

Abstract
The oxidative phosphorylation (OXPHOS) system is the only structure in animal cells with components encoded by two genomes, maternally transmitted mitochondrial DNA (mtDNA), and biparentally transmitted nuclear DNA (nDNA). MtDNA‐encoded genes have to physically assemble with their counterparts encoded in the nucleus to build together the functional respiratory complexes. Therefore, structural and functional matching requirements between the protein subunits of these molecular complexes are rigorous. The crosstalk between nDNA and mtDNA needs to overcome some challenges, as the nuclear‐encoded factors have to be imported into the mitochondria in a correct quantity and match the high number of organelles and genomes per mitochondria that encode and synthesize their own components locally. The cell is able to sense the mito‐nuclear match through changes in the activity of the OXPHOS system, modulation of the mitochondrial biogenesis, or reactive oxygen species production. This implies that a complex signaling cascade should optimize OXPHOS performance to the cellular‐specific requirements, which will depend on cell type, environmental conditions, and life stage. Therefore, the mitochondria would function as a cellular metabolic information hub integrating critical information that would feedback the nucleus for it to respond accordingly. Here, we review the current understanding of the complex interaction between mtDNA and nDNA.
Keywords: cytoplasmic communication, intergenomic coadaptation, mitochondrial DNA, mitochondrial haplotypes, mito‐nuclear interactions, nucleo‐mitochondrial mismatch, respiratory complexes and supercomplexes, retrograde responses
1. CO‐EVOLUTION OF GENOMES
The acquisition of mitochondria is certainly one of the most important events in the early evolution of the eukaryotes. It was what allowed cells to achieve the bioenergetic capability to reach eukaryotic cell complexity. 1 This development is explained by the endosymbiotic theory of the origin of mitochondria. 2 Lynn Margulis then suggested that the origin of eukaryotes was related to the increasing production of free oxygen by photosynthetic prokaryotes and the rising levels of oxygen in the atmosphere at the Archaean‐Proterozoic transition. 3 However, different studies proved that eukaryotes were already around long before that event. 4 Therefore, it was suggested that the rise of atmospheric oxygen was closely tied to Earth's tectonic evolution. 5 Furthermore, the importance of oxygen in the process is also questioned by the ability of some sponges to control their anaerobic microbial population, 6 which may imply that sponges already lived when oxygen levels were lower and unstable.
Regardless of the driving force, the current view is that mitochondria arise from a prokaryotic cell (whose host was an archaeon) and this symbiosis affords mutual advantages. The benefit for the endosymbiont that originated the mitochondria was enormous and explained the evolutionary tendency to lose genes and become genetically more streamlined due to the constant internal environment compared to the outside world. 7 This phenomenon ended up in transforming the ancestor of the mitochondria into an organelle. The reduction of the genome was accompanied by the folding up of their osmotic membrane generating excess energy production capability that allowed the proto‐eukaryotic cell to increase its size and, more importantly, the complexity of its genome. 1 In addition, the host cell not only guarantees an excess of ATP, but it also gained substrate versatility (e.g., H2). 8
Despite of the massive gene loss, mitochondrial DNA (mtDNA) retained a non‐random core of genes that encode the same group of membrane‐integral respiratory proteins, and the ribosomal and transfer RNAs needed to be expressed locally. 9 This fact is the basis of the need for a crosstalk between the two genomes to achieve the balance of the cellular energy requirements based on two different and independent genomes in most eukaryotes. Particularly, the mammalian mitochondrial genome encodes 13 mRNA codifying the 13 polypeptides of the mitochondrial electron transport chain (mtETC) that have to physically interact with almost 73 nuclear‐encoded peptides thoroughly for effective electron transfer and ATP production 10 (Figure 1, Table 1). The mtDNA also encodes 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs (rRNAs), assisting with the translation of mitochondrial mRNAs, and contains one non‐coding region, known as the displacement loop (D‐loop). 11 Another particularity of their structure is the absence of introns and the lack of histones. Finally, all genes are transcribed as large polycistrons, so all the 37 mitochondrial‐encoded genes are expressed regardless of the cell type. 12
FIGURE 1.
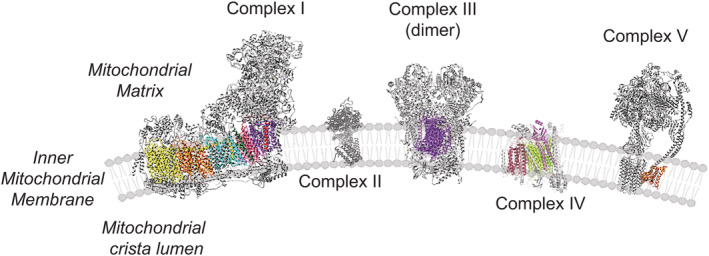
OXPHOS System: Overview of nuclear and mitochondrial‐encoded proteins. In color, mitochondrial‐encoded peptides (Complex I: mt‐ND1, mt‐ND2, mt‐ND3, mt‐ND4, mt‐ND4L, mt‐ND5 and mt‐ND6; Complex III: mt‐CYB; Complex IV: mt‐CO1, mt‐CO2, mt‐CO3; ATP Synthase (Complex V): mt‐ATP6 and mt‐ATP8). In grey, all the nuclear‐encoded subunits
TABLE 1.
Structural subunits of the different mitochondrial OXPHOS complexes to which both genomes (mtDNA and nDNA) contribute with the indication of the chromosomal location in the human genome
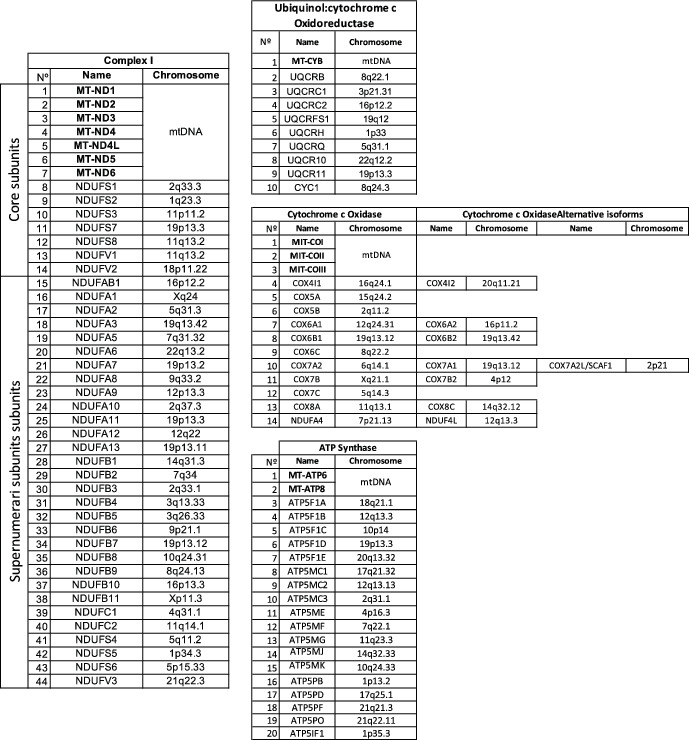
|
Note: In the case of Complex IV, there are alternative isoforms that may be incorporated under specific physiological conditions and cell types.
Abbreviations: NADH, ubiquinol oxidoreductase or Complex I; Ubiquinol, cytochrome c oxidoreductase or Complex III; Cytochrome c Oxidase or Complex IV and ATP Synthase or Complex V.
The evolution of the mitochondrial genome to their actual size, ~20 kb in vertebrates, was not achieved at a slow rate during metazoan evolution. It was proposed that mtDNA evolves 5 to 10 times faster than nuclear DNA (nDNA) in higher primates 13 and that progress is 55‐fold higher in copepods. 14 However, in corals 15 and plants 16 changes happen at lower rate than in nDNA. Three factors are proposed as promoters of a high mutation rate in mitochondrial genomes. Firstly, mitochondria generate free oxygen radicals, producing a highly‐mutagenic internal environment 17 ; secondly, mtDNA is continuously replicated 18 ; and thirdly, mtDNA repairing capacity could be less efficient than that in the nucleus. 19 MtDNA can be repaired by an excision activity in which the mitochondrial uracil‐DNA glycosylase and the 8‐oxoguanine DNA glycosylase (OGG1) play an important role in hydrolyzing N‐glycosidic bonds, and the posterior incision is mediated by the AP endonuclease 1 (APE1). 20 , 21 Although the homologous recombination is infrequent in the mammalian cell, evidences have shown double‐strand breaks can be repaired by nonhomologous end joining. 22 Furthermore, the organelle presents the capacity to repair O 6‐methyl‐2′‐deoxyguanosine and O 6‐ethyl‐2′‐deoxyguanosine 23 and single‐strand breaks, to induce the removal of unpaired nucleotides by mtDNA mismatch repair and base excision repair (base deamination, hydrolyzation, alkylation, and oxidation), and to hydrolyze molecules that the DNA polymerase y is not able to use during the replication of the genome (8‐oxo‐2′‐deoxyguanosine, 8‐oxo‐2′‐deoxyadenosine and 2‐hydroxy‐2′‐deoxtadenosine triphosphates) using the MTH1 activity. 24 In case of unrepairable lesions in the mtDNA, molecules harboring abasic 25 sites or DNA double‐stranded breaks 26 , 27 are degraded. 28
The final outcome is that critical proteins required for the physiological maintenance of the cell fitness, for example, growth, homeostasis, and survival, are encoded in two genomes that are inherited through different rules. MtDNA are transmitted to progeny by one of the gametes making them likely to present clonal expansion, which increases the variance across the population, while nuclear genes are recombined by sex every generation. Therefore, these two genomes generate variability through different mechanisms and, despite this they must still remain compatible. This is why the higher mutation rate of mtDNA in different species may have evolved to provide the required variability of a segment of the genome that lacks sexual transmission and, therefore, does not suffer recombination during meiosis.
