excess fatty acid or astrocyte-conditioned media on purified astrocyte, oligodendrocyte or neuron cultures in vitro. RNA was isolated from neural cells that were either untreated or stimulated with 100 μM PA and/or 100 μM metformin, for 24 or 72 h using RNA STAT-60 (Tel-Test; Friendswood, TX, USA). The relative amount of RNA in each case was determined in 100 ng of RNA using an iCycler iQ5 system (Bio-Rad Laboratories; Hercules, CA, USA) and normalized to the constitutively expressed gene Rn18S. Mean expression levels under each condition were expressed as a percent of that seen under untreated conditions. The probes (Thermo Fisher Scientific; Waltham, MA, USA and Integrated DNA Technologies; Coralville, IA, USA) and primers (Integrated DNA Technologies; Coralville, IA, USA) utilized are described in Table 3.
Table 3.
Primers used for quantitative real-time PCR.
| Gene | Accession number | Probe and primer information |
|---|---|---|
| ALDH1L1 | NM_027406.1 | Applied Biosystems, Assay ID: Mm03048957_m1 |
| GAP43 | NM_008083.2 | Applied Biosystems, Assay ID: Mm00500404_m1 |
| GFAP | NM_010277.2 | GCAGATGAAGCCACCCTGG/ GAGGTCTGGCTTGGCCAC |
| Iba-1 | NM_019467.2 | Applied Biosystems, Assay ID: Mm00479862_g1 |
| IGF-1 |
NM_001111274.1, NM_001111275.1, NM_001111276.1, NM_010512.4, NM_184052.3 |
Applied Biosystems, Assay ID: Mm00439560_m1 |
| IGF-1 receptor | NM_010513 | Applied Biosystems, Assay ID: Mm00802831_m1 |
| IGF-2 | NM_001122736.1, NM_001122737.1, NM_010514.3 | Applied Biosystems, Assay ID: Mm00439564_m1 |
| IL-6 | NM_031168.1 | Applied Biosystems, Assay ID: Mm00446190_m1 |
| Insulin | NM_008386.3 | Applied Biosystems, Assay ID: Mm01950294_s1 |
| Insulin receptor | NM_010568.2 | Applied Biosystems, Assay ID: Mm01211875_m1 |
| MBP | NM_001025251 | CCAGTAGTCCATTTCTTCAAGAACAT/GCCGATTTATAGTCGGAAGCTC |
| NFH | NM_010904.3 | CATTGAGATTGCCGCTTACAG/ TTATGTGCGTGGATATGGAGG |
| Olig2 | NM_016967 | IDT, Assay ID: Mm.PT.58.42319010 |
| PGC1 alpha | NM_008904.2 | Applied Biosystems, Assay ID: Mm01208835_m1 |
| PLP | NM_011123.2 | TCTTTGGCGACTACAAGACCAC/CACAAACTTGTCGGGATGTCCTA |
| Rn 18S | NR_003278.3 | Applied Biosystems, Assay ID: Mm03928990_g1 |
| SIRT1 | NM_001159589.1, NM_019812.2 | Applied Biosystems, Assay ID: Mm00490758_m1 |
| Synapsin-1 | NM_001110780.1 | Applied Biosystems, Assay ID: Mm00449772_m1 |
2.8. TCA cycle profiling in the spinal cord
Targeted liquid chromatography with tandem mass spectrometry (LC-MS-MS) was used to quantify TCA cycle metabolites using previously described methods (Dutta et al., 2016). Briefly, spinal cord samples were snap-frozen at −80 °C at the time of harvest. Before use, samples were pulverized in liquid nitrogen and weighed prior to being diluted in 1× PBS to 100 mg/ml. 13C6-phenylalanine (2 μl at 250 ng/ μl) was added as an internal standard and samples were deprotonated with cold acetonitrile: methanol (1:6 ratio) at 4 °C. The supernatant was isolated by centrifugation and divided into two aliquots, prior to drying using a stream of nitrogen gas for final analysis on a Quadruple Time-of-Flight Mass Spectrometer (6550 Q-TOF; Agilent Technologies; Santa Clara, CA, USA) coupled with an Ultra High-Pressure Liquid Chromatograph (1290 Infinity UHPLC; Agilent Technologies). All measurements were performed against 12-point calibration curves that underwent the same derivatization with internal standard. Data alignment, filtering, univariate, multivariate statistical and differential analysis was performed using Mass Profiler Professional (Agilent Technologies, Inc.).
2.9. Statistical analysis
Data are expressed as the mean ± SEM. For tissue staining in experimental spinal cord injury studies, quantification of all counts and staining densities were made considering measurements from 5 tissue sections encompassing the injury epicenter and the two, 2 mm blocks of spinal cord above and below the epicenter for each animal. For statistical analysis, a mean for each animal (including all 5 sections) was made and used to create a group mean (n = 5 per dietary condition, per time point). Alternatively, to investigate any differences in the injury epicenter from those above and below, a mean was made for each animal reflecting the 2 segments above, the epicenter, or the 2 segments below, and these used to create a group mean for each individual level for statistical analysis.
One- or Two-way multifactorial analysis of variance (ANOVA) was carried out using the SigmaStat statistical program version 13.0 (Systat Software; San Jose, CA, USA). To evaluate the impact of diet on sensorimotor outcomes after injury over time, BMS scores and subscores, inclined plane, and bladder function test results were analyzed using a Two-way repeated measures ANOVA with fixed effects and the Newman-Keuls (NK) post-hoc test. Quantitative measurements of neuropathology from non-spinal cord injury controls represent combined mean ± SEM of results obtained from 30 dpi experimental controls. The histological, RNA and cell culture analysis across multiple groups were determined using a One-way ANOVA with the NK post-hoc test. However, in instances where the Shapiro-Wilk failed, multiple groups were determined using a non-parametric ANOVA on RANKS with Dunn’s test. Statistical significance was set at P < .05. Quantification of results in all histological and cell culture experiments was performed without knowledge of the treatment group.
3. Results
3.1. Western diet impairs insulin signaling systemically and in the adult spinal cord
Signs of impaired insulin signaling were observed both systemically and in the spinal cord of mice consuming a diet rich in saturated fat and sucrose (Fig. 1 and Fig. S1). First, consumption of a HFHS diet for 7 (Fig. 1A–C) or 12 wk. (Fig. S1, A–B) promoted expected increases in body weight and fasting glucose levels relative to mice consuming a RD and. In addition, we observed 1.3-fold reductions in the expression of IGF-1 RNA, a key component of the insulin signaling system, in the spinal cord of mice consuming HFHS (Fig. 1D, P < .05, Student’s t-test). The trend towards reductions in IGF-2 and Insulin in the same samples were not statistically significant (Fig. 1E–H). In addition, 1.4-fold reductions in immunoreactivity for IGF-1R in GM were observed in the spinal cord of mice consuming HFHS compared to a RD P < .05, Two-way ANOVA, NK (Fig. 1I–N and Fig. S1, C–D). The loss of IGF-1R protein seen with HFHS consumption prior to SCI closely resembled the loss observed at 14 and 30 after SCI in white matter and gray matter, irrespective of diet. By 30 dpi, IGF-1R levels were higher in gray matter of mice consuming HFHS (P < .01, Two-way ANOVA, NK). These findings suggest that obesity induced by a Western diet not only impairs the insulin signaling system systemically, but also concurrently alters key components of the insulin-IGF-1 signaling system within the parenchyma of the adult spinal cord.
Fig. 1.

Systemic insulin resistance is reflected in the spinal cord of mice consuming a Western diet before and after SCI. (A, C) Mice consuming a HFHS diet have significant increases in body weight compared to those consuming a RD (RD, n = 14 and HFHS, n = 15, female mice). (B) Mice consuming HFHS for 7 wk. also have higher fasting blood glucose and impaired glucose tolerance (RD, n = 5 and HFHS, n = 5, female mice). (A, B Two-way ANOVA, *P < .05, **P < .01, ***P < .001). (D–H) Expression of IGF-1 RNA was reduced in mice consuming HFHS for 12 wk. (RD, n = 4 and HFHS, n = 4 male mice, *P < .05, Student’s t-test). (I–N) Representative images and histograms showing immunoreactivity for IGF-1R was reduced in the spinal cord white matter of mice consuming HFHS at 30 dpi (RD, uninjured n = 5, 14 dpi n = 4, 30 dpi n = 5 and HFHS, uninjured n = 5, 14 dpi n = 5, 30 dpi n = 5 female mice, *P < .05, **P < .01, ***P < .001, Two-way ANOVA, NK). (L) Cartoon shows the 2 mm of the SCI epicenter, and the 4 mm regions of the spinal cord above and below that were quantified. (J–N) Histograms show findings for all regions combined (J and M) and the individual SCI regions, that is +4 (Above), 0 (Epicenter) and −4 (Below) (K and N). (O–U) Reductions in spinal cord IGF-1 and IGF1-R in the intact spinal cord of mice consuming HFHS were accompanied by reductions in TCA cycle intermediates, Oxaloacetate, αKetoglutarate, Aspartate, 2-Hydroxyglutarate, Glutamate and cis-Aconitic Acid (RD, n = 4 and HFHS, n = 4 at 12 wk., male mice, *P < .05, Student’s t-test). Scale bar = 100 μm (I). RD = regular diet, HFHS = high-fat high-sucrose.
