

Abstract
Dinitrogen (N2) is the most abundant gas in Earth's atmosphere, but its inertness hinders its use as a nitrogen source in the biosphere and in industry. Efficient catalysts are hence required to ov. ercome the high kinetic barriers associated to N2 transformation. In that respect, molecular complexes have demonstrated strong potential to mediate N2 functionalization reactions under mild conditions while providing a straightforward understanding of the reaction mechanisms. This Review emphasizes the strategies for N2 reduction and functionalization using molecular transition metal and actinide complexes according to their proposed reaction mechanisms, distinguishing complexes inducing cleavage of the N≡N bond before (dissociative mechanism) or concomitantly with functionalization (associative mechanism). We present here the main examples of stoichiometric and catalytic N2 functionalization reactions following these strategies.
Keywords: catalysis, coordination complexes, functionalization, mechanism, nitrogen
Dinitrogen (N2) is the most abundant gas in the Earth's atmosphere, but its inertness hinders its use as a nitrogen source in the biosphere and in industry. This review emphasizes the strategies for N2 reduction and functionalization using molecular transition metal and actinide complexes according to their proposed reaction mechanisms and presents the main examples of stoichiometric and catalytic N2 functionalization reactions.
1. Introduction
Atmospheric N2 is the primary source of all nitrogen atoms present on earth, but its assimilation by most living organisms requires its initial transformation into more reactive nitrogen species, all originating from ammonia. The conversion of molecular dinitrogen into ammonia is one of the most important chemical processes from both an industrial and a biological perspective. The biological fixation of dinitrogen occurs in the nitrogenase enzymes, which can effectively reduce N2 to NH3 under ambient conditions. However, the amount of dinitrogen fixed in such biological systems is not sufficient to replenish the reserves of nutrients needed in current agriculture practices. [1] The ground‐breaking discovery of artificial N2 reduction by Fritz Haber and its industrialization and optimization by Carl Bosch and Alwin Mittasch at the beginning of the 20th century,[ 2 , 3 ] allows to currently transform about half of the total reactive nitrogen available on Earth and is crucial to sustain the current 7.8 billion population. [4]
This process however requires reactors operating at high temperature and pressure and using highly purified N2 and H2. The use of H2 (mainly derived from fossil fuels steam reforming) as a reductant and the harsh conditions required result in a high CO2 footprint accounting for about 2 % of the world's CO2 emissions. [5] Industrial ammonia production can be carried out at very high energy efficiency, but the high temperature and pressure and the complex management of thermodynamic losses required render it only profitable at large scale, and therefore requires substantial capital investment. Consequently, a distribution infrastructure to ship from centralized sources to the local users is required, further increasing the overall environmental and energetic footprint of the process. More sustainable, delocalized and energy efficient alternatives to synthetic dinitrogen fixation are hence highly desirable in this context. [6] Nonetheless, energy‐efficient processes to reduce N2 under ambient conditions still face major challenges due to its chemical inertness, resulting from the thermodynamic strength of the N≡N bond (bond dissociation enthalpy of 944 kJ mol−1), the low proton affinity of N2 and its high kinetic stability towards reduction and oxidation. This high reduction stability of N2 originates from a poor electron affinity (calculated value of −1.903 eV) and a large LUMO‐HOMO gap (10.82 eV) resulting from strongly antibonding π* lowest unoccupied molecular orbitals (Scheme 2). [7] To overcome such high kinetic barriers requires energy along with an efficient catalyst.
Scheme 2.
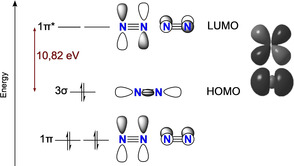
Molecular orbital diagram of free dinitrogen along with DFT‐computed HOMO and LUMO (B3LYP).
The Mo‐nitrogenase enzyme meets these challenges utilizing the energy of 16 equiv of ATP to transform one dinitrogen molecule, using water as the proton source (Scheme 1). The ability to mediate this reaction under mild conditions relies in that nitrogenases are effective promoters of multi‐electron and multi‐proton transfers, but the complete reaction mechanism is complex and still not completely uncovered today.[ 8 , 9 , 10 , 11 ] The Fe‐based heterogeneous catalysts currently used in the Haber–Bosch process rely on a very different strategy, using H2 as a source of electrons and protons (Scheme 1). [12] However, these require harsh conditions (>400 °C, >200 bar) to overcome the severe kinetic barriers to the dissociative nitrogen chemisorption on the catalyst surface. [13]
Scheme 1.
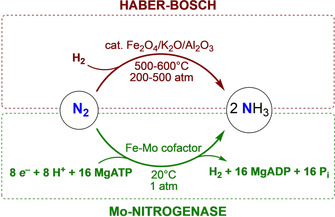
Comparative dinitrogen reduction to ammonia by the Haber–Bosch process (top) and in the Mo‐nitrogenase enzyme (bottom).
By providing a straightforward understanding of the reaction mechanisms and by allowing for the fine management of electron and proton transfer steps via the tuning of the active sites properties, molecular complexes have shown a strong potential to promote catalytic nitrogen reduction under mild conditions.[ 12 , 14 , 15 ] To date, dinitrogen metal complexes have been isolated with almost all metals from first to third row to rare‐earth metals,[ 16 , 17 ] but very few have led to the stoichiometric functionalization of dinitrogen and only a handful have been reported to enable catalytic functionalization.[ 18 , 19 , 20 ] Although recent reviews have surveyed dinitrogen activation by polynuclear complexes, [20] bond‐forming reactions from activated dinitrogen [19] and catalytic dinitrogen to ammonia formation using molecular complexes, [15] this Review will explore a complementary approach, encompassing aspects from all these reviews. We aim to provide an overview of the successful strategies utilizing molecular complexes to promote nitrogen reduction and functionalization with H, C and Si sources under mild conditions according to their proposed reaction mechanisms. For this purpose, we provide here two tools for a simple overview of the reactivity and performances of all complexes reported throughout this Review: the diversity of products obtained are compiled in Schemes 15, 19 and 47, while the performances of the complexes to mediate these transformations are summarized in Tables S1–S7 (Supporting Information). Other bond‐forming reactions with elements such as boron, phosphorous or with other Lewis acids are excluded from the scope of the current Review as their scarcity complicates the possibility to draw mechanistic conclusions. The interested reader may refer to the recent reviews from Chirik and co‐workers [19] and Simmoneau and Etienne, [21] which provide a good overview on the recent research in the field.
Scheme 15.
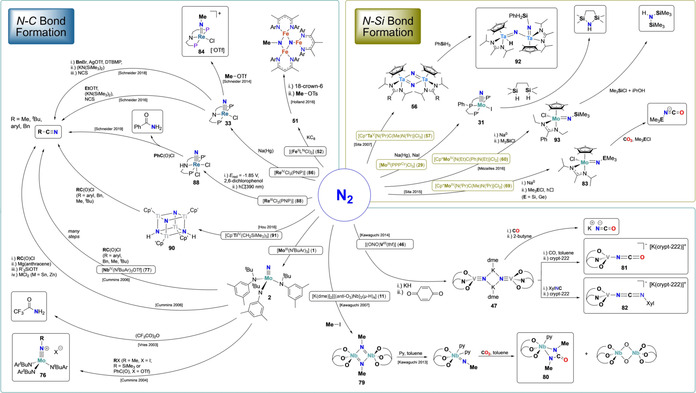
Stoichiometric functionalization of metal‐nitrides originating from N2 (N‐C and N−Si bond formation).
Scheme 19.
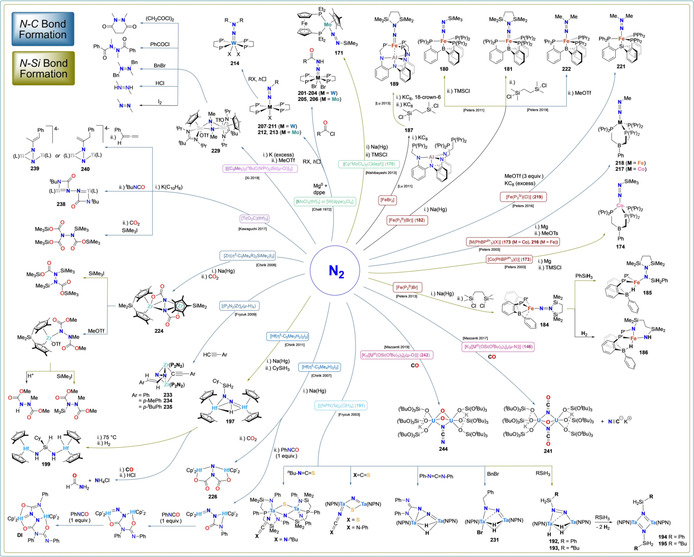
Stoichiometric functionalization of metal‐bound N2 (N−C and N−Si bond formation).
Scheme 47.
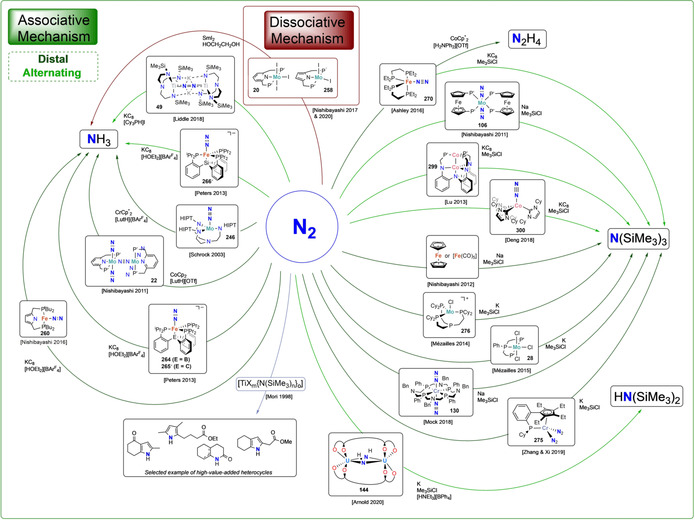
Catalytic systems for N2 functionalization (N−H, N−Si and N−C bond formation) and associated mechanism.
1.1. Molecular orbital considerations
Understanding the N2–metal interaction is key in rationalizing strategies for activating and functionalizing dinitrogen. Simple molecular orbitals models are a convenient way to simply illustrate and understand structural details and reactivity, and complementary to more in‐depth computational studies. [22] The free dinitrogen molecular orbital diagram is shown in Scheme 2.
Most mononuclear d‐block metal complexes coordinate N2 in an end‐on fashion. Side‐on N2 bonding has been rarely observed in isolated mononuclear complexes,[ 23 , 24 , 25 ] but may occur for f‐block metal complexes or during the end‐to‐end rotation of mononuclear dinitrogen complexes. [26] In an end‐on coordination mode, the σ‐character of the HOMO and the π*‐character of the LUMO of N2 enable its coordination to the metal via a σ‐donation/π‐backbonding binding scheme, yet much less effective than for the more polarized isoelectronic CO molecule (Scheme 3). Such an interaction can weaken the triple bond of N2 by populating the π* molecular orbitals and by removing electron density from the bonding s‐orbital.
Scheme 3.
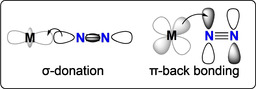
σ‐donation/π‐backbonding scheme of metal‐bound dinitrogen.