The dependency of all mitochondrial functions on the nuclear genome, besides the uniparental and haploid transmission of the mtDNA, generates an asymmetry of the capability of adaptability between these two genomes. Therefore, nDNA is exposed to an intense selection to maintain the fitness of the cell. One example of this asymmetry is on COX genes of anthropoid primates, where the liver isoform of COX8 (nuclear‐encoded) present a high rate of amino‐acid substitution in the part of the protein in contact with COX I (mitochondrial‐encoded). 29 Furthermore, a wide variety of polymorphism can be witnessed in all metazoan mtDNA. These variations distinguish species but are also extensive between populations of the same species and influence their interaction with the nuclear genome. Accordingly, mtDNA of different species are incompatible with their nucleus. In addition, it was found that during maternal‐offspring mitochondrial transmission, mtDNA inheritance is more likely to match with the nuclear genome ancestry to ensure consistency between these two independent genetic systems. 30 Due to the haploid nature of mtDNA, every de novo mutation is exposed to selection forces that fixed the beneficial ones and eliminated the pathological ones, and this choice was dependent on the sex‐specific selection, 31 , 32 or sex linkage of the interacting nuclear genes. 33
1.1. Haplotypes and haplogroups
The extranuclear genome of the cell was used as a powerful tool for populational genetic studies. Due to the high sequence variability within species, mutations have been accumulated historically and fixed in the population allowing the classification of different sequences of the genome into haplotypes. The main source of variance between them is the single nucleotide polymorphisms (SNPs). In humans, the control region (CR) of the mitochondrial genome, containing the origin of replication and transcriptional regulation loci, has been one of the most studied regions due to the high variability of substitutions. 34 , 35 The different mtDNA sequences have been segregated during evolution and exposed to the global migration flow, resulting in them being grouped into mtDNA lineages or haplogroups. Human haplogroups defined different populations in Africa (L haplogroups). As they migrated out of Africa, M and N haplogroups derived from haplogroup L3). N is the Eurasian haplogroup, giving rise in Europe, the root haplogroup (R) for H, J, T, and U haplogroups. Today, 10 different haplogroups explain most of the mtDNA European population (H, I, J, K, M, T, U, V, W, and X). Haplogroups A, B, C, D, F, and G emerged, after humans moved to Asia, from N and M mtDNA lineages. 36 The pioneer studies of mtDNA in human samples from different geographic locations were performed analyzing the conserved polymorphisms by restriction fragment length polymorphisms (RFLPs). These studies led to the classification of the genome sequence between ethnic groups. 37 , 38 More than 70% of the mtDNA from sub‐Saharan African population can be addressed using HpaI ER (position 3,592) 37 ; AluI for Asian Mongoloid variants (position 10,397, not present in European either African haplogroups) 39 ; HaeIII (position 663) combined with AluI (positions 5,176 and 13,262) in the case of Native American mtDNA sequences. 40 The different mtDNA haplotypes have been maintained and selected in nature due to mito‐nuclear interactions.
1.2. Adaptation to environment and association studies
The effort to understand which genetic forces are responsible for sustaining the selection pressure that fix the different mtDNA polymorphisms in nature has been the focus of several studies in recent decades. Adaptive selection of the different mtDNA genomes to environmental cues was described in a cohort of 1,125 human samples where the complete coding regions were analyzed. 41 The enrichment of certain variants in specific regions allowed investigators to conclude that the selection of haplotypes could relay in differences in energy metabolism linked to mtDNA variants and associated to exposure for long periods of time to different environmental conditions such as latitude and temperature variations. 41 , 42
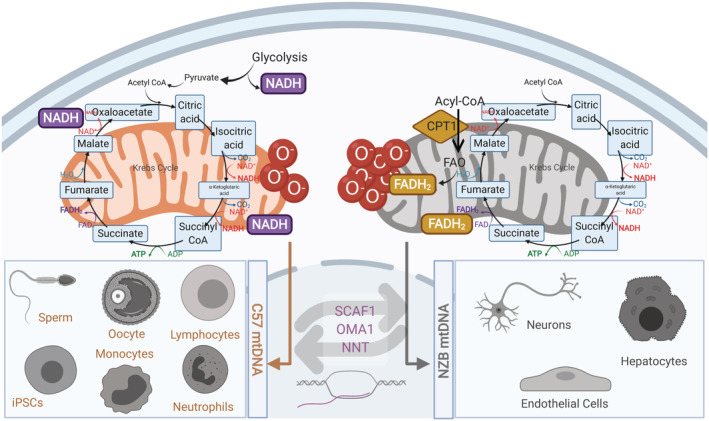
Association studies supporting the link between non‐pathological mtDNA variants with a growing variety of diseases are overwhelming. Furthermore, mtDNA variants in humans can modulate the organismal metabolism and modulate the severity of diseases. Meta‐analysis of the different databases encompassing more than 50,000 patients reported the association of different haplotypes with diseases such as Alzheimer, 43 Huntington disease, 44 Parkinson, 45 multiple sclerosis 46 , 47 type 2 diabetes, 48 , 49 , 50 and cancer. 51 , 52 The phenotypical relevance of H, J, T, and K haplotypes were previously described in clinics. Thus, phenotypical differences between human mtDNA haplotypes and spermatozoa mobility were revealed after the analysis of the major European variants (H, I, J, K, M, T, U, V, W, and X), with the best and worst performing ones being H and T mtDNAs, respectively. 53 Similar studies revealed the relevance of mtDNA for survival after sepsis. 54 The results obtained in the study determined that enhanced oxidative phosphorylation (OXPHOS) performance of haplotype H promoted an increase in the maximum temperature (higher heat generation capacity) and improved the clinical outcome of patients with severe infections. 54 While haplotype H presents lower likelihood of AIDS 55 and severe sepsis, haplogroup J was associated with a worse prognosis. Haplogroup J was demonstrated to lead to increased levels of LHON disease (Leber's hereditary optic neuropathy) associated mutations 56 and presents susceptibility to Multiple Sclerosis. 57 Haplotype K offers protection against thyroid cancer, 52 Parkinson disease 58 and Alzheimer 59 and is associated, together with haplotype J, with increased longevity. 60 , 61 However, studies correlating different haplotypes in diseases and aging showed contradictory results. 48 Despite the observation that J haplogroup was associated with longevity in European populations and D haplogroup among the Japanese population, 62 contradictory results were published underlining the importance of factors such as the living environment, ethnic background and the variability between the prevalent diseases in each population. 63 This is also the case of the correlation between haplotypes and sporadic breast cancer risk. Thus, haplotype K was reported to have more incidence of breast cancers in comparison with haplogroup U lineages, 64 but later evidence to this association could not be replicated. 65 The limitation to study the phenotypic characterization of the different haplotypes is the specific nDNA–mtDNA interaction. As highlighted in the Parkinson disease, haplogroups J, T, K, and U are postulated to decrease and H to increase the risk of the pathology. However, the interaction of the mtDNA sequences with nuclear variants for PARK2 and TFAM are suggested to increase the risk of Parkinson in the haplogroup cluster HV. 66 Co‐evolution and interaction between both genomes make it difficult to consolidate the association studies. The effects of polymorphic mtDNA excluding the divergence effects from the nDNA were first described in transmitochondrial cybrid cell lines 67 , 68 , 69 , 70 , 71 and then using mouse models carrying different mtDNA species under a common nuclear background. 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 The use of transmitochondrial cybrids has been extensively used to understand mtDNA‐nDNA compatibility as an in vitro model to study the effects of having different mtDNA species under the same nuclear background. 80 The divergence in OXPHOS function caused by the sequence variation, rather than the absolute number of differences in sequence, has a greater impact on the overall phenotype. This fact has been assessed in transmitochondrial cybrid cell lines harboring different mtDNA haplotypes in human 32 , 53 and mouse variants. 67 Using cybrids with different mouse haplotypes, it was shown that cells harboring NZB mtDNA contain more mitochondria that those harboring C57 mtDNA under the same L292 nuclear background. Equalizing reactive oxygen species (ROS) production reduce mitochondrial content only in cells with NZB mtDNA, confirming the previous hypothesis. It was later shown that this is also confirmed in vivo, since the livers and hearts of conplastic animals respond, as cultured cells, adjusting to the tissue metabolic demands under different combinations of nDNA–mtDNA 75 (Figure 2a).
FIGURE 2.
