

Abstract
A series of 4d/4f‐polyarsenides, ‐polyarsines and ‐polystibines was obtained by reduction of the Mo‐pnictide precursor complexes [{CptMo(CO)2}2(μ,η 2:2‐E2)] (E=As, Sb; Cpt=tBu substituted cyclopentadienyl) with two different divalent samarocenes [Cp*2Sm] and [(CpMe4nPr)2Sm]. For the reductive conversion of the Mo‐stibide only one product was isolated, featuring a planar tetrastibacyclobutadiene moiety as an unprecedented ligand for organometallic compounds. For the corresponding Mo‐arsenide a tetraarsacyclobutadiene and a second species with a side‐on coordinated As2 2− anion was isolated. The latter can be considered as reaction intermediate for the formation of the tetraarsacyclobutadiene.
Keywords: lanthanides, molybdenum, polyarsenides, polystibides, reduction
Ring up: By reduction of [{CptMo(CO)2}2(μ,η 2:2‐E2)] (E=As, Sb; Cpt=tBu substituted cyclopentadienyl) with different samarocenes, new d/f‐polyarsenides and unprecedented 4d/4f‐polypnictogens were obtained. The latter feature for the first time a planar Sb4‐ring, which shows a rare analogy to cyclobutadiene, as ligand in organometallic chemistry.
Polypnictides are well known examples for Zintl ions. [1] Due to the broad scope of potential applications (e.g. in solar cells), the solid state chemistry of rare earth polypnictides has been investigated intensively. [2] The heavier congeners of nitrogen form numerous ring and cage motifs in the solid state. [1] Due to the isoelectronic relationship between C−H and E (E=group 15 element) polypnictides such as cyclo‐P4 2− or cyclo‐P5 − (isoelectronic to the cyclobutadiene dianion or cyclopentadienyl anion) are versatile ligands in d‐block chemistry. [3]
Based on this, the chemistry of molecular f‐element complexes ligated by polypnictides is a current and growing research topic. The first f‐element polyphosphide [(Cp*2Sm)4P8] was synthesized in 2009. [4] It was obtained by a reductive approach, in which a divalent samarium complex was reacted with elemental P4. In a comparable reductive pathway d/f‐element polyphosphides were synthesized starting from d‐element (poly)phosphide complexes as precursor compounds. [5] While these molecular polyphosphide f‐element compounds and to a minor extent the analogue polyarsenides have already been reported, [6] the heavier congeners are still rare and only few 4f‐polystibides are known. The first compound in this area was published by Evans et al. in 1992. They reported the reductive cleavage of Sb−C bonds by using Sb(nBu)3 and [Cp*2Sm] to obtain [{(Cp*)2Sm}3(μ‐η 2:2:1Sb3)(thf)]. [7] Other polystibides of the f‐elements are [(η 5‐CpMe 2Dy)3 {μ‐(SbMes)3Sb}], [K([2.2.2]crypt)]3[Ln(η 4‐Sb4)3]⋅4py (py=pyridine; Ln=La, Y, Ho, Er, Lu) and [(Cp*2Sm)4Sb8]. [8] Additionally, only one d/f‐polystibide [{(Cp*2Sm)3(μ 4,η 1:2:2:2‐Sb4)}2Hg] is known. It was isolated as an intermediate in the reaction of [Cp*2Sm] with Sb/Hg alloy just recently without any further characterization. [8c]
However, it is not known if the isoelectronic relation between polystibides and hydrocarbon based ligands in f‐element chemistry is still valid as seen for P and As and whether it is possible to use such isoelectronic moieties as bridging ligands in d/f‐compounds.
Considering this lack of knowledge, we felt challenged to further expand the class of d/f‐element polystibides and compare them to their lighter homologues. In earlier investigations, we reported the reduction of [{CpMo(CO)2}2(μ,η 2:2‐P2)] with [Cp*2Sm], which resulted in 4d/4f‐element polyphosphides. [5a] [Cp*2Sm] acts here as single‐electron‐transfer reagent (SET) [9] and thus induces the reduction of [{CpMo(CO)2}2(μ,η 2:2‐P2)]. This reaction resulted in a product mixture of three different products (Scheme 1, A as main product as well as B and C as minor products). Due to the fact that the analogous Mo‐arsenides and Mo‐stibides [{CpMo(CO)2}2(μ,η 2:2‐E2)] (E=As, Sb) are available, [10] we decided to use these compounds for accessing d/f‐element polypnictides of the heavier group 15 elements. For solubility reasons, the tBu substituted cyclopentadienyl (Cpt) was employed as a ligand on the molybdenum precursor. Reduction of the previously unknown [{CptMo(CO)2}2(μ,η 2:2‐As2)] with solvent free [Cp*2Sm] in hot n‐hexane resulted in a straightforward formation of the mixed d/f‐metal species [(Cp*2Sm)2As2(CptMo(CO)2)2] (1) (Scheme 2 and Figure 1).
