

Abstract
Objective
Recent clinical trials targeting amyloid beta (Aβ) and tau in Alzheimer's disease (AD) have yet to demonstrate efficacy. Reviewing the hypotheses for AD pathogenesis and defining possible links between them may enhance insights into both upstream initiating events and downstream mechanisms, thereby promoting discovery of novel treatments. Evidence that in Down syndrome (DS), a population markedly predisposed to develop early onset AD, increased APP gene dose is necessary for both AD neuropathology and dementia points to normalization of the levels of the amyloid precursor protein (APP) and its products as a route to further define AD pathogenesis and discovering novel treatments.
Background
AD and DS share several characteristic manifestations. DS is caused by trisomy of whole or part of chromosome 21; this chromosome contains about 233 protein‐coding genes, including APP. Recent evidence points to a defining role for increased expression of the gene for APP and for its 99 amino acid C‐terminal fragment (C99, also known as β‐CTF) in dysregulating the endosomal/lysosomal system. The latter is critical for normal cellular function and in neurons for transmitting neurotrophic signals.
New/updated hypothesis
We hypothesize that the increase in APP gene dose in DS initiates a process in which increased levels of full‐length APP (fl‐APP) and its products, including β‐CTF and possibly Aβ peptides (Aβ42 and Aβ40), drive AD pathogenesis through an endosome‐dependent mechanism(s), which compromises transport of neurotrophic signals. To test this hypothesis, we carried out studies in the Ts65Dn mouse model of DS and examined the effects of Posiphen, an orally available small molecule shown in prior studies to reduce fl‐APP. In vitro, Posiphen lowered fl‐APP and its C‐terminal fragments, reversed Rab5 hyperactivation and early endosome enlargement, and restored retrograde transport of neurotrophin signaling. In vivo, Posiphen treatment (50 mg/kg/d, 26 days, intraperitoneal [i.p.]) of Ts65Dn mice was well tolerated and demonstrated no adverse effects in behavior. Treatment resulted in normalization of the levels of fl‐APP, C‐terminal fragments and small reductions in Aβ species, restoration to normal levels of Rab5 activity, reduced phosphorylated tau (p‐tau), and reversed deficits in TrkB (tropomyosin receptor kinase B) activation and in the Akt (protein kinase B [PKB]), ERK (extracellular signal‐regulated kinase), and CREB (cAMP response element–binding protein) signaling pathways. Remarkably, Posiphen treatment also restored the level of choline acetyltransferase protein to 2N levels. These findings support the APP gene dose hypothesis, point to the need for additional studies to explore the mechanisms by which increased APP gene expression acts to increase the risk for AD in DS, and to possible utility of treatments to normalize the levels of APP and its products for preventing AD in those with DS.
Major challenges for the hypothesis
Important unanswered questions are: (1) When should one intervene in those with DS; (2) would an APP‐based strategy have untoward consequences on possible adaptive changes induced by chronically increased APP gene dose; (3) do other genes present on chromosome 21, or on other chromosomes whose expression is dysregulated in DS, contribute to AD pathogenesis; and (4) can one model strategies that combine the use of an APP‐based treatment with those directed at other AD phenotypes including p‐tau and inflammation.
Linkage to other major theories
The APP gene dose hypothesis interfaces with the amyloid cascade hypothesis of AD as well as with the genetic and cell biological observations that support it. Moreover, upregulation of fl‐APP protein and products may drive downstream events that dysregulate tau homeostasis and inflammatory responses that contribute to propagation of AD pathogenesis.
Keywords: Alzheimer's disease, APP, Down syndrome, Posiphen, early endosome, neurotrophin signaling, Ts65Dn mouse
1. OBJECTIVE
A wealth of recent evidence points to Alzheimer's disease (AD) pathogenesis as arising from toxic oligomeric species of amyloid beta (Aβ) and tau and to a role for inflammation, including that mediated by microglial cells. 1 , 2 , 3 Despite compelling evidence for contributions of Aβ and tau to pathogenesis, clinical studies targeting them, including immune approaches, β‐secretase inhibitors, and γ‐secretase inhibitors, have yet to demonstrate efficacy. 4 , 5 , 6 , 7 There is urgent need to consider why trials have failed and to reexamine the premises that guided their design. Moreover, continuing investment is needed to explore the basic biology of AD pathogenesis, to define critical molecular and cellular events, and to discover treatments that target them. This effort will benefit by considering both existing and new hypotheses and by addressing possible intersections. The insights derived could elucidate both initiating events and those that result from dysregulation of downstream pathways, many of which may combine in as‐yet poorly defined ways to cause neurodegeneration. Such studies might elucidate treatments specific to stages in the evolution of AD and suggest both stage‐specific and combination treatments. They might also point to the need for personal and precision‐based approaches. 8
Success in deciphering pathogenesis is predictably increased if studies address a population in which a known genetic variation causes AD. Down syndrome (DS) appears to serve in this regard. DS, the most common genetic cause of AD, is due to trisomy of whole or part of Homo sapiens chromosome 21 (HSA21); this chromosome harbors approximately 233 protein‐coding genes, including that for the amyloid precursor protein (APP). 9 , 10 Substantial evidence points to the necessity for increased APP gene copy number for the emergence of AD neuropathology and dementia in DS, 11 , 12 , 13 thus providing a theoretical basis for exploring APP gene dose‐based studies of AD pathogenesis and for treatments for preventing AD in this population. Herein we pursued studies testing the APP gene dose hypothesis. Our success using one approach to reduce fl‐APP and its products to rescue AD‐relevant endosomal phenotypes in a mouse model of DS supports the validity of the hypothesis and encourages pursuit of additional approaches to address the impact of increased APP gene expression on cellular events, including dysregulation of the endosomal pathway, that contribute to AD pathogenesis in DS. Whether or not this same approach proves relevant to AD in other populations should be explored.
2. BACKGROUND
2.1. Historical evolution and current perspective
AD, the most commonly diagnosed dementia in the elderly, has several clinical manifestations including memory loss, cognitive decline, behavioral disorders, and inability to maintain activities of daily living. 14 , 15 , 16 As knowledge of AD clinical and neuropathological manifestation increased it became clear that other populations were affected at much younger ages by a very similar or identical process. Autosomal dominant transmission of AD (ie, familial AD [FAD]) has been demonstrated to be due to mutations in the gene for APP and in the genes for Presenilin 1 and 2 (PSEN1 and PSEN2), which encode the catalytic subunits of the γ‐secretase enzyme complex that participates in APP processing. Cases of FAD are also rarely caused by duplication of the gene for APP, thus resulting in three copies in the genome. 17 , 18 , 19 Increased risk for AD in the general population has been linked to a number of gene variants, with up to 29 identified to date. 20 , 21 The most prominent risk variant is the apolipoprotein E (APOE) ɛ4 allele. 22 As for FAD, in the DS population a genetic factor plays a defining role. Indeed, increased APP gene copy number markedly increases AD risk in DS. 10 , 23 Thus although children, adolescents, and young adults experience a common, albeit variable, set of clinical and cognitive challenges, 24 in a large fraction of the aging DS population a dementia syndrome ensues that shows many similarities to AD. 23 , 25 , 26 Beyond age 40, increasing deficits in recall, explicit memory, and receptive language function are the precursors of dementia. Dementia in DS is often marked by changes in behavior and personality, and by executive dysfunction. 10 , 25 , 27 Although studies of genetic risk variants for AD in DS has received little attention, the APOE ɛ4 allele is associated with increased accumulation of Aβ 28 and increased risk of earlier onset of dementia. 29 In summary, the remarkable correspondence between the general and DS populations in clinical manifestations, biomarkers, 30 , 31 , 32 , 33 and neuropathological features 34 , 35 , 36 justifies the designation of AD in DS (AD‐DS). 23 It is notable that the necessary role played by increased APP gene dose in AD‐DS motivates, and may simplify, approaches for exploring AD pathogenesis and treatment of AD‐DS.
In both AD and AD‐DS, neuropathology manifests in several characteristic features; neuritic amyloid plaques and neurofibrillary tangles (NFTs) are hallmarks. 34 , 35 , 36 Amyloid plaques mark the extracellular accumulation and deposition of Aβ species derived from APP processing, whereas NFTs consist of aberrantly folded and abnormally phosphorylated forms of the microtubule‐binding protein tau. Although these common neuropathological signatures characterize AD and AD‐DS, it has become increasingly clear that amyloid plaques and NFTs may not in themselves be directly linked to AD pathogenesis. Indeed, growing evidence points to AD pathogenesis as more closely linked to toxic assemblies of Aβ and tau. Recent studies strongly support an important role for such oligomeric Aβ and tau species as conferring disease‐relevant toxicities in both AD and AD‐DS. 2 , 3 , 27 , 37 We recently provided a speculative synthesis of the events underlying the initiation and progression of AD pathogenesis in which Aβ oligomers and tau oligomers play key, likely interacting roles. 27 Although propagation and spread of oligomers has been convincingly documented, the molecular definition of toxic species and the mechanisms by which they induce pathogenesis require further study. A pervasive and characteristic feature of AD neuropathology shared with AD‐DS is enlargement of early endosomes, as marked by immunostaining for Rab5. Indeed, endosomal enlargement appears before the evolution of plaques and tangles in AD and in AD‐DS. 38 A compelling need for informing pathogenesis is understanding whether or not and how changes in Aβ, tau, and endosomes initiate these processes and how they result in downstream changes in synaptic structure and function leading to neuronal dysfunction and degeneration. Integrating neuropathological, cell biological, and genetic evidence will serve to define ever more cogent hypotheses for understanding and treating AD and AD‐DS.
RESEARCH IN CONTEXT
Systemic review: The failure of Alzheimer's disease (AD) clinical trials targeting amyloid beta (Aβ) and tau motivates reconsideration of the mechanisms underlying AD pathogenesis and innovations in defining and pursuing novel targets. Increased APP gene dose is necessary for AD in those with Down syndrome (AD in DS). Targeting the amyloid precursor protein (APP) and its products to prevent AD in DS may provide unique insights into pathogenesis in this population and inform pathogenic mechanisms for AD in the general population and in those with familial AD (FAD).
Interpretation: Using Posiphen, an orally available small molecule, in studies in vitro and in vivo we normalized the levels of fl‐APP and its C‐terminal fragments and in vivo lowered the levels of Aβ42 in the Ts65Dn mouse model of DS. These changes were correlated with reversal of endosomal changes characteristic of AD and AD in DS. Our studies are evidence that targeting increased expression of APP in DS may impact pathogenesis.
Future directions: Various approaches targeting increased APP levels may be pursued at the level of APP mRNA, APP protein, APP processing and clearance of Aβ.
2.2. Rationale
Although late‐onset AD, FAD, and AD‐DS share genetic, clinical, and neuropathological features, 25 , 26 , 39 an identified genetic factor plays an evident, defining role in those with FAD and AD‐DS: mutations in APP, PSEN1, and PSEN2 in FAD, and an extra full or partial copy of chromosome 21 harboring APP in AD‐DS. Thus for AD‐DS there is compelling evidence in both humans and in mouse models for the necessity of increased APP gene dose. 11 , 12 , 13 , 40 , 41 This realization, derived from converging lines of evidence and insights, have motivated studies to explore mechanisms and possible treatments for events due to increased APP gene dose. 42
Regarding the pathogenetic events induced by increased APP gene dose, a number of mechanistic questions are raised: (1) whether or not increased APP gene dose acts through increased levels of fl‐APP and/or its products; (2) which APP product(s) play a role; (3) whether changes in the responsible product(s) are due to the level of production, to clearance, or both; (4) to what extent the cellular locus of the product(s) influences its actions; and (5) what molecular events are likely to initiate neurodegeneration, as suggested by their impact on cellular processes known to support neuronal structure, function, and survival. Answers to these questions are only now being addressed but recent findings speak to the importance of APP and its 99 amino acid C‐terminal fragment (C99, also known as β‐CTF) as markedly dysregulating a fundamental aspect of cell biology—the endosomal/lysosomal system whose role in transporting and degrading a large variety of cellular proteins and organelles is vital for neuronal function. 41 , 43 , 44 , 45 Increased levels of each of these APP gene products have been shown to activate the early endosomal small GTPase Rab5, with subsequent enlargement and dysfunction of early endosomes as well as downstream compartments in the endosomal/lysosomal system. 41 , 43 , 45 , 46 , 47 , 48 Especially noteworthy is the role that early endosomes play in communicating and regulating trophic signals within neurons and that mediate trophic information exchanged between neurons connected in circuits, including those that support cognition. 40 , 42 , 47 An important attribute of the APP gene dose hypothesis is that it integrates the effect of increased APP gene dose to robust genetic evidence for mutations in the PSENs causing FAD and thus speaks informatively to the hypothesis that γ‐secretase insufficiency serves a root cause of AD. 49 , 50 , 51 Thus a more general strategy, one suggested by the necessary role of increased APP gene dose in AD‐DS and possible contributions of fl‐APP and its β‐CTF and Aβ products, would be to target increased APP gene expression.