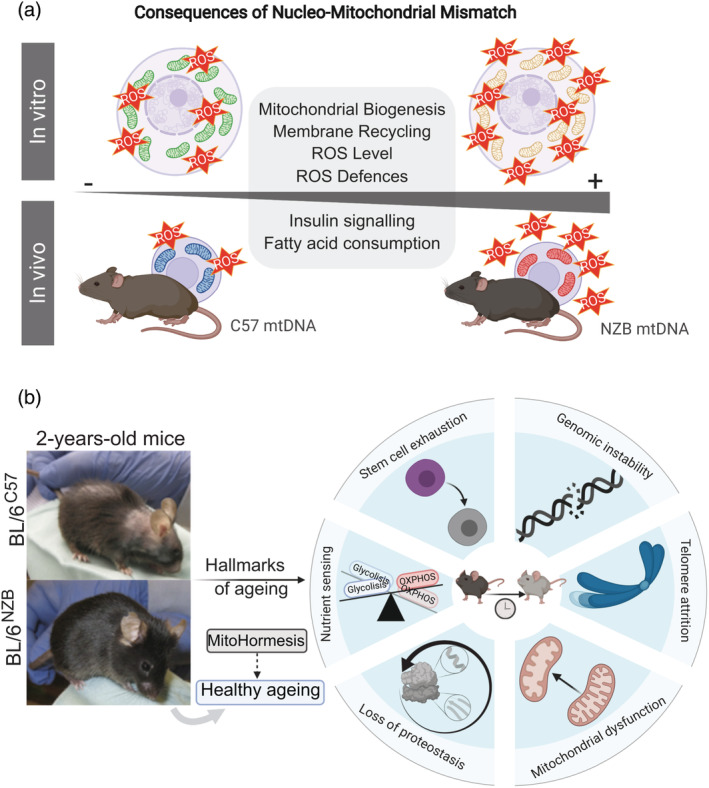
(a) The consequences of nucleo‐mitochondrial mismatch on basal ROS production in culture cells 59 and animal models. 67 (b) Mitohormesis contributes to healthy ageing. An increase in non‐pathological basal mtROS levels induce regular mild mitochondrial stress, priming the mitochondrial adaptive response to elevated levels of cellular stress. BL/6NZB conplastic mice present a delay in the hallmarks of aging detailed in the schematic
Furthermore, the cybrid cell lines have allowed the study of the crosstalk between nuclear and mitochondrial genes in vitro to understand the contribution of mtDNA species in different pathologies. Retinal degeneration was studied using human cybrid cell lines harboring haplotype H or J, protective and high risk, respectively, in age‐related macular degeneration (AMD). 81 Haplogroup J presented a downregulation of mtDNA‐encoded genes, mostly involving complex I (ND1, ND2, ND3, and ND4/ND4L), complex IV (CO2, CO3) and CV (ATP6) subunits and ‐subsequently‐ lower ATP generation via OXPHOS. 81 , 82 Differential expression of pathways as the complement (CFH, C3, MYO7A, or EFEMP1) and inflammation (TGFA, IL‐33, and IL‐6) was also found. 81 Osteoarthritis (OA) has also been associated with mtDNA polymorphisms in humans. 70 , 83 , 84 Biochemical differences in osteoarthritis were outlined using transmitochondrial cybrid cell lines for different mtDNA haplotypes. Cybrids with swapped haplotypes J or H were used to evaluate the biochemical differences in osteoarthritis from both mtDNAs. Cell lines harboring haplotype J showed lower mitochondrial respiratory capacity, and ATP and ROS generation. Better cell survival under mtDNA haplotype J was associated with a downregulation of the expression of the proapoptotic gene BBC3. 70 Further analyses using a supplementary haplotype N showed that fatty acid oxidation decreased under mtDNA variants J and H in comparison with N in OA cybrid models. 71 For decades, efforts to validate mtDNA variants and their role in different diseases were made to identify‐specific treatment and diagnosis approaches. Recent advances in cancer showed a differential response to cisplatin in human transmitochondrial cybrid cell lines harboring either H or J mtDNA haplotypes. 85 This study pointed out the possible role of the retrograde response between mitochondria and nucleus to drug‐derived cell death. 85 The regulation of mitochondrial function through mtDNA/nDNA variable match adjusted by compensatory mechanisms introduces a critical and new element in association studies linking human diseases and mtDNA variants. This concept was first highlighted with the development of conplastic mouse strains. 72 The analyses performed in 16 mtDNA species, with different levels of sequence variations, under the nuclear background of the C57BL/6 strain reported phenotypical differences and susceptibility to experimental autoimmune encephalomyelitis and neurophysiological conditions like anxiety. 72 The exchange of mtDNA variants (C57 and NZB mtDNAs) under the same nuclear background (C57BL/6) was the first proof in vivo that haplotypes alter, in a tissue‐specific way, the reorganization of the assembly and super‐assembly of respiratory complexes in the mtETC. 75 The MtDNA haplotype has a deep influence on OXPHOS performance, ROS generation, and energy homeostasis metabolism (Figure 2a), resulting in different healthy longevities between conplastic strains 75 (Figure 2b). The effort involved in extrapolating the results observed in cell lines and mice models to humans underscores the limitations of the current approaches. The biological differences between species and different living habits and environment as well as the mixture of the nuclear genome in humans made difficult to understand the molecular mechanism involved in the adaptation toward different environmental pressures. The impact of mtDNA variants in cell fate decision presents a high potential in therapeutics at a populational level.
1.3. Connection between OXPHOS genetic complexity, retrograde response, and cytoplasmic communication
Different studies demonstrate that the phenotypic expression linked to mitochondrial genome depends on the nuclear background. This is the case for human cells where the mitochondria were replaced either by chimpanzee or gorilla mitochondria 86 and the result was a 40% reduction in the activity of complex I. Similarly, a decline in OXPHOS capacity was reported after backcrosses between Drosophila simulans and Drosophila mauritiana 87 that entail a decrease in fertility and offspring viability. Another example comes from hybrid models using three different species of Saccharomyces where they documented the inability to regulate the translation of different mitochondrial‐encoded‐proteins affecting their sterility. 88 These findings indicate that functional incompatibilities between the mitochondrial and nuclear genomes play a role in the generation of reproductive barriers due to its effect in mitochondrial function described above. Then, mito‐nuclear interaction can act as significant modulator of evolutionary processes as speciation, because each population is exposed to differing forces of natural selection as a result of inhabiting distinct spatial and temporal environments. 89
Besides the mtDNA evolution in terms of animal speciation, the specific role of mtDNA in bioenergetics and nuclear gene expression modulation and their link with different longevities and pathological phenotypes remains to be understood. The fact that different haplotypes might led to variable generation of ROS can affect the development or progression of different diseases. One example is the correct assembly and stabilization of complex I and the role of superoxide‐derived levels when the match between interacting proteins is not well fitted. In the activation‐induced T‐cell death (AICD), the ROS levels produced by CI trigger the expression of CD95L, crucial during AICD progression 90 and CI inhibition in human T‐cells prevents AIDC. 90 It was proposed that the functional epistasis between mtDNA polymorphisms and their interaction with the nuclear alleles might promote different phenotypes through additive effects. Integrated analyses of genetic data revealed a large number of nuclear genetic variants that modulate mitochondrial‐encoded genes in humans (~21%, multiple testing). 91 Hence, underlining the mechanisms of functional epistasis is relevant to understand the penetrance of mtDNA mutations and diseases.
1.4. Physical match constraint between nDNA/mtDNA OXPHOS‐encoded genes
The OXPHOS system is composed of four respiratory complexes, named complex I (NADH–ubiquinone oxidoreductase), complex II (succinate:ubiquinone oxidoreductase), complex III (ubiquinol‐cytochrome c reductase) and complex IV (cytochrome c oxidase); two electron carrier molecules, ubiquinone and cytochrome c, and the H + ‐ATP synthase. In mammals, mtDNA encodes a reduced number of genes: 13 mRNAs, 22 tRNAs and 2 rRNAs. All proteins encoded in the mtDNA are structural components of the multiprotein mitochondrial respiratory complexes (Figure 1). To build functional respiratory complexes, proteins encoded in mtDNA need to match structurally and functionally in a rigorous way with their counterparts encoded in the nuclear genome (Figure 1). This imposes a close‐fitting co‐evolution of both genomes challenged by the hugely different mechanism to generate variability for nDNA and mtDNA. As we noted above, genetic diversity in nDNA‐encoded OXPHOS genes is achieved between individuals by sexual reproduction ‐meiosis and recombination‐ by mixing and selecting different combinations of alleles for nuclear‐encoded subunits of the OXPHOS system available within the population gene pool. This does not cause diversity in mtDNA encoded genes since it is maternally inherited. 92 In addition, genetic diversity may also be achieved within the same individual. The diversity in nDNA can arise from cell differentiation, where the subset of genes of a given cell type is different from the subset of genes expressed in a different cell type, while all the mtDNA genes are expressed regardless the cell type. This may generate a particular genetic‐defined environment that could modify the requirements of OXPHOS demands and therefore be more or less permissive as to minor mismatches. Moreover, some structural subunits of the OXPHOS system encoded by the nDNA have tissue‐specific variants 93 and some isoforms are expressed only upon a functional defined stimulus (e.g., hypoxia inducible subunits). 94 , 95 Heterozygosity in nDNA implies that two alleles for OXPHOS structural proteins with differences in sequence can promote a variable match with the mtDNA partners. Most of the nuclear‐encoded OXPHOS genes are biparentally transmitted both in mice and humans, offering simultaneously alternative nuclear structural subunits of nuclear‐encoded genes. Therefore, the co‐expression of alternative alleles could generate a high number of structurally different respiratory complexes. As for mtDNA diversity, it can arise from natural (e.g., higher mutation rate in mtDNA, 13 , 96 mtDNA mutations in somatic cells followed by somatic segregation and amplification 97 and/or artificial processes, including the manipulation of oocytes for fertility treatments or to prevent transmission of mtDNA pathological mutations. 98
As we said before, interactions between proteins encoded in the two different cellular genomes were first described in transmitochondrial cybrid cell lines. 99 OXPHOS performance fails when confronting ‐in the same cytoplasm‐ mtDNA and nDNA from closely related species, suggesting a rapidly acquired match between both genomes in evolution. 100 , 101 , 102 This implies that different functional OXPHOS system may arise as a result of heterogeneous combination between mtDNA variants and the nuclear genome 67 , 75 (Figure 2a), being the mismatch between the gene products encoded by the mtDNA and the nDNA rapidly acquired during evolution. 103 The fact that there is a mismatch between them is hypothesized to be related to differences in the interaction between proteins that form the mitochondrial respiratory complexes as observed in neurons. 104 The balance between free respiratory complexes (RCs) and supercomplexes (SCs) allows adaptation to stress and nutrient conditions. 75 , 105 , 106 The role of the mito‐nuclear crosstalk and role of RCs and SCs by handling OXPHOS structural heterogeneity and metabolic plasticity remains to be elucidated.
1.5. Regulation of mitochondrial and nuclear gene expression
There is a complex and integrated crosstalk between the nucleus and mitochondria to reach the required expression programs in response to different stimuli. At the same time, a retrograde response between mitochondria and the rest of the organelles needs to interact and respond in coordination to the different cellular signals. 107 , 108 The way nDNA damage can induce mitochondrial loss of fitness led to identifying novel mechanisms to target after stochastic cellular damage. One example is the ERCC1‐XPF DNA endonuclease, which protects nuclear genome integrity by nucleotide excision repair, inter‐strand crosslink and double‐strand breaks. The first studies using genome‐wide RNAi screening in C. elegans linked genes critical for nDNA repair with mitophagy (PINK1, DPR1, ATM, or p53, among others). In mice, the Ercc1−/Δ model shows the capacity of increasing mitochondrial‐derived ROS levels and subsequent oxidative damage, thereby accelerating cell senescence and the aging phenotype. 109 This was ultimately demonstrated, showing that the rate of accumulation of senescence cells with age via this pathway is tissue‐specific. 110
Mitochondrial biogenesis and mtDNA replication are processes that require the interaction between nDNA and mtDNA (Figure 3). Different mtDNA variants with the same nuclear background can induce differences in the expression of the nuclear transcription factors responsible for mtDNA replication. The inducible mtDNA loss promotes lower acetylation marks of histone H3, H4 and H2B. 111 These results indicate that respiration regulates the nuclear epigenetics profiles through changes in the TCA (tricarboxylic acid) cycle metabolites like the α‐ketoglutarate (histone demethylation) and acetyl‐CoA (histone acetylation). But how do different mtDNA sequences alter the epigenetics profile? Changes in the chromosomal methylation patterns were found between the different mouse embryonic stem cell lines harboring different mtDNA haplotypes (CC9mus, CC9spretus, CC9dunni, and CC9pahari). Variations in the mitochondrial genome also regulate the global DNA methylation patterns by modifying the flux of the TCA by differential activity of enzymes as the malate dehydrogenase 2 (MDH2) or metabolite levels (i.e., α‐ketoglutarate). 112 Covalent modifications in the genome such as 5mC (5‐methylcytosine) and 5hmC (5‐hydroxymethylcytosine) are well studied in mammals. However, little is known about other epigenetic marks like the 6 mA (N6‐methyladenine). Six milliamperes have been described as being present in mice brains, accumulated in the prefrontal cortex and negatively correlated with the neural gene expression upon environmental stress. 113 Recently, the presence of 6 mA has been shown to be enriched in mtDNA from human HepG2 cells, with more than a 1,300‐fold higher incidence than the whole DNA cellular content. The presence of 6 mA led to a decrease in mitochondrial‐encoded genes transcription, mtDNA replication and decreased mitochondrial performance, suggesting its role in repressing the mitochondrial transcription factor TFAM binding. Moreover, upon mitochondrial stress promoted by hypoxia, overexpression of METTL4 methyltransferase and higher accumulation of 6 mA was reported. 114 Two issues that should be investigated further are whether the mtDNA variants presents different epigenetic mechanisms under different stress situations and if 6 mA in mtDNA promotes changes in the nuclear gene expression profile.
