

Abstract
The combination of multiple neurodegenerative proteinopathies is increasingly recognized. Together they can potentiate neuronal dysfunction and contribute to complex neurological symptoms. We report an octogenarian female case of multiple extraneural metastases of a rectal carcinoma. She attempted suicide, which ultimately led to cardiorespiratory failure nine days after hospital admission. Apart from the suicide attempt and late‐onset depression, other psychiatric or neurological symptoms were not reported. Unexpectedly, histopathologic examination revealed prominent aging‐related tau astrogliopathy (ARTAG) of all five types (subpial, subependymal, grey and white matter, and perivascular) affecting cortical and subcortical brain regions. This pathology was associated with intermediate Alzheimer's disease neuropathologic change (A2B2C2 score), cerebral amyloid angiopathy, Lewy body‐type α‐synuclein proteinopathy (Braak stage 4), and a multiple system transactivation response DNA‐binding protein of 43 kDa (TDP‐43) proteinopathy also involving the astroglia. In summary, we report a complex and extensive combination of multiple proteinopathies with widespread ARTAG of all five types in a patient who had attempted suicide. Although longitudinal psychometric tests and neuropsychological evaluations were not performed, this report poses the question of thresholds of cognition and pathology load, describes ARTAG affecting unusually widespread brain regions, and supports the notion that complex proteinopathies should be regarded as a frequent condition in the elderly.
Keywords: ARTAG, Astroglia, Lewy‐body, Tau, TDP‐43
INTRODUCTION
Neurodegenerative diseases are characterized by progressive dysfunction and loss of neurons and deposition of pathologically altered proteins in different brain regions. The clinical symptomatology of the different neurodegenerative diseases is mostly related to the anatomical involvement of neuronal damage. 1 Classification of neurodegenerative disease is currently protein‐based and takes into account the particular cell types that accumulate a specific protein. 1 Furthermore, the clinical phenotype and neuropathological changes can be influenced by concomitant neurodegenerative pathologies and other conditions, such as vascular lesions. 2 , 3 , 4 Other systemic and local factors, including inflammation and metabolic changes, have been shown to also influence the brain's microenvironment, potentially further modifying disease. 5 , 6 , 7 The interaction of different pathologies as an aggravating factor of neurodegeneration has been proposed. 8 , 9 , 10 There is, however, no well‐defined threshold leading to manifest neurological impairment; furthermore, pathological changes including mixed pathologies have also been described in non‐demented elderly probands. 11
The focus of the evaluation of solely neuronal pathology in neurodegenerative diseases has changed with the recognition of the involvement of glial cells and their potential roles in neurodegeneration. 12 , 13 , 14 Astrocytes, with their important role in the brain's homeostasis and microenvironment, are cells of special interest. 12 , 13 , 15 Astroglial tau pathology has been increasingly recognized not only in tau proteinopathies but also in aging, 16 which facilitated the introduction of the umbrella term aging‐related tau astrogliopathy (ARTAG). 17 ARTAG is characterized by the presence of so‐called thorn‐shaped astrocytes (TSAs) and granular/fuzzy astrocytes (GFAs), and is mostly seen in the elderly. 18 Different types of ARTAG in a white matter, grey matter, subpial, subependymal, and perivascular allocation can be distinguished. 19 Moreover, a sequential distribution for the different types of ARTAG has been recognized. 18 The clinical significance of ARTAG is still not fully elucidated. In a cohort of old patients with concurrent Alzheimer's disease (AD)‐related pathology, cortical ARTAG was identified to be associated with dementia, while limbic and brainstem ARTAG was not. 20 Atypical clinical phenotypes combining dementia and psychiatric symptoms and/or parkinsonism have been described in patients with widespread grey matter ARTAG. 21 ARTAG seems to be present in cases of AD with both atypical and typical clinical presentation. 22 Following the description of clusters of argyrophilic thorn‐shaped astrocytes in combination with AD pathology and with the clinical manifestation of primary progressive aphasia, 23 a recent study suggested a negative impact of white matter ARTAG pathology to language and possibly visuospatial networks. 24 Recently, astroglial transactivation response DNA‐binding protein of 43 kDs (RDP‐43) proteinopathy has been also identified in atypical tau astrogliopathy. 25 , 26 The relationship between TDP‐43 and tau is, therefore, an increasing matter of interest.
Herein we present a case of widespread ARTAG of all types in an elderly patient who had attempted suicide with concurrent widespread deposition of pathological tau, TDP‐43, amyloid‐β (Aβ), and α‐synuclein without clinically striking neurological manifestation of disease.
CLINICAL SUMMARY
The present case was examined in the frame of the Vienna Trans‐Danube Aging (VITA) study. The patient was, however, not systematically evaluated longitudinally for neurological or psychiatric symptoms. The clinical information was retrospectively extracted from available reports, and no additional details on neurological and/or psychiatric data than those reported below were available. Neuroradiological examination results were not available.
The 84‐year‐old female patient suffered from stenosing rectal cancer with a suspected metastasis to the left adrenal gland. She was capable of living alone and taking care of herself. Cancer was treated with laser surgery in a palliative setting only, mainly because of age and comorbidities, such as adiposity, diabetes mellitus, arterial hypertension, and severe extracerebral atherosclerosis. Stenting of the stenosis was planned. Seven months later, at the age of 85, she attempted suicide with benzodiazepines and was admitted to the hospital. The nursing report described her being oriented and communicative without symptoms of dementia. She had no signs or symptoms of insanity. Before and during admission no validated neurological or psychiatric examinations were conducted. Her general condition worsened, and the nursing report described a depressed mood. An acute ileus had to be treated surgically by transversostomy. Peritoneal carcinomatosis and metastasis to the liver, spleen, and adrenal glands were identified during the surgical procedure, from which she did not recover and died a few days later of cardiorespiratory failure. In summary, the clinical phenotype was compatible with late‐onset depression without apparent dementia.
Neuropathological findings
Neuropathological examination method
Formalin‐fixed, paraffin‐embedded tissue blocks (2.5 × 2.0 cm) were evaluated. Postmortem delay was 48 h. The formalin fixation time was six weeks. The left brain hemisphere was used for histological examination and the right brain hemisphere was fresh‐frozen and stored at −80°C. Tissue paraffin blocks comprised the frontal, cingular, temporal, parietal, occipital cortices and white matter, anterior and posterior portions of the hippocampus, caudate nucleus, accumbens nucleus, putamen, globus pallidus, thalamus, mesencephalon, pons, medulla oblongata, cerebellar anterior vermis, cerebellar hemisphere, and dentate nucleus. Sections were stained with hematoxylin and eosin (HE) and Klüver‐Barrera (KB) and in selected areas with Bielschowsky or Gallyas silver impregnation methods. The following monoclonal antibodies were used for immunohistochemistry: anti‐hyperphosphorylated tau (clone AT8, pS202/Thr205, 1:200; Thermo Fisher Scientific, Rockford, IL, USA), anti‐four repeat (4R) tau (clone RD4, 1:200; Upstate, Charlottesville, VA, USA), anti‐three repeat (3R) tau (clone RD3, 1:2000; Upstate, Charlottesville, VA, USA), anti‐phosphorylated TDP‐43 (clone pS409/410, 1:2000; Cosmo Bio, Tokyo, Japan), anti‐ubiquitin (clone Ubi‐1, 1:50000; Millipore, Temecula, CA, USA), anti‐α‐synuclein (clone 5G4, 1:2000; Roboscreen, Leipzig, Germany), anti‐Aβ (clone 6F/3D, 1:50; Dako, Glostrup, Denmark), and anti‐p62 (clone 3/p62 lck ligand, 1:500; BD Transduction Laboratories, Franklin Lakes, NJ, USA) antibodies. The DAKO EnVision Detection Kit, peroxidase/DAB and rabbit/mouse (Dako) were used for visualization of antibody binding reaction. Neuropathological alterations, including astrogliosis, neuronal loss, and degree of various abnormal protein depositions, were evaluated in all examined anatomical regions. The different pathological protein depositions were evaluated semiquantitatively as follows: none (−), no immunoreactivity; mild (+), just a few immunoreactive cells, estimated under 10%; moderate (++), estimated 10–50% of cells; and severe (+++), estimated more than 50% of cells. Double immunolabeling was performed in selected areas using the monoclonal anti‐hyperphophorylated tau (clone AT8; 1:100; Thermo Fisher Scientific), polyclonal anti‐glial fibrillary acidic protein (GFAP) (1:500; Dako), polyclonal anti‐TDP‐43 (1:100; ProteinTech Group, Chicago, Il, USA), and polyclonal anti‐phosphorylated TDP‐43 (1:2500; Cosmo Bio, Tokyo, Japan) antibodies. The secondary antibodies were Alexa Fluor (AF) 555‐labeled donkey anti‐mouse IgG (1:200; Molecular Probes, Eugene, OR, USA), AF 488‐labeled goat anti‐rabbit IgG (1:200; Molecular Probes), and Cy3‐labeled goat anti‐mouse IgG (1:1000; Jackson Immuno Research, Cambridgeshire, UK). We evaluated double immunofluorescence labeling with a Zeiss LSM 780 Confocal Laser Microscope at the core facilities of the Medical University of Vienna.
Macroscopic findings
The formalin‐fixed brain weighed 1183 g. A mild atherosclerosis without luminal stenosis of the basal arteries was noted. Gross examination (Fig. 1A, B) revealed narrowing of the ambient gyrus. The substantia nigra and locus coeruleus showed relatively preserved pigmentation.
Fig. 1.
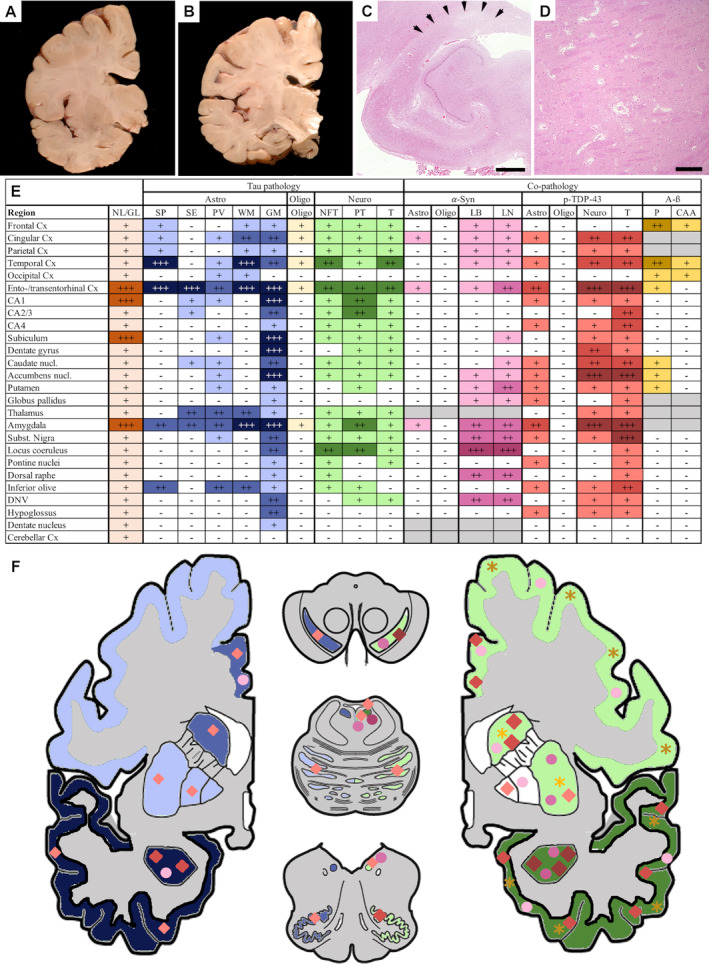
Macroscopic and microscopic neuropathological alterations. Coronal sections of one brain hemisphere show gross subregional hippocampal atrophy (A, B). Histological sections show segmental hippocampal sclerosis (C). Arrows indicate area of sclerosis. Arteriolosclerosis with enlargement of perivascular spaces is observed (D). Summary of neuropathological changes, including semiquantitative scoring of neuronal loss and gliosis (NL/GL) and semiquantitative scoring of immunohistochemistry, results in different brain regions (E). Left side of the image (F, hemibrain) indicates astrocytic pathology illustrated in blue (phosphorylated tau), red rhombi (phosphorylated TDP‐43), and pink spheres (α‐synuclein). Right side of the image (F, hemibrain) indicates neuronal tau pathology illustrated in green, α‐synuclein pathology illustrated by pink spheres, phosphorylate TDP‐43 pathology illustrated by red rhombi, and Aβ pathology illustrated by yellow asterisks. Semiquantitative assessment (table and color gradient) is defined as follows: none (–/white) no pathology/immunoreactivity; mild pathology (+/light color) few immunoreactive cells estimated under 10%; moderate pathology (++/medium color) estimated 10–50% of cells; severe pathology (+++/dark color) estimated more than 50% of cells; not evaluated (grey color). Aβ, Aβ pathology; *, dorsal tier of the substantia nigra; Astro, astrocytic pathology; α‐Syn, α‐synuclein pathology; CAA, cerebral amyloid angiopathy; Cx, cortex; DNV, dorsal nucleus of the vagus nerve; GM, grey matter ARTAG; LB, Lewy bodies; LN, Lewy neurites; Neuro, neuronal pathology; NFT, neurofibrillary tangles; NL/GL, neuronal loss/gliosis; nucl., nucleus; Oligo, oligodendrocytic pathology; P, parenchyma; PT, pretangles; p‐TDP‐43, phosphorylated TDP‐43 pathology; PV, perivascular ARTAG; SE, subependymal ARTAG; SP, subpial ARTAG; Subst. nigra, substantia nigra; T, threads; WM, white matter ARTAG; ARTAG, aging‐related tau astrogliopathy. Scale bars: 1 mm (C), 350 μm (D).