3.2. Function of the tricarboxylic acid cycle is impaired in the spinal cord of mice consuming a Western diet
Whether the changes observed in insulin signaling partners in the spinal cord of mice consuming a diet rich in saturated fat and sucrose impair energy homeostasis was evaluated using a targeted LC-MS-MS approach to quantify the TCA cycle intermediates. Relative to mice consuming a RD, the spinal cord of mice consuming HFHS for 12 wk. showed significant reductions in the abundance of cis-Aconitic Acid (2-fold reduced), α-Ketoglutarate (4-fold reduced), Glutamate (1.5-fold reduced), 2-Hydroxyglutarate (2-fold reduced), Aspartate (1.5-fold reduced) and Oxaloacetate (5-fold reduced) (Fig. 1O–U, P < .05, Student’s t-test). Collectively, perturbations in the insulin signaling system and TCA cycle function with Western diet consumption point to potential deficits in ATP production and energy homeostasis that may leave the adult spinal cord vulnerable to degenerative changes and limit repair responses occurring after SCI.
3.3. Recovery of sensorimotor and bladder function were impaired after SCI in mice with diet-induced obesity
Having established that consumption of a HFHS diet promotes insulin resistance systemically and signs on impaired insulin signaling and energy homeostasis in the spinal cord, we next sought to determine how this affects recovery of function after experimental SCI. Mice consuming a HFHS diet for 7 wk. prior to SCI, showed reductions in recovery of coordinated stepping in the BMS open field test, in hindlimb strength evaluated by the Inclined plane test and in recovery of spontaneous bladder voiding (Fig. 2). Mice consuming a RD or HFHS were equally impaired on the BMS at 1 dpi and both groups showed significant improvements by 7 dpi (P < .01, Two-way ANOVA), with progressive increases thereafter. Recovery in BMS scores in mice consuming a RD were significantly greater than those consuming HFHS at 14 dpi (P < .001) and at 30 dpi (P < .05, Two-way ANOVA). At the 30 dpi endpoint, BMS scores of RD mice averaged 0.7 points higher than those of mice consuming HFHS (Fig. 2A), reflecting improvements in occasional plantar stepping. BMS subscores of mice consuming a RD were also significantly higher than HFHS mice from 21 dpi onward (P < .001, Two-way ANOVA). At the 30 dpi endpoint, BMS subscores in mice consuming a RD were an average of 2.6 points higher than that of parallel mice consuming HFHS (P < .001, Two-way ANOVA) (Fig. 2B). This difference in BMS subscore reflects improvements in parallel paw placement, ankle rotation and coordination. The inclined plane test was used to evaluate hind limb strength and showed improvements in both groups by 7 dpi, however the magnitude of recovery was impaired in mice consuming HFHS relative to a RD at all time points post injury (P < .001). Notably, higher mean maximal angles were achieved in mice consuming a RD compared to HFHS at 7, 14, 21 and 30 dpi (P < .001, Two-way ANOVA) (Fig. 2C). Assessment of bladder fullness showed impairment of recovery of spontaneous voiding in mice consuming HFHS compared to a RD at the 1, 3 and 4 wk. recovery intervals (P < .001, Two-way ANOVA) (Fig. 2D). Spared tissue volume showed significant reductions in spinal segments above the injury epicenter in mice consuming HFHS at 30 dpi (P < .05, Two-way ANOVA) (Fig. 2G).
Fig. 2.

Consumption of a Western diet impairs recovery of sensorimotor and bladder function after SCI and is associated with reductions in spared spinal cord tissue volume. (A–C) Recovery of sensorimotor function assessed by the Basso Mouse Scale (BMS) score (A) and BMS subscore (B), and the Inclined Plane test (C) were reduced in mice consuming HFHS compared to those consuming a RD. (D) Signs of spontaneous bladder release also showed delayed recovery in mice consuming HFHS compared to RD at wk. 1, 3 and 4 after SCI (RD, n = 9 and HFHS, n = 10, Two-way ANOVA, *P < .05, **P < .01, ***P < .001). (E–G) Application of the Cavalieri estimator probe to Eriochrome and neurofilament dual stained spinal cord sections taken from 2 mm blocks encompassing the injury epicenter (0), and 4 mm above and below, showed no differences in the volume of white matter damage or in the volume of spared white matter at 30 dpi across groups (RD, uninjured n = 5, 14 dpi n = 4, 30 dpi n = 5 and HFHS, uninjured n = 5, 14 dpi n = 5, 30 dpi n = 5, *P < .05, **P ≤ .01, ***P < .001, Two-way ANOVA). RD = regular diet, HFHS = high-fat high-sucrose.
3.4. Consumption of a Western diet promotes degenerative changes in the adult spinal cord, exacerbates neural damage after injury and impairs signs of recovery
To gain insights into the potential molecular drivers of impaired functional recovery in mice consuming HFHS, we next quantified immunoreactivity for key markers of neural injury and repair, including myelin integrity by assessment of myelinating cells (Olig2 and CC-1), the major myelin proteins (MBP and PLP), astrogliosis using GFAP and ALDH1L1 and microgliosis with Iba1. In addition, we quantified any changes in ventral horn neurons by counts of NeuN+ cells, and quantified the appearance of serotonergic (5-HT+) axons and GAP43+ growth cones. The results of this analysis suggest that diet-induced obesity driven by HFHS consumption drives inflammatory astrogliosis and a loss of mature myelinating cells in both the intact and injured adult spinal cord and impairs signs of neural repair (Figs. 3–5 and Figs. S2 and S4).
Fig. 3.

Consumption of a Western diet decreases mature myelinating cells before and after SCI. (A–H) At 30 dpi reductions in the number of CC-1+ mature oligodendrocytes were observed across the gray and white matter of mice consuming HFHS. Olig2+ oligodendrocyte progenitor cells were increased in gray matter at 14 and 30 dpi in HFHS mice. (I–P) The major myelin protein PLP was reduced in GM and WM before and after SCI in mice consuming HFHS while MBP levels were enhanced in WM at 30 dpi (RD, uninjured n = 5, 14 dpi n = 4, 30 dpi n = 5 and HFHS, uninjured n = 5, 14 dpi n = 5, 30 dpi n = 5, female mice, *P < .05, **P < .01, ***P < .001, Two-way ANOVA, NK). Boxed area on cartoon of spinal cord shows area quantified. All images are representative with scale bar = 200 μm (A and B, I and J). RD = regular diet, HFHS = high-fat high-sucrose.
Fig. 5.

Consumption of a Western diet promotes astrogliosis and microgliosis. (A–E) Astroglial reactivity was investigated in the uninjured spinal cord (C), and at 14 and 30 dpi in spinal cord sections through the injury epicenter, and in 2 mm blocks for 4 mm above and 4 mm below, by quantifying immunoreactivity for GFAP and ALDH1L1 in the gray matter (GM) or white matter (WM). Mice consuming a HFHS diet showed increases in GFAP-immunoreactivity (C) and GFAP/ALDH1L1 ratios (E) in white matter prior to SCI. (F–J) The spinal cord of mice consuming HFHS showed increased numbers of Iba-1+ microglia at 14 and 30 dpi with soma size being higher at 30 dpi in mice consuming HFHS. Skeletal analysis of microglial morphology showed a reductions in the number of end points associated with Iba1+ microglia in mice consuming HFHS prior to SCI (RD, uninjured n = 5, 14 dpi n = 4, 30 dpi n = 5 and HFHS, uninjured n = 5, 14 dpi n = 5, 30 dpi n = 5, female mice, *P < .05, **P < .01, ***P < .001, Two-way ANOVA, NK). Boxed area in cartoons shows area quantified in each case. Representative images are shown with scale bar = 200 μm (A, B and F) and scale bar = 50 μm (G). RD = regular diet, HFHS = high-fat high-sucrose.
3.5. Consumption of a diet rich in saturated fat and sucrose differentially regulates the integrity of myelin before and after SCI
Myelin plays an essential role in facilitating nerve impulse conduction and in the trophic and metabolic support of axons (Funfschilling et al., 2012; Saab and Nave, 2017; Stassart et al., 2018). Myelinating cells are highly vulnerable to degeneration after SCI (Alizadeh et al., 2019; Almad et al., 2011; Giacci et al., 2018). To determine how consumption of a diet rich in saturated fat and sucrose influences myelin integrity after SCI, we quantified the number of cells immunoreactive for Olig2 - a marker of oligodendrocyte progenitor and young oligodendrocytes in addition to CC-1 - a mature oligodendrocyte marker (Fig. 3A-H). The area immunostained for the major myelin proteins PLP or MBP was also quantified (Fig. 3 I–P). The number of Olig2+ cells in the spinal cord total or gray matter of mice consuming HFHS was increased at 14 (1.3-fold) and 30 dpi (1.2-fold) (Fig. 3C and D, 14 and 30 dpi; P < .05, P < .01 and P < .001, Two-way ANOVA, NK). The number of Olig2+ cells in spinal cord white matter of mice consuming HFHS was decreased at 30 dpi (Fig. 3E, P < .05, Two-way ANOVA, NK). Expression of Olig2 RNA was already reduced in the intact spinal cord after 12 wk. of HFHS (Fig. S4 E, P < .05, Student’s t-test). The number of CC-1+ oligodendrocytes was reduced in mice consuming HFHS compared to mice consuming a RD by 1.6-fold at 30 dpi across the gray and white matter (Fig. 3F–H, 30 dpi, P < .001 Two-way ANOVA, NK).