FIGURE 3.
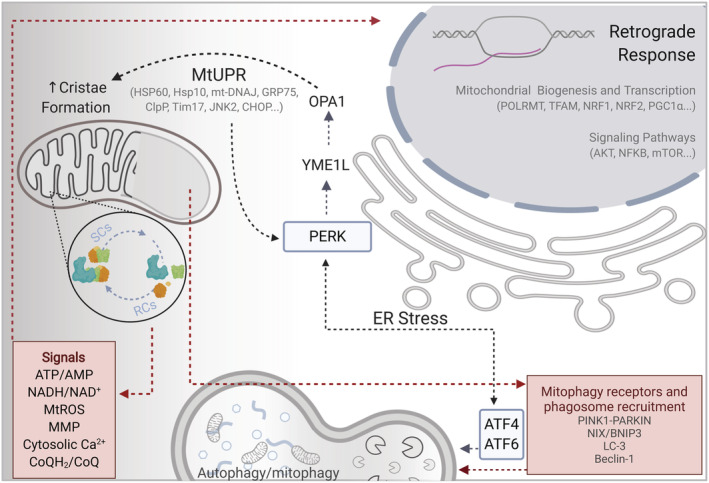
Summary of the proposed regulatory signals and pathways involved in the integration of the OXPHOS genetic complexity, retrograde response, and cytoplasmic communication
1.6. Retrograde response, cytoplasmic communication, and mitochondrial quality control
The role of the nucleo‐mitochondrial crosstalk, the so‐called retrograde response, remains to be fully elucidated. This complex signaling involves the activation of several nuclear‐encoded transcription factors required for mitochondrial maintenance by handling OXPHOS structural heterogeneity and metabolic plasticity. Furthermore, inter‐organelle contact sites can spatially regulate the trafficking of molecules and production and turnover of lipids under stress conditions like cell starvation or cell growth. One example is the interaction between mitochondria and lysosomes, regulating intracellular calcium homeostasis mediated by the lysosomal/late‐endosomal cation channel TRPML1. 115 Moreover, the mitochondria‐associated membranes (MAMs), connection sites between mitochondria and the endoplasmic reticulum (ER), are involved in the regulation of calcium homeostasis, ROS generation and autophagy (Figure 3). Recent studies show how autophagy and the fatty acid metabolism can be conditioned by the ER‐mitochondria interactions. 116 , 117 Moreover, ER stress induces assembly of the respiratory supercomplexes through the PERK‐eIF2α pathway. 105
The advantage of having mitochondria for eukaryotic cells not only bring up the energy enhanced, the new symbiosis results in the basis for the evolution of active cell death, best known as apoptosis. 118 , 119 At least three mechanisms involve mitochondria as the orchestrators of apoptosis 120 , 121 : (a) release of proteins that prompt activation of caspases family proteases, for example, cytochrome c release from mitochondria and Bax (Bcl‐2 associated X protein) translocation from cytosol to mitochondria are regarded as two key upstream molecular events of apoptosis. 122 , 123 The first one is a key component of the apoptosome complex for activation of caspase‐9 124 while the second is a pro‐apoptotic Bcl‐2 protein; (b) alteration of redox potential, which means that anything that decreases coupling efficiency triggers an increase in superoxide production that can activate caspases via interaction with proteins of the mitochondrial permeability transition complex 125 or oxidant‐induced DNA damage, 126 and was recently described as a mechanism of caspase‐independent cell‐death pathway activated by detrimental levels of ROS, 127 and (c) even if it is not a mechanism that trigger apoptosis, disruption of mtETC and the consequent decrease in OXPHOS and ATP production could kill a cell, indeed ATP appears to be required for downstream events in apoptosis. 128 Furthermore, it has been reported that dynamics of mitochondrial fusion and fission can regulate cell health. Specifically, increased fission, decreased fusion, or both of these events, cause a stage of apoptosis upstream of caspase activation and close to Bax translocation and cytochrome c release. 129 Interestingly, downregulating specific proteins involved in mitochondrial morphology like Drp1 and Fis1 prevents cytochrome c release and Bax translocation is lost, respectively. 130
To avoid cell death mitochondria, need strict regulation of their homeostasis. To prevent cellular damage while preserving a population of healthy mitochondria, several quality control mechanisms have evolved and all of them require the interaction between the nuclear and organelle genome. Among them, the more relevant are the translation attenuation process, mitochondrial unfolded protein response (mtUPR) and mitophagy and autophagy (Figure 3). Alterations in mitochondrial performance activate the GCN2 kinase (GCN2/EIF2AK4), which phosphorylates the eukaryotic initiation factor alpha (eIF2α) and promotes the inhibition of the protein synthesis and protein imported into mitochondria. PKR/EIF2AK2 and HRI/EIF2AK1 trigger translation attenuation upon different conditions as heme deficiency or viral infections. Furthermore, PERK/eIF2α K3 axis modulates ATF4 expression to adapt the cell to stress situations (Figure 3). 131 The mtUPR promotes the transcription of proteins involved in mitochondrial dynamics (e.g., DRP‐1) several mitochondrial proteases and chaperones (e.g., mtHSP70, m‐AAA/i‐AAA) and components of the mitochondrial protein import machinery (e.g., TIMM17, TIMM23). 132 The overall unfolded protein response and the unusual location of mitochondrial proteins in the cytoplasm activate the proteasome. 133 The selective removal of mitochondria was first described in 2005, 134 and its crucial role in the organelle quality control has been pointed out during the last decade. How mitochondria might be specifically selected for mitophagy remains an open question. It can be speculated that an unequal balance between mitochondrial fragmentation and fusion may give rise to formation of mitochondrial clusters where separate mitochondria are unable to fuse with the mitochondrial reticulum causing alteration in membrane potential, pH or loss of mtDNA. 135 Elucidating the mechanism of selective mitochondrial removal as a way of OXPHOS optimization and adaptability is quite challenging (Figure 3). Changes in the respiratory complex (RC) assemblies during stress conditions promote differences in the mitochondrial membrane fluidity and composition, as this is the case of cardiolipin content, which provides a dimeric cross‐linked phospholipid structure needed to avoid destabilization of RCs and supercomplexes (SCs) and improving bioenergetic functions. 136 CL is highly sensitive to oxidative damage due to its proximity to the ROS production sites and can be redistributed into the outer and inner mitochondrial membrane (OMM and IMM, respectively) under mitochondrial stress, affecting the selective degradation of mitochondria. Upon interaction of CL with LC3 and Beclin‐1, 137 , 138 the organelle is recognized and recruited into autophagosomes. In mammals, mitochondrial autophagy can be also mediated by the mitochondrial kinase PINK1 and the recruitment of the cytoplasmic E3 ubiquitin ligase PARKIN 139 or other mitophagy receptors from the BH3‐only family (NIX/BNIP3‐BNIP3L). 140 , 141 In MEFs, mitochondria are removed by mitophagy in response to hypoxia, which is dependent on HIF‐1 and the induction of its downstream target BNIP3. 142 Oxidative stress can also induce mitophagy, and since mitochondria may be both the major source and target for ROS damage, it could be selectively degraded to maintain cell survival in yeast and in mammalian cells. 143 , 144 Intriguingly, the same levels of ROS can induce autophagy or cell death in cancer cell lines, but primary mouse astrocytes fail to stimulate autophagy suggesting that normal cell lines tolerate higher levels of ROS through antioxidant proteins such as SOD2 and catalase. 145 , 146
2. INTRA‐INDIVIDUAL MTDNA VARIABILITY: NUCLEAR CONTROL OF MTDNA SEGREGATION
Recent results demonstrate that different combinations of nDNA/mtDNA promote differences in OXPHOS performance and impact on organismal metabolism. 72 , 75 , 76 , 77 , 78 , 79 However, the differences in the mitochondrial genome sequence emerge from (a) inter‐individual variability at a populational level or (b) intra‐individual variations, with the possibility of co‐existence of different mtDNA species in the same cell, also known as heteroplasmy. 147 As we explained above, natural generation of heteroplasmy is actively combated by several mechanisms during maternal transmission, including the degradation of the paternal mtDNA upon fertilization, and the existence of a genetic mtDNA bottleneck in oocyte development. Nevertheless, heteroplasmy is the most common feature in mtDNA‐linked diseases, where mutated copies of mtDNA are inherited from maternal oocytes. 12 , 92 Mutator mice (deficient in the proofreading capacity of the mitochondria DNA polymerase 148 , 149 ) and deletor mice (deficient in the mitochondrial Twinkle helicase 150 ) were both especially useful in studying the impact of random mtDNA mutations on the individual to animal models. Both mice lead to late‐onset RC deficiency but only the mutator mice present an early‐ageing phenotype 151 which implies that the association between mtDNA mutations and ageing must be clarified. However, heteroplasmy is nowadays recognized as a physiological condition, 30 , 152 most of the cells in healthy individuals present low‐level mtDNA heteroplasmy; what seems to be less physiological is a high load of heteroplasmy. Importantly, the level of somatic mtDNA mutations increases with age, 97 with infections 153 , 154 and during cancer progression. 155 However, what are the potential metabolic consequences for the body if more than one option of nDNA/mtDNA is available simultaneously at high levels? Since heteroplasmy is more common than expected and de novo somatic mutations rise with age, 154 , 156 understanding its implications in physiology become a medical necessity. Heteroplasmy is defined by divergence in sequence irrespective of the extension or the functional consequences of this divergence. However, discarding the direct pathological mutations in the mitochondrial genome, the expected tendency should be if differences in sequence are smaller, then so should be the consequences. Mathematical models support the hypothesis that random drift and different replication times can account for the accumulation and fixation of initial coexistence between polymorphic mtDNAs. 157 , 158 , 159 However, different studies strongly support the idea of competitions between mtDNAs, which can promote an intracellular selection of mitochondria with specific mtDNA. 160 , 161 , 162 Therefore, the adaptive response of the cell might modulate these differences to reach the required performance of the OXPHOS system and metabolic demands.
