

Abstract
The etiology of autism spectrum disorder (ASD) is multifactorial and complex and likely involves interactions among genetic, epigenetic and environmental factors. With respect to environmental influences, a growing literature implicates intrauterine experiences in the origin of this pervasive developmental disorder. In this prospective longitudinal design, we examine the hypothesis that fetal exposure to maternal cortisol may confer ASD risk. In addition, because ASD is four times more prevalent in males than females and because sexually dimorphic responses to intrauterine experiences are commonly observed, we examine whether or not any associations differ by fetal sex. Maternal plasma cortisol was measured at 15, 19, 25, 31, and 37 weeks’ gestation in a sample of 84 pregnant women. ASD symptoms were assessed in their 5-year old children with the Social Communication Questionnaire (SCQ). Fetal exposure to lower levels of maternal cortisol was associated with higher levels of ASD symptoms among boys only. The observed hypocortisolemic profile exhibited by these mothers may indicate a risk factor that precedes the stress of caregiving for a child with ASD and may not be solely a consequence of the stress of caregiving as previously thought. Further, these findings confirm the value of examining prenatal hormone exposures as predictors of ASD risk and support the premise that altered prenatal steroid exposures may play a role in the etiology of ASD.
Keywords: prenatal, pregnancy, cortisol, autism spectrum disorder (ASD), HPA axis, caregiver stress
The Centers for Disease Control and Prevention (CDC) estimates that 1 in 68 children has been identified with Autism Spectrum Disorder (ASD), a developmental disability characterized by social and communication impairments and repetitive or restricted behaviors and interests (American Psychiatric Association, 2013; Centers for Disease Control and Prevention, 2014). ASD is common in all racial, ethnic, and socioeconomic groups, and evidence suggests that autism symptoms are continuously distributed across the population (Baron-Cohen, Wheelwright, Skinner, Martin, & Clubley, 2001; Constantino & Todd, 2003; Mulligan, Richardson, Anney, & Gill, 2009; Posserud, Lundervold, & Gillberg, 2006; Spiker, Lotspeich, Dimiceli, Myers, & Risch, 2002). Although significant advances have been made in identifying genetic contributions to this disorder (Abrahams & Geschwind, 2008; Hallmayer et al., 2011; Risch et al., 2014; Robinson, Neale, & Hyman, 2015; Sebat et al., 2007; Trottier, Srivastava, & Walker, 1999), there also is accumulating support for significant non-genetic or environmental influences (Durkin et al., 2008; Janecka et al., 2017; Lai, Lombardo, & Baron-Cohen, 2014; Landrigan, 2010) and for gene x environment interactions (Abbott, Gumusoglu, Bittle, Beversdorf, & Stevens, 2018; Hecht et al., 2016; Schaafsma et al., 2017; Tordjman et al., 2014). Among these environmental influences, a growing literature implicates intrauterine experiences in the etiology of ASD, including obstetric complications and adverse birth phenotype (Atladottir et al., 2010; Schendel & Bhasin, 2008), prenatal steroid profiles (Baron-Cohen et al., 2015; Knickmeyer & Baron-Cohen, 2006) and prenatal stress exposures (Beversdorf et al., 2005; Class et al., 2014; Jones et al., 2010; Kinney, Munir, Crowley, & Miller, 2008; Rodriguez & Bohlin, 2005; Sjaarda et al., 2017; Varcin, Alvares, Uljarevic, & Whitehouse, 2017).
A Potential Role Prenatal Cortisol Exposures in ASD
Here we examine the hypothesis that fetal exposure to maternal cortisol may confer ASD risk (Gitau, Adams, Fisk, & Glover, 2005; Gore, Martien, Gagnidze, & Pfaff, 2014; Matthews, 2000; Rose’meyer, 2013; Rose’meyer, 2014; Whitaker-Azmitia, Lobel, & Moyer, 2014). The plausibility of this hypothesis is supported not only by the documentation of links between intrauterine stress exposures and ASD risk described above, but also because cortisol (the primary glucocorticoid (GC) in humans) has been implicated broadly as a principal effector of fetal programming due to its critical role in fetal organ and brain development and the fact that it is modulated by stress exposures and environmental conditions (Moisiadis & Matthews, 2014; Sandman & Glynn, 2009; Sarkar, Bergman, O’Connor, & Glover, 2008; Seckl, 2004; Welberg & Seckl, 2001).
Cortisol in human pregnancy.
Cortisol is a steroid hormone that plays a critical role in normal development and is the end-product of the hypothalamic-pituitary-adrenal (HPA) axis, one of the body’s major stress response systems. HPA axis activity is regulated by the release of hypothalamic corticotrophin-releasing hormone (CRH), which stimulates the synthesis and release of adrenocorticotropic hormone (ACTH). Release of ACTH from the pituitary into the blood stream triggers cortisol production and release from the adrenal cortex. Circulating cortisol has effects on nearly every organ and tissue in the body (Munck, Guyre, & Holbrook, 1984). During human pregnancy, regulation of the maternal HPA axis changes dramatically with the synthesis of CRH from the placenta, beginning as early as the seventh week of gestation (Davis & Sandman, 2010; McLean et al., 1995; Sandman & Glynn, 2009). In contrast to the role of cortisol in the negative feedback regulation of the HPA axis, cortisol stimulates placental CRH production, resulting in a positive feedback loop that allows for the simultaneous increase of CRH, ACTH, and cortisol in both the maternal and fetal compartments (King, Nicholson, & Smith, 2001; Petraglia, Florio, Nappi, & Genazzani, 1996). Over the course of normal gestation, maternal cortisol increases two- to four-fold (Mastorakos & Ilias, 2003; Sandman et al., 2006).
Fetal exposure to increasing concentrations of maternal cortisol is regulated by a placental enzyme, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which oxidizes cortisol to cortisone (Beitins, Bayard, Ances, Kowarski, & Migeon, 1973; Brown et al., 1996). Activity of placental 11β-HSD2 increases throughout most of gestation, but because it is only a partial barrier, some active maternal cortisol passes through the placenta, resulting in significant concordance between cortisol levels in the maternal and fetal compartments (Gitau, Cameron, Fisk, & Glover, 1998; Gitau, Fisk, Teixeira, Cameron, & Glover, 2001). Prior to parturition, the activity of the 11β-HSD2 decreases, further increasing fetal exposure to maternal cortisol (Giannopoulos, Jackson, & Tulchinsky, 1982; Murphy, Smith, Giles, & Clifton, 2006). The normative increase in maternal cortisol during gestation coupled with the decrease in placental 11β-HSD2 activity at the end of pregnancy ensures that the fetus is exposed to sufficient levels of cortisol during the third trimester, which is important for maturation of the fetal lungs and other organs (including the brain) and for preparation of the fetus for labor and delivery (Austin & Leader, 2000; Hacking, Watkins, Fraser, Wolfe, & Nolan, 2001). In addition, these late cortisol exposures have been characterized as comprising a critical “switch” necessary for normal brain development (Matthews, 2000). We have demonstrated that these heightened exposures late in gestation may be associated with salutary effects on brain development (Davis, Head, Buss, & Sandman, 2017; Davis & Sandman, 2010).
Altered HPA-axis function among mothers of children with ASD.
Our proposal that fetal exposure to dysregulated maternal cortisol trajectories may confer risk for ASD is supported by the fact that aberrant HPA-axis functioning is observed among mothers of individuals with ASD. For example, mothers of adolescents and adults with ASD exhibit a profile of HPA hypoactivity characterized by a blunted cortisol awakening response when compared to mothers of typically developing individuals (Fecteau et al., 2017; Seltzer et al., 2010; Wong et al., 2012). Interestingly, this hyporesponsive profile may be more pronounced among mothers of children with ASD compared to mothers of children with other developmental disabilities. Dykens and Lambert (2013), using group-based trajectory analyses, identified two distinctive diurnal cortisol profiles among mothers caring for children with ASD and other developmental disabilities − typical and blunted. Eighty-nine percent of the mothers of children with ASD fell into the group exhibiting the blunted profile, whereas mothers of children with Down syndrome, Prader-Willi syndrome, and William’s syndrome were roughly evenly distributed between the two trajectory groups (53 vs 47%). Because chronic stress has been implicated in dysregulated HPA-axis function (Miller, Chen, & Zhou, 2007; Tsigos & Chrousos, 2002), the widely accepted interpretation of these findings is that these altered maternal HPA profiles are a consequence of parenting a child with a developmental disorder (Bitsika, Sharpley, Andronicos, & Agnew, 2017; Davis & Carter, 2008; Rivard, Terroux, Parent-Boursier, & Mercier, 2014; Theule, Wiener, Tannock, & Jenkins, 2010). However, the existing literature has yet to consider the possibility that maternal hypocortisolism may represent a biological profile that could confer risk for ASD in offspring.