This binding scheme allows identifying optimal scenarios for N2 activation. Electron rich ligands have the potential to increase the electron density at the metal center and strengthen π‐backbonding. Following the same reasoning, metal complexes with a large number of valence electrons should lead to stronger activation. This effect may yet be limited by the decrease in energy of the d‐orbitals from left to right in the periodic table, lowering the overlap with dinitrogen's antibonding orbitals. For that reason, middle and group 8 transition metals often present the best compromise, as highlighted by the numerous complexes from these groups in N2 activation literature and the present review. [15]
However, binding N2 to a single metal center in ambient conditions and in the absence of other reagents is not sufficient to induce N2 cleavage. The presence of a second metal center or of a Lewis or Brønsted acid is required to induce the scission of the N≡N bond. Two mechanistic paths can then be observed, involving either the initial cleavage of the triple bond to form metal nitrides functionalized in a second step by proton sources or electrophiles or the concomitant reduction and functionalization of the coordinated N2 molecule with proton sources or electrophiles. By analogy with the terms typically used to describe N2 mechanisms on heterogeneous catalysts, in this Review we will use the terms associative and dissociative to describe these two reaction paths. The terms may be over simplistic when describing activity of molecular complexes but are effective to identify common features in series of complexes. Within associative paths, the site of exogeneous substrate binding differentiates two mechanistic pathways depending on the distal or alternating functionalization of N2, as depicted in Scheme 4.
Scheme 4.
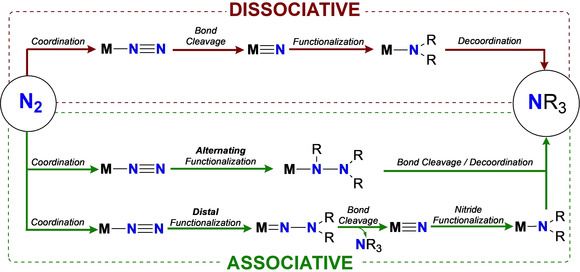
Dissociative and associative mechanisms for dinitrogen functionalization at a metal center M.
This Review will cover the most representative examples of N2 functionalization according to their proposed reaction mechanisms, highlighting first complexes reacting via a dissociative mechanism followed by associative pathway examples, to end on a brief exploration of reported catalytic systems.
2. Dissociative Mechanism
The major driving force in a dissociative mechanism is the formation of thermodynamically stable nitride complexes, while the later functionalization of the formed nitrides is the key parameter to envision a catalytic reduction of N2. Most complexes mediating N2 cleavage necessitate the use of external reducing agents and only a small number of compounds have been reported to cleave N2 without using reducing equivalents originating from an exogeneous electron source. Both possibilities will be distinguished here, further differentiating complexes forming terminal or bridging nitrides, summarized in Table S1. When reported, the functionalization of the formed nitrido ligands will be presented. An overview of all dissociative functionalizations is provided in Scheme 15 at the end of this section and summarized in Table S2.
2.1. Cleavage without external reducing agents
To date, only a limited number of complexes have been reported to split the dinitrogen triple bond without the use of exogeneous electron sources. In such a case, the complex is overall oxidized, the electrons being either provided by the metal center or from a concomitant reductive elimination step.
To our knowledge, only two examples of mononuclear metal complexes have been shown to cleave N2 into terminal nitrides, both being MoIII complexes. The first example is a MoIII‐trisamide complex introduced by Cummins in 1995. [27] Stirring a toluene solution of [MoIII(N(tBu)Ar)3] (1, Ar=3,5‐(CH3)2C6H3) under a dinitrogen atmosphere resulted in the successive binding and cleavage of the dinitrogen molecule to form the mononuclear MoVI‐nitride complex 2 (Scheme 5 a). The end‐on bridging intermediate (μ‐N2)[Mo(N(tBu)Ar)3]2 was observed and characterized by EXAFS [28] and later on by single crystal XRD. [29]
Scheme 5.
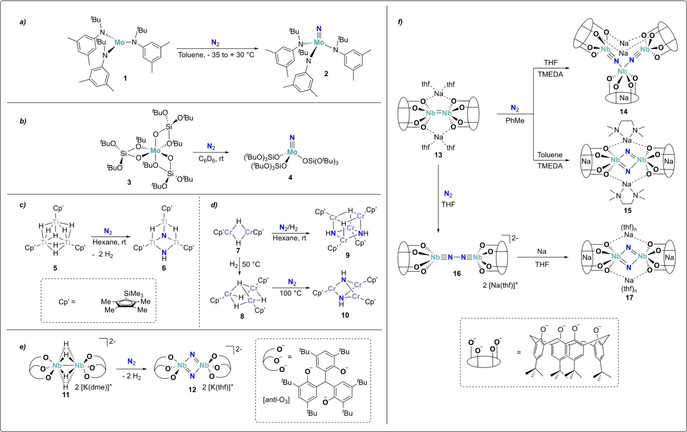
Dissociative N2‐splitting by mononuclear molybdenum complexes 1 (a) and 3 (b), trinuclear TiIII and CrIII hydride complexes 5 (c) and 7/8 (d) and dinuclear NbIII complexes 11 (e) and 13 (f).
This work, constituting the first example of full dinitrogen cleavage by a well‐defined transition metal complex, opened new perspectives for designing strategies for N2 reduction. The key feature for N2 activation is the presence of a highly reactive unsaturated trivalent MoIII species. In the case of C 3 symmetric [MoIII(N(tBu)Ar)3] (1), the three bulky amide ligands help to stabilize such species for example, by preventing them from dimerization. In addition, the MoL3 fragment is isolobal to a nitrogen atom by its three unpaired electrons populating the mainly d , dxz and dyz molecular orbitals, providing a significant thermodynamic stability to the corresponding Mo nitrides. A second MoIII complex displaying similar reactivity towards dinitrogen was reported more than 20 years later. [30] When the octahedral complex [MoIII(OSi(OtBu)3)3] (3) was exposed to dinitrogen, the corresponding mononuclear nitride complex (4) could be formed (Scheme 5 b). At the difference of the tris‐amide complex 1, the tris‐silanolate complex 3, is coordinatively saturated, but was shown to form a three‐coordinate MoIII complex in situ from the parent distorted octahedral complex thanks to the hemilabile character of the κ 2 coordinated silanolate ligands.
In addition to these two mononuclear examples, a few multimetallic complexes that react with dinitrogen in the absence of external reducing agents have been reported. In that case, N2 splitting results in the formation of multinuclear bridging nitride complexes. The trinuclear titanium(III) polyhydride complex 5 was shown to react with dinitrogen to form the bridging dinitride complex 6 with concomitant evolution of two equivalents of dihydrogen (Scheme 5 c). [31] In that complex, the electrons required for the reaction are formally not only delivered by the metal centers but also resulted from the reductive elimination of two equivalents of H2 per dinitrogen molecule. This system is relevant in the understanding of the biological dinitrogen fixation process, since bridging hydride ligands and analogous H2 elimination prior to N2 binding have been shown to be of key importance in the nitrogenase enzyme.[ 11 , 32 , 33 ] The cleavage of N2 by the related chromium(II) and chromium(III) hydride complexes 7 and 8 was demonstrated very recently (Scheme 5 d). [34] These compounds, which are the first well‐defined chromium hydride complexes capable of fixing N2, were shown to generate the corresponding bridging imide complexes 9 and 10 upon treatment with N2(/H2).
Reductive elimination of H2 as a strategy to provide electrons for the reduction of dinitrogen was also found for the niobium(IV) tridentate aryloxide bridging tetrahydride dimer [((anti‐O3)Nb)2(μ‐H)4][K(dme)]2 (11, anti‐O3, see Scheme 5 e). [35] Complex 11 was reported to react with N2 to afford the bridging dinitride, dinuclear NbV complex [((anti‐O3)Nb)2(μ‐N)2][K(thf)]2 (12). Among the six electrons required for the reduction of N2, four of them result from the reductive elimination of two equivalents of H2, while two electrons originate from the one‐electron oxidation of the two niobium centers. A very recent study demonstrated that the corresponding tetrahydride complexes with Na+ and Li+ counter cations showed a very different behavior. In both cases, reaction with N2 did not result in its full cleavage but lead to the elimination of only one equivalent of H2 and to the formation of corresponding side‐on/end‐on bridging dinitrogen complexes. [36]
Another low‐valent dinuclear NbIII complex 13, supported by a tetra‐anionic calix[4]arene ligand (Scheme 5 f), had been earlier reported by Floriani and co‐workers.[ 37 , 38 ] In toluene, this highly reducing species readily cleaved the N2 triple bond to yield a dimeric or a trimeric complex (14 and 15) with two bridging nitrido ligands. However, in more polar solvents such as THF, the corresponding dinitrogen bridged complex 16 was formed. The [N–N]4− unit could be further reduced for example, with sodium, again leading to full cleavage of the dinitrogen moiety and the formation of the bridging nitride complex 17.
2.2. In situ reduction under dinitrogen
The vast majority of complexes mediating dinitrogen reduction via a dissociative mechanism necessitate employing an external reducing agent concomitantly to N2 addition. This strategy avoids the challenging isolation of highly reactive reduced metal species, but implies that in most cases the reaction mechanism can only be proposed based on theoretical calculations and in situ spectroscopy. This should nevertheless not be seen as a drawback when targeting a catalytic system which requires dinitrogen and reducing agent to be present simultaneously.
2.2.1. In situ reduction under dinitrogen using alkali reducing agents
Frequently employed reducing agents such as sodium naphthalenide, sodium mercury amalgam, potassium graphite etc., contain alkali metal cations which may significantly contribute to the activation and cleavage of dinitrogen. Lewis acidic alkali metal cations can interact with coordinated dinitrogen molecules and further weaken their N−N bonds, as comprehensively reviewed by Holland in 2017. [39] We will focus here in two subsections on terminal and bridging nitride complexes formed by in situ N2 reduction using alkali‐metal reducing agents.
Formation of terminal nitrides: A significant number of transition metal complexes with pincer ligands were shown to cleave dinitrogen via both associative or dissociative mechanisms, depending on the ligand steric bulk and transition metal used. These bulky planar three‐coordinate neutral or mono‐anionic ligands are particularly suited for N2 activation: The T‐shaped geometry and tunable steric bulk of pincer ligands prevent the dimerization and deactivation of the corresponding complexes, of key importance to maintain reactivity of the formed nitride compounds. Molybdenum has been the most frequently studied metal in pincer complexes mediating dinitrogen activation, and Mo‐pincer catalytic systems with outstanding performances have been developed. Schrock and co‐workers reported dinitrogen cleavage by the MoIII PCP‐pincer complex 18 (Scheme 6 a). [40] Reduction with sodium mercury amalgam under dinitrogen afforded the corresponding terminal nitride complex [MoIVI(N)(PCP)]− (PCP=1,3‐[OP(tBu)2]2C6H3, 19) which was isolated as an ‐ate complex with a sodium‐crown ether counter cation. Surprisingly, attempts to selectively protonate the formed nitride with a variety of different acids only led to the undesired protonation of the phosphinite ligands.
Scheme 6.
