

Abstract
The amyloid precursor protein (APP) is a transmembrane protein mostly found in neurons. Cleavage of this protein by β-secretase can lead to the formation of amyloid-β (Aβ) peptide plaque, which leads to Alzheimer’s disease. Genomic analysis of an Icelandic population that did not show symptoms of Alzheimer’s at an advanced age led to the discovery of the A673T mutation. This mutation can reduce β-secretase cleavage by 40%. We hypothesized that the insertion of this mutation in patients’ neurons could be an effective and sustainable method of slowing down or even stopping the progression of Alzheimer’s disease. We modified the APP gene in HEK293T cells and in SH-SY5Y neuroblastoma using a Cas9 nickase (Cas9n)-deaminase enzyme to convert the alanine codon to a threonine. Several Cas9n-deaminase variants were tested to compare their efficiency of conversion. The results were characterized and quantified by deep sequencing. We successfully introduced the A673T mutation in 53% of HEK293T cells alongside a new mutation (E674K), which seemed to further reduce Aβ peptide accumulation. Our approach aimed to provide a new strategy for the treatment of Alzheimer’s and in so doing, demonstrate the capacity of base editing techniques for treating genetic diseases.
Keywords: CRISPR/Cas9, Alzheimer's disease, A673T, Islandic mutation, FAD, Base editing
Graphical abstract
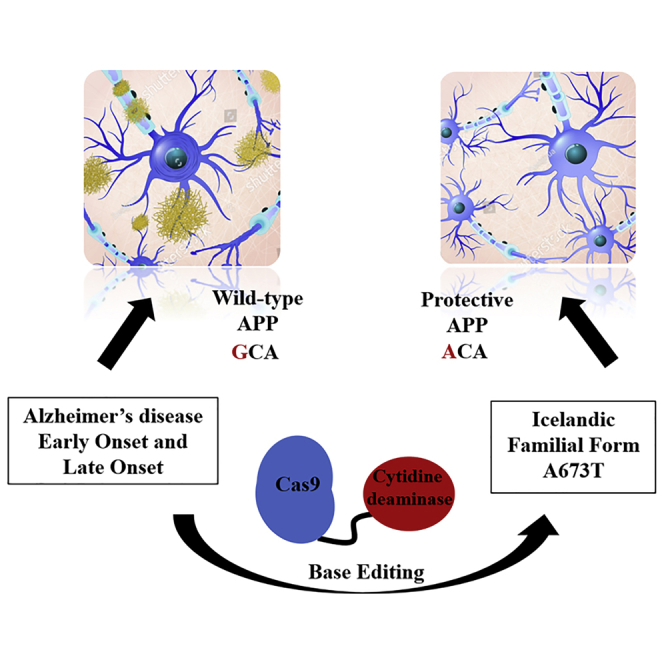
The A673T mutation is associated with neuroprotection against Aβ peptide secretion, leading to plaque buildup in Alzheimer’s disease. Base editing allows this mutation to be introduced into the human cell genome and has shown a decrease in Aβ peptides in vitro.
Introduction
Alzheimer’s disease (AD) is one of the most well-known diseases in the world due to its prevalence and the lack of effective treatment options. It is responsible for 70% of the reported cases of dementia worldwide. There are currently about 47.5 million cases with an estimated progression to 75.6 million in 2030, according to the World Health Organization.1,2 Patients experience memory loss and temporal and spatial confusion and have difficulty planning tasks.3 This eventually results in the deaths of patients.4 This disease is the result of the accumulation of amyloid plaques between neurons.
These plaques are generally the result of the excessive cleavage of the amyloid precursor protein (APP) by β-secretase 1 (BACE1). APP is a membrane protein that is preferentially cleaved by α-secretase. When the protein is cleaved by BACE1, amyloid-β (Aβ) peptides are formed. High concentrations of these peptides promote the formation of aggregates, which accumulate as plaques between neurons in the brains of patients.5,6
Numerous mutations in the APP gene have been shown to favor cleavage of the protein by BACE. They are known as familial AD (FAD) mutations, since they favor the development of AD. All APP FAD mutations are located between exons 16 and 17 where the protein cleavage sites are located. The majority of these FADs are named based on where in the world they were found, as well as the amino acid mutation for which they are responsible (e.g., London V717I mutation).7,8 Recently, the Icelandic APP mutation (A673T) was discovered in a Scandinavian population.9 Individuals carrying this mutation present almost no accumulation of Aβ peptides in their brains even at the age of 95. This mutation reduces BACE1 cleavage in the APP by 40% and reduces the aggregation of β-peptides.9,10 The mutation of the alanine codon into a threonine is due to a single base pair (bp) mutation in exon 16. Moreover, the A673T mutation has been linked to a greater life expectancy, as carriers of this mutation are 50% more likely to reach the age of 85 when compared to non-carriers. Further substantiating this claim, Kero et al.11 identified a 104-year-old person carrying the A637T gene who later died with little Aβ pathology.