Scheme 1.
Earlier work: Observed mixture of products in the reduction of [{CpMo(CO)2}2(μ,η 2:2‐P2)]. [5a]
Scheme 2.
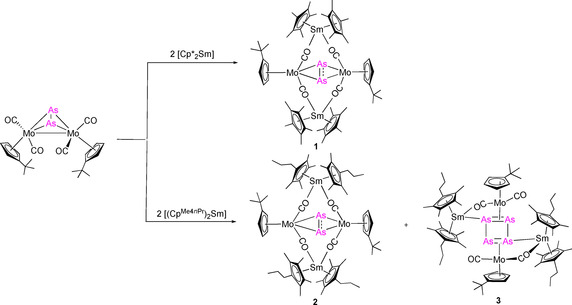
Reduction of [{CptMo(CO)2}2(μ,η 2:2‐As2)] with [Cp*2Sm(thf)2] and [(CpMe4nPr)2Sm], resulting in [(Cp*2Sm)2As2(CptMo(CO)2)2] (1) [((CpMe4nPr)2Sm)2As2(CptMo(CO)2)2] (2), and [((CpMe4nPr)2Sm)2As4(CptMo(CO)2)2] (3).
Figure 1.
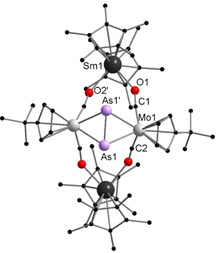
Molecular structure of 1 in the solid state. [22] Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sm1−O1 2.378(4), Sm1−O2′ 2.391(4), Mo1−As1 2.6847(9), Mo1−As1′ 2.6997(9), Mo1−C1 1.892(6), As1−As1′ 2.238(2), C1−O1 1.212(3), O1‐Sm‐O2′ 77.6(2), Mo1‐As1‐Mo1′ 103.92(3).
Despite the lack of direct Sm–As contacts, compound 1 is a remarkable species because it represents the formally expected product upon a two‐electron reduction of [{CptMo(CO)2},2(μ,η 2:2‐As2)]. The Mo−Mo bond is cleaved upon the double reduction, resulting in a [{CptMo(CO)2}2(μ,η 2:2‐As2)]2− dianion.
This is charged balanced by two [Cp*2Sm]+ cations, which bind via isocarbonyl species to the complex core (Sm1−O1 2.378(4), Sm1−O2′ 2.391(4) Å). In the corresponding P–complex A (Scheme 1) and Mo coordination compounds featuring a N=N2− unit, [11] the central E2 2− unit is end‐on coordinated to the Mo atoms. In contrast, the As2 2− unit in 1 is side‐on bound. The As−As bond length of 2.238(2) Å is in the range of As−As double bonds known in the literature and only slightly shortened in comparison to [{CpMo(CO)2}2(μ,η 2:2‐As2)].[ 10a , 12 ] However, quantum chemical calculations suggest a weakened double bond (see below).