Classical histopathological features
Basic histopathology revealed neuronal loss and reactive astrogliosis, appearance of amyloid plaques, and congophilic angiopathy with thickened vessel walls, as well as segmental hippocampal sclerosis affecting the CA1 sector and the subiculum. Neuronal loss and astrogliosis were particularly prominent in limbic regions, including the amygdala, the entorhinal cortex, the hippocampal CA1 sector, and the subiculum (Fig. 1C). The other evaluated regions showed mild neuronal loss and gliosis (Fig. 1E). Bielschowsky silver staining revealed a moderate density of neuritic plaques. 27 In addition, a moderate amount of arteriosclerotic vessels as cerebrovascular disease, expanded Virchow–Robin spaces, and pigment laden macrophages around vessels was found (Fig. 1D).
Immunohistochemical findings of phophorylated tau
Tau pathology consisted of neuronal, astroglial, and oligodendroglial cytoplasmatic deposits. The anatomical distribution of tau immunoreactivity was widespread, predominating in the limbic system but affecting subcortical regions as well (Fig. 1E, F). In general, astrocytes were the most severely affected cell type, followed by neurons and only mildly involved oligodendrocytes (Fig. 1E).
Neuronal tau pathology consisted of neurofibrillary tangles (NFT), pretangles, and neuropil threads, and was most distinct in the temporal and entorhinal/transentorhinal cortex, CA1 and CA2/3 sectors of the hippocampus, the amygdala, and the locus coeruleus (Figs 1E; 2G, H, M). These were immunoreactive for both 3R and 4R tau isoforms. NFT pathology was stage III according to Braak and Braak. 28 Only single pretangles and small NFTs could be found in neocortical areas, and subcortical areas exhibited only mild to moderate neuronal tau pathology, mostly in the form of diffuse cytoplasmic tau staining with only a few NFTs (Figs. 1E; 2M, N). No argyrophilic grain pathology was identified. Only a few oligodendroglial coiled bodies were seen in the frontal, cingulate and, temporal gyri, hippocampal white matter, and capsula interna, as well as in the amygdala and the corpus callosum.
Fig. 2.
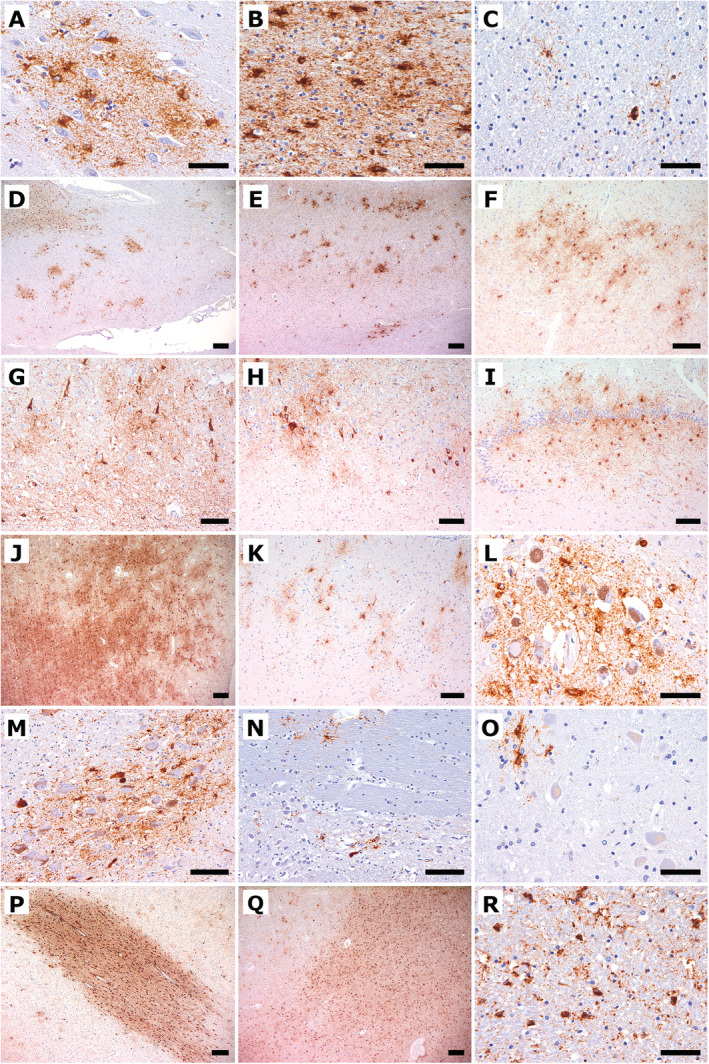
Immunohistochemical findings of hyperphosphorylated tau using the antibody AT8. (A) The cingulate gyrus shows a cluster of GFA tau deposits. (B) The temporal ccortex shows severe white matter ARTAG. (C) The temporal white matter shows focal granular astroglial tau deposits. (D) The cingulate gyrus shows patchy clustering of GFAs in the grey matter and additionally white matter ARTAG. (E) The temporal cortex shows grey matter ARTAG. (F) The ambient gyrus shows grey matter ARTAG. (G) CA1 shows neuronal tau pathology, including NFTs, pretangles, neuropil threads, and additional of grey matter ARTAG. (H) The CA2/3 sectors show GFAs and neuronal tau pathology. (I) The dentate gyrus shows prominent grey matter ARTAG. (J) The amygdala shows extensive grey matter ARTAG. (K) The accumbens nucleus shows patchy clusters of GFA. (L) The substantia nigra shows pronounced astrocytic tau pathology. (M) The locus ceruleus shows neuronal and more astroglial tau pathology. (N) Pontine base shows a single NFT and astroglial tau immunoreactivity. (O) The dentate nucleus shows astroglial tau immunoreactivity. (P) The temporal white matter shows extensive white matter ARTAG as well as perivascular ARTAG. (Q) The amygdala shows severe white matter ARTAG including perivascular ARTAG. (R) The pyramidal tract shows white matter ARTAG. Scale bars: 200 μm (D, E, J, P, Q), 100 μm (F‐I, K, M, N), 50 μm (A‐C, L, O, R).