The Current Study
Here with a prospective longitudinal design in which maternal prenatal cortisol profiles during gestation were characterized and ASD symptoms in a sample of 5-year old children were assessed, we examine whether or not fetal exposures to maternal cortisol are associated with child ASD symptoms. Further, because ASD is overrepresented among males (Baron-Cohen et al., 2011; Lai, Lombardo, Auyeung, Chakrabarti, & Baron-Cohen, 2015; Werling & Geschwind, 2013), and sex differences exist in fetal responses to adversity (Bale, 2011, 2016; Manson, 2008; Sandman, Glynn, & Davis, 2013), including specifically to prenatal GC exposures (Adibi et al., 2015; Bale & Epperson, 2015; Glynn & Sandman, 2012; Kim, Bale, & Epperson, 2015; Sandman, Davis, Buss, & Glynn, 2011), we test whether the relations between prenatal cortisol and ASD symptoms differ based on fetal sex.
Method
Study Overview
Study participants included mother-infant pairs from a longitudinal study of prenatal psychobiological risk and development. Women with singleton pregnancies less than 16 gestational weeks were recruited from obstetric clinics and a large university medical center in Southern California. Maternal cortisol was assessed 5 times during pregnancy and child ASD symptoms were assessed at 5 years of age.
Participants
The sample comprised 84 mothers and their children (Mage = 5.13, 51.2% female). Initial prenatal recruitment criteria included: singleton pregnancy, English speaking, non-smoker, over 18 years of age, no use of steroid medication, and no evidence for drug or alcohol use during pregnancy. Additional inclusion criteria for this study were that the child had reached age 5 during the funded study period. Mothers gave informed consent for all aspects of the protocol, which was approved by the Institutional Review Board. The mothers were 27 percent Latina, 50 percent non- Hispanic white, and 88 percent were married to or cohabitating with the child’s father. Fifty percent of the children were first born and the mean gestational age at birth was 39.4 weeks (range 35.3 – 42.6 weeks’ gestation). Additional descriptive information for the study sample is shown in Table S1 of the Supplemental Material available online.
Procedures
Maternal plasma samples were obtained for cortisol analysis at 15 (M = 15.47 ± 0.95), 19 (M = 19.56 ± 1.09), 25 (M = 25.71 ± 1.08), 31 (M = 31.16 ± 0.88), and 36+ (M = 36.81 ± 0.90) weeks’ gestation. Mothers completed the Social Communication Questionnaire when children were 5 years of age. Maternal reports of ethnicity, age, educational level, income, and marital status were collected by structured interview.
Cortisol assessment.
Maternal blood samples (20 ml/draw) were withdrawn by antecubital venipuncture into EDTA (purple top) vacutainers and then immediately chilled on ice. Aprotinin (Sigma Chemical, St. Louis, MO) was added at 500 KlU/ml blood. The mean sample collection time across study visits was 13:39 hours (range across gestational visits: 13:32 to 13:40) and the mean standard deviation of collection time across visits was 85 minutes (range: 73 to 108 minutes). Samples were centrifuged at 2000 × g for 15 min, decanted into polypropylene tubes, and stor2ed at −80°C until assayed.
Plasma cortisol levels were determined by a competitive binding solid-phase enzyme- linked immunosorbent assay (IBL Immuno Biological Laboratories America, Minneapolis, MN) with reported sensitivity of .22 μg/dl. Plasma samples (20 μl) and enzyme conjugate (200 μl) were thoroughly mixed in antibody-coated microtiter wells and incubated at room temperature for 60 minutes. Each well was then washed three times with wash solution (400 μl per well), followed by a 15-min incubation at room temperature with substrate solution (100 μl). Absorbance units were measured at 450 nm within 10 minutes of adding stop solution. This assay has less than 9% cross-reactivity with progesterone and less than 2% cross-reactivity with other naturally-occurring steroid hormones (e.g., testosterone, estradiol). All samples were assayed in duplicate. Interassay and intra-assay coefficients of variance were less than 8% with a minimum detectable level of .25 μg/dl.
Assessment of autism spectrum disorder symptoms.
Child ASD symptoms were assessed using the Social Communication Questionnaire (SCQ) Lifetime version, a widely-used, validated parent-report questionnaire based on the Autism Diagnostic Interview − Revised (ADI-R; Lord, Rutter, & Le Couteur, 1994). The ADI-R is one of the “gold standard” instruments for use in the assessment of ASD. Because the ADI-R is time-consuming, typically taking 1.5 to 2 hours, and requires a highly trained clinician, the SCQ was developed for use as a brief screening tool. The SCQ Lifetime version consists of 40 binary-scaled questions that evaluate communication skills, social relating, and range of interests in children. Item 1 is not scored but is used to determine whether the child has enough language to evaluate abnormalities in language. Therefore, scores range from 0–39 and scores above the cutoff of 15 (sensitivity of 0.85 and specificity of 0.75) suggest the individual is likely to have ASD or another neurodevelopmental condition (Rutter, 2003).
Studies examining the distribution of core features of ASD (communication, social reciprocal interaction, and restricted/repetitive/stereotyped patterns of behavior) in general populations demonstrate that the social deficits characteristic of ASD are common and the distribution of these traits continuous (Baron-Cohen et al., 2001; Constantino & Todd, 2003; Mulligan et al., 2009; Posserud et al., 2006; Spiker et al., 2002). For use in this study’s general community sample, SCQ scores were treated as a continuous variable, an approach which is consistent with current emphases on dimensionality and the view that symptom-based approaches are necessary for clarifying definition and classification of mental illnesses (e.g. NIMH Research Domain Criteria (RDoC) project; Calamari, Wiegartz, & Janeck, 1999; Constantino, 2011; Cuthbert, 2014; Insel et al., 2010).
Determination of obstetric risk and birth phenotype.
Maternal and infant medical records were reviewed to assess pregnancy complications, medication use during gestation and birth outcome. An obstetric risk score accounted for prenatal infection, pregnancy-induced hypertension, gestational diabetes, oligohydramnios, polyhydramnios, preterm labor, vaginal bleeding, placenta previa, and anemia. A cumulative score assessing prenatal obstetric risk was derived from the sum of all present risk variables (Hobel, 1982). Gestational age at birth (GAB) was calculated using patient report of last menstrual period and confirmed with early pregnancy ultrasound according to American Congress of Obstetrician and Gynecologists guidelines (American Congress of Obstetricians and Gynecologists, 2014).
Statistical analyses.
For primary analyses to assess cumulative fetal exposure to maternal cortisol across gestation, an index of maternal prenatal cortisol levels was calculated by averaging maternal cortisol levels from each of the five gestational assessments. Because cortisol production is affected both by advancing gestation and by diurnal rhythms, before averaging, cortisol values were residualized within collection timepoint for gestational week and time of day of sample collection. A multiple linear regression was performed to examine whether maternal prenatal cortisol levels were associated with child ASD symptoms. Demographic (maternal age, maternal race/ethnicity, cohabitation with the child’s father, socioeconomic status), pregnancy (obstetric risk, gestational age at birth), and child (age, sex, birth order) variables were considered as potential covariates (See Table S2 for bivariate correlations with maternal prenatal cortisol and child ASD symptoms). Gestational age at birth, socioeconomic status and obstetric risk were associated with child ASD symptoms at p < .10 and were therefore included in the model. Given well-documented sex differences in the prevalence of ASD (Baron-Cohen, Knickmeyer, & Belmonte, 2005; Baron-Cohen et al., 2011; Ingudomnukul, Baron-Cohen, Wheelwright, & Knickmeyer, 2007; Knickmeyer & Baron-Cohen, 2006; Nakayama, Takahashi, Wakabayashi, Oono, & Radford, 2007) and evidence for sex differences in fetal programming (Bale, 2011, 2016; Clifton, 2010; Manson, 2008; Sandman et al., 2013), we evaluated fetal sex as a potential moderator of the association between maternal prenatal cortisol and child ASD symptoms.
Overall, medication use during gestation in the study sample was low and did not impact study findings (See Supplement for details of these analyses).