This suggests that the introduction of the A673T mutation in the neurons of AD patients and pre-AD individuals could be beneficial in treating the disease. To accomplish this, we are proposing the use of base editing technology.12,13 Base editors are a new type of fusion protein that uses a cytidine deaminase (CDA) enzyme, an amino acid linker sequence ranging from 3 to 21 amino acids, and a CRISPR-associated protein nickase (Cas9n; containing a D10A mutation to prevent the cut of one DNA strand). This technology was based on CRISPR. CRISPR evolved in bacteria, such as Streptococcus pyogenes (Sp)14 and Staphylococcus aureus (Sa),15 as an adaptive immune response to resist bacteriophages. Recently, researchers worldwide have successfully adapted this system to modify or remove specific nucleotides. The original CRISPR system used a Cas and a single guide RNA (sgRNA) to selectively cleave a specific nucleic acid sequence.16 The Cas9n in base editors also uses a sgRNA; however, it is only able to introduce single-stranded breaks in the DNA. After binding to the protospacer adjacent motif (PAM) and confirming the sequence with the sgRNA, the Cas9n nicks the DNA and liberates an R-loop. This exposes a small section of single-stranded genomic DNA, and some cytosines within a deamination window at 12 to 17 nucleotides from the PAM (between the 4th and 8th nucleotides upstream of the guide) are preferentially modified by the cytosine deaminase (APOBEC1) and converted to uracil, forming a U:G pair of nucleotides.12 The cell then engages its base excision repair mechanism to solve the U:G mismatch. This is initiated by the excision of the uracil by uracil N-glycosylase (UNG). The uracil glycosylase inhibitor (UGI) that is fused to the C-terminal of the Cas9n protects this intermediate and increases the conversion of the U:G to U:A and finally T:A by 50%.12 The final result is a fusion protein that allows for the direct, programmable, and irreversible conversion of a C:G bp into T:A pair without directly inducing indels. The combination of these elements forms the complex BE3 (Base Editing version 3) (Figure 1). This technique was subsequently improved by Kim, Komor and coworkers17, 18, 19 with the release of the Base Editing version 4 (BE4) system. Another frequently used base editor is Target-activation-induced CDA (AID), which uses a different lamprey-derived deaminase (PmCDA1). This base editor has a different deamination window (between the 2nd and 6th nucleotides upstream of the guide) and demonstrates a different efficacy depending on the DNA sequence targeted13,20 (Figure 1).
Figure 1.

Structures of BE3, BE4, and Target-AID variants
Various cytidine deaminases (CDAs; APOBEC1, YE1_APOBEC1, or Target-AID [CDA1]) were fused with various Cas9 nickases (Cas9ns; SpCas9nVQR, SaCas9nKKH, SpCas9nEQR).
Since they were invented, base editors have evolved at a phenomenal speed and have already been used in research, medicine, and even agriculture. Base editors could theoretically correct about 60% of all existing genetic diseases to date.21 An impediment to this, however, is their delivery to the diseased cell in vivo. Their large size precludes them from simple packaging in adeno-associated viruses (AAVs) or other common viral vectors. For the moment, they are primarily being used to create cell or animal models. Base editing was used in agriculture to give resistance to a pesticide on a variety of rice. The most notable medical applications were, among others, the correction of the p53 gene involved in breast cancer or the modification of the risk factor of Alzheimer APOE4 into a safer one.12,22
Overall, this system is limited in its scope due to the Cas9 protein’s requirement for a PAM. For the Cas9 to target a designated sequence, it must first bind to an invariable, pre-defined set of nucleotides.23 Only after the Cas9 interacts with its PAM can its guide bind.24,25 After the sgRNA confirms that it has bound to the right sequence, the protein can induce a double-strand break (DSB). As a result, researchers are often limited in their selection of their Cas9 protein based on the presence of a specific PAM. Since the base editing system relies on a Cas9 for targeting, it is likewise constrained by the presence of an adequate PAM.
In the present study, we used the CRISPR-Cas9 base editing method to mutate the APP gene, allowing a seamless and efficient A673T editing in human cells. We tested several Cas9/deaminase variants in order to find the most efficient enzyme to induce the Icelandic mutation.
Results
Deaminase design
BE3_SpCas9n (henceforth designated as BE3) was the original SpCas9n-deaminase made by Komor et al.12 (Figure 1). This fusion protein contained three previously described protein sequences: the APOBEC1 deaminase, a 16-amino acids linker; the SpCas9n, a second 4-amino acids linker; and the UGI. This version of the enzyme recognized a 5′-NGG-3′ PAM sequence. Unfortunately, there is no such 5′-NGG-3′ motive near codon 673 in exon 16 of the APP gene (Figure 2).
Figure 2.

Amino acid modifications produced by base editing of the various cytidines
(A) Part of exon 16 of the wild-type APP (APPWT) gene is illustrated. In the antisense strand, there are five cytidines in red (C1 to C5), which are potentially in the editing window of each base editor. The 673 alanine codon is in green. (B) The anticipated APP gene after all five cytidines in the targeting window is deaminated, resulting in the change of four amino acids. Note the C5 nucleotide is a silent mutation that does not change the resulting amino acid. (C) The sequence of the APP gene when only cytidine C2 has been deaminated to create the A673T mutation (in blue).
As such, it was necessary to use a new base editor to induce the cytosine to thymine conversion required for the creation of the A673T mutation. Our selection was comprised of the different variants available on Addgene at the beginning of this study.
Mutant SpCas9 nucleases have previously been produced to react with alternative PAMs.26 Of them, Cas9nVQR and Cas9nEQR were selected for the purposes of this study, The VQR variant has been shown to robustly bind to sequences with 5′-NGAN-3′ PAMs, whereas the EQR variant is more specific and thus anneals to an 5′-NGAG-3′ PAM. These mutant Cas9 molecules were used as a base to create the following base editors: BE3_SpCas9nVQR, BE3_SpCas9nEQR, BE3_SaCas9nKKH, BE4-Gam_SpCas9n, YE1-BE3_SpCas9n, and Target-AID SpCas9n (purchased from Addgene). BE4-Gam_SpCas9nVQR, BE4-Gam_SpCas9nEQR, YE1-BE3_SpCas9nVQR, YE1-BE3_SpCas9nEQR, YE1-BE3_SaCas9nKKH, Target-AID SpCas9nVQR, Target-AID SpCas9nEQR, and Target-AID SaCas9nKKH were made in our laboratory. Several sgRNA lengths, from 17 bp to 22 bp, were tested in order to influence the deamination window (list available in Table S1). One or two sgRNA copies were inserted in a modified pBSU6 plasmid.