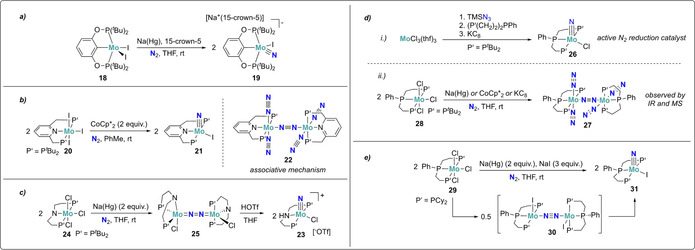
Dissociative N2‐splitting by molybdenum complexes 18 (a), 20 (b), 24 (c), 28 (d), and 29 (e), all bearing pincer ligands.
Furthermore, a variety of molybdenum pincer complexes with PNP and PPP scaffolds were shown to be able to cleave the dinitrogen molecule into nitrides. A pyridine‐based PNP‐pincer ligand scaffold was employed by Nishibayashi and co‐workers for the catalytic transformation of dinitrogen into ammonia. Stoichiometric reduction of the MoIII trisiodide complex 20 with either decamethylcobaltocene (CoCp*2) [41] or samarium diiodide [42] under dinitrogen yielded the corresponding terminal MoIV nitride complex 21 (Scheme 6 b). An end‐on bridging dinitrogen MoI intermediate was postulated as an intermediate based on synthetic experiments and theoretical calculations. Interestingly, a distinct mechanism was proposed for the corresponding Mo0 dinitrogen complex 22 supported by the same pincer ligand. This mechanism will be discussed in further details in Section 4.1.[ 43 , 44 ]
Molybdenum complexes bearing PNP‐pincer ligands for dinitrogen cleavage were studied by the group of Schneider. [45] In 2017, the formation of a MoV nitride complex [Mo(HPNP)(N)Cl][OTf] (HPNP=NH(CH2CH2PtBu2)2, 23) from the corresponding MoIV complex 24 and N2 was reported (Scheme 6 c). The two‐electron reduction of 24 with sodium mercury amalgam under a dinitrogen atmosphere yielded the end‐on bridging dinitrogen complex 25, in which, according to its N−N bond length and vibrational frequency, the N–N double bond was still intact. However, treatment of 25 with a Brønsted acid such as HOTf (−OTf=−OS(O)2CF3), induced full splitting of the dinitrogen unit into the corresponding nitride 23 (Scheme 6 d). While the reaction scheme may suggest an associative mechanism, the structure of 23 revealed that protonation took place at the amide ligand backbone. This proton‐assisted dinitrogen splitting was assigned to a lowering of the energy of the N–N anti‐bonding σ* orbital upon protonation of the amine in the pincer ligand backbone. Population of this N–N antibonding orbital subsequently promotes full dinitrogen cleavage. Dinitrogen scission was also demonstrated by the analogous tungsten complex. [46]
A catalytic system for dinitrogen reduction to ammonia based on PPP‐type Mo‐pincer complexes was developed. [47] There are two main advantages in the modulation of the ligand scaffold from PNP to PPP. Tridentate phosphine ligands are less Brønsted basic and thus more stable towards protonation, disfavoring the competitive hydrogen evolution reaction. The presence of acidic protons on the ligand scaffold may lead to undesired hydrogen evolution reaction under the reducing conditions applied during catalysis. Furthermore, their π‐accepting properties can stabilize a variety of molybdenum oxidation states during the catalytic cycle. Although independently synthesized from the corresponding azide, the MoIV‐nitride complex [Mo(N)Cl(PPP)] (PPP=PhP(CH2CH2PtBu2)2, 26) served as an active catalyst for nitrogen reduction, implying that catalysis proceeds through a dissociative mechanism (Scheme 6 d i). A bridging dinitrogen Mo0 complex 27 was identified as an intermediate species during catalysis and as a byproduct in the stoichiometric reduction of 28 under dinitrogen by IR spectroscopy and mass spectrometry (Scheme 6 d ii).
Another PPP Mo nitride complex [Mo(N)Cl(PPP)] (PPP=PhP(CH2CH2PCy2)2, 29) was prepared recently from dinitrogen cleavage by Mézailles and co‐workers. [48] The triphosphino molybdenum(I) iodide complex 30, generated in situ from the parent MoIII trischloride precursor, readily split N2 at room temperature to the corresponding terminal MoIV‐nitride 31 (Scheme 6 e).
Schneider and co‐workers investigated PNP pincer ligands as supporting scaffolds for rhenium complexes. [49] Reduction of [ReIIICl2(PNP)] (PNP=N(CH2CH2PtBu2)2, 32) with sodium‐mercury amalgam under dinitrogen led to the cleavage of the dinitrogen triple bond and to the formation of the ReV‐nitride complex [ReVCl(N)(PNP)] (33) (Scheme 7 a). [50] Complex 33 could also be prepared by using an outer‐sphere reducing agent or by electrochemical means, and will be discussed in Section 2.2.2. Mechanistic studies revealed that dinitrogen is activated via the formation of a bridging μ‐N2 dinuclear intermediate 34, which was isolated at low temperature and characterized by XRD. [50] The influence of backbone unsaturation on the pincer ligand was investigated in a follow‐up study (Scheme 7 b). [51] The ReIII complex 35 was shown to cleave N2 into the corresponding ReV nitride 36 either by chemical or electrochemical reduction, at less cathodic potentials but yet with lower yields than reported for complex 32 with a saturated ligand backbone.
Scheme 7.
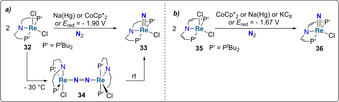
(Electro)chemically induced N2‐splitting by rhenium complexes 32 (a) and 35 (b).
Formation of bridging nitrides: The high nucleophilicity of metal nitrides strongly favors their behavior as bridging ligands, resulting in the formation of di‐ or multinuclear complexes, reviewed in this section. Formation of such multinuclear assemblies was often shown to be further promoted by the presence of alkali cations originating from the reducing agents and acting as bridging groups between the nitride moieties and ligands scaffolds.
Reported in 1999, the vanadium(III) silylamino(disilylamido) complex 37 constituted of the earliest reported complexes capable of cleaving N2. The presence of one equivalent of potassium graphite afforded the bridging bis(μ‐nitrido) VV complex 38 upon N2 cleavage (Scheme 9 a). [52] When two equivalents of potassium graphite were used, the mixed valent VIV–VV complex 39 was obtained. Structural analysis of 39 revealed that one potassium cation was bridging between a supporting amide‐ and a nitride ligand, highlighting its potential role in the reaction of the calixpyrrole uranium(III) complex 40 with one equivalent of potassium naphthalenide under a dinitrogen atmosphere led to the formation of the dinuclear, μ‐nitrido UIV/UV mixed‐valent complex 41 (Scheme 8). X‐ray structural analysis revealed that each of the bridging nitride ligands of 41 were here also additionally coordinated to a potassium cation.
Scheme 9.
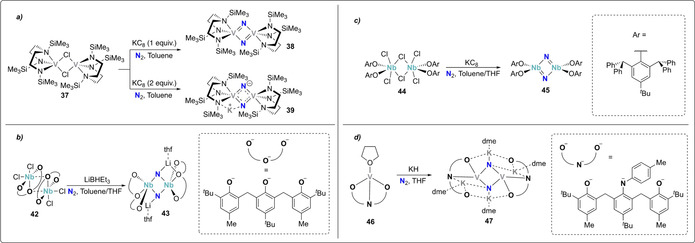
Dissociative N2‐splitting by group 5 transition metal complexes 37 (a), 42 (b), 44 (c), and 46 (d).
Scheme 8.
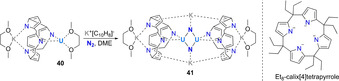
Dissociative N2‐splitting by uranium complex 40.
Analogous alkali ion incorporation was observed upon treatment of the tris‐aryloxide (chelating OOO ligand) NbV complex 42 with excess LiBHEt3 under a dinitrogen atmosphere, affording the bridging dinitride complex 43 (Scheme 9 b). [53] Two lithium cations are there bridging between a nitride and an aryloxide ligand. An analogous reactivity was observed using instead a monodentate but sterically more encumbered aryloxide ligand. The NbV dimeric complex 44 can cleave the N2 triple bond in presence of potassium graphite to form the dimeric niobium nitride complex 45 (Scheme 9 c). Protonolysis of 45 with anhydrous HCl resulted in the formation of ammonium chloride. [54]
Incorporation of four potassium ions coordinated to the nitride ligands and the ligand framework was observed upon treatment of the bis‐aryloxy‐amide (chelating ONO ligand) vanadium(III) complex 46 with potassium hydride under dinitrogen affording the bis(μ‐nitride) VIV dimer 47 (Scheme 9 d). [55]
Using a triamidoamine ligand scaffold, Liddle and co‐workers further illustrated the influence of alkali or alkali‐earth reducing agents mediating the cleavage of dinitrogen by a TiIV complex 48.[ 56 , 57 ] When potassium graphite was used as the reductant, four‐electron reduction of N2 was observed (Scheme 10, left arrow). The dinuclear TiIV complex 49 with two potassium cations coordinating to the bridging [N–N]4− unit was then characterized. When performing the reduction with three equivalents of Mg0 instead, six‐electron reduction of N2 took place and afforded the bridging nitride complex 50 (Scheme 10, right arrow). The structure of 50 revealed that transmetallation from titanium to magnesium occurred. This work underlines the importance of cooperativity between the transition metal and the (earth‐)alkali cation used for dinitrogen reduction, since Mg2+ ions appeared to be necessary to induce full N2 cleavage. Treatment of the bridging nitride complex either with a strong acid (HCl) or with H2 in combination with a frustrated Lewis base pair (PtBu3+B(C6F6)3) led to the release of ammonia.
Scheme 10.
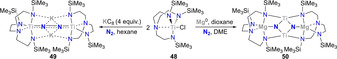
Dissociative N2‐splitting by titanium complex 48.
A whole series of low‐valent, three‐coordinate FeII complexes with β‐diketiminate supporting ligands were reported by Holland and co‐workers to bind and cleave N2 upon treatment with a variety of alkali metal based reducing agents.[ 58 , 59 , 60 , 61 ] Trinuclear and tetranuclear iron dinitrogen and nitride complexes were isolated and characterized. The degree of dinitrogen activation was observed to depend on the type and the amount of the reducing agent used as well as on the steric demand of the β‐diketiminate ligand substituents. The dinitrogen molecule was proposed to be cooperatively activated by π donation from the low‐valent iron centers as well as by the Lewis acidic activation of the alkali ions. Scheme 11 a displays the synthesis of the mixed‐valent tetranuclear FeII/FeIII nitride complex 51 from the corresponding dimeric FeII chloride complex 52. Interestingly, the solid‐state structure of 51 revealed that the potassium ions coordinate to the nitride and chloride ligands as well as to the aryl rings of the β‐diketiminate ligands. A fine tuning of the acid used to protonate the nitride ligands allowed identifying weak acids such as tBu3C6H2OH to selectively protonate the nitride to form ammonia in high yields. [61] When metallic Na as an external reducing agent was used instead of KC8, the formation of the trinuclear iron nitride complex 53 was observed. [62]
Scheme 11.