Secondary analyses were then conducted to examine whether any associations between maternal prenatal cortisol levels and child ASD symptoms detected in the regression model were dependent on gestational timing. Multilevel modeling (MLM) techniques were utilized to examine week-by-week associations between maternal prenatal cortisol levels and child ASD symptoms (Raudenbush & Bryk, 2002). HLM offers several advantages over other OLS statistical methods for the evaluation of variation over time. First, standard regression or ANOVA models are limited to one component of variability, the deviation of the individual from the group mean. In comparison, HLM also takes into account the within-person variability assessed over time. Second, estimates of the lack of fit in modeling each individual’s data are derived and the less reliable data are weighted less heavily. Third, HLM produces robust estimates despite missing values for the repeated dependent measure. Cases with complete data are weighted more heavily, but all cases are included in the estimation of effects. Initial testing indicated that a quadratic model best fit maternal prenatal cortisol trajectories and that child ASD symptoms were not associated with linear or quadratic change in maternal prenatal cortisol levels over gestation. Therefore, a series of two-level models were computed to test differences in maternal prenatal cortisol levels (intercepts) centered at 1-week intervals within the range of assessments available (15–37 weeks). Level 1 (time-variant, within-dyad) variables were maternal prenatal cortisol levels, gestational week, gestational week squared, and time of day of sample collection. Level-2 (timeinvariant, between-dyad) variables were child ASD symptoms and covariates (gestational age at birth, socioeconomic status, and obstetric risk).
Results
As expected, a repeated measures ANOVA indicated that maternal plasma cortisol levels increased over the course of pregnancy (F(1, 71) = 127.88, p < .001). Average maternal gestational cortisol levels did not differ as a function of fetal sex (t = −0.43 – 1.45, p = .15 − .67). Male (M = 6.73, SD = 3.58) and female (M = 6.47, SD = 4.18) children also did not differ in their ASD symptom scores (t = −0.31, p = .76), which is consistent with the literature examining distribution of these symptoms and traits in the general population (Constantino, 2011; Constantino & Todd, 2003; Mulligan et al., 2009; Ruzich et al., 2015). Three participants scored at the clinical cut point for this questionnaire (see Figure S1). Further, one male child in the sample had received a clinical diagnosis of ASD and his SCQ score was 27. Regression and MLM analyses reported here exclude this individual. See the Supplemental Material available online for regression and MLM analyses that do include this case.
Regression analyses did not reveal a main effect of maternal prenatal cortisol levels; however, the interaction between prenatal maternal cortisol and child sex was statistically significant (Table 1). As shown in Figure 1, lower prenatal maternal cortisol levels were associated with higher levels of ASD symptoms in boys (slope = −0.45, t = −2.48, p = .01), but not in girls (slope = 0.10, t = 0.71, p = .48).
Table 1.
Multiple regression model predicting child ASD symptoms
| B (SE) | β | Partial r | |
|---|---|---|---|
| Gestational Age at Birth | −0.42 (0.30) | −.14 | −.16 |
| Obstetric Risk | 1.26 (0.69) | .19† | .20 |
| SES | −1.18 (0.43) | −.28** | −.30 |
| Prenatal Cortisol | 0.10 (0.13) | .10 | .09 |
| Child Sex | 0.14 (0.77) | .02 | .02 |
| Prenatal Maternal Cortisol × Child Sex | −0.55 (0.21) | −.32* | −.28 |
Note: Child sex coded 0 = female. Prenatal cortisol was residualized for gestational week of collection and for time of day of collection.
p < .10
p < .05
p < .01
Figure 1.
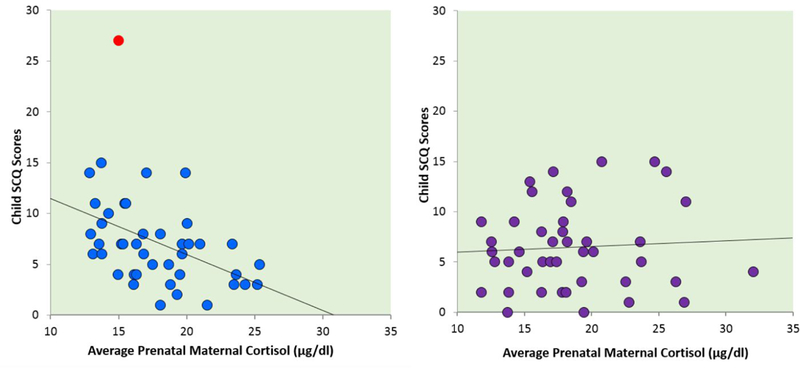
Association between average prenatal maternal plasma cortisol and child SCQ scores. The graph on the left represents this correlation for male children (r = −.45, p = .003). The graph on the right represents this correlation for female children (r = .06, p = .70). One male child had an ASD diagnosis (left panel, red data point). Including this case in the analyses did not alter the association between prenatal cortisol and SCQ scores among males (r = −.42, p = .006). Correlations account for both gestational week of sample collection and for time of day of collection.
Multilevel modeling indicated that the association between prenatal maternal cortisol levels on ASD symptoms in boys did not differ as a function of gestational timing, with statistically significant intercept differences present across gestation (Coefficient: −0.35, SE = 0.13, t = −2.78, p = .01; see Figure 2).
Figure 2.
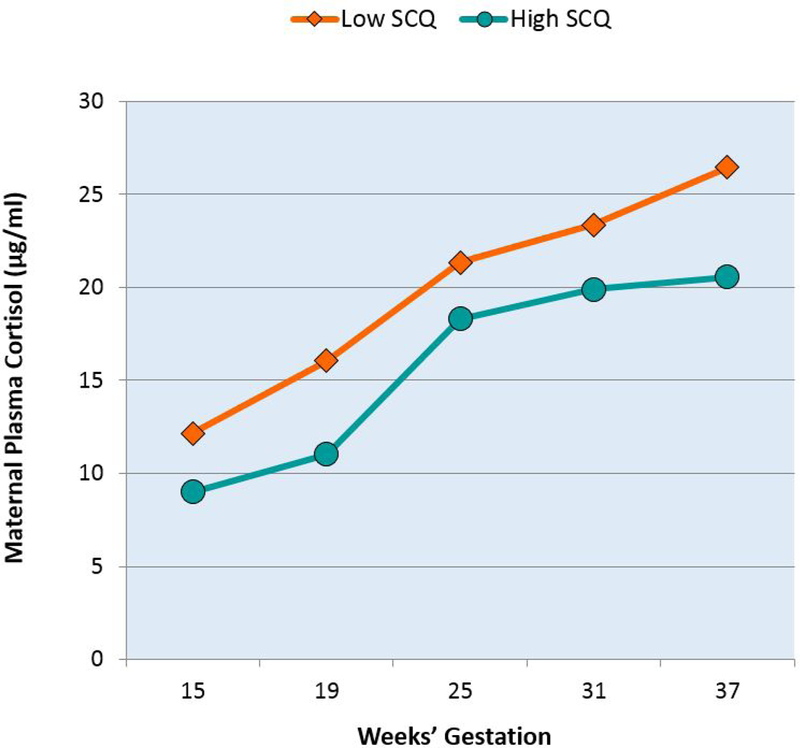
Maternal gestational plasma cortisol profiles in pregnancies with male fetuses. Mothers of boys with low SCQ scores exhibit lower levels of cortisol across pregnancy compared to mothers of boys with high SCQ scores. Trajectories of upper and lower quartiles of male child SCQ scores derived from the multilevel models are shown for illustrative purposes only. All analyses were conducted with SCQ scores as a continuous outcome and with adjustment for time of day of sample collection.
Discussion
Here we document a link between exposure to lower levels of prenatal maternal cortisol and increased manifestation of ASD symptoms in early childhood among boys. Consistent with the majority of studies examining prenatal influences on ASD risk, these effects were not observed among girls (Baron-Cohen et al., 2011; Harrington, Lee, Crum, Zimmerman, & Hertz- Picciotto, 2014; Werling & Geschwind, 2013). These findings also are consistent with a number of previous studies documenting hyporesponsive HPA-axis function among mothers of individuals with ASD (Dykens & Lambert, 2013; Foody, James, & Leader, 2015; Seltzer et al., 2010). Because chronic stress exposures contribute to altered HPA-axis function (Kinlein, Wilson, & Karatsoreos, 2015; Miller et al., 2007; Naughton, Dinan, & Scott, 2014), it is not surprising that the prevailing interpretation of the hyporesponsive profile exhibited by mothers of children with ASD is that it is due to the significant and documented stress associated with caring for a child with a developmental disability (De Andres-Garcia, Moya-Albiol, & Gonzalez-Bono, 2012; Dykens & Lambert, 2013; Fecteau et al., 2017; Foody et al., 2015; Wong, Mailick, Greenberg, Hong, & Coe, 2014; Wong et al., 2012). However, the findings presented here raise the possibility of an alternate explanation specifically, that causality may run the other way, or at least not be solely due to the stress of caregiving. It is important to note that we are not suggesting that among these mothers such caregiving does not represent a significant stressor, nor that this chronic stressor does not have the potential to influence the HPA-axis function, but rather that altered HPA-axis function among these individuals also may represent a marker of offspring ASD risk worthy of further consideration. In fact, given that there is significant stability in a woman’s endocrine profile (including cortisol) across pregnancies (Fox, Sandman, Davis, & Glynn, 2015), it is possible, even likely, that this may be one contributing environmental factor that promotes concordant sibling phenotypic development. More specifically, the findings here, coupled with the intraindividual stability in gestational physiology (Fox et al., 2015), may shed light on the observation that there is a higher recurrence risk for ASD among maternal compared to paternal half siblings (Gr0nborg, Schendel, & Parner, 2013; Risch et al., 2014).