As shown in Figure 2, the antisense DNA strand was targeted using three different BE3s. These base editors differed in their Cas9n enzymes, with each possessing different PAMs: 5′-NGAN-3′ (for SpCas9nVQR),26 5′-NGAG-3′ (for SpCas9nEQR),26 or 5′-AGAGAT-3′ (for SaCas9nKKH).27 This was done to induce the deamination of the cytidine in position 2 (C2) in the editing window in the antisense strand into a thymine, thereby changing the alanine codon (GCA) into a threonine codon (ACA) in the sense strand, i.e., the A673T mutation.
APP deamination: A673T editing
Deep sequencing was used to determine the deamination percentage of each cytosine (C1 to C5) present in the target window of the deaminase (analysis examples available in Table S2). C2 is the cytidine of interest in this study, corresponding to A673T. Its percentage on the figures is calculated by summing the deep-sequencing reads containing C2 only, C1 + C2, C2 + C3, etc. All of the efficiencies presented below for C1−C5 are, unless otherwise specified, overall deamination efficiencies, i.e., the percentage of reads possessing the mutation, in the presence of other mutations or not. This counting is done to underline the specificity of some models.
The editing efficiency in HEK293T cells was greater (by 40%) than in SH-SY5Y cells throughout the study (Figure 3A). The three variants of Cas9 used with the APOBEC1 deaminase (BE3_SpCas9nEQR, BE3_SpCas9nVQR, and BE3_SaCas9nKKH) showed a similar editing profile; however, BE3_SpCas9nVQR showed a greater overall deamination efficiency compared to BE3_SpCas9nEQR and BE3_SaCas9nKKH of 70% and 98%, respectively (Figures 3B for HEK293T and S1 for SH-SY5Y). BE3_SpCas9nVQR enzyme with a sgRNA targeting 19 nucleotides exhibited the highest deamination rate (C1: 6.8%, C2: 3.9%, C3: 15.2%, C4: 2.2%, C5: 2.1%). As a result, SpCas9nEQR and SaCas9nKKH were no longer used in subsequent experiments. Inclusion of two sgRNA cassettes in the plasmid increased the deamination by up to 84% (Figure 3C). All subsequent experiments were thus carried out by transfecting two sgRNA cassettes. Unexpectedly, the BE4 model did not show the expected efficiency in this experiment. BE4_SpCas9nVQR enzyme with a sgRNA targeting 19 nucleotides was 22% less efficient than its BE3 counterpart. As for BE4_SpCas9nVQR with a sgRNA targeting 20 nucleotides, it was 44% less efficient than its BE3 counterpart. As illustrated in Figure 3D, the best global and C2-specific deamination using APOBEC1 was obtained with the BE3_SpCas9nVQR enzyme with a sgRNA targeting 20 nucleotides in both cell models (SH-SY5Y available in Figure S2). C1 and C3 nucleotides were deaminated more frequently than the C2 nucleotide with every base editor using the APOBEC1 enzyme (present in all of the BE3 and BE4 constructs). From these experiments, it was concluded that the SpCas9nEQR and the SaCas9nKKH base editor variants were showcasing poor deamination rates and were less effective than the BE3_SpCas9nVQR variant (Figure S3). Thus, the BE3_SpCas9nEQR and SaCas9nKKH were not used in the subsequent experiments.
Figure 3.

Percentages of CDA produced by various enzymes and sgRNAs
Plasmids coding for the various base editors and for one or two sgRNAs were transfected in HEK293T and SH-SY5Y cells. The number of nucleotides targeted by the sgRNAs is indicated after the names of the enzymes. DNA was extracted 3 days post-transfection, APP exon 16 was PCR amplified and sent for deep sequencing. (A) Significant differences between SH-SY5Y and HEK2093T editing efficacy are shown with BE3_SpCas9nVQR. Transfected with two copies of sgRNA. The figure illustrates the mean ± SEM (n = 3). (B) BE3_SpCas9nEQR, BE3_SpCas9nVQR, and BE3_SaCas9nKKH enzymes were tested in HEK293T cells. They were transfected with two copies of sgRNA. The figure illustrates the mean ± SEM (n = 4). (C) The comparison between the use of one copy sgRNA versus two copies during base editing transfection in HEK293T cells is demonstrated. The figure illustrates the mean ± SEM (n = 3). (D) The BE4_SpCas9nVQR and BE3_SpCas9nVQR enzymes were tested in HEK293T cells. The figure illustrates the mean ± SEM (n = 3). SH-SY5Y results available in complementary results Figures S1 and S2. Statistical test: two-way ANOVA Tukey’s multiple comparisons test, (A) n = 3, (B) n = 4, (C) n = 3, and (D) n = 3. p value style: ∗p < 0.0332, ∗∗∗∗p < 0.0001.
When the initial experiments were performed, the conversion window was quite wide. That is, the targeted cytidine (C2) was not deaminated frequently enough compared to the other adjacent cytidines C1 (38% less on average) and C3 (68% less on average). In response, new CDA enzymes were designed (YE1-BE3_SpCas9nVQR, YE1-BE3_SpCas9nEQR, YE1-BE3_SaCas9nKKH, Target-AIDSpCas9nVQR, Target-AIDSpCas9nEQR, Target-AIDSaCas9nKKH). The enzymes were tested with new sgRNAs targeting 17, 18, 19, or 20 nucleotides. In this context the Target-AID-SpCas9nVQR deaminated a higher percentage of cytidines, especially C1 and C2, resulting in a narrower target window (Figures 4A for SH-SY5Y and S4 for HEK293T cells).
Figure 4.