Posiphen, (+)‐phenserine tartrate, is the optically pure positive enantiomer of (−)‐phenserine, an acetyl‐cholinesterase (AChE) inhibitor. 52 Posiphen is a much less potent AChE inhibitor (≈1000‐fold) than Phenserine 53 and acts as a somewhat selective translational inhibitor to reduce the levels of APP and Aβ42 in human neuroblastoma cell cultures, rodent primary neurons, and in the brains of wild‐type mice. 53 , 54 It also reduced Aβ42 in the cerebral cortex of transgenic mice (Tg2576) over‐expressing the human APP gene with the Swedish mutation K670N/M671L (APPSWE), a model of early onset AD. 55 Beneficial effects due to decreased APP and Aβ42 levels have been described. Posiphen treatment resulted in an increase in hippocampal neurogenesis in both young and aged APPSWE mice. 55 Posiphen was reportedly neurotrophic and neuroprotective in neural cell cultures under conditions that mimic AD. 56 Posiphen induced a significant decrease of fl‐APP in APP23 mice (over‐expressing human APP with the Swedish double mutation). 57 Recently Posiphen was shown to reduce APP and related products, and fully restore memory, learning, and long‐term potentiation in an APP/PS1 mouse model of Alzheimer's disease. 58 The human relevance of Posiphen treatment was addressed in a Phase I clinical trial in which Posiphen was well tolerated and reduced the level of soluble APP (sAPP) fragments and tau species with a trend to decrease Aβ42 in the cerebrospinal fluid (CSF) of subjects with mild cognitive impairment (MCI). 59 Posiphen is currently in a Phase I trial for early stage AD. Of note, Posiphen undergoes N‐demethylation in the N1 and N8 positions to generate the respective metabolites N1‐Norposiphen and N8‐Norposiphen. These metabolites share with Posiphen in the ability to reduce APP levels. 54
3. NEW OR UPDATED HYPOTHESIS
We hypothesize that increased APP gene dose acts through increased levels of APP and its product to dysregulate endosomal transport of neurotrophic signals. 42 , 47 Accordingly, measures to reduce the levels of fl‐APP protein and its products in models of DS will act to prevent or reduce endosomal dysfunction and restore trophic signaling. We begin by detailing our studies on Posiphen. Posiphen negatively regulates APP translation. 53 , 58 The proposed mechanism of action of Posiphen builds on a regulatory role for iron in APP expression. The APP 5′‐UTR contains an iron‐response element (IRE) stem loop that mediates translational control of APP expression. 60 The IREs are 30‐nucleotide RNA motifs containing the classic 5′‐CAGUGX‐3′ (X = U, C, or A) sequence. The APP IRE with 5′‐CAGAGC motif is homologous with the canonical IRE RNA stem‐loop that binds the iron regulatory proteins (IRP1 and IRP2) to control iron‐dependent translation. 61 , 62 Among them, IRP1, but not IRP2, binds to the APP IRE. 63 IRP binding to the IRE prevents the release of the messenger RNA (mRNA) and, therefore, its association with the ribosome, thus suppressing translation. In the presence of increased cellular iron levels, iron binds to IRP to induce a conformational change leading to dissociation of IRP1 from APP mRNA, thus promoting translation. 62 , 64 Studies of 5′‐UTR IRE stem loop in the mRNA of SNCA encoding α‐synuclein motivates a model of Posiphen in which the compound increases the affinity of the IRP for the IRE, leading to decreased translation of APP mRNA. 54 The result of Posiphen treatment is reduced levels of fl‐APP and its products. We amplified the 5′‐UTRs of App mRNAs from the brains of both 2N and Ts65Dn mice and aligned with the corresponding sequences of mouse and human APP mRNAs from the National Center for Biotechnology Information Entrez data base. The predicted IRE sequences and conserved CAGAGC loop from 2N and Ts65Dn mice were identical with the mouse sequence in the database (Supplementary Figure S1), providing a theoretical basis for manipulation of APP expression by Posiphen in Ts65Dn mice. Our hypothesis argues that by using Posiphen to normalize fl‐APP and its products in DS we will also normalize the structure and function of early endosomes with a reduction in AD‐DS relevant phenotypes. Herein we address this through studies of Posiphen in a DS mouse model.
3.1. Early experimental data
3.1.1. In vitro studies on Ts65Dn cortical neurons
Posiphen reduced translation of App mRNA in vitro to normalize APP protein
To study the effect of Posiphen treatment on endosomal pathology in Ts65Dn neurons, we first asked if Posiphen would reduce the level of fl‐APP. In vitro studies were carried out in primary cortical neurons from Ts65Dn mice, a well‐established and much studied genetic model of DS. 65 , 66 Ts65Dn mice are segmentally trisomic for 90 mouse genes with homologues on the long arm of human chromosome 21 67 ; the gene for mouse APP is present within the segment, thus increasing mouse App gene dose to three copies in these mice. 40 , 68 Euploid (2N) cortical neurons served as controls. Embryonic (E18) cortical neurons were dissected and cultured in vitro for 5 days (DIV5) followed by treatment for an additional 48 hours with a series of concentrations of Posiphen (0.5–10.0 µM). Immunoblotting was used to analyze the level of fl‐APP as well as the protein products of other selected genes either triplicated in the Ts65Dn mouse or genes, which when mutated, result in neurodegenerative disorders (Figure 1A‐G). Posiphen concentration‐dependently reduced the level of APP in Ts65Dn neurons. At 5 µM and 10 µM, the levels were significantly reduced compared to vehicle‐treated Ts65Dn neurons; at these concentrations the levels were not statistically different from those in vehicle‐treated 2N neurons. A statistically insignificant trend to reduced fl‐APP was seen in 2N neurons at the same concentrations (Figure 1A, B). Although Posiphen reduced APP in Ts65Dn neurons it had no effect on the protein products of other genes present in three copies in Ts65Dn, including DYRK1A and SOD1, which encode dual specificity tyrosine‐phosphorylation‐regulated kinase 1A (Dyrk1a) and superoxide dismutase‐1 (SOD1), respectively (Figure 1A, C, D). Nor were significant effects observed on the levels of tau or huntingtin (Figure 1A, E, F). In agreement with earlier findings, 54 Posiphen reduced α‐synuclein in both 2N and Ts65Dn neurons by about 30% at 10 µM (Figure 1A, G).
FIGURE 1.
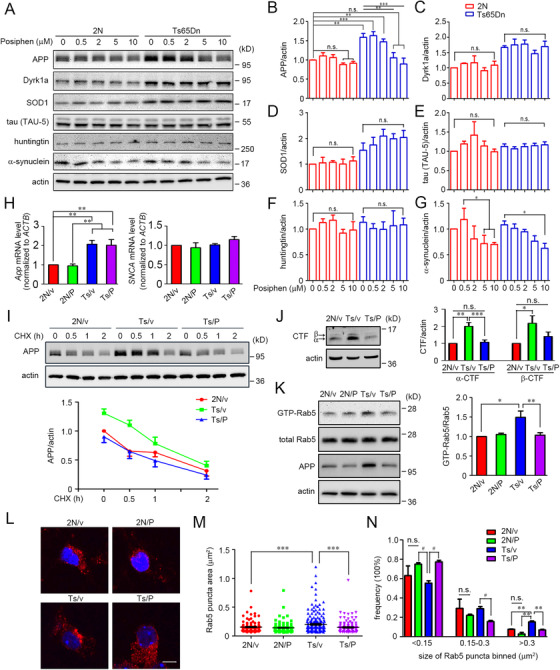
Posiphen normalized fl‐APP and reversed enlargement of early endosomes and increased activation of Rab5 in primary Ts65Dn cortical neurons. (A) Primary cortical neurons from 2N or Ts65Dn mice at DIV5 were treated with different concentrations of Posiphen, as indicated, for another 48 hours, followed by immunoblotting to analyze the levels of APP, Dyrk1a, SOD1, tau, huntingtin, and α‐synuclein. β‐actin served as an internal loading control. The results of statistical analysis for each protein in B‐G are shown (N = 4). (H) The levels of App and SNCA mRNAs from the primary 2N or Ts65Dn cortical neurons treated with vehicle or Posiphen at 5 µM for 48 hours were assessed via real‐time PCR (N = 5). (I) The turnover rates of the fl‐APP protein level in vehicle or Posiphen‐treated primary 2N or Ts65Dn neurons were measured by CHX treatment for the durations indicated with the levels of fl‐APP normalized to β‐actin (N = 4). (J) As in panel A, the levels of α‐CTF and β‐CTF in 2N or Ts65Dn neurons treated with vehicle or Posiphen (5 µM) were quantitated and normalized against β‐actin (N = 5). (K) The levels of GTP‐Rab5 and total Rab5 were measured in primary 2N or Ts65Dn cortical neurons treated at DIV5 with vehicle or Posiphen (5 µM) for 48 hours. The level of GTP‐Rab5 in vehicle‐treated 2N neurons was set at 100% (N = 4). (L) Primary 2N or Ts65Dn cortical neurons were cultured on poly‐d‐lysine–coated glass coverslips and treated at DIV5 with Posiphen (5 µM) for 48 hours. Representative images for each case are shown. Nuclei were stained with Hoechst 33342 (blue). (M) The size of Rab5‐positive early endosomes (135‐218 endosomes in each group) was quantified. (N) The size distribution of the Rab5‐positive early endosomes are shown. Scale bar: 5 µm. In all panels: *P < .05, **P < .01, ***P < .001, n.s., non‐significant, one‐way ANOVA followed by Newman‐Keuls multiple comparison test; for panel N, # P < .05, n.s., non‐significant, paired Student t‐test
Posiphen has been shown to reduce translation of APP mRNA. 53 To define the mechanism by which Posiphen acted in Ts65Dn neurons we measured the levels of the mRNAs for APP and α‐synuclein in Ts65Dn and 2N neurons treated under the conditions used to examine fl‐APP. We failed to detect a significant change in the levels of these mRNAs (Figure 1H). To rule out increased degradation of APP protein in response to Posiphen, we examined its effects on the turnover of fl‐APP in studies in which cycloheximide was used to prevent synthesis. Posiphen had little or no effect on the rate of APP degradation in Ts65Dn neurons (Figure 1I). A 50% reduction in fl‐APP was achieved in both vehicle‐ and Posiphen‐treated neurons at≈1.5 hours (Ts65Dn‐vehicle: 1.4 hours; Ts65Dn‐Posiphen: 1.3 hours). The small difference would not explain the decrease in fl‐APP registered in Ts65Dn neurons to 2N levels. Degradation of fl‐APP in vehicle‐treated 2N neurons reached 50% at 1.4 hours, a value comparable to those in Ts65Dn neurons. Finally, as for fl‐APP, its CTF products (both α‐CTF and β‐CTF, the former is the 83 amino acid C‐terminal fragment, also known as C83) were reduced to 2N values in Ts65Dn neurons treated with Posiphen (Figure 1J). Accordingly, and consistent with earlier findings, 53 we conclude that Posiphen acted to reduce APP protein levels in Ts65Dn neurons via reduced translation of App mRNA.
Posiphen reversed the increase in Rab5 activation and enlargement of early endosomes
Earlier in vivo studies showed increased activation of Rab5 and increased size of early endosomes in Ts65Dn mice. 40 , 41 Increased levels of fl‐APP or its β‐CTF are known to induce Rab5 hyperactivation and early endosome enlargement. 41 , 43 Consistent with a role for increased APP gene dose as responsible for increased GTP‐Rab5 levels, that is, the activated form of the GTPase, in the brains of Ts65Dn mice, the increase relative to 2N mice was absent when App gene copy number was reduced to 2 (ie, Ts65Dn:App +/+/−)(Supplementary Figure S2). Increased activation of Rab5 was shown to increase the size of early endosomes. 41 To test if Posiphen would impact Rab5 activity and early endosome structure in Ts65Dn neurons we used a concentration (5 µM) shown to normalize APP protein in these neurons (Figure 1A, B). In Ts65Dn neurons, as compared with 2N controls, there was an ≈50% increase in the level of GTP‐Rab5 (Figure 1K), a finding consistent with our earlier findings on Ts65Dn neurons in vivo. 41 Posiphen treatment reduced Rab5 activity in Ts65Dn neurons to the levels in 2N neurons; there was no significant effect of Posiphen on GTP‐Rab5 levels in 2N neurons (Figure 1K). Next, to assess the effect of Posiphen on the size of early endosomes, we used immunostaining to examine Rab5. Posiphen treatment significantly reduced the average size of early endosomes in Ts65Dn neurons (2N‐vehicle: 0.155 ± 0.008 µm2; Ts65Dn‐vehicle: 0.200 ± 0.011 µm2; Ts65Dn‐Posiphen: 0.149 ± 0.008 µm2)(Figure 1L, M). To further explore Posiphen effects, the size distribution of early endosomes was examined. Although in vehicle‐treated Ts65Dn neurons there was a relative increase in the frequency of endosomes in the largest category (>0.3 µm2), treatment with Posiphen largely restored this measure to 2N levels (Figure 1N). Posiphen also increased the frequency of small endosomes (<0.15 µm2) and reduced the frequency of intermediate‐sized endosomes (0.15–0.3 µm2) in Ts65Dn neurons (Figure 1N). These findings for Posiphen mirrored the effect of reducing App gene expression on endosome size in Ts65Dn neurons in vitro. 41 They give evidence that Posiphen‐mediated reductions in the levels of APP protein in Ts65Dn neurons restored normal levels of Rab5 activity and reversed enlargement of early endosomes.