Mitonuclear epistatic interactions are widespread and make a significant contribution to the variability in disease penetrance. 32 , 163 Pathological mtDNA mutations can be found in homoplasmy, for example, mutations in ND genes associated with LHON 164 , 165 or m.1555A>G mutation in the 12S rRNA associated with deafness, 166 but it is much more frequent to find them in heteroplasmy. Regarding homoplasmy, it is common that the mutations have variable penetrance. For instance, the very same mutation can produce a fatal disease in some patients and no apparent symptoms in others, 167 while the molecular defect is the same in both cases. 32 This has been explained by a potentially different mechanism. It was reported that m.4290T>C mutation in humans and m.3739G>A in mice that affect mt‐Ti gene can reach homoplasmy because both allowed alternative folding of tRNAIle, the correct and the non‐functional one. Thus, in some individuals the insufficient amount of the functionally folded tRNA affects the mitochondrial protein capacity and ends up in abnormal elevation in ROS production. To compensate that mitochondrial biogenesis is increased leading to a sufficient quantity of the correctly folded tRNA and allowing fertile and healthy individuals. However, in some individuals the antioxidant capacity is efficient enough to blunt the mitochondrial biogenetic response, thereby preventing the rescue of the mitochondrial protein synthesis and causing the neurological disease observed in some individuals. 67 Another mechanism relies on the interaction between nuclear and mitochondrial variants. Thus, one‐point mutation affects the nuclear variant (KAL1 c.146G>T) that does not cause pathogenicity by itself and the other affects mtDNA (tRNA cys, m.5800A>G) that synergistically causes Kallman syndrome with different penetrance on progeny. 163 The pathogenicity of heteroplasmic mutation is more complex and depends on its segregation, mutation load and interactions with nuclear and mtDNA genetic contexts and the environment.
The recent discovery that in human populations, a dominant influence of the mtDNA over the nuclear genome can become manifest within one generation 30 strongly supports the belief that diverse mtDNA haplotypes may demand different responses from the nucleus also in humans, that may induce a malfunction if they are present together in the same cell. The study performed by Wei et al. (2019) corrected the long‐lasting belief that non‐pathological mtDNA variability in human populations was accumulated following a neutral equilibrium model without any relevance of potential interactions with nuclear genes, demonstrating the co‐evolution of nuclear and mitochondrial genomes. 30 The identification of nuclear genes whose products interact with mtDNA sequence variants, and the elucidation of their mode of action, will ultimately increase our understanding of mtDNA segregation and its role in complex genetic disorders, in which the description of all the genes coding for the structural components of the OXPHOS system as well as the protein synthesis apparatus of mitochondria, the mitochondrial ribosomal proteins, rRNAs and tRNAs do not allow us to understand why the very same mutation can cause different disease phenotypes. This is exemplified by the 3243A>G mutation in mtDNA, affecting the tRNALeuUUR gene that has been associated with the MELAS syndrome 168 in some patients, and with Type II diabetes mellitus and deafness in others. 169
The selection of mtDNAs during germ‐line transmission was first described as non‐existent 170 but, however, later it was actually observed. 161 Both observations can be compatible because mtDNA segregation is dependent on the nuclear background. The nuclear genetic control under the co‐existence of more than one species of non‐pathological mtDNA was pointed out for first time in 2003 in the work of Battersby et al. 171 The positive selection towards one haplotype over another was modulated by the nuclear genome, with the selection trend being stronger under a similar nuclear background 171 (Figure 4). Jokinen et al. 172 showed that differences in Gimap3 gene expression appear to be a critical factor for the segregation of mitochondrial genome only in mouse hematopoietic tissues. Since Gimap3 localizes to the ER and not mitochondria, is not in direct contact with mitochondrial nucleoids. Therefore, the segregation process is unlikely to be dependent on the identification and removal of the different mtDNA species at the level of the mtDNA molecule. It rather suggests that the selection is a consequence of organelle functional differences caused by the different mtDNAs. Jokinen et al. 172 also showed that the expression of a related protein, Gimap5, is involved in the mtDNA segregation within the hematopoietic compartment. Gimap5 has been shown to regulate T‐cell proliferation, 173 with clear evidence of its role in T‐cell autophagy and apoptosis. 174 Mechanisms and principles that govern mtDNA segregation using mouse models harboring both haplotypes co‐existing in the same cell have been studied for the last two decades. 160 , 161 , 162 , 171 , 175 , 176 , 177 , 178 Wai et al. analyzed the changes in the proportion of heteroplasmy (Balb/c vs. NZB mtDNA haplotypes) between primary follicles and secondary follicles (between post‐natal day 1 and post‐natal day 29) and found a significant increase in NZB mtDNA in more mature oocytes. 179 At that time, the authors did not attribute any functional role to the proportion of heteroplasmy because it was assumed that those haplotypes were phenotypically interchangeable, calling the phenomenon a neutral heteroplasmy. An assumption that was demonstrated to be inaccurate years later. In 2016, the exchange of polymorphic mtDNAs (C57 and NZB mtDNA) under the same nuclear background (C57BL/6JOlaHsd), revealed that both mtDNA variants do not behave as neutral. They impact on OXPHOS performance, organismal metabolism and healthy ageing 75 (Figure 2b). In 2007, Acton et al. proposed that heteroplasmy has by itself functional consequences. 180 They reported how heteroplasmic mice harboring mtDNA sequences with natural polymorphisms (BALB/cByJ and NZB mtDNA) developed systemic hypertension at an early age with alterations in cardiac function. 180 The systemic hypertension was corrected during the adulthood of the mice, but heteroplasmic animals also showed increased body and fat mass and hematological differences at almost all the stages through the mouse lifespan. 180 Years later, the study headed by D. C. Wallace showed that heteroplasmic mice harboring NZB and 129S6 mtDNAs under the BL/6C57J background presented reduced food intake and respiratory exchange ratio, and differences in behavior (i.e., lower locomotor activity) accompanied by cognitive impairment and increased stress response. 161 The heteroplasmy between mtDNA haplotypes is modulated during oocyte formation and additionally during early embryo development. This condition modulates overall embryo metabolism, the metabolic cell fitness and the formation induced pluripotent stem cells (iPSCs). 177 Genetic and pharmacological interventions were able to modulate OXPHOS performance and regulate competition among mtDNA haplotypes during oocyte development, early embryonic stages and during the post‐natal live of the individuals 177 , 178 (Figure 4). Differential crosstalk between mitochondrial and nuclear genomes can make heteroplasmy to behave as a co‐dominant phenomenon. Therefore, each mtDNA haplotype requests a proper and different nuclear response to reach the optimum efficiency with the minimum ROS production. 178 To resolve the mechanisms and principles that govern mtDNA segregation in germline transmission and mitotic and postmitotic segregation, different heteroplasmic mice models were challenged by inducing genetic modifications or pharmacological and nutritional interventions. 177 , 178 The kinetics of segregation and the preferred mtDNA variant depend on the nuclear background of the strain 177 , 178 (Figure 4). Differential crosstalk between mitochondrial and nuclear genomes can make heteroplasmy to behave as a co‐dominant phenomenon. Thus, each mtDNA haplotype requires a proper and different nuclear response to reach the optimum efficiency with the minimum ROS production. 178
FIGURE 4.
Heteroplasmic mouse models harboring in the same cytoplasm, different non‐pathological mtDNA variants‐haplotypes. MtDNA preference is cell‐type‐specific and not tissue‐specific. Also, the metabolic differentiation program determines the preferred mtDNA haplotype and the mtDNA segregation is driven by functional selection and strongly modulated by the crosstalk between the nucleus and mitochondria
3. CONCLUSIONS AND PERSPECTIVES
The genetic determination of the OXPHOS system function is the outcome of multiple nDNA–mtDNA matches, which impose an intrinsic and unprecedented multivariable characteristic. The mitochondrial OXPHOS system is regulated by novel systemic mechanisms; the understanding of how these mechanisms shape the overall organismal physiology is quite challenging. The accumulated data until date demonstrates that diverse nDNA–mtDNA matches are detected by the cell through feedback mechanisms that differ depending on the tissue, environment, or even organismal age. The conditions in which certain mtDNA types are selected, and the mechanisms for specific selection of some nDNA–mtDNA matches under different stresses and physiological processes (germline transmission, mitosis, changes in nutrient availability, aging, etc.), and whether these mechanisms are tissue‐specific remain to be fully elucidated. Such investigations will greatly improve our understanding of how mitochondrial bioenergetics regulate cellular processes, ranging from apoptosis to autophagy. Moreover, this will refine knowledge of related pathologies, including: mtDNA‐linked diseases, metabolic syndrome, obesity, cardiovascular disease, cancer, and aging. The regulation of mitochondrial function through variable nDNA–mtDNA matching, adjusted by compensatory mechanisms, introduces a critical new element in association studies linking human diseases and mtDNA variants. It is likely that in some context these associations would be manifested, while in others, they would be hidden. However, in all cases the intergenomic match would be a critical contributing factor to health and disease.
Recent advances demonstrate a previously undiscovered paradigm that mitochondrial therapy can alter nDNA–mtDNA matching, with wide biological implications. One potential field of interest would be tissue transplantation; for example, introduction of new nDNA–mtDNA pairs may accelerate the appearance of metabolic disorders in the acceptor organism. We would also benefit from a clearer understanding of competitive mtDNA segregation and which tissues can tolerate heteroplasmy. It is vital to investigate whether transcellular mtDNA migration can induce intercellular heteroplasmy, a potential consequence of aging through the accumulation of somatic mtDNA variants. Defining the mechanisms that control mtDNA segregation may help address the puzzling observation that during heteroplasmy, pathological mtDNA variants are often present and amplified, triggering disease with worsening symptoms.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
All the authors wrote the manuscript.
