

Summary
Background
Patients with plaque psoriasis treated with biologic therapies need more efficacious, safe and convenient treatments to improve quality of life. Risankizumab and secukinumab inhibit interleukin‐23 and interleukin‐17A, respectively, and are effective in adult patients with moderate‐to‐severe plaque psoriasis but have different dosing regimens.
Objectives
To compare directly the efficacy and safety of risankizumab vs. secukinumab over 52 weeks.
Methods
IMMerge was an international, phase III, multicentre, open‐label, efficacy–assessor‐blinded, active‐comparator study, in which adult patients with chronic, moderate‐to‐severe plaque psoriasis were randomized in a 1 : 1 ratio to treatment with risankizumab 150 mg or secukinumab 300 mg. Primary efficacy endpoints were the proportions of patients achieving ≥ 90% improvement from baseline in Psoriasis Area and Severity Index (PASI 90) at week 16 (noninferiority comparison with margin of 12%) and week 52 (superiority comparison).
Results
In total 327 patients from nine countries were treated with risankizumab (n = 164) or secukinumab (n = 163). Risankizumab was noninferior to secukinumab in the proportion of patients achieving PASI 90 at week 16 [73·8% vs. 65·6%; difference of 8·2%, 96·25% confidence interval (CI)−2·2 to 18·6; within the 12% noninferiority margin] and superior to secukinumab at week 52 (86·6% vs. 57·1%; difference of 29·8%, 95% CI 20·8–38·8; P < 0·001), thus meeting both primary endpoints. All secondary endpoints (PASI 100, static Physician's Global Assessment 0 or 1, and PASI 75) at week 52 demonstrated superiority for risankizumab vs. secukinumab (P < 0·001). No new safety concerns were identified.
Conclusions
At week 52, risankizumab demonstrated superior efficacy and similar safety with less frequent dosing compared with secukinumab.
Short abstract
What is already known about this topic?
The need remains for treatments with sustained efficacy and a more convenient dosing schedule in moderate‐to‐severe psoriasis.
Risankizumab and secukinumab are indicated for the treatment of adults with moderate‐to‐severe plaque psoriasis and target interleukin‐23 and interleukin‐17, respectively.
To date, risankizumab and secukinumab have not been directly compared.
What does this study add?
IMMerge directly compared the safety and efficacy of risankizumab and secukinumab in patients with moderate‐to‐severe plaque psoriasis using ≥ 90% improvement in Psoriasis Area and Severity Index at weeks 16 (noninferiority) and 52 (superiority) as primary endpoints.
In terms of efficacy risankizumab was noninferior to secukinumab at week 16 and superior to secukinumab at week 52 of treatment based on primary endpoint analyses. The two medications had a similar safety profile.
Linked Comment: Schmitt-Egenolf. Br J Dermatol 2021; 184: 3–4.
Plain language summary available online
Plaque psoriasis is a chronic inflammatory immune‐mediated skin disorder with an approximate prevalence of 1–4% globally. 1 , 2 , 3 Psoriasis is associated with increased morbidity and mortality, and may lead to disability while negatively affecting patient quality of life. 4 , 5 , 6 , 7 Interleukin (IL)‐23 contributes to psoriasis by stimulating proliferation, differentiation and maintenance of T helper 17 cells and innate immune cells, which produce proinflammatory cytokines such as IL‐17. 8 , 9 Importantly, discovery of the IL‐23/IL‐17 immunological pathway was key to our expanding knowledge about the pathogenesis of psoriasis, and in the development of new targeted therapeutic agents for psoriasis. 10 , 11 Although approved biologics are effective for the treatment of plaque psoriasis, there is a need for more efficacious therapy that will achieve and maintain higher response rates in the long term. Sustained skin clearance and less frequent dosing intervals are important to control moderate‐to‐severe plaque psoriasis in patients and improve quality of life. 12 , 13 , 14
Risankizumab is a humanized IgG1 monoclonal antibody that inhibits IL‐23 by binding to its p19 subunit. 15 , 16 , 17 Results from four multicentre, randomized, double‐blinded studies (UltIMMa‐1, UltIMMa‐2, IMMhance and IMMvent) that enrolled 2109 adult patients with moderate‐to‐severe plaque psoriasis, along with an open‐label extension study, supported the approval of risankizumab in the USA, Canada, Europe and Japan in 2019. 18 , 19 , 20 Results from active‐comparator studies have shown that risankizumab has greater efficacy in patients with moderate‐to‐severe plaque psoriasis than the IL‐12/IL‐23 inhibitor ustekinumab (UltIMMa‐1 and UltIMMa‐2) and the tumour necrosis factor‐α inhibitor adalimumab (IMMvent). 18 , 21 Secukinumab is a human IgG1 monoclonal antibody targeting IL‐17A that was approved in 2015 for the treatment of plaque psoriasis, and has demonstrated greater efficacy in clearing skin than ustekinumab. 22 , 23 Here, we compared the efficacy and safety of risankizumab vs. secukinumab in patients with moderate‐to‐severe plaque psoriasis. Details of the availability of the data for researchers are provided in Appendix 2.
Patients and methods
Patients
This study recruited adult patients with a diagnosis of chronic, moderate‐to‐severe plaque psoriasis with or without psoriatic arthritis for at least 6 months before the baseline visit and who were candidates for systemic therapy including secukinumab. Patients had to demonstrate at least 10% body surface area covered with psoriasis plaques, a static Physician’s Global Assessment (sPGA) score of ≥ 3, and a Psoriasis Area and Severity Index (PASI) ≥ 12 at screening and baseline. Patients were not eligible to participate if they had a history of erythrodermic psoriasis, generalized or localized pustular psoriasis, medication‐induced or medication‐exacerbated psoriasis, or new‐onset guttate psoriasis (or any other active skin disease) that might interfere with the study assessments. Other exclusion criteria included a history of inflammatory bowel disease (based on warnings and precautions provided in the secukinumab prescribing information), chronic infections, active systemic infection (except common cold) during the last 2 weeks preceding the baseline visit, and history of malignancy, except for successfully treated nonmelanoma skin cancer or localized carcinoma in situ of the cervix, within the last 5 years. Previous exposure to risankizumab or secukinumab was not permitted.