PLP is a hydrophobic transmembrane lipoprotein that plays a key role in the formation and maintenance of the multilamellar myelin sheath (Jahn et al., 2009; Schneider et al., 2005). PLP was significantly reduced (1.2-fold) in the spinal cord of mice consuming HFHS prior to SCI in total or gray matter of spinal cord (Fig. 3K and L, P < .05, Two-way ANOVA, NK). PLP levels were also reduced by 1.3-fold in the spinal cord gray matter of mice consuming HFHS at 30 dpi compared to mice consuming a RD (Fig. 3L). MBP is a hydrophilic protein that is essential for maintenance of myelin structure through interactions with lipids in the myelin membrane (Fulton et al., 2010; White and Kramer-Albers, 2014). MBP was not impacted by HFHS consumption prior to SCI and was equally reduced across the HFHS and RD groups at 14 dpi (Fig. 3N–P and Fig. S4 C). By 30 dpi, mice consuming HFHS had recovered white matter MBP levels to a greater extent than RD (1.3-fold) (GM + WM, P < .001; WM, P < .01, Two-way ANOVA, NK). Collectively, these results point to a Western diet as a risk factor for the loss of myelin prior to and after SCI with these cellular changes reflected in decrements in PLP, but not MBP abundance.
3.6. Signs of neural repair after SCI are impaired in mice consuming a diet high in fat and sucrose
Injury to the spinal cord results in neuron and axon degeneration with axonal sprouting highlighted by GAP43+ growth cones that can foster neural plasticity and repair (Fig. 4 and Fig. S4). Serotoninergic (5-HT+) axons show an unusual propensity for sprouting and regeneration after SCI (Hawthorne et al., 2011; Jin et al., 2016). As expected, 5-HT+ axons were reduced across groups at 14 dpi; however, greater losses were observed in spinal segments above in mice consuming HFHS (1.5-fold) compared to those consuming a RD (Fig. 4C). Also, increases in GAP43+ growth cones observed in mice consuming a RD by 30 dpi were not observed in mice consuming HFHS (Fig. 4H–I). Also as anticipated, there was a prominent loss of NeuN+ neurons in the injury epicenter at 14 and 30 dpi; however, this was not impacted by diet (Fig. 4E–F). The expression of neurofilament RNA was already reduced in the intact spinal cord after 12 wk. of HFHS compared to RD (Fig. S4 G, P < .01, Student’s t-test). By contrast, neurofilament immunoreactivity was not impacted by HFHS consumption prior to SCI (Fig. 4K–L). Neurofilament immunoreactivity was equally reduced across groups at 14 dpi (Fig. 4K, P < .05, Two-way ANOVA, NK). By 30 dpi, mice consuming HFHS had recovered neurofilament levels to a greater extent than RD (1.7-fold higher) (P < .05, Two-way ANOVA, NK). These findings suggest that consumption of a diet high in fat and sucrose exacerbates loss or slows repair of 5-HT axons after SCI and limits growth cone regenerative responses.
Fig. 4.

Signs of neural repair after SCI are impaired in mice consuming a Western diet. (A–C) Serotonergic 5-HT+ axons were reduced in spinal segments above the injury epicenter at 14 dpi in mice consuming HFHS compared to a RD. (D–F) The number of NeuN+ neurons in the ventral horn did not differ between groups. (G–I) GAP43-immunoreactive growth cones were reduced at 30 dpi the injury epicenter of mice consuming HFHS relative to a RD. (J–L) NFH-immunoreactivity was higher in mice consuming HFHS at 30 dpi (RD, uninjured n = 5, 14 dpi n = 4, 30 dpi n = 5 and HFHS, uninjured n = 5, 14 dpi n = 5, 30 dpi n = 5, female mice, *P < .05, **P < .01, ***P < .001, Two-way ANOVA, NK). Boxed area on cartoon of spinal cord shows area quantified. Representative images are shown in each case with scale bar = 100 μm (A, D and J), 200 μm (G). RD = regular diet, HFHS = high-fat high-sucrose.
3.7. Diet-induced obesity drives astrogliosis in the intact adult spinal cord
To address the impact of consumption of a HFHS diet on astrogliosis after SCI, we quantified immunoreactivity for GFAP and used ALDH1L1 to estimate total astrocyte abundance (Fig. 5A–E, Fig. S2, A–F). Unexpectedly, we observed that mice consuming a HFHS diet already had significant increases in GFAP in the spinal cord white and gray matter, approximately 2-fold, prior to SCI (Fig. 5C and Fig. S2 A–B, Two-way ANOVA, NK). The expression of GFAP RNA was also already increased by 1.5-fold in the intact spinal cord after 12 wk. of HFHS (Fig. S4 A, **P < .01, Student’s t-test). Notably, increases in GFAP immunoreactivity in the otherwise intact spinal cord of mice consuming HFHS were similar in magnitude to those observed in the spinal cord at 14 dpi. There was a progressive and parallel increase in GFAP by 30 dpi across the HFHS ans RD groups.
Increases in the pan-astrocyte marker ALDH1L1 were observed in a parallel manner across RD and HFHS groups after SCI consistent with expected astroglial proliferation (Fig. 5D, S2 C and D). GFAP/ALDH1L1 ratios in the white matter of mice consuming HFHS prior to SCI reached 2.5-fold higher levels compared to mice consuming a RD (Fig. 5 E). Increases in GFAP/ALDH1L1 ratios suggest that increases in GFAP in the intact spinal cord of mice consuming HFHS are likely to reflect increases in GFAP expression per astrocyte (Fig. 5E). These results highlight the idea that a Western diet alone can increase astroglial reactivity in the intact adult spinal cord.
3.8. Signs of microglial activation in the spinal cord are exacerbated by diet-induced obesity prior to and after SCI
To determine any impact of HFHS consumption on microglial reactivity prior to and after SCI, we quantified the density of Iba-1 immunoreactive cells and assessed their shape using skeletal analysis (Morrison et al., 2017) (Fig. 5F–J and Fig. S2, G–L). First, HFHS consumption did not have a significant impact on the abundance of Iba-1 immunoreactivity prior to SCI (Fig. S2 G-I), although there was a significant reduction in the number of branch end points (Fig. 5J, P < .05, Two-way ANOVA, NK) and a corresponding trend towards increased soma size (Fig. 5I). As expected, Iba-1 immunoreactivity increased several-fold in response to SCI, with Iba-1 soma size being higher in mice consuming HFHS at 30 dpi (Fig. 5I, P < .05, Two-way ANOVA, NK). These findings suggest that HFHS consumption increases microglial reactivity in the intact and chronically injured adult spinal.
3.9. Neural injury is exacerbated by high fat and high sucrose culture conditions and is prevented by an insulin-sensitizing agent
To explore the link between HFHS consumption and changes in insulin sensitivity, we modeled HFHS conditions in an organotypic spinal cord slice culture model and determined if effects could be prevented by the insulin-sensitizing agent, metformin (Figs. 6–8 and Fig. S3). First, culture of spinal cord slices in high glucose media with the addition of the long chain fatty acid, palmitate (100 μM) for 7 DIV resulted in a loss of MBP- (2.1-fold) and neurofilament- (5-fold) (P < .01, One-way ANOVA, NK) (Fig. 6C–E and Fig. S3). Linking these changes to insulin resistance, signs of neural injury were completely prevented when the insulin-sensitizer metformin was included in the cell culture media. Of interest, while the increases in GFAP immunoreactivity with the addition of palmitate to the culture media did not reach the level of statistical significance, inclusion of metformin in the media resulted in 1.4-fold reductions (Fig. 6B, P < .05, One-way ANOVA, NK). In addition, in the presence of metformin, excess of the long chain fatty acid palmitate promoted a 1.8-fold increase in Olig2 cell numbers (Fig. 6D, P < .05, One-way ANOVA, NK).
Fig. 6.
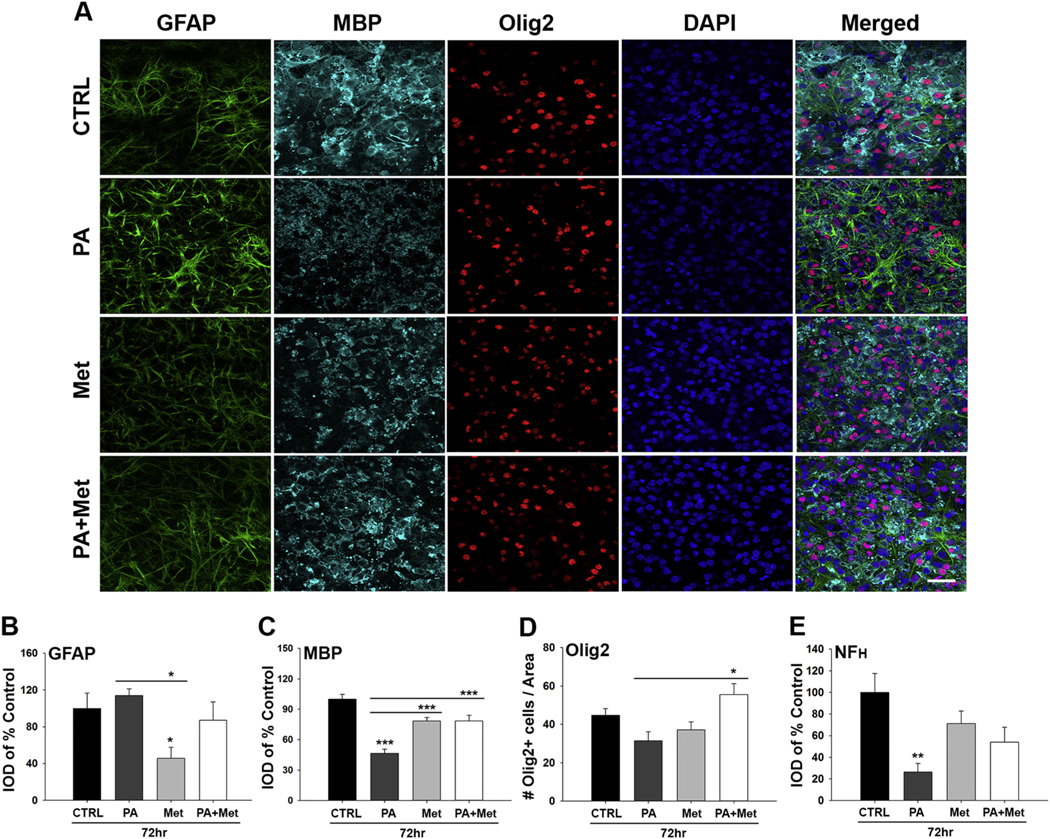
Spinal cord slice cultures grown in media containing high levels of saturated fat and sucrose show signs of neural injury that are prevented by the insulin-sensitizer metformin. (A–E) Representative images and associated histograms show that the addition of palmitate (PA) to 7 DIV organotypic spinal cord slice cultures results in reduced MBP immunoreactivity (C), Olig2+ cell counts and (D) NFH immunoreactivity (E) (see Fig. S3 for NFH images). Notably, metformin alone reduced the level of GFAP immunoreactivity in spinal cord slice cultures (B). Co-treatment of cultures with PA and metformin improved MBP levels and Olig2+ cell counts (*P < .05, **P < .01, ***P < .001, One-way ANOVA, NK). Scale bar = 50 μm. IOD = Integrated optical density.
Fig. 8.

Hypothetical model by which dietary HFHS and an insulin-sensitizing agent (metformin) influence astrocyte activation and myelinating cells in the adult spinal cord. We highlight two possible interactive mechanisms by which HFHS consumption promotes dysmyelination and how metformin may overcome these effects. (1) Our data suggest that consuming HFHS can directly impair oligodendrocyte IGF-1 responsiveness and PGC-1α expression each of which may contribute to the loss of oligodendrocyte progenitor cells (OPCs) and mature PLP expressing oligodendrocytes observed. (2) HFHS also promotes astrocyte activation, including increases in IL-6 and decreases in IGF-1 expression, that we hypothesize contribute to a disruption of oligodendrocyte metabolism, including IGF-1R expression through an indirect mechanism. (3) The ability of the insulin-sensitizing agent metformin to protect oligodendrocytes from the direct and indirect dysmyelinating effects of HFHS is coupled to improvements in IGF-1R, SIRT1 and PGC-1α expression.
3.10. Neural injury driven by high-fat high-sucrose conditions occurs by direct and indirect cellular mechanisms that are linked to insulin signaling and PGC-1α expression
To distinguish potential pathogenic effects of HFHS on individual neural cell types and a link to insulin resistance, we examined the impact of HFHS conditions applied to dissociated cultures of purified astrocytes, oligodendrocytes or neurons (Fig. 7), alone or in combination with the insulin-sensitizing agent metformin. Paralleling in vivo observations, cultures of astrocytes grown under HFHS conditions showed increased expression of GFAP (1.3-fold, P < .05) and IL-6 (2.3-fold, P < .001, One-way ANOVA, NK) (Fig. 7A–B). Astrocytes are a significant source of IGF-1 and notably IGF-1 levels were reduced by 1.5-fold in the presence of HFHS (P < .05, One-way ANOVA, NK). Reductions in astrocyte IGF-1R or in the InsR (insulin-R) under HFHS conditions did not reach the level of significance (Fig. 7C–E).
Fig. 7.

HFHS impairs oligodendrocyte function through direct and indirect mechanisms, both of which can be reversed by metformin. (A–G) Primary astrocytes grown in high sucrose culture media containing palmitate (PA, 100 uM) to model HFHS conditions, show increased signs of pro-inflammatory astrogliosis (GFAP and IL-6 expression), effects which were prevented by inclusion of metformin (Met, 100 μM in the media. HFHS culture conditions also reduced astrocyte IGF-1). (H–U) HFHS culture conditions promoted reductions in PLP, IGF-1R and PGC-1α in Olineu oligodendrocyte cultures (H-N) and reductions in neurofilament (NF(H)), GAP43 and Synapsin 1 (Syn-1) in primary cortical neuron cultures (O-U). The addition of MET to the media prevented these effects and was linked to preserving or increasing expression of SIRT1 and PGC-1α. (V-ΑΒ) Application of the < 100 and > 10 kDa concentrated fraction of conditioned media from astrocytes (ACM) grown in HFHS alone reduced the expression of Olig2, MBP, PLP, IGF-1R and PGC-1α in primary oligodendrocyte cultures after 24 h, effects that were largely prevented when the astrocytes were also treated with Met (*P < .05, **P < .01, ***P < .001, One-way ANOVA, NK).
The in vitro impact of HFHS on oligodendrocyte differentiation also mirrored in vivo findings, resulting in reduced expression of PLP (1.5-fold reduction) and IGF-1R (1.8-fold reduction) (P < .001) (Fig. 7H–K). HFHS-elicited reductions in PLP expression were attenuated by the addition of metformin to the media. In the presence of metformin, HFHS promoted MBP, Olig2 and IGF-1R expression by differentiating OPCs. ~1.8-fold (P < .05) (Fig. 7I–K). The significant increases in myelin production and IGF-1R in the presence of HFHS and metformin were also accompanied by significant elevations in both SIRT1 (4.7-fold) and PGC-1α (2.3-fold). As expected (Cuyas et al., 2018), metformin alone also promoted small, but significant increases in SIRT1 (1.5-fold) and PGC-1α (1.9-fold) (P < .05, One Way ANOVA) (Fig. 7M–N).
Primary cortical neurons cultured under HFHS conditions showed ~1.7-fold reductions in neurofilament, GAP43 and Synapsin-1 expression (Fig. 7O–Q), in addition to ~2.5-fold reductions in SIRT1 and PGC-1α (P < .05, One-way ANOVA, NK) (Fig. 7T–U), effects that were completely prevented by the addition of metformin to the media. Of additional interest, neurons cultured in control media with the addition of metformin alone showed ~1.7-fold increases in the expression neurofilament, GAP43 and Synapsin-1 (P < .05, One-way ANOVA, NK).
To determine if HFHS-elicited changes in astrogliosis in vivo and in vitro play key roles in the dysmyelinating effects of HFHS observed, we examined the impact of conditioned media from astrocytes grown in HFHS alone, or in the presence of metformin (Fig. 7V-AB). Compared to media from astrocytes grown in high sucrose alone, media from astrocytes with added palmitate promoted a loss of expression of Olig2+ (2.8-fold), MBP (1.3-fold) and PLP (1.6-fold) (Fig. 7V–X). Linking reductions in the expression of markers of myelin health to astrocyte-induced perturbations in insulin singling intermediates, conditioned media from HFHS treated astrocytes also elicited a reduction in IGF-1R and PGC-1α in oligodendrocytes, effects that were completely prevented by metformin (Fig. 7Y-AB).
4. Discussion
A diet high in fat and sucrose, also referred to as a “Western diet”, promotes systemic metabolic dysfunction and is a risk factor for neurodegenerative conditions; however, there is limited information regarding mechanisms of action in the intact or injured CNS. Since individuals with SCI are particularly vulnerable to the development of insulin resistance, obesity and cardiovascular dysfunction - all hall-marks of metabolic syndrome, we investigated the impact of a Western diet on the adult spinal cord and its response to traumatic injury. Findings show, for the first time, that systemic insulin resistance is reflected in abnormalities in IGF-1 and IGF-1R expression in the spinal cord, even prior to SCI, and that this is accompanied by impaired TCA cycle function. These significant Western diet-related impairments in the potential for insulin signaling and metabolic energy homeostasis were accompanied by reductions in PLP and increases in signs of astrocyte and microglial reactivity in the intact spinal cord. After SCI, loss of mature myelin-producing cells was exacerbated, growth cones were stunted and recovery of sensorimotor and bladder function were impaired. Although the systemic influences of a Western diet on impairments in neural recovery are not excluded, complementary cell culture approaches suggest that insulin resistance at the level of the CNS has the potential to contribute to neurotoxicity through direct and indirect astrocyte-related mechanisms and that these effects can be overcome by treatment with metformin, a clinically useful insulin-sensitizing agent.
4.1. Western diet impairs neurobehavioral recovery after experimental SCI
The present study demonstrates that consumption of a Western diet impairs recovery of coordinated stepping, strength and bladder function in an experimental model of SCI. Prior studies also link a Western diet to neuropathology, including the development of Alzheimer’s disease (Emmerzaal et al., 2015; Haan, 2006), an exacerbation of functional decline after TBI (Lee et al., 2016; Mowery et al., 2009; Sherman et al., 2016) and reduced function after SCI (Harris et al., 2019). Obesity is also associated with a significant increase in the risk of the development of MS (Hedstrom et al., 2014), including increased disability (Oliveira et al., 2014). Despite the clinical and experimental links between systemic metabolic dysfunction and neurological decline the neural mechanisms underpinning these events remain poorly understood.