Deamination efficiencies using various Cas9n-deaminases and sgRNAs targeting various numbers of nucleotides
(A) The difference of deamination in SH-SY5Y for cytidines C1 to C5 produced by the Target-AID-SpCas9nVQR and BE3_SpCas9nVQR enzymes and two copies of a sgRNA targeting 17 to 20 nucleotides. Error bars represent mean with SEM (n = 3). Statistical test showing prevalence of C2 compared to others, C1 and C3−C5: ordinary one-way ANOVA Dunnett’s multiple comparisons test (n = 3). p value style: ∗p < 0.0001. (B) Difference between YE1-BE3_SpCas9nVQR and BE3_SpCas9nVQR in SH-SY5Y cells. The figure illustrates the mean ± SEM (n = 4). (C) The percentage of deamination in HEK293T cells transfected with Target-AID-SpCas9nVQR and two copies of a sgRNA targeting 17 to 20 nucleotides after optimization. The figure illustrates the mean ± SEM (n = 5). Statistical test: two-way ANOVA Tukey’s multiple comparisons test, (A) n = 3, (B) n = 4, and (C) n = 5. p value style: ∗∗∗∗p < 0.0001.
With the use of a sgRNA targeting either 17 or 18 bp, C2 was preferentially deaminated up to ∼10% reaching statistical significance. The highest deamination of the C2 nucleotide was obtained with a sgRNA hybridizing with 19 nucleotides for SH-SY5Y (16.8%) and for HEK293T (14%). A similar deamination profile was observed in the SH-SY5Y and HEK293T cells. A 40% higher deamination rate was noted in SH-SY5Y for the first time in the study, indicating that Target-AID SpCas9nVQR could work better in these cells.
The YE1-BE3 variant was also tested. This variant contained two mutations W90Y and R126E in the APOBEC1 gene and has been reported to create a narrower conversion window.17 This enzyme was tested with sgRNAs targeting 19, 20, 21, or 22 nucleotides. This variant converted an insignificant percentage (<1%) of cytidine C2 into thymine in exon 16 of the APP gene with the three Cas9 variants (expected for SpCas9nEQR and SaCas9KKH) (Figures 4B for SH-SY5Y and S5 for HEK293T).
The experiment was repeated in HEK293T with Target-AID-SpCas9nVQR combined with sgRNA targeting 19, 20, 21, or 22 nucleotides to test for differences in the deamination window. The deamination window, which best targeted C2, was formed with a 19-bp guide and allowed 53.3% editing in HEK293T cells (Figure 4C). However, when C2 was deaminated using this guide, C1 was deaminated 90% of the time to create the E674K mutation (global C2 deamination: 53.3% = C2 only [3.8%]; C1 + C2 [36.3%]; C1 + C2 + C5 [5.5%]; C1 + C2 + C3 [2.3%]; C2 + C4 + C5 [1.8%]; other combinations [3.6%]) (Table S2). Although a guide of 17 bp made it possible to better isolate C2, we favored a greater deamination. We therefore decided to use Target-AID-SpCas9nVQR with a sgRNA targeting 19 nucleotides for the subsequent experiment.
Aβ peptides’ concentration decreases in APP SH-SY5Y cell lines
The next experiment was based on our previous work in which we observed that the A673T mutation could reduce Aβ peptides concentrations even in the presence of other codominant FAD mutations.28 The best FAD mutation responding to the treatment was the London mutation V717I (observed decrease of Aβ peptides’ concentrations in Table S3). The aim was to deaminate nucleotide C2 in plasmids coding for APP containing a FAD mutation and attesting an Aβ40/42 concentration decrease as proof of principle.
Different SH-SY5Y cell lines that constitutively overexpressed wild-type (WT) APP, V717I, or C1 + C2 mutations (E674K, A673T) were produced through lentiviral transduction of an APP695 cDNA plasmid. These cell lines were created, in part, to account for low basal peptide secretion in the SH-SY5Y cells. Under in vitro conditions, the detection of Aβ42 was impossible due to low secretion levels (Figure 5B). The overexpression of APPWT in the SH cell line increased Aβ40 secretion by 34% and allowed Aβ42 to be detected.
Figure 5.

Deamination and Aβ peptide quantification in SH-APPWT and SH-APPV717I cell supernatant
SH-SY5Y cells were transfected (identified as treated in the figure) with a lentivirus plasmid containing Target-AID-SpCas9nVQR and two copies of the sgRNA targeting 19 nucleotides. (A) Target-AID-SpCas9nVQR deamination profile in SH-SY5Y cell lines containing either the APPWT gene or the APP gene with the London mutation (V717I). (B) Aβ peptides’ concentration in the SH-SY5Y supernatant. The figure illustrates the mean ± SEM (n = 8). Statistical test: two-way ANOVA Sidak’s multiple comparisons test (n = 8, 4 biological replicates and 2 technical replicates each). p value style: ∗p < 0.0332, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001.
These cells were subsequently transfected with a lentivirus plasmid EF1-Target-AID-SpCas9nVQR-2U6 and sgRNA binding to 19 nucleotides. Interestingly, as observed in Figure 5A, the SH-SY5Y APP cell lines demonstrated different deamination profiles after treatment with the same base editor. C1 was deaminated by 3.3% in APP WT and 3.5% in the APP containing V717I. Likewise, C2 was deaminated by 6.6% in SH-APPWT and 7.6% in SH-APPV717I. Prior experiments demonstrated roughly equal deamination between C1 and C2; however, in this condition, there was a reduction in total deamination for C1 of approximately 50% in both WT and V717I APP cell lines.
Treatment with the base editor led to a reduction of Aβ40 and Aβ42 peptides in the cell culture medium (Figure 5B). With only 7% of the C2 deaminated, SH-APPWT experienced a reduction of 23% in Aβ40 and 6.7% in Aβ42. The SH-APPV717I cell line had a deamination in C2 of 8% and demonstrated a reduction of 26% in Aβ40 and 31% in Aβ42. In comparison, the SH-APPC1 + -C2 cell line had a 100% conversion of C:T in positions C1 and C2 and saw a reduction of 52% in Aβ40 and 56% in Aβ42 compared to SH-APPWT. Due to the low secretion of Aβ42 peptides during these experiments, only reductions in Aβ40 peptide secretions yielded statistically significant results (Figure 5B). This is in spite of the experiments having been performed in quadruplicate.