Study design and treatment
IMMerge was a phase III, international, multicentre, randomized, open‐label, efficacy–assessor‐blinded, active‐comparator study of up to 88 weeks’ total duration (Clinicaltrials.gov identifier: NCT03478787). The study included a 30‐day screening period, and eligible patients were randomized in a 1 : 1 ratio via a centralized Interactive Response Technology system to open‐label treatment with risankizumab or secukinumab for up to 64 weeks (Figure 1). Leading up to study treatment initiation, patients were not allowed to use phototherapy or any systemic nonbiologic treatment for 4 weeks, or systemic biologic treatment for 6 weeks or longer, depending on the biologic. Risankizumab was administered as two subcutaneous injections of 75 mg (150 mg total) at weeks 0 and 4, and every 12 weeks thereafter until the last dose at week 40, except for patients in France, who received additional doses at weeks 52 and 64 to allow for continuous treatment until it was commercially available for patients in France. Secukinumab was administered as two subcutaneous injections of 150 mg (300 mg total) at weeks 0, 1, 2, 3 and 4, and every 4 weeks thereafter until the last dose at week 48. All study treatments were administered by healthcare professionals. The final efficacy assessment was performed at week 52. Approximately 20 weeks after the final dose, study patients were called by phone for a safety follow‐up.
Figure 1.
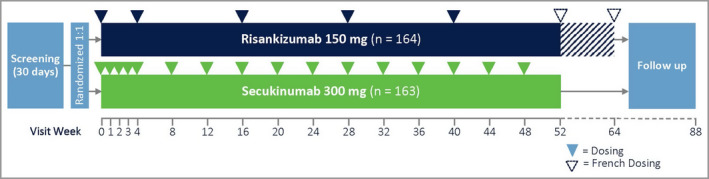
Study design. The hatched area from week 52 to week 64 corresponds to two additional doses administered to patients in France. The additional doses did not affect efficacy assessments performed at week 52.
This study was performed at hospitals, academic medical centres, clinical research units and/or private practices under the supervision of certified dermatologists who had significant experience in conducting clinical trials. The study was conducted in accordance with the Good Clinical Practice guideline as defined by the International Conference on Harmonisation, the Declaration of Helsinki and/or all applicable federal and local regulations and an institutional review board. All patients provided written informed consent. The protocol, informed consent forms and recruitment materials, and all participant materials were reviewed and approved by institutional ethics committees and/or institutional review boards.
Assessments
Efficacy assessments
Efficacy assessments were performed by a qualified physician or designee at each study site at all appropriate study visits. The efficacy assessor was fully trained on the protocol and could not perform efficacy assessments prior to having completed all necessary training. The efficacy assessor remained blinded to each patient’s treatment and clinical laboratory results, and all safety data during the course of the study. The efficacy assessor was instructed to document the dermatological assessments on paper worksheets and was not allowed access to patient electronic case report forms.
Efficacy endpoints
This study assessed two primary efficacy endpoints: (i) the proportion of patients achieving an improvement in PASI of at least 90% from baseline (PASI 90) at week 16 to assess the noninferiority of risankizumab vs. secukinumab (noninferiority margin of 12%), and (ii) the proportion of patients achieving PASI 90 at week 52 to assess the superiority of risankizumab vs. secukinumab. Efficacy endpoints were assessed prior to study drug administration at study visits when both events occurred. To control for the overall type I error at alpha = 0·05, we predistributed alpha to primary endpoints as follows: week 16 alpha = 0·0375, week 52 alpha = 0·0125. If the primary endpoint at week 16 was achieved, the remaining alpha would be passed to week 52 such that the alpha at week 52 would be increased to 0·05. 24 , 25 Secondary endpoints were assessed at week 52, and were ranked and tested in sequential order, requiring statistically significant results before testing the subsequent endpoint. In rank order, the secondary endpoints were the proportion of patients who achieved PASI 100, the proportion of patients who achieved an sPGA score of 0 or 1, and the proportion of patients who achieved PASI 75, all evaluating the superiority of risankizumab vs. secukinumab.
Safety assessment
Safety was assessed via treatment‐emergent adverse events (TEAEs), serious adverse events (AEs), AEs, and AEs of special interest that led to discontinuation of study treatment. Major adverse cardiovascular events (MACEs) and events of anaphylaxis were to be confirmed by adjudication committees blinded to the study drug. Vital signs, laboratory tests, physical examinations and electrocardiogram measurements were also performed.
Statistical analysis
Study size determination
Study size was determined by considering that approximately 310 patients (155 per treatment group) would provide > 90% power to detect the difference between treatment groups in PASI 90 at week 52 using a two‐sided alpha level of 0·0125, and 90% power to determine the noninferiority of risankizumab relative to secukinumab using a tolerance limit of 12% and a two‐sided alpha level of 0·0375 at week 16. 24 , 25
Endpoint analyses and imputation
Efficacy endpoints were determined using the intent‐to‐treat population, defined as all patients randomized at baseline. Categorical variables were assessed using the Cochran–Mantel–Haenszel test adjusting for stratification by weight (≤ 100 kg vs. > 100 kg) and prior systemic biologic use for psoriasis (none vs. at least one). The noninferiority margin of 12% for the rate of PASI 90 at week 16 was chosen as it was expected to preserve 80% of the treatment effect of secukinumab over placebo.
Missing efficacy data were accounted for using nonresponder imputation, whereby any patient who had a missing value at a study visit was categorized as a nonresponder for that visit, unless the patient was a responder both before and after a specific visit window. Safety analyses were performed on all intent‐to‐treat patients who received at least one dose of study drug (safety population). Statistical analyses were performed using SAS version 9·4 (SAS Institute, Inc., Cary, NC, USA) using the Unix operating system.