The impairments we observe in recovery of function after SCI in mice consuming a Western diet were associated with pathogenic changes across glial and neuron compartments. In addition to obesity and insulin resistance, a cardinal feature of metabolic syndrome is the establishment of a pro-inflammatory state. That a Western diet drives a pro-inflammatory state in the otherwise intact spinal cord is suggested by increases in signs of astroglial and microglial reactivity, each a key hallmark of CNS inflammation. Increases in GFAP and signs of microglial reactivity in the CNS with high fat consumption have been previously documented (Byrne et al., 2015; Gzielo et al., 2017; Valdearcos et al., 2014). Microglial activation with high fat consumption is linked to pro-inflammatory cytokine production and neural injury in hippocampus (Spencer et al., 2017) and hypothalamus (Valdearcos et al., 2014). Remarkably, the HFHS-induced elevations in GFAP we observe in the uninjured spinal cord equaled that seen at 14 d after SCI with GFAP/ALDH1L1 ratios in white matter suggesting increases represent elevated GFAP per astrocyte. Moreover, microglial soma size was increased by HFHS at 30 dpi, pointing to increased activation. Increases in signs of astrogliosis and microgliosis in the spinal cord of mice consuming HFHS support an emerging hypothesis that systemic metabolic dysfunction is reflected in increased CNS inflammatory responses and that this can promote and exacerbate neural injury and in addition, impede recovery of function.
Myelination is a dynamic process that can be influenced by experience and other environmental factors with our current and prior findings suggesting a negative impact of HFHS consumption. We recently reported that that myelin-producing oligodendrocytes and their progenitors are lost in the spinal cord of otherwise uninjured adult mice consuming a Western diet (Yoon et al., 2016). Here we observe a HFHS-induced loss of PLP using a similar albeit not identical diet, although reductions in myelinating cells were not statistically significant. Prior studies document a loss of MBP in cortical and hippocampal white matter in mice consuming a high fat high fructose diet with associated with cognitive decline (Graham et al., 2019). Together, these findings point to the potential for demyelinating effects in the adult CNS with consumption of excess saturated fat and sugars. Notably, both the Yoon et al., 2016 and Graham et al., 2019 studies showed that exercise completely prevented the demyelinating effects of a Western diet. New findings also suggest that high saturated fat consumption alone may be sufficient to produce oligodendrocyte loss in both the adult brain and spinal cord (Langley et al., 2020).
Oligodendroglia are vulnerable to apoptosis after SCI with degeneration initiated early after injury and continuing into the chronic period (Almad et al., 2011). Loss of myelinating cells disrupts K+ and Na + channels resulting in conduction failure and leaves spared axons vulnerable to ongoing injury and degeneration (Alizadeh et al., 2019; Pukos et al., 2019). In spinal cord white matter at the chronic 30 dpi time point, we observe that HFHS exacerbates reductions in CC-1+ mature oligodendrocytes and impairs signs of oligodendrocyte replacement, that is Olig2+ OPCs/young oligodendrocytes. In spinal cord gray matter by contrast, we observed increased Olig2+ OPCs in mice consuming HFHS at 14 and 30 dpi. Since prior studies have shown that the central canal region is a potential source of OPCs (Gao et al., 2015), it is possible that the increase in Olig2+ OPCs in spinal cord gray matter after SCI, but the reduction in white matter, reflects an impairment in the migration of newly generated OPCs. Also, we cannot rule out any differential impact of HFHS on oligodendrocyte loss or proliferation across the gray and white matter after SCI (Tripathi and McTigue, 2007; Vigano et al., 2013). Notably, despite increased loss of Olig2+ and CC-1+ oligodendrocytes in the spinal cord white matter at 30 dpi in mice consuming HFHS, no reductions in PLP and an increase in MBP closer to baseline were observed. These findings suggest that in the context of SCI, HFHS consumption may enhance myelin protein production by remaining oligodendrocytes. Given the lipid rich nature of the myelin membranes, further documentation of the contribution of dietary lipids to myelin regeneration after SCI, as has been noted in experimental models of demyelinating disease (Berghoff et al., 2017), will be of interest. Together, the current findings point to the need to better understand the source of replacement OPCs after SCI, any differential OPC loss across the gray and white matter and the potential impact of HFHS on these events and on the ability of oligodendrocytes to build myelin membranes.
Although HFHS consumption did not significantly exacerbate the loss of spinal cord neurons or 5-HT axons after SCI, greater losses in neurofilament 200 was observed at 14 dpi. Interestingly, somewhat higher levels of neurofilament were seen by 30 dpi in mice consuming HFHS, although the significance of these findings will require further study. Suggesting that HFHS contributes to impairments in the capacity for neural repair after SCI, while GAP43+ growth cones were increased in spinal cord gray matter of mice consuming a RD by 30 dpi, such increases were not observed in mice consuming HFHS.
4.2. Western diet triggers signs of CNS insulin resistance and impaired energy homeostasis
4.2.1. Link between Western diet and insulin resistance
While the impact of a Western diet on the CNS and its response to injury is no doubt multifactorial, evidence from the current study, taken with that in the literature (Agrawal et al., 2014; Greenwood and Winocur, 2005; McNay et al., 2010; Song et al., 2015), point to a central role for increased CNS insulin resistance. In addition to the expected HFHS-triggered increase in systemic insulin resistance, we observed abnormalities in insulin signaling intermediates in the spinal cord of otherwise intact mice, including reductions in IGF-1 and IGF-1R expression. These findings support the concept that a Western diet not only promotes signs of insulin resistance systemically, but by reducing IGF-1 and IGF-1R in the spinal cord, also impairs the capacity for CNS IGF-1 signaling prior to and after SCI.
In the CNS, IGF-1/Insulin signaling plays critical roles in oligodendrocyte survival and differentiation (Barres et al., 1992; Bibollet-Bahena and Almazan, 2009; Carson et al., 1993; Chesik et al., 2007; Frederick and Wood, 2004; Lin et al., 2005; McMorris and DuboisDalcq, 1988; Min et al., 2012; Ness et al., 2002; Zeger et al., 2007). Therefore, HFHS-associated reductions in IGF-1 signaling intermediates could contribute to the demyelinating effects observed here and in our prior studies (Yoon et al., 2016). Indeed, brain insulin resistance has already been linked to neurodegenerative disease (de la Monte, 2012; Salkovic-Petrisic et al., 2013) and cognitive decline (Greenwood and Winocur, 2005). IGF-1 delivery also promotes neurogenesis and attenuates neurobehavioral deficits after experimental TBI (Saatman et al., 1997). Indeed, IGF-1 is also an important mediator of axon spouting after SCI (Liu et al., 2017). The potential neuropathic effects of perturbations in IGF-1 signaling have potential to be widespread given that CNS insulin signaling both reduces inflammation and promotes neuron survival (Adzovic et al., 2015; Bateman and McNeill, 2006; Carlson et al., 2014; Duarte et al., 2008; Madathil and Saatman, 2015; Suh et al., 2013).
The significance of understanding the development of insulin resistance to SCI relates in part to reports that individuals with SCI are frequently glucose intolerant and have significantly elevated insulin levels, resulting in overt insulin resistance (Duckworth et al., 1980). Overall the prevalence of diabetes in individuals with SCI is upwards of 66% with diabetes-related complications affecting eye function, wound healing, and overall quality of life (Farkas and Gater, 2018; Lavela et al., 2006). Insulin resistance is also a recognized feature of TBI pathology disrupting cerebral energy homeostasis (Kim and Feldman, 2012). Notably, Karelina et al., 2016 showed that TBI itself promotes acute changes in brain insulin sensitivity, increasing inflammation and neurodegenerative consequences (Karelina et al., 2016). Insulin and IGF-1 are also altered in MS with decreased insulin sensitivity and postprandial hyperinsulinemia (Penesova et al., 2015). Hyperinsulinemia is associated with cognitive impairment in both Alzheimer’s and in MS (Watson and Craft, 2006). The current findings suggest systemic insulin resistance also puts the intact adult spinal cord at risk for astroglial reactivity, microgliosis and a loss of myelinating cells that together exacerbate responses to traumatic injury and impede signs of repair.
4.2.2. Link between Western diet and impaired TCA cycle function
Due to its high-energy demands under normal conditions and particularly after injury, the CNS is exceptionally vulnerable to disturbances in energy homeostasis. For example, in the case of acute TBI, the brain enters a period of hyperglycolysis, in which glucose utilization increases to meet increased metabolic demands (Bergsneider et al., 1997; Oddo et al., 2008). To address the potential impact of a Western diet on energy homeostasis in the spinal cord, we quantified function of the TCA cycle using targeted mass-spectroscopy. Mice with signs of insulin resistance systemically and in the CNS due to HFHS consumption showed reductions in key components of the TCA cycle, including oxaloacetate, α-ketoglutarate and aspartate. The perturbations in TCA cycle function elicited by a Western diet highlight potential deficits in ATP production and energy homeostasis. These metabolic impairments may in turn leave the spinal cord vulnerable to degenerative changes and impede repair prior to and in the aftermath of SCI. Although further investigation is needed, a mechanistic link between the perturbations in TCA cycle function elicited by HFHS and myelin loss prior to injury, and an exacerbation of demyelinating effects after SCI is supported by studies in other neuropathologies. For example, in TBI and Alzheimer’s, glycolysis and conversion of lactate to pyruvate for use in the TCA cycle are inhibited, contributing to the development of cerebral metabolic depression (Bergsneider et al., 2000; Dunn-Meynell and Levin, 1997; Hovda et al., 1991; Prins and Hovda, 2009; Yoshino et al., 1991). Even in mild TBI, impairments in availability of energy metabolites has severe and long-lasting consequences (Prins et al., 2013; Prins et al., 2010; Weil et al., 2014).