Abbreviations
- AICD
activation‐induced T‐cell death
- CR
control region
- D‐loop
displacement loop
- ER
endoplasmic reticulum
- iPSCs
induced pluripotent stem cells
- LHON
Leber's hereditary optic neuropathy
- MAM
mitochondria‐associated membrane
- MtDNA
mitochondrial DNA
- MtETC
mitochondrial electron transport chain
- mtUPR
mitochondrial unfolded protein response
- nDNA
nuclear DNA
- OXPHOS
oxidative phosphorylation
- RFLPs
restriction fragment length polymorphisms
- ROS
reactive oxygen species
- rRNA
ribosomal RNA
- SNPs
single nucleotide polymorphisms
- RCs
respiratory complexes
- SCs
respiratory supercomplexes
- TCA cycle
tricarboxylic acid cycle
- tRNA
transfer RNA
ACKNOWLEDGMENTS
The authors thank the whole GENOXPHOS group for suggestions and discussions. Figures are created with BioRender.com. This study was supported by MINECO: SAF2015‐65633‐R, RTI2018‐099357‐B‐I00 and HFSP (RGP0016/2018). A. V. L.‐V. was supported by fellowship SVP‐2013‐068089 from MINECO to J. A. E. In addition, A. V. L.‐V. was supported by a Postdoctoral Fellowship from the Fundación Alfonso Martin Escudero (Spain).
Lechuga‐Vieco AV, Justo‐Méndez R, Enríquez JA. Not all mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life. 2021;73:511–529. 10.1002/iub.2434
Funding information Fundación Alfonso Martin Escudero (Spain), Grant/Award Number: Postdoctoral Fellowship; Human Frontier Science Program, Grant/Award Number: RGP0016/2018; Ministerio de Ciencia e Innovación, Grant/Award Numbers: RTI2018‐099357‐B‐I00, SAF2015‐65633‐R; Ministerio de Economía y Competitividad, Grant/Award Number: RGP0016
Contributor Information
Ana Victoria Lechuga‐Vieco, Email: ana.lechuga-vieco@kennedy.ox.ac.uk.
José Antonio Enríquez, Email: jaenriquez@cnic.es.
REFERENCES
- 1. Martin WF, Garg S, Zimorski V. Endosymbiotic theories for eukaryote origin. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14:255–274. [DOI] [PubMed] [Google Scholar]
- 3. Kopp RE, Kirschvink JL, Hilburn IA, Nash CZ. The Paleoproterozoic snowball earth: A climate disaster triggered by the evolution of oxygenic photosynthesis. Proc Natl Acad Sci U S A. 2005;102:11131–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brocks JJ, Logan GA, Buick R, Summons RE. Archean molecular fossils and the early rise of eukaryotes. Science. 1999;285:1033–1036. [DOI] [PubMed] [Google Scholar]
- 5. Kump LR, Barley ME. Increased subaerial volcanism and the rise of atmospheric oxygen 2.5 billion years ago. Nature. 2007;448:1033–1036. [DOI] [PubMed] [Google Scholar]
- 6. Hoffmann F, Larsen O, Thiel V, et al. An anaerobic world in sponges. Geomicrobiol J. 2005;22:1–10. [Google Scholar]
- 7. Lane CE, Archibald JM. The eukaryotic tree of life: Endosymbiosis takes its TOL. Trends Ecol Evol. 2008;23:268–275. [DOI] [PubMed] [Google Scholar]
- 8. Martin W, Müller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392:37–41. [DOI] [PubMed] [Google Scholar]
- 9. Gray MW, Lang BF, Burger G. Mitochondria of protists. Annu Rev Genet. 2004;38:477–524. [DOI] [PubMed] [Google Scholar]
- 10. McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Analysis of mitochondrial subunit assembly into respiratory chain complexes using blue native polyacrylamide gel electrophoresis. Anal Biochem. 2007;364:128–137. [DOI] [PubMed] [Google Scholar]
- 11. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. [DOI] [PubMed] [Google Scholar]
- 12. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown WM, George M Jr, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 1979;76:1967–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Willett CS. Quantifying the elevation of mitochondrial DNA evolutionary substitution rates over nuclear rates in the intertidal copepod Tigriopus californicus . J Mol Evol. 2012;74:310–318. [DOI] [PubMed] [Google Scholar]
- 15. Hellberg ME. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 2006;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmer JD, Herbon LA. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 1988;28:87–97. [DOI] [PubMed] [Google Scholar]
- 17. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. [DOI] [PubMed] [Google Scholar]
- 18. Johnson AA, Johnson KA. Exonuclease proofreading by human mitochondrial DNA polymerase. J Biol Chem. 2001;276:38097–38107. [DOI] [PubMed] [Google Scholar]
- 19. Mason PA, Lightowlers RN. Why do mammalian mitochondria possess a mismatch repair activity? FEBS Lett. 2003;554:6–9. [DOI] [PubMed] [Google Scholar]
- 20. Anderson CT, Friedberg EC. The presence of nuclear and mitochondrial uracil‐DNA glycosylase in extracts of human KB cells. Nucleic Acids Res. 1980;8:875–888. [PMC free article] [PubMed] [Google Scholar]
- 21. de Souza‐Pinto NC, Eide L, Hogue BA, et al. Repair of 8‐oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8‐oxoguanine accumulates in the mitochondrial dna of OGG1‐defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 22. Coffey G, Lakshmipathy U, Campbell C. Mammalian mitochondrial extracts possess DNA end‐binding activity. Nucleic Acids Res. 1999;27:3348–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Myers KA, Saffhill R, O'Connor PJ. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis. 1988;9:285–292. [DOI] [PubMed] [Google Scholar]
- 24. Nakabeppu Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat Res. 2001;477:59–70. [DOI] [PubMed] [Google Scholar]
- 25. Xu W, Boyd RM, Tree MO, Samkari F, Zhao L. Mitochondrial transcription factor a promotes DNA strand cleavage at abasic sites. Proc Natl Acad Sci U S A. 2019;116:17792–17799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moretton A, Morel F, Macao B, et al. Selective mitochondrial DNA degradation following double‐strand breaks. PLoS One. 2017;12:e0176795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peeva V, Blei D, Trombly G, et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun. 2018;9:1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Furda AM, Marrangoni AM, Lokshin A, Van Houten B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair (Amst). 2012;11:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldberg A, Wildman DE, Schmidt TR, et al. Adaptive evolution of cytochrome c oxidase subunit VIII in anthropoid primates. Proc Natl Acad Sci U S A. 2003;100:5873–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei W, Tuna S, Keogh MJ, et al. Germline selection shapes human mitochondrial DNA diversity. Science. 2019;364:eaau6520. [DOI] [PubMed] [Google Scholar]
- 31. Gregorius HR, Ross MD. Selection with gene‐cytoplasm interactions. I. Maintenance of cytoplasm polymorphisms. Genetics. 1984;107:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreno‐Loshuertos R, Ferrín G, Acín‐Pérez R, et al. Evolution meets disease: Penetrance and functional epistasis of mitochondrial tRNA mutations. PLoS Genet. 2011;7:e1001379–e1001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rand DM, Clark AG, Kann LM. Sexually antagonistic cytonuclear fitness interactions in Drosophila melanogaster . Genetics. 2001;159:173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parsons TJ, Muniec DS, Sullivan K, et al. A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet. 1997;15:363–368. [DOI] [PubMed] [Google Scholar]
- 35. Bronstein O, Kroh A, Haring E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol Biol. 2018;18:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Torroni A, Huoponen K, Francalacci P, et al. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144:1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denaro M, Blanc H, Johnson MJ, et al. Ethnic variation in Hpa 1 endonuclease cleavage patterns of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1981;78:5768–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Merriwether DA, Clark AG, Ballinger SW, et al. The structure of human mitochondrial DNA variation. J Mol Evol. 1991;33:543–555. [DOI] [PubMed] [Google Scholar]
- 39. Ballinger SW, Schurr TG, Torroni A, et al. Southeast Asian mitochondrial DNA analysis reveals genetic continuity of ancient mongoloid migrations. Genetics. 1992;130:139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Torroni A, Neel JV, Barrantes R, Schurr TG, Wallace DC. Mitochondrial DNA "clock" for the Amerinds and its implications for timing their entry into North America. Proc Natl Acad Sci U S A. 1994;91:1158–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ruiz‐Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. [DOI] [PubMed] [Google Scholar]
- 42. Camus MF, Wolff JN, Sgrò CM, Dowling DK. Experimental support that natural selection has shaped the latitudinal distribution of mitochondrial haplotypes in Australian Drosophila melanogaster . Mol Biol Evol. 2017;34:2600–2612. [DOI] [PubMed] [Google Scholar]
- 43. Bi R, Zhang W, Yu D, et al. Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer's disease in Han Chinese. Neurobiol Aging. 2015;36(1604):e1607–e1616. [DOI] [PubMed] [Google Scholar]
- 44. Mousavizadeh K, Rajabi P, Alaee M, Dadgar S, Houshmand M. Usage of mitochondrial D‐loop variation to predict risk for Huntington disease. Mitochondrial DNA. 2015;26:579–582. [DOI] [PubMed] [Google Scholar]
- 45. Hudson G, Nalls M, Evans JR, et al. Two‐stage association study and meta‐analysis of mitochondrial DNA variants in Parkinson disease. Neurology. 2013;80:2042–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ban M, Elson J, Walton A, et al. Investigation of the role of mitochondrial DNA in multiple sclerosis susceptibility. PLoS One. 2008;3:e2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tranah GJ, Santaniello A, Caillier SJ, et al. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology. 2015;85:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chinnery PF, Mowbray C, Patel SK, et al. Mitochondrial DNA haplogroups and type 2 diabetes: A study of 897 cases and 1010 controls. J Med Genet. 2007;44:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liao WQ, Pang Y, Yu CA, Wen JY, Zhang YG, Li XH. Novel mutations of mitochondrial DNA associated with type 2 diabetes in Chinese Han population. Tohoku J Exp Med. 2008;215:377–384. [DOI] [PubMed] [Google Scholar]
- 50. Fang H, Hu N, Zhao Q, et al. mtDNA Haplogroup N9a increases the risk of type 2 diabetes by altering mitochondrial function and intracellular mitochondrial signals. Diabetes. 2018;67:1441–1453. [DOI] [PubMed] [Google Scholar]
- 51. Canter JA, Kallianpur AR, Parl FF, Millikan RC. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African‐American women. Cancer Res. 2005;65:8028–8033. [DOI] [PubMed] [Google Scholar]