Results
Patients
Of 409 patients screened, in total 327 from Canada, France, Germany, Italy, the Netherlands, Poland, Spain, the UK and the USA were randomized to risankizumab 150 mg (n = 164) or secukinumab 300 mg (n = 163) (Figure 2). Totals of 162 (98·8%) and 150 (92·0%) patients randomized to risankizumab and secukinumab, respectively, completed week 16 of the study, while 151 (92·1%) and 135 (82·8%), respectively, completed week 52. The study was conducted from May 2018 (first patient screened) through March 2020 (last patient follow‐up).
Figure 2.
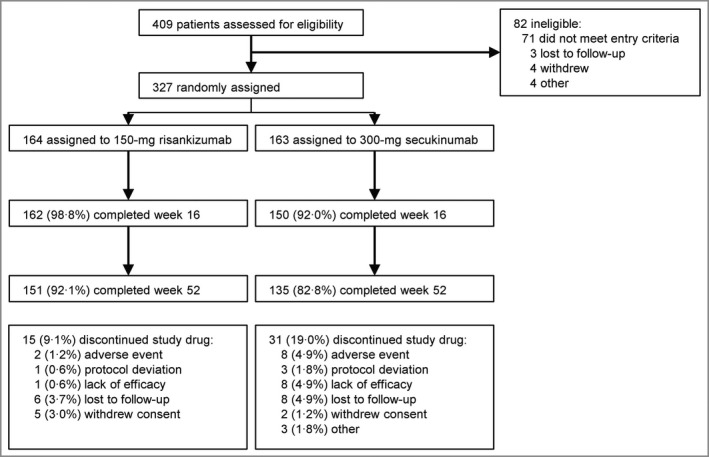
Patient disposition. aSome patients had discontinued the study drug as described below.
The patient demographics and disease characteristics are summarized in Table 1. The mean ± SD patient age was 47·1 ± 14·1 years and 65·1% were male. At baseline, patients had a mean ± SD PASI of 19·9 ± 7·2, and 84·7% of patients had an sPGA score of 3.
Table 1.
Baseline demographics and disease characteristics
| Characteristic | Risankizumab 150 mg (n = 164) | Secukinumab 300 mg (n = 163) |
|---|---|---|
| Age (years), mean ± SD | 47·3 ± 13·4 | 46·8 ± 14·9 |
| Male, n (%) | 112 (68·3) | 101 (62·0) |
| Race, n (%) | ||
| White | 151 (92·1) | 144 (88·3) |
| Black/African American | 6 (3·7) | 6 (3·7) |
| Asian | 6 (3·7) | 11 (6·7) |
| Other | 1 (0·6) | 2 (1·2) |
| Hispanic or Latino ethnicity, n (%) | 37 (22·6) | 34 (20·9) |
| Bodyweight category, n (%) | ||
| ≤ 100 kg | 112 (68·3) | 109 (66·9) |
| > 100 kg | 52 (31·7) | 54 (33·1) |
| Duration of plaque psoriasis (years), mean ± SD | 18·6 ± 12·6 | 17·4 ± 13·2 |
| sPGA category, n (%) | ||
| 3 | 140 (85·4) | 137 (84·0) |
| 4 | 24 (14·6) | 25 (15·3) |
| < 3 or missing | 0 | 1 (0·6) |
| Body surface area (%), mean ± SD | 23·8 ± 13·8 | 26·0 ± 16·1 |
| PASI, mean ± SD | 19·8 ± 6·3 | 20·1 ± 8·1 |
| Previously used biologics for psoriasis, n (%) | 62 (37·8) | 58 (35·6) |
| IL‐17 inhibitor | 13 (7·9) | 12 (7·4) |
| IL‐23 inhibitor | 3 (1·8) | 2 (1·2) |
| Tumour necrosis factor inhibitor | 38 (23·2) | 38 (23·3) |
| IL‐12/IL‐23 inhibitor | 15 (9·1) | 22 (13·5) |
IL, interleukin; PASI, Psoriasis Area and Severity Index; sPGA, static Physician’s Global Assessment.
Efficacy assessments
Primary efficacy endpoints
The results of the two primary efficacy analyses showed that 73·8% (n = 121) of patients randomized to risankizumab achieved PASI 90 at week 16 compared with 65·6% (n = 107) of patients randomized to secukinumab. The difference in proportions of PASI 90 responders between groups (adjusted for stratification factors) was 8·2% [96·25% confidence interval (CI) −2·2 to 18·6], which fell within the 12% noninferiority margin; thus, the primary endpoint of noninferiority of risankizumab to secukinumab at week 16 was met (Figure 3). At week 52, 86·6% (n = 142) of patients randomized to risankizumab achieved PASI 90 compared with 57·1% (n = 93) of patients randomized to secukinumab. The adjusted difference was 29·8% (95% CI 20·8–38·8, P < 0·001) (Figure 3); thus, the primary endpoint of superiority of risankizumab to secukinumab at week 52 was met.
Figure 3.