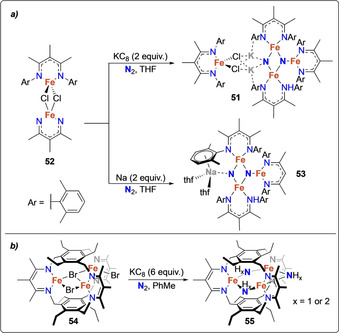
Dissociative N2‐splitting by iron complexes 52 (a) and 54 (b) bearing β‐diketiminate ligands.
Bridging of three β‐diketiminate moieties by two phenyl rings gave access to the trinuclear FeII bromide complex 54 which enabled reductive cleavage of the dinitrogen triple bond. This afforded the trinuclear complex 55 containing μ‐imide and/or μ‐amide bridging groups (abbreviated as NHx, x=1 or 2) between the iron centers (Scheme 11 b). [63] Mössbauer spectroscopy revealed that the iron centers in 55 were present as FeII and FeIII. In comparison to the iron nitride complexes supported by non‐bridging β‐diketiminate ligands (vide supra), no potassium cations were incorporated into the structure of 55. The origin of the protons bound to the bridging N‐containing ligands could however not be unambiguously assigned.
Sita and co‐workers reported the synthesis of the dinitride‐bridged tantalum dimer [{Cp*Ta[N(iPr)C(R)N(iPr)](μ‐N)}2] (56, Cp*=η 5‐C5Me5, R=Me, NMe2, Ph) obtained by reduction of the tantalum amidinate trichloride complexes [Cp*Ta[N(iPr)C(R)N(iPr)]Cl3] (57, Cp*=η 5‐C5Me5) with four equivalents of potassium graphite under a dinitrogen atmosphere (Scheme 12 a). [64]
Scheme 12.
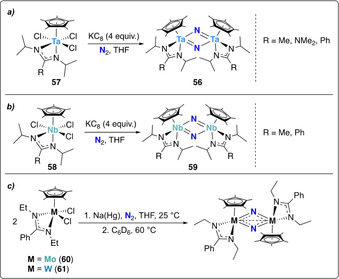
Dissociative N2‐splitting by group 5 and 6 amidinate complexes, 57 (a), 58 (b), 60 and 61 (c).
The same ligand set was used to prepare a series of group 5 and 6 transition metals complexes.[ 65 , 66 , 67 ] Among those, only the niobium complex [Cp*Nb[N(iPr)C(R)N(iPr)]Cl3] (58, R=Me, Ph) presented the same dinitrogen cleavage activity in presence of potassium graphite, forming the bridging nitride complex 59 (Scheme 12 b). [67] Reducing the steric demand on the N substituents of the amidinate ligand (Et vs. iPr) allowed for homolytic cleavage of N2 promoted by the corresponding group 6 transition metal complexes 60 and 61 in presence of sodium amalgam (Scheme 12 c). [68]
2.2.2. N2 splitting induced by light or electrochemistry
As highlighted above, the use of alkali‐metal reducing agents may complexify the evaluation of their individual roles in the reaction mechanism, as potentially acting both as reducing agents and Lewis acids. In addition, the highly negative redox potential of alkali ions restrains their use for the development of catalytic systems, as large thermodynamic costs would be associated. Strategies allowing to separately investigate N2 cleavage and functionalization while being able to easily tune the energy provided to the system are hence highly desirable for both mechanistic analysis and the discovery of new catalytic systems. The following section aims at discussing the formation of metal nitride complexes from dinitrogen triggered by photo‐ or electrochemical means.
Photochemical activation of dinitrogen was described in few reports, taking advantage of potential MLCT transitions to the N–N π* orbital to promote N2 splitting.[ 69 , 70 ] The first example of photoactivation of N2 was reported by Floriani and co‐workers. The three coordinate complex [Mo(Mes)3] (Mes=2,4,6‐Me3(C6H2)), which was generated in situ by treatment of [MoCl4⋅dme] with four equivalents of mesitylene‐Grignard, reacted with dinitrogen by a four‐electron reduction under the formation of a dimeric MoV complex 62 with a bridging [N–N]4− unit (Scheme 13 a). [71] Compared to the tris‐amide complex 1 developed by Cummins (Scheme 5), [27] the mesitylene ligands in 62 are less electron donating than the anionic amide ligands and could not induce full cleavage of the dinitrogen moiety. 62 was reported to be stable in refluxing benzene, however, the formation of the bridging nitride complex 63 was observed upon exposure to UV light (λ=365 nm).
Scheme 13.
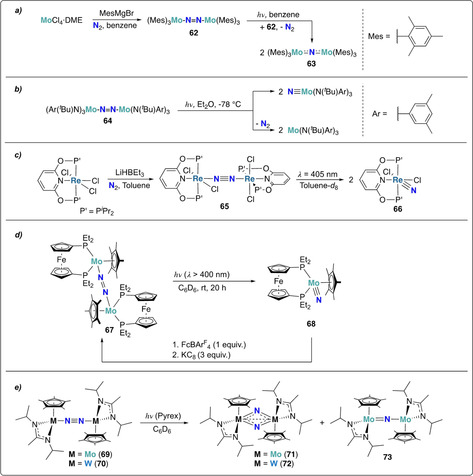
Photolytic N2‐splitting of dinitrogen bridged group 6 and 7 metal complexes 62 (a), 64 (b), 65 (c), 67 (d), 69 and 70 (e).
Transient absorption spectroscopy experiments revealed that the bridging dinitrogen complex 64 prepared by Cummins et al. could also be photoactivated, resulting in both Mo−N and N−N bond cleavage (Scheme 13 b).[ 29 , 72 ]
The photolytic cleavage of the rhenium bridging dinitrogen complex 65 was reported very recently (Scheme 13 c). [73] Similar to complex 62 (vide supra), 65 was found to be thermally stable, whereas irradiation with visible light (λ=405 nm) led to photolytic cleavage of the dinitrogen unit into the corresponding terminal nitride complex 66. Nitride functionalization to ammonia was achieved by a PCET‐type mechanism using SmI2 and H2O as reducing agent and proton source, respectively.
An additional example of photochemically triggered dinitrogen splitting was uncovered by Nishibayashi et al. [74] The ferrocenyl‐diphosphine and pentamethyl‐cyclopentadienyl supported dinitrogen bridged Mo‐complex 67, obtained from a molybdenum hydride complex and dinitrogen in two steps, was transformed to the corresponding Mo‐nitride 68 by irradiation with visible light (λ>400 nm) (Scheme 13 d). Note that the original dinitrogen‐bridging dinuclear Mo‐complex 67 could be regenerated by sequential oxidation with [FeCp2][BArF 4] and reduction with potassium graphite. Treatment of Mo‐nitride 68 with excess decamethylcobaltocene (CoCp*2) and [LutH][BArF 4] led to the formation of ammonia (0.37 equiv based on 68).
Sita and co‐workers disclosed the light mediated dinitrogen cleavage of group VI (M=Mo or W) bridging dinitrogen complexes (69 and 70) (Scheme 13 e). [75] Upon photolysis of 69 or 70, the respective bridging dinitride complexes 71 and 72 were obtained, together with small amounts of mono‐nitride complex 73 when Mo complex 69 was used. As mentioned in the previous section, modifying the steric demand of the supporting amidinate ligand from isopropyl to ethyl (Scheme 12 c) allowed for the conversion of the photochemically driven cleaving step into a thermally driven reaction. [68]
Electrochemical (or outer‐sphere) dinitrogen reduction to nitrides may represent a key step to the development of catalytic systems, however very few complexes have been reported to mediate dinitrogen cleavage in the absence of external functionalization agents such as protons or electrophiles. The first of such an example was reported by Schneider and co‐workers, showing that dinitrogen splitting and subsequent preparation of complex 32 could also be accomplished by controlled potential electrolysis under an N2 atmosphere (E red=−1.90 V vs. Fc/Fc+, 0.2 m nBu4N+PF6 − in THF) (Scheme 7 a), or by using an outer‐sphere reducing agent (CoCp*2). [50] The same behavior was observed for the analogous complex 35. [51]
The one‐electron oxidation induced dinitrogen splitting reported by Masuda shortly after represents another rare example of nitride formation enabled by electrochemistry, yet with a very different strategy. [76] This approach relied on the oxidation of the Mo0 complex trans‐[Mo(depe)2(N2)2] (74, depe=Et2PCH2CH2PEt2) by chemical ([FeCp2][BArF 4]) or electrochemical means (E ox=+0.5 V vs. Pt‐wire), generating in situ [(μ‐N2)(MoII(depe)2)2]2+ with a MoII−N=N−MoII core followed by the formation of the MoIV nitride complex 75 (Scheme 14). Nevertheless, this protocol could not be easily exploited for catalytic N2 reduction as it operates under oxidative conditions, whereas the conversion of dinitrogen to ammonia requires an overall reduction of the substrate.
Scheme 14.
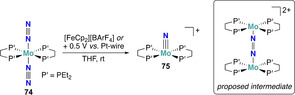
Electrochemical N2‐splitting by molybdenum complex 74.
2.3. Nitride functionalization
The previous section provided a significant number of metal complexes able to fully cleave dinitrogen to nitrides, but to date only very few catalytic systems were identified with such complexes (see Scheme 47 in Section 4). One of the main challenges to establish such catalytic system is to functionalize the formed nitrides. Transition metal nitrides that are formed upon dissociative N2 cleavage are often very stable and their functionalization hence requires strong electrophiles/proton sources. These harsh conditions could interfere with the low‐valent metal precursor. To overcome this limitation, a few synthetic cycles were developed, providing the reducing agents and functionalizing groups in a stepwise manner. [77] Following this strategy, most of the formed nitrides react with strong proton sources to generate ammonia, but in many cases destruction of the metal complex is observed simultaneously. These examples were treated together with the description of the nitride formation in the previous sections. The following section focuses on synthetic cycles enabling the formation of N−C and N−Si bonds from nitrides that originate from N2. Such reactions allow for the synthesis of fine chemicals with the N atom originating from dinitrogen. This can be especially interesting for the manufacture of value‐added products from N2 as well as for the synthesis of 15N labelled compounds (if using 15N2 as the nitrogen source). All nitride functionalization reactions reported here are summarized in Scheme 15 and Table S2. The reader is asked to refer to this scheme for all structures mentioned in this section.
2.3.1. N−C bond formation
Many investigations have been performed on the MoVI trisamide nitride complex 2. [27] The weak nucleophilic character of this nitride allowed for functionalization with a variety of strong electrophiles such as methyl iodide, Me3SiOTf or PhC(O)OTf yielding substituted imido complexes 76 (Scheme 15). [78] Cummins and co‐workers developed a synthetic cycle consisting of the molybdenum‐ and niobium trisamide complex 1 and 77, which converted dinitrogen into nitriles. [79] The N−C bond forming reaction took place in between a NbV nitride and an acyl chloride species. The first well defined transfer of the nitride ligand from [MoVI(NtBuAr)(15N)] (2–15 N) to an organic molecule was achieved using trifluoroacetic anhydride as an acceptor. [80] Quantitative formation of CF3CO15NH2 was observed with concomitant decomposition of the molybdenum complex. A full synthetic cycle that converted dinitrogen into organic nitriles was later disclosed by Cummins (Scheme 16 a). Key step was the use of SnCl2 or ZnCl2 as strong Lewis acids and chloride donors to release the free nitrile and to form the MoIV trisamide chloride complex 78 which could be reduced back to the initial MoIII trisamide complex 1 and thus close the synthetic cycle. [81]
Scheme 16.