- 52. Cocoş R, Schipor S, Badiu C, Raicu F. Mitochondrial DNA haplogroup K as a contributor to protection against thyroid cancer in a population from Southeast Europe. Mitochondrion. 2018;39:43–50. [DOI] [PubMed] [Google Scholar]
- 53. Ruiz‐Pesini E, Lapeña AC, Díez‐Sánchez C, et al. Human mtDNA haplogroups associated with high or reduced spermatozoa motility. Am J Hum Genet. 2000;67:682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baudouin SV, Saunders D, Tiangyou W, et al. Mitochondrial DNA and survival after sepsis: A prospective study. Lancet. 2005;366:2118–2121. [DOI] [PubMed] [Google Scholar]
- 55. Guzmán‐Fulgencio M, Berenguer J, Micheloud D, et al. European mitochondrial haplogroups are associated with CD4+ T cell recovery in HIV‐infected patients on combination antiretroviral therapy. J Antimicrob Chemother. 2013;68:2349–2357. [DOI] [PubMed] [Google Scholar]
- 56. Brown MD, Starikovskaya E, Derbeneva O, et al. The role of mtDNA background in disease expression: A new primary LHON mutation associated with Western Eurasian haplogroup J. Hum Genet. 2002;110:130–138. [DOI] [PubMed] [Google Scholar]
- 57. Kalman B, Lublin FD, Alder H. Characterization of the mitochondrial DNA in patients with multiple sclerosis. J Neurol Sci. 1996;140:75–84. [DOI] [PubMed] [Google Scholar]
- 58. van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carrieri G, Bonafè M, De Luca M, et al. Mitochondrial DNA haplogroups and APOE4 allele are non‐independent variables in sporadic Alzheimer's disease. Hum Genet. 2001;108:194–198. [DOI] [PubMed] [Google Scholar]
- 60. Bonafè M, Barbi C, Olivieri F, et al. An allele of HRAS1 3'variable number of tandem repeats is a frailty allele: Implication for an evolutionarily‐conserved pathway involved in longevity. Gene. 2002;286:121–126. [DOI] [PubMed] [Google Scholar]
- 61. Niemi AK, Hervonen A, Hurme M, Karhunen PJ, Jylhä M, Majamaa K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Hum Genet. 2003;112:29–33. [DOI] [PubMed] [Google Scholar]
- 62. Tanaka M, Gong JS, Zhang J, Yoneda M, Yagi K. Mitochondrial genotype associated with longevity. Lancet. 1998;351:185–186. [DOI] [PubMed] [Google Scholar]
- 63. Pinos T, Nogales‐Gadea G, Ruiz JR, et al. Are mitochondrial haplogroups associated with extreme longevity? A study on a Spanish cohort. Age (Dordr). 2012;34:227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007;67:4687–4694. [DOI] [PubMed] [Google Scholar]
- 65. Mosquera‐Miguel A, Alvarez‐Iglesias V, Carracedo A, et al. Is mitochondrial DNA variation associated with sporadic breast cancer risk? Cancer Res. 2008;68:623–625. author reply 624. [DOI] [PubMed] [Google Scholar]
- 66. Gaweda‐Walerych K, Zekanowski C. The impact of mitochondrial DNA and nuclear genes related to mitochondrial functioning on the risk of Parkinson's disease. Curr Genomics. 2013;14:543–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moreno‐Loshuertos R, Acín‐Pérez R, Fernández‐Silva P, et al. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat Genet. 2006;38:1261–1268. [DOI] [PubMed] [Google Scholar]
- 68. Ishikawa K, Hayashi J. Generation of mtDNA‐exchanged cybrids for determination of the effects of mtDNA mutations on tumor phenotypes. Methods Enzymol. 2009;457:335–346. [DOI] [PubMed] [Google Scholar]
- 69. Lee WT, Cain JE, Cuddihy A, et al. Mitochondrial DNA plasticity is an essential inducer of tumorigenesis. Cell Death Discov. 2016;2:16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fernández‐Moreno M, Soto‐Hermida A, Vázquez‐Mosquera ME, et al. Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta‐analysis and functional study. Ann Rheum Dis. 2017;76:1114–1122. [DOI] [PubMed] [Google Scholar]
- 71. Dalmao‐Fernández A, Lund J, Hermida‐Gómez T, et al. Impaired metabolic flexibility in the osteoarthritis process: A study on Transmitochondrial Cybrids. Cells. 2020;9:809–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu X, Gimsa U, Wester‐Rosenlöf L, et al. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 2009;19:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kumarasamy S, Gopalakrishnan K, Yerga‐Woolwine S, Farms P, Joe B. Novel conplastic strains reveal direct and independent effects of mitochondrial genomic variants on intrinsic aerobic fitness. FASEB J. 2012;26:1098.1097. [Google Scholar]
- 74. Houštěk J, Vrbacký M, Hejzlarová K, et al. Effects of mtDNA in SHR‐mtF344 versus SHR conplastic strains on reduced OXPHOS enzyme levels, insulin resistance, cardiac hypertrophy, and systolic dysfunction. Physiol Genomics. 2014;46:671–678. [DOI] [PubMed] [Google Scholar]
- 75. Latorre‐Pellicer A, Moreno‐Loshuertos R, Lechuga‐Vieco AV, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–565. [DOI] [PubMed] [Google Scholar]
- 76. Hirose M, Künstner A, Schilf P, et al. Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci Rep. 2017;7:15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Niemann J, Johne C, Schröder S, et al. An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle. Free Radic Biol Med. 2017;102:174–187. [DOI] [PubMed] [Google Scholar]
- 78. Tourmente M, Hirose M, Ibrahim S, et al. mtDNA polymorphism and metabolic inhibition affect sperm performance in conplastic mice. Reproduction. 2017;154:341–354. [DOI] [PubMed] [Google Scholar]
- 79. Hirose M, Künstner A, Schilf P, et al. A natural mtDNA polymorphism in complex III is a modifier of Healthspan in mice. Int J Mol Sci. 2019;20:2359–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chomyn A. Platelet‐mediated transformation of human mitochondrial DNA‐less cells. Methods in enzymology, Volume 264 New York; London: Academic Press, 1996; p. 334–339. [DOI] [PubMed] [Google Scholar]
- 81. Kenney MC, Chwa M, Atilano SR, et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: Insights into mitochondrial‐nuclear interactions. Hum Mol Genet. 2014;23:3537–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kenney MC, Chwa M, Atilano SR, et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: Implications for age‐related macular degeneration. PLoS One. 2013;8:e54339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shen JM, Feng L, Feng C. Role of mtDNA haplogroups in the prevalence of osteoarthritis in different geographic populations: A meta‐analysis. PLoS One. 2014;9:e108896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Soto‐Hermida A, Fernández‐Moreno M, Oreiro N, Fernández‐López C, Rego‐Pérez I, Blanco FJ. mtDNA haplogroups and osteoarthritis in different geographic populations. Mitochondrion. 2014;15:18–23. [DOI] [PubMed] [Google Scholar]
- 85. Patel TH, Norman L, Chang S, et al. European mtDNA variants are associated with differential responses to Cisplatin, an anticancer drug: Implications for drug resistance and side effects. Front Oncol. 2019;9:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Barrientos A, Kenyon L, Moraes CT. Human xenomitochondrial cybrids. Cellular models of mitochondrial complex I deficiency. J Biol Chem. 1998;273:14210–14217. [DOI] [PubMed] [Google Scholar]
- 87. Sackton TB, Haney RA, Rand DM. Cytonuclear coadaptation in Drosophila: Disruption of cytochrome c oxidase activity in backcross genotypes. Evolution. 2003;57:2315–2325. [DOI] [PubMed] [Google Scholar]
- 88. Chou JY, Hung YS, Lin KH, Lee HY, Leu JY. Multiple molecular mechanisms cause reproductive isolation between three yeast species. PLoS Biol. 2010;8:e1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK. Mitonuclear interactions: Evolutionary consequences over multiple biological scales. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kaminski M, Kiessling M, Süss D, Krammer PH, Gülow K. Novel role for mitochondria: Protein kinase Ctheta‐dependent oxidative signaling organelles in activation‐induced T‐cell death. Mol Cell Biol. 2007;27:3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ali AT, Boehme L, Carbajosa G, Seitan VC, Small KS, Hodgkinson A. Nuclear genetic regulation of the human mitochondrial transcriptome. Elife. 2019;8:e41927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1980;77:6715–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lomax MI, Grossman LI. Tissue‐specific genes for respiratory proteins. Trends Biochem Sci. 1989;14:501–503. [DOI] [PubMed] [Google Scholar]
- 94. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF‐1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. [DOI] [PubMed] [Google Scholar]
- 95. Tello D, Balsa E, Acosta‐Iborra B, et al. Induction of the mitochondrial NDUFA4L2 protein by HIF‐1α decreases oxygen consumption by inhibiting complex I activity. Cell Metab. 2011;14:768–779. [DOI] [PubMed] [Google Scholar]
- 96. Eyre‐Walker A, Awadalla P. Does human mtDNA recombine? J Mol Evol. 2001;53:430–435. [DOI] [PubMed] [Google Scholar]
- 97. Payne BA, Wilson IJ, Yu‐Wai‐Man P, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Herbert M, Turnbull D. Progress in mitochondrial replacement therapies. Nat Rev Mol Cell Biol. 2018;19:71–72. [DOI] [PubMed] [Google Scholar]
- 99. Giles RE, Stroynowski I, Wallace DC. Characterization of mitochondrial DNA in chloramphenicol‐resistant interspecific hybrids and a cybrid. Somatic Cell Genet. 1980;6:543–554. [DOI] [PubMed] [Google Scholar]
- 100. De Francesco L, Attardi G, Croce CM. Uniparental propagation of mitochondrial DNA in mouse‐human cell hybrids. Proc Natl Acad Sci U S A. 1980;77:4079–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kenyon L, Moraes CT. Expanding the functional human mitochondrial DNA database by the establishment of primate xenomitochondrial cybrids. Proc Natl Acad Sci U S A. 1997;94:9131–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ellison CK, Burton RS. Disruption of mitochondrial function in interpopulation hybrids of Tigriopus californicus . Evolution. 2006;60:1382–1391. [PubMed] [Google Scholar]
- 103. Ma H, Marti Gutierrez N, Morey R, et al. Incompatibility between nuclear and mitochondrial genomes contributes to an interspecies reproductive barrier. Cell Metab. 2016;24:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hevner RF, Wong‐Riley MT. Mitochondrial and nuclear gene expression for cytochrome oxidase subunits are disproportionately regulated by functional activity in neurons. J Neurosci. 1993;13:1805–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Balsa E, Soustek MS, Thomas A, et al. ER and nutrient stress promote assembly of respiratory chain Supercomplexes through the PERK‐eIF2α Axis. Mol Cell. 2019;74:877–890.e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. García‐Poyatos C, Cogliati S, Calvo E, et al. Scaf1 promotes respiratory supercomplexes and metabolic efficiency in zebrafish. EMBO Rep. 2020;21:e50287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rand DM, Haney RA, Fry AJ. Cytonuclear coevolution: The genomics of cooperation. Trends Ecol Evol. 2004;19:645–653. [DOI] [PubMed] [Google Scholar]