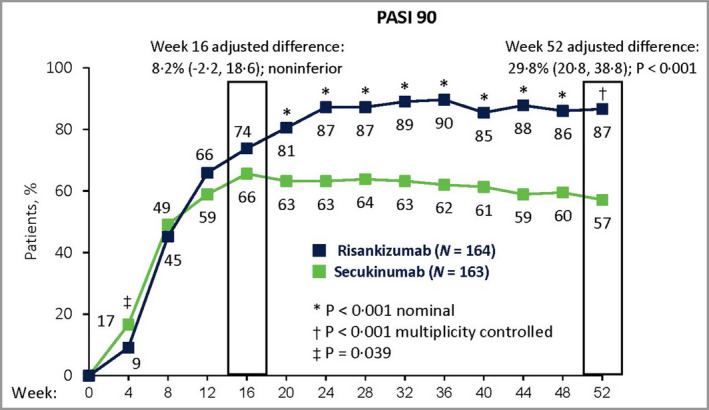
Primary efficacy results. Proportions of patients with ≥ 90% reduction in Psoriasis Area and Severity Index (PASI 90) at week 16 and week 52. Risankizumab was noninferior to secukinumab at week 16 based on a noninferiority margin of 12%, and superior to secukinumab at week 52 (P < 0·001). Data were assessed for the intent‐to‐treat population. The adjusted‐difference confidence interval (CI) values were 96·25% CI at week 16 and 95% CI at week 52. P‐values were calculated from the Cochran–Mantel–Haenszel test, stratified by weight (≤ 100 kg vs. > 100 kg) and prior systemic biologic use for psoriasis. Nonresponder imputation was used for missing data.
Secondary efficacy endpoints
At week 52, 65·9% of patients randomized to risankizumab vs. 39·9% randomized to secukinumab achieved PASI 100, resulting in an adjusted difference of 26·2% (95% CI 15·9–36·5, P < 0·001; Figure 4a). An sPGA score of 0 or 1 was achieved in 87·8% of patients treated with risankizumab and 58·3% of patients treated with secukinumab at week 52, an adjusted difference of 29·8% (95% CI 20·9–38·8, P < 0·001; Figure 4b). PASI 75 was achieved by 89·6% of patients treated with risankizumab compared with 69·9% of patients treated with secukinumab, an adjusted difference of 20·0% (95% CI 11·7–28·3, P < 0·001; Figure 4c). Thus, all secondary endpoints demonstrated superiority for risankizumab vs. secukinumab at week 52.
Figure 4.
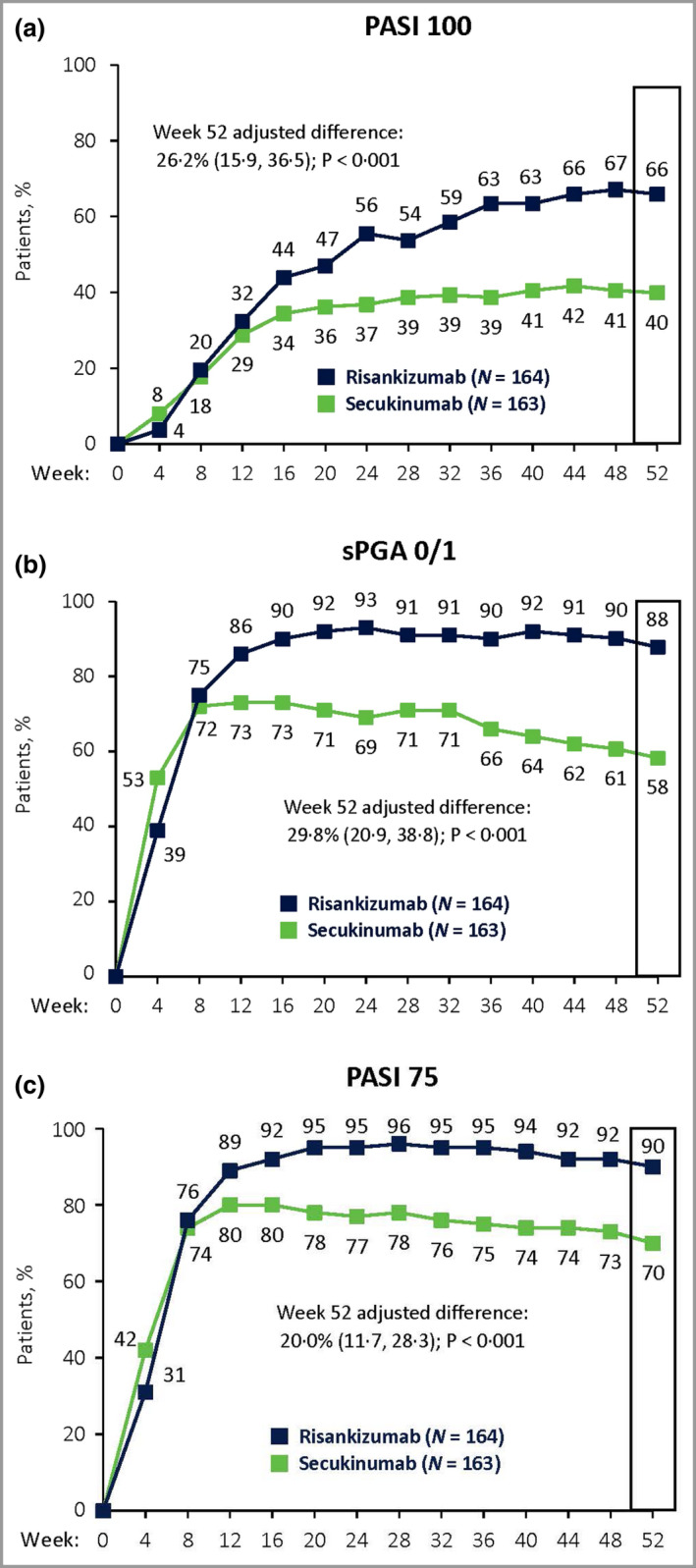
Secondary efficacy results. Proportions of patients achieving (a) 100% reduction in Psoriasis Area and Severity Index (PASI 100), (b) static Physician’s Global Assessment (sPGA) of 0 or 1 and (c) PASI 75 at week 52. Significantly more patients treated with risankizumab achieved all secondary endpoints than did patients treated with secukinumab (P < 0·001), demonstrating superiority of risankizumab over secukinumab. Secondary endpoints (week 52) are indicated by the boxed regions. Data were assessed for the intent‐to‐treat population. The adjusted‐difference confidence interval was set at 95%. P‐values were calculated from the Cochran–Mantel–Haenszel test, stratified by weight (≤ 100 kg vs. > 100 kg) and prior systemic biologic use for psoriasis. Nonresponder imputation was used for missing data.