Since the isolated yields of 1 are moderate, we anticipate that further reactions as seen for the Mo‐phosphide (Scheme 1) take place. [5a] However, further reaction products could not be isolated from the synthesis of 1 under the chosen reaction conditions. Therefore, the solubility of the samarium reagent was altered by formally replacing one methyl group by a n‐propyl group on the Cp ring. The resulting reagent [(CpMe4nPr)2Sm] was treated with [{CptMo(CO)2}2(μ,η 2:2‐As2)] in n‐heptane at elevated temperature. As result a product mixture of two species was isolated by crystallization (Scheme 2). The first one, [(CpMe4nPr)2Sm)2As2(CptMo(CO)2)2] (2), is similar to 1 (see Supporting Information for details, Figure S13), whereas the second one [((CpMe4nPr)2Sm)2As4(CptMo(CO)2)2] (3) represents the anticipated second reduction product and thus gives a first insight into the reaction pathway. Compound 3 is the As‐analogue of compound B (Scheme 1), which is observed in the reduction of the corresponding Mo‐phosphide. Despite the use of different reaction conditions and solvents, we were not able to separate the two compounds. Therefore, both compounds are not discussed by spectroscopic details. However, it can be concluded that compound 3 (Figure 2), which features a planar four‐membered As4 unit, seems to be the result of further reaction and defragmentation of 2. In 3, each {CptMo(CO)2} fragment coordinates in a η 2‐mode to the central As4 ring, which is built upon the formation of two new As−As bonds, merging two As2 units together. The resulting [As4(CptMo(CO)2)2]2− building block is further stabilized by coordination of two [Cp*2Sm]+ cations. In contrast to 1 and 2, the samarocene moieties are directly bound to As (Sm1−As1 3.0300(8) Å). Furthermore, an isocarbonyl bridge (Sm1−O1 2.380(2) Å), which is in the range of 1 and 2, is formed.
Figure 2.
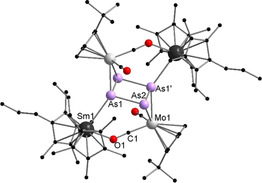
Molecular structure of 3 in the solid state. [22] Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sm1−As1 3.0300(8), Sm1−O1 2.380(2), Mo1−As2 2.6498(7), Mo1−As1′ 2.6261(7), Mo1−C1 1.873(3), As1−As2 2.4862(7), As1′−As2 2.3503(6), C1−O1 1.206(4), O1‐Sm‐As1 76.99(6), As1′‐Mo1‐As2 52.91(2), Mo1‐As1′‐Sm1′ 142.35(2), As1′‐As2‐Mo1 63.03(2), As1‐As2‐Mo1 105.01(3), As2‐As1‐As2′ 91.16(2), As1‐As2‐As1′ 88.84(2).
In contrast to the highly symmetric aromatic As4 2− ring, which is found for example, in [(Cptt 2Sm)(μ,η 4:η 4‐As4)(Cp*Fe)] or [(DippForm)2Sm}2(μ 2‐η 4:η 4‐As4)] (DippForm={(2,6‐iPr2C6H3)NC(H)=N(2,6‐iPr2C6H3)}−), the As−As bond lengths in 3 differ from each other.[ 6a , 13 ]
There are two longer bonds (As1−As2 2.4862(7) Å), which are in the range of As−As single bonds (e.g. ca. 2.44 Å in carbene stabilized diarsenic) [14] and two shorter ones (As1′−As2 2.3503(6) Å). However, these are still longer than expected for As−As double bonds due to the side‐on coordination to the {CptMo(CO)2} fragments in a η 2‐mode. The angles within the ring are close to 90° (91.16(2) and 88.84(2)°), confirming the formation of an As4 unit in the solid state.
Thus, the obtained As4 unit is comparable to [(LCo)2As4] (L=bis(2,6‐diisopropylphenyl)‐β‐dialdiminate) [15] and can be best described as a neutral tetraarsacyclobutadiene, while the negative charge should be strongly distributed over the whole complex core and especially the Sm‐OC‐Mo moieties. This is quite remarkable, because the pnictogen analogues of cyclobutadiene should be, like the latter, very reactive and unstable.[ 15 , 16 ] To the best of our knowledge, compound 3 is only the second example of a metal‐coordinated/stabilized tetraarsacyclobutadiene and therefore expands the group of heavier group 15 compounds which show an isoelectronic relation towards hydrocarbon based ligands. In contrast to the previous example, [15] it was not generated from yellow arsenic but by merging two As2 units.