There are a number of theories that implicate prenatal steroid hormone exposures in the etiology of ASD, with a predominate focus on the role of testosterone (Knickmeyer & Baron-Cohen, 2006; Pfaff, Rapin, & Goldman, 2011; Rose’meyer, 2013; Whitaker-Azmitia et al., 2014). We are aware of only one study that considered prenatal cortisol levels and risk for ASD. Baron-Cohen et al. (2015) identified a latent steroidogenic factor in amniotic fluid, derived from shared variance in progesterone, 17 α-hydroxy-progesterone, androstenedione, testosterone and cortisol, which was elevated among pregnancies resulting in offspring with ASD. Because individual hormone levels were not tested, it is not possible to discern the unique contributions of cortisol to ASD risk in this cohort. However, our findings, coupled with those of Baron-Cohen et al. (2015), confirm the value of examining prenatal hormone exposures as predictors of ASD risk and support the premise that altered prenatal steroid exposures may contribute to the etiology of ASD.
GCs play a critical role in normal brain development and exert persisting effects on lifespan HPA axis function (Davis, Waffarn, & Sandman, 2011; Howland, Sandman, & Glynn, 2017; Kapoor, Petropoulos, & Matthews, 2008; Matthews, 2002), which is one reason why they have been widely proposed as a central mechanism for programming the fetus (Matthews, 2000; Trejo, Cuchillo, Machin, & Rua, 2000; Welberg & Seckl, 2001). Most regions of the CNS rely on GCs for normal maturation (Challis et al., 2001), and GCs exert these effects by initiating terminal maturation, remodeling axons and dendrites and affecting cell survival (Gelman, Flores-Ramos, Lopez-Martinez, Fuentes, & Grajeda, 2015; Zunszain, Anacker, Cattaneo, Carvalho, & Pariante, 2011). Further, late gestational GC exposures, which are facilitated by the drop in 11β-HSD2, provide critical developmental ‘switching’ in the fetal brain (Fowden, Li, & Forhead, 1998; Moisiadis & Matthews, 2014).
A prevalent hypothesis suggests that the overrepresentation of neurodevelopmental disorders, including ASD, among males may be due in part to sex-specific differences in placental function (Bale, 2011; Davis & Pfaff, 2014; Gabory, Roseboom, Moore, Moore, & Junien, 2013; Sandman et al., 2013). The placenta, a transient endocrine organ of fetal origin, continually responds to changes in the maternal milieu, with dynamic implications for the intrauterine environment. Placental tissue is sex specific, and sexually dimorphic responses most likely operate through alterations in gene expression (Graves, 2010; Osei-Kumah, Smith, Jurisica, Caniggia, & Clifton, 2011), changes in energy mobilization and oxygen transport (O’Connell, Moritz, Walker, & Dickinson, 2013) and inflammatory responses (Clifton & Murphy, 2004; Reynolds, Vickers, Harrison, Segovia, & Gray, 2015). Most directly relevant to the findings here, sex-specific placental responses to GCs have been documented (Stark, Wright, & Clifton, 2009). In response to synthetic GC treatment for preterm labor, female fetuses exhibit larger increases in placental 11P-HSD2 activity compared to males. Male fetuses also may be more vulnerable to a relative lack of cortisol because during the third trimester, testosterone concentrations are higher in male pregnancies and this could inhibit binding of GCs to their receptors (Da Silva, 1999; Simmons, France, Keelan, Song, & Knox, 1994).
Limitations
It is widely accepted that autism has a significant genetic component and it is the case that both mothers and individuals with ASD manifest dysregulated HPA-axis function (Dykens & Lambert, 2013; Hill, Wagner, Shedlarski, & Sears, 1977; Marinovic-Curin et al., 2008; Taylor & Corbett, 2014); therefore, we cannot rule out the possibility that our findings reflect a shared genetic vulnerability rather than the consequences of prenatal GC exposures. Further, and perhaps more interestingly, emerging research has documented synergistic effects between certain genetic profiles and exposures to prenatal stress in determining ASD risk (Hecht et al., 2016; Schaafsma et al., 2017). Our study focused narrowly on prenatal GCs, and so did not allow consideration of important gene x environment interactions. The relatively small sample size comprises a third limitation to the current study. However, our confidence in the results of this prospective study is enhanced by the fact that in this sample we document three associations that are consistent with other, more well-established findings: 1) an association between a hyporesponsive maternal HPA-axis profile and ASD among offspring (Dykens & Lambert, 2013; Foody et al., 2015; Seltzer et al., 2010), 2) positive associations between adverse birth phenotype, obstetric risk conditions and ASD symptoms (Atladottir et al., 2010; Schendel & Bhasin, 2008), and 3) observed sex differences revealing a male vulnerability, which is consistent with both the empirical and theoretical literature (Baron-Cohen et al., 2011; Werling & Geschwind, 2013). A fourth limitation relates to our use of a parent-report screening instrument as the measure of ASD symptoms. However, the use of a dimensional instrument to assess symptoms is consistent with an RDoC approach and current views advocating the use of dimensional measures to advance understanding of ASD (Calamari et al., 1999; Constantino, 2011; Cuthbert, 2014; Insel et al., 2010). Further, the distribution of SCQ scores was normal and the observed variation is consistent with what would be expected based on use of this and other similar instruments administered in the general population (See Figure S1; Constantino & Todd, 2003; Hoekstra, Bartels, Verweij, & Boomsma, 2007; Mulligan et al., 2009; Posserud, Lundervold, & Gillberg, 2009; Wigham, McConachie, Tandos, & Le Couteur, 2012). Relatedly, the importance and relevance of these findings for ASD should be considered with caution in light of the fact that we tested our hypotheses in a general community sample. Future research in clinical populations is required to confirm whether prenatal cortisol exposures might contribute to the development of this disorder.
Implications
ASD is a heterogeneous condition, characterized by marked variability in clinical presentation and biological and behavioral phenotypes (Amaral, 2011; Beversdorf & Missouri Autism Summit, 2016; Ellegood et al., 2015; Loth, Murphy, & Spooren, 2016). With respect to identification of biomarkers, this unique heterogeneity demands identification of ASD subgroups that are more biologically homogeneous, a goal requiring novel stratification approaches that are sensitive to developmental stages and timing of exposures (Beversdorf & Missouri Autism Summit, 2016; Loth, Spooren, et al., 2016). Taken together, our new findings suggest that it might prove fruitful to incorporate prenatal GC exposures into approaches aimed at addressing the heterogeneity of this condition and also have several broader implications for advancing understanding of the origins of ASD. First, they are consistent with the premise that the prenatal period plays a role in determining risk for ASD, and that sex differences in the prevalence of developmental disorders such as ASD may be due in part to the sexually dimorphic placenta. Second, the fact that we observed hyporesponsive HPA-axis profiles among the mothers prior to the birth of their children, provides a plausible alternative view of existing literature documenting blunted HPA-axis activity in parents of children with ASD. The observed hypocortisolemic profile in these mothers may represent a risk factor that precedes the stress of caregiving for a child with ASD and not solely a consequence of the stress of caregiving. Further, the findings presented here suggest that additional focus should be on pathways related to HPA-axis function as well as on the developmental origins of this pervasive developmental disorder.
Supplementary Material
Acknowledgements
This research was supported by National Institutes of Health grants NS41298 and MH- 96889. The authors thank the families who participated in these projects. We also thank the dedicated staff at the Early Human and Lifespan Development Program and the Women and Children’s Health and Well-Being Project.