Safety
In the safety analysis, TEAEs were reported for 117 (71·3%) patients treated with risankizumab and 116 (71·2%) patients treated with secukinumab (Table 2). Serious AEs were reported for nine (5·5%) and six (3·7%) patients treated with risankizumab and secukinumab, respectively; no patients died during the study. The most commonly reported TEAEs for risankizumab were nasopharyngitis, upper respiratory tract infection, headache, arthralgia, diarrhoea and bronchitis (Table 2).
Table 2.
Summary of treatment‐emergent adverse events (TEAEs) a
| Parameter | Patients, n (%) | |
|---|---|---|
| Risankizumab 150 mg (n = 164) | Secukinumab 300 mg (n = 163) | |
| Any TEAE a | 117 (71·3) | 116 (71·2) |
| SAEs | 9 (5·5) | 6 (3·7) |
| Severe (grade ≥ 3) TEAE | 11 (6·7) | 7 (4·3) |
| TEAE possibly related to study drug | 49 (29·9) | 46 (28·2) |
| SAE possibly related to study drug | 1 (0·6) | 1 (0·6) |
| TEAE leading to drug discontinuation | 2 (1·2) | 8 (4·9) |
| Deaths | 0 | 0 |
| TEAEs of special interest | ||
| Adjudicated MACE | 2 (1·2) | 0 |
| Serious infection | 3 (1·8) | 0 |
| Tuberculosis | 0 | 0 |
| Malignant tumours | 1 (0·6) | 3 (1·8) |
| Malignant tumours (non‐NMSC) | 0 | 0 |
| Serious hypersensitivity | 0 | 1 (0·6) |
| Any TEAE with ≥ 5% frequency | ||
| Nasopharyngitis | 35 (21·3) | 27 (16·6) |
| Upper respiratory tract infection | 21 (12·8) | 14 (8·6) |
| Headache | 9 (5·5) | 15 (9·2) |
| Arthralgia | 9 (5·5) | 10 (6·1) |
| Diarrhoea | 9 (5·5) | 9 (5·5) |
| Bronchitis | 3 (1·8) | 11 (6·7) |
MACE, major adverse cardiovascular event; NMSC, nonmelanoma skin cancer; SAE, serious adverse event.
Defined as adverse events occurring with an onset within 20 weeks after the last dose of study drug administration.
In total eight (4·9%) patients in the secukinumab arm discontinued study treatment because of AEs, compared with two (1·2%) patients treated with risankizumab. Three patients discontinued treatment with secukinumab because of events considered by the study investigators to be related to study treatment. These events included arthralgia in one patient; ulcerative colitis in one patient; and chest discomfort, lethargy, rash and throat tightness in one patient. The two events that led to discontinuation in the risankizumab group (hepatomegaly and suicidal ideation) were not considered related to study treatment.
Two patients treated with risankizumab experienced nonfatal myocardial infarctions that were confirmed as MACEs by the adjudication committee. The first event occurred on study day 105 in a 49–year‐old man who was a smoker at the time of the event with a 35‐year smoking history (15 cigarettes per day), was overweight with poor dietary habits, and had a strong family history of early myocardial infarction (both parents had cardiac disease in their early 40s). The second event occurred on study day 311 in a 72–year‐old man who was a smoker at the time of the event with a 20‐year smoking history (10 cigarettes per day), and had a medical history of hyperlipidaemia and a family history of heart failure. Neither event was considered to have a causal relationship with risankizumab treatment and both patients remained in the study without treatment interruption. No MACEs occurred in patients treated with secukinumab.
Other TEAEs of special interest are presented in Table 2. Three patients in the risankizumab arm experienced serious infections (two experienced urinary tract infections, and one experienced histiocytic necrotizing lymphadenitis). One case of new‐onset inflammatory bowel disease (ulcerative colitis) occurred in a patient treated with secukinumab, which was considered serious and related to treatment, and led to treatment discontinuation. There were no cases of new‐onset inflammatory bowel disease among patients treated with risankizumab. Candida infections (including oral and vulvovaginal candidiasis) were reported for seven patients overall and were evenly distributed between groups (three and four patients treated with risankizumab and secukinumab, respectively).
Discussion
In this phase III trial of adult patients with moderate‐to‐severe plaque psoriasis, risankizumab was noninferior to secukinumab at week 16 (difference of 8·2% in PASI 90 response rate between groups, which fell within the predetermined noninferiority margin), and superior at week 52 based on PASI 90 (primary endpoint), as well as PASI 100, sPGA 0/1 and PASI 75 (secondary endpoints). The safety profile of risankizumab in this study is comparable with that of secukinumab, and no new safety concerns were identified during the study.
Overall, the efficacy and safety results observed in this trial are generally consistent with those seen in the other phase III risankizumab efficacy and safety studies, further supporting its benefit‐to‐risk profile for the treatment of moderate‐to‐severe plaque psoriasis. 18 , 23 , 26 , 27 , 28 , 29 , 30 , 31 The efficacy and safety of secukinumab observed in this study are also consistent with efficacy and safety findings noted in previous phase III trials, validating its consistent performance in controlled clinical trials. 20 , 23 , 27 , 31 , 32 , 33 The trend we observe in this study of a smaller difference in PASI 90 response between IL–23– and IL‐17A‐targeted treatments at week 16 followed by larger differences favouring the IL‐23 inhibitor after 52 weeks of treatment are consistent with results from a previous phase III study. 31
Recent clinical trials with biologic agents, including those with risankizumab and secukinumab where psoriasis treatment was assessed for improvements in quality of life, 18 , 23 , 33 highlight the importance of patients’ experiences when they undergo treatment for this condition. The results from our study demonstrate that, under the indicated treatment regimens, risankizumab provides efficacy that is noninferior to secukinumab at week 16 (after seven doses of secukinumab vs. two doses of risankizumab), and superior efficacy at week 52 (16 doses of secukinumab vs. five doses of risankizumab).