Synthetic cycles promoted by transition metal complexes 2 (a), 46 (b) and 68 (c) for the synthesis of isocyanates and nitriles utilizing N2 as a nitrogen source.
Similarly, Kawaguchi et al. reported the alkylation of the bridging μ‐nitrido niobium dimer 12 with methyl iodide to afford the corresponding imide bridged complex 79 (overview Scheme 15). [35] Subsequent addition of pyridine and reaction with CO2 afforded the NbV‐ureate complex 80. [82]
N−C bond formation, integrated into a synthetic cycle could also be accomplished from the VV nitride complex 47, which was obtained from the VIII complex 46 by reductive cleavage of N2 (Scheme 16 b). The nitride ligand of 47 was functionalized with CO to the corresponding isocyanate complex 81. Free potassium isocyanate precipitated upon addition of dimethylacetylene and dissolving in THF recovered the original complex 46. [55] Additional reactivity of 5 was observed with 2,6‐xylylisocyanide, which formed a carbodiimide complex 82.
Formation of isocyanates was also observed using the end‐on dinitrogen bridged dimer [(Cp*Mo[N(iPr)C(Me)N(iPr)])2(μ‐N2)] (69). Complex 69 was used in a chemical cycle producing isocyanates (R3ENCO) from N2, CO2 and R3ECl (R3E=Me3Si, Ph3Si, Me3Ge or Me3C) (Scheme 16 c). [75] Treatment of 69 with R3ECl under UV light afforded the corresponding terminal imido complexes 83. Reaction with CO2 and excess R3ECl followed by reduction under N2 led to the release of substituted isocyanate (R3ENCO) and regeneration of the original complex 69.
Organic nitriles were likewise obtained from PNP‐type pincer rhenium nitrido complexes in high yields. [83] While the nitride moiety in complex [ReV(PNP)Cl(N)] (33, PNP=((tBu2PCH2CH2)2N) could not be protonated with HOTf (which led to protonation of the ligand backbone), it could be functionalized to the methylimide complex 84 using MeOTf (Scheme 17 a). [49] The very same outcome was reported for functionalization of the nitrido complex 35 bearing a pincer ligand with an unsaturated backbone (Scheme 17 b). [51] Methylation of the nitrido ligand of the iron complex 51 was also achieved upon reaction with methyl tosylate (MeOTs, TsO−=p‐CH3‐C6H4‐SO3 −). [84]
Scheme 17.
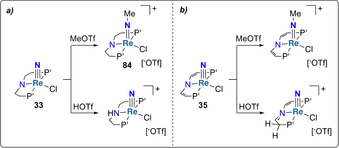
Functionalization of metal‐nitride complexes 33 (a) and 35 (b).
Analogously, reaction of 33 with ethyl triflate yielded a rhenium ethyl imido complex which was deprotonated with the strong base (KN(SiMe3)2) to afford the corresponding ReIII ketimide complex 85 (Scheme 18 a). Treatment with N‐chlorosuccinimide (NCS) led to the release of free acetonitrile and the rhenium(IV) trichloride complex 86. Reduction of this trichloride complex with sodium mercury amalgam under dinitrogen regenerated the putative rhenium nitride 33 and closed the chemical cycle which produced acetonitrile from ethyl triflate and N2. The very same protocol was applied for the synthesis of benzonitrile with in situ generated benzyl triflate as the carbon source. [85] Use of a sterically less demanding pincer ligand (isopropyl instead of tertiary butyl substituents on the phosphorous) enabled the formation of benzonitrile and benzamide from dinitrogen and benzoyl chloride (Scheme 18 b). [86] Interestingly, this synthetic cycle exploited for each step a different activation strategy: i) photochemical splitting of Re‐dinitrogen complex 87, ii) thermally induced reaction of benzoyl chloride with the formed nitride complex 88 and iii) electrochemical reduction of 89 under N2 to regenerate the dinitrogen bridged Re‐complex 87.
Scheme 18.
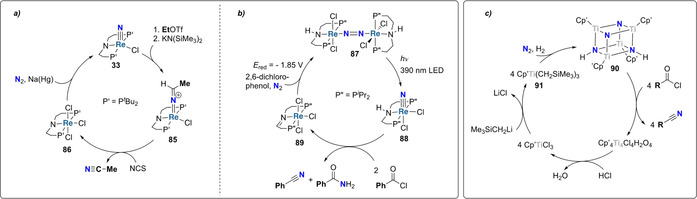
Synthetic cycles promoted by transition metal complexes 33 (a), 89 (b) and 91 (c) for the synthesis of organic nitriles utilizing N2 as a nitrogen source.
Multinuclear titanium complexes were also used as a platform to split dinitrogen followed by functionalization of the formed nitrides into organic nitriles (Scheme 18 c).[ 31 , 87 ] The tetranuclear bisimido/bisnitrido complex [(Cp′Ti)4(μ3‐NH)2(μ3‐N)2] (90, Cp′=C5Me4SiMe3) was obtained from [Cp′Ti(CH2SiMe3)3] (91) by hydrogenation under dinitrogen and heating under dinitrogen at 180 °C. Reaction with a variety of acyl chlorides yielded the corresponding organic nitrile compounds. The original titanium complex 90 could be obtained again upon treatment with HCl followed by Me3SiCH2Li.
2.3.2. N−Si bond formation
Alternatively, a few examples of electrophilic functionalization of metal nitride obtained by N2 reduction have been reported using silylation reagents such as silanes of silyl halides.
Mézailles and co‐workers used the bis(silane) HSiMe2(CH2)2SiMe2H to functionalize the PPP‐type pincer supported MoIV nitride complex 31. The use of a bis(silane) functionalizing group allowed the release of the N‐containing product as a cyclic silylamine in good yields (see overview Scheme 15). [48]
A different behavior had been disclosed by Sita and co‐workers using PhSiH3 to functionalize the bridging bis(μ‐nitrido)‐tantalum complex [{Cp*Ta[N(iPr)C(Me)N(iPr)](μ‐N)}2] (56). The functionalization product could not be released, affording the silyl substituted Ta‐imido complex 92. [64] However, as depicted in overview Scheme 15, using the Mo analogue [Cp*Mo[N(Et)C(Ph)N(Et)]Cl2] (60), quantitative formation of hexamethyldisilazane (HN(SiMe3)2) could be obtained upon reacting the terminal imido complex 93 with an alcohol X−OH (X=iPr, Me3Si) and Me3SiCl, allowing for the full cleavage of the metal nitride bond and release of the N‐containing product. [88]
The proposed mechanism for the generation of HN(SiMe3)2 involves the formal addition of HCl (generated from the reaction of X−OH with Me3SiCl) across the Mo=N double bond of the imido complex 93, generating a transient MoIV amido complex. This intermediates further reacts with a second equivalent of Me3SiCl to yield HN(SiMe3)2 and regenerate the initial MoIV dichloride complex 60.
2.4. Summary
The review of the complexes above highlights that, despite a significant number of molecular complexes are able to mediate the full cleavage of dinitrogen, only a few of them have been identified to mediate this cleavage in the absence of external reducing agents. In addition, the high stability of the formed nitrides is generally a key driving force of dissociative mechanisms. However, this high stability represents a challenge to be overcome when considering the functionalization of metal nitrides, which requires to utilize strong electrophiles and proton sources. The common incompatibility of such reagents with the metal complex precursors nevertheless constitutes one of the main limiting factors for the use of such complexes in catalytic conditions. Milder homolytic bond formation mechanisms, notably via PCET, may provide a promising strategy to utilize these metal complexes in a catalytic way, without the need of stepwise synthetic cycles.
3. Associative Pathways
The major driving force of associative reaction pathways relies on the activation of M−N2 species through the functionalization of the coordinated N2 moiety with electron deficient substituents, facilitating the transfer of electron density onto the N2 moiety and its subsequent reductive cleavage. We will distinguish in this section key associative N2 cleavage strategies based on the stepwise protonation and silylation of N2 or on the functionalization of N2 with carbon‐based electrophiles. This section will mainly focus on the stoichiometric transformation of N2, while systems enabling catalytic conversion of dinitrogen will be discussed in Section 4. Associative mechanisms typically involve the initial coordination of N2 without full cleavage: for most examples presented here we will hence not specifically focus on the N2 binding step but mainly on subsequent functionalization steps. An overview of all functionalization strategies towards N−H, N−Si and N−C bond formation reported in this section are compiled in Scheme 19 and Tables S3–S5.
3.1. N−H bond formation
Hydrogenation strategies of N2 require the provision of protons and electrons. Several strategies have been explored and will be reported in this section, differentiating mechanisms involving the separate supply of electrons and protons using Brønsted acid sources and reducing agents (that could be the complex itself) and reaction pathways providing protons and electrons via a sole source, namely dihydrogen or hydrides.
3.1.1. Protonation pathways
Direct protonation of the metal bound dinitrogen is one of the predominant criteria towards its conversion to ammonia. The key requirement for such protonation to occur relies on providing sufficient electron density to the bound dinitrogen moiety. [32] This often necessitates the use of low‐valent metal centers, which however typically display low Lewis acidity and N2 binding affinity while enhancing the risk of competitive proton reduction to dihydrogen. The Lewis acidity of the metal centers can yet be modulated by the ligand used. This allows for an advantageous N2 over H+ binding as well as furnishing substantial electron density on the N2 moiety for directed protonation via concomitant electron transfer in so‐called proton‐coupled electron transfer steps. Such strategies towards the protonation of metal ligated N2 species will be addressed in this section.
Bis‐diphosphine dinitrogen complexes of molybdenum and tungsten were early synthesized by the reduction under a N2 atmosphere of [MoCl3(thf)3] or [WCl4(thf)4] with sodium amalgam or metallic Mg [89] or of molybdenum tris‐acetylacetonate complexes with AlEt3. [90] This series of dinitrogen complexes enabled an extensive number of studies on N2 functionalization utilizing these species as starting N2 synthons. Chatt and co‐workers demonstrated as early as 1972 that such bis‐diphosphine dinitrogen complexes can undergo direct protonation using strong acids. Bis‐diphosphine complexes of tungsten, trans‐[(dppe)2W(N2)2] (94) (dppe=Ph2PCH2CH2PPh2), [91] and molybdenum, trans‐[(dppe)2Mo(N2)2] (95), trans‐[(depe)2Mo(N2)2] (96) can be protonated using strong inorganic acids (e.g. HBr or HCl) in toluene or THF to produce the corresponding diazene complexes [MX2(dppe)2NH=NH] (M=W (97), Mo (98), and X=Cl or Br) and [MoBr2(depe)2NH=NH] (99) in good yields (Scheme 20).[ 91 , 92 ] These seven coordinated diazene complexes further underwent proton reorganization to generate the corresponding hydrazido complexes, for example, 100, upon dissociation of an anionic halide ligand. The enhanced cationic character of the metal resulting from halide dissociation was attributed as a possible reason for this reorganization. Interestingly, Pickett and Talarmin demonstrated in 1985 that such hydrazido complexes could also be generated electrochemically but with a p‐TsO counterion. [93]
Scheme 20.
