

Abstract
Photothermal therapy (PTT) attracted tremendous attention due to its non-invasiveness and localized treatment advantages. However, heat shock proteins (HSPs) associated self-preservation mechanism bestows cancer cells thermoresistance to protect them from the damage of PTT. To minimize the thermoresistance of cancer cells and improve the efficacy of PTT, we developed an integrated on-demand nanoplatform composed of a photothermal conversion core (gold nanorod, GNR), a cargo of a HSPs inhibitor (triptolide, TPL), a mesoporous silica based nano-reservoir, and a photothermal and redox di-responsive polymer shell. The nanoplatform can be enriched in the tumor site, internalized into cancer cells, release the encapsulated TPL under the trigger of intracellular elevated glutathione and near-infrared laser irradiation. Ultimately, the liberated TPL could diminish thermoresistance of cancer cells by antagonizing the PTT induced heat shock response via multiple mechanisms to maximize PTT effect for cancer treatment.
Keywords: Photothermal therapy, gold nanorod, heat shock protein, mesoporous silica nanoparticle, cancer
Graphical Abstarct
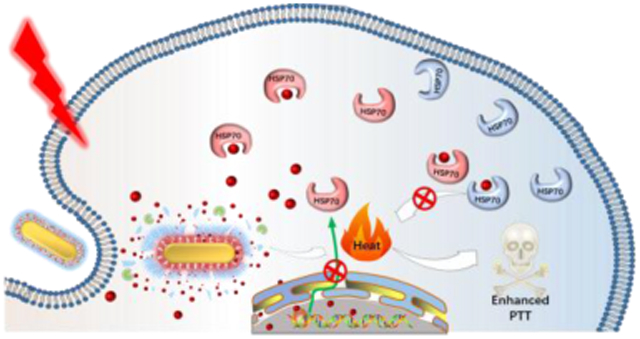
Triptolide nano-reservoir, which can be in-situ activated by NIR laser irradiation and intracellular high redox potential, has been developed to overcome the thermoresistance of cancer cells. Thanks to the inhibited PTT induced heat shock response, the photothermal therapy efficacy of the gold nanorod/mesoporous nanocomplex is greatly improved.
1. Introduction
Photothermal therapy (PTT), a localized therapeutic modality in cancer treatment induced by the irradiation of a near-infrared light (NIR), has recently attracted considerable attention because of its non-invasiveness, deep tissue penetration, and spatiotemporal selectivity [1]. PTT generally employs photothermal conversion agents, such as NIR resonant nanomaterials (gold nanorod [2], carbon nanotubes [3], graphene [4]), semiconductor [5], and indocyanine green (ICG) [6], to convert NIR light to hyperthermia (above 42 °C), which subsequently cook and kill cancer cells, and ultimately realizing effective tumor ablation or even total tumor eradication. Unfortunately, as a self-preservation mechanism, the hyperthermia-treated cancer cells can activate their anti-apoptotic and cytoprotective pathways to acquire tolerance to heat stress, termed as thermoresistance [7], especially when the heat is within “hyperthermia range” (42–47 °C) [8]. In order to minimize the thermoresistance of cancer cells, high dose of photothermal conversion agents, high laser intensity, or repeated laser irradiation was introduced to achieve a harsh photothermal heating effect that cancer cells cannot handle. However, those approaches may induce dose-dependent toxicity, inflammation, and inevitable heat diffusion that threaten the cells and healthy tissues nearby. An alternative strategy is to integrate PTT with other treatment modalities of different mechanisms, such as photodynamic therapy (PDT) [9], chemotherapy [10], gene therapy [11], and immunotherapy [12] in a single nano-platform (all in one), which exhibits great potential to achieve synergistic antitumor efficacy. However, the elevated tolerance to heat is still in suspense. Moreover, those multi-modular systems require complicated construction and tediously spatiotemporal accommodation, which have the potential of causing confusing intermission and diming their superiority.
Heat shock proteins (HSPs) are known as a family of ATP-dependent chaperone molecules that play diverse roles in the regulation of signal transduction and protective effect from adverse conditions, such as heat [13]. In many types of cancer cells, the expression of HSPs is significantly elevated, which can serve as a biomarker [14], involving in various processes such as tumor cell proliferation, invasion, metastasis. Moreover, its overexpression is positively correlated with the poor prognosis and tolerance to multiple therapies. It has been proven that HSPs play a critical role in initiating the defense mechanism of tumor in thermoresistance [15]. Upon hyperthermia, tumor cells would overexpress HSPs rapidly to repair the thermally damaged proteins and initiate relative anti-apoptosis mechanisms, such as inhibiting caspase-3 activation [16], thus compromising the efficiency of PTT. With the expectation of elevating tumor cell’s susceptibility to heat, the combined application of HSP inhibitors or specific siRNA with PTT would be a promising approach to maximize the efficiency of PTT. Several HSPs inhibitors have been exploited to reduce the thermoresistance in PTT [17]. For instance, Chen and co-workers [18] fabricated a mutually synergistic micelle co-loaded with Cypate and a targeted therapeutic molecule 17-allylamino-17-demethoxygeldanamycin (17-AAG), a specific heat shock protein 90 ( HSP90 ) inhibitor. 17-AAG could specifically bind with heat-induced HSP90, thereby inhibiting anti-apoptotic p-ERK1/2 proteins and thermoresistant associated p-Akt to boost PTT efficiency. Gu and co-workers [17f] integrated small interfering RNAs against heat shock protein 70 with a hollow gold nanoshell to construct a light-controlled dual-functional nanosystem. The NIR irradiation not only manipulated hyperthermia mediated PTT, but also triggered the siRNAs detaching from the nanosystem specifically and escaping from endosomes for HSP70 silencing, which was one of the main drawbacks for gene therapy [19]. The smart nanosystem delicately realized localized PTT with controlled HSP70 silencing, showing great potential for clinical translation. However, in these cases, small molecule inhibitors against HSPs combined with hyperthermia lacked a synchronized release profile. Furthermore, the effect of small molecule inhibitors against HSPs was usually hysteretic. They only bind specific sites of HSPs during hyperthermia, reducing the antagonism to heat, while having a limited effect on their stressful expression. Moreover, for siRNA delivery, a series of barriers should be solved before it shows efficacy, such as serum instability in circulation system, poor cell membrane penetrability due to its negative charge, and inability of endosomal escape. Although these barriers were overcome by the virtue of some delicate nanosystems, their complicated design would increase the complication and decrease repeatability for clinical translation. Thus, there is an urgent need to develop a desirable nano-platform that integrates PTT with HSPs silencing or inhibiting effect in a general strategy.
Triptolide (TPL), a diterpenoid triepoxide extracted from Chinese herb Tripterygium wilfordii Hook F, has been used for more than centries in traditional Chinese medicine. TPL exerts a myriad of pharmacological activity against different diseases, including inflammatory diseases [20], antifertility [21], autoimmune diseases [20a, 20b] and cancer [22]. Many studies have reported that TPL exhibited super anti-proliferation and anti-metastasis effects, which were comparable or superior to those of some conventional antitumor drugs, such as doxorubicin, mitomycin, paclitaxel, and cisplatin. Its potential mechanisms include inducing autophagy [23], cell cycle arrest [24], causing lysosomal-mediated programmed cell death [25], activating caspase-dependent cell death [26], inhibiting transcription factor NF-kappaB and AP-1 transcriptional activity [27], especially involving in blocking heat shock response [22a, 28]. It was revealed that a likely target of TPL is in the assembly of functional transcriptionally active heat shock transcription factors 1 (HSF1) complexes on the HSP70 promoter [29]. TPL abrogates the transactivation function of HSF1 at a low nanomolar level, thus suppresses the HSP70 promoter-reporter construct and endogenous HSP70 gene expression, but not interferes in the early events of trimer formation, hyperphosphorylation, and DNA binding, which renders treated cell more sensitive to various stress-induced cell apoptosis. Another study validated the regulatory function of TPL in heat shock response is independent of HSF1. It further revealed that TPL can upregulate miR-142-3p in PDAC cells, which directly bound to the 3’UTR of HSP70 and decreased HSPA1B (HSP70) mRNA and protein levels [28a]. However, the underlying mechanism of miR-142-3p upregulation by TPL remains to be unclarified. Besides inhibiting the expression of HSPs at transcription level, TPL also abolish existing HSPs chaperone activity by binding its specific domain. Given the super and general inhibition effect of TPL on HSPs, integration TPL with PTT is expected to overcome heat endurance and maximize PTT efficiency.