Posiphen rescued the deficit in the retrograde axonal transport of brain‐derived neurotrophic factor (BDNF)
Neurotrophic factor signals must be transmitted by retrograde transport to neuronal somas over long distances from presynaptic domains in axons as well as in dendrites. 47 , 69 Reduced retrograde axonal transport of nerve growth factor (NGF) signaling is linked to basal forebrain cholinergic neuron (BFCN) atrophy in animal and cell models of DS. 40 , 41 Evidence of reduced retrograde transport of brain‐derived neurotrophic factor (BDNF)/tropomyosin receptor kinase B (TrkB) signaling endosomes has also been documented in the cortex of Ts65Dn mice. 70 To determine if Posiphen treatment impacted neurotrophin signaling and trafficking we examined the retrograde transport of BDNF in primary cultures of E18 Ts65Dn or 2N cortical neurons maintained in microfluidic culture chambers (Figure 2A, B) in which axons are separated from their corresponding cell bodies. 71 Quantum dot–labeled BDNF (QD‐BDNF) was used to track axonal transport of BDNF by live cell imaging 72 (Figure 2B). Compared with 2N neurons, Ts65Dn neurons displayed slower instantaneous (ie, moving) velocity and an increase in endosome pausing (Figure 2C‐E). Posiphen treatment resulted in an increase in instantaneous velocity from 1.50 ± 0.12 µm/s (≈70% of the 2N value) to 1.90 ± 0.14 µm/s in Ts65Dn neurons, a value comparable to that in 2N neurons (2.16 ± 0.15 µm/s)(Figure 2C, D); it also decreased the percentage of time that endosome movement was paused (44 ± 5% to 24 ± 5%)(Figure 2C, E), again resulting values comparable to 2N neurons (29 ± 5%). There was a non‐significant trend to an increase for average pause duration in Ts65Dn neurons; whereas Posiphen reduced this, the change was not significant (Figure 2F). As an overall measure of transport, the average retrograde velocity in Posiphen‐treated Ts65Dn neurons was 1.51 ± 0.16 µm/s, significantly faster than that in vehicle‐treated Ts65Dn neurons (0.94 ± 0.14 µm/s), and comparable to that in 2N neurons (1.70 ± 0.19 µm/s) (Figure 2C, G). Finally, although Posiphen had no significant effect on retrograde BDNF transport in 2N neurons, there were trends toward an increase in the instantaneous and average velocity and a decrease in percent of time paused (Supplementary Figure S3). We conclude that Posiphen reversed impaired retrograde axonal transport of BDNF‐containing endosomes in Ts65Dn neurons. Consistent with earlier in vivo evidence showing that increased App gene expression in Ts65Dn mice contributes significantly to reduced neurotrophin axonal transport, 40 and these in vitro data for improved BDNF transport in Ts65Dn neurons in the context of Posiphen‐mediated reductions in APP and its products, we note also that deficits in axonal BDNF transport in Ts65Dn neurons were partially rescued when App gene copy number was reduced to two (ie, Ts65Dn:App +/+/−)(Supplementary Figure S4). Taken together, the evidence argues that Posiphen treatment effects on axonal transport are mediated at least in part through its actions on APP.
FIGURE 2.
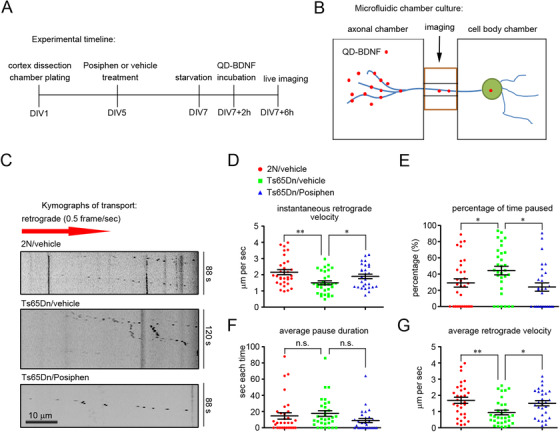
Posiphen rescued the deficit in the retrograde axonal transport of BDNF in cortical neurons. (A) Experimental design. Primary cultures of 2N and Ts65Dn cortical neurons were treated at DIV5 with Posiphen (5 µM) for 48 hours, followed by live imaging. (B) Diagram of microfluidic chamber in which primary cultures of 2N and Ts65Dn neurons were maintained; the distal axons of neurons are fluidically isolated from neuron cell bodies. QD‐BDNF was added to the axonal chamber in preparation for tracking axonal transport of BDNF by live cell imaging. (C) Representative kymographs of QD‐BDNF retrograde transport in 2N and Ts65DN neurons treated with vehicle or Posiphen. Scale bar, 10 µm. The data for retrograde instantaneous velocity, percentage of time paused, average pause duration, and average velocity of axonally transported QD‐BDNF were quantitated and presented in D‐G. Results are shown as mean ± SEM. Data were obtained from at least 15 axons for each condition. *P < .05 and **P < .01, n.s., non‐significant, one‐way ANOVA test followed by Newman‐Keuls multiple comparison test
Posiphen reversed deficits in neurotrophic signal transduction
Increased APP gene dose has been shown to impact neurotrophin‐induced mitogen‐activated protein kinase (MAPK) pathway activation. 41 Given the important role ascribed to endosomal signaling for neurotrophins 47 and Posiphen effects on axonal transport of BDNF‐containing endosomes, we asked if Posiphen would impact BDNF signaling. We examined activation of the TrkB receptor and two downstream signaling pathways—Akt (protein kinase B [PKB]) (ie, a key upstream element in Akt/PI3K [phosphoinositide 3‐kinase] signaling) and ERK 1/2 (extracellular signal‐regulated kinase 1/2) (ie, a key upstream element in MAPK [mitogen‐activated protein kinase kinase] signaling). Mass cultures of 2N or Ts65Dn E18 cortical neurons were treated with Posiphen or the vehicle for 48 hours before adding BDNF (20 ng/mL). Early and sustained phosphorylation of TrkB, Akt, and ERK (pTrkB, pAkt and pERK) was observed in all groups, as measured at 5 and 30 minutes after BDNF exposure. However, as compared with 2N neurons, vehicle‐treated Ts65Dn neurons responded less robustly to BDNF with respect to activation of TrkB, Akt, and ERK (Figure 3). The changes were present after 5 minutes and 30 minutes for all measures (Figure 3A‐D). Posiphen treatment had no significant effect on signaling in 2N neurons. In contrast, a marked effect was seen in Ts65Dn neurons with increased activation of TrkB, Akt, and ERK. The changes relative to vehicle‐treated Ts65Dn neurons were significant at both time points for TrkB, and at 30 minutes for ERK; Posiphen treatment demonstrated a trend to increased signaling at 5 minutes for ERK and at 5 minutes and 30 minutes for Akt. In view of the absence of changes in the total levels of TrkB, Akt, and ERK, Posiphen increased the specific activity of BDNF signaling through its TrkB receptor to downstream pathways. It is notable that we failed to detect a significant difference in signaling between Posiphen‐treated Ts65Dn neurons and vehicle‐treated 2N neurons (Figure 3). These finding are evidence that in Ts65Dn neurons Posiphen reversed deficits in BDNF signaling.
FIGURE 3.
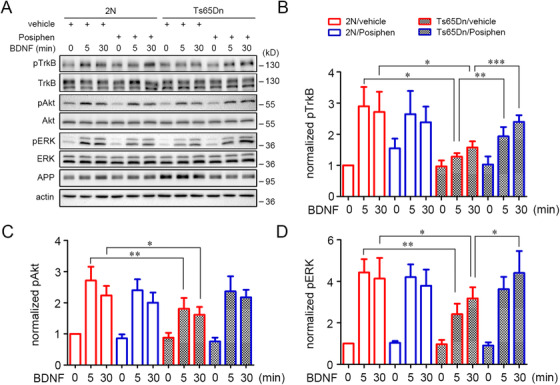
Posiphen reversed deficits in neurotrophic signal transduction in Ts65Dn cortical neurons in vitro. (A) Primary 2N and Ts65Dn cortical neurons were cultured in 24‐well plate (mass cultures) through DIV5 and then treated with Posiphen (5 µM) for 48 hours, starved for 2 hours in neurobasal media containing the same concentration of Posiphen, and finally stimulated with 20 ng/mL BDNF for the periods indicated. Equal amounts of protein lysates were analyzed by SDS/PAGE and immunoblotting with specific antibodies as indicated. Quantitation of the relative levels of protein species are shown for activated TrkB (B), activated Akt (C), and activated Erk1/2 (D) (N = 5). *P < .05, **P < .01, ***P < .001, one‐way ANOVA test followed by Newman‐Keuls multiple comparison test
3.1.2. In vivo studies in Ts65Dn mice
Posiphen treatment was well tolerated
Posiphen is hydrophobic and penetrates brain‐blood barrier by lipid solubility/transmembrane diffusion. 59 , 73 Earlier studies explored the pharmacokinetic (PK)/pharmacodynamic (PD) characteristics of Posiphen and defined effective doses of Posiphen in mouse models of AD. 58 As reported, intraperitoneal (i.p.) injection of Posiphen reduced APP in AD mice, but induced apparent side effects, including tremors, at a dose of 75 mg/kg, i.p. 58 In preliminary studies in 3‐month‐old 2N and Ts65Dn mice we used doses of 25 mg/kg/d and 50 mg/kg/d. Posiphen levels were measured in mice treated i.p. for 3 weeks. Mice tolerated treatment well and showed no changes in mobility or behavior; all mice demonstrated weight gain. At exactly 90 minutes after the last injection, Posiphen levels were measured in both plasma and brain (Figure 4A, B). Dose‐dependent changes were detected. In brain, Posiphen levels greatly exceeded (by 8 – 9‐fold) those in plasma in both 2N and Ts65Dn mice (at 50 mg/kg/d, 2N brain: 1900 ng/g; 2N plasma: 220 ng/mL; Ts65Dn brain: 1700 ng/g; Ts65Dn plasma: 200 ng/mL)(Figure 4A, B). The significantly higher levels in brain versus plasma and the brain levels of Posiphen were similar to those achieved in an earlier study. 58 , 74 It is notable that only at 50 mg/kg/d did Posiphen reach a concentration in brain (3.8 µM) shown to be effective in reducing fl‐APP in vitro. As expected, Posiphen at 50 mg/kg/d reduced fl‐APP in hippocampus to 2N levels; no significant reduction was detected using 25 mg/kg/d (Figure 4C). Thus in Ts65Dn mice comparable effects on APP expression and PK characteristics were achieved with the same dose of Posiphen as in APP/PS1 transgenic mice. 58
FIGURE 4.
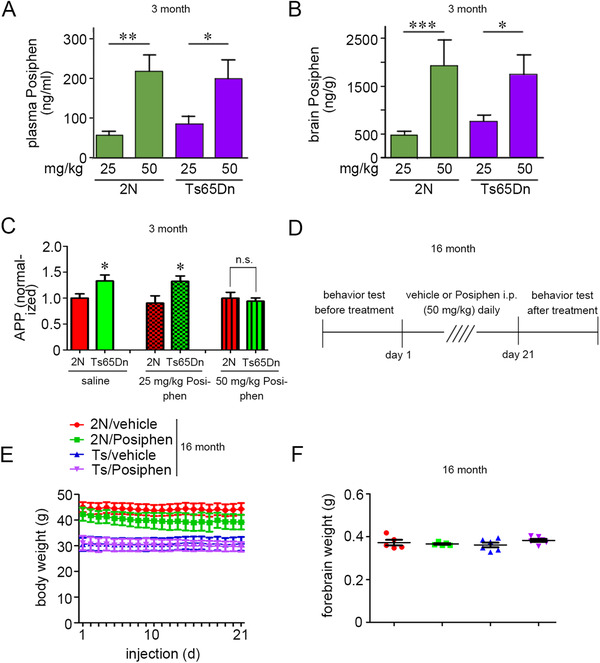
Posiphen treatment was well tolerated in vivo. (A, B) Distribution of Posiphen in plasma and brain of 3‐month‐old 2N and Ts65Dn mice. The concentration of Posiphen in plasma and brain of mice treated i.p. with 25 mg/kg/d or 50 mg/kg/d Posiphen for 3 weeks was determined by LC‐MS/MS. Mean ± SEM are shown (N = 15 per group). (C) 3‐month‐old 2N and Ts65Dn male mice were treated i.p. with vehicle or 25 mg/kg/d or 50 mg/kg/d Posiphen for 3 weeks. The fl‐APP levels were assayed in the hippocampus. N = 10‐17, *P < .05, n.s., non‐significant, unpaired Student t‐test. (D) Experimental design for vehicle or 50 mg/kg/d i.p. Posiphen treatment in 16‐month 2N and Ts65Dn male mice. (E) Body weight of each mouse was measured daily during treatment (N = 7‐9). (F) Weight of the dissected forebrain was determined at sacrifice (N = 5 for each group)
Accordingly, we initiated a second series of studies and elected to study aged Ts65Dn mice using 50 mg/kg/d i.p. for 3 weeks and during behavioral testing for an additional 5 days to examine effects at 16 months, an age at which dysfunction and degeneration are present. 40 , 68 Indeed, at this age neurodegenerative changes linked to App gene dose are well advanced. Behavioral testing was conducted both before and after treatment (Figure 4D). At 16 month of age, Ts65Dn mice displayed a significantly lower body weight compared with 2N mice (Figure 4E). Treatment was well‐tolerated. We noted no changes in behavior or mobility, and no tremors were detected. Measuring body weight of each group daily showed no significant effects of i.p. injection on body weight (Figure 4E). Mouse brains were dissected 4‐6 hours post the last injection of Posiphen. Posiphen treatment had no effect on brain weight in either 2N or Ts65Dn mice (Figure 4F). We also measured the brain contents of both Posiphen and its metabolites and found that at 4‐6 hours after the last treatment of Posiphen, that Posiphen and N8‐Norposiphen were more abundant as compared with N1‐Norposiphen in both 2N and Ts65Dn mice (Supplementary Figure S5). There was no difference for Posiphen and its metabolites between 2N and Ts65Dn mice, pointing to a comparable rate and pattern of metabolism.