References
- Abbott PW, Gumusoglu SB, Bittle J, Beversdorf DQ, & Stevens HE (2018). Prenatal stress and genetic risk: How prenatal stress interacts with genetics to alter risk for psychiatric illness. Psychoneuroendocrinology, 90, 9–21. 10.1016/j.psyneuen.2018.01.019 [DOI] [PubMed] [Google Scholar]
- Abrahams BS, & Geschwind DH (2008). Advances in autism genetics: On the threshold of a new neurobiology. Nature Reviews Genetics, 9(5), 341–355. 10.1038/nrg2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibi JJ, Lee MK, Saha S, Boscardin WJ, Apfel A, & Currier RJ (2015). Fetal sex differences in human chorionic gonadotropin fluctuate by maternal race, age, weight and by gestational age. Journal of Developmental Origins of Health and Disease, 6(6), 493–500. 10.1017/s2040174415001336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG (2011). The promise and the pitfalls of autism research: An introductory note for new autism researchers. Brain Research, 1380, 3–9. http://dx.doi.org/10.1016Zj.brainres.2010.11.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Congress of Obstetricians and Gynecologists. (2014). Committee Opinion No. 611. Method for estimating due date. Obstetrics & Gynecology, 124(4), 863–866. 10.1097/01.AOG.0000454932.15177.be [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders: DSM-5. Washington, D.C.: American Psychiatric Association. [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, & Parner ET (2010). Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. Journal of Autism and Developmental Disorders, 40(12), 1423–1430. 10.1007/s10803-010-1006-y [DOI] [PubMed] [Google Scholar]
- Austin MP, & Leader L (2000). Maternal stress and obstetric and infant outcomes: Epidemiological findings and neuroendocrine mechanisms. Australian and New Zealand Journal of Obstetrics and Gynaecology, 40(3), 331–337. http://dx.d0i.0rg/l0.1111/j.1479-828X.2000.tb03344.x [DOI] [PubMed] [Google Scholar]
- Bale TL (2011). Sex differences in prenatal epigenetic programming of stress pathways. Stress, 14(4), 348–356. 10.3109/10253890.2011.586447 [DOI] [PubMed] [Google Scholar]
- Bale TL (2016). The placenta and neurodevelopment: Sex differences in prenatal vulnerability. Dialogues in Clinical Neuroscience, 18(4), 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, & Epperson CN (2015). Sex differences and stress across the lifespan. Nature Neuroscience, 18(10), 1413–1420. 10.1038/nn.4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Auyeung B, N0rgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L, . . . Lombardo MV (2015). Elevated fetal steroidogenic activity in autism. Molecular Psychiatry, 20(3), 369. 10.1038/mp.2014.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Knickmeyer RC, & Belmonte MK (2005). Sex differences in the brain: Implications for explaining autism. Science, 310(5749), 819–823. 10.1126/science.1115455 [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, & Knickmeyer R (2011). Why are autism spectrum conditions more prevalent in males? PLoS Biology, 9(6), e1001081. 10.1371/journal.pbio.1001081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Wheelwright S, Skinner R, Martin J, & Clubley E (2001). The autism- spectrum quotient (AQ): Evidence from Asperger syndrome/high-functioning autism, males and females, scientists and mathematicians. Journal of Autism and Developmental Disorders, 31(1), 5–17. 10.1023/A:1005653411471 [DOI] [PubMed] [Google Scholar]
- Beitins IZ, Bayard F, Ances IG, Kowarski A, & Migeon CJ (1973). The metabolic clearance rate, blood production, interconversion and transplacental passage of cortisol and cortisone in pregnancy near term. Pediatric Research, 7(5), 509–519. 10.1203/00006450-197305000-00004 [DOI] [PubMed] [Google Scholar]
- Beversdorf DQ, Manning SE, Hillier A, Anderson SL, Nordgren RE, Walters SE, . . . Bauman ML (2005). Timing of prenatal stressors and autism. Journal of Autism and Developmental Disorders, 35(4), 471–478. 10.1007/s10803-005-5037-8 [DOI] [PubMed] [Google Scholar]
- Beversdorf DQ, & Missouri Autism Summit C (2016). Phenotyping, etiological factors, and biomarkers: Toward precision medicine in autism spectrum disorders. Journal of Development and Behavioral Pediatrics, 37(8), 659–673. 10.1097/DBP.0000000000000351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitsika V, Sharpley CF, Andronicos NM, & Agnew LL (2017). What worries parents of a child with Autism? Evidence from a biomarker for chronic stress. Research in Developmental Disabilities, 62, 209–217. 10.1016/j.ridd.2017.02.003 [DOI] [PubMed] [Google Scholar]
- Brown RW, Diaz R, Robson AC, Kotelevtsev YV, Mullins JJ, Kaufman MH, & Seckl JR (1996). The ontogeny of 11 beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor gene expression reveal intricate control of glucocorticoid action in development. Endocrinology, 137(2), 794–797. 10.1210/endo.137.2.8593833 [DOI] [PubMed] [Google Scholar]
- Calamari JE, Wiegartz PS, & Janeck AS (1999). Obsessive-compulsive disorder subgroups: A symptom-based clustering approach. Behavioral Research and Therapy, 37(2), 113–125. 10.1016/S0005-7967(98)00135-1 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. (2014). Prevalence of autism spectrum disorders among children 8 years − autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveillance Summaries. [PubMed] [Google Scholar]
- Challis JR, Sloboda D, Matthews SG, Holloway A, Alfaidy N, Patel FA, . . . Newnham J (2001). The fetal placental hypothalamic-pituitary-adrenal (HPA) axis, parturition and post natal health. Molecular and Cell Endocrinology, 185(1–2), 135–144. 10.1016/S0303-7207(01)00624-4 [DOI] [PubMed] [Google Scholar]
- Class QA, Abel KM, Khashan AS, Rickert ME, Dalman C, Larsson H, . . . D’Onofrio BM (2014). Offspring psychopathology following preconception, prenatal and postnatal maternal bereavement stress. Psychological Medicine, 44(1), 71–84. 10.1017/S0033291713000780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton VL (2010). Review: Sex and the human placenta: Mediating differential strategies of fetal growth and survival. Placenta, 31 Suppl, S33–39. [DOI] [PubMed] [Google Scholar]
- Clifton VL, & Murphy VE (2004). Maternal asthma as a model for examining fetal sex-specific effects on maternal physiology and placental mechanisms that regulate human fetal growth. Placenta, 25 Suppl A, S45–52. 10.1016/j.placenta.2004.01.004 [DOI] [PubMed] [Google Scholar]
- Constantino JN (2011). The quantitative nature of autistic social impairment. Pediatric Research, 69(5 Pt 2), 55r–62r. 10.1203/PDR.0b013e318212ec6e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, & Todd RD (2003). Autistic traits in the general population: A twin study. Archives of General Psychiatry, 60(5), 524–530. 10.1001/archpsyc.60.5.524 [DOI] [PubMed] [Google Scholar]
- Cuthbert BN (2014). The RDoC framework: Facilitating transition from ICD/DSM to dimensional approaches that integrate neuroscience and psychopathology. World Psychiatry, 13(1), 28–35. 10.1002/wps.20087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva JA (1999). Sex hormones and glucocorticoids: interactions with the immune system. Annals of the New York Acadamy of Sciences, 876, 102–117; discussion 117–108. 10.1111/j.1749-6632.1999.tb07628.x [DOI] [PubMed] [Google Scholar]
- Davis EP, Head K, Buss C, & Sandman CA (2017). Prenatal maternal cortisol concentrations predict neurodevelopment in middle childhood. Psychoneuroendocrinology, 75, 56–63. http://dx.doi.org/10.1016Zj.psyneuen.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EP, & Pfaff D (2014). Sexually dimorphic responses to early adversity: Implications for affective problems and autism spectrum disorder. Psychoneuroendocrinology, 49, 11–25. 10.1016/j.psyneuen.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EP, & Sandman CA (2010). The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child Development, 81(1), 131–148. 10.1111/j.1467-8624.2009.01385.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EP, Waffarn F, & Sandman CA (2011). Prenatal treatment with glucocorticoids sensitizes the HPA axis response to stress among full-term infants. Developmental Psychobiology, 53(2), 175–183. 10.1002/dev.20510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis NO, & Carter AS (2008). Parenting stress in mothers and fathers of toddlers with autism spectrum disorders: Associations with child characteristics. Journal of Autism and Developmental Disorders, 38(7), 1278–1291. 10.1007/s10803-007-0512-z [DOI] [PubMed] [Google Scholar]
- De Andres-Garcia S, Moya-Albiol L, & Gonzalez-Bono E (2012). Salivary cortisol and immunoglobulin A: Responses to stress as predictors of health complaints reported by caregivers of offspring with autistic spectrum disorder. Hormones and Behavior, 62(4), 464–474. 10.1016/j.yhbeh.2012.08.003 [DOI] [PubMed] [Google Scholar]
- Durkin MS, Maenner MJ, Newschaffer CJ, Lee LC, Cunniff CM, Daniels JL, . . . Schieve LA (2008). Advanced parental age and the risk of autism spectrum disorder. American Journal of Epidemiology, 168(11), 1268–1276. 10.1093/aje/kwn250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens EM, & Lambert W (2013). Trajectories of diurnal cortisol in mothers of children with autism and other developmental disabilities: Relations to health and mental health. Journal of Autism and Developmental Disorders, 43(10), 2426–2434. 10.1007/s10803-013-1791-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M, . . . Lerch JP (2015). Clustering autism: Using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Molecular Psychiatry, 20(1), 118–125. 10.1038/mp.2014.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecteau SM, Boivin L, Trudel M, Corbett BA, Harrell FE Jr., Viau R, . . . Picard F (2017). Parenting stress and salivary cortisol in parents of children with autism spectrum disorder: Longitudinal variations in the context of a service dog’s presence in the family. Biological Psychology, 123, 187–195. http://dx.doi.org/10.10167j.biopsycho.2016.12.008 [DOI] [PubMed] [Google Scholar]
- Foody C, James JE, & Leader G (2015). Parenting stress, salivary biomarkers, and ambulatory blood pressure: a comparison between mothers and fathers of children with autism spectrum disorders. Journal of Autism and Developmental Disorders, 45(4), 1084–1095. 10.1007/s10803-014-2263-y [DOI] [PubMed] [Google Scholar]
- Fowden AL, Li J, & Forhead AJ (1998). Glucocorticoids and the preparation for life after birth: Are there long-term consequences of the life insurance? Proceedings of the Nutrition Society, 57(1), 113–122. 10.1079/PNS19980017 [DOI] [PubMed] [Google Scholar]
- Fox M, Sandman CA, Davis EP, & Glynn LM (2015). Intra-individual consistency in endocrine profiles across successive pregnancies. The Journal of Clinical Endocrinology & Metabolism, 100(12), 4637–4647. 10.1210/jc.2015-2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabory A, Roseboom TJ, Moore T, Moore LG, & Junien C (2013). Placental contribution to the origins of sexual dimorphism in health and diseases: Sex chromosomes and epigenetics. Biology of Sex Differences, 4(1), 5. 10.1186/2042-6410-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman PL, Flores-Ramos M, Lopez-Martinez M, Fuentes CC, & Grajeda JP (2015). Hypothalamic-pituitary-adrenal axis function during perinatal depression. Neuroscience Bulletin, 31(3), 338–350. 10.1007/s12264-014-1508-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulos G, Jackson K, & Tulchinsky D (1982). Glucocorticoid metabolism in human placenta, decidua, myometrium and fetal membranes. The Journal of Steroid Biochemisty and Molecular Biology, 17(4), 371–374. 10.1016/0022-4731(82)90628-8 [DOI] [PubMed] [Google Scholar]
- Gitau R, Adams D, Fisk NM, & Glover V (2005). Fetal plasma testosterone correlates positively with cortisol. Archives of Disease in Childhood Fetal and Neonatal Edition, 90(2), F166–169. 10.1136/adc.2004.049320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitau R, Cameron A, Fisk NM, & Glover V (1998). Fetal exposure to maternal cortisol. The Lancet, 352(9129), 707–708. 10.1016/s0140-6736(05)60824-0 [DOI] [PubMed] [Google Scholar]
- Gitau R, Fisk NM, Teixeira JM, Cameron A, & Glover V (2001). Fetal hypothalamic- pituitary-adrenal stress responses to invasive procedures are independent of maternal responses. The Journal of Clinical Endocrinology & Metabolism, 86(1), 104–109. 10.1210/jcem.86.1.7090 [DOI] [PubMed] [Google Scholar]
- Glynn LM, & Sandman CA (2012). Sex moderates associations between prenatal glucocorticoid exposure and human fetal neurological development. Developmental Science, 15(5), 601–610. 10.1111/j.1467-7687.2012.01159.x [DOI] [PubMed] [Google Scholar]
- Gore AC, Martien KM, Gagnidze K, & Pfaff D (2014). Implications of prenatal steroid perturbations for neurodevelopment, behavior, and autism. Endocrine Reviews, 35(6), 961–991. 10.1210/er.2013-1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JA (2010). Review: Sex chromosome evolution and the expression of sex-specific genes in the placenta. Placenta, 31 Suppl, S27–32. http://dx.doi.org/10.1016Zj.placenta.2009.12.029 [DOI] [PubMed] [Google Scholar]
- Grønborg TK, Schendel DE, & Parner ET (2013). Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: A population-based cohort study. JAMA Pediatrics, 167(10), 947–953. 10.1001/jamapediatrics.2013.2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacking D, Watkins A, Fraser S, Wolfe R, & Nolan T (2001). Respiratory distress syndrome and antenatal corticosteroid treatment in premature twins. Archives of Disease in Childhood Fetal and Neonatal Edition, 85(1), F77–78. 10.1136/fn.85.lF75g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, . . . Risch N (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry, 68(11), 1095–1102. 10.1001/archgenpsychiatry.2011.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington RA, Lee LC, Crum RM, Zimmerman AW, & Hertz-Picciotto I (2014). Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics, 133(5), e1241–1248. 10.1542/peds.2013-3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht PM, Hudson M, Connors SL, Tilley MR, Liu X, & Beversdorf DQ (2016). Maternal serotonin transporter genotype affects risk for ASD with exposure to prenatal stress. Autism Research, 9(11), 1151–1160. 10.1002/aur.1629 [DOI] [PubMed] [Google Scholar]
- Hill SD, Wagner EA, Shedlarski JG Jr., & Sears SP (1977). Diurnal cortisol and temperature variation of normal and autistic children. Developmental Psychobiology, 10(6), 579–583. 10.1002/dev.420100612 [DOI] [PubMed] [Google Scholar]
- Hobel C (1982). Identification of the patient at risk. In Perinatal medicine: Management of the high risk fetus and neonate (pp. 3–28): Williams & Wilkins, Baltimore. [Google Scholar]
- Hoekstra RA, Bartels M, Verweij CJ, & Boomsma DI (2007). Heritability of autistic traits in the general population. Archives of Pediatrics and Adolescent Medicine, 161(4), 372–377. 10.1001/archpedi.1614.372 [DOI] [PubMed] [Google Scholar]
- Howland MA, Sandman CA, & Glynn LM (2017). Developmental origins of the human hypothalamic-pituitary-adrenal axis. Expert Review of Endocrinology & Metabolism, 12(5), 321–339. 10.1080/17446651.2017.1356222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingudomnukul E, Baron-Cohen S, Wheelwright S, & Knickmeyer R (2007). Elevated rates of testosterone-related disorders in women with autism spectrum conditions. Hormones and Behavior, 51(5), 597–604. 10.1016/j.yhbeh.2007.02.001 [DOI] [PubMed] [Google Scholar]
- Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, . . . Wang P (2010). Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. American Journal of Psychiatry, 167(7), 748–751. 10.1176/appi.ajp.2010.09091379 [DOI] [PubMed] [Google Scholar]
- Janecka M, Haworth CMA, Ronald A, Krapohl E, Happe F, Mill J, . . . Rijsdijk F (2017). Paternal age alters social development in offspring. Journal of the American Academy of Child and Adolescent Psychiatry, 56(5), 383–390. http://dx.doi.Org/10.1016/j.jaac.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Smith RM, Edwards KS, Givens B, Tilley MR, & Beversdorf DQ (2010). Combined effect of maternal serotonin transporter genotype and prenatal stress in modulating offspring social interaction in mice. International Journal of Developmental Neuroscience, 28(6), 529–536. 10.1016/j.ijdevneu.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Petropoulos S, & Matthews SG (2008). Fetal programming of hypothalamic- pituitary-adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Research Reviews, 57(2), 586–595. http://dx.doi.org/10.1016Zj.brainresrev.2007.06.013 [DOI] [PubMed] [Google Scholar]
- Kim DR, Bale TL, & Epperson CN (2015). Prenatal programming of mental illness: Current understanding of relationship and mechanisms. Current Psychiatry Reports, 17(2), 5. 10.1007/s11920-014-0546-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King BR, Nicholson RC, & Smith R (2001). Placental corticotrophin-releasing hormone, local effects and fetomaternal endocrinology. Stress, 4(4), 219–233. 10.3109/10253890109014747 [DOI] [PubMed] [Google Scholar]
- Kinlein SA, Wilson CD, & Karatsoreos IN (2015). Dysregulated hypothalamic-pituitary- adrenal axis function contributes to altered endocrine and neurobehavioral responses to acute stress. Frontiers in Psychiatry, 6, 31. 10.3389/fpsyt.2015.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney DK, Munir KM, Crowley DJ, & Miller AM (2008). Prenatal stress and risk for autism. Neuroscience & Biobehavioral Reviews, 32(8), 1519–1532. 10.1016/j.neubiorev.2008.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickmeyer RC, & Baron-Cohen S (2006). Fetal testosterone and sex differences in typical social development and in autism. Journal of Child Neurology, 21(10), 825–845. http://dx.doi.org/101177/08830738060210101601 [DOI] [PubMed] [Google Scholar]
- Lai MC, Lombardo MV, Auyeung B, Chakrabarti B, & Baron-Cohen S (2015). Sex/gender differences and autism: Setting the scene for future research. Journal of the American Academy of Child and Adolescent Psychiatry, 54(1), 11–24. 10.1016/jjaac.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Lombardo MV, & Baron-Cohen S (2014). Autism. The Lancet, 383(9920), 896–910. 10.1016/s0140-6736(13)61539-1 [DOI] [PubMed] [Google Scholar]
- Landrigan PJ (2010). What causes autism? Exploring the environmental contribution. Current Opinion in Pediatrics, 22(2), 219–225. 10.1097/M0P.0b013e328336eb9a [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, & Le Couteur A (1994). Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders, 24(5), 659–685. [DOI] [PubMed] [Google Scholar]
- Loth E, Murphy DG, & Spooren W (2016). Defining precision medicine approaches to autism spectrum disorders: Concepts and challenges. Frontiers in Psychiatry, 7, 188. 10.3389/fpsyt.2016.00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loth E, Spooren W, Ham LM, Isaac MB, Auriche-Benichou C, Banaschewski T, . . . Murphy DG (2016). Identification and validation of biomarkers for autism spectrum disorders. Nature Reviews Drug Discovery, 15(1), 70–73. 10.1038/nrd.2015.7 [DOI] [PubMed] [Google Scholar]
- Manson JE (2008). Prenatal exposure to sex steroid hormones and behavioral/cognitive outcomes. Metabolism, 57 Suppl 2, S16–21. http://dx.doi.org/10.1016Zj.metabol.2008.07.010 [DOI] [PubMed] [Google Scholar]
- Marinović-Curin J, Marinovic-Terzic I, Bujas-Petkovic Z, Zekan L, Skrabic V, Dogas Z, & Terzic J (2008). Slower cortisol response during ACTH stimulation test in autistic children. European Child & Adolescent Psychiatry, 17(1), 39–43. 10.1007/s00787-007-0632-1 [DOI] [PubMed] [Google Scholar]
- Mastorakos G, & Ilias I (2003). Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Annals of the New York Academy of Sciences, 997, 136–149. 10.1196/annals.1290.016 [DOI] [PubMed] [Google Scholar]
- Matthews SG (2000). Antenatal glucocorticoids and programming of the developing CNS. Pediatric Research, 47(3), 291–300. 10.1203/00006450-200003000-00003 [DOI] [PubMed] [Google Scholar]
- Matthews SG (2002). Early programming of the hypothalamo-pituitary-adrenal axis. Trends in Endocrinology & Metabolism, 13(9), 373–380. 10.1016/S1043-2760(02)00690-2 [DOI] [PubMed] [Google Scholar]
- McLean M, Bisits A, Davies J, Woods R, Lowry P, & Smith R (1995). A placental clock controlling the length of human pregnancy. Nature Medicine, 1(5), 460–463. 10.1038/nm0595-460 [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, & Zhou ES (2007). If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychological Bulletin, 133(1), 25–45. 10.1037/0033-2909.133.125 [DOI] [PubMed] [Google Scholar]
- Moisiadis VG, & Matthews SG (2014). Glucocorticoids and fetal programming part 1: Outcomes. Nature Reviews Endocrinology, 10(7), 391–402. 10.1038/nrendo.2014.73 [DOI] [PubMed] [Google Scholar]
- Mulligan A, Richardson T, Anney RJ, & Gill M (2009). The Social Communication Questionnaire in a sample of the general population of school-going children. Irish Journal of Medical Science, 178(2), 193–199. 10.1007/s11845-008-0184-5 [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, & Holbrook NJ (1984). Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrine Reviews, 5(1), 25–44. 10.1210/edrv-5-1-25 [DOI] [PubMed] [Google Scholar]
- Murphy VE, Smith R, Giles WB, & Clifton VL (2006). Endocrine regulation of human fetal growth: The role of the mother, placenta, and fetus. Endocrine Reviews, 27(2), 141–169. 10.1210/er.2005-0011 [DOI] [PubMed] [Google Scholar]
- Nakayama Y, Takahashi T, Wakabayashi A, Oono H, & Radford MH (2007). Sex differences in the relationship between cortisol levels and the Empathy and Systemizing Quotients in humans. Neuroendocrinology Letters, 28(4), 445–448. [PubMed] [Google Scholar]
- Naughton M, Dinan TG, & Scott LV (2014). Corticotropin-releasing hormone and the hypothalamic-pituitary-adrenal axis in psychiatric disease. Handbook of Clinical Neurology, 124, 69–91. 10.1016/b978-0-444-59602-4.00005-8 [DOI] [PubMed] [Google Scholar]
- O’Connell BA, Moritz KM, Walker DW, & Dickinson H (2013). Treatment of pregnant spiny mice at mid gestation with a synthetic glucocorticoid has sex-dependent effects on placental glycogen stores. Placenta, 34(10), 932–940. http://dx.doi.org/10.1016Zj.placenta.2013.06.310 [DOI] [PubMed] [Google Scholar]
- Osei-Kumah A, Smith R, Jurisica I, Caniggia I, & Clifton VL (2011). Sex-specific differences in placental global gene expression in pregnancies complicated by asthma. Placenta, 32(8), 570–578. 10.1016/j.placenta.2011.05.005 [DOI] [PubMed] [Google Scholar]
- Petraglia F, Florio P, Nappi C, & Genazzani AR (1996). Peptide signaling in human placenta and membranes: Autocrine, paracrine, and endocrine mechanisms. Endocrine Reviews, 17(2), 156–186. 10.1210/edrv-17-2-156 [DOI] [PubMed] [Google Scholar]
- Pfaff DW, Rapin I, & Goldman S (2011). Male predominance in autism: Neuroendocrine influences on arousal and social anxiety. Autism Research, 4(3), 163–176. 10.1002/aur.191 [DOI] [PubMed] [Google Scholar]
- Posserud MB, Lundervold AJ, & Gillberg C (2006). Autistic features in a total population of 7–9-year-old children assessed by the ASSQ (Autism Spectrum Screening Questionnaire). Journal of Child Psychology and Psychiatry, 47(2), 167–175. 10.1111/j.1469-7610.2005.01462.x [DOI] [PubMed] [Google Scholar]
- Posserud MB, Lundervold AJ, & Gillberg C (2009). Validation of the autism spectrum screening questionnaire in a total population sample. Journal of Autism and Developmental Disorders, 39(1), 126–134. 10.1007/s10803-008-0609-z [DOI] [PubMed] [Google Scholar]
- Raudenbush SW, & Bryk AS (2002). Hierarchical linear models: Applications and data analysis methods (Vol. 1): Sage. [Google Scholar]
- Reynolds CM, Vickers MH, Harrison CJ, Segovia SA, & Gray C (2015). Maternal high fat and/or salt consumption induces sex-specific inflammatory and nutrient transport in the rat placenta. Physiological Reports, 3(5). 10.14814/phy2.12399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Hoffmann TJ, Anderson M, Croen LA, Grether JK, & Windham GC (2014). Familial recurrence of autism spectrum disorder: Evaluating genetic and environmental contributions. The American Journal of Psychiatry, 171(11), 1206–1213. 10.1176/appi.ajp.2014.13101359 [DOI] [PubMed] [Google Scholar]
- Rivard M, Terroux A, Parent-Boursier C, & Mercier C (2014). Determinants of stress in parents of children with autism spectrum disorders. Journal of Autism and Developmental Disorders, 44(7), 1609–1620. 10.1007/s10803-013-2028-z [DOI] [PubMed] [Google Scholar]
- Robinson EB, Neale BM, & Hyman SE (2015). Genetic research in autism spectrum disorders. Current Opinion in Pediatrics, 27(6), 685–691. 10.1097/mop.0000000000000278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, & Bohlin G (2005). Are maternal smoking and stress during pregnancy related to ADHD symptoms in children? Journal of Child Psychology and Psychiatry, 46(3), 246–254. 10.1111/j.1469-7610.2004.00359.x [DOI] [PubMed] [Google Scholar]
- Rose’meyer R (2013). A review of the serotonin transporter and prenatal cortisol in the development of autism spectrum disorders. Molecular Autism, 4(1), 37. 10.1186/2040-2392-4-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose’meyer R (2014). Is cortisol the underlying mediator of prenatal risk factors associated with autism spectrum disorders? OA Autism, 2(1), 2. [Google Scholar]
- Rutter M, Anthony B, Lord C (2003). The Social Communication Questionnaire Manual: Los Angeles: Western Psychological Services. [Google Scholar]
- Ruzich E, Allison C, Smith P, Watson P, Auyeung B, Ring H, & Baron-Cohen S (2015). Measuring autistic traits in the general population: A systematic review of the Autism- Spectrum Quotient (AQ) in a nonclinical population sample of 6,900 typical adult males and females. Molecular Autism, 6, 2. 10.1186/2040-2392-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman CA, Davis EP, Buss C, & Glynn LM (2011). Prenatal programming of human neurological function. International Journal of Peptides, 2011, 837596. 10.1155/2011/837596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman CA, Glynn L, Schetter CD, Wadhwa P, Garite T, Chicz-DeMet A, & Hobel C (2006). Elevated maternal cortisol early in pregnancy predicts third trimester levels of placental corticotropin releasing hormone (CRH): priming the placental clock. Peptides, 27(6), 1457–1463. 10.1016/j.peptides.2005.10.002 [DOI] [PubMed] [Google Scholar]
- Sandman CA, & Glynn LM (2009). Corticotropin-releasing hormone (CRH) programs the fetal and maternal brain. Future Neurology, 4(3), 257–261. 10.2217/fnl.09.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman CA, Glynn LM, & Davis EP (2013). Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. Journal of Psychosomatic Research, 75(4), 327–335. 10.1016/jjpsychores.2013.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Bergman K, O’Connor TG, & Glover V (2008). Maternal antenatal anxiety and amniotic fluid cortisol and testosterone: Possible implications for foetal programming. Journal of Neuroendocrinology, 20(4), 489–496. 10.1111/j.1365-2826.2008.01659x [DOI] [PubMed] [Google Scholar]
- Schaafsma SM, Gagnidze K, Reyes A, Norstedt N, Mansson K, Francis K, & Pfaff DW (2017). Sex-specific gene-environment interactions underlying ASD-like behaviors. Proceedings of the National Academy of Sciences of the United States of America, 114(6), 1383–1388. 10.1073/pnas.1619312114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendel D, & Bhasin TK (2008). Birth weight and gestational age characteristics of children with autism, including a comparison with other developmental disabilities. Pediatrics, 121(6), 1155–1164. http://dx.doi.org/.1542/peds.2007-1049 [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, … Wigler M (2007). Strong association of de novo copy number mutations with autism. Science, 316(5823), 445–449. 10.1126/science.1138659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seckl JR (2004). Prenatal glucocorticoids and long-term programming. European Journal of Endocrinology, 151 Suppl 3, U49–62. 10.1530/eje.0.151U049 [DOI] [PubMed] [Google Scholar]
- Seltzer MM, Greenberg JS, Hong J, Smith LE, Almeida DM, Coe C, & Stawski RS (2010). Maternal cortisol levels and behavior problems in adolescents and adults with ASD. Journal of Autism and Developmental Disorders, 40(4), 457–469. 10.1007/s10803-009-0887-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D, France JT, Keelan JA, Song L, & Knox BS (1994). Sex differences in umbilical cord serum levels of inhibin, testosterone, oestradiol, dehydroepiandrosterone sulphate, and sex hormone-binding globulin in human term neonates. Biology of the Neonate, 65(5), 287–294. 10.1159/000244074 [DOI] [PubMed] [Google Scholar]
- Sjaarda CP, Hecht P, McNaughton AJM, Zhou A, Hudson ML, Will MJ, … Liu X (2017). Interplay between maternal Slc6a4 mutation and prenatal stress: A possible mechanism for autistic behavior development. Scientific Reports, 7(1), 8735. 10.1038/s41598-017-07405-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiker D, Lotspeich LJ, Dimiceli S, Myers RM, & Risch N (2002). Behavioral phenotypic variation in autism multiplex families: Evidence for a continuous severity gradient. American Journal of Medical Genetics, 114(2), 129–136. [DOI] [PubMed] [Google Scholar]
- Stark MJ, Wright IM, & Clifton VL (2009). Sex-specific alterations in placental 11beta- hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 297(2), R510–514. 10.1152/ajpregu.00175.2009 [DOI] [PubMed] [Google Scholar]
- Taylor JL, & Corbett BA (2014). A review of rhythm and responsiveness of cortisol in individuals with autism spectrum disorders. Psychoneuroendocrinology, 49, 207–228. http://dx.doi.org/10.10167j.psyneuen.2014.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theule J, Wiener J, Tannock R, & Jenkins JM (2010). Parenting stress in families of children with ADHD. Journal of Emotional and Behavioral Disorders, 21(1), 3–17. http://dx.doi.org/10.117771063426610387433 [Google Scholar]
- Tordjman S, Somogyi E, Coulon N, Kermarrec S, Cohen D, Bronsard G, … Xavier J (2014). Gene x environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Frontiers in Psychiatry, 5, 53. 10.3389/fpsyt.2014.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo JL, Cuchillo I, Machin C, & Rua C (2000). Maternal adrenalectomy at the early onset of gestation impairs the postnatal development of the rat hippocampal formation: Effects on cell numbers and differentiation, connectivity and calbindin-D28k immunoreactivity. Journal of Neuroscience Research, 62(5), 644–667. [DOI] [PubMed] [Google Scholar]
- Trottier G, Srivastava L, & Walker CD (1999). Etiology of infantile autism: A review of recent advances in genetic and neurobiological research. Journal of Psychiatry & Neuroscience, 24(2), 103–115. [PMC free article] [PubMed] [Google Scholar]
- Tsigos C, & Chrousos GP (2002). Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. Journal of Psychosomatic Research, 53(4), 865–871. 10.1016/S0022-3999(02)00429-4 [DOI] [PubMed] [Google Scholar]
- Varcin KJ, Alvares GA, Uljarevic M, & Whitehouse AJO (2017). Prenatal maternal stress events and phenotypic outcomes in Autism Spectrum Disorder. Autism Research. 10.1002/aur.1830 [DOI] [PubMed] [Google Scholar]
- Welberg LA, & Seckl JR (2001). Prenatal stress, glucocorticoids and the programming of the brain. Journal of Neuroendocrinology, 13(2), 113–128. http://dx.doi.org/10.1111Zj.1365-2826.2001.00601.x [DOI] [PubMed] [Google Scholar]
- Werling DM, & Geschwind DH (2013). Sex differences in autism spectrum disorders. Current Opinion in Neurology, 26(2), 146–153. 10.1097/WCO.0b013e32835ee548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Lobel M, & Moyer A (2014). Low maternal progesterone may contribute to both obstetrical complications and autism. Medical Hypotheses, 82(3), 313–318. 10.1016/j.mehy.2013.12.018 [DOI] [PubMed] [Google Scholar]
- Wigham S, McConachie H, Tandos J, & Le Couteur AS (2012). The reliability and validity of the Social Responsiveness Scale in a UK general child population. Research in Developmental Disabilities, 33(3), 944–950. 10.1016/j.ridd.2011.12.017 [DOI] [PubMed] [Google Scholar]
- Wong JD, Mailick MR, Greenberg JS, Hong J, & Coe CL (2014). Daily work stress and awakening cortisol in mothers of individuals with autism spectrum disorders or Fragile X Syndrome. Family Relations, 63(1), 135–147. 10.1111/fare.12055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JD, Seltzer MM, Greenberg JS, Hong J, Almeida DM, & Coe CL (2012). Stressful life events and daily stressors affect awakening cortisol level in midlife mothers of individuals with autism spectrum disorders. Aging & Mental Health, 16(8), 939–949. 10.1080/13607863.2012.688191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunszain PA, Anacker C, Cattaneo A, Carvalho LA, & Pariante CM (2011). Glucocorticoids, cytokines and brain abnormalities in depression. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 35(3), 722–729. 10.1016/j.pnpbp.2010.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.