Two patients who experienced MACEs – both in the risankizumab group – had pre‐existing risk factors for cardiovascular disease. Neither event was considered drug related, which is consistent with findings from other phase III trials. 18 , 20 , 21 Ulcerative colitis is another event of interest based on epidemiological reports, suggesting a potential relationship between inflammatory bowel disease and psoriasis, and there are reports indicating that IL‐17 inhibitors may be associated with new cases or exacerbation of existing cases of inflammatory bowel disease. 34 , 35 , 36 , 37 , 38 Only one event of ulcerative colitis was observed in the current study, but it was considered serious and related to treatment with secukinumab, and resulted in the patient’s discontinuation from study treatment. In general, our safety findings were consistent with the overall safety profile of risankizumab as demonstrated by 2673 patients investigated in the pivotal clinical trial programme. 18 , 20 , 21 , 39
We also observed that a larger proportion of patients discontinued treatment with secukinumab than risankizumab (17·2% and 9.1%, respectively). The data demonstrate that this difference is largely driven by differences in efficacy and AEs between groups. Specifically, eight patients in the secukinumab arm vs. one patient treated with risankizumab discontinued treatment due to lack of efficacy, while an additional eight patients in the secukinumab arm vs. two in the risankizumab arm discontinued due to AEs (Figure 2).
As reported in clinical trials and registries, there is a loss of efficacy of biologic agents over time, and this is an important consideration when treating patients with plaque psoriasis. Long‐term studies of the tumour necrosis factor‐α inhibitors adalimumab, etanercept and infliximab all demonstrate there is a loss of treatment response over time, and that this loss of efficacy may lead to more than half of all discontinuations from studies in which tumour necrosis factor‐α inhibitors are analysed. 40 , 41 , 42 , 43 Agents that target the IL‐23/IL‐17 immunological pathway, such as risankizumab, secukinumab and the IL‐12/IL‐23 inhibitor ustekinumab, appear to have improved long‐term efficacy. 20 , 23 , 34 , 44 , 45 , 46 , 47 , 48 , 49 Our data show that there is superior efficacy of risankizumab to secukinumab over 52 weeks. Formal comparisons evaluating longer‐term maintenance of response and duration of treatment are needed. 50
This study’s limitations include the open‐label design; however, a blinded efficacy assessor was used to determine efficacy and the observed results were within those reported from other double‐blinded studies evaluating these compounds. This study design was also used in recent active‐comparator studies in plaque psoriasis and psoriatic arthritis. 51 , 52 Another possible limitation is that differences in time between the last dose and the last efficacy assessment may have impacted the results because the half‐lives of the treatments are similar. 22 , 53
In conclusion, in this phase III, active‐comparator clinical trial, risankizumab demonstrated noninferior efficacy to secukinumab after 16 weeks, and superior efficacy after 52 weeks of treatment. These data may help inform practitioners when selecting a biologic therapy for their patients with moderate‐to‐severe plaque psoriasis.
Supporting information
Powerpoint S1 Journal Club Slide Set.
Acknowledgments
The authors thank AbbVie for support of this manuscript. AbbVie and the authors thank the patients who participated in this clinical trial and all study investigators for their contributions. The authors also thank Nannette Englehardt and Lawrence McNamee, both from AbbVie, for their contributions to the study. Medical writing support, funded by AbbVie, was provided by Nate Connors, PhD, Kersten Reich, MPH, CMPP™ and Lamara D. Shrode, PhD, CMPP™, of JB Ashtin, who developed the first draft based on an author‐approved outline and assisted in implementing author revisions throughout the editorial process. JB Ashtin adheres to Good Publication Practice (GPP3) guidelines and International Committee of Medical Journal Editors recommendations. R.B.W. is supported by the Manchester NIHR Biomedical Research Centre.
Appendix 1. Conflicts of interest
R.B.W. has received research grants from and leads clinical trials for AbbVie, Almirall, Amgen, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, LEO Pharma, Novartis, Pfizer and UCB Pharma; and has received consulting fees from AbbVie, Almirall, Amgen, Arena Pharmaceuticals, Avillion, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Janssen, LEO Pharma, Eli, Lilly, Novartis, Pfizer, Sanofi and UCB Pharma. A.B. has served as a scientific adviser and/or clinical study investigator for AbbVie, Aclaris, Almirall, Arena, Pharmaceuticals, Athenex, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Dermira, Eli Lilly and Company, Forte, Galderma, Janssen, LEO, Novartis, Ortho, Pfizer, Rapt, Regeneron, Sandoz, Sanofi Genzyme, Sun Pharma and UCB Pharma; and as a paid speaker for AbbVie. Y.P. has received grant funding and honoraria for services as an investigator, speaker and member of advisory boards from AbbVie, Amgen, Bausch, Janssen‐Ortho and UCB Pharma; and has received grant funding as an investigator from Baxter, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eli Lilly, Galderma, Genentech, GlaxoSmithKline, Incyte, LEO Pharma, MedImmune, Merck, Novartis, Pfizer, Regeneron, Sanofi, Serono and Takeda. C.P. has received grants from and has been a consultant for AbbVie, Almirall, Amgen, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen, LEO Pharma, Merck, Novartis, Pfizer, Sandoz and UCB Pharma. S.B., M.K., T.W. and Z.G. are full‐time employees of AbbVie Inc. and may hold AbbVie stock and/or stock options.