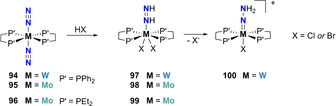
Associative N2 protonation using group 6 bis‐diphosphine complexes, 94–96 resulting in hydrazine complexes 97–99 and hydrazido complex 100.
This reactivity was shown to be maintained for a broad range of bis‐diphosphine complexes [M(N2)2(PP)2] that include redox active moieties such as ferrocenyl‐, ruthenocenyl‐, and chromobenzene‐based bis‐diphosphine ligands (PP) where the redox active moieties were incorporated in place of the ‐CH2CH2‐ group, as highlighted in Scheme 21 a. The formation of diazene intermediates trans‐[M(depf)2(=N‐NH2)(OTf)]OTf (M=W (101) or Mo (102), depf=bis‐diethylphosphinoferrocene) was also identified upon treatment with triflic acid (Scheme 21 c). At the difference of ethylene(bis‐diphoshine) complexes 94–96 presented above, formation of NH3 was observed upon treatment of complexes 103–107 with H2SO4.[ 94 , 95 , 96 ] This different behavior regarding the formation of NH3 was suggested to originate from the hemilabile character of the nonflexible metallocene ligands. Analogous complexes in mixed ligand environments with both bidentate and monodentate ligands such as 108–110 also showed similar reaction patterns, yet with an overall lower activity towards ammonia formation (Scheme 21 b). [96]
Scheme 21.
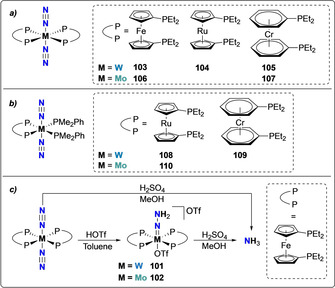
Associative N2 protonation using group 6 metal complexes, bearing metallocene diphosphine ligands 103–110 (a) and (b), and resulting hydrazido complexes 101–102 (c).
This behavior appeared to be characteristic of a broad range of bis‐diphosphine dinitrogen complexes, notably based on other transition metals such as Cr and Fe. Trans‐[Cr(dmpe)2(N2)2] (dmpe=Me2P‐CH2CH2‐PMe2) could be obtained by reduction of trans‐[Cr(dmpe)2Cl2] using sodium‐mercury amalgam, [97] Rieke magnesium [98] or nBuLi. [99] While ammonia or hydrazine were not released by protonation using a variety of proton sources, [100] the Cr hydrazido complex [Cr(OTf)(dmpe)2(=N‐NH2)][OTf], similar to the Mo and W analogues described before, was isolated. [101] Leigh et al. reported the stoichiometric activation of dinitrogen to ammonia using the iron(0)‐dinitrogen complex [Fe(dmpe)2N2] (111) (Scheme 22 a).[ 102 , 103 , 104 ] Interestingly, different reaction products could be observed depending on the protonation condition used: 0.12 equiv of NH3 and trace amounts of hydrazine were obtained upon reaction with HCl in THF whereas only hydrazine was formed when MgCl2 was added to the reaction mixture in presence of HCl.
Scheme 22.
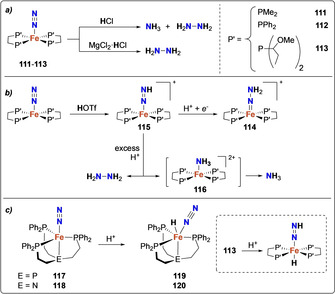
Associative N2 protonation using iron complexes 111–113 supported by chelating diphosphines (a), and resulting hydrazido complex 114, diazenido complex 115 and ammonia complex 116 (b). Iron complexes 117–118 supported by chelating triphosphines and hydrides 119–120 (c).
Very similar type of reactivity was also encountered for other iron‐diphosphine complexes such as [(dppe)2Fe0‐N2] (112) [104] and water soluble trans‐[Fe(DMeOPrPE)2(N2)] (113). Formation of the [Fe=N‐NH2] species 114 in the presence of weaker acids, for example, HOTf or [Ph2NH2][OTf], and excess of [CoCp*2] further suggested the occurrence of a distal associative mechanism for N2 reduction. [105] It should be noted that in addition of the hydrazine complex 114 mentioned above, the other potential intermediates formed en route to NH3, for example, diazine 115 and ammonia complex 116 were isolated by ex situ reaction with the corresponding N‐containing synthons,[ 106 , 107 , 108 ] allowing to propose the reaction mechanism shown in Scheme 22 b.
The overall six electron reduction yielding to ammonia formation was proposed to occur via the participation of multiple Fe0 complexes acting as sacrificial reducing agents to provide the six electrons, as further suggested by the isolation of the bridging N2 dimer [{Fe(dmpe)2}2(μ‐N2)]. [109] In addition, product distribution was observed to be highly solvent dependent, as a consequence of the modulation of the acidity of the proton sources in different solvents: Complex 111 produced N2H4 in pentane, only NH3 in THF and a mixture of both N2H4 and NH3 in Et2O when treated with [H(OEt2)2][OTf]. [109]
The use of tetradentate tripodal phosphine ligands allowed to identify that iron hydrides were key reaction intermediates obtained upon protonation of the Fe dinitrogen complex: the protonation of tris‐(2‐diphenylphosphino)‐ethylene)phosphine (PPPh 3) iron dinitrogen complex [Fe(PPPh 3)(N2)] (117) [110] or tris‐(2‐diphenylphosphino)‐(ethylene)amine (NPPh 3) iron dinitrogen complex [Fe(NPPh 3)(N2)] (118) [111] resulted in the formation of stable iron hydride complexes 119 and 120 (Scheme 22 c). DFT calculations [107] and experimental [106] studies by Tyler and co‐workers allowed to demonstrate that such iron‐hydride species were also formed with the bis‐diphosphine complex 113 (Scheme 22 a). These studies suggested that protonation of the iron centre occurred first to generate hydrides trans to the N2 ligand, followed by protonation of the latter. The complete series of trans‐hydride dinitrogen, hydrazido‐, hydrazine‐ and amido‐complexes was characterized, highlighting the stability of these trans‐hydride species at all steps of ammonia generation. [106]
Essentially analogous reactivity to that observed with bidentate phosphine ligands was noticed with Mo and W complexes bearing monodentate phosphines. Protonation of trans‐[W(N2)2(MePPh2)4] (121) with HCl lead to the formation of the diazene complex trans‐[WCl(MePPh2)4NH=NH]Cl [112] (122). However, the cis‐isomer of [W(N2)2(Me2PPh)4] (123) showed significantly higher activity, leading to the quantitative formation of ammonia upon treatment with H2SO4 in methanol (Scheme 23).[ 113 , 114 ] Mixtures of ammonia and hydrazine were observed for the molybdenum analogue, cis‐[Mo(N2)2(Me2PPh)4] (124) under the same conditions.[ 115 , 116 ] These studies were key to identify the relatively lower reactivity of trans isomers towards NH3 formation [114] and constituted the first example of the use of H2SO4 in methanol as a proton source for ammonia generation, which inspired many following studies. [112] Direct coordination of methanol to the metal centers in these reaction conditions was early proposed as a key step for ammonia formation. [117] The difference of activity of complexes 123 and 124 with respect to their bidentate analogues 94 and 95 (Scheme 20, vide supra) towards NH3 formation (the latter two showing essentially no activity) was attributed to the easier dissociation of one of the monodentate PMe2Ph ligand upon protonation of N2. [114]
Scheme 23.
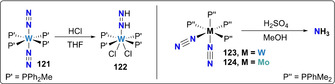
Associative N2 protonation using group 6 monodentate phosphine complexes 121, 123, 124 and resulting hydrazine complex 122.
This behavior was exploited to design complexes bearing both tridentate and monodentate phosphine ligands. Protonation of the complex cis‐[Mo(N2)2(PPP)(PPh3)] (125) (PPP=PhP(CH2CH2PPh2)2) under milder conditions (HBr in Toluene) resulted in the quantitative formation of NH3 (Scheme 24 a).[ 118 , 119 ] This complex allowed the identification of the origin of the electrons required for N2 reduction: isolation of [Mo(PPP)Br3] (126) after NH3 formation suggested that each Mo center provided three electrons. The easier dissociation of monodentate PPh3 ligand provided access to the anionic Br−, leading to a further increase in electron density on the reaction center.[ 96 , 120 ] To circumvent the use of one equivalent of complex acting as a sacrificial electron donor, Mo complexes bearing redox non‐innocent ferrocenyl diphosphine ligands in combination with tridentate PPP ligands were investigated for N2 activation (Scheme 24 b) and exhibited the highest catalytic activity towards NH3 formation among a series of PPP Mo complexes with bidentate diphosphine ligands. [119]
Scheme 24.
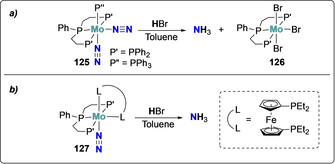
Associative N2 protonation by molybdenum complexes 125 and 127 bearing chelating triphosphines.
Inspired by active catalysts for H2 production replicating the proton relays found in the second coordination sphere of hydrogenase enzymes,[ 121 , 122 ] Mock and Bullock incorporated pendant amines to polydentate phosphine ligands for Cr dinitrogen complexes. Such ligands can act as proton relays and could mediate the challenging multiple protons transfers required for N2 reduction to ammonia. While Cr dinitrogen complexes bearing diphosphine [123] and triphosphine [98] ligands [Cr(N2)2(PBn 2NPh 2)2] (128) and [Cr(N2)(PBn 3NPh 3)] (129) with pendant amines were not reported to mediate N2 protonation (Scheme 25 a), the macrocyclic P4N4 chromium complex [Cr(N2)(PBn 4NPh 4)] (130) generates a mixture of N2H5 + and NH4 +, in presence of excess acid and reducing agent. The ratio between the two products being dependent of the reaction conditions: hydrazine is favored at low temperature while ammonia is obtained in quantitative yield at room temperature.[ 124 , 125 ] DFT calculations suggested that upon protonation, 130 generates the diazenido complex [Cr(N=NH)(PBn 4NPh 4)]+ (131), together with the hydride complex [CrH(N2)(PBn 4NPh 4)]+ (132) (Scheme 25 b, inset). [124] Most importantly, this complex was used to illustrate the first functionalization by hydrogen atom transfer of an end‐on bound dinitrogen ligand: NH3 was quantitively obtained from the reaction of 130 with TEMPOH (2,2,6,6‐tetramethylpiperidine‐1‐ol) (Scheme 25 b). [125]
Scheme 25.
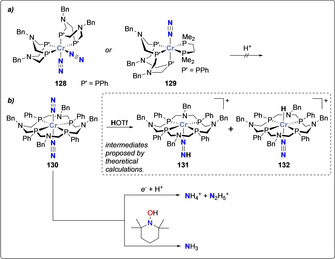
N2 protonation by chromium complexes bearing macrocyclic phosphines ligands: no reactivity towards protonation of complexes 128–129 (a) and protonation of complex 130 using proton or hydrogen atom donors (b). Hydrazido complex 131 and hydride complex 132 proposed by DFT calculations are provided in inset.
In addition to above described end‐on coordinated mononuclear N2 complexes, a few studies explored the protonation of complexes containing bridging end‐on M‐N2‐M cores (Scheme 26). Bercaw and co‐worker reported the preparation of [{(Cp*)2Zr(N2)}2‐(μ2‐N2)] (133) upon reduction of [Zr(Cp*)2Cl2] under a N2 atmosphere.[ 24 , 126 , 127 ] Addition of Brønsted acids to this N2‐bridged complex resulted in the formation of hydrazine and the release of two equivalents of dinitrogen. [126] End‐on bridging N2 dithioguadinate niobium and tantalum complexes 134 and 135 were reported,[ 128 , 129 , 130 , 131 , 132 ] and analogously afforded hydrazine as the main product upon stoichiometric protonation, whereas in presence of excess additional reducing agent, NH3 was generated instead.[ 133 , 134 , 135 , 136 ] Quantitative generation of NH3 and N2H4 was also observed for a series of dinitrogen bridged, phosphine (P′) supported dimeric gold clusters 136‐P′. [135] The presence of hydrazine as the main reduction product for this series of complexes suggested that N2 reduction occurred via an alternating associative pathway.
Scheme 26.
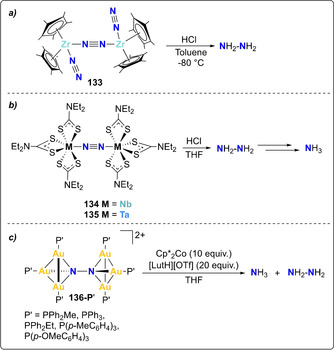
Associative N2 protonation by complexes 133 (a), 134 and 135 (b) and gold clusters 136‐P′ (c).
In 2011, Evans and co‐workers reported the first example of reductive protonation of a rare‐earth dinitrogen bridged complex using the bis‐yttrium complex {[(Me3Si)2N]2(thf)Y}2(μ‐η 2:η 2‐N2) (137). [137] Reduction of complex 137 with KC8 afforded the N2 3− bridging complex {[(Me3Si)2N]2(thf)Y}2(μ‐η 2:η 2‐N2[K(thf)6] (138). Upon treatment of 137 with one equiv of [Et3NH][BPh4], disproportionation to a 1:1 mixture of 138 and complex {[(Me3Si)2N]2(thf)Y}2(μ‐N2H2) (139) occurred (Scheme 27). Complex 139 had also been isolated in lower yields via the direct reduction of the precursor complex Y{[N(SiMe3)2}3 with excess KC8 in THF under N2. The hydrogen atoms in the latter case were hypothesized to originate from the methyl groups of the N(SiMe3)2 − ligand.
Scheme 27.
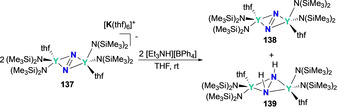
Protonation of the bridging N2 3− moiety in bis‐yttrium complex 137 to produce the corresponding hydrazido complex 139 via disproportionation.
The potential of actinide complexes to mediate the reductive protonation of dinitrogen was first demonstrated using the thorium bis‐phenoxide complex 140, which formed the amide complex 141 upon treatment with excess potassium naphthalenide under a dinitrogen atmosphere (Scheme 28 a). [138] However, reaction intermediates in this reaction and the source of the protons could not be identified in this seminal study. Very recently, Arnold et al. further illustrated the potential of poly‐phenoxide actinide complexes for dinitrogen activation. The m‐tetraphenoxide‐arene ligated binuclear complex of UIV and ThIV (142 and 143) readily react with dinitrogen in presence of 4 equiv of KC8 to generate corresponding hydrazido (N2H2 2−) bridged dimeric dianionic complexex (144 and 145) (Scheme 28 b). [18] The source of protons was identified as the ligand benzylic C−H bond, resulting in the formation of a new metal‐carbon bond to each metal center. Subsequent protonation of the of the bridging hydrazido complex with weak acids (e.g. PyHCl or [HNEt3][BPh4]) afforded ammonia in yields up to 1.1 equiv with respect to complex 144 and 145.
Scheme 28.
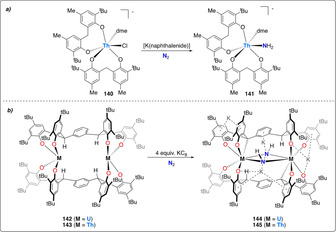
(a) Activation of dinitrogen by mononuclear Th complex (140) with diphenoxide ligand. (b) Associative activation of dinitrogen with polyphenoxide complex of U (144) and Th (145) via ligand deprotonation.
Last, the recent work of Mazzanti and co‐workers has illustrated the potential of actinides to mediate dinitrogen cleavage and hydrogenation. [139] The strongly reducing dinuclear UIII complex 146, which incorporates a bridging nitride ligand and the coordination of three potassium cations, was shown to react even in the solid state with one equivalent of dinitrogen (Scheme 29). Four electron reduction (2× UIII→UV) led to the formation of complex 147 with a side‐on bridging [N–N]4− unit. This side‐on bridging N2 4− moiety could be cleaved upon protonation with excess acid (e.g. HCl) to afford ammonia in 42 % yield. Higher ammonia yields of 77 % could be obtained upon initial treatment with H2 followed by protonation.
Scheme 29.
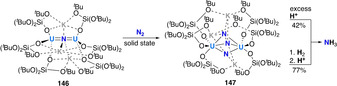
Associative N2 splitting and protonation/hydrogenation by uranium siloxide complex 146.
3.1.2. Hydrogenation pathways
Reduction of N2 to hydrazine or ammonia necessitates the use of protons and electrons. All examples presented so far in this section involved the use of protons from Brønsted acids, the electrons being provided by the complexes themselves. Alternatively, protons and electrons can be both supplied using H2 or hydrides to hydrogenate N2, furnishing one or two electrons per proton added. In direct analogy with the Haber–Bosch process, early attempts of direct hydrogenation of end‐on bound terminal dinitrogen complexes were not successful.[ 90 , 140 ] Nevertheless, acidic transition metal hydrides [(η5‐C5H5)2ZrHCl] [141] (148), [H2Fe(CO)4] (149), [HFeCo3(CO)12] (150) or [HCo(CO)4] [142] (151) were shown to be effective at promoting the hydrogenation of cis‐[W(N2)2(PPh2Me)4] (123) to NH3 and N2H4 (Scheme 30). Morris and Hidai showed separately that the Ru‐H2 complex [η5‐C5H5Ru (η 2‐H2)(dtfpe)]BF4 (152) and in situ generated trans‐[RuCl(η 2‐H2)(dppe)2]X (153) are capable of hydrogenating the coordinated N2 molecule in trans‐[W(N2)2(dppe)2] (94) to produce the hydrazido complex 100F [143] and subsequently NH3.[ 143 , 144 ] Following the same strategy, hydrogen activation and transfer steps could also be realized via the coupling of polysulfido bridged iron, iridium or rhodium complexes, able to activate H2 to form polyhydrosulfido complexes 154–156, which were used to hydrogenate the dinitrogen tetraphosphine W complex 123 to yield NH3. [145]
Scheme 30.
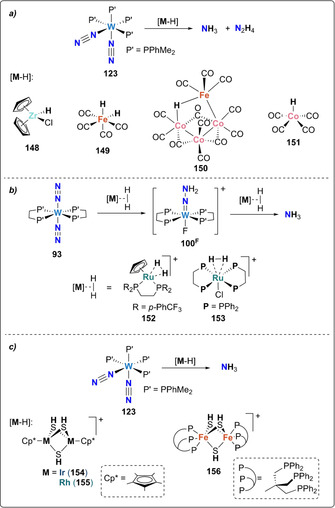
Associative N2 hydrogenation by tungsten complexes 123 (a and c) and 93 (b) using various metal hydrides 148–156.
Budzelaar and co‐workers proposed an alternative strategy for N2 hydrogenation based on the use of sodium hydride as reducing agent and proton source and promoted by a Cr complex with redox non‐innocent diiminepyridine ligands. [146] Treatment of the formally monovalent CrI complex 157 with an excess of sodium hydride under a dinitrogen atmosphere afforded the high spin CrII imide complex 158 (Scheme 31). Interestingly, when sodium hydride was added in a stepwise manner, bridging dinitrogen intermediates either bearing a [N=N]2− or a [N−N]4− bridging unit could be isolated and fully characterized, indicative of an associative mechanism.
Scheme 31.
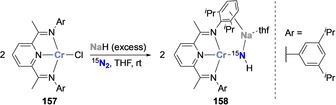
Associative N2 functionalization with sodium hydride mediated by the diiminepyridine Cr complex 157 and resulting amido complex 158.
The first example of direct hydrogenation of dinitrogen was reported by Fryzuk and co‐workers upon treatment of a side‐on bridging N2 zirconium complex [(P2N2)Zr‐(μ‐N2)‐Zr(P2N2)] (159) (P2N2={(Ph‐PCH2SiMe2)2N}2) with dihydrogen, generating complex 160 with a singly protonated dinitrogen moiety [N‐NH]3− (Scheme 32 a).[ 147 , 148 ] Applying this strategy to the side‐on bridged N2 zirconocene complex [(CpMe4H)2Zr‐N2‐Zr(CpMe4H)2] (161) allowed the hydrogenative activation of N2 at 1 atmosphere of H2 at room temperature to afford complex [(CpMe4H)2Zr‐(μ‐NH‐NH)‐Zr(CpMe4H)2] (162) (Scheme 32 b). Further heating of complex 162 under H2 led to the cleavage of the N−N bond and formation of NH3.[ 149 , 150 ]
Scheme 32.
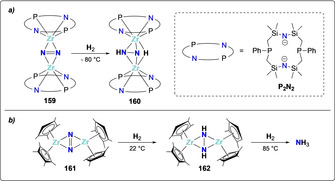
Associative N2 hydrogenation by zirconium complexes, 159 (a), 161 (b) and resulting hydrazine complexes 160 (a), 162 (b).
Alternatively, hydrogenation of N2 can be carried out using pre‐formed metal hydride complexes. Hou and co‐workers showed that the polyhydride bridged titanium PNP complex 163 readily coordinated N2 by releasing one equivalent of H2 and forming the side‐on/end‐on dinitrogen bridging complex 164. [151] Upon heating to 60 °C, complex 164 was transformed to the μ2‐imido/μ2‐nitrido/hydrido bis‐titanium complex 165. The same product was also isolated from the direct reaction of [(PNP)Ti(L)2] (166, X=CH2SiMe3) with H2 and N2 at room temperature (Scheme 33).
Scheme 33.
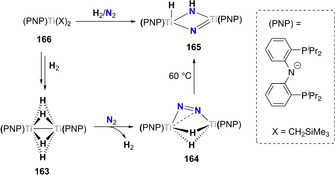
Associative N2 hydrogenation by titanium hydride complex 163 and resulting dinitrogen complex 164 and nitrido‐imido complex 165.
The molecularly‐defined silica‐supported tantalum bisneopentyl neopentylidene complex 167 showed analogous reactivity after initial treatment at 150 °C with molecular H2 to generate the tantalum hydride species 168 (Scheme 34). Upon exposure to N2, these complexes afforded the amido‐imido complex [(≡OSi)2Ta(=NH)(NH2)] (169). [152] In situ spectroscopic studies and theoretical analysis allowed proposing an associative distal mechanism and identifying several reaction intermediates (Scheme 34, dashed insert).[ 152 , 153 ]
Scheme 34.