Unequivocal tufted astrocytes or astrocytic plaques were not observed. Instead, astrocytic tau pathology consisted of TSAs and GFAs as seen in ARTAG, 19 including grey matter (Fig. 2A, D, E–O), white matter (Fig. 2B–D, J, N, P‐R), subpial, subependymal, and perivascular (Fig. 2D, P, Q) types; GFAs predominated in the amygdala (Fig. 2J), the accumbens nucleus (Fig. 2K), and the hippocampus (Fig. 2G‐I) but were noted in the brainstem nuclei as well, such as the substantia nigra (Fig. 2L), locus coeruleus (Fig. 2M), periaquaeductal central grey matter, hypoglossal nucleus, dorsal nucleus of the vagal nerve, dorsal raphe, and inferior olives. AT8‐identified astroglial pathology was exclusively immunoreactive for 4R tau isoform. Phosphorylated tau immunoreactivity colocalized with GFAP‐immunoreactive astrocytes (Fig. 4A–C). The pattern was compatible with widespread grey matter ARTAG. 18 White matter ARTAG was most abundant in the basal brain regions, the amygdala (Figs. 1E; 2Q), and the inferior temporal gyrus (Fig. 2B, C, P). Although lobar and brainstem regions were affected to a lesser extent, white matter ARTAG corresponded to stage 3 out of three of either patterns. 18 Subpial ARTAG was most prominent in basal brain regions but also affected lobar and brainstem regions (Fig. 1E), according to subpial ARTAG stage 3 out of three of either patterns. 18 Perivascular ARTAG was most prominent in the parahippocampal gyrus, the thalamus, the amygdala, and the inferior olive (Fig. 1E, 2Q). Subependymal ARTAG was most prominent at the inferior horn of the lateral ventricle and around the third ventricle. For a detailed mapping of astroglial tau pathologies, see Figure 1.
Fig. 4.
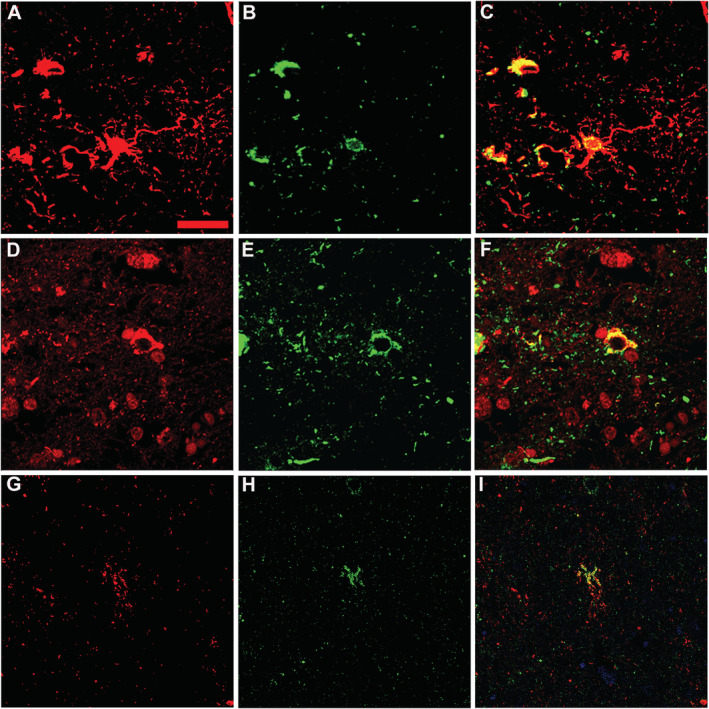
Findings of doube immunofluorescence staining in the brain. (A–C) Double immunofluorescence of the amygdala using antibodies against GFAP (red, A) and hyperphosphorylated tau (green, B) reveals accumulation of hyperphosphorylated tau in astrocytes (yellow, C). (D–F) Double immunofluorescence staining of the amygdala using antibodies against TDP‐43 (red, D) and hyperphosphorylated tau (green, E) shows codistribution of both proteins within the cytoplasm of some cells with neuronal morphology (yellow, F). (G–I) Double immunofluorescence staining of the entorhinal cortex using antibodies against phosphorylated TDP‐43 (green, H) and hyperphosphorylated tau (red, G) shows codistribution of both proteins within the cytoplasm of some cells with astroglial morphology (yellow, I). Scale bar: 10 μm (A‐I).