Hereby, we designed an integrated nanoplatform composed of a photothermal conversion core (gold nanorod, GNR), a cargo of a HSPs inhibitor (TPL), a mesoporous silica based nano-reservoir, and a photothermal and redox di-responsive polymer shell (poly[(2-(pyridin-2-yldisulfanyl)-co-[poly(ethylene glycol)]-co-poly(N-Isopropyl methacrylamide)]) (PDA-PEG-PNiPMA, 3P) [30] was designed and fabricated. Given the strong surface plasmon resonance, outstanding light to heat conversion efficiency and high heat stability [31], GNR as photothermal conversion agents was chosen to be the core of nanocomplex. The GNR core was further coated with mesoporous silica (GRN@MSN) for loading TPL (TPL-GRN@MSN) in a silica shell. The polymer nanogel of 3P, has outstanding thermal and redox potential responsive expansile property [30], was decorated on GNR via electrostatic interaction [32] and further crosslinked by disulfide bonds (TPL-GRN@MSN-3P) as responsive gatekeepers to avoid undesirable premature drug leakage during the circulation. After the nanocomplex was internalized into cancer cells, the hyperthermia generated from NIR irradiation and intracellular high level of GSH would lead to the polymer gatekeeper rapidly expand and disassemble to boost TPL release. Subsequently, the liberated TPL would antagonize the PTT induced heat shock response via multiple mechanisms to maximize PTT (Scheme 1).
Scheme 1.

Schematic illustration of A) the preparation process of GSH and photothermal diresponsive of TPL-GNR@MSN-3P and B) the use of TPL-GNR@MSN-3P for enhanced photothermal therapy efficacy.
2. Results and Discussion
2.1. Synthesis and characterization of TPL-AuNR@MSN-3P
The thermo-responsive polymer (PDA-PEG-PNiPMA, 3P) was synthesized according our previously published method [30] and its synthetic route was shown in Figure S1. The chemical structure of PDS-OH, PDA monomer, and PDA-PEG-PNiPMA were confirmed by proton nuclear magnetic resonance (1H NMR) (Figure S2). The average molecular weight of PDA-PEG-PNiPMA was around 30,000 Da calculated by gel permeation chromatography (Figure S3) using a polystyrene standard calibration curve. PNiPMA is one of the most intensively investigated thermal-responsive polymer, which would undergo a reversible phase transition from hydrophilic to hydrophobic when environment temperature rises higher than its Low Critical Solution Temperature (LCST, 43 °C) [35]. By taking advantage of this property, many PNiPMA polymer-based micro/nanoparticles have been developed as thermo-responsive smart drug delivery systems [36]. Recently, we reported a PNiPMA based triple-responsive expansile nanogel, which was fabricated from the triblock copolymer PDA-PEG-PNiPMA. Different from the conventional shrunken property, PDA-PEG-PNiPMA based triple responsive nanogel (TRN) showed obvious expanding property when the temperature was higher than its transition temperature. Traditional PNiPMA containing nanoparticle shrinks due to the collapse of PNiPMA layers during the transition from hydrophilic to hydrophobic state. Similarly, for the PDA-PEG-PNiPMA nanogel, when the temperature rise to higher than its transition temperature, the PNiPMA segments will become hydrophobic and phase separate from the originally codissolved PEG segments. Due to the restrain of the disulfide bonds, the hydrophobic PNiPMA segment can not join the hydrophobic PDA layer, while forming individual local hydrophobic pocket instead. Therefore, the size of the PDA-PEG-PNiPMA nanogel increases when the temperature is higher than its transition temperature. It was revealed that the transition temperature of TRN can be regulated by simply tuning its crosslinking density (CD) during the fabrication process. Based on our preliminary investigation of DLS size (Figure S4), the transition temperature of TRN with 25 % CD was around 37 °C, while the transition temperature shifted to around 42 °C when CD increased to 30 %, which is slightly higher than physiological temperature. Therefore, in the following experiments, CD was fixed at 30 % unless otherwise specified.
GNRs were synthesized via a seed-mediated surfactant-directed method. Transmission electron microscope (TEM) images (Figure 1A) showed that the average length and width of GNRs were 42.3 ± 2.1 nm and 10.8 ± 1.3 nm, respectively. The aspect ratio is around 3.9, which was critical for near-infrared (NIR) light to induce photothermal conversion effect. A mesoporous silica shell was coated on GNRs surface by a modified sol–gel method to prepare GNR@MSN. The TEM image (Figure 1B) showed GNR@MSN possessed a homogeneous shell with a thickness of 20 nm and ordered pores of around 3 nm, which offered a high capacity for drug loading. The loading efficiency of TPL into GNR@MSN (TPL-GNR@MSN) via hydrophobic interaction and absorption effect was 50 %. Moreover, the efficiency could be easily tuned by changing the feeding ratio of TPL to GNR@MSN.
Figure 1.

TEM images of GNR A), GNR@MSN B) and GNR@MSN-3P C). D) UV-vis absorption spectra of GNR@MSN-3P and its components. E) Zeta potential of GNR, GNR@MSN, and GNR@MSN-3P. F) The dynamic light scattering size of GNR@MSN-3P in various temperatures.
To prevent TPL from premature releasing in the circulatory system and realize expected photothermal responsive release in the tumor site, PDA-PEG-PNiPMA synthesized in advance was coated onto TPL-GNR@MSN via electrostatic interaction between the pyridine of PDA-PEG-PNiPMNA and silanol group on the surface of GNR@MSN, and further strengthened by intermolecular crosslinking through the formation of disulfide bonds to yield TPL- GNR@MSN-3P nanoparticles. The appearance of dim pore and smooth surface in TEM image (Figure 1C) indicated successful polymer coating. The longitudinal surface plasmon resonance (SPR) spectrum (Figure 1D) and zeta potential of GNR, GNR@MSN and GNR@MSN-3P were characterized. The typical SPR peak of as-prepared GNR was at 770 nm. The addition of a mesoporous silica shell induced the red shift of SRP peak to 784 nm owing to the change of local refractive index [37]. Finally, the coating of PDA-PEG-PNiPMA made the SPR peak further shifted to 800 nm, which located in the NIR region and displayed great potential for light-mediated therapy. Meanwhile, two additional peaks appeared at 240 nm and 280 nm in GNR@MSN-3P, which were coincident to ones of PDA-PEG-PNiPMA nanogel, further confirmed the successful coating of PDA-PEG-PNiPMA polymer in GNR@MSN-3P. The zeta potentials (Figure 1E) of GNR, GNR@MSN, and GNR@MSN-3P were +3.8 mV, −30.5 mV, and −25.6 mV, respectively. This inverted and fluctuated zeta potential change provided strong evidence on the successful construction of nanosystem. Subsequently, we investigated the thermal-responsive (expanding) property of GNR@MSN-3P. Similar to TRN, GNR@MSN-3P was relatively stable at room temperature (25 °C) and physiological temperature (37 °C), its DLS size (Figure 1F) was 151.1 nm and 152.4 nm, respectively. However, GNR@MSN-3P size rapidly expanded to 295 nm, 385 nm and 455 nm (Figure 1F) when the temperature rose to 42 °C, 45 °C, and 50 °C, respectively, which suggests an opportunity for boosted drug release. It is also worth noting that the addition of GSH could induce slight expanding of the nanoparticle (Figure S5).
2.2. Photothermal effect
To evaluate the photothermal transformation efficiency and capacity of GNR@MSN-3P, different concentrations of GNR@MSN-3P was subjected to NIR laser irradiation at 808 nm around its longitudinal SPR wavelength and the temperature change was recorded. As shown in Figure 2A, a sharp temperature increase was observed in GNR@MSN-3P solution in a short period of time and plateaued eventually after exposure to the irradiation. As expected, the temperature increase was concentration-dependent. For instance, when the concentration of GNR@MSN-3P was 12.5 μg/mL, the solution temperature risen by 22.3 °C within 8 min at the irradiation intensity of 2 W/cm2, which was sufficient to induce irreversible hyperthermia in tumor cells. A similar conclusion could also be drawn by analyzing infrared thermal photographs (Figure 2B). Furthermore, a laser intensity-dependent temperature rise was also observed (Figure 2C). These results indicated that the photothermal effect of GNR@MSN-3P could be finely tuned by regulating either nanoparticles concentration or laser intensity. To evaluate the photothermal stability of GNR@MSN-3P, the nanocomposite was challenged with 6 cycles of heating-cooling. The temperature-time curve and peak shape (Figure 2D) were of no obvious change after the recurrent heating-cooling procedure, which suggests that nanocomposite had super photothermal stability. GNR@MSN displayed a similar temperature-time curve (Figure S6a-b) and photothermal stability property (Figure 2D) to GNR@MSN-3P, suggesting there was no influence of the coating of PDA-PEG-PNiPMA polymer on GNR@MSN photothermal property.
Figure 2.

The temperature change A) and infrared thermal images B) of GNR@MSN during 808 nm laser irradiation (2 W/cm2). C) Temperature change of GNR@MSN (25 μg/mL) under 808 nm laser irradiation with different laser power. D) Photothermal stability of GNR and GNR@MSN under 808 nm laser irradiation (2 W/cm2). E) TPL release profiles from TPL-GNR@MSN at pH 5.0 and 7.4 with or without GSH and 808 nm laser (2 W/cm2). The release study was carried out at 37 °C unless otherwise noted.
2.3. Drug release kinetics
To validate the gatekeeper function of the coated PDA-PEG-PNiPMA polymer, which prevents premature leakage of drug during the circulation and realizes on-demand release at a targeted site, the TPL release profile was studied in vitro. As shown in Figure 2E, a little amount of TPL was released from TPL- GNR@MSN-3P in PBS buffer (pH 7.4), and the 24 h cumulative release of TPL was less than 10 %, indicating the favorable shield effect of PDA-PEG-PNiPMNA. However, upon laser irradiation, TPL displayed an obvious burst release behavior along with the laser irradiation, and the cumulative release of TPL was ladder-like up to 42% after 3 cycles of irradiation ON/OFF alternating treatment within 24 h. Considering the co-exist of high GSH concentration in cancer cell cytoplasm and the disulfide crosslink in the PDA-PEG-PNiPMA shell, we added 10 mM of GSH in PBS buffer to simulate a reducing cytoplasm environment to further study its drug release behavior. It was revealed that GSH could promote TPL sustained and slow-release, and the accumulated release was around 30% in 24 h, mainly attributing to the break of disulfide bond by GSH. As expected, the combination of laser irradiation and GSH could trigger drug release in cooperation, and the accumulated release was steeply gradient up to 60 % in 24 h. These results indicate the nanocomposite could respond to intrinsic elevated redox GSH and extrinsic flexible NIR to programmed release loaded TPL.
2.4. Cellular uptake and cell-based photothermal effect
The internalization of nanocomposite into targeted cells is essential for a drug delivery system, especially when the drug target is located in an intracellular region. To improve cancer cell internalization of nanoparticles, one common strategy is decorating a targeting ligand on the surface of the nanoparticles, which endows high affinity for the nanoparticles to bind with the receptor specific or over-expressed on the membrane of cancer cells [38]. However, surface decorations may change the physicochemical property of the nanoparticle, especially its surface charge and hydration shell, and subsequently affecting its pharmacokinetic property [39]. Thus, no surface decoration was adopted in this system. The uptake behavior of 4T1 cells to the GNR@MSN-3P was investigated by a confocal fluorescence microscope. Red fluorescence signal was presented throughout the entire 4T1 cell cytoplasm after 6 h of incubation (Figure 3A), suggesting that GNR@MSN-3P can effectively enter the cancer cells.
Figure 3.

A) The fluorescence images of 4T1 cells incubated with Cy3 labeled GNR@MSN-3P for different times. The infrared thermal images B) and temperature change C) of 4T1 cells under 808 nm laser irradiation (2 W/cm2) after incubation with GNR@MSN-3P for 6 h.
After confirming the successful internalization of GNR@MSN-3P into cancer cells, we further investigated its photothermal effect in killing cancer cells. 4T1 cells were incubated with GNR@MSN-3P of different concentrations for 6 h and then exposed to irradiation for 5 min, and the temperature change was recorded. As shown in Figure 3B and 3C, there was nearly no change in temperature for the untreated cells. However, the temperature of the culture medium rose rapidly (within 3 min) upon exposure to the laser irradiation in the GNR@MSN-3P pre-incubated cells in a concentration-depend manner. The plateaued temperature were 40.5, 42.5, 46.3, 48.2, and 53.2 °C with the concentration of 6.3, 12.5, 25.0, 50, and 100 μg/mL, respectively, reaching “hyperthermia range” [40] (39-42 °C, called mild hyperthermia; > 45 °C, called ablation) to cook and kill cancer cells directly.
2.5. HSP70 expression
During a hyperthermia treatment, cancer cells become tolerant to heat stress, which is termed as thermoresistance. It was demonstrated that heat shock proteins (HSPs), acting as chaperone proteins, are the critical factors for cancer thermoresistance [14]. HSPs as multifunctional intracellular chaperone proteins, which are typically being expressed to maintain cellular structural and functional integrity in a low level [41]. Once undergoing heat stress, HSPs expression in cancer cells would rapidly elevate to a relatively high level to exhibit cytoprotective and anti-apoptotic effect. Herein, we expected to minimize this thermoresistance, and consequently endow cancer cells susceptibility to hyperthermia by incorporating triptolide (TPL) as a universal HSPs inhibitor. Recently, some studies reported that TPL could inhibit cancer cell proliferation and metastases by down-regulating HSP70 via inactivation of glycosylation of the transcription factor Sp1 [42] and upregulating negative gene regulators miR-142-3p [43]. Therefore we used HSP70 as a representative HSP to study the effect of TPL on HSPs. First, we used western blotting to investigate the influence of free TPL on HSP70 expression. As shown in Figure 4A, the expression of HSP70 in 4T1 cells treated with various concentrations (25, 50, and 100 nM) of TPL for 24 h were of no obvious difference compared with control cells. However, when the treatment time was prolonged to 48 h, HSP70 protein expression was significantly decreased and the decrease exhibited a distinct dose-dependent manner (Figure S7). Those results indicate that TPL had a limited effect on HSP70 physiological expression in 4T1 cells within a short time, but significantly reduced its physiological expression in a relatively long time, which may be one of the primary mechanism of inducing cancer cell death. Next, we investigated the effect of TPL on heat stress-induced HSP70 expression. As shown in Figure 4A, in comparison to unheated cells, a boosted expression of HSP70 in 4T1 cells incubated at defined mild temperature (42 °C) was observed, validating that heat could trigger a heat shock response to elevate HSP70 expression in cells. For the cells pretreated with TPL, the heat-induced HSP70 upregulation was suppressed in a dose-dependent manner. At the concentration of 100 nM, the HSP70 expression was suppressed to its physiological basal level. Thereafter, we further verified whether TPL-GNR@MSN-3P could exhibit a similar effect. The western blotting result in Figure 4B indicated that similarly to free TPL, TPL loaded nanoparticles also did not affect physiologic HSP70 expression in unheated cells, but it could restore HSP70 expression to initial state in the pretreated cell post-subjecting to hyperthermia, which would make cancer cells more vulnerable to heat, and therefore enhance the efficiency of PTT induced hyperthermia therapy.
Figure 4.

HSP70 expression in 4T1 cells pretreated with free TPL: A) and TPL-GNR@MSN-3P; B) under heat or not by western blotting analysis, β-actin was used as an internal control. C) Fluorescence images of live (green) and dead (red) 4T1 cells co-stained with Calcein-AM and EthD-1 after different formations treatment with or without 808 nm laser (2 W/cm2, 5 min). D) Normalized cell viability of 4T1 cells treated with free TPL, GNR@MSN-3P and TPL-GNR@MSN-3P under 808 nm laser irradiation or not (2 W/cm2, 5 min).
2.6. In vitro cell killing evaluation
Based on the superior photothermal effect of GNR@MSN-3P in cancer cells and the antagonism effect of TPL on HSP70 acute hyperexpression induced by hyperthermia, we inferred that a synergistic antitumor effect may be achieved through the combination of PTT and TPL. We first utilized live/dead cell staining assay to directly visualize cell death after receiving different treatments, where live cells and dead cells were differentiated by staining with calcein-AM (green) and ethD-1 (red), respectively. As shown in Figure 4C, most cells in control with or without laser exposure, and GNR@MSN-3P without laser exposure were alive as evidenced by the green fluorescence. A moderate number of cells with red fluorescence in free TPL treated cells with or without laser exposure, and GNR@MSN-3P treated cells with laser exposure were observed. Although some cells with red fluorescence were presented in the TPL-GNR@MSN-3P without irradiation treated group, nearly all the cells in TPL-GNR@MSN-3P coupled with NIR irradiation treated group were dead and exhibited red fluorescence, confirming the strong synergetic killing effect of PTT and TPL for TPL-GNR@MSN-3P.
The in vitro anticancer activity TPL-GNR@MSN-3P was further quantitatively investigated in 4T1 cells by methylthiazolyl tetrazolium (MTT) assay. As shown in Figure 4D, more than 85 % of 4T1 cells treated with GNR@MSN-3P were alive, even at the concentration of 200 μg/mL, indicating the excellent biocompatibility of the nanoparticle. 4T1 cells were sensitive to free TPL, and its cytotoxicity to 4T1 cells presented a dose and time-dependent manner (Figure 4D and Figure S8). Compared with GNR@MSN-3P treated cells, the cell viability of TPL-GNR@MSN-3P treated cells was a little bit lower due to the appearance of toxic TPL released from the nanoparticle, while was higher than that in free TPL treated cells. This difference may be attributed to that intracellular gradual release of TPL triggered by GSH compared to swarming exposure of free TPL (Figure 2E). Upon NIR laser irradiation, GNR@MSN-3P generated heat and killed cancer cells in a dose-dependent manner, with only 32 % of cells alive at the GNR concentration of 50 μg/mL. Dramatically, an enhanced cell-killing effect was observed in TPL-GNR@MSN-3P treated cells coupled with NIR irradiation, more than 85 % cells were killed at GNR and TPL concentration up to 50 μg/mL and 200 nM, respectively, which was 2.53 times and 1.35 times higher than that in TPL-GNR@MSN-3P treated cells without exposure to laser and GNR@MSN-3P treated cell with NIR irradiation, respectively. However, exposure to NIR irradiation was of no influence on cell viability in free TPL treated cells. With the consideration of above TPL release profiles (Figure 2E) from the nanoparticle and its antagonism effect on HSP70 (Figure 4A, 4B), the enhanced therapeutic effect in TPL-GNR@MSN-3P treated cells under irradiation could be attributed to TPL burst release triggered by heat and redox dual triggers significantly increase tumor cell sensitivity to hyperthermia. Based on the results showing in Figure 4A and 4B, TPL, both free one and TPL-GRN@MSN-3P, downregulates the expression of HSP70 in a dose-dependent manner, which subsequently sensitizes the 4T1 cells to PTT treatment. The boosted cell killing effect of PTT from the TPL-GRN@MSN-3P treatment was evidenced by the significantly fewer alive cells as shown in Figure 4C and 4D.
2.7. Biodistribution and in vivo photothermal effect
Encouraged by the exciting in vitro results, we further investigated the in vivo properties and explore the feasibility of GNR@MSN-3P for cancer therapy in a 4T1 cell based xenograft breast cancer model. The in vivo distribution of the cyanine5 (Cy5)-labeled nanoparticle at the prescribed time (2, 4, 6, 8, 24 h post-injection) was examined with an IVIS small animal imaging system, which can provide the optimal irradiation window to realize an optimal photothermal effect. As shown in Figure 5A, it was found that the fluorescence signals in the tumor region gradually increased over the first 6 h post-injection, indicating the accumulation process of nanoparticles in the tumor due to the enhanced permeability and retention (EPR) effect [44]. At 6 h post-injection, the fluorescence intensity in the tumor reached the maximum, demonstrating the max accumulation of nanoparticles in the tumor. Subsequently, the fluorescence signals fainted slowly over time. After 24 h post-injection, the mice were sacrificed, and the tumor and main organs were harvested for ex vivo imaging (Figure 5B). Obviously, the fluorescence intensity in the tumor was still higher than other organs except for the reticuloendothelial system, which caused relatively high distribution in the liver, spleen, and lung.
Figure 5.

A) In vivo fluorescence images of 4T1 tumor-bearing mice at indicated time point following iv injection with Cy5 labeled GNR@MSN-3P. B) Ex vivo fluorescence images of the tumor and main organs at 24 h post-injection. C) In vivo thermal images and D) temperature rising curves of 4T1 tumor-bearing mice at 6 h post-injection of PBS, free TPL, GNR@MSN-3P, and TPL-GNR@MSN-3P) with a 808 nm laser-irradiation (2 W/cm2).
Based on the above in vivo imaging observation, 6 h post-injection was selected as the optimal time point for laser irradiation. Thus, we further investigated the laser-triggered photothermal effect of GNR@MSN-3P in vivo 6 h after intravenous administration of the nanoparticle. As shown in the photothermal images (Figure 5C) and time-temperature curves (Figure 5D), the local tumor temperature elevated rapidly to 52.2 and 50.9 °C within 2 min in GNR@MSN-3P and TPL-GNR@MSN-3P treated mice, respectively, which were high enough to ablate malignant cells. In marked contrast, the same irradiation only resulted a little temperature increase of the tumor in the free TPL and PBS treated mice, suggesting negligible hyperthermia. These results highlighted the promising photothermal performance of GNR@MSN-3P in vivo.
2.8. In vivo antitumor therapy
Encouraged by the enhanced cytotoxicity of TPL in GNR@MSN-3P coupled with NIR irradiation in vitro, effective accumulation of nanoparticles in the tumor, and favorable NIR-induced photothermal conversion in vivo, we further investigated the in vivo antitumor effect of TPL-GNR@MSN-3P in 4T1 tumor-bearing mice. After the tumor volume reached around 100 mm3, the mice were randomly divided into 8 groups: (1) PBS, (2) PBS with 808 nm laser irradiation, (3) Free TPL, (4) Free TPL with 808 nm laser irradiation, (5) GNR@MSN-3P, (6) GNR@MSN-3P with 808 laser irradiation, (7) TPL-GNR@MSN-3P, and (8) TPL-GNR@MSN-3P with 808 laser irradiation.
As shown in Figure 6A and 6C, the relative tumor volume in PBS treated control group rapidly increased throughout the experiment. In comparison with the control, no obvious tumor growth inhibition was observed in PBS coupled with laser irradiation treatment group, indicating that NIR irradiation alone was nearly no effect on suppressing the growth of the tumor. Although TPL exhibited superior and a broad spectrum of antitumor efficacy through multiple mechanisms [15a, 22a, 29, 43, 45], the severe toxicity to healthy tissues and frequent multiple dosing were the main obstacles that hinder the clinical application and translation of TPL in oncotherapy [46]. To minimize the potential side effect of TPL, a single dose of TPL at 1 mg/kg was adopted for the in vivo study. Owning to the single and low dose drug administration, only a negligible tumor suppression effect was observed for the free TPL treated group, whether coupled with NIR irradiation or not. As expected, the non-irradiated mice in TPL-GNR@MSN-3P treated group and GNR@MSN-3P treated group did not exhibit noticeable tumor suppression effect. Upon NIR irradiation, GNR@MSN-3P could retard tumor growth to some extent due to the PTT effect, as evidenced by a slight increase in tumor volume. In striking contrast, TPL-GNR@MSN-3P coupled with NIR irradiation exhibited excellent tumor growth inhibitory efficacy, as evidenced by the significant tumor regression and even eradication. Reasonably, NIR irradiation not only induced the photothermal therapy but also triggered the burst release of TPL (Figure 2E) and subsequently inhibited the synthesis of HSPs (Figure 4B), resulting in reduced thermoresistance and improved sensitivity to hyperthermia. Immunofluorescence staining results (Figure 6F and 6G) revealed that HSP70 (green fluorescence) was widely expressed in the control group due to the disordered tumor microenvironment, which could maintain tumor aggressiveness and heterogeneity and protect the tumor from various treatments [14]. Upon single hyperthermia treatment in the GNR@MSN-3P coupled with 808 laser irradiation group, the stressed-induced protective HSP70 was markedly upregulated, weakening the thermal ablation induced damage. By contrast, the integration of TPL and hyperthermia in the TPL-GNR@MSN-3P coupled with NIR irradiation substantially prevented the upregulation of HSP70, sensitizing cancer cells to hyperthermia treatment, and thereby enhancing the PTT efficacy. These results were consistent with the observed HSP70 expression upon different treatments in vitro. The final tumor size (Figure 6B) and the average tumor weight (Figure 6D) also validated that TPL-GNR@MSN-3P coupled with NIR irradiation yielded the best antitumor activity. In addition, no significant body weight change was observed (Figure 6E) in all groups during the treatment, suggesting all treatments had no significant systemic toxicity to the animals. Moreover, hematoxylin and eosin (H&E) stained tissue sections (Figure S9) of major organs (heart, liver, spleen, lung, and kidney) also confirmed that no apparent tissue damage was observed among all groups, demonstrating the favorable biocompatibility and systemic safety of the TPL-GNR@MSN-3P nanoparticle.
Figure 6.

A) Representative photographs of tumor-bearing mice after various treatments during the 12-d of post treatment. B) Photographs of excised tumors of each group on day 12. C) Tumor growth profiles of 4T1 tumor-bearing the mice treated with various formulations. D) Tumor weight obtained on day 12. E) Body weight changes of 4T1 tumor-bearing mice in different groups. F) Immunofluorescence staining of HSP70 of tumors after different treatments. G) Quantitative analysis of HSP70 in the tumor tissue.
3. Conclusion
An integrated on-demand nanoplatform, TPL-GNR@MSN-3P, which is composed of a photothermal conversion core, a cargo of a HSPs inhibitor, a mesoporous silica based nano-reservoir, and a temperature and redox dual-responsive polymer shell, has been developed. The nanoplatform can be enriched in the tumor site, internalized into cancer cells. Upon NIR light irradiation, the gold nanorod generates heat to yield photothermal therapy effect for killing cancer cells and trigger the expansion of the polymer shell. The temperature and redox dual-responsive polymer serves as a gatekeeper, which will allow the encapsulated triptolide be released intracellularly due to the elevated endogenous glutathione and external NIR light-induced in-situ heat production. The on-demand released triptolide reduces the photothermal therapy induced thermoresistance by down-regulating the production of heat shock proteins. Consequently, the TPL-GNR@MSN-3P nanoplatform coupled with NIR irradiation alleviates tumor burden by 90% while not causing systemic toxicity. Thus, the on-demand TPL-GNR@MSN-3P nanoplatform provides a novel tool to maximize the benefit of photothermal therapy in cancer treatment.
4. Experimental Section
Chemicals
Tris(2-carboxyethyl)phosphine (TCEP), 2, 2-Azobisisobutyronitrile (AIBN), Poly(ethylene glycol)methacrylate (Mn= 360 Da), N-isopropyl methacrylamide (NiPMN), sodium borohydride (NaBH4), ascorbic acid, tetrachloroauric acid (HAuCl4·3H2O), silver nitrate (AgNO3), Tetraethylorthosilicate (TEOS), N-cetyltrimethylammonium bromide (CTAB) , and glutathione (GSH) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Cyanine5 NHS ester was purchased from Lumiprobe Corporation (Hallandale Beach, FL, USA). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetra-zoliumbromide (MTT), fetal bovine serum (FBS), 0.25% trypsin-EDTA, penicillin-streptomycin (PS), phosphate buffered saline (PBS), Hoechst 33342, calcein AM, ethidium Homodimer-1 (EthD-1), and HSP70 Monoclonal Antibody (5A5) were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). All the other solvents used in this research were purchased from Sigma-Aldrich and used without any further purification unless otherwise noted.
Synthesis of Synthesis of thermal and redox dual-responsive PDA-PEG-PNiPMA polymer
PDA monomer was prepared as our previous method [33], and then PDA-PEG-PNiPMA polymer was synthesized by radical polymerization of PDA monomer (PDA), Poly (ethylene glycol) methacrylate (PEG) and N-isopropyl methacrylamide (NiPMA) [30]. Briefly, 1 mmol of PDA (241.3mg), 1 mmol of PEG (Mw 360), and 0.5 mmol of NiPMN were dissolved in 5 ml anisole, degassed for 15 minutes by purging with nitrogen, and then heated to 65 °C under stirring. After the temperature was stable for half an hour, 0.085 mmol of AIBN in 1 ml of degassed anisole was added dropwise. The polymerization was continued for 24 hours at 65 °C. The final polymer was collected and purified by precipitation using ice-cold ether. The structure of the PDA-PEG-PNiPMA polymer was confirmed by 1H NMR and its molecular weight was measured by gel permeation chromatography (GPC, Tosoh, Japan).
Synthesis of Synthesis of gold nanorods and mesoporous silica-coated gold nanorod
The gold nanorods and mesoporous silica-coated gold nanorods were prepared using seed-mediated growth and modified Stöber method with some little modifications [34]. All glassware used was rinsed by aqua regia to ensure their cleanness. For gold nanorods preparation, first, 0.25 ml of 0.01 M HAuCl4.3H2O was added into 9.75 ml of 0.1 M CTAB under vigorously stirring and kept the stirring for 10 min. After that, 0.6 ml of 0.01 M NaBH4 in ice-cold deionized water was injected into mixture quickly and keep the stir for 10 min forming gold seeds. The gold seeds solution was aged for 1 hour at room temperature before used. The growth solution of gold nanorods was prepared by mixing the solution of 475 ml of 0.1 M CTAB, 25 ml of 0.01 M HAuCl4.3H2O and 6.5 ml of 0.01 M AgNO3. Thereafter, 2.75 ml of 0.1 M ascorbic acid was added dropwise to the growth solution under gently stirring. Upon the solution turning to colorless, 0.6 ml of gold seed solution was added to growth solution dropwise under vigorously stirring. The color of the solution would change within 15-20 min, then slowed down the stirring and aged the growth solution for 16 h at 28 °C. The prepared gold nanorod was purified by centrifugation at 13,500 rcf. for 30 min to remove excess CTAB, and then re-dispersed in deionized water for further use. Modified Stöber method was applied to prepare mesoporous silica-coated gold nanorod. Typically, 0.1 M of CTAB was added into the above gold nanorod stock solution to adjust the final CTAB concentration to 0.6 mM and stirred for 30 min at 28°C. Then, a moderate amount of 0.1 M NaOH was added to the solution to adjust the pH value to 10.6~10.8. Next, 200 μl of 20 % TEOS in method was added into the solution dropwise under general stirring, and the reaction was carried on for 20 h at 28 °C. The silica-coated gold nanorod was obtained via centrifugation at 12000 rcf. for 20 min and rinsed with ethanol for 3 times. The precipitate was dispersed in 50 ml of 10 mg/mL NH4NO3 ethanol solution and washed with ultrasound for 1 h, this process was repeated for 3 times to remove the CTAB template in mesoporous silica completely, the final product was denoted as GNR@MSN and suspended in deionized water for further use.
Preparation of TPL-GNR@MSN and TPL-GNR@MSN-3P
One milliliter of TPL methanol solution (2 mg/mL) was added to 4 ml of 400 μg/mL GNR@MSN in methanol, the mixture was treated with sonication for 1 min and gently stirred for 24 h at RT. Then the TPL loaded GNR@MSN (TPL-GNR@MSN) was centrifuged at 12,000 rcf for 20 min and washed with deionized water for 3 times. The supernatant was collected and the unloaded TPL was determined by HPLC (1260 Infinity II, Agilent Technologies, Palo Alta, USA) equipped with a UV-Vis detector at 245 nm. The TPL loading efficiency was calculated as follows: (feeding amount of TPL - the amount of TPL in GNR@MSN)/feeding amount of TPL x 100%. Different loading efficiencies were achieved by adjusting the feed ratio of TPL and GNR@MSN. To prepare PDA-PEG-PNiPMA polymer-coated TPL-GNR@MSN (defined as TPL-GNR@MSN-3P), 80 mg of PDA-PEG-PNiPMA polymer was dissolved in 400 μl of DMSO. TCEP (6.16 mg in 100 μl DMSO), which can induce 30 % of crosslinking density of the polymer, was added to the polymer solution and incubated for 15 min. Then the polymer mixture was added into TPL-GNR@MSN solution under stirring and kept stirring for aerial oxidation crosslinking overnight in the dark. The final TPL-GNR@MSN-3P was purified by centrifugation. The yielded nanoparticles were washed with distilled water for 3 times and stored at 4 °C before further use.
Size and surface charge characterizations
The UV-Vis spectra of different nanoparticles were measured by a multifunctional automatic microplate reader (SpectraMax® i3x, Molecular Devices, Sunnyvale, USA). The morphology of prepared nanoparticles was characterized by transmission electron microscope (Hitachi HT7800 TEM, Hitachi High Technologies, Tokyo, Japan). The DLS sizes and zeta potentials of GNR@MSN-3P and TPL-GNR@MSN-3P at different temperatures (25, 37, 42, 45 °C) were measured by Nano ZS Zetasizer (Malvern Instruments, UK).
Photothermal measurement and photothermal stability assay
For time and concentration-dependent photothermal study, 500 μl of GNR@MSN and GNR@MSN-3P nanoparticles with different concentrations (6.25, 12.5, 25, 50 μg/mL, referred to GNR) were placed in plastic tubes and irradiated with a 808 laser (Model LSR-PS-II, Lasever Inc., China) at a power of 500 mW (2 W/cm2, 8 min), the changes in temperature and infrared thermal image were monitored and captured by a FLIR i7 thermal imaging camera every 0.5 min. For the photothermal stability assay, GNR@MSN and GNR@MSN-3P with 50 μg/mL were subjected to 6 cycles of NIR irradiation (500 mW, 2 W/cm2, irradiating for 5 min, cooling for 5 min), the temperature changes were recorded as aforementioned photothermal measurement.
Evaluation the NIR triggered TPL release
To study the NIR induced TPL release, 0.5 ml of 50 μg/mL TPL-GNR@MSN-3P suspended in PBS (0.01 M, pH7.4, 0.1 % Tween 80) containing 10 mM GSH or not was added to 1.5 ml Eppendorf tube and placed the tubes in a 37 °C incubator with gentle agitation. At specific intervals, the samples were irradiated by 808 nm laser at the output power of 500 mW (2 W/cm2) for 5 min. After that, the samples were centrifuged at 12,000 rcf. for 10 min. The supernatants were withdrawn to quantify the amount of released drug using a HPLC and the deposits were re-suspended by the same volume of fresh medium.
Cell culture
Murine mammary carcinoma 4T1 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10 % of fetal bovine serum (FBS, Gibco), 100 U/mL of penicillin and 100 mg/mL of streptomycin under a humidified atmosphere of 5 % CO2 at 37°C. The culture medium was replaced with a fresh one every two days.
Cellular internalization and intracellular photothermal effect
4T1 cells were seeded in 35 mm glass-bottom dishes at a density 4×104 cells/well. After 24 h of incubation at 37°C, the old medium was replaced with a fresh complete medium containing 50 μg/mL of Cy3 labeled GNR@MSN-3P nanoparticles. After incubating for different time periods (2, 4, 6 h), the cells were washed by PBS and fixed with 4 % paraformaldehyde for 15 min. The nuclei were stained with 10 μg/mL of Hoechst 33254 for 10 min at room time. The uptake of nanoparticles was observed with a Carl Zeiss LSM700 confocal microscope.
To investigate the in vitro photothermal effect of the nanoparticle, 4T1 cells were cultured in 96 wells plate at a density of 6,000 per well. After 24 h of culture, the medium was replaced with complete media containing various concentrations of GNR@MSN-3P (6.2, 12.5, 25.0, 50.0, 100.0 μg/mL). After incubation for 6 h, the medium was discarded and replaced with a fresh complete medium. The plate was plated in a 37 °C incubator and cells were irradiated by a 808 nm laser at the output power of 500 mW (2 W/cm2) for 5 min, the temperatures and infrared thermal images were monitored and captured by a FLIR i7 thermal imaging camera every 30 s.
Cell viability assay
The cytotoxicity of free TPL in 4T1 cells was estimated by MTT assay. In brief, 4T1 cells were seeded in 96-well plates at a density of 7500 cells per well in 100 μl of complete culture medium. After incubation for 24 h, the medium was replaced with a fresh medium containing free TPL with different concentrations. After incubation for 24 h or 48 h, MTT solution (10 μl, 5 mg/mL in PBS) was added to each well and incubated for another 4 h, then the medium was discarded and replaced with 100 μl of DMSO. The absorbance at 570 nm of each well was measured by using a microplate reader. To evaluate the NIR induced photothermal cytotoxicity and TPL enhanced cytotoxicity, 4T1 cells were seeded in 96-well plate at a density of 7,500 cells per well. After being incubated for 24 h, the medium was replaced with fresh medium containing GNR@MSN-3P, TPL-GNR@MSN-3P and free TPL with different concentrations. Six hour later, the medium was discarded and the cells were rinsed with PBS following adding 100 μl of fresh medium. The designated cells were subjected to a 808 laser irradiation at the output power of 500 mW (2 W/cm2) for 5 min in a 37 °C incubator. The cells were further cultured for another 24 h followed with MTT assay as mentioned above.
Live/Dead Cell Staining Assay
4T1 cells were seeded in 96-well plate at a density of 7,500 cells per well. After 24 h of incubation, the old medium was replaced with fresh one containing GNR@MSN-3P, TPL-GNR@MSN-3P, or free TPL at corresponding dose of 25 μg/mL of gold nanorod and/or 100 nM of TPL. After additional 6 h of incubation, the culture medium was refreshed, and the cells were irradiated by a 808 nm laser at power of 500 mW (2 W/cm2) for 5 min in a 37 °C incubator. After 24 h of incubation, cells were treated with calcein-AM and EthD-1 in PBS buffer solution for 15 min at 37 °C with 5% CO2 according to the manufacturer's instructions to stain live/dead cells.
Western blotting analysis of HSP70
In brief, 4T1 cells were seeded in 6-well plate at a density of 4×105 cells per well for 24 h. The cells were treated with free TPL, TPL-GNR@MSN-3P and corresponding TPL-GNR@MSN of different concentrations of. After 6 h of incubation, the culture medium was refreshed, and the cells were immersed in 42 °C water bath for 5 min. After 24 h of incubation, the cells were lysed, and the proteins were collected for western blotting. In brief, proteins were separated by 10 % of SDS-PAGE electrophoresis and blotted on PVDF membrane. After the membrane was blocked with 5 % of BSA for 1 h, it was incubated with HSP70 primary antibody overnight at 4 °C, the membranes were washed with TBST and incubated with the secondary antibody membraned for 1 h. Enhanced chemiluminescence was used to detect the targeted protein expression. β-actin was used as an internal reference protein.
Animals and tumor model
All animal experiments were carried on following the protocols approved by the Institutional Animal Care and Use Committee (IACUC). Female BALB/c mice (5-6 weeks old) were provided by the Jackson laboratory. The xenograft tumor model was established by subcutaneous injection of 2 x 106 4T1 cells on the back region of the left and right hind legs. Tumor growth was monitored every two days by measuring with a digital caliper. The volume of tumor was calculated using the formula: volume of tumor = (length x width2)/2.
In vivo fluorescence imaging and photothermal imaging
When tumor volume reached a size of around 100 mm3, 200 μl of Cy5 labeled GNR@MSN-3P (containing 150 μg GRN) was injected into the tumor-bearing mice via tail vein. At a specific time point post-injection, the mice were anesthetized and imaged using an IVIS Lumina III whole-body imaging system (PerkinElmer Inc., Waltham, USA) at with the filter setting of excitation: 630 nm; emission: 650-670 nm. After 24 h of observation, mice were sacrificed, and the main organs (heart, lung, liver, spleen, and kidney) and tumors were collected for ex vivo imaging to study nanoparticles tissue distribution.
Photothermal imaging was carried following a similar procedure. At 6 h post-injection of GNR@MSN-3P (containing 150 μg GRN), the mice were anesthetized by 2 % isoflurane and the tumors were exposed to 808 nm laser irradiation at the output power of 500 mW (2 W/cm2) for 5 min. At the same time, temperatures and infrared thermal images were recorded and captured by FLIR i7 thermal imaging camera every 30 s.
In vivo antitumor effect evaluation
When bilateral tumors grew to around 100 mm3 in volume, the tumor-bearing mice were randomly divided into 4 groups (4-5 mice per group) and injected with 1) PBS, 2) GNR@MSN-3P (7.5 mg/kg of GNR), 3) TPL-GNR@MSN-3P (7.5 mg/kg of GNR and 1 mg/kg of TPL), and 4) Free TPL (1 mg/kg of TPL) via the tail vein. At 6 h post-injection, one side tumor in each mouse was exposed to 808 nm laser irradiation at the output power of 500 mW (2 W/cm2) for 5 min and the contralateral tumor did not expose laser as a non-irradiation control for its corresponding treatment. The tumor volume and body weight were monitored every day. At day 12, all mice were sacrificed, tumors were excised, weighed, and photographed. Subsequently, major organs (heart, liver, spleen, lung, and kidney of mice were also collected for histopathology and immunohistochemistry analysis.
Histological Examinations
Analysis of three tumors excised from each group was carried out with immunofluorescence (HSP70) assay and hematoxylin and eosin (H&E) staining to verify the therapeutic mechanism and effect. After the treatment, all major organs were collected for H&E to evaluate the biocompatibility and biosafety of all treatment..
Statistical Analysis
All experiments were repeated at least three times. Statistical significance was analyzed by a Student’s test and data are displayed as mean ± standard deviation (SD). Statistical significance was inferred at a value of p < 0.05.
Supplementary Material
Acknowledgements
The authors thank National Institutes of Health (1R15CA188847-01A1, 1R21CA252360-01, and 1R01AG054839-01A1) for the financial support of the research. They also thank the University of South Carolina Center for Targeted Therapeutics Microscopy and Flow Cytometry Core Facility (National Institutes of Health 5P20GM109091) for technical support.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].a) Wan H, Zhang Y, Liu Z, Xu G, Huang G, Ji Y, Xiong Z, Zhang Q, Dong J, Zhang W, Nanoscale 2014, 6, 8743–8753; [DOI] [PubMed] [Google Scholar]; b) Guo B, Sheng Z, Hu D, Liu C, Zheng H, Liu B, Advanced Materials 2018, 30, 1802591. [DOI] [PubMed] [Google Scholar]
- [2].a) Dickerson EB, Dreaden EC, Huang X, El-Sayed IH, Chu H, Pushpanketh S, McDonald JF, El-Sayed MA, Cancer letters 2008, 269, 57–66; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Choi WI, Kim J-Y, Kang C, Byeon CC, Kim YH, Tae G, ACS nano 2011, 5, 1995–2003. [DOI] [PubMed] [Google Scholar]
- [3].a) Liang C, Diao S, Wang C, Gong H, Liu T, Hong G, Shi X, Dai H, Liu Z, Advanced materials 2014, 26, 5646–5652; [DOI] [PubMed] [Google Scholar]; b) Wang C, Xu L, Liang C, Xiang J, Peng R, Liu Z, Advanced materials 2014, 26, 8154–8162. [DOI] [PubMed] [Google Scholar]
- [4].Yang K, Hu L, Ma X, Ye S, Cheng L, Shi X, Li C, Li Y, Liu Z, Advanced materials 2012, 24, 1868–1872. [DOI] [PubMed] [Google Scholar]
- [5].a) Shi H, Sun Y, Yan R, Liu S, Zhu L, Liu S, Feng Y, Wang P, He J, Zhou Z, Nano letters 2019, 19, 937–947; [DOI] [PubMed] [Google Scholar]; b) Berciaud S, Cognet L, Lounis B, Nano letters 2005, 5, 2160–2163. [DOI] [PubMed] [Google Scholar]
- [6].Sharker SM, Lee JE, Kim SH, Jeong JH, In I, Lee H, Park SY, Biomaterials 2015, 61, 229–238. [DOI] [PubMed] [Google Scholar]
- [7].a) Jin Y, Liang L, Sun X, Yu G, Chen S, Shi S, Liu H, Li Z, Ge K, Liu D, NPG Asia Materials 2018, 10, 373–384; [Google Scholar]; b) Wang Z, Li S, Zhang M, Ma Y, Liu Y, Gao W, Zhang J, Gu Y, Advanced science 2017, 4, 1600327; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao G, Jiang YW, Sun W, Guo Y, Jia HR, Yu XW, Pan GY, Wu FG, Small 2019, 15, 1900501. [DOI] [PubMed] [Google Scholar]
- [8].a) Harmon B, Corder A, Collins R, Gobe G, Allen J, Allan D, Kerr J, International journal of radiation biology 1990, 58, 845–858; [DOI] [PubMed] [Google Scholar]; b) Calderwood SK, in Tumor Ablation, Springer, 2013, pp. 29–37. [Google Scholar]
- [9].a) Chen Q, Wang C, Cheng L, He W, Cheng Z, Liu Z, Biomaterials 2014, 35, 2915–2923; [DOI] [PubMed] [Google Scholar]; b) Liu T, Wang C, Cui W, Gong H, Liang C, Shi X, Li Z, Sun B, Liu Z, Nanoscale 2014, 6, 11219–11225; [DOI] [PubMed] [Google Scholar]; c) Guo M, Mao H, Li Y, Zhu A, He H, Yang H, Wang Y, Tian X, Ge C, Peng Q, Biomaterials 2014, 35, 4656–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Li W, Peng J, Tan L, Wu J, Shi K, Qu Y, Wei X, Qian Z, Biomaterials 2016, 106, 119–133; [DOI] [PubMed] [Google Scholar]; b) Liu H, Chen D, Li L, Liu T, Tan L, Wu X, Tang F, Angewandte Chemie International Edition 2011, 50, 891–895. [DOI] [PubMed] [Google Scholar]
- [11].a) Kim J, Kim J, Jeong C, Kim WJ, Advanced drug delivery reviews 2016, 98, 99–112; [DOI] [PubMed] [Google Scholar]; b) Huang S, Duan S, Wang J, Bao S, Qiu X, Li C, Liu Y, Yan L, Zhang Z, Hu Y, Advanced Functional Materials 2016, 26, 2532–2544. [Google Scholar]
- [12].a) Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z, Nature communications 2016, 7, 1–13; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peng J, Xiao Y, Li W, Yang Q, Tan L, Jia Y, Qu Y, Qian Z, Advanced science 2018, 5, 1700891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Lindquist S, Craig E, Annual review of genetics 1988, 22, 631–677; [DOI] [PubMed] [Google Scholar]; b) Parsell D, Lindquist S, Annual review of genetics 1993, 27, 437–496. [DOI] [PubMed] [Google Scholar]
- [14].a) Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR, Trends in biochemical sciences 2006, 31, 164–172; [DOI] [PubMed] [Google Scholar]; b) Jego G, Hazoumé A, Seigneuric R, Garrido C, Cancer letters 2013, 332, 275–285; [DOI] [PubMed] [Google Scholar]; c) Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S, Trends in pharmacological sciences 2017, 38, 226–256. [DOI] [PubMed] [Google Scholar]
- [15].a) Oh HJ, Chen X, Subjeck JR, Journal of Biological Chemistry 1997, 272, 31636–31640; [DOI] [PubMed] [Google Scholar]; b) Khoei S, Goliaei B, Neshasteh-Riz A, Deizadji A, FEBS letters 2004, 561, 144–148; [DOI] [PubMed] [Google Scholar]; c) Evgen’ev M, Garbuz D, Zatsepina O, Russian Journal of Developmental Biology 2005, 36, 218–224. [Google Scholar]
- [16].Rocchi P, Jugpal P, So A, Sinneman S, Ettinger S, Fazli L, Nelson C, Gleave M, BJU international 2006, 98, 1082–1089. [DOI] [PubMed] [Google Scholar]
- [17].a) Ali MR, Ali HR, Rankin CR, El-Sayed MA, Biomaterials 2016, 102, 1–8; [DOI] [PubMed] [Google Scholar]; b) Chen W-H, Luo G-F, Lei Q, Hong S, Qiu W-X, Liu L-H, Cheng S-X, Zhang X-Z, ACS nano 2017, 11, 1419–1431; [DOI] [PubMed] [Google Scholar]; c) Tang X, Tan L, Shi K, Peng J, Xiao Y, Li W, Chen L, Yang Q, Qian Z, Acta pharmaceutica sinica B 2018, 8, 587–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang S, Tian Y, Tian W, Sun J, Zhao S, Liu Y, Wang C, Tang Y, Ma X, Teng Z, ACS nano 2016, 10, 8578–8590; [DOI] [PubMed] [Google Scholar]; e) Liu D, Ma L, Liu L, Wang L, Liu Y, Jia Q, Guo Q, Zhang G, Zhou J, ACS Appl Mater Interfaces 2016, 8, 24455–24462; [DOI] [PubMed] [Google Scholar]; f) Wang Z, Li S, Zhang M, Ma Y, Liu Y, Gao W, Zhang J, Gu Y, Adv Sci (Weinh) 2017, 4, 1600327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luo H, Wang Q, Deng Y, Yang T, Ke H, Yang H, He H, Guo Z, Yu D, Wu H, Chen H, Advanced Functional Materials 2017, 27. [Google Scholar]
- [19].a) Dominska M, Dykxhoorn DM, Journal of cell science 2010, 123, 1183–1189; [DOI] [PubMed] [Google Scholar]; b) Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J, Nature biotechnology 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Chen BJ, Leukemia & lymphoma 2001, 42, 253–265; [DOI] [PubMed] [Google Scholar]; b) Qiu D, Kao PN, Drugs in R & D 2003, 4, 1–18; [DOI] [PubMed] [Google Scholar]; c) Lin N, Liu C, Xiao C, Jia H, Imada K, Wu H, Ito A, Biochemical pharmacology 2007, 73, 136–146. [DOI] [PubMed] [Google Scholar]
- [21].LUE Y, HIKIM APS, WANG C, LEUNG A, BARAVARIAN S, REUTRAKUL V, SANGSAWAN R, CHAICHANA S, SWERDLOFF RS, Journal of andrology 1998, 19, 479–486. [PubMed] [Google Scholar]
- [22].a) Phillips PA, Dudeja V, McCarroll JA, Borja-Cacho D, Dawra RK, Grizzle WE, Vickers SM, Saluja AK, Cancer research 2007, 67, 9407–9416; [DOI] [PubMed] [Google Scholar]; b) Johnson SM, Wang X, Evers BM, Journal of Surgical Research 2011, 168, 197–205; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng C, Zhu H, Song H, Wang Z, Huang G, Li D, Ma Z, Ma J, Qin Q, Sun X, Chinese Journal of Cancer Research 2014, 26, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mujumdar N, Mackenzie TN, Dudeja V, Chugh R, Antonoff MB, Borja–Cacho D, Sangwan V, Dawra R, Vickers SM, Saluja AK, Gastroenterology 2010, 139, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li J, Zhu W, Leng T, Shu M, Huang Y, Xu D, Qiu P, Su X, Yan G, Oncology reports 2011, 25, 979–987. [DOI] [PubMed] [Google Scholar]
- [25].Owa C, Messina ME Jr, Halaby R, International journal of women's health 2013, 5, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Carter BZ, Mak DH, Schober WD, McQueen T, Harris D, Estrov Z, Evans RL, Andreeff M, Blood 2006, 108, 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiang X-H, Wong BC-Y, Lin MC-M, Zhu G-H, Kung H-F, Jiang S-H, Yang D, Lam S-K, Oncogene 2001, 20, 8009–8018. [DOI] [PubMed] [Google Scholar]
- [28].a) MacKenzie TN, Mujumdar N, Banerjee S, Sangwan V, Sarver A, Vickers S, Subramanian S, Saluja AK, Molecular cancer therapeutics 2013, 12, 1266–1275; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Westerheide SD, Kawahara TL, Orton K, Morimoto RI, Journal of biological chemistry 2006, 281, 9616–9622. [DOI] [PubMed] [Google Scholar]
- [29].Westerheide SD, Kawahara TL, Orton K, Morimoto RI, J Biol Chem 2006, 281, 9616–9622. [DOI] [PubMed] [Google Scholar]
- [30].He H, Cattran AW, Nguyen T, Nieminen AL, Xu P, Biomaterials 2014, 35, 9546–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu L, Ouyang S, Ye J, Angewandte Chemie International Edition 2013, 52, 6689–6693. [DOI] [PubMed] [Google Scholar]
- [32].Palanikumar L, Choi ES, Cheon JY, Joo SH, Ryu J-H, Advanced Functional Materials 2015, 25, 957–965. [Google Scholar]
- [33].Bahadur KCR, Xu P, Adv Mater 2012, 24, 6479–6483. [DOI] [PubMed] [Google Scholar]
- [34].Abadeer NS, Brennan MR, Wilson WL, Murphy CJ, ACS nano 2014, 8, 8392–8406. [DOI] [PubMed] [Google Scholar]
- [35].von Nessen K, Karg M, Hellweg T, Polymer 2013, 54, 5499–5510. [Google Scholar]
- [36].Pernia Leal M, Torti A, Riedinger A, La Fleur R, Petti D, Cingolani R, Bertacco R, Pellegrino T, ACS nano 2012, 6, 10535–10545. [DOI] [PubMed] [Google Scholar]
- [37].Liu J, Detrembleur C, De Pauw-Gillet MC, Mornet S, Jérôme C, Duguet E, Small 2015, 11, 2323–2332. [DOI] [PubMed] [Google Scholar]
- [38].Blanco M, Teijón C, Olmo R, Teijón J, in Recent advances in novel drug carrier systems, IntechOpen, 2012. [Google Scholar]
- [39].a) Wang J, Sui M, Fan W, Current drug metabolism 2010, 11, 129–141; [DOI] [PubMed] [Google Scholar]; b) Yamashita F, Hashida M, Advanced drug delivery reviews 2013, 65, 139–147. [DOI] [PubMed] [Google Scholar]
- [40].Zhang Z, Wang J, Chen C, Advanced Materials 2013, 25, 3869–3880. [DOI] [PubMed] [Google Scholar]
- [41].Morrow G, Tanguay RM, The international journal of biochemistry & cell biology 2012, 44, 1613–1621. [DOI] [PubMed] [Google Scholar]
- [42].Banerjee S, Sangwan V, McGinn O, Chugh R, Dudeja V, Vickers SM, Saluja AK, Journal of Biological Chemistry 2013, 288, 33927–33938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].MacKenzie TN, Mujumdar N, Banerjee S, Sangwan V, Sarver A, Vickers S, Subramanian S, Saluja AK, Mol Cancer Ther 2013, 12, 1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jiang JX, Keating JJ, De Jesus EM, Judy RP, Madajewski B, Venegas O, Okusanya OT, Singhal S, American journal of nuclear medicine and molecular imaging 2015, 5, 390. [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang FZ, Ho DH-H, Wong RH-F, Oncotarget 2018, 9, 22301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xiong F, Chen H, Chang X, Yang Y, Xu H, Yang X, in 2005 IEEE Engineering in Medicine and Biology 27th Annual Conference, IEEE, 2006, pp. 4966–4969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.