Posiphen normalized the levels of fl‐APP and its products and reversed the increase in Rab5 activity
As in vitro, Posiphen treatment of Ts65Dn mice significantly reduced fl‐APP (Figure 5A, B); the resulting decrease was to a level insignificantly different from that in vehicle‐treated 2N mice. Posiphen had a modest but statistically insignificant effect on fl‐APP in 2N mice. The CTF products (both α‐CTF and β‐CTF) of fl‐APP in Ts65Dn mice were also reduced to 2N values; no significant reduction was seen in 2N mice (Figure 5A, B). We used Meso Scale Discovery (MSD) assay to assess the levels of Aβ species. 75 There was a marked increase of Aβ42 and Aβ40 in Ts65Dn mice compared with 2N mice; Posiphen treatment in Ts65Dn mice induced a decrease in Aβ42 to a level not significantly different from that in the vehicle‐treated 2N mice. However, Posiphen had a small, statistically insignificant effect on the level of Aβ40, a result consistent with a previous report 58 (Figure 5C, D). Mirroring its actions in vitro, Posiphen had no effect on the level of App mRNA in the cortex of either 2N or Ts65Dn mice (Figure 5E). The effect on fl‐APP was selective because there was no effect in the Ts65Dn or 2N brain on Dyrk1a, SOD1, or huntingtin (Figure 5A, F). Total tau was detected using the TAU‐5 antibody. As in vitro, two bands were detected and quantified together. Although no statistically significant difference was detected comparing Ts65Dn and 2N or comparing vehicle with Posiphen treatment, we noted an apparent decrease in the intensity of the upper band with Posiphen (Figure 5A). Not evident in vitro, this change is unexplained but could reflect a difference in the level of phosphorylated tau species. Consistent with this suggestion, as assessed by immunoblotting for phosphorylation of tau on Thr231 and PHF1 epitopes, we discovered that Posiphen reduced phosphorylation of tau in Ts65Dn mice with a smaller statistically insignificant effect in 2N mice (Figure 5A, G, H). Changes in tau phosphorylation in Ts65Dn may be a function of the presence in one extra copy of other genes, including DYRK1A and RCAN1. As indicated, Posiphen had no effect on the level of Dyrk1a. The gene for regulator of calcineurin 1 (Rcan1) is present on HSA21 and the segmentally trisomic fragment in Ts65Dn, shows increased expression in DS, 76 and has been linked to tau phosphorylation. 77 To examine a role for Rcan1 we also assessed Rcan1 expression. The protein was significantly increased in the brains of Ts65Dn mice compared with 2N mice but Posiphen had no effect on the level of Rcan1 in either Ts65Dn or 2N mice (Figure 5A, I). In contrast to in vitro findings, we detected no significant effect of Posiphen on α‐synuclein (Figure 5A, F).
FIGURE 5.
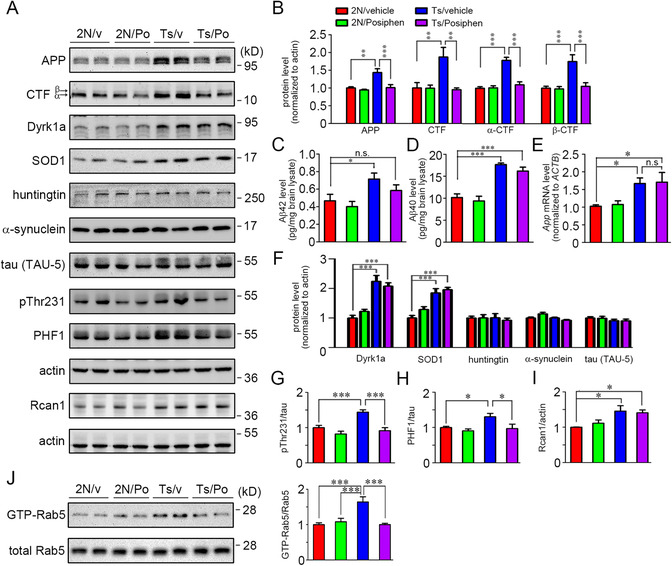
Posiphen reduced fl‐APP and its products and reversed increased Rab5 activity in Ts65Dn mice. (A) 16‐month‐old 2N and Ts65Dn male mice were treated i.p. for 3 weeks and during behavioral testing for an additional 5 days with vehicle or 50 mg/kg/d Posiphen. The levels of selected proteins in forebrain were measured. The levels of Aβ species were evaluated using MSD assays. A portion of brain lysates was incubated with GTP‐agarose beads to pull down activated GTP‐Rab5. A portion of cortex was used for whole mRNA extraction followed by cDNA synthesis and real‐time PCR to examine the App mRNA level. (B) The levels of fl‐APP and its CTFs (α‐CTF and β‐CTF) were analyzed. (C, D) The levels of Aβ42 and Aβ40 for each group were assessed using MSD assays. (E) The levels of App mRNA for each group were analyzed. (F) The levels of Dyrk1a, SOD1, huntingtin, α‐synuclein, and tau for each group were analyzed. The levels of p‐tau including pThr231 and PHF1 were also examined in (G‐H). (I) The protein levels of Rcan1 for each group were analyzed. (J) The levels of GTP‐Rab5 were quantitated and normalized against total Rab5. N = 5 for all panels, *P < .05, **P < .01 and ***P < .001, n.s., non‐significant, one‐way ANOVA test followed by Newman‐Keuls multiple comparison test
It is important to note that as predicted from the literature 41 and in vitro studies, normalization of fl‐APP and its products was accompanied by normalization of Rab5 activity in Ts65Dn mice. There was no effect of Posiphen on GTP‐Rab5 in 2N mice (Figure 5J). Taken together with earlier findings, and the consistent linkage between increased levels of APP gene expression and Rab5 activity, we conclude that Posiphen‐mediated normalization of fl‐APP and its products restored normal activity of Rab5 in vivo.
Posiphen restored Trk signaling and the level of choline acetyltransferase (ChAT) protein
We asked if Posiphen‐mediated normalization of Rab5 activity would positively impact measures of neurotrophin signaling in the Ts65Dn brain. The levels of pTrkB, pAkt, and pERK were significantly lower in the Ts65Dn brain as compared to the 2N brain (Figure 6A‐D); the reductions ranged from 25% to 50%. We also examined pCREB (the phosphorylated form of cAMP response element‐binding protein [CREB]), a species that is axonally transported on endosomes that carry neurotrophin/Trk signals. 78 pCREB was demonstrated in earlier studies to be lower in Ts65Dn mice, 79 a finding that we confirmed showing reductions of ≈40% (Figure 6A, E). Posiphen treatment restored the levels of pTrkB, pAkt, pERK, and pCREB in Ts65Dn brains (Figure 6A‐E). No significant changes were registered in the 2N brain. The expression of ChAT, the neurotransmitter synthetic enzyme for acetylcholine (Ach), is regulated by neurotrophin signaling induced by NGF and BDNF. 80 , 81 ChAT is present in cholinergic neurons and dysfunction of the cholinergic system contributes to the pathogenesis of AD and AD‐DS. 42 , 82 , 83 We found that ChAT protein was reduced in the Ts65Dn brain in comparison to the 2N brain. As with the neurotrophin‐responsive markers, Posiphen treatment restored the ChAT level in the Ts65Dn brain to that in 2N (Figure 6A, F).
FIGURE 6.
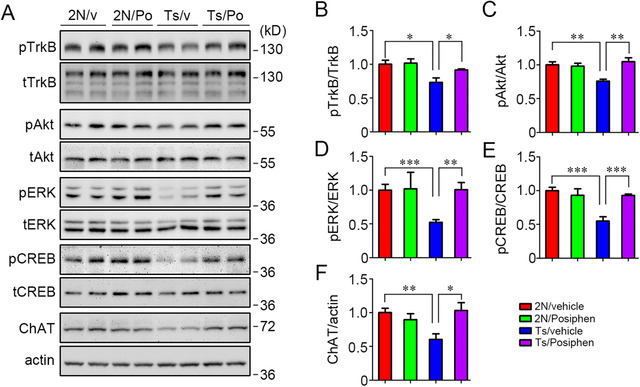
Posiphen reversed reductions in activation of TrkB, Akt, ERK, and CREB, and restored ChAT level in Ts65Dn mice. (A) The activation of TrkB, Akt, ERK, and CREB was examined in the forebrain of 2N and Ts65Dn mice treated i.p. with either vehicle or 50 mg/kg/d Posiphen for 3 weeks and during behavioral testing for an additional 5 days. The levels of ChAT protein were also measured. Quantitation of the relative levels is shown for activated TrkB (B), activated Akt (C), activated ERK1/2 (D), and activated CREB (E). The results for ChAT are also shown (F). N = 5, *P < .05, **P < .01, and ***P < .001, unpaired Student t‐test
To ask if Posiphen impacted the number of cholinergic neurons in the medial septum, we used unbiased stereology and immunostaining for ChAT and estimated the number of ChAT‐positive neurons in the brains of 2N or Ts65Dn treated with Posiphen or vehicle. There was no difference between groups (one‐way ANOVA followed by Turkey multiple comparison test; P = .1720). Although the average number of ChAT‐positive neurons was greater in 2N than Ts65Dn mice treated with vehicle, the difference was not statistically significant (P = .8030), and there was no statistically significant difference between Posiphen‐treated 2N and Ts65Dn neuron (P = .9106). Finally, a trend toward decreased numbers in Posiphen‐ versus vehicle‐treated Ts65Dn mice was also not significant (P = 0.7901). The average numbers of ChAT‐positive neurons were as follows (mean ± SEM): vehicle‐treated 2N (2436 ± 576; N = 4); Posiphen‐treated 2N (1260 ± 241; N = 4); vehicle‐treated Ts65Dn (1992 ± 225; N = 6); Posiphen‐treated Ts65Dn (1584 ± 320; N = 6). Taken together, the findings show that normalizing APP protein is correlated with restoration of signaling pathways and rescue of ChAT protein levels, but did not impact the number of ChAT immunopositive neurons in the aged Ts65Dn brain.
Posiphen had no effect on deficits in open field activity, nest building, and Y‐maze in Ts65Dn mice
Finally we investigated whether or not Posiphen would impact anxiety‐ and locomotion/exploration‐related behaviors and working memory. To address this question studies were carried out before and after Posiphen treatment in 16‐month‐old Ts65Dn and age‐matched 2N mice. The open‐field test measures locomotor and anxiety‐like behaviors. 84 In this test, there was a significant difference in total time spent in the center square between 2N and Ts65Dn mice; the decrease in Ts65Dn mice was documented in earlier studies 85 , 86 , 87 and may be interpreted as evidence of increased anxiety (Supplementary Figure S6A, B). Posiphen treatment had no effect on Ts65Dn mice in this parameter (Supplementary Figure S6A, B). In contrast, in 2N mice there were comparable decreases for both the vehicle‐ and Posiphen‐treated groups in time spent in the center (Supplementary Figure S6A, B). The absence of a change in Ts65Dn mice may reflect the very small amount of time that even vehicle‐treated mice spent in the center (Supplementary Figure S6A). There was a trend to increased total distance moved (≈20%) in Ts65Dn mice versus 2N mice, but the difference was not statistically significant. Treatment with either vehicle or Posiphen had a small but significant effect on reducing total distance moved in 2N mice, but no effect on Ts65Dn mice (Supplementary Figure S6C, D).
Nest building is a hippocampus and prefrontal cortex–dependent non‐learned behavior widely observed throughout the animal kingdom. 88 This test has been reported to be deficient in Ts65Dn mice. 68 , 89 We confirmed this, finding that Ts65Dn mice registered smaller nest scores and higher percentages of remaining nestlet material, as measured by weight (Supplementary Figure S6E, F). Posiphen treatment had no effect on nest‐building behavior in Ts65Dn mice (Supplementary Figure S6E, F). This finding is consistent with previous reports that deficits in nest building in these mice were independent of increased App gene dose. 68 , 89 The absence of differences in these behaviors comparing Posiphen‐treated and vehicle‐treated Ts65Dn mice is further evidence that Posiphen treatment was well tolerated.
Ts65Dn mice have been demonstrated to have deficient hippocampal function, 90 changes that may to some extent reflect those in AD 65 , 91 but that also report the effects of changes during development. 85 , 87 , 92 Deficient working memory contributes to cognitive impairment in DS. 93 Thus we also tested whether Posiphen could affect working memory in Ts65Dn mice, and the rate of spontaneous alternations in Y‐maze was measured. The alternation rate trended to lower values in Ts65Dn mice versus 2N mice, but the difference was not statistically significant, reflecting impaired working memory (Supplementary Figure S7) and consistent with previous findings. 85 , 90 Note that the average value in these old 2N mice (55%) was less than that in younger animals (ages 4 to 11 months = 65%), 85 , 87 , 90 as was that for Ts65Dn mice (55% in earlier younger mice vs 50% in this study). Posiphen treatment had no effect on either 2N or Ts65Dn mice (Supplementary Figure S7).
3.2. Future experiments and validation studies
3.2.1. Our findings motivate renewed attention to the mechanisms by which APP and its products induce AD pathogenesis
The current work addresses the use of Posiphen, which has been shown to reduce APP level by reducing translation of APP mRNA. 53 In our studies Posiphen acted in vitro on Ts65Dn neurons to reduce translation of App mRNA, as demonstrated by examining App mRNA levels and the turnover of fl‐APP protein. These findings were consistent with a previous report. 53 Whether the changes in Posiphen‐treated mice were due solely to the reduction in APP levels and normalization of Rab5 activity is an open question, but taken together our data support the conclusion that both effects were significant. The question arises as to how normalizing APP and reducing increased Rab5 activity could restore signaling. Although additional studies will be needed, existing evidence points to increased Rab5 activity as resulting in increased internalization of surface receptors for BDNF. 70 Thus reducing APP and its effects on Rab5 activity may restore the levels of surface receptors, including TrkB, to normal levels to support more robust signaling. The effects on p‐tau are further evidence of the positive impact of Posiphen treatment, but again, one can only speculate as to the underlying mechanism(s). In this context it is interesting that Aβ has been shown to induce phosphorylation of tau, suggesting that reduction in this product of APP could contribute to a reduction in p‐tau levels. Given the important correlation between tau pathology and disease progression in both AD and AD‐DS, this facet of Posiphen effects could prove to play a therapeutic role. The excellent safety profile for Posiphen in three, Phase I trials and evidence for lowering APP products in human CSF of patients with MCI 59 motivate consideration of evaluating this drug in future clinical trials in adults with DS.