Immunohistochemical findings of α‐synuclein, Aβ, and phosphorylated TDP‐43
Immnohistochemistry for α‐synuclein revealed widespread Lewy‐type pathology consisting of Lewy bodies, Lewy neurites, and diffuse neuronal cytoplasmic staining (Fig. 3A–F, 1E) consistent with stage 4 according to Braak et al., with only single Lewy bodies in cortical areas. 29 No oligodendroglial Papp–Lantos inclusions typical of multiple system atrophy were observed. Instead, mild astroglial α‐synuclein pathology was observed, being restricted to the amygdala and entorhinal region (Fig. 1E, F). Aβ‐ immunoreactive plaques (diffuse, primitive, and cored) were observed in the neocortex, the entorhinal cortex, and the basal ganglia corresponding to Thal phase 3 (Fig. 1E). 30 In addition, a mild to moderate degree of cerebral amyloid angiopathy type 2, without involvement of capillaries according to Thal, was detected in neocortical leptomeninges, without macrohemorrhagic or microhemorrhagic complications (Fig. 1E). 31 Overall, Alzheimer's disease neuropathological change corresponded to an intermediate severity score (A2B2C2) according to current NIA/AA criteria. 32
Fig. 3.
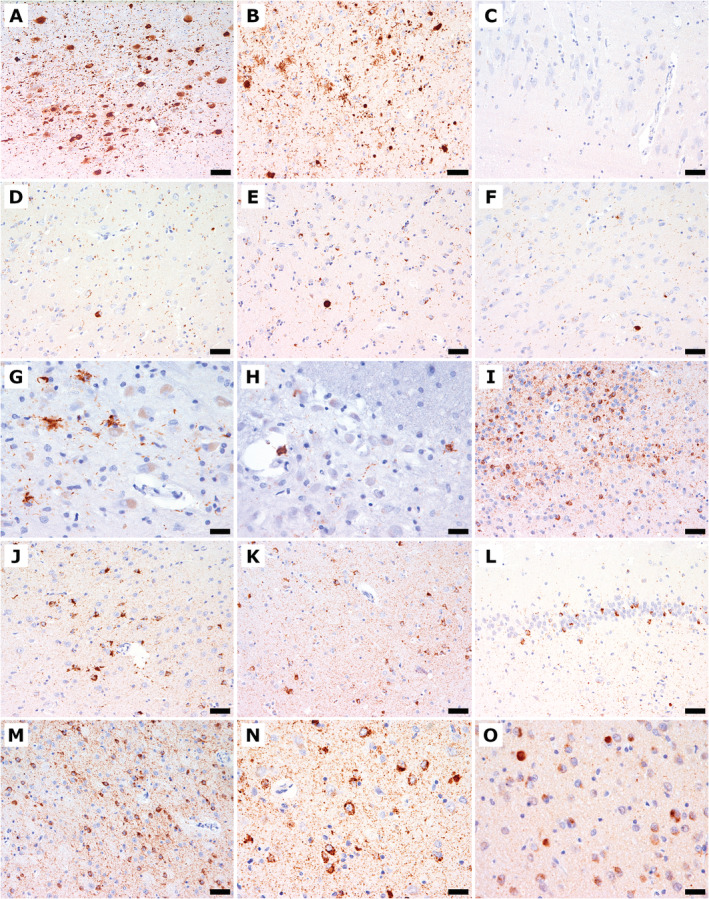
Immunohistochemical findings of α‐synuclein (A–F), phosphorylated TDP‐43 (G–N), and TDP‐43 (O). (A) The locus coeruleus shows Lewy bodies, Lewy neurites, and diffuse granular cytoplasmic immunoreactivity. (B) The amygdala shows Lewy bodies, Lewy neurites, and astroglial α‐synuclein immunoreactivity. (C) The CA2/3 sectors show single delicate dot‐like α‐synuclein‐immunoreactive neurites. (D‐F) The entorhinal cortex (D), putamen (E), and gyrus cinguli (F) show Lewy bodies and Lewy neurites. (G) The inferior olive shows phosphorylated TDP‐43 immunoreactivity with astroglial cytoplasmic deposits and threads. (H) The pontine nucleus shows astroglial cytoplasmic phosphorylated TDP‐43 immunoreactivity and fine threads. (I) The septum nucleus shows marked diffuse neuronal pathology and fine threads and granular deposits in the neuropil. (J) The cingulate gyrus shows thread pathology, neuronal and astroglial phosphorylated TDP‐43 immunoreactivity. (K) The eEntorhinal cortex shows thread pathology and neuronal and astrocytic phosphorylated TDP‐43 immunoreactivity. (L) The dentate gyrus shows threads, and compact, neuronal, and phosphorylated TDP‐43 immunoreactivity. (M) The accumbens nucleus shows marked diffuse cytoplasmic neuronal pathology and fine threads and granular neuropil deposits. (N) The amygdala shows severe neuronal and here only few astrocytic cytoplasmic immunoreactivity as well as abundant fine threads. (O) The amygdala shows loss of neuronal nuclear physiological TDP‐43 immunoreactivity and compact cytoplasmic inclusions. Scale bars: 100 μm (A); 50 μm (B‐F, I‐M); 25 μm (G, H, N, O).
Phosphorylated TDP‐43 immunoreactivity was extensive and consisted of a high density of fine neuropil threads, compact and more frequently diffuse granular or dash‐like neuronal, and less frequently astroglial cytoplasmic deposits (see Figs. 1E, F; 3G, H, J, K, N). There were no intranuclear inclusions and no phosphorylated TDP‐43‐negative/p62‐positive inclusions suggestive of C9orf72 expansion mutation. 33 TDP‐43 pathology was most prominent in limbic areas, including the amygdala, the entorhinal/transentorhinal cortex, and the accumbens nucleus (Fig. 1E, 3G–N). The substantia nigra showed a marked involvement of the dorsal tier. The cingulate and temporal cortices, the dentate gyrus, the pyramidal layer of the hippocampus, the caudate nucleus, and the septal nuclei were moderately involved. The thalamus and brainstem nuclei showed minor involvement. On double immunofluorescence, occasional co‐distribution of non‐phosphorylated TDP‐43 and phosphorylated at detected by AT8 was noted in some neuronal cytoplasmic inclusions (Fig. 4D–F) and a fraction of phosphorylated tau‐positive astroglia was also positive for phosphorylated TDP‐43 (Fig. 4G–I).