Next we drew our attention to antimony compounds and performed analogue reactions with [{CptMo(CO)2}2(μ,η 2:2‐Sb2)] (Scheme 3). Again, both samarocenes [Cp*2Sm] and [(CpMe4nPr)2Sm] were used as SET reagents at elevated temperatures. This resulted in [(Cp*2Sm)2Sb4(CptMo(CO)2)2] (4) and [((CpMe4nPr)2Sm)2Sb4(CptMo(CO)2)2] (5), which could be isolated by crystallization, in moderate to good yields (67 % for 4; 31 % for 5) (Scheme 3). In contrast to the reduction of the Mo‐phosphide and Mo‐arsenide, no further products could be isolated. Compounds 4 and 5 (Figure 3 and Figure 4) feature a planar Sb4 unit similar to those of the lighter congeners observed in B and 3. In general, only few examples for perfectly planar Sb4 rings have been reported to date. These are the ionic inorganic species [K([2.2.2]crypt)]2 +[Sb4]2− [17] and [PPh4]+ 2[S6Sb6]2−. [18] Until now, the metalorganic chemistry of Sb4 units features mostly butterfly‐type structures and configurations which can formally be derived from the neutral Sb4 tetrahedron. [19] Therefore, a planar Sb4 ring as ligand for an organometallic or coordination compound is, to the best of our knowledge, unique.
Scheme 3.
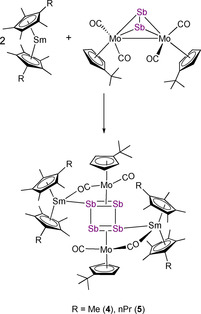
Synthesis of [(Cp*2Sm)2Sb4(CptMo(CO)2)2] (4) and [((CpMe4nPr)2Sm)2Sb4(CptMo(CO)2)2] (5) with R=Me (4), n‐propyl (5).
Figure 3.
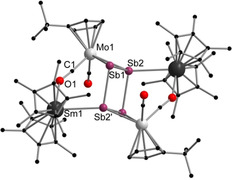
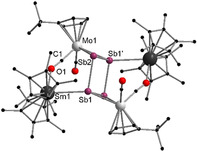
Molecular structure of 4 in the solid state. [22] Hydrogen atoms are omitted for clarity. Selected bond lengths [Å], angles [°]: Sm1−Sb2′ 3.2375(7), Sm1−O1 2.381(6), Mo1−Sb2 2.8146(9), Mo1−Sb1 2.8549(9), Mo1−C1 1.894(8), Sb1−Sb2 2.7313(8), Sb1−Sb2′ 2.8608(7), C1−O1 1.209(10), O1‐Sm‐Sb2′ 76.73(14), Sb1‐Mo1‐Sb2 57.59(2), Mo1‐Sb2‐Sm1′ 153.18(2), Sb1‐Sb2‐Mo1 61.94(2), Sb2′‐Sb1‐Mo1 102.32(2), Sb2‐Sb1‐Sb2′ 87.73(2), Sb1‐Sb2‐Sb1′ 92.28(2).
Figure 4.
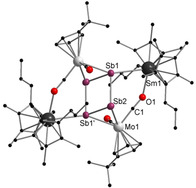
Molecular structure of 5 in the solid state. [22] Hydrogen atoms and solvent molecules (toluene) are omitted for clarity. Selected bond lengths [Å] and angles [°]: Sm1−Sb1 3.2794(8), Sm1−O1 2.386(7), Mo1−Sb2 2.8464(11), Mo1−Sb1′ 2.8315(11), Mo1−C1 1.888(10), Sb1−Sb2 2.8618(10), Sb1′−Sb2 2.7254(10), C1−O1 1.221(12), O1‐Sm‐Sb1 76.8(2), Sb1′‐Mo1‐Sb2 57.37(3), Mo1‐Sb1′‐Sm1′ 152.00(3), Sb1′‐Sb2‐Mo1 61.59(3), Sb1‐Sb2‐Mo1 103.46(3), Sb2‐Sb1‐Sb2′ 90.16(3), Sb1‐Sb2‐Sb1′ 89.84(3).