Enlargement of early endosomes is extensive in neurons from the brains of both DS patients and animal models. 38 , 41 The significance of dysregulation of early endosome size for pathogenesis is not yet fully defined 46 but a compelling case can be made for disruption of retrograde neurotrophin signaling in causing the dysfunction and loss of neurons. First, increased levels of APP and β‐CTF are responsible for increased activation of Rab5 and for enlargement of early endosomes. 41 , 43 , 45 Indeed, together with these changes, overexpression of fl‐APP or β‐CTF in BFCNs decreased retrograde axonal transport of the neurotrophin NGF, decreased NGF signaling, and induced atrophy of BFCNs. 41 Second, in the Ts65Dn mouse, increased App gene dose was linked to reduced endosome‐mediated transport of NGF and degeneration of BFCNs. 40 Given the evidence in Ts65Dn mice for the presence of NGF in enlarged early endosomes that contain APP and CTFs in cholinergic axons, 40 together with the evidence that NGF signals through its receptors in signaling endosomes undergoing retrograde axonal transport to neuron cell bodies, 42 , 47 , 94 it is likely that disrupted transport of NGF in signaling endosomes plays a critical role in BFCN degeneration. Third, the scenario just outlined for NGF signaling is likely to be shared by other neurotrophic factors. Thus, in the Ts65Dn cortex, enlarged Rab5‐positive endosomes containing activated TrkB receptors were seen in synapses and there was a corresponding decrease in TrkB‐positive endosomes in neuron cell bodies. 70 It is important to note that studies continue to explore the mechanisms by which increased APP gene dose induces increased Rab5 activation. Although increasing evidence supports a role for APP and β‐CTF 41 , 43 , 45 , 95 it is noteworthy that Aβ is present in early endosomes in early AD and in the young DS brain 96 suggesting that Aβ may also contribute to the initiation of pathogenetic events linked to endosomal dysfunction. Our findings encourage increased attention to preclinical studies of the mechanisms responsible for changes in early endosomes as well as downstream elements in the endosomal/lysosomal system, including a focus on the impact on neurotrophic factor signaling. Studies to evaluate the status of vulnerable neurons whose degeneration is linked to increased APP gene dose and physiological and behavioral phenotypes shared between AD and AD‐DS can also be recommended. Studies in human neuronal model systems based on induced pluripotent stem cell (iPSC)–derived or induced neuron approaches would serve to validate observations in mouse model systems.
Future studies will benefit from the knowledge that there are two pathways by which Aβ is secreted from axons. 97 In the first, endocytosis of APP in the soma results in the formation of sAPP fragments and Aβ that are then moved anterogradely, probably in a Rab11‐positive endosomal compartment, to the axons for release. 97 , 98 , 99 Of interest, the β‐CTF, but not the α‐CTF, is present within the compartment; increased levels of β‐CTF were reported to reduce the relative amount of anterograde transport. 100 In the second pathway, fl‐APP is moved anterogradely to the axon before processing, with local processing leading to local release of Aβ and presumably other APP products. 101 , 102 This pathway depends on endocytosis of APP in the axon. In earlier studies, we found that APP and its CTFs were present within early endosomes in the axons of cholinergic neurons in vivo; their presence in enlarged early endosomes and colocalization with Rab5 demonstrated that this compartment was impacted. 40 This localization is consistent with the participation of the Rab5 pathway in retrograde transport; we also found that increased expression of β‐CTF, but not α‐CTF, resulted in the presence in axons of enlarged varicosities that were positive for mCherry‐Rab5WT. Increased β‐CTF expression decreased retrograde velocity of endosomes containing NGF and reduced NGF retrograde signaling in the cell body. 41 It is notable that fl‐APP and β‐CTF both drive Rab5 activation and early endosome enlargement, 41 , 43 β‐CTF through an APPL1 (the adaptor protein containing a pleckstrin‐homology [PH] domain, phosphotyrosine binding [PTB] domain, and leucine zipper motif 1)‐dependent mechanism. 43 In turn, the increase in GTP‐Rab5 levels is highly correlated with reduced retrograde transport of endosomes. 41 In addition, increases in Aβ may affect axonal transport. 103 , 104 Thus the endocytic pathway, and its various subcompartments, appear to be regulated by APP and its products, and these changes may contribute to pathogenesis.
It was reported that Posiphen could upregulate BDNF expression and exert neurotrophic action. 56 It is not clear whether this neurotrophic effect contributes to Posiphen's effect on Trk signaling in Ts65Dn mice described here. It is worth further study to explore whether there is any change of neurotrophin levels due to Posiphen treatment. However, the lack of effect by Posiphen in 2N mice argues that a direct neurotrophic effect for Posiphen mediated by changes in APP would be limited to Ts65Dn neurons.
Three unexpected results deserve mention. One was the very modest effect of Posiphen on the levels of fl‐APP in 2N mice, both in vitro and in vivo. The relative differences between the responses of 2N and Ts65Dn mice are unexplained, but we suggest that differences in iron loading in 2N and Ts65Dn neurons may contribute, as iron was required for the Posiphen effects in enhancing the binding of IRP1 to APP IRE in an in vitro system (unpublished data, Maria Maccecchini). Indeed, increased iron loading may be characteristic of neurons in the Ts65Dn model and possibly also in people Down syndrome. In support of this suggestion, the gene for Intersectin 1 (ITSN1), which is trisomic in Ts65Dn and Down syndrome patients displays elevated expression. 105 , 106 It is notable that ITSN1 is involved in receptor‐mediated endocytosis, including that of the transferrin/transferrin receptor complex that mediates iron internalization. 107 Transferrin loading was increased in the brains of Down syndrome compared with controls, 108 thus the extra copy of ITSN1 may contribute to increased transferrin and possibly iron transport in those with Down syndrome. It was also noted that iron was significantly increased in the globus pallidus and frontal cortex of AD patients and increased with Braak stage; in addition, transferrin was increased in the frontal cortex of AD, as compared with elderly controls, 109 , 110 although the mechanism by which these increases were produced is unknown. It is of interest that ITSN1 was found to localize on Rab5‐positive early endosomes, 111 raising the possibility that increased Rab5 activity in Ts65Dn brains may interact with ITSN1 to increase iron loading. We note that Posiphen also reduced APP levels in the brains of APP23, but not in wild‐type mouse. 57 This raises the speculation that increased levels of APP alone can increase iron loading and thereby enable responses to Posiphen, but additional studies are needed to prove this. Given the proposed mechanism for Posiphen action referenced above, the possibility exists that iron levels are increased in Ts65Dn neurons, rendering them more susceptible to Posiphen effects in reducing App gene expression.
It should be noted that the IRE of APP mRNA in the 5′‐UTR was found recently to overlap with the active site for microRNA miR‐346, which was found to upregulate APP translation. 112 How this regulatory element may interact with Posiphen is yet to be defined, but under conditions of low cellular iron levels miR‐346 appears to inhibit IRP1 binding to the IRE, thus enhancing APP translation. 112 In the setting of increased iron levels, as are suggested by studies in DS and supported in AD. 109 , 110 IRP1 binding to the IRE is reduced, resulting in increased translation 112 , 113 ; Posiphen would oppose the impact of increased iron by recruiting the IRP1 to the IRE. Clearly, the level of cellular iron would influence not just the translation of APP but also the potential effects of miR‐346 and Posiphen. The active site for miR‐346 also overlaps with an interleukin‐1 (IL‐1) acute box element. 112 As AD is accompanied by neuroinflammation and ferroptosis, which may implicate changes in both IL‐1 and iron, 1 , 114 Posiphen effects could therefore be influenced by these factors.
Second, in 2N mice we saw pre‐ to post‐treatment changes in the open field test with respect to time in the center of the platform and to total distance moved. In view of the fact that at the concentrations achieved in vivo a small degree of inhibition of AChE was possible, 53 one must consider this as contributing. However, the changes detected were present to the same extent in both the vehicle and Posiphen‐treated mice, ruling out a role for AChE inhibition. Possibly, then, increased anxiety due to manipulation and repeated injections was responsible; if so, the absence of a significant change in Ts65Dn mice may reflect reduced sensitivity to such influences. Third, we noted a difference between the in vitro and in vivo responses of α‐synuclein to Posiphen; while Posiphen reduced α‐synuclein levels in vitro it did not do so in vivo. That Posiphen actions may well differ in such different environments is likely, but we note that others have shown Posiphen‐induced reductions in mice transgenically expressing α‐synuclein in vivo. 74 Although conceivably the different genetic environment present in these mice and those tested herein may explain the differences, the underlying explanation for the lack of efficacy in our mice is unexplained.
Beyond Posiphen, other approaches targeting the APP gene dose hypothesis should be pursued. Accordingly, studies to reduce the levels of APP mRNA (eg, antisense oligonucleotides [ASOs] directed specifically at APP) 115 , 116 , 117 and the translation of APP mRNA (as for small molecules like Posiphen and miRNAs) can be envisioned. Various miRNAs were reported to impact expression of either APP or the β‐secretase 1 (β‐site Aβ precursor protein cleaving enzyme 1[BACE1]). 118 Of interest, in vitro miR‐298 suppresses APP, BACE1, and some tau moieties in a 3′‐UTR dependent manner. 119 Thus, studies of miRNA‐mediated targeting of APP levels in DS may be considered. In addition, approaches to reduce the processing of β‐CTF and Aβ by γ‐secretase (eg, using γ‐secretase modulators) 75 , 120 and enhance clearance of Aβ peptides (active and passive immunotherapies) 42 , 121 can also be suggested to reduce levels in brain interstitial fluid as well in endosomes wherein Aβ may act to dysregulate early endosome function. In this context it can be asked whether or not and how increased levels of Aβ due to increased APP gene dose could intersect with the biology of endosomes. One possibility is suggested by the observations for the effect of increased β‐CTF on excessive Rab5 activation. 41 , 43 Noting that both Aβ and β‐CTF are substrates of γ‐secretase raise the possibility that competition for processing of Aβ and β‐CTF by this enzyme complex could increase the levels of β‐CTF as well as Aβ42. Thus increased Aβ42 could exacerbate increases in β‐CTF thereby enhancing β‐CTF–mediated increases in Rab5 activity would then lead to dysfunction of early endosomes. 47 It is notable that our studies also point to the possibility that targeting downstream events that regulate endosomal/lysosomal function, including the increased activation of Rab5, could be considered. 42 Targeting of Rab5 using ASOs to reduce translation, with small molecule inhibitors of Rab5 activity, and using small molecules that inhibit Rab5 GEFs (guanine nucleotide exchange factors) or activate Rab5 GAPs (GTPase activating proteins) could be considered. 122 But whether or not Rab5‐based approaches rescue manifestations of APP gene dose downstream of Rab5 is an open question.
3.2.2. Models of AD‐DS: defining the optimal context for translation to humans
Ideally model systems would precisely recapitulate events known to drive the pathological and functional signatures characteristic of human neurodegenerative disorders. Absent such a model system, there is no assurance that interventions proven effective in model will translate to humans. Indeed, some would argue that effective translation is unlikely. Unfortunately, no existing rodent model of AD or AD‐DS perfectly recapitulates the human disease. This is not surprising when one considers the many differences between the human and rodent brain in size, the diversity of cell types present, the number and patterns of genes expressed, the differences in brain structure, the different time courses of developmental and age‐related events, and the influence of other bodily systems, including the immune system and gut. Given these limitations investigators have created AD models informed by pathological features and genes demonstrated to cause or greatly increase risk of AD. Thus, most are due to introduction into the mouse genome of one or more human transgenes encoding mutant APP and PSEN; recent models also incorporate transgenes for mutant human tau to produce neurofibrillary pathology. 123 , 124 Remarkably, with few exceptions, 125 , 126 amyloid plaque and tau neurofibrillary pathology are not present together in AD models. 123 , 124 Genetic findings have focused interest on the participation of microglial cells in pathogenesis, promising models in which this facet of pathology is captured. Accordingly, one can expect additional models based on new findings for AD risk factors. Finally, an important advance has been the creation of models in which human mutations have been knocked into the mouse genome, such as those of Saito et al 127 ; this approach allows for gene expression driven by endogenous promoters and may well bring additional benefits. However, even in the case that one could precisely replicate pathological findings, the models may prove insufficient for modeling molecular events critical for pathogenesis, such as those induced by the presence of oligomeric assemblies of Aβ and tau, 27 some of which may occur only in a human cellular context. Given the limitations just enumerated, it is important to aim for models in which a mutant (or variant‐harboring) human gene(s) of interest is expressed under endogenous promoters at wild‐type levels and to ask if this model recapitulates age‐related changes in transcriptomic, biochemical, pathological, and cognitive phenotypes found in AD. It is reasonable to assert that the more robustly these goals are satisfied the more likely is translation to be successful.