DISCUSSION
We present the neuropathological findings of a supposedly oligosymptomatic octogenarian with an extensive and complex proteinopathy combining widespread tau astrogliopathy compatible with all types of ARTAG with additional moderate neuronal and mild oligodendroglial tau pathology, widespread TDP‐43 proteinopathy with segmental hippocampal sclerosis, Lewy body‐type synuclein proteinopathy, and Aβ deposition. The constellation of tau pathologies together with the widespread TDP‐43 proteinopathy, including astrocytic involvement, and additional proteinopathies could suggest a distinct disease entity of the elderly. Tau pathology was most extensive in astrocytes with prominent subependymal, subpial, and perivascular grey matter, and white matter showing ARTAG. Tau pathology consisted mainly of 4R‐tau isoforms without meeting the morphological criteria of 4R tau proteinopathies, such as progressive supranuclear palsy (PSP), argyrophilic grain disease, corticobasal degeneration (CBD), or globular glial tauopathies (GGT). On the one hand, neuronal tau pathology was considerably less than tau pathology in astrocytes, with only few tangles and pretangles in subcortical structures and GFA, but with no NFTs in the subthalamic nucleus, distinguishing the case from PSP. No typical argyrophilic grains were detected. On the other hand, astrocytic tau pathology with GFAs and TSAs was abundant, but no typical tufted astrocytes, astrocytic plaques, or globular glial inclusions indicative for PSP, CBD, or GGT, respectively, were detected. Spherical neuronal 3R tau‐immunoreactive inclusions as seen in Pick's disease were lacking. 34
The parallel distribution of the pathologies with an accentuation in the medial temporal lobe and basal brain structures suggests that the limbic system, and in particular the amygdala, is a hotpot for proteinopathies and likely for a synergistic interaction of abnormal protein deposits. 8 , 35 In general, so‐called mixed pathologies are more prevalent than previously thought, especially in the brain of older individuals. 4 , 36 In our case, phospho‐TDP‐43 pathology and tau‐astrogliopathy seemed to overlap in specific brain areas; furthermore, we detected neuronal and also astroglial colocalization of phospho‐tau and phosphorylated TDP‐43 (Fig. 4H, I). In particular, compact phosphorylated TDP‐43‐positive neuronal cytoplasmic inclusions showed occasional codistribution with phosphorylated tau, while diffuse dash‐like phosphorylated TDP‐43 immunoreactivity did not seem to codistribute with phosphoyrlated tau. Some astroglial cells co‐expressed both proteins. Interestingly, Kim et al. (2018) described combined tau and TDP‐43 proteinopathies in a frontotemporal lobar degeneration (FTLD) cohort with less elderly patients, reinforcing the concept that tau and TDP‐43 may share pathogenic pathways in neurodegenerative conditions. 37 If we assess the TDP‐43 pathology in the context of a potential FTLD‐TDP, the predominance of dash‐like cytoplasmic neuronal pathology and fine‐granular thread pathology would be more consistent with type B. 38 However, the topographical distribution showed a mediotemporal/limbic predominance with a marked involvement of the amygdala, hippocampus, entorhinal/transentorhinal cortex, and temporal lobe, as well as of the accumbens nucleus, septal nuclei, anterior caudate nucleus, and cingulum, with moderate pathology in the dorsal tier of the substantia nigra and inferior olives. Other cortical areas, including the frontal cortex, were only mildly or not affected at all. Therefore, the distribution of phosphorylated TDP‐43 pathology eventually would correspond to early stage 6 according to Josephs et al., as described for AD‐related TDP‐43 pathology, 39 , 40 or to stage 2 limbic‐predominant age‐related TDP‐43 encephalopathy (LATE) 41 and would not suggest a classical FTLD‐TDP. The distribution of Lewy‐type pathology and Alzheimer's disease neuropathological change did not have an atypical distribution.
Further studies are needed to shed light on novel genetic aspects involved in common pathogenic events. Indeed, widespread TSAs and GFAs have also been described in a novel GRN nonsense mutation associated with TDP‐43 pathology, 42 a mixture of bushy and tufted astrocytes in a TARDBP mutation, 43 and astrocytic plaques and ARTAG in 17q21.31 duplication. 44 , 45
Because astrocytes have been shown to perform divergent actions, the predominance of astroglial tau pathology could be interpreted in two different ways: the astrocytic tau pathology could represent the brain's effort to remove pathological protein deposits, or it could mark a primary astroglial tau immunoreactive pathology, eventually in the frame of a response to a yet unidentified neurodegenerative event affecting a particular anatomical region. 12 , 13 , 15 Indeed, it has been shown that astroglial tau‐immunoreactive pathology can precede local accumulation of neuronal tau‐immunoreactive pathology. 16 Either way, the resulting astroglial dysfunction may contribute to a disruption in neuronal homeostasis and protein processing and eventually facilitate further abnormal protein aggregation. Thus, the abundant tau astrogliopathy observed in the case presented here could indicate an exaggerated astroglial response to a primary or secondary neurodegenerative event and eventually trigger further neurodegenerative alterations resulting in this complex combined proteinopathy, including astroglial TDP‐43. Therefore, while in a single descriptive case report we cannot draw firm conclusions on the relationship between astroglial and neuronal tau proteinopathy, we speculate that the extensive tau astrogliopathy is a driving force capable of marking a distinct neurodegenerative phenotype. As a limitation of our study, we cannot address the question of how this contributes to any clinical phenotype; however, some studies do suggest that certain forms of astroglial tau pathologies might reflect distinct clinical syndromes. 16
Interestingly, some of the morphological alterations are reminiscent of amyotrophic lateral sclerosis and parkinsonism–dementia complex in the Kii Peninsula (ALS/PDC) in Japan and the ALS/PDC on Guam, in particular the combination of pronounced tau pathology with TDP‐43 deposition and alpha‐synuclein accumulation. 46 , 47 , 48 However, the cases of ALS/PDC of Guam and Kii exhibit extensive NFT pathology without Aβ plaques, and a prominent involvement of the motor system. 46 In our case, NFT pathology was less pronounced. In addition, our case clinically seems to be markedly different, with a relative lack of dementia, parkinsonism, and ALS symptoms. Although the neurodegeneration pattern could be considered within the FTLD spectrum without obvious motor neurone involvement, no systematic neurological or psychiatric evaluation was performed in our patient. We cannot state, therefore, that our patient lacked any signs of cognitive decline. However, notably, the patient was independent and living alone before admission to hospital, which could indicate a certain level of preserved cognitive function. The mechanisms behind cognitive resilience in patients of old age remain unknown. 20 , 49 , 50 , 51 The combination of resistance to pathology, maintenance of functional networks, metabolism, and brain structure have been postulated as cardinal factors for so‐called successful aging. 51 However, a high burden of non‐AD changes as seen in our case has been described as a critical factor for increased vulnerability to dementia. 20
The distinct involvement of the limbic system could underpin the neuropathological changes being a contributing factor to depression in this patient. Considering that the amygdala was the most severely affected structure in the brain, it might be presumed to be the point of origin of the different neurodegenerative changes, which would be in line with the recently proposed sequential stages of ARTAG. 18 Interestingly, the amygdala has been highlighted as a potentially significant locus of misfolding of different neurodegeneration‐associated proteins. 35 Moreover, the amygdala, with its pivotal role in emotion processing, has been linked to mood disorders. 52 Several neuroimaging studies, postmortem histological studies, 53 , 54 as well as studies examining the amygdala network function describe abnormalities in depressive patients. 55 , 56 It is, therefore, likely that the combination of neurodegenerative pathologies in this region has lowered the threshold for the development of depressive symptoms. Experimental studies have also showed that chronic stress and depression per se may influence and aggravate abnormal protein aggregation, such as of alpha‐synuclein, and, in turn, influence mood/emotional states. 57
The multimorbid state of this elderly patient could be a reason for neurological symptoms being overlooking as the patient's cancer may have deviated the focus of clinical evaluations. This case highlights the importance of systematic and detailed neurological and psychiatric examination in elderly persons, as they tend to exhibit several systemic multimorbidities, which could lead to the underrecognition of subtle neuropsychiatric symptoms and, consequently, to suboptimal treatment.
In summary, this unusual case of extensive and widespread ARTAG with a complex multiproteinopathy may represent an independent disease entity in the elderly with tau astrogliopathy as the leading force. While we cannot definitely state that our patient did not show cognitive decline, the presence of late‐onset depression suggests that psychiatric symptoms in the elderly might be indicative of an underlying neurodegenerative process. This highlights the importance of a detailed neuropathological work‐up including the screening of the most important brain regions for pathological changes to allow further understanding of the complex interactions in neurodegeneration. 11 , 58 Finally, the interaction between tau astrogliopathies and other proteinopathies, particularly TDP‐43, merits further study.
DISCLOSURE
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
GGK is supported by the Rossy Foundation, the Edmond J. Safra Foundation, and the Bishop Karl Golser Award. SK received a grant from the City of Vienna/Austria (“Hochschuljubiläumsfonds” grant number H‐283459/2019). The funding source had no role in the design, practice, or analysis of this study.
REFERENCES
- 1. Kovacs GG. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine. Int J Mol Sci 2016; 189: 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology 2005; 25: 111–124. [DOI] [PubMed] [Google Scholar]
- 3. Kovacs GG, Alafuzoff I, Al‐Sarraj S et al. Mixed brain pathologies in dementia: The BrainNet Europe consortium experience. Dement Geriatr Cogn Disord 2008; 26: 343–350. [DOI] [PubMed] [Google Scholar]
- 4. Rahimi J, Kovacs GG. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014; 6: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science 2016; 353: 777–783. [DOI] [PubMed] [Google Scholar]
- 6. Heneka MT, Carson MJ, El Khoury J et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015; 14: 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van der Velpen V, Teav T et al. Systemic and central nervous system metabolic alterations in Alzheimer's disease. Alzheimers Res Ther 2019;11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moussaud S, Jones DR, Moussaud‐Lamodière EL, Delenclos M, Ross OA, McLean PJ. Alpha‐synuclein and tau: Teammates in neurodegeneration? Mol Neurodegener 2014; 9: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morales R, Estrada LD, Diaz‐Espinoza R et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer's and prion diseases. J Neurosci 2010; 30: 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Aβ, tau, and α‐synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 2010; 30: 7281–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wennberg AM, Whitwell JL, Tosakulwong N et al. The influence of tau, amyloid, alpha‐synuclein, TDP‐43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging 2019; 77: 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heales SJR, Lam AAJ, Duncan AJ, Land JM. Neurodegeneration or neuroprotection: The pivotal role of astrocytes. Neurochem Res 2004; 29: 513–519. [DOI] [PubMed] [Google Scholar]
- 13. Pekny M, Pekna M, Messing A et al. Astrocytes: A central element in neurological diseases. Acta Neuropathol 2016; 131: 323–345. [DOI] [PubMed] [Google Scholar]
- 14. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 2017; 35: 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verkhratsky A, Zorec R, Parpura V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol 2017; 27: 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovacs GG. Astroglia and tau: New perspectives. Front Aging Neurosci 2020; 12: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kovacs GG, Lee VM, Trojanowski JQ. Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol 2017; 27: 675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kovacs GG, Xie SX, Robinson JL et al. Sequential stages and distribution patterns of aging‐related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun 2018; 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovacs GG, Xie SX, Lee EB et al. Multisite assessment of aging‐related tau astrogliopathy (ARTAG). J Neuropathol Exp Neurol 2017; 76: 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson JL, Corrada MM, Kovacs GG et al. Non‐Alzheimer's contributions to dementia and cognitive resilience in the 90+ study. Acta Neuropathol 2018; 136: 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovacs GG, Molnár K, László L et al. A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 2011; 122: 205–222. [DOI] [PubMed] [Google Scholar]
- 22. Nolan A, De Paula Franca Resende E, Petersen C et al. Astrocytic tau deposition is frequent in typical and atypical Alzheimer disease presentations. J Neuropathol Exp Neurol 2019; 78: 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munoz DG, Woulfe J, Kertesz A. Argyrophilic thorny astrocyte clusters in association with Alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol 2007; 114: 347–357. [DOI] [PubMed] [Google Scholar]
- 24. Resende E de PF, Nolan AL, Petersen C et al. Language and spatial dysfunction in Alzheimer disease with white matter thorn‐shaped astrocytes. Neurology 2020; 94: e1353–e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferrer I, Legati A, García‐Monco JC et al. Familial behavioral variant frontotemporal dementia associated with astrocyte‐predominant tauopathy. J Neuropathol Exp Neurol 2015; 74: 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gelpi E, Aldecoa I, Lopez‐Villegas D et al. Atypical astroglial phTDP43 pathology in astroglial predominant tauopathy. Submitted. [DOI] [PMC free article] [PubMed]
- 27. Mirra SS, Heyman A, McKeel D et al. The consortium to establish a registry for Alzheimer's disease (CERAD) – Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479–486. [DOI] [PubMed] [Google Scholar]
- 28. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging 1995; 16: 271–278. [DOI] [PubMed] [Google Scholar]
- 29. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 2004; 318: 121–134. [DOI] [PubMed] [Google Scholar]
- 30. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–1800. [DOI] [PubMed] [Google Scholar]
- 31. Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003; 62: 1287–1301. [DOI] [PubMed] [Google Scholar]
- 32. Montine TJ, Phelps CH, Beach TG et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: A practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al‐Sarraj S, King A, Troakes C et al. P62 positive, TDP‐43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72‐linked FTLD and MND/ALS. Acta Neuropathol 2011; 122: 691–702. [DOI] [PubMed] [Google Scholar]
- 34. Dickson DW. Pick's disease: A modern approach. Brain Pathol 2006; 8: 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nelson PT, Abner EL, Patel E et al. The amygdala as a locus of pathologic misfolding in neurodegenerative diseases. J Neuropathol Exp Neurol 2018; 77: 2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kovacs GG. Are comorbidities compatible with a molecular pathological classification of neurodegenerative diseases? Curr Opin Neurol 2019; 32: 279–291. [DOI] [PubMed] [Google Scholar]
- 37. Kim E‐J, Brown JA, Deng J et al. Mixed TDP‐43 proteinopathy and tauopathy in frontotemporal lobar degeneration: Nine case series. J Neurol 2018; 265: 2960–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackenzie IR, Neumann M. Reappraisal of TDP‐43 pathology in FTLD‐U subtypes. Acta Neuropathol 2017; 134: 79–96. [DOI] [PubMed] [Google Scholar]
- 39. Josephs KA, Murray ME, Whitwell JL et al. Updated TDP‐43 in Alzheimer's disease staging scheme. Acta Neuropathol 2016; 131: 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Josephs KA, Murray ME, Tosakulwong N et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP‐43 types in non‐FTLD brains. Acta Neuropathol 2019; 137: 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nelson PT, Dickson DW, Trojanowski JQ et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): Consensus working group report. Brain 2019; 142: 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gómez‐Tortosa E, Baradaran‐Heravi Y, González Alvarez V et al. Presence of tau astrogliopathy in frontotemporal dementia caused by a novel Grn nonsense (Trp2*) mutation. Neurobiol Aging 2019; 76: 214.e11–214.e15. [DOI] [PubMed] [Google Scholar]
- 43. Gelpi E, van der Zee J, Turon Estrada A, Van Broeckhoven C, Sanchez‐Valle R. TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol 2014; 40: 225–230. [DOI] [PubMed] [Google Scholar]
- 44. Alexander J, Kalev O, Mehrabian S et al. Familial early‐onset dementia with complex neuropathologic phenotype and genomic background. Neurobiol Aging 2016; 42: 199–204. [DOI] [PubMed] [Google Scholar]
- 45. Le Guennec K, Quenez O, Nicolas G et al. 17q21.31 duplication causes prominent tau‐related dementia with increased MAPT expression. Mol Psychiatry 2017; 22: 1119–1125. [DOI] [PubMed] [Google Scholar]
- 46. Yase Y, Yoshida S, Kihira T, Wakayama I, Komoto J. Kii ALS dementia. Neuropathology 2001; 21: 105–109. [DOI] [PubMed] [Google Scholar]
- 47. Oyanagi K, Wada M. Neuropathology of parkinsonism‐dementia complex and amyotrophic lateral sclerosis of Guam: An update. J Neurol 1999; 246: II19–II27. [DOI] [PubMed] [Google Scholar]
- 48. Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y. Amyotrophic lateral sclerosis and parkinsonism‐dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018; 38: 98–107. [DOI] [PubMed] [Google Scholar]
- 49. Jiskoot LC, Dopper EGP, Den HT et al. Presymptomatic cognitive decline in familial frontotemporal dementia: A longitudinal study. Neurology 2016; 87: 384–391. [DOI] [PubMed] [Google Scholar]
- 50. Rittman T, Borchert R, Jones S et al. Functional network resilience to pathology in presymptomatic genetic frontotemporal dementia. Neurobiol Aging 2019; 77: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease. Neurology 2018; 90: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct Funct 2008; 213: 93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hamilton JP, Siemer M, Gotlib IH. Amygdala volume in major depressive disorder: A meta‐analysis of magnetic resonance imaging studies. Mol Psychiatry 2008; 13: 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bowley MP, Drevets WC, Öngür D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 2002; 52: 404–412. [DOI] [PubMed] [Google Scholar]
- 55. Li W, Ward BD, Xie C et al. Amygdala network dysfunction in late‐life depression phenotypes: Relationships with symptom dimensions. J Psychiatr Res 2015; 70: 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guo Z, Liu X, Xu S et al. Abnormal changes in functional connectivity between the amygdala and frontal regions are associated with depression in Alzheimer's disease. Neuroradiology 2018; 60: 1315–1322. [DOI] [PubMed] [Google Scholar]
- 57. Burtscher J, Copin J‐C, Rodrigues J et al. Chronic corticosterone aggravates behavioral and neuronal symptomatology in a mouse model of alpha‐synuclein pathology. Neurobiol Aging 2019; 83: 11–20. [DOI] [PubMed] [Google Scholar]
- 58. Kovacs GG, Milenkovic I, Wöhrer A et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: A community‐based autopsy series. Acta Neuropathol 2013; 126: 365–384. [DOI] [PubMed] [Google Scholar]