4.3. Direct and indirect neurotoxic effects of a Western diet including link to insulin resistance
To investigate the molecular mechanisms linking a Western diet to insulin resistance, astrogliosis, myelin loss and axon injury, we modeled these conditions in cell culture using either organotypic spinal cord slices or purified neural cultures. Culture conditions with high glucose and high saturated lipid palmitate, promoted reductions in myelin proteins and neurofilament expression suggestive of neural injury. Expression of GAP43 and synapsin-1 were also reduced by HFHS in primary neuron cultures pointing to impairments in growth cones and synapse function. These deleterious effects were accompanied by increases in astrocyte IL-6 and perturbations in IGF-1 and IGF-1R signaling partners. A role for insulin resistance triggered by culture under HFHS conditions directly at the level of astrocytes, oligodendrocytes and neurons is suggested by the prevention of negative effects by the insulin-sensitizing agent, metformin. Metformin is a first-line drug for the treatment of type 2 diabetes (Bosi, 2009). Findings here demonstrate that metformin prevents HFHS-elicited decreases in MBP and PLP, as well as neurofilament, GAP43 and synapsin-1 expression, both in dissociated cell cultures and in organotypic spinal cord slices. Metformin also restored IGF-1R and insulin receptor expression in oligodendrocyte and neuron cultures, thereby linking the beneficial effects of metformin directly to an improved potential for insulin signaling.
The results presented suggest that within the CNS, HFHS has effects across glial and neuron compartments, including direct effects on astrocytes, oligodendrocytes and neurons. Furthermore, in vivo results point to pro-inflammatory changes in microglia. To test the possibility that the signs of astroglial reactivity observed in vivo and in vitro after HFHS treatment contribute to myelin injury and are linked to insulin resistance, we next determined the impact of conditioned media fractions collected from astrocytes grown under HFHS conditions alone, or in the presence of metformin. Supporting the idea that HFHS conditions alter levels of astrocyte-secreted factors that promote oligotoxicity, media from HFHS-treated astrocytes promoted reductions in Olig2, MBP and PLP, along with IGF-1R and PGC-1α. Linking the HFHS-induced changes in astrocytes to reduced insulin sensitivity, our data revealed that conditioned media from HFHS astrocytes grown with the addition of metformin no longer suppressed oligodendrocyte differentiation or components of the insulin signaling pathway. Given these intriguing findings, additional studies are warranted to identify the factors secreted by HFHS-treated astrocytes that may contribute to reductions in oligodendrocyte differentiation, with reductions in IGF-1 secretion and increases in IL-6 identified as potential key contributors.
To gain additional insight into the molecular mechanisms by which HFHS, metformin or the combination impacts neural function we quantified changes in SIRT1 a key metabolic sensor and regulator of metabolic homeostasis and stress. SIRT1 is a histone/protein deacetylase that regulates the activity of a variety of transcription factors and co-regulators, including PGC-1α to increase mitochondrial function, energy metabolism and gluconeogenesis (Ng et al., 2015). Culture conditions consisting of HFHS, but not HFHS + metformin, directly promoted reductions in SIRT1 and PGC-1α expression in purified cultures of neurons or oligodendrocytes. Since SIRT1 upregulates antioxidant genes through PGC-1α deacetylation (Brunet et al., 2004; Pardo and Boriek, 2012; Pardo et al., 2011; St-Pierre et al., 2006), it is possible that the maintenance of SIRT1 expression in HFHS neural cultures treated with metformin provided neuroprotective benefits.
PGC-1α is a transcriptional co-activator that also enhances mitochondrial biogenesis, fatty acid oxidation and oxidative metabolism (Johri et al., 2013; Liang and Ward, 2006). In addition, and of particular relevance to the current study, PGC-1α also plays key roles in myelinogenesis (Camacho et al., 2013; Kiebish et al., 2012; Xiang et al., 2011). For example, prior studies provide strong links between PGC-1α, MBP expression and myelin health, with PGC-1α knockout mice showing pronounced white matter abnormalities (Kiebish et al., 2012; Leone et al., 2005; Xiang et al., 2011). The current studies strengthen the link between SIRT1, PGC-1α and myelin by demonstrating that under the influence of HFHS directly, or when grown with media fractions from HFHS treated astrocytes, oligodendrocytes not only show reductions in myelin related gene expression, but also coordinate reductions in PGC-1α. Moreover, restoration of myelin-related gene expression was improved by metformin and co-ordinate increases in PGC1α were observed.
5. Conclusions
Collectively, the in vivo and in vitro neurotoxic effects we observe to be elicited by HFHS along with the reversal of these effects by metformin, at least in vitro, indicate that an important future direction will be to further elucidate any therapeutic benefits in vivo. At a systemic level metformin exerts multifactorial effects that include not only insulin-sensitization and control of glucose metabolism, but also an influence on lipids, cholesterol and cytokine production (van Stee et al., 2018). Of particular significance to the current study, metformin preconditioning improved neuron preservation in a rat model of SCI (Guo et al., 2018; Wang et al., 2016) and reduced blood spinal cord barrier disruption (Zhang et al., 2017). Metformin was also shown to improve remyelination in an experimental perinatal hypoxia-ischemia brain injury model (Qi et al., 2017), to provide neuroprotection after experimental TBI (Tao et al., 2018), to prevent and reverse microglial reactivity and neuropathic pain in a spared nerve injury model (Inyang et al., 2019), and to reduce peripheral nerve inflammatory injury in the streptozotocin model of type 2 diabetes (Los et al., 2019). In individuals with severe TBI, metformin improves serum S100b and neutrophil/ lymphocyte ratios (Taheri et al., 2019). Of interest to the demyelinating effects of HFHS consumption, both metformin and thiazolidinediones, reduce MS relapse rate (Negrotto et al., 2016). Here we have established efficacy of metformin across multiple cell culture platforms, providing new insights into direct and indirect neural mechanisms of action, in addition to the identification of potential molecular mediators. Taken with the urgent need to define new treatment strategies to prevent the secondary consequences of a Western diet and to improve outcomes after neural injury, including SCI, we expect the results of the current study will inspire a comprehensive in vivo analysis of the potential beneficial effects of metformin in the intact and injured CNS and the possibility that this will confer additional benefits when combined with existing neural repair strategies.
Supplementary Material
Acknowledgements
The work was supported by a grant from the Minnesota State Spinal Cord Injury and Traumatic Brain Injury Research Program, the Mayo Clinic Center for Biomedical Discovery and the Mayo Clinic Metabolomics Core (U24DK100469 and UL1TR000135). Portions of this work were also supported by R01NS052741, RG4958 from the National Multiple Sclerosis Society, a grant from the Craig H. Neilsen Foundation and the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology.
Abbreviations:
- 5-HT
5-hydroxytryptamine receptors
- ALDH1L1
aldehyde dehydrogenase 1 family member L1
- ATP
Adenosine triphosphate
- CC-1
Adenomatous polyposis coli clone
- CNS
Central nervous system
- DAPI
4′,6-Diamidino-2-phenylindole
- DIV
Day in vitro
- DPI
Day post injury
- GAP43
Growth Associated Protein 43
- GFAP
Glial fibrillary acidic protein
- GM
Gray matter
- HFHS
High in fat and sucrose
- Iba-1
Ionized calcium binding adaptor molecule 1
- IGF-1
Insulin-like growth factor 1
- IGF-1R
Insulin-like growth factor 1 receptor
- IL-6
Interleukin 6
- IsnR
Insulin receptor
- MBP
Myelin Basic Protein
- MS
Multiple sclerosis
- NeuN
Neuronal nuclei
- NFH
Neurofilament
- Olig2
Oligodendrocyte Transcription Factor 2
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator 1 alpha
- PLP
Myelin proteolipid protein
- RD
Regular diet
- SCI
Spinal cord injury
- SIRT1
Sirtuin 1
- TBI
Traumatic brain injury
- TCA
Tricarboxylic acid cycle
- WM
White matter
Footnotes
Declaration of Competing Interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.104934.