Additionally, data from a previous study performed in our lab were compared to the cell lines produced in this study (Table S3).28 In that study, SH-SY5Y cells were transfected with plasmids coding for the APP gene containing the A673T mutation. This condition was considered a positive control for A673T insertion (100% insertion) and similarly produced quantifiable Aβ40 and Aβ42 peptides. Cells transfected with the A673T-APP had a reduction of 63% in Aβ40 and a reduction of 46% in Aβ42 when compared to APPWT. Cells transfected with a V717I/A673T-APP plasmid had a reduction of 81% of Aβ40 and 65% of Aβ42 compared to cells transfected with V717I-APP. Cells transfected with an A673T/E674K-APP (C1 + C2 plasmid) had a reduction of Aβ40 peptides of 44% and Aβ42 peptides of 53% compared to cells transfected with APPWT plasmids (Table S3). When comparing the C1 + C2 data obtained in the present study in mixed cell line population (52% reduction in Aβ40 and 56% reduction in Aβ42) to the previous study with plasmids (44% reduction in Aβ40 and 53% reduction in Aβ42), it is apparent that the data are extremely similar.
Discussion
This study has focused on the development of a base editing technique to introduce the A673T mutation in cultured cells. The effects of this mutation on Aβ accumulation were demonstrated through its introduction into mixed SH-SY5Y cells lines containing either the APP WT gene or the APP gene with the London mutation V717I (London). WT and London mixed cell lines that were treated with the base editors had a reduction of 23% and 26% in Aβ40 peptides and 6.7% and 31.8% in Aβ42 peptides in vitro. This was considered promising when accounting for the low transfection efficiencies during this study. A significant reduction in C1 deamination (approximately 50% less deaminated than C2) was also noted when using the Target-AID-SpCas9nVQR (Figure 5A). Although this reduction was not anticipated, one possible explanation was that the genome-integrated cDNA did not create the same environment as the native APP gene. This could have led to the creation of a different deamination window. It was noteworthy that the Target-AID-SpCas9nVQR base editor still demonstrated some unwanted C3−C5 deamination. The strongest total C2 deamination seen in this study (53.3%) demonstrated a C2 deamination of 42.6% when excluding all combinations containing either C3 or C4 (C5 being silent) (Table S2).
In a previous experiment, in which the results are illustrated in Table S3, we noted that the addition of E674K seemed to have a positive synergistic effect with A673T. We therefore created an SH-SY5Y cell line containing the APP gene with deaminated C1 and C2 positions to further investigate this. The cell line served to better assess the effects of the E674K (C1) off-target mutation in a uniform population. Each time a base editor was used, a heterogenous population of cells was created with mutations occurring anywhere along C1−C5. The Aβ readings were therefore the result of a mixture of cells. It was noted that this cell line experienced a stronger reduction of Aβ peptides compared to APPWT treated (Figure 5B). In addition, it demonstrated roughly one-half of the Aβ42 peptide concentration compared to APPWT treated. Since this peptide is the leading contributor to the formation of plaque in AD, this was considered relevant for the purposes of creating a therapy for this disease. One hypothesis to explain why the deamination of C1 leads to decreased Aβ peptide concentration is that BACE1 might have more affinity basic amino acids.29 The artificial E674K mutation, being very close to the cutting site, exchanges a negatively charged glutamate (E) for a positively charged lysine (K), which could reduce cleavage by BACE1.
Interestingly, it was also noted that the reduction in Aβ peptide secretions in SH-Y5Y cells containing the APPC1 + -C2 gene inserted in the genome was extremely similar to the reduction percentage seen with transfected APPC1 + -C2 plasmids (Figure 5B; Table S3). This similarly suggests that the decrease in Aβ peptide concentrations shown in Figure 5B for SH-APPWT and SH-APPV717I might be influenced by their levels of C1 and C2 deamination (Figure 5A). Since these cells have a notable deamination in C1, and Table S3 demonstrates different reductions in Aβ peptides between APP-A673T and APPC1 + -C2, it is likely that the genomic C1 mutation in these cells is influencing Aβ peptide secretion. At the present time, there is no data suggesting that the additional cytosine mutation can alter the protective effects of the A673T mutation. In the future it will be necessary to verify whether these additional mutations affect the aggregation of the peptides or have any other effects in vivo.
More recent base editors have been developed, such as XBe3 and evoCDA1-BE4max, since the beginning of this study.22,30 However, as the first used an APOBEC1 deaminase, and the other seemed to have a wider conversion window than our Target-Aid, both were deemed unusable for this study. The presence of several cytosines in our area of interest obliged us to select the narrowest editing window available. To this end, both the CRISPR-Cas9 and deaminase sections of the base editor had to be optimized to induce the A673T mutation.
Regarding the CRISPR-Cas9 section of the base editor, the final choice (Target-AID_SpCas9VQR) was the most efficient Cas9n in this study. However, whereas it did preferentially deaminate the targeted cytosine, it also weakly deaminated the other four nearby cytosines. As each Cas9n has its own PAM, the ideal choice for a base editor is often locus dependent. Looking at our study, SpCas9VQR was the most effective of the three Cas9 variants. Concerning the indels, it was shown that Target-AID also generated less off target than the BE3 model, which was already only 5%.13,20 An example of these indels was calculated in Table S2 using the Script presented in Figure S7. However, this could be different for another gene.
Looking at the deaminase component of the base editor, after communicating with Dr. Komor’s team, it was explained that the reduced deamination of the C2 nucleotide with APOBEC1 was the result of inhibition. This CDA is inhibited when a G precedes the target C (our C2 target is preceded by a G). The BE3 and BE4 base editors were therefore deaminating C1 and C3 predominantly, which was not beneficial toward our proposed treatment. Conversely, Target-AID showed a much higher deamination rate with a narrower conversion window around C1 and C2. We therefore designed a new base editor called Target-AID-SpCas9nVQR using Target-AID-SpCas9n as a model. This new base editor demonstrated a greater deamination rate for our desired mutation compared to the other readily available base editors.