In 4 and 5, the central Sb4 unit forms a planar rectangle, with angles close to 90° (87.73(2)° and 92.28(2)° (4), 89.84(3)° and 90.16(3)° (5)). The Sb4 unit consists of two short (Sb1−Sb2 2.7313(8) Å (4) and Sb1′−Sb2 2.7254(10) Å (5)) and two longer (Sb1−Sb2′ 2.8608(7) Å (4) and Sb1−Sb2 2.8618(10) Å (5)) Sb−Sb bonds. The two longer bonds are in the range of a Sb−Sb single bond as found in Ph4Sb2 (2.837 Å). [20] However, the shorter bonds are still longer than in an anticipated Sb−Sb double bond, for example, in the distibene [TbtSb=SbTbt] (Tbt=2,4,6‐tris‐[bis(trimethyl‐silyl)methyl]phenyl) (2.642(1) Å), [21] which is a result of the η 2‐coordination to the {CptMo(CO)2} fragments (analogous to 3). Therefore, the remarkable analogy to hydrocarbon based cyclobutadiene can even be seen for the second‐heaviest group 15 homolog, forming—to the best of our knowledge—the first metal‐coordinated tetrastibacyclobutadiene. For further stabilization of the latter, both samarium atoms coordinate to one Sb atom of the central Sb4 ring each with bond distances of 3.2375(7) Å (4) and 3.2794(8) Å (5). These are in agreement with samarium stibide compounds such as [(Cp*2Sm)2Sb2] or [{(Cp*2Sm)3(μ 4,η 1:2:2:2‐Sb4)}2Hg] and are slightly shorter than in [{(Cp*2Sm)4(μ 4,η 2:2:2:2‐Sb8)] (3.31–3.41 Å). [8c] In addition, a Sm isocarbonyl bridge is formed (Sm1−O1 2.381(6) Å (4) and 2.386(7) Å (5)). Therefore, both compounds are the first examples of a Sb4 moiety, which is isostructural towards hydrocarbon based ligands and additionally serves as a bridging ligand between a lanthanide and a transition metal. Compounds 4 and 5 were additionally characterized by IR spectroscopy. Two separated bands for the CO ligands are observed. Whereas the terminal CO ligands are detected as strong bands at frequencies of 1898 cm−1 (4) and 1896 cm−1 (5), the slightly weaker bands of the bridging CO moieties exhibit stretching frequencies at lower energies (1633 cm−1 (4), 1630 cm−1 (5), (Figures S4 and S5). The data is in agreement with the literature. [20]
Attempting to investigate the bonding properties of these novel systems with the aid of quantum chemical calculations is difficult due to the low molecular symmetries, as it was the case with the analogue phosphorus compounds before. [5a] Therefore, we focused on a comparative energy analysis of the pnictogen systems based on theoretical data (technical details given in the Supporting Information). Figure 5 easily explains that the formation of [(Cp2*Sm)2E2(CpMo(CO)2)4] (E=As, Sb; type A, Scheme 1) among the pnictogen model compounds can be excluded. From the analysis of the molecular structures (data given in the Supporting Information) a distinct difference regarding the E2 units can explain this fact: P2 fits—as can be seen from the small torsion angle Mo‐P‐P‐Mo—particularly well into the 14‐membered „Sm2(CO)4Mo4“ ring system, while As2 and Sb2 protrude clearly from the plane of the 14‐link ring of type A‐systems due to their sizes (tors(Mo‐E‐E‐Mo): P2: 20.7, As2: 34.9, Sb2: 41.7°). By comparing E2 bond distances and shared electron numbers (SEN) from Ahlrichs‐Heinzmann population analyses in these compounds with theoretical data on E2, H2E2 and E2H4 as reference systems for formal triple, double or single E2 bonds, [21] its bond strength can be convincingly estimated. We conclude that in [(Cp2*Sm)2P2(CpMo(CO)2)4] (A) a double bond is present, whereas in the theoretical As and Sb homologues of A the E−E bond would only be of strong single bond character. In contrast, in the As compounds 1 and 2 a weakened double bond of the As2 unit is found. Both findings are in line with the energetic findings.
Figure 5.
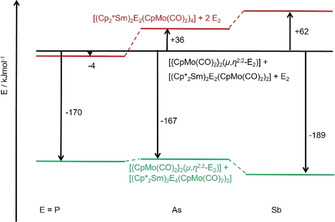
Energetic relationship of the compounds under discussion. The species „[{CpMo(CO)2}2(μ,η 2:2‐E2)]+[(Cp2*Sm)2P2(CpMo(CO)2)2]+E2“ were taken as reference systems. They were arbitrarily set to identical energies.