Appendix 2. Data sharing
AbbVie is committed to responsible data sharing regarding the clinical trials they sponsor. This includes access to anonymized, individual and trial‐level data (analysis datasets), as well as other information (e.g. protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan (SAP) and execution of a data sharing agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.AbbVie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
Funding sources AbbVie Inc. funded this study, and participated in the study design, research, analysis, data collection, interpretation of data, reviewing and approval of the publication. All authors had access to the data and participated in the development, review, critique and approval of the manuscript throughout the editorial process, and approved the final manuscript draft submitted for publication. All authors agree to be accountable for all aspects of the work, ensuring the accuracy and integrity of the publication. Medical writing support was paid for by AbbVie.
Conflicts of interest statements can be found in Appendix 1.
Plain language summary available online
References
- 1. Parisi R, Symmons DPM, Griffiths CEM et al. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 2013; 133:377–85. [DOI] [PubMed] [Google Scholar]
- 2. Lebwohl MG, Bachelez H, Barker J et al. Patient perspectives in the management of psoriasis: results from the population‐based Multinational Assessment of Psoriasis and Psoriatic Arthritis Survey. J Am Acad Dermatol 2014; 70:871–81. [DOI] [PubMed] [Google Scholar]
- 3. Chandran V, Raychaudhuri SP. Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun 2010; 34:J314–21. [DOI] [PubMed] [Google Scholar]
- 4. Ni C, Chiu MW. Psoriasis and comorbidities: links and risks. Clin Cosmet Investig Dermatol 2014; 7:119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mehta NN, Yu Y, Pinnelas R et al. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med 2011; 124:775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boehncke W‐H, Schön MP. Psoriasis. Lancet 2015; 386:983–94. [DOI] [PubMed] [Google Scholar]
- 7. Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med 2014; 4:a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL‐23–IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014; 14:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol 2017; 13:525–34. [DOI] [PubMed] [Google Scholar]
- 10. Blauvelt A, Chiricozzi A. The immunologic role of IL‐17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 2018; 55:379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol 2017; 140:645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Carvalho AVE, Duquia RP, Horta BL, Bonamigo RR. Efficacy of immunobiologic and small molecule inhibitor drugs for psoriasis: a systematic review and meta‐analysis of randomized clinical trials. Drugs R D 2017; 17:29–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boehncke W‐H, Brembilla NC. Unmet needs in the field of psoriasis: pathogenesis and treatment. Clin Rev Allergy Immunol 2018; 55:295–311. [DOI] [PubMed] [Google Scholar]
- 14. Puig L, Thom H, Mollon P et al. Clear or almost clear skin improves the quality of life in patients with moderate‐to‐severe psoriasis: a systematic review and meta‐analysis. J Eur Acad Dermatol Venereol 2017; 31:213–20. [DOI] [PubMed] [Google Scholar]
- 15. Papp KA, Blauvelt A, Bukhalo M et al. Risankizumab versus ustekinumab for moderate‐to‐severe plaque psoriasis. N Engl J Med 2017; 376:1551–60. [DOI] [PubMed] [Google Scholar]
- 16. Krueger JG, Ferris LK, Menter A et al. Anti‐IL‐23A mAb BI 655066 for treatment of moderate‐to‐severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single‐rising‐dose, randomized, double‐blind, placebo‐controlled trial. J Allergy Clin Immunol 2015; 136:116–24. [DOI] [PubMed] [Google Scholar]
- 17. Singh S, Kroe‐Barrett RR, Canada KA et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high‐affinity humanized anti‐IL23A antibody. MAbs 2015; 7:778–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gordon KB, Strober B, Lebwohl M et al. Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 2018; 392:650–61. [DOI] [PubMed] [Google Scholar]
- 19. Reddy V, Yang EJ, Myers B, Liao W. Clinical evaluation of risankizumab‐rzaa in the treatment of plaque psoriasis. J Inflam Res 2020; 13:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blauvelt A, Leonardi CL, Gooderham M et al. Efficacy and safety of continuous risankizumab therapy versus treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol 2020; 156:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reich K, Gooderham M, Thaçi D et al. Risankizumab compared with adalimumab in patients with moderate‐to‐severe plaque psoriasis (IMMvent): a randomised, double‐blind, active‐comparator‐controlled phase 3 trial. Lancet 2019; 394:576–86. [DOI] [PubMed] [Google Scholar]
- 22. Sanford M, McKeage K. Secukinumab: first global approval. Drugs 2015; 75:329–38. [DOI] [PubMed] [Google Scholar]
- 23. Blauvelt A, Reich K, Tsai T‐F et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate‐to‐severe plaque psoriasis up to 1 year: results from the CLEAR study. J Am Acad Dermatol 2017; 76:60–9. [DOI] [PubMed] [Google Scholar]
- 24. United States Food and Drug Administration . Multiple endpoints in clinical trials: guidance for industry. Available at: https://www.fda.gov/media/102657/download (last accessed 30 June 2020).