The off-target deamination from base editing is localized to exposed single-stranded DNA or RNA (ssDNA or ssRNA). In our study, this meant that off-target events were generally located in the conversion window. However, it has been found that ssDNA and ssRNA throughout the genome could be the target of the deaminase leading to undesirable mutations in coding and non-coding nucleic acid sequences.31,32 This would effectively allow the deaminase to modify random nucleic acids independent from Cas9n binding. In response, research groups have begun to engineer new deaminase enzymes that demonstrated fewer off-target events.31 In the future, it will be worthwhile to attempt this experiment again with base editors, such as evoAPOBEC1-BE4max and BE4-SsAPOBEC3B models, which have been designed with these new enzymes (allowing high and precise deamination and reducing off targets), or NG-Cas9, SpG, and SpRY (having a non-specific PAM).22,33, 34, 35 These enzymes have also been designed to have a narrower conversion window, which could be used to favor C2 editing. We hope that the recent development of the Prime editing technology might allow us to modify only the C2 nucleotide.36
We have investigated possible off-target deamination due to mistargeting by the Cas9n using an in silico model. The Benchling.com interface using the Crispr.mit.edu algorithm was used to calculate possible off-target events for our sgRNA.37 No notable off-target events (Figure S6) were found. No off targets were found when the search was expanded to identify off target with one mismatch. One possible off-target site was predicted after the search was widened to two mismatches. However, this off target was located in a non-coding DNA sequence.
For this type of gene editing, one of the most important factors is the safe and efficient delivery of the therapeutic agents. For the in vivo experiments and for an eventual clinical application, a dual AAV vector delivery system may prove the only option to introduce the CDA transgene in the neurons. In fact, a single virus could not package a base editor given its size. Base editing could be used to treat many hereditary diseases, even though some specific optimizations will be required to adjust for each different target in the genome.
The next part of this project will aim to deliver our best base editor in mouse model NL/F/G and V717I neurons, as well as in the neurons of Alzheimer patients (obtained by direct conversion of fibroblasts) in vitro.38,39 This will be accomplished by using a dual AAV for packaging as well as an intein system to reassemble the base editor after delivery.40 The next step will be to transduce the mouse models mentioned above with a low immunogenic AAV-PHP.eB serotype that specifically targets the CNS in vivo.41 Although these results are still only preliminary, we hope to develop a potential treatment that will slow down the development of amyloid plaques in Alzheimer’s patients.
Materials and methods
The enzymes that we have tested are shown in Box 1 (Deaminase variants description and construction).
Box 1. Deaminase variants description and construction.
The enzymes that we have tested are the following:
-
(1)
BE3_SpCas9nVQR, produced by Kleinstiver et al.26 is a variant of BE3, which contains an SpCas9n protein with 3 mutated amino acids D1135V/R1335Q/T1337R. The gene for this enzyme was available at Addgene as pBK-VQR-BE3 (#85171).
-
(2)
BE3_SpCas9nEQR, also produced by Kleinstiver et al.,26 is another variant of BE3, which contains an SpCas9n protein with 3 mutated amino acids D1135E/R1335Q/T1337R. The gene for this enzyme was available at Addgene as pBK-EQR-BE3 (#85172).
-
(3)
BE3_SaCas9nKKH, which was also produced by Kleinstiver et al.,27 is a variant of BE3, which contains an SaCas9n protein (from Sa) with 3 mutant codons E782K/N968K/R1015H. The gene for this enzyme was available at Addgene as pJL-SaKKH-BE3 (#85170).
-
(4)
YE1-BE3_SpCas9n is a construct variant of BE3, which contains 2 mutant codons (W90Y/R126E) in the rAPOBEC1 sequence. The gene for this enzyme was available at Addgene as pBK-YE1-BE3 (#85174). We used this plasmid to construct all other variants containing the YE1 rAPOBEC1 deaminase.
-
(5)
YE1-BE3_SpCas9nVQR is a variant of the YE1-BE3_SpCas9n, which was made in our laboratory. The rAPOBEC1 deaminase sequence in plasmid pBK-VQR-BE3 was replaced with restriction enzyme digestion/ligation by the YE1 variant from plasmid YE1-BE3.
-
(6)
YE1-BE3_SpCas9nEQR is a variant of the YE1-BE3_SpCas9n, which was also made in our laboratory. The rAPOBEC1 deaminase sequence in plasmid pBK-EQR-BE3 was replaced by the YE1 sequence from plasmid YE1-BE3.
-
(7)
YE1-BE3_SaCas9nKKH is a variant of the BE3_SaCas9nKKH, which was made in our laboratory. The rAPOBEC1 deaminase sequence in plasmid BE3_SaCas9nKKH was replaced by the YE1 variant from plasmid YE1-BE3.
-
(8)
BE4-Gam_SpCas9nVQR is a variant of the BE4-Gam (Addgene; #100806), which was made in our laboratory by PCR mutagenesis to create the VQR mutation in the SpCas9n gene.
-
(9)
BE4-Gam_SpCas9nEQR is a variant of the BE4-Gam, available at Addgene (#100806). This variant was made in our laboratory by PCR mutagenesis to create the EQR mutation in the SpCas9n gene.
-
(10)
Target-AID SpCas9nVQR is a variant of the Target-AID enzyme described by Komor et al.19 The original Target-AID enzyme contains the SpCas9n(D10A), a 105-amino acid linker, CDA1 (i.e., the AID that was modified by Nishida et al.13), an 11-amino acid linker, and the UGI. The original plasmid, named nCas9-PmCDA1-UGI, available at Addgene (#76620), was modified by PCR mutagenesis to create the VQR version of the SpCas9n.