References
- Adzovic L, et al. , 2015. Insulin improves memory and reduces chronic neuroinflammation in the hippocampus of young but not aged brains. J. Neuroinflammation 12, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R, et al. , 2014. Deterioration of plasticity and metabolic homeostasis in the brain of the UCD-T2DM rat model of naturally occurring type-2 diabetes. Biochim. Biophys. Acta 1842, 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh A, et al. , 2019. Traumatic spinal cord injury: an overview of pathophysiology, models and acute injury mechanisms. Front. Neurol 10, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almad A, et al. , 2011. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics 8, 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, et al. , 1992. Cell death in the oligodendrocyte lineage. J. Neurobiol 23, 1221–1230. [DOI] [PubMed] [Google Scholar]
- Basso DM, et al. , 2006. Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma 23, 635–659. [DOI] [PubMed] [Google Scholar]
- Bateman JM, McNeill H, 2006. Insulin/IGF signalling in neurogenesis. Cell. Mol. Life Sci 63, 1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff SA, et al. , 2017. Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat. Commun 8, 14241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsneider M, et al. , 1997. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg 86, 241–251. [DOI] [PubMed] [Google Scholar]
- Bergsneider M, et al. , 2000. Dissociation of cerebral glucose metabolism and level of consciousness during the period of metabolic depression following human traumatic brain injury. J. Neurotrauma 17, 389–401. [DOI] [PubMed] [Google Scholar]
- Bibollet-Bahena O, Almazan G, 2009. IGF-1-stimulated protein synthesis in oligodendrocyte progenitors requires PI3K/mTOR/Akt and MEK/ERK pathways. J. Neurochem 109, 1440–1451. [DOI] [PubMed] [Google Scholar]
- Bosi E, 2009. Metformin–the gold standard in type 2 diabetes: what does the evidence tell us? Diabetes Obes. Metab 11 (Suppl. 2), 3–8. [DOI] [PubMed] [Google Scholar]
- Brunet A, et al. , 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 303, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Burda JE, et al. , 2013. Critical role for PAR1 in kallikrein 6-mediated oligodendrogliopathy. Glia 61, 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne FM, et al. , 2015. Characterisation of pain responses in the high fat diet/streptozotocin model of diabetes and the analgesic effects of antidiabetic treatments. J. Diabetes Res 2015, 752481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho A, et al. , 2013. Peroxisome proliferator-activated receptor gamma-coactivator1 alpha coordinates sphingolipid metabolism, lipid raft composition and myelin protein synthesis. Eur. J. Neurosci 38, 2672–2683. [DOI] [PubMed] [Google Scholar]
- Carlson SW, et al. , 2014. Conditional overexpression of insulin-like growth factor-1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J. Neuropathol. Exp. Neurol 73, 734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, et al. , 1993. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 10, 729–740. [DOI] [PubMed] [Google Scholar]
- Chesik D, et al. , 2007. The insulin-like growth factor system in multiple sclerosis. Int. Rev. Neurobiol 79, 203–226. [DOI] [PubMed] [Google Scholar]
- Cuyas E, et al. , 2018. Metformin is a direct SIRT1-activating compound: computational modeling and experimental validation. Front. Endocrinol 9, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AI, et al. , 2008. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3beta signaling pathways and changes in protein expression. Biochim. Biophys. Acta 1783, 994–1002. [DOI] [PubMed] [Google Scholar]
- Duckworth WC, et al. , 1980. Glucose intolerance due to insulin resistance in patients with spinal cord injuries. Diabetes 29, 906–910. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Levin BE, 1997. Histological markers of neuronal, axonal and astrocytic changes after lateral rigid impact traumatic brain injury. Brain Res. 761, 25–41. [DOI] [PubMed] [Google Scholar]
- Dutta T, et al. , 2016. Impact of long-term poor and good glycemic control on metabolomics alterations in type 1 diabetic people. J. Clin. Endocrinol. Metab 101, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerzaal TL, et al. , 2015. 2003–2013: a decade of body mass index, Alzheimer’s disease, and dementia. J. Alzheimers Dis 43, 739–755. [DOI] [PubMed] [Google Scholar]
- Farkas GJ, Gater DR, 2018. Neurogenic obesity and systemic inflammation following spinal cord injury: a review. J. Spinal Cord Med 41, 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick TJ, Wood TL, 2004. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol. Cell. Neurosci 25, 480–492. [DOI] [PubMed] [Google Scholar]
- Fulton D, et al. , 2010. The multiple roles of myelin protein genes during the development of the oligodendrocyte. ASN Neuro. 2, e00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funfschilling U, et al. , 2012. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, et al. , 2015. Nicotine modulates neurogenesis in the central canal during experimental autoimmune encephalomyelitis. Neuroscience 297, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gater DR Jr., et al. , 2019. Prevalence of metabolic syndrome in veterans with spinal cord injury. J. Spinal Cord Med 42, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacci MK, et al. , 2018. Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo. J. Neurosci 38, 6491–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert O, et al. , 2014. Serum lipid concentrations among persons with spinal cord injury - a systematic review and meta-analysis of the literature. Atherosclerosis. 232, 305–312. [DOI] [PubMed] [Google Scholar]
- Graham LC, et al. , 2016. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep 6, 21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LC, et al. , 2019. Exercise prevents obesity-induced cognitive decline and white matter damage in mice. Neurobiol. Aging 80, 154–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood CE, Winocur G, 2005. High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging 26 (Suppl. 1), 42–45. [DOI] [PubMed] [Google Scholar]
- Groah SL, et al. , 2011. Cardiometabolic risk in community-dwelling persons with chronic spinal cord injury. J. Cardiopulm. Rehabil. Prev 31, 73–80. [DOI] [PubMed] [Google Scholar]
- Guo Y, et al. , 2018. Metformin protects against spinal cord injury by regulating autophagy via the mTOR signaling pathway. Neurochem. Res 43, 1111–1117. [DOI] [PubMed] [Google Scholar]
- Gzielo K, et al. , 2017. Long-term consumption of high-fat diet in rats: effects on microglial and astrocytic morphology and neuronal nitric oxide synthase expression. Cell. Mol. Neurobiol 37, 783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan MN, 2006. Therapy insight: type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat. Clin. Pract. Neurol 2, 159–166. [DOI] [PubMed] [Google Scholar]
- Harris KK, et al. , 2019. Energy balance following diets of varying fat content: metabolic dysregulation in a rodent model of spinal cord contusion. Phys. Rep 7, e14207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawthorne AL, et al. , 2011. The unusual response of serotonergic neurons after CNS injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. J. Neurosci 31, 5605–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom AK, et al. , 2014. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. 82, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoane MR, et al. , 2011. The effects of a high-fat sucrose diet on functional outcome following cortical contusion injury in the rat. Behav. Brain Res 223, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovda DA, et al. , 1991. Diffuse prolonged depression of cerebral oxidative metabolism following concussive brain injury in the rat: a cytochrome oxidase histochemistry study. Brain Res. 567, 1–10. [DOI] [PubMed] [Google Scholar]
- Huitinga I, et al. , 1995. Macrophages in T cell line-mediated, demyelinating, and chronic relapsing experimental autoimmune encephalomyelitis in Lewis rats. Clin. Exp. Immunol 100, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inyang KE, et al. , 2019. The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol. Res 139, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn O, Tenzer S, Werner HB, et al. , 2009. Myelin proteomics: molecular anatomy of an insulating sheath. Mol. Neurobiol 40 (1), 55–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, et al. , 2019. Obesity promotes EAE through IL-6 and CCL-2-mediated T cells infiltration. Front. Immunol 10, 1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, et al. , 2016. Regrowth of serotonin axons in the adult mouse brain following injury. Neuron. 91, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, et al. , 2013. PGC-1alpha, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med 62, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M, Fehlings M, 2002. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the mouse: part 1. Clip design, behavioral outcomes, and histopathology. J. Neurotrauma 19, 175–190. [DOI] [PubMed] [Google Scholar]
- Karelina K, et al. , 2016. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci 43, 1034–1043. [DOI] [PubMed] [Google Scholar]
- Katoh H, et al. , 2019. Regeneration of spinal cord connectivity through stem cell transplantation and biomaterial scaffolds. Front. Cell. Neurosci 13, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, et al. , 2012. Chronic caloric restriction attenuates a loss of sulfatide content in PGC-1alpha−/− mouse cortex: a potential lipidomic role of PGC-1alpha in neurodegeneration. J. Lipid Res 53, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Feldman EL, 2012. Insulin resistance in the nervous system. Trends Endocrinol. Metab 23, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley MR, et al. , 2020. High fat diet consumption results in mitochondrial dysfunction, oxidative stress, and oligodendrocyte loss in the central nervous system. Biochim. Biophys. Acta Mol. basis Dis 1866, 165630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavela SL, et al. , 2006. Diabetes mellitus in individuals with spinal cord injury or disorder. J. Spinal Cord Med 29, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, et al. , 2016. Obesity and neurocognitive recovery after sports-related concussion in athletes: a matched cohort study. Phys. Sportsmed 44, 217–222. [DOI] [PubMed] [Google Scholar]
- Leone TC, et al. , 2005. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Ward WF, 2006. PGC-1alpha: a key regulator of energy metabolism. Adv. Physiol. Educ 30, 145–151. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, et al. , 2005. IGF-1 protects oligodendrocyte progenitor cells and improves neurological functions following cerebral hypoxia-ischemia in the neonatal rat. Brain Res. 1063, 15–26. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. , 2017. A sensitized IGF1 treatment restores corticospinal axon-dependent functions. Neuron. 95 (817–833), e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los DB, et al. , 2019. Preventive role of metformin on peripheral neuropathy induced by diabetes. Int. Immunopharmacol 74, 105672. [DOI] [PubMed] [Google Scholar]
- Madathil SK, Saatman KE, 2015. IGF-1/IGF-R signaling in traumatic brain injury: impact on cell survival, neurogenesis, and behavioral outcome. In: Kobeissy FH (Ed.), Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, Boca Raton (FL). [Google Scholar]