- 25. Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med 2009; 28:586–604. [DOI] [PubMed] [Google Scholar]
- 26. McKeage K, Duggan S. Risankizumab: first global approval. Drugs 2019; 79:893–900. [DOI] [PubMed] [Google Scholar]
- 27. Langley RG, Elewski BE, Lebwohl M et al. Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med 2014; 371:326–38. [DOI] [PubMed] [Google Scholar]
- 28. Okubo Y, Ohtsuki M, Morita A et al. Long‐term efficacy and safety of secukinumab in Japanese patients with moderate to severe plaque psoriasis: 3‐year results of a double‐blind extension study. J Dermatol 2019; 46:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu N‐L, Hsu C‐J, Sun F‐J, Tsai T‐F. Efficacy and safety of secukinumab in Taiwanese patients with moderate to severe plaque psoriasis: subanalysis from ERASURE phase III study. J Dermatol 2017; 44:1129–37. [DOI] [PubMed] [Google Scholar]
- 30. Lacour JP, Paul C, Jazayeri S et al. Secukinumab administration by autoinjector maintains reduction of plaque psoriasis severity over 52 weeks: results of the randomized controlled JUNCTURE trial. J Eur Acad Dermatol Venereol 2017; 31:847–56. [DOI] [PubMed] [Google Scholar]
- 31. Reich K, Armstrong AW, Langley RG et al. Guselkumab versus secukinumab for the treatment of moderate‐to‐severe psoriasis (ECLIPSE): results from a phase 3, randomized controlled trial. Lancet 2019; 394:831–9. [DOI] [PubMed] [Google Scholar]
- 32. Blauvelt A, Prinz JC, Gottlieb AB et al. Secukinumab administration by pre‐filled syringe: efficacy, safety and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol 2015; 172:484–93. [DOI] [PubMed] [Google Scholar]
- 33. Thaçi D, Blauvelt A, Reich K et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol 2015; 73:400–9. [DOI] [PubMed] [Google Scholar]
- 34. Reich K, Warren RB, Coates LC, Di Comite G. Long term efficacy and safety of secukinumab in the treatment of the multiple manifestations of psoriatic disease. J Eur Acad Dermatol Venereol 2020; 34:1161–73. [DOI] [PubMed] [Google Scholar]
- 35. Augustin M, Reich K, Glaeske G et al. Co‐morbidity and age‐related prevalence of psoriasis: analysis of health insurance data in Germany. Acta Derm Venereol 2010; 90:147–51. [DOI] [PubMed] [Google Scholar]
- 36. Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population‐based study. Gastroenterology 2005; 129:827–36. [DOI] [PubMed] [Google Scholar]
- 37. Wolf N, Quaranta M, Prescott NJ et al. Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J Med Genet 2008; 45:114–16. [DOI] [PubMed] [Google Scholar]
- 38. Hohenberger M, Cardwell LA, Oussedik E, Feldman SR. Interleukin‐17 inhibition: role in psoriasis and inflammatory bowel disease. J Dermatolog Treat 2018; 29:13–18. [DOI] [PubMed] [Google Scholar]
- 39. Gordon KB, Bachelez H, Blauvelt A et al. Pooled long‐term safety analysis of risankizumab in patients with moderate‐to‐severe psoriasis. Presented at the 8th Annual Meeting of the American Academy of Dermatology, Denver, CO, 20–24 March 2020; Poster 16332.
- 40. Papp K, Menter A, Poulin Y et al. Long‐term outcomes of interruption and retreatment vs. continuous therapy with adalimumab for psoriasis: subanalysis of REVEAL and the open‐label extension study. J Eur Acad Dermatol Venereol 2013; 27:634–42. [DOI] [PubMed] [Google Scholar]
- 41. Esposito M, Gisondi P, Cassano N et al. Survival rate of antitumour necrosis factor‐α treatments for psoriasis in routine dermatological practice: a multicentre observational study. Br J Dermatol 2013; 169:666–72. [DOI] [PubMed] [Google Scholar]
- 42. Tyring S, Gordon KB, Poulin Y et al. Long‐term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol 2007; 143:719–26. [DOI] [PubMed] [Google Scholar]
- 43. Menter A, Feldman SR, Weinstein GD et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate‐to‐severe plaque psoriasis. J Am Acad Dermatol 2007; 56:31. [DOI] [PubMed] [Google Scholar]
- 44. Kimball AB, Gordon KB, Fakharzadeh S et al. Long‐term efficacy of ustekinumab in patients with moderate‐to‐severe psoriasis: results from the PHOENIX 1 trial through up to 3 years. Br J Dermatol 2012; 166:861–72. [DOI] [PubMed] [Google Scholar]
- 45. Langley RG, Lebwohl M, Krueger GG et al. Long‐term efficacy and safety of ustekinumab, with and without dosing adjustment, in patients with moderate‐to‐severe psoriasis: results from the PHOENIX 2 study through 5 years of follow‐up. Br J Dermatol 2015; 172:1371–83. [DOI] [PubMed] [Google Scholar]
- 46. Leonardi CL, Kimball AB, Papp KA et al. Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 76‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 1). Lancet 2008; 371:1665–74. [DOI] [PubMed] [Google Scholar]
- 47. Menter A, Papp K, Gooderham M et al. Drug survival of biologic therapy in a large, disease‐based registry of patients with psoriasis: results from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). J Eur Acad Dermatol Venereol 2016; 30:1148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papp KA, Langley RG, Lebwohl M et al. Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 52‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 2). Lancet 2008; 371:1675–84. [DOI] [PubMed] [Google Scholar]
- 49. Warren RB, Smith CH, Yiu ZZ et al. Differential drug survival of biologic therapies for the treatment of psoriasis: a prospective observational cohort study from the British Association of Dermatologists Biologic Interventions Register (BADBIR). J Invest Dermatol 2015; 135:2632–40. [DOI] [PubMed] [Google Scholar]
- 50. Sawyer LM, Malottki K, Sabry‐Grant C et al. Assessing the relative efficacy of interleukin‐17 and interleukin‐23 targeted treatments for moderate‐to‐severe plaque psoriasis: a systematic review and network meta‐analysis of PASI response. PLOS ONE 2019; 14:e0220868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Papp KA, Barber K, Bissonnette R et al. A randomized, blinded assessor study to evaluate the efficacy and safety of etanercept 50 mg once weekly plus as needed topical agent vs. etanercept 50 mg twice weekly in patients with moderate to severe plaque psoriasis (REFINE). J Eur Acad Dermatol Venereol 2015; 29:361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mease PJ, Smolen JS, Behrens F et al. A head‐to‐head comparison of the efficacy and safety of ixekizumab and adalimumab in biological‐naive patients with active psoriatic arthritis: 24‐week results of a randomised, open‐label, blinded‐assessor trial. Ann Rheum Dis 2020; 79:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pang Y, Khatri A, Suleiman AA, Othman AA. Clinical pharmacokinetics and pharmacodynamics of risankizumab in psoriasis patients. Clin Pharmacokinet 2020; 59:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Powerpoint S1 Journal Club Slide Set.