-
(11)
Target-AID SpCas9nEQR is a variant of the Target-AID enzyme described by Komor et al.19 The original Target-AID enzyme contains the SpCas9n(D10A), a 105-amino acid linker, CDA1 (i.e., the AID that was modified by Nishida et al.13), an 11-amino acid linker, and the UGI. The original plasmid, named nCas9-PmCDA1-UGI and sgRNA (hypoxanthine-guanine phosphoribosyl transferase [HPRT]; Target-AID), available at Addgene (#76620), was modified by PCR mutagenesis to create the EQR version of the SpCas9n.
-
(12)
Target-AID SaCas9nKKH is a variant of the original plasmid called Target-AID, available at Addgene (#76620). The SpCas9n was replaced by SaCas9nKKH with Gibson assembly. Briefly, the SpCas9n was removed by cutting the plasmid with two restriction enzymes: Nhe1 and BsiW1.
Co-transfection in HEK293T and SH-SY5Y cells of the Cas9n/CDA plasmid and pBSU6 sgRNA
The transfection reagent (Lipofectamine 2000) and Opti-MEM-1 culture media were purchased from Life Technologies (Carlsbad, CA, USA). The HEK293T and SH-SY5Y cells were transfected with a plasmid coding for one Cas9n-deaminase and a sgRNA. The day before the transfection, 100,000 cells were seeded per well in a 24-well plate in DMEM (DMEM/F12 for SH), supplemented with 10% fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin 100 μg/mL). The following morning, the culture medium was changed for 500 μL of fresh medium. The plate was maintained at 37°C for the time required to prepare the transfection solution. For the transfection, solutions A and B were first prepared. Solution A contained 48 μL of Opti-MEM and 2 μL of Lipofectamine 2000 for a final volume of 50 μL. Solution B was prepared as follows: 1 vol of DNA solution containing 800 ng of DNA was mixed (400 ng of base editor plasmid and 400 ng of pBSU6 sgRNA or GFP plasmid for negative controls) with 1 vol of Opti-MEM to obtain a final volume of 50 μL. Solutions A and B were then mixed by up and down movements and incubated at room temperature for 20 min. 100 μL of the mixed solution was then added to each well. The plate was left in the CO2 incubator for a period of 4 to 6 h before a fresh medium change. The plate was incubated for 72 h in the CO2 incubator before extracting the genomic DNA.
Cell harvesting
Cells were detached 72 h post-transfection by performing up and down movements in 1 mL culture medium with a pipette. These cells were transferred to an Eppendorf tube and centrifuged at 8,000 rpm for 10 min. The supernatant medium was carefully removed without disturbing the cell pellets. These cell pellets were washed once with 1 mL of Hank’s balanced salt solution (HBSS) solution and centrifuged at 8,000 rpm for 10 min. The HBSS was then carefully removed without disturbing the cell pellets.
DNA extraction
The cells were lysed with 100 μL of lysis buffer containing 1% Sarkosyl and 0.5 M EDTA (pH 8), supplemented with 10 μL of proteinase K solution (20 mg/mL). These tubes were incubated at 50°C for 15 min. 400 μL of 50 mM Tris (pH 8) was then added to each tube. Next, 500 μL of a mixture of phenol:chloroform:isoamyl alcohol (respectively, 25:24:1) was added. The tubes were centrifuged at 16,000 rpm for 2 min. The aqueous upper phase was transferred to a new tube. 50 μL of NaCl 5 M was added to each tube and mixed thoroughly. 1 mL of ice-cold ethanol 100% was added to each tube and mixed for genomic DNA precipitation. The tubes were centrifuged at 16,000 rpm for 7 min, and ethanol was carefully removed to avoid disturbing the DNA pellets. These DNA pellets were washed once with 400 μL ethanol 70%. The tubes were centrifuged at 16,000 rpm for 5 min under a vacuum (Speedvac) to remove the ethanol and the DNA pellet rapidly. The DNA was solubilized in 50−100 μL of sterile water and stored at −20°C until quantification was performed. The DNA solutions were dosed at 260 nm with a spectrophotometer.
Stable SH-SY5Y APP cell lines’ generation
A lentivirus with cytomegalovirus promotor (CMV)-APP-P2A-puromycin-woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) was produced in a 10-cm Petri dish containing 4 million HEK293T cells. 40 μg of four lentiviral plasmids was transfected (15 μg APP plasmid, 15 μg Gag-pol, 5 μg REV, 5 μg vesicular stomatitis virus G glycoprotein [VSVG]) using the calcium phosphate method. The medium was replaced by 6 mL of fresh medium, 16 h later. The medium was harvested 72 h post-transfection, filtered, and applied to 6-well plates, each containing 1 million SH-SY5Y. The medium was renewed the morning after. The cells were selected with 8 μg/mL of puromycin, 72 h post-transduction.
Supernatant analysis
At 72 h post-transfection, 100 μL of the culture medium of each SH-SY5Y APP cell line was harvested and filtered at 0.4 μm to remove cell debris. Protease inhibitors (1 mM PMSF + 1× complete tabs from Roche) were added. The media were then stored at −80°C. The concentrations of Aβ peptides 42 and 40 (most common AD biomarkers) were measured with the Meso Scale Discovery (MSD; Rockville, MD, USA) Neurodegenerative Disease Assay 6E10 Kit. Standards and samples were prepared according to the manufacturer’s protocols and always tested in technical duplicates.
Deep sequencing analysis
Deep sequencing samples were prepared by a PCR reaction with special primers containing a bar code sequence (BCS) to aid the subsequent sequencing (i.e., BCS1: 5′-ACACTGACGACATGGTTCTACAGGTAGGCTTTGTCTTACAGTGTTA-3′ and BCS2: 5′-TACGGTAGCAGAGACTTGGTCTTGGTAATCCTATAGGCAAGCATTG-3′). DNA sequences were analyzed with the Illumina sequencer. Roughly 10,000 reads were obtained per sample.