In conclusion, the product formation upon reduction of [{CpMo(CO)2}2(μ,η 2:2‐P2)] and [{CptMo(CO)2}2(μ,η 2:2‐E2)] (E=As, Sb) depends strongly on the pnictide. As seen from the formation of 1, 2, and A, the Mo−Mo bond is cleaved first. The coordination of the central E2 2− unit (end‐on vs. side‐on) and the bond order within this unit depends on the pnictide. Subsequent rearrangements lead to rectangular planar central E4 units with alternating short and long bond distances. Whereas this ring structure is only a minor byproduct in the case of phosphorus containing compound, it is the main product for the analogue Sb complex. The resulting compounds feature for the first time a tetrastibacyclobutadiene ligand in organometallic and coordination chemistry. Moreover, they show that the remarkable and rare isoelectronical analogy to cyclobutadiene, which was already observed for phosphorus and arsenic, can still be obtained for the heavier pnictogen antimony.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft (DFG) (No. 266153560, Ro 2008/17‐2 and Sche 384/33‐2) and to the Russian Foundation for Basic Research (RFBR, grant 19‐03‐00568) for financial support. The authors acknowledge computational support by the state of Baden‐Württemberg through bwHPC and the Deutsche Forschungsgemeinschaft (DFG) through grant No INST 40/467‐1 FUGG. N.R.’s Ph.D. study is additionally funded by the Fonds der Chemischen Industrie (102431). We also thank Maria Haimerl for the support in the synthesis of yellow arsenic solutions. Open access funding enabled and organized by Projekt DEAL.
N. Reinfandt, C. Schoo, L. Dütsch, R. Köppe, S. N. Konchenko, M. Scheer, P. W. Roesky, Chem. Eur. J. 2021, 27, 3974.
Dedicated to Professor Wolfgang Kaim on the occasion of his 70th birthday.
References
- 1. Faessler T. F., Structure and Bonding, Vol. 140 (Ed.: Mingos M. P.), Springer Verlag, Berlin, 2011. [Google Scholar]
- 2. Zide J. M. O., Kleiman-Shwarsctein A., Strandwitz N. C., Zimmerman J. D., Steenblock-Smith T., Gossard A. C., Forman A., Ivanovskaya A., Stucky G. D., Appl. Phys. Lett. 2006, 88, 162103. [Google Scholar]
- 3.
- 3a. Detzel M., Mohr T., Scherer O. J., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1994, 33, 1110–1112; [Google Scholar]; Angew. Chem. 1994, 106, 1142–1144; [Google Scholar]
- 3b. Scherer O. J., Winter R., Wolmershäuser G., Z. Anorg. Allg. Chem. 1993, 619, 827–835. [Google Scholar]
- 4. Konchenko S. N., Pushkarevsky N. A., Gamer M. T., Köppe R., Schnöckel H., Roesky P. W., J. Am. Chem. Soc. 2009, 131, 5740–5741. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Arleth N., Gamer M. T., Köppe R., Pushkarevsky N. A., Konchenko S. N., Fleischmann M., Bodensteiner M., Scheer M., Roesky P. W., Chem. Sci. 2015, 6, 7179–7184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Li T., Gamer M. T., Scheer M., Konchenko S. N., Roesky P. W., Chem. Commun. 2013, 49, 2183–2185; [DOI] [PubMed] [Google Scholar]
- 5c. Li T., Wiecko J., Pushkarevsky N. A., Gamer M. T., Köppe R., Konchenko S. N., Scheer M., Roesky P. W., Angew. Chem. Int. Ed. 2011, 50, 9491–9495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9663–9667; [Google Scholar]
- 5d. Li T., Kaercher S., Roesky P. W. Chem. Soc. Rev. 2014, 43, 42—57. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Arleth N., Gamer M. T., Köppe R., Konchenko S. N., Fleischmann M., Scheer M., Roesky P. W., Angew. Chem. Int. Ed. 2016, 55, 1557–1560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1583–1586; [Google Scholar]
- 6b. Schoo C., Bestgen S., Schmidt M., Konchenko S. N., Scheer M., Roesky P. W., Chem. Commun. 2016, 52, 13217–13220; [DOI] [PubMed] [Google Scholar]
- 6c. Schoo C., Bestgen S., Egeberg A., Seibert J., Konchenko S. N., Feldmann C., Roesky P. W., Angew. Chem. Int. Ed. 2019, 58, 4386–4389; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4430–4434; [Google Scholar]
- 6d. Huang W., Diaconescu P. L., Eur. J. Inorg. Chem. 2013, 4090–4096; [Google Scholar]
- 6e. Selikhov A. N., Mahrova T. V., Cherkasov A. V., Fukin G. K., Kirillov E., Alvarez Lamsfus C., Maron L., Trifonov A. A., Organometallics 2016, 35, 2401–2409; [Google Scholar]
- 6f. Li T., Kaercher S., Roesky P. W. Chem. Soc. Rev. 2014, 43, 42–57. [DOI] [PubMed] [Google Scholar]
- 7. Evans W. J., Gonzales S. L., Ziller J. W., J. Chem. Soc. Chem. Commun. 1992, 0, 1138–1139. [Google Scholar]
- 8.