Models of AD‐DS are, of course, subject to the same limitations. A key feature for AD‐DS models is that in the setting of trisomy 21 an increased dose of the wild‐type APP gene appears to be necessary for the pathogenesis of AD. The upshot is that securing endogenous levels of expression of wild‐type APP in a model system that harbors many if not all of the other genes on HSA21 is an important first step in creating a valid genetic model. The extent to which such a model recapitulates disease phenotypes then becomes the criterion by which it can be judged as potentially useful for translational studies. No existing model of AD‐DS is perfect. The Ts65Dn model of DS employed here is that most often used in studies of DS 67 but is limited by these features of its genome: mouse gene homologues of those present on HSA21, rather than human genes, are present in three copies; the number of these genes (90) is about 40% of those on HSA21; and there are extra copies of about 35 genes not present on HSA21. 67 Nevertheless, these mice show many of the features of AD‐DS, including neuron loss, 40 pre‐tangle‐like collections of p‐tau, 128 activation of microglia 129 and, as demonstrated herein, changes in the endosomal pathway. In addition, as discussed herein, reducing mouse App gene dose to two copies prevents neurodegeneration. 40 The ability to recapitulate the necessity of increased APP gene dose for degenerative phenotypes powerfully supports the validity of the model and, as we argue, rationally focuses attention on strategies targeting APP and its products. The Dp16 mouse model harbors a duplicate copy of mouse chromosome 16 with about 115 of the genes with homologues on HSA21, 130 and none on other chromosomes; however, the number of genes present in three copies (≈115) is still only about 50% of that in humans. Ongoing studies of this mouse are expected to demonstrate AD‐relevant phenotypes (unpublished observations). Nevertheless, there is a clear need for additional models of AD‐DS. For example, amyloid plaques, although not necessarily directly contributing to pathogenesis, may serve as a reservoir to sequester toxic Aβ species, buffer their levels, or otherwise serve to mediate aspects of dysfunction of nearby axons and dendrites. 131 A model of AD‐DS that includes plaques would help to understand what possible role(s) is played. Creating a model with amyloid plaques will almost certainly require humanizing the Aβ sequence in the mouse App gene. A new model for DS is the transchromosomic model in which the mouse genome has been modified by addition of a human chromosome 21 containing 93% of expressed and regulatable HSA21 protein‐coding genes. These mice thus have one copy of human APP. They do not have plaques but by humanizing the mouse Aβ sequence in the endogenous mouse alleles it may be possible to induce plaque formation. 132 Additional studies will be required to show whether and to what extent other degenerative features are present. Although incorporating neurofibrillary tau pathology would be important, this may or may not be possible by knocking in wild‐type human tau, as existing models employ transgenes encoding mutant human tau. In summary, mouse models of AD‐DS have enabled much progress in understanding a role for APP gene dose in pathogenesis but they as yet imperfectly replicate the human disease. Accordingly, AD‐DS models can be useful for studying pathogenesis and testing interventions targeting disease pathways, but the extent to which preclinical findings can be used to predict clinical efficacy is an open question.
By replicating key features of the human genetic and cellular context, human models of AD‐DS are likely to play an especially important role in studies of both pathogenesis and treatment. Especially useful may be those in which the 3D environment of AD is recapitulated. Recent success in using iPSC‐derived cells to create 3D models of AD is encouraging as is the use of organoid technology. 133 , 134 , 135 With respect to AD‐DS, relatively few studies using this technology have been published, but recent advances encourage the view that they will add significantly to our understanding. 133 , 136 However, one cannot discount that the in vivo environment confers many advantages to the biology of AD‐DS and therefore attempts to translate in vitro human models to a brain‐like environment may prove very important. Transplantation of iPSC‐derived neurons to the mouse brain serves as one avenue to achieve this goal, as recently highlighted. 137 The ability to detect developmental and age‐related changes in synaptic structure and function and to characterize neurodegenerative phenotypes may prove incisive in a new generation of studies aimed at translating preclinical studies to the clinic.
3.2.3. Posiphen as a possible treatment for AD‐DS: Next steps
Posiphen treatment in DS is rational. Future studies will build upon evidence from trials in the non‐DS AD population. In a Phase I trial in MCI patients the drug was well tolerated, and in CSF reduced the level of sAPP fragments, tau, and p‐tau, and demonstrated a trend toward lower Aβ42. 59 An ongoing Phase II trial measures the synthesis and degradation of Aβ42 by the stable isotope labeling kinetics method 138 at different doses of Posiphen. The drug's safety and its inhibition of APP, its fragments, tau, and p‐tau do suggest that it should be tested in Phase I trials in DS. The trial population could include those with and without dementia, between the ages of 35 and 55 years, and in addition to safety considerations could explore the impact of Posiphen on plasma biomarkers (Aβ levels, p‐tau181 and 217, neurofilament light chain [NFL]) and amyloid and tau imaging using positron emission tomography. Although those recruited are highly likely to harbor three alleles of APP, an important inclusion criterion would be evidence that this is the case. Evidence of safety and target engagement would presage later stage trials to evaluate efficacy and a reduction in decline on measures of cognition and function specific for the DS population, as are now being developed under the Longitudinal Investigation for Enhancing Down Syndrome Research Study (LIFE‐DSR), an observational study of adults with DS.
4. MAJOR CHALLENGES TO THE HYPOTHESIS
4.1. Reversing changes in neurons and other cells mediated by chronically increased APP gene dose
The data pointing to a necessary role for increased APP gene dose for AD neuropathology and dementia in AD‐DS are strong. 11 , 12 , 13 , 40 , 41 Although it relies on the findings from only two cases in which detailed clinical data and postmortem findings have been reported, 12 , 13 it is further substantiated by studies of APP gene locus duplication in rare families with early onset AD. 139 , 140 Nevertheless, the hypothesis is challenged by noting that those with partial trisomy for APP have never been exposed to increased levels of the protein and its products. A question is whether or not it will be possible to reverse the years‐long presence of increased APP and its products in those with full trisomy for chromosome 21, which constitute 95% of the DS population. Our studies suggest that this may be possible because we found that we could reverse endosomal phenotypes in even quite old Ts65Dn mice. Nevertheless, we failed to find an effect of Posiphen on cognition or the number of cholinergic neurons. Moreover, age‐related changes not due to increased APP gene dose could intervene to reduce the effects of treatments directed at APP. Future studies using younger mice or treating for longer periods will be needed to test for benefits in behavioral measures and in preventing neuron dysfunction and death in mouse models of DS. Because to our knowledge no one has yet demonstrated an APP gene dose effect in cognition in rodents, demonstrating a change may require discovery of such measures. Possibly markers sensitive to endosomal function in cholinergic and other vulnerable populations would also prove useful. Future studies would be further enhanced by linking changes in models to humans by employing biomarkers used in human trials, such as plasma levels of Aβ species and p‐tau as well as NFL.
4.2. The significance of endosomal dysfunction for AD pathogenesis
An important question is whether the changes in endosomal biology contribute significantly to pathogenesis in AD. Another is whether or not what is learned in AD in DS will translate to other types of AD. There is need for an open mind, but one can point to these findings: (1) impaired endosomal morphology and function is one of the early manifestations in both AD and DS humans and in mouse models 42 , 46 ; (2) APP, along with elevated β‐CTF, drives enlargement of early endosome and dysfunction of the endosome system 41 , 43 , 45 ; (3) enlarged endosomes convey trophic signals less effectively in vitro and in vivo 40 , 41 ; and (4) there is a close correlation between neurodegeneration and APP gene dose mediated increases in endosomal dysregulation. 40 , 41 Studies using siRNA in cell culture, in the Ts65Dn:App +/+/− mouse, and using APP/β‐CTF overexpression all support the contribution of APP gene dose in inducing early endosome phenotypes along with cholinergic degeneration. 40 , 41 Thus while increased APP gene dose may well have effects beyond changes in endosomes that contribute to degeneration, existing data argue strongly that the two phenomena are linked and deserve careful study, especially in view of the evidence that continuing neurotrophic support is required for maintenance of mature neurons, including neurons of the basal forebrain. 47 Indeed, we view the possibility that the cumulative effect over many years of reduced neurotrophin signaling contributes significantly to neurodegeneration. 42 Thus given the involvement of endosomes in both AD and AD‐DS we think the focus on endosomes may provide insights relevant to both disorders, although not necessarily with respect to the treatments used to address them.
4.3. When to intervene using APP‐directed treatments for AD‐DS
It is unknown when to target increased APP gene dose. Ideally, one would initiate treatment to prevent AD. But as for other genetic disorders of aging, it is unclear why increased APP gene expression requires more than 30 years to be expressed pathologically and more than 40 years to be expressed as dementia. Should one consider AD‐DS as a developmental disorder and treat beginning during childhood; conversely, should treatment be delayed until the onset of changes that can be identified as pathognomonic of the presence of AD pathogenesis? One may argue for either. For example, given endosomal changes already in the prenatal brain 38 it could be argued that treatment should begin in childhood or even prenatally. Although one can speculate that abnormal endosome size might induce changes in neuronal function in children, to our knowledge there is no evidence that increased APP gene dose is responsible for intellectual deficits during childhood when FAD is due to duplication of the APP gene locus. 140 In contrast, FAD due to mutations in PSENs may result in functional changes in affected children. 141 Additional clinical studies in those with APP duplications will be needed to indicate whether to target APP in DS during childhood. To intercept the onset of AD pathogenesis, the availability of increasingly informative AD biomarkers may prove helpful. 32 In addition, the discovery of biomarkers sensitive to endosomal dysfunction could inform as to the initiation of this feature of pathogenesis. Whether one treats in children or adults, studies of efficacy carried out over prolonged periods will require novel clinical trial designs and agents with very low or no toxicity.
4.4. Possible off‐target effects of APP‐targeting treatments
Small molecule inhibitors are prone to off‐target effects. Evidence for such effects is routinely sought in both preclinical and clinical studies. Herein we saw that Posiphen reduced α‐synuclein in both primary 2N and Ts65Dn neurons in vitro. Whether the effect on α‐synuclein in vitro was due to the same mechanism as for APP is uncertain, but this conclusion is supported by other findings. 54 , 142 , 143 Nevertheless, our finding of an effect in vitro but not in vivo raises the possibility that Posiphen may act by more than one mechanism. Limiting off‐target effects may be favored by treatment approaches inherently more specific, as can be practiced using ASOs and others targeting specific RNA sequences.
4.5. Reducing APP must be carefully regulated and may impact expression and actions of other genes and their products
The finding that increased APP levels are responsible for disease manifestations points to reducing APP to normal levels as possibly preventing or reducing these manifestations. To achieve this goal one will need to tailor treatments whose effect is normalization and not elimination of APP and its products. Dosing strategies and use of peripheral biomarker monitoring will support this goal—for example, the measurement of plasma Aβ levels. However, we do not know if adaptive cellular and genetic changes occur in response to increased APP and what impact reducing APP levels will have. Such changes can be forecast in preclinical studies, as was practiced in a limited fashion herein.
4.6. APP may not be the only chromosome 21 gene target to prevent AD in DS
It will be important to address the impact of other chromosome 21 genes in impacting the pathogenesis of AD‐DS. Genes of interest include: DYRK1A, whose multifold actions include effects on tau phosphorylation 144 , 145 ; and RCAN1 146 and SYNJ1 (the gene encoding Synaptojanin 1), 147 which play a role in membrane trafficking and transmission of neurotrophic signals. Of interest, increased BACE2 (the gene encoding β‐site Aβ precursor protein cleaving enzyme 2 [BACE2]) expression was recently reported to modulate APP processing to reduce the accumulation of Aβ in human organoid culture. 133
4.7. Using APP‐based treatments together with treatments directed at non‐APP targets
The ability to combine treatments may enable therapeutic interventions at different disease stages. For example, although very early intervention would target APP, in the context of advanced disease, the use of treatments targeting tau pathology and/or inflammation may be appropriate. At present there is no evident contraindication for concurrent use of APP‐directed and non–APP‐directed therapies, but combined treatments will require preclinical testing and careful monitoring in clinical trials.
5. LINKAGE TO OTHER MAJOR THEORIES
Our hypothesis interfaces with the amyloid cascade hypothesis by providing a novel context in which to consider it and a mechanistic basis by which to test it. Thus the cell biology of APP, the enzyme systems that mediate its processing, the actions of its products and the gene variants that create increased risk and modulate onset can now be viewed in the context of the role played by increased APP gene dose in AD‐DS. This invites a new attempt to integrate a role for APP and its products in the events that initiate and propagate pathogenesis over decades, leading to degeneration of neuronal structure and function.
In addition to the amyloid cascade hypothesis, 2 our synthesis allows for reconsideration of the “cholinergic hypothesis” and “deficient neurotrophic hypothesis.” The former speaks to the vulnerability of cholinergic neurons of the basal forebrain, 148 whereas the latter was motivated by the nearly concurrent discovery of central nervous system neurotrophic factors, including the discovery that NGF was one such factor and acted on basal forebrain cholinergic neurons, 47 and that disrupted NGF signaling plausibly contributed to the atrophy and death of cholinergic neurons.
An equally significant point of intersection is with FAD due to APP mutation and gene duplication and with mutations in PSEN1 and PSEN2. The processing of APP in endosomes places the protein and its products in precisely the compartment known to be dysregulated in studies in which increases in APP and β‐CTF lead to endosomal dysfunction including disrupted transport of NGF signaling in basal forebrain cholinergic neurons. 41 Strikingly, in mouse models of DS, reducing App gene dose partially rescues changes in endosomal transport of NGF and prevents degeneration of basal forebrain cholinergic neurons, thus linking App gene dose effects to both endosomal dysfunction and degeneration. 40 How changes in tau might intersect with the APP gene dose hypothesis is uncertain except to note that tau‐stabilized microtubules are the track upon which endosomes are transported. A more incisive role for tau in mediating neurodegeneration is now emerging 149 , 150 and we may discover other points at which the APP/endosome story interact with tau. Finally, inflammation mediated by microglia and astrocytes in AD pathogenesis may be in part explained by the Aβ products and perhaps other products of APP. Sustained microglial activation results in defective Aβ phagocytosis and sustained release of proinflammatory cytokines 1 ; other insights may emerge in which closer links between endosomal function and inflammation are forthcoming.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was performed strictly according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experiments involving the use of animals have been approved by the University of California San Diego Institutional Animal Care and Use Committee (Protocol#: S09315). All animals were maintained and bred according standard procedures. The best efforts were made to minimize the suffering of animals.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
X.‐Q. C. and W.C.M. designed the studies. X.‐Q.C., A.S., M.L.P., C.O., P.D.N., and A.M.K performed the experiments and analyzed the data. X.‐Q. C., M.M., and W.C.M. wrote the article. All authors reviewed the article.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We thank all the Mobley lab members for the helpful comments and fruitful discussions. We thank Dr. Moses Chao (New York University) for providing pTrkB antibody, and Dr. Peter Davies (Albert Einstein University) for PHF1 antibody.
Chen X‐Q, Salehi A, Pearn ML, et al. Targeting increased levels of APP in Down syndrome: Posiphen‐mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimer's Dement. 2021;17:271–292. 10.1002/alz.12185
[Correction added on September 28, 2020, after first online publication: The copyright line and license information for this article were updated.]
Contributor Information
Xu‐Qiao Chen, Email: q0chen@ucsd.edu.
William C. Mobley, Email: wmobley@ucsd.edu.
REFERENCES
- 1. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 2018;4:575‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 2016;8(6):595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shafiei SS, Guerrero‐Munoz MJ, Castillo‐Carranza DL. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Dyck CH. Anti‐Amyloid‐beta monoclonal antibodies for Alzheimer's disease: pitfalls and promise. Biol. Psychiatry. 2018;83(4):311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Medina M. An overview on the clinical development of tau‐based therapeutics. Int. J. Mol. Sci. 2018;19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar D, Ganeshpurkar A, Kumar D, Modi G, Gupta SK, Singh SK. Secretase inhibitors for the treatment of Alzheimer's disease: long road ahead. Eur. J. Med. Chem. 2018;148:436‐452. [DOI] [PubMed] [Google Scholar]
- 7. Selkoe DJ. Alzheimer disease and aducanumab: adjusting our approach. Nat. Rev. Neurol. 2019;15(7):365‐366. [DOI] [PubMed] [Google Scholar]
- 8. Ho D, Quake SR, McCabe ERB, et al. Enabling technologies for personalized and precision medicine. Trends Biotechnol. 2020;38(5):497‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gardiner K, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003;318:137‐147. [DOI] [PubMed] [Google Scholar]
- 10. Antonarakis SE, Skotko BG, Rafii MS, et al. Down syndrome. Nat Rev Dis Primers. 2020;6(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Korbel JO, Tirosh‐Wagner T, Urban AE, et al. The genetic architecture of Down syndrome phenotypes revealed by high‐resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U. S. A. 2009;106(29):12031‐12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Doran E, Keator D, Head E, et al. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer's Disease: the Role of APP. J Alzheimers Dis. 2017;56(2):459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prasher VP, Farrer MJ, Kessling AM, et al. Molecular mapping of Alzheimer‐type dementia in Down's syndrome. Ann. Neurol. 1998;43(3):380‐383.9506555 [Google Scholar]
- 14. Scheltens P, Blennow K, Breteler MM, et al. Alzheimer's disease. Lancet. 2016;388(10043):505‐517. [DOI] [PubMed] [Google Scholar]
- 15. Dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M, et al. Alzheimer's disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018;25(26):3141‐3159. [DOI] [PubMed] [Google Scholar]
- 16. Apostolova LG. Alzheimer Disease. Continuum. 2016;22(2 Dementia):419‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W. Molecular basis of familial and sporadic Alzheimer's disease. Curr. Alzheimer Res. 2016;13(9):952‐963. [DOI] [PubMed] [Google Scholar]
- 18. Shea YF, Chu LW, Chan AO, Ha J, Li Y, Song YQ. A systematic review of familial Alzheimer's disease: differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formosan Med. Assoc. 2016;115(2):67‐75. [DOI] [PubMed] [Google Scholar]
- 19. Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther. 2011;3(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry. 2015;77(1):43‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bertram L, Tanzi RE. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019;15(4):191‐192. [DOI] [PubMed] [Google Scholar]
- 22. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921‐923. [DOI] [PubMed] [Google Scholar]
- 23. Rafii MS, Kleschevnikov AM, Sawa M, Mobley WC. Down syndrome. Handb. Clin. Neurol. 2019;167:321‐336. [DOI] [PubMed] [Google Scholar]
- 24. Saenz RB. Primary care of infants and young children with Down syndrome. Am. Fam. Physician. 1999;59(2):381‐390. 392, 395‐386. [PubMed] [Google Scholar]
- 25. Ballard C, Mobley W, Hardy J, Williams G, Corbett A. Dementia in Down's syndrome. Lancet Neurol. 2016;15(6):622‐636. [DOI] [PubMed] [Google Scholar]
- 26. Wiseman FK, Al‐Janabi T, Hardy J, et al. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16(9):564‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen XQ, Mobley WC. Alzheimer disease pathogenesis: insights from molecular and cellular biology studies of oligomeric abeta and tau species. Front. Neurosci. 2019;13:659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hyman BT, West HL, Rebeck GW, Lai F, Mann DM. Neuropathological changes in Down's syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol. 1995;52(4):373‐378. [DOI] [PubMed] [Google Scholar]
- 29. Schupf N, Kapell D, Lee JH, et al. Onset of dementia is associated with apolipoprotein E epsilon4 in Down's syndrome. Ann. Neurol. 1996;40(5):799‐801. [DOI] [PubMed] [Google Scholar]
- 30. Rafii MS, Lukic AS, Andrews RD, et al. PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down syndrome: results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis. 2017;60(2):439‐450. [DOI] [PubMed] [Google Scholar]
- 31. Mak E, Padilla C, Annus T, et al. Delineating the topography of amyloid‐associated cortical atrophy in Down syndrome. Neurobiol. Aging. 2019;80:196‐202. [DOI] [PubMed] [Google Scholar]
- 32. Fortea J, Carmona‐Iragui M, Benejam B, et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet Neurol. 2018;17(10):860‐869. [DOI] [PubMed] [Google Scholar]
- 33. Matthews DC, Lukic AS, Andrews RD, et al. Dissociation of Down syndrome and Alzheimer's disease effects with imaging. Alzheimers Dement. 2016;2(2):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 1985;82(12):4245‐4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology. 1985;35(7):957‐961. [DOI] [PubMed] [Google Scholar]
- 36. Hyman BT. Down syndrome and Alzheimer disease. Prog. Clin. Biol. Res. 1992;379:123‐142. [PubMed] [Google Scholar]
- 37. Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid‐beta oligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64(s1):S567‐S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cataldo AM, Peterhoff CM, Troncoso JC, Gomez‐Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157(1):277‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Megarbane A, Ravel A, Mircher C, et al. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet Med. 2009;11(9):611‐616. [DOI] [PubMed] [Google Scholar]
- 40. Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29‐42. [DOI] [PubMed] [Google Scholar]
- 41. Xu W, Weissmiller AM. Amyloid precursor protein‐mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016;126(5):1815‐1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen XQ, Mobley WC. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: converging insights from alternative hypotheses. Front. Neurosci. 2019;13:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim S, Sato Y, Mohan PS, et al. Evidence that the rab5 effector APPL1 mediates APP‐betaCTF‐induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol. Psychiatry. 2016;21(5):707‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lauritzen I, Pardossi‐Piquard R, Bourgeois A, et al. Intraneuronal aggregation of the beta‐CTF fragment of APP (C99) induces Abeta‐independent lysosomal‐autophagic pathology. Acta Neuropathol. 2016;132(2):257‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer's‐related endosome dysfunction in Down syndrome is Abeta‐independent but requires APP and is reversed by BACE‐1 inhibition. Proc. Natl. Acad. Sci. U. S. A. 2010;107(4):1630‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nixon RA. Amyloid precursor protein and endosomal‐lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017;31(7):2729‐2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen XQ, Sawa M, Mobley WC. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radical Biol. Med. 2018;114:52‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA. Dysfunction of autophagy and endosomal‐lysosomal pathways: roles in pathogenesis of Down syndrome and Alzheimer's disease. Free Radical Biol. Med. 2018;114:40‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chavez‐Gutierrez L, Bammens L, Benilova I, et al. The mechanism of gamma‐Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31(10):2261‐2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen J. The presenilin hypothesis of Alzheimer's disease: evidence for a loss‐of‐function pathogenic mechanism. Proc. Natl. Acad. Sci. U. S. A. 2007;104(2):403‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwart D, Gregg A, Scheckel C, et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP beta‐CTFs, not abeta. Neuron. 2019;104(2):256‐270. e255. [DOI] [PubMed] [Google Scholar]
- 52. Greig NH, Sambamurti K, Yu QS, Brossi A, Bruinsma GB, Lahiri DK. An overview of phenserine tartrate, a novel acetylcholinesterase inhibitor for the treatment of Alzheimer's disease. Curr. Alzheimer Res. 2005;2(3):281‐290. [DOI] [PubMed] [Google Scholar]
- 53. Lahiri DK, Chen D, Maloney B, et al. The experimental Alzheimer's disease drug posiphen [(+)‐phenserine] lowers amyloid‐beta peptide levels in cell culture and mice. J Pharmacol Exp Ther. 2007;320(1):386‐396. [DOI] [PubMed] [Google Scholar]
- 54. Mikkilineni S, Cantuti‐Castelvetri I, Cahill CM, Balliedier A, Greig NH, Rogers JT. The anticholinesterase phenserine and its enantiomer posiphen as 5'untranslated‐region‐directed translation blockers of the Parkinson's alpha synuclein expression. Parkinsons Dis. 2012;2012:142372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lilja AM, Rojdner J, Mustafiz T, et al. Age‐dependent neuroplasticity mechanisms in Alzheimer Tg2576 mice following modulation of brain amyloid‐beta levels. PLoS One. 2013;8(3):e58752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lilja AM, Luo Y, Yu QS, et al. Neurotrophic and neuroprotective actions of (‐)‐ and (+)‐phenserine, candidate drugs for Alzheimer's disease. PLoS One. 2013;8(1):e54887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marutle A, Ohmitsu M, Nilbratt M, Greig NH, Nordberg A, Sugaya K. Modulation of human neural stem cell differentiation in Alzheimer (APP23) transgenic mice by phenserine. Proc. Natl. Acad. Sci. U. S. A. 2007;104(30):12506‐12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Teich AF, Sharma E, Barnwell E, et al. Translational inhibition of APP by Posiphen: efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement (N Y). 2018;4:37‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid‐beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2012;83(9):894‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rogers JT, Randall JD, Cahill CM, et al. An iron‐responsive element type II in the 5'‐untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277(47):45518‐45528. [DOI] [PubMed] [Google Scholar]
- 61. Rogers JT, Xia N, Wong A, Bakshi R, Cahill CM. Targeting the iron‐response elements of the mRNAs for the Alzheimer's amyloid precursor protein and ferritin to treat acute lead and manganese neurotoxicity. Int. J. Mol. Sci. 2019;20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou ZD, Tan EK. Iron regulatory protein (IRP)‐iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017;12(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cho HH, Cahill CM, Vanderburg CR, et al. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein‐1. J Biol Chem. 2010;285(41):31217‐31232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anderson CP, Shen M, Eisenstein RS, Leibold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta. 2012;1823(9):1468‐1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res. 1990;360:263‐280. [PubMed] [Google Scholar]
- 66. Davisson MT, Schmidt C, Reeves RH, et al. Segmental trisomy as a mouse model for Down syndrome. Prog Clin Biol Res. 1993;384:117‐133. [PubMed] [Google Scholar]
- 67. Gupta M, Dhanasekaran AR, Gardiner KJ. Mouse models of Down syndrome: gene content and consequences. Mamm Genome. 2016;27(11‐12):538‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Salehi A, Faizi M, Colas D, et al. Restoration of norepinephrine‐modulated contextual memory in a mouse model of Down syndrome. Sci Transl Med. 2009;1(7):7ra17. [DOI] [PubMed] [Google Scholar]
- 69. Harrington AW, Ginty DD. Long‐distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci. 2013;14(3):177‐187. [DOI] [PubMed] [Google Scholar]
- 70. Nosheny RL, Belichenko PV, Busse BL, et al. Increased cortical synaptic activation of TrkB and downstream signaling markers in a mouse model of Down syndrome. Neurobiol Dis. 2015;77:173‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao X, Chen XQ, Han E, et al. TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington's disease. Proc. Natl. Acad. Sci. U. S. A. 2016;113(38):E5655‐5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen XQ, Fang F, Florio JB, et al. T‐complex protein 1‐ring complex enhances retrograde axonal transport by modulating tau phosphorylation. Traffic. 2018;19(11):840‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Banks WA. Drug delivery to the brain in Alzheimer's disease: consideration of the blood‐brain barrier. Adv Drug Deliv Rev. 2012;64(7):629‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuo YM, Nwankwo EI, Nussbaum RL, Rogers J, Maccecchini ML. Translational inhibition of alpha‐synuclein by Posiphen normalizes distal colon motility in transgenic Parkinson mice. Am J Neurodegener Dis. 2019;8(1):1‐15. [PMC free article] [PubMed] [Google Scholar]
- 75. Kounnas MZ, Danks AM, Cheng S, et al. Modulation of gamma‐secretase reduces beta‐amyloid deposition in a transgenic mouse model of Alzheimer's disease. Neuron. 2010;67(5):769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fuentes JJ, Genesca L, Kingsbury TJ, et al. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin‐mediated signaling pathways. Hum Mol Genet. 2000;9(11):1681‐1690. [DOI] [PubMed] [Google Scholar]
- 77. Lloret A, Badia MC, Giraldo E, et al. Amyloid‐beta toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer's disease. J Alzheimers Dis. 2011;27(4):701‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra‐axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10(2):149‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kazim SF, Blanchard J, Bianchi R, Iqbal K. Early neurotrophic pharmacotherapy rescues developmental delay and Alzheimer's‐like memory deficits in the Ts65Dn mouse model of Down syndrome. Sci Rep. 2017;7:45561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Giordano T, Pan JB, Casuto D, Watanabe S, Arneric SP. Thyroid hormone regulation of NGF, NT‐3 and BDNF RNA in the adult rat brain. Brain Res Mol Brain Res. 1992;16(3‐4):239‐245. [DOI] [PubMed] [Google Scholar]
- 81. Li Y, Holtzman DM, Kromer LF, et al. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15(4):2888‐2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain. 2018;141(7):1917‐1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ferreira‐Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. Alzheimer's disease: targeting the cholinergic system. Curr Neuropharmacol. 2016;14(1):101‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Seibenhener ML, Wooten MC. Use of the open field maze to measure locomotor and anxiety‐like behavior in mice. J Vis Exp. 2015(96):e52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kleschevnikov AM, Yu J, Kim J, et al. Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2017;103:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Belichenko PV, Kleschevnikov AM, Becker A, et al. Down syndrome cognitive phenotypes modeled in mice trisomic for all HSA 21 homologues. PLoS One. 2015;10(7):e0134861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lysenko LV, Kim J, Henry C, et al. Monoacylglycerol lipase inhibitor JZL184 improves behavior and neural properties in Ts65Dn mice, a model of down syndrome. PLoS One. 2014;9(12):e114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1(3):1117‐1119. [DOI] [PubMed] [Google Scholar]
- 89. Heller HC, Salehi A, Chuluun B, et al. Nest building is impaired in the Ts65Dn mouse model of Down syndrome and rescued by blocking 5HT2a receptors. Neurobiol Learn Mem. 2014;116:162‐171. [DOI] [PubMed] [Google Scholar]
- 90. Faizi M, Bader PL, Tun C, et al. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down syndrome: activation of beta1‐adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol Dis. 2011;43(2):397‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Salehi A, Faizi M, Belichenko PV, Mobley WC. Using mouse models to explore genotype‐phenotype relationship in Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(3):207‐214. [DOI] [PubMed] [Google Scholar]
- 92. Kleschevnikov AM, Belichenko PV, Faizi M, et al. Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J Neurosci. 2012;32(27):9217‐9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10(4):411‐413. [DOI] [PubMed] [Google Scholar]
- 94. Delcroix JD, Valletta J, Wu C, et al. Trafficking the NGF signal: implications for normal and degenerating neurons. Prog. Brain Res. 2004;146:3‐23. [DOI] [PubMed] [Google Scholar]
- 95. Kwart D, Gregg A, Scheckel C, et al. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP beta‐CTFs, not abeta. Neuron. 2019. [DOI] [PubMed] [Google Scholar]
- 96. Cataldo AM, Petanceska S, Terio NB, et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25(10):1263‐1272. [DOI] [PubMed] [Google Scholar]
- 97. Niederst ED, Reyna SM, Goldstein LS. Axonal amyloid precursor protein and its fragments undergo somatodendritic endocytosis and processing. Mol Biol Cell. 2015;26(2):205‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid‐beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009;29(11):3565‐3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Woodruff G, Reyna SM, Dunlap M, et al. Defective transcytosis of APP and lipoproteins in human iPSC‐Derived neurons with familial Alzheimer's disease mutations. Cell Rep. 2016;17(3):759‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rodrigues EM, Weissmiller AM, Goldstein LS. Enhanced beta‐secretase processing alters APP axonal transport and leads to axonal defects. Hum Mol Genet. 2012;21(21):4587‐4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kamal A, Almenar‐Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin‐mediated axonal transport of a membrane compartment containing beta‐secretase and presenilin‐1 requires APP. Nature. 2001;414(6864):643‐648. [DOI] [PubMed] [Google Scholar]
- 102. Cirrito JR, Kang JE, Lee J, et al. Endocytosis is required for synaptic activity‐dependent release of amyloid‐beta in vivo. Neuron. 2008;58(1):42‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid‐beta peptide oligomers disrupt axonal transport through an NMDA receptor‐dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30(27):9166‐9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ramser EM, Gan KJ, Decker H, et al. Amyloid‐beta oligomers induce tau‐independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neurons. Mol Biol Cell. 2013;24(16):2494‐2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Siddiqui A, Lacroix T, Stasko MR, Scott‐McKean JJ, Costa AC, Gardiner KJ. Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK‐801. Genes Brain Behav. 2008;7(7):810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hunter MP, Nelson M, Kurzer M, et al. Intersectin 1 contributes to phenotypes in vivo: implications for Down's syndrome. Neuroreport. 2011;22(15):767‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Malakooti N, Pritchard MA, Adlard PA, Finkelstein DI. Role of metal ions in the cognitive decline of Down syndrome. Front Aging Neurosci. 2014;6:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Leveugle B, Spik G, Perl DP, Bouras C, Fillit HM, Hof PR. The iron‐binding protein lactotransferrin is present in pathologic lesions in a variety of neurodegenerative disorders: a comparative immunohistochemical analysis. Brain Res. 1994;650(1):20‐31. [DOI] [PubMed] [Google Scholar]
- 109. Loeffler DA, Connor JR, Juneau PL, et al. Transferrin and iron in normal, Alzheimer's disease, and Parkinson's disease brain regions. J Neurochem. 1995;65(2):710‐724. [DOI] [PubMed] [Google Scholar]
- 110. van Duijn S, Bulk M, van Duinen SG, et al. Cortical iron reflects severity of Alzheimer's disease. J Alzheimers Dis. 2017;60(4):1533‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wong KA, Wilson J, Russo A, et al. Intersectin (ITSN) family of scaffolds function as molecular hubs in protein interaction networks. PLoS One. 2012;7(4):e36023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Long JM, Maloney B, Rogers JT, Lahiri DK. Novel upregulation of amyloid‐beta precursor protein (APP) by microRNA‐346 via targeting of APP mRNA 5'‐untranslated region: implications in Alzheimer's disease. Mol Psychiatry. 2019;24(3):345‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. 2014;5:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Han C, Liu Y, Dai R, Ismail N, Su W, Li B. Ferroptosis and its potential role in human diseases. Front Pharmacol. 2020;11:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Erickson MA, Niehoff ML, Farr SA, et al. Peripheral administration of antisense oligonucleotides targeting the amyloid‐beta protein precursor reverses AbetaPP and LRP‐1 overexpression in the aged SAMP8 mouse brain. J Alzheimers Dis. 2012;28(4):951‐960. [DOI] [PubMed] [Google Scholar]
- 116. Kumar VB, Farr SA, Flood JF, et al. Site‐directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21(12):1769‐1775. [DOI] [PubMed] [Google Scholar]
- 117. Farr SA, Erickson MA, Niehoff ML, Banks WA, Morley JE. Central and peripheral administration of antisense oligonucleotide targeting amyloid‐beta protein precursor improves learning and memory and reduces neuroinflammatory cytokines in Tg2576 (AbetaPPswe) mice. J Alzheimers Dis. 2014;40(4):1005‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Schonrock N, Matamales M, Ittner LM, Gotz J. MicroRNA networks surrounding APP and amyloid‐beta metabolism–implications for Alzheimer's disease. Exp Neurol. 2012;235(2):447‐454. [DOI] [PubMed] [Google Scholar]
- 119. Chopra Nipun, Wang Ruizhi, Maloney Bryan, Nho Kwangsik, Beck John S., Pourshafie Naemeh, Niculescu Alexander, Saykin Andrew J., Rinaldi Carlo, Counts Scott E., Lahiri Debomoy K. (2020) MicroRNA‐298 reduces levels of human amyloid‐β precursor protein (APP), β‐site APP‐converting enzyme 1 (BACE1) and specific tau protein moieties. Molecular Psychiatry, 10.1038/s41380-019-0610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wagner SL, Rynearson KD, Duddy SK, et al. Pharmacological and toxicological properties of the potent oral gamma‐Secretase Modulator BPN‐15606. J Pharmacol Exp Ther. 2017;362(1):31‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2018. Alzheimers Dement. 2018;4:195‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Muller MP, Goody RS. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases. 2018;9(1‐2):5‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Myers A, McGonigle P. Overview of transgenic mouse models for Alzheimer's disease. Curr. Protoc. Neurosci. 2019;89(1):e81. [DOI] [PubMed] [Google Scholar]
- 124. Mullane K, Williams M. Preclinical Models of Alzheimer's Disease: relevance and Translational Validity. Curr. Protoc. Pharmacol. 2019;84(1):e57. [DOI] [PubMed] [Google Scholar]
- 125. Oddo S, Caccamo A, Shepherd JD, et al. Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409‐421. [DOI] [PubMed] [Google Scholar]
- 126. Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487‐1491. [DOI] [PubMed] [Google Scholar]
- 127. Saito T, Matsuba Y, Mihira N, et al. Single App knock‐in mouse models of Alzheimer's disease. Nat Neurosci. 2014;17(5):661‐663. [DOI] [PubMed] [Google Scholar]
- 128. Di Domenico F, Tramutola A, Barone E, et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: focus on HNE‐modified proteins in a mouse model of Down syndrome. Redox Biol. 2019;23:101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann. Neurol. 2004;56(5):675‐688. [DOI] [PubMed] [Google Scholar]
- 130. Yu T, Li Z, Jia Z, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19(14):2780‐2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sadleir KR, Kandalepas PC, Buggia‐Prevot V, Nicholson DA, Thinakaran G, Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer's disease. Acta Neuropathol. 2016;132(2):235‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kazuki Y, Gao FJ, Li Y, et al. A non‐mosaic transchromosomic mouse model of down syndrome carrying the long arm of human chromosome 21. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Alić Ivan, Goh Pollyanna A., Murray Aoife, Portelius Erik, Gkanatsiou Eleni, Gough Gillian, Mok Kin Y., Koschut David, Brunmeir Reinhard, Yeap Yee Jie, O’Brien Niamh L., Groet Jürgen, Shao Xiaowei, Havlicek Steven, Dunn N. Ray, Kvartsberg Hlin, Brinkmalm Gunnar, Hithersay Rosalyn, Startin Carla, Hamburg Sarah, Phillips Margaret, Pervushin Konstantin, Turmaine Mark, Wallon David, Rovelet‐Lecrux Anne, Soininen Hilkka, Volpi Emanuela, Martin Joanne E., Foo Jia Nee, Becker David L., Rostagno Agueda, Ghiso Jorge, Krsnik Željka, Šimić Goran, Kostović Ivica, Mitrečić Dinko, Francis Paul T., Blennow Kaj, Strydom Andre, Hardy John, Zetterberg Henrik, Nižetić Dean (2020) Patient‐specific Alzheimer‐like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose‐sensitive AD suppressor in human brain. Molecular Psychiatry, 10.1038/s41380-020-0806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329‐2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Choi SH, Kim YH, Hebisch M, et al. A three‐dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014;515(7526):274‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xu R, Brawner AT, Li S, et al. OLIG2 drives abnormal neurodevelopmental phenotypes in human iPSC‐Based organoid and chimeric mouse models of Down syndrome. Cell Stem Cell. 2019;24(6):908‐926. e908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Real R, Peter M, Trabalza A, et al. In vivo modeling of human neuron dynamics and Down syndrome. Science. 2018;362(6416). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Paterson RW, Gabelle A, Lucey BP, et al. SILK studies ‐ capturing the turnover of proteins linked to neurodegenerative diseases. Nat Rev Neurol. 2019;15(7):419‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sleegers K, Brouwers N, Gijselinck I, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129(Pt 11):2977‐2983. [DOI] [PubMed] [Google Scholar]
- 140. Rovelet‐Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early‐onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24‐26. [DOI] [PubMed] [Google Scholar]
- 141. Quiroz YT, Schultz AP, Chen K, et al. Brain imaging and blood biomarker abnormalities in children with autosomal dominant Alzheimer disease: a cross‐sectional study. JAMA Neurol. 2015;72(8):912‐919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Olivares D, Huang X, Branden L, Greig NH, Rogers JT. Physiological and pathological role of alpha‐synuclein in Parkinson's disease through iron mediated oxidative stress; the role of a putative iron‐responsive element. Int J Mol Sci. 2009;10(3):1226‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rogers JT, Mikkilineni S, Cantuti‐Castelvetri I, et al. The alpha‐synuclein 5'untranslated region targeted translation blockers: anti‐alpha synuclein efficacy of cardiac glycosides and Posiphen. J Neural Transm (Vienna). 2011;118(3):493‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Ryoo SR, Jeong HK, Radnaabazar C, et al. DYRK1A‐mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;282(48):34850‐34857. [DOI] [PubMed] [Google Scholar]
- 145. Garcia‐Cerro S, Martinez P, Vidal V, et al. Overexpression of Dyrk1A is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of Down syndrome. PLoS One. 2014;9(9):e106572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Patel A, Yamashita N, Ascano M, et al. RCAN1 links impaired neurotrophin trafficking to aberrant development of the sympathetic nervous system in Down syndrome. Nat Commun. 2015;6:10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Cremona O, Di Paolo G, Wenk MR, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99(2):179‐188. [DOI] [PubMed] [Google Scholar]
- 148. Bartus RT, Dean RL, 3rd BeerB, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408‐414. [DOI] [PubMed] [Google Scholar]
- 149. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5‐21. [DOI] [PubMed] [Google Scholar]
- 150. Morris M, Knudsen GM, Maeda S, et al. Tau post‐translational modifications in wild‐type and human amyloid precursor protein transgenic mice. Nat Neurosci. 2015;18(8):1183‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information