Bioinformatics analysis of the deep sequencing results
Deamination profiles were estimated by BAM file extraction and analysis, using a bash script described in Figure S7. All of the results have been verified with CRISPResso2 and showed similar results.
Statistical analysis
All statistic tests and graphs were performed as recommended by GraphPad Prism 7.0. Two-way ANOVA Tukey’s multiple comparisons test was used to test significance with at least three biological replicas for Figures 3, 4, and S1–S5. Ordinary one-way ANOVA Dunnett’s multiple comparisons test was used in Figure 4A with three biological replicas. Two-way ANOVA Sidak’s multiple comparisons test was used to test significance with four biological replicas (two technical replicas each) for Figure 5B; ∗p < 0.0332, ∗∗p < 0.0021, ∗∗∗p < 0.0002, and ∗∗∗∗p < 0.0001.
Acknowledgments
We would like to thank the Quebec Cell, Tissue and Gene Therapy (THÉCELL) Network for its support. This research project was supported by grants from the Weston Brain Institute.
Author contributions
A.G. designed the experiments, performed the experiments, and wrote the manuscript. J.R. assisted with the design of the experiments and provided technical assistance for molecular biology. F.-G.B. and T.B. assisted with base editors’ construction. G.L. revised and corrected the manuscript. J.P.T. conceived the experiments and corrected the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.02.032.
Supplemental information
References
- 1.2015). World Alzheimer Report 2015. The global impact of dementia: An analysis of prevalence, incidence, cost and trends (Alzheimer’s Disease International).
- 2.2019). World Alzheimer Report 2019. Attitudes to dementia (Alzheimer’s Disease International).
- 3.2021). Dementia (World Health Organization), https://www.who.int/news-room/fact-sheets/detail/dementia.
- 4.Reitz C., Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014;88:640–651. doi: 10.1016/j.bcp.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masters C.L., Multhaup G., Simms G., Pottgiesser J., Martins R.N., Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alonso Vilatela M.E., López-López M., Yescas-Gómez P. Genetics of Alzheimer’s disease. Arch. Med. Res. 2012;43:622–631. doi: 10.1016/j.arcmed.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 8.(2021). Mutations. APP. (Alzforum), https://www.alzforum.org/mutations/app.
- 9.Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 10.Maloney J.A., Bainbridge T., Gustafson A., Zhang S., Kyauk R., Steiner P., van der Brug M., Liu Y., Ernst J.A., Watts R.J., Atwal J.K. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014;289:30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kero M., Paetau A., Polvikoski T., Tanskanen M., Sulkava R., Jansson L., Myllykangas L., Tienari P.J. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol. Aging. 2013;34:1518.e1–1518.e3. doi: 10.1016/j.neurobiolaging.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 12.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 14.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 17.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komor A.C., Badran A.H., Liu D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell. 2017;169:559. doi: 10.1016/j.cell.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Komor A.C., Zhao K.T., Packer M.S., Gaudelli N.M., Waterbury A.L., Koblan L.W., Kim Y.B., Badran A.H., Liu D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017;3:eaao4774. doi: 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasaguri H., Nagata K., Sekiguchi M., Fujioka R., Matsuba Y., Hashimoto S., Sato K., Kurup D., Yokota T., Saido T.C. Introduction of pathogenic mutations into the mouse Psen1 gene by Base Editor and Target-AID. Nat. Commun. 2018;9:2892. doi: 10.1038/s41467-018-05262-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molla K.A., Yang Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019;37:1121–1142. doi: 10.1016/j.tibtech.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Thuronyi B.W., Koblan L.W., Levy J.M., Yeh W.H., Zheng C., Newby G.A., Wilson C., Bhaumik M., Shubina-Oleinik O., Holt J.R., Liu D.R. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019;37:1070–1079. doi: 10.1038/s41587-019-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology (Reading) 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 24.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z., Gonzales A.P., Li Z., Peterson R.T., Yeh J.R. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleinstiver B.P., Prew M.S., Tsai S.Q., Nguyen N.T., Topkar V.V., Zheng Z., Joung J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guyon A., Rousseau J., Lamothe G., Tremblay J.P. The protective mutation A673T in amyloid precursor protein gene decreases Aβ peptides production for 14 forms of Familial Alzheimer’s Disease in SH-SY5Y cells. PLoS ONE. 2020;15:e0237122. doi: 10.1371/journal.pone.0237122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S., Wang Z., Cai F., Zhang M., Wu Y., Zhang J., Song W. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J. Neurosci. 2017;37:6915–6925. doi: 10.1523/JNEUROSCI.0340-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–893. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin S., Zong Y., Gao Q., Zhu Z., Wang Y., Qin P., Liang C., Wang D., Qiu J.L., Zhang F., Gao C. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science. 2019;364:292–295. doi: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 33.Yu Y., Leete T.C., Born D.A., Young L., Barrera L.A., Lee S.J., Rees H.A., Ciaramella G., Gaudelli N.M. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 2020;11:2052. doi: 10.1038/s41467-020-15887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu Z., Kuang Y., Ren B., Yan D., Yan F., Spetz C., Sun W., Wang G., Zhou X., Zhou H. SpRY greatly expands the genome editing scope in rice with highly flexible PAM recognition. Genome Biol. 2021;22:6. doi: 10.1186/s13059-020-02231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., Saido T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 39.Drouin-Ouellet J., Pircs K., Barker R.A., Jakobsson J., Parmar M. Direct Neuronal Reprogramming for Disease Modeling Studies Using Patient-Derived Neurons: What Have We Learned? Front. Neurosci. 2017;11:530. doi: 10.3389/fnins.2017.00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy J.M., Yeh W.H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.