- Martino Adami PV, et al. , 2017. Worsening of memory deficit induced by energy-dense diet in a rat model of early-Alzheimer’s disease is associated to neurotoxic Abeta species and independent of neuroinflammation. Biochim. Biophys. Acta Mol. basis Dis 1863, 731–743. [DOI] [PubMed] [Google Scholar]
- McMorris FA, Dubois-Dalcq M, 1988. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J. Neurosci. Res 21, 199–209. [DOI] [PubMed] [Google Scholar]
- McNay EC, et al. , 2010. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem 93, 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, et al. , 2012. Insulin-like growth factor I regulates G2/M progression through mammalian target of rapamycin signaling in oligodendrocyte progenitors. Glia. 60, 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, 2012. Early intranasal insulin therapy halts progression of neurodegeneration: progress in Alzheimer’s disease therapeutics. Aging Health 8, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CD, et al. , 2018. Does muscle atrophy and fatty infiltration plateau or persist in chronic spinal cord injury? J. Clin. Densitom 21, 329–337. [DOI] [PubMed] [Google Scholar]
- Morrison H, et al. , 2017. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep 7, 13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowery NT, et al. , 2009. Stress insulin resistance is a marker for mortality in traumatic brain injury. J. Trauma 66, 145–151 (discussion 151–3). [DOI] [PubMed] [Google Scholar]
- Muir E, et al. , 2019. Recent advances in the therapeutic uses of chondroitinase ABC. Exp. Neurol 321, 113032. [DOI] [PubMed] [Google Scholar]
- Nash MS, et al. , 2016. Cardiometabolic syndrome in people with spinal cord injury/disease: guideline-derived and nonguideline risk components in a pooled sample. Arch. Phys. Med. Rehabil 97, 1696–1705. [DOI] [PubMed] [Google Scholar]
- National Spinal Cord Injury Statistical Center Spinal Cord Injury Facts and Figures at a Glance [Internet]. University of Alabama at Birmingham. 2017. Available from: https://msktc.org/lib/docs/Data_Sheets_/SCIMS_Facts_and_Figures_2017_August_FINAL.pdf. [Google Scholar]
- Negrotto L, et al. , 2016. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol. 73, 520–528. [DOI] [PubMed] [Google Scholar]
- Ness JK, et al. , 2002. IGF-I and NT-3 signaling pathways in developing oligodendrocytes: differential regulation and activation of receptors and the downstream effector Akt. Dev. Neurosci 24, 437–445. [DOI] [PubMed] [Google Scholar]
- Ng F, et al. , 2015. SIRT1 in the brain-connections with aging-associated disorders and lifespan. Front. Cell. Neurosci 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo M, et al. , 2008. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit. Care Med 36, 3233–3238. [DOI] [PubMed] [Google Scholar]
- Oliveira SR, et al. , 2014. Disability in patients with multiple sclerosis: influence of insulin resistance, adiposity, and oxidative stress. Nutrition 30, 268–273. [DOI] [PubMed] [Google Scholar]
- Pardo PS, Boriek AM, 2012. An autoregulatory loop reverts the mechanosensitive Sirt1 induction by EGR1 in skeletal muscle cells. Aging (Albany NY) 4, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo PS, et al. , 2011. Induction of Sirt1 by mechanical stretch of skeletal muscle through the early response factor EGR1 triggers an antioxidative response. J. Biol. Chem 286, 2559–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penesova A, et al. , 2015. Hyperinsulinemia in newly diagnosed patients with multiple sclerosis. Metab. Brain Dis. 30, 895–901. [DOI] [PubMed] [Google Scholar]
- Plemel JR, et al. , 2008. A graded forceps crush spinal cord injury model in mice. J. Neurotrauma 25, 350–370. [DOI] [PubMed] [Google Scholar]
- Prins ML, Hovda DA, 2009. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J. Neurotrauma 26, 1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, et al. , 2010. Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev. Neurosci 32, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, et al. , 2013. Repeated mild traumatic brain injury: mechanisms of cerebral vulnerability. J. Neurotrauma 30, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukos N, et al. , 2019. Myelin status and oligodendrocyte lineage cells over time after spinal cord injury: what do we know and what still needs to be unwrapped? Glia. 67, 2178–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi B, et al. , 2017. Metformin attenuates cognitive impairments in hypoxia-ischemia neonatal rats via improving remyelination. Cell. Mol. Neurobiol 37, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Radulovic M, et al. , 2013. Kallikrein cascades in traumatic spinal cord injury: in vitro evidence for roles in axonopathy and neuron degeneration. J. Neuropathol. Exp. Neurol 72, 1072–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic M, et al. , 2016. Targeting the thrombin receptor modulates inflammation and astrogliosis to improve recovery after spinal cord injury. Neurobiol. Dis. 93, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Nave KA, 2017. Myelin dynamics: protecting and shaping neuronal functions. Curr. Opin. Neurobiol 47, 104–112. [DOI] [PubMed] [Google Scholar]
- Saatman KE, et al. , 1997. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp. Neurol 147, 418–427. [DOI] [PubMed] [Google Scholar]
- Salkovic-Petrisic M, et al. , 2013. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm. (Vienna) 120, 233–252. [DOI] [PubMed] [Google Scholar]
- Scarisbrick IA, et al. , 1999. Differential expression of brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 in the adult rat spinal cord: regulation by the glutamate receptor agonist kainic acid. J. Neurosci 19, 7757–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MJ, et al. , 2016. Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes. 65, 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR, 2005. Design-based stereology in neuroscience. Neuroscience. 130, 813–831. [DOI] [PubMed] [Google Scholar]
- Schneider A, Lander H, Schulz G, Wolburg G, Nave KA, Schulz JB, Simons M, 2005. Palmitoylation is a sorting determinant for transport to the myelin membrane. J Cell Sci. 118 (Pt 11), 2415–2423 [DOI] [PubMed] [Google Scholar]
- Sherman M, et al. , 2016. Adult obese mice suffer from chronic secondary brain injury after mild TBI. J. Neuroinflammation 13, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, et al. , 2015. Impairment of insulin receptor substrate 1 signaling by insulin resistance inhibits neurite outgrowth and aggravates neuronal cell death. Neuroscience. 301, 26–38. [DOI] [PubMed] [Google Scholar]
- Spann RA, et al. , 2017. Chronic spinal cord changes in a high-fat diet-fed male rat model of thoracic spinal contusion. Physiol. Genomics 49, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SJ, D’Angelo H, Soch A, Watkins LR, Maier KA, Barrientos RM, 2017. High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol Aging. 58, 88–101. 10.1016/j.neurobiolaging.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassart RM, et al. , 2018. The axon-myelin unit in development and degenerative disease. Front. Neurosci 12, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stee MF, et al. , 2018. Actions of metformin and statins on lipid and glucose metabolism and possible benefit of combination therapy. Cardiovasc. Diabetol 17, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberger J, et al. , 2003. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation 107, 1448–1453. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, et al. , 2006. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 127, 397–408. [DOI] [PubMed] [Google Scholar]
- Stubbs CO, Lee AJ, 2004. The obesity epidemic: both energy intake and physical activity contribute. Med. J. Aust 181, 489–491. [DOI] [PubMed] [Google Scholar]
- Suh HS, et al. , 2013. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J. Neuroinflammation 10, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri A, et al. , 2019. A randomized controlled trial on the efficacy, safety, and pharmacokinetics of metformin in severe traumatic brain injury. J. Neurol 266, 1988–1997. [DOI] [PubMed] [Google Scholar]
- Tao L, et al. , 2018. Neuroprotective effects of metformin on traumatic brain injury in rats associated with NF-kappaB and MAPK signaling pathway. Brain Res. Bull 140, 154–161. [DOI] [PubMed] [Google Scholar]
- Terson de Paleville DGL, et al. , 2019. Epidural stimulation with locomotor training improves body composition in individuals with cervical or upper thoracic motor complete spinal cord injury: a series of case studies. J. Spinal Cord Med 42, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault P, et al. , 2016. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget. 7, 67808–67827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans S, et al. , 2014. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the Renin Angiotensin system. J. NeuroImmune Pharmacol 9, 209–217. [DOI] [PubMed] [Google Scholar]
- Tripathi R, McTigue DM, 2007. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia. 55, 698–711. [DOI] [PubMed] [Google Scholar]
- Tyagi E, et al. , 2013. Vulnerability imposed by diet and brain trauma for anxiety-like phenotype: implications for post-traumatic stress disorders. PLoS One 8, e57945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdearcos M, et al. , 2014. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 9, 2124–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, et al. , 2019. Spinal cord injury: pathophysiology, treatment strategies, associated challenges, and future implications. Cell Tissue Res. 377, 125–151. [DOI] [PubMed] [Google Scholar]
- Vigano F, et al. , 2013. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat. Neurosci 16, 1370–1372. [DOI] [PubMed] [Google Scholar]
- Wang C, et al. , 2016. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun 477, 534–540. [DOI] [PubMed] [Google Scholar]
- Watson GS, Craft S, 2006. Insulin resistance, inflammation, and cognition in Alzheimer’s disease: lessons for multiple sclerosis. J. Neurol. Sci 245, 21–33. [DOI] [PubMed] [Google Scholar]
- Weil ZM, et al. , 2014. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol. Dis 70, 108–116. [DOI] [PubMed] [Google Scholar]
- White R, Kramer-Albers EM, 2014. Axon-glia interaction and membrane traffic in myelin formation. Front. Cell. Neurosci 7, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, et al. , 2011. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. J. Neurosci 31, 9544–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahiro AM, et al. , 2019. Classification of obesity, cardiometabolic risk, and metabolic syndrome in adults with spinal cord injury. J. Spinal Cord Med 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, et al. , 2015. The thrombin receptor is a critical extracellular switch controlling myelination. Glia. 63, 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, et al. , 2016. Interplay between exercise and dietary fat modulates myelinogenesis in the central nervous system. Biochim. Biophys. Acta 1862, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, et al. , 2017. Protease activated receptor 2 controls myelin development, resiliency and repair. Glia. 65, 2070–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino A, et al. , 1991. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 561, 106–119. [DOI] [PubMed] [Google Scholar]
- Zeger M, et al. , 2007. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia. 55, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, et al. , 2017. Metformin ameliorates BSCB disruption by inhibiting neutrophil infiltration and MMP-9 expression but not direct TJ proteins expression regulation. J. Cell. Mol. Med 21, 3322–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.