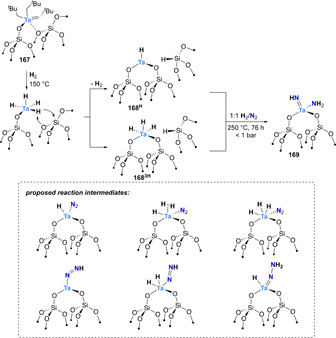
Associative N2‐splitting and protonation by silica‐supported tantalum complex 167. The structures in the inset describe intermediates observed by in situ spectroscopy and theoretical studies.
3.2. N−Si bond formation
A significant number of complexes have been shown to mediate N2 silylation via associative pathways. Yet, the vast majority of silylation reactions were demonstrated to be catalytic in the presence of excess reducing agents and silylation reagents and will be reported in Section 4 on catalytic systems. Only examples of stoichiometric functionalization will be discussed in the present section.
Stoichiometric distal N‐silylation was reported for the very stable complex [MoCp*(depf)(N2)] (170). [154] Reduction with excess sodium amalgam followed by silylation with trimethylsilyl choride afforded the corresponding [Mo−N=N−SiMe3] species (171) (Scheme 35 a).
Scheme 35.
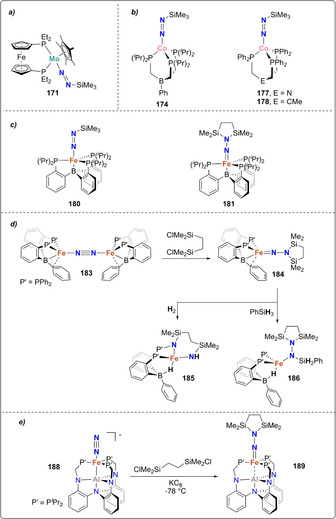
Mono‐ and bis‐silylated N2 complexes: mono‐silylated complexes of molybdenum (171) (a), cobalt (174) and iron (180–181) (b), bis‐silylation of N2 coordinated by the iron dimer 183 (c), mono‐silylated complexes 177–178 (d) and of the bis‐silylated alumatrane complex 189 (e).
In 2003, Peters et al. showed that a cobalt‐dinitrogen complex bearing a phosphine borate ligand Mg(thf)4[Co(PhBPiPr 3)(N2)]2 (172) [PhBPiPr 3=PhB(CH2PiPr 2)3 ‐] could be silylated at the terminal nitrogen of the coordinated N2 ligand (Scheme 35 b). [155] Stepwise reduction of the complex [Co(PhBPiPr 3)I] (173) with metallic Mg in THF under N2 atmosphere allowed generating complex 172. Addition of trimethylsilyl chloride to 172 led to the formation of [(PhBPiPr 3)CoN=N‐SiMe3] (174). This activity was demonstrated to be preserved upon variation of the heteroatom in the triphos ligand. Reduction of the cobalt complexes [Co(EPPh 3)Cl] (175, E=N; or 176, E=CMe; EPPh 3=E(CH2CH2PPh2)3) under a dinitrogen atmosphere with excess reducing agent (e.g. Li, Na, Mg) followed by reaction with Me3SiCl also afforded the terminally monosilylated complexes [Co(XPPh 3)(N=N‐SiMe3)] (177, E=N; 178 E=CMe) (Scheme 35 b). [156] Analogous monosilylation was also shown later on for [Fe(P3 B)(N2)] (179) (P3 B=tris‐[2‐(diisopropylphosphino)‐phenyl]borane) which resulted in the formation of the N‐silylated complex 180 (Scheme 35 c). [157] The bis‐silylated product [(P3 B)Fe=N‐N(Me2SiCH2CH2SiMe2)] (181) could also be obtained from complex [Fe(P3 B)Br] (182) upon treatment with 1,2‐bis(chlorodimethylsilyl)ethane in presence of excess sodium mercury amalgam (see overview Scheme 19). Observation of the dissociation of one of the phosphines in presence of exogenous ligands (e.g. MeCN) motivated the investigation of diphosphino complexes such as [Fe2(P2 B)(μ‐1,2‐N2)]Fe2(P2 B)] (183) (P2 B=bis‐diphenylphosphinophenylborane) (Scheme 35 d). Complex 183 similarly showed the formation of [(P2 B)Fe=N‐N(Me2SiCH2CH2SiMe2)] (184), but also allowed for further functionalization using H2 or phenylsilane to afford complexes 185 and 186, respectively. [158]
An original approach involving an alumatrane ligand was investigated by Lu and co‐workers. [159] The ligand AltraPhos (AltraPhos=Al[N(o‐C6H4NCH2PiPr2)3]) allowed for the isolation of the zero‐valent iron complex [Fe(N2)AltraPhos] (187). Reduction of complex 187 with potassium graphite in the presence of 18‐crown‐6 yielded the reduced complex [K(18‐crown‐6)][Fe(N2)AltraPhos] (188) (Scheme 35 e). Treatment of complex 188 with 1,2‐bis(chlorodimethylsilyl)ethane at −78 °C afforded the FeII complex [Fe{N2(SiMe2CH2)2})AltraPhos] (189). Despite the difference in size between alumina and boron, 189 displays a similar iron‐imide bond distance to that of Peters’ metallaboratrane complex 181.
Fryzuk and co‐workers investigated the silylation of the Ta–N2 complex [(μ‐η1:η2‐N2){Ta(NPN)H}2] (190) obtained upon treatment of the tetrahydrido Ta dimer [{(NPN)Ta}2(μ‐H)4] (191) with N2. Unlike for previously presented complexes, the authors here used a reductive strategy utilizing silanes instead of chlorosilanes to functionalize the N2 synthon. Treatment of complex 190 with one equivalent of silane RSiH3 (R=Ph, nBu) led to the formation of the mono‐silylated complexes 192 (R=Ph) and 193 (R=nBu) respectively. In presence of excess silane, these monosilylated complexes reacted further with a second molecule of silane to produce the bis‐silylated complexes 194 and 195 via an unprecedented Si−H bond activation on a N2 complex (Scheme 36).[ 160 , 161 ]
Scheme 36.
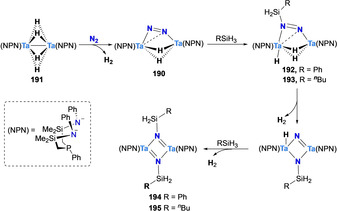
Associative N2 silylation by tantalum hydride complex 191.
Chirik and co‐workers reported an original example of a stepwise dinitrogen functionalization via the intermediacy of a silylilated dinitrogen moiety. [162] Treatment of the dinitrogen complex [{(η 5‐C5H2‐1,2,4‐Me3)2Hf}2(μ 2,η2,η2‐N2)] (196) with cyclohexyl silane generated the mono silylated dinitrogen complex 197 (Scheme 37). Complete N−N bond cleavage was observed upon heating complex 197, inducing the isomerization to complex 198. Further hydrogenation of complex 198 afforded the ‐NH‐Si(HCy)‐NH‐ bridged dimetallic hafnocene hydride complex 199. This pre‐activation of the dinitrogen unit by silylation in complex 197 also enabled its reactivity with carbon monoxide to produce the amide complex 200, generating formamide and ammonia with excess hydrochloric acid.
Scheme 37.
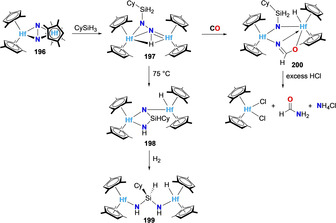
Silylation of the bridging dinitrogen moiety in substituted dinuclear hafnocene complex 196 and further functionalization of the dinitrogen ligand.
3.3. N−C bond formation
Chatt and co‐workers early reported the functionalization of bis‐diphosphino tungsten and molybdenum complexes 94 and 95 with acyl chlorides (RCOCl; R=Me, Et, Ph, or p‐MeOC6H4) in THF to form trans‐[WCl2(dppe)2(N‐NHC(O)R)] [163] (201–204) and trans‐[MoCl2(dppe)2(N‐NHC(O)R)] [164] (205–206) complexes (Scheme 38 a). The surprising presence of protons in the product was proposed to result from the presence of HCl generated by the reaction of acyl chlorides with traces of moisture. Using the same starting complexes 94 and 95, photochemical alkylation with various alkyl halides R‐X (R=Me, Et, nPr, iPr, tBu, ‐(CH2CH2‐)2, X=Cl, Br) led to the preparation of monoalkylated diazenido complexes [(dppe)2M‐N=NR] ((207–211, M=W) and (212 and 213), M=Mo)) [165] as well as of dialkylated hydrazido complexes [(dppe)2M=N‐NR2] (214–215), M=W) (R=Me, ‐(CH2CH2‐)2), depending of the alkyl moiety used (Scheme 38 b).[ 166 , 167 ] Transfer of the alkyl groups was suggested to occur in a stepwise manner on the distal nitrogen atom, and both diazenido [(dppe)2M−N=NR] and hydrazido [(dppe)2M=N‐NR2] species were isolated. [169] Similarly, acylation of the Re complex [(PhMe2P)3Re(N2)Cl2] with acetyl‐ and benzoylchloride resulted in the formation of the monoacylated diazenido complexes [(PhMe2P)3Re(N2COR)Cl2]+ (R=Me, Ph ). [165]
Scheme 38.
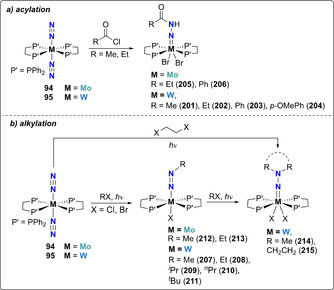
Associative N2 acylation (a) and alkylation (b) by group 6 diphosphine complexes.
Peters and co‐workers further explored N−C bond formation on N2 using iron and cobalt complexes with phosphino borane and silane ligand scaffolds. The reaction of complex 173 and its iron analogue [Fe(PhBPiPr 3)(N2)][Cl] (216) with 1 equiv of MeOTs and in presence of excess Mg as a reducing agent afforded the monomethylated diazenido complexes [(PhBPiPr 3)CoN=NMe] (217) and [(PhBPiPr 3)FeN=NMe] (218) (Scheme 39 a). [155] Modification of the ligand, leading to a trigonal pyramidal coordination, allowed for bis‐methylation of the dinitrogen unit. Silylide (P3 Si=tris(2‐(diisopropylphosphino)phenyl)silylide)) and borane (P3 B) supported iron complexes 219 and 220 were used as starting materials to obtain the bis‐methylated diazenido complexes 221 and 222 upon treatment with excess MeOTf (Scheme 39 b).[ 169 , 170 ]
Scheme 39.
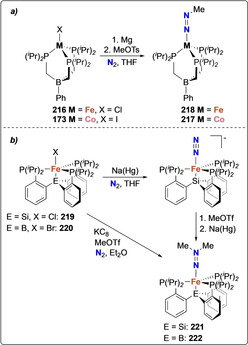
Associative N2 alkylation by cobalt complex 173 (a), and iron complexes 216 (a) and 219/220 (b).
This early work triggered a significant interest for the functionalization of activated dinitrogen moieties with alkyl groups. Metallocene N2 complexes demonstrated a rich reactivity in that context. The complex [Zr((η 