- 108. Osada N, Akashi H. Mitochondrial‐nuclear interactions and accelerated compensatory evolution: Evidence from the primate cytochrome C oxidase complex. Mol Biol Evol. 2012;29:337–346. [DOI] [PubMed] [Google Scholar]
- 109. Robinson AR, Yousefzadeh MJ, Rozgaja TA, et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018;17:259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yousefzadeh MJ, Zhao J, Bukata C, et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell. 2020;19:e13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Martínez‐Reyes I, Diebold LP, Kong H, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016;61:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lee WT, Sun X, Tsai TS, et al. Mitochondrial DNA haplotypes induce differential patterns of DNA methylation that result in differential chromosomal gene expression patterns. Cell Death Discov. 2017;3:17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yao B, Cheng Y, Wang Z, et al. DNA N6‐methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat Commun. 2017;8:1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hao Z, Wu T, Cui X, et al. N6‐Deoxyadenosine methylation in mammalian mitochondrial DNA. Mol Cell. 2020;78:382–395.e388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Peng W, Wong YC, Krainc D. Mitochondria‐lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc Natl Acad Sci U S A. 2020;117:19266–19275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bosc C, Broin N, Fanjul M, et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria‐endoplasmic reticulum contact sites. Nat Commun. 2020;11:4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ibrahim A, Yucel N, Kim B, Arany Z. Local mitochondrial ATP production regulates endothelial fatty acid uptake and transport. Cell Metab. 2020;32:309–319.e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kroemer G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997;4:443–456. [DOI] [PubMed] [Google Scholar]
- 119. Redza‐Dutordoir M, Averill‐Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2016;1863:2977–2992. [DOI] [PubMed] [Google Scholar]
- 120. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 121. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell‐free extracts: Requirement for dATP and cytochrome c. Cell. 1996;86:147–157. [DOI] [PubMed] [Google Scholar]
- 123. Hsu YT, Wolter KG, Youle RJ. Cytosol‐to‐membrane redistribution of Bax and Bcl‐X(L) during apoptosis. Proc Natl Acad Sci U S A. 1997;94:3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. [DOI] [PubMed] [Google Scholar]
- 125. Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. [DOI] [PubMed] [Google Scholar]
- 126. Grishko V, Pastukh V, Solodushko V, Gillespie M, Azuma J, Schaffer S. Apoptotic cascade initiated by angiotensin II in neonatal cardiomyocytes: Role of DNA damage. Am J Physiol Heart Circ Physiol. 2003;285:H2364–H2372. [DOI] [PubMed] [Google Scholar]
- 127. Holze C, Michaudel C, Mackowiak C, et al. Oxeiptosis, a ROS‐induced caspase‐independent apoptosis‐like cell‐death pathway. Nat Immunol. 2018;19:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 129. Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. [DOI] [PubMed] [Google Scholar]
- 132. Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS‐1 promotes OXPHOS recovery during the UPR(mt). Mol Cell. 2015;58:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wrobel L, Topf U, Bragoszewski P, et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015;524:485–488. [DOI] [PubMed] [Google Scholar]
- 134. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. [DOI] [PubMed] [Google Scholar]
- 135. Saito T, Sadoshima J. Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res. 2015;116:1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Li XX, Tsoi B, Li YF, Kurihara H, He RR. Cardiolipin and its different properties in mitophagy and apoptosis. J Histochem Cytochem. 2015;63:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Antón Z, Landajuela A, Hervás JH, et al. Human Atg8‐cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy. 2016;12:2386–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differentiation. 2009;16:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang H, Bosch‐Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 143. Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005;12:1613–1621. [DOI] [PubMed] [Google Scholar]
- 144. Rodríguez‐Hernández A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. [DOI] [PubMed] [Google Scholar]
- 145. Chen Y, McMillan‐Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron‐transport‐chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–4166. [DOI] [PubMed] [Google Scholar]
- 146. Chen Y, McMillan‐Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–182. [DOI] [PubMed] [Google Scholar]
- 147. Røyrvik EC, Burgstaller JP, Johnston IG. mtDNA diversity in human populations highlights the merit of haplotype matching in gene therapies. Mol Hum Reprod. 2016;22:809–817. [DOI] [PubMed] [Google Scholar]
- 148. Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. [DOI] [PubMed] [Google Scholar]
- 149. Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. [DOI] [PubMed] [Google Scholar]
- 150. Tyynismaa H, Mjosund KP, Wanrooij S, et al. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late‐onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ahlqvist KJ, Hamalainen RH, Yatsuga S, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15:100–109. [DOI] [PubMed] [Google Scholar]
- 152. Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat Rev Genet. 2015;16:530–542. [DOI] [PubMed] [Google Scholar]
- 153. Li M, Foli Y, Liu Z, et al. High frequency of mitochondrial DNA mutations in HIV‐infected treatment‐experienced individuals. HIV Med. 2017;18:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ziada AS, Lu MY, Ignas‐Menzies J, et al. Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell. 2019;18:e13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Yuan Y, Ju YS, Kim Y, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet. 2020;52:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Aryaman J, Bowles C, Jones NS, Johnston IG. Mitochondrial network state scales mtDNA genetic dynamics. Genetics. 2019;212:1429–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chinnery PF, Samuels DC. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am J Hum Genet. 1999;64:1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Brown DT, Samuels DC, Michael EM, Turnbull DM, Chinnery PF. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am J Hum Genet. 2001;68:533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jenuth JP, Peterson AC, Shoubridge EA. Tissue‐specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet. 1997;16:93–95. [DOI] [PubMed] [Google Scholar]
- 161. Sharpley MS, Marciniak C, Eckel‐Mahan K, et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Burgstaller JP, Johnston IG, Jones NS, et al. MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 2014;7:2031–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wang F, Huang GD, Tian H, et al. Point mutations in KAL1 and the mitochondrial gene MT‐tRNA(cys) synergize to produce Kallmann syndrome phenotype. Sci Rep. 2015;5:13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. [DOI] [PubMed] [Google Scholar]
- 165. Vilkki J, Savontaus ML, Nikoskelainen EK. Genetic heterogeneity in Leber hereditary optic neuroretinopathy revealed by mitochondrial DNA polymorphism. Am J Hum Genet. 1989;45:206–211. [PMC free article] [PubMed] [Google Scholar]
- 166. Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic‐induced and non‐syndromic deafness. Nat Genet. 1993;4:289–294. [DOI] [PubMed] [Google Scholar]
- 167. Lamantea E, Carrara F, Mariotti C, Morandi L, Tiranti V, Zeviani M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome <em>b</em> gene associated with a combined deficiency of complexes I and III. Neuromuscul Disord. 2002;12:49–52. [DOI] [PubMed] [Google Scholar]
- 168. Goto Y‐i, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. [DOI] [PubMed] [Google Scholar]
- 169. van den Ouweland JM, Lemkes HH, Ruitenbeek W, et al. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–371. [DOI] [PubMed] [Google Scholar]
- 170. Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14:146–151. [DOI] [PubMed] [Google Scholar]
- 171. Battersby BJ, Loredo‐Osti JC, Shoubridge EA. Nuclear genetic control of mitochondrial DNA segregation. Nat Genet. 2003;33:183–186. [DOI] [PubMed] [Google Scholar]
- 172. Jokinen R, Lahtinen T, Marttinen P, et al. Quantitative changes in Gimap3 and Gimap5 expression modify mitochondrial DNA segregation in mice. Genetics. 2015;200:221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Patterson AR, Endale M, Lampe K, et al. Gimap5‐dependent inactivation of GSK3β is required for CD4(+) T cell homeostasis and prevention of immune pathology. Nat Commun. 2018;9:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Serrano D, Ghobadi F, Boulay G, Ilangumaran S, Lavoie C, Ramanathan S. GTPase of the immune‐associated nucleotide protein 5 regulates the Lysosomal calcium compartment in T lymphocytes. Front Immunol. 2017;8:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Johnston IG, Jones NS. Evolution of cell‐to‐cell variability in stochastic, controlled, Heteroplasmic mtDNA populations. The American Journal of Human Genetics. 2016;99:1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Burgstaller JP, Kolbe T, Havlicek V, et al. Large‐scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations. Nat Commun. 2018;9:2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Latorre‐Pellicer A, Lechuga‐Vieco AV, Johnston IG, et al. Regulation of mother‐to‐offspring transmission of mtDNA Heteroplasmy. Cell Metab. 2019;30:1120–1130.e1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Lechuga‐Vieco AV, Latorre‐Pellicer A, Johnston IG, et al. Cell identity and nucleo‐mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci Adv. 2020;6:eaba5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2008;40:1484–1488. [DOI] [PubMed] [Google Scholar]
- 180. Acton BM, Lai I, Shang X, Jurisicova A, Casper RF. Neutral mitochondrial Heteroplasmy alters physiological function in Mice1. Biol Reprod. 2007;77:569–576. [DOI] [PubMed] [Google Scholar]