- 8a. Pugh T., Chilton N. F., Layfield R. A., Chem. Sci. 2017, 8, 2073–2080; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Min X., Popov I. A., Pan F.-X., Li L.-J., Matito E., Sun Z.-M., Wang L.-S., Boldyrev A. I., Angew. Chem. Int. Ed. 2016, 55, 5531–5535; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5621–5625; [Google Scholar]
- 8c. Schoo C., Bestgen S., Egeberg A., Klementyeva S., Feldmann C., Konchenko S. N., Roesky P. W., Angew. Chem. Int. Ed. 2018, 57, 5912–5916; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6015–6019. [Google Scholar]
- 9. Kefalidis C. E., Essafi S., Perrin L., Maron L., Inorg. Chem. 2014, 53, 3427–3433. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Sullivan P. J., Rheingold A. L., Organometallics 1982, 1, 1547–1549; [Google Scholar]
- 10b. Harper J. R., Rheingold A. L., J. Organomet. Chem. 1990, 390, c36–c38. [Google Scholar]
- 11. Margulieux G. W., Turner Z. R., Chirik P. J., Angew. Chem. Int. Ed. 2014, 53, 14211–14215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14435–14439. [Google Scholar]
- 12. Tuscher L., Helling C., Wölper C., Frank W., Nizovtsev A. S., Schulz S., Chem. Eur. J. 2018, 24, 3241–3250. [DOI] [PubMed] [Google Scholar]
- 13. Schoo C., Bestgen S., Köppe R., Konchenko S. N., Roesky P. W., Chem. Commun. 2018, 54, 4770–4773. [DOI] [PubMed] [Google Scholar]
- 14. Abraham M. Y., Wang Y., Xie Y., Wei P., H. F. Schaefer III , v. R. Schleyer P., Robinson G. H., Chem. Eur. J. 2010, 16, 432–435. [DOI] [PubMed] [Google Scholar]
- 15. Spitzer F., Balázs G., Graßl C., Keilwerth M., Meyer K., Scheer M., Angew. Chem. Int. Ed. 2018, 57, 8760–8764; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8896–8900. [Google Scholar]
- 16. Yao S., Lindenmaier N., Xiong Y., Inoue S., Szilvási T., Adelhardt M., Sutter J., Meyer K., Driess M., Angew. Chem. Int. Ed. 2015, 54, 1250–1254; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1266–1270. [Google Scholar]
- 17. Critchlow S. C., Corbett J. D., Inorg. Chem. 1984, 23, 770–774. [Google Scholar]
- 18. Martin T. M., Schimek G. L., Pennington W. T., Kolis J. W., Dalton Trans. 1995, 501–502. [Google Scholar]
- 19.
- 19a. Ganesamoorthy C., Krüger J., Wölper C., Nizovtsev A. S., Schulz S., Chem. Eur. J. 2017, 23, 2461–2468; [DOI] [PubMed] [Google Scholar]
- 19b. Tuscher L., Ganesamoorthy C., Bläser D., Wölper C., Schulz S., Angew. Chem. Int. Ed. 2015, 54, 10657–10661; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10803–10807. [Google Scholar]
- 20.
- 20a. Hillier A. C., Liu S. Y., Sella A., Zekria O., Elsegood M. R. J., J. Organomet. Chem. 1997, 528, 209–215; [Google Scholar]
- 20b. Scherer O. J., Sitzmann H., Wolmershäuser G., J. Organomet. Chem. 1984, 268, C9–C12. [Google Scholar]
- 21. Köppe R., Schnöckel H., Z. Anorg. Allg. Chem. 2000, 626, 1095–1099. [Google Scholar]
- 22.CCDC 2015996, 2015997, 2015998, 2015999 and 2016000 for compounds 1, 2, 3, 4 and 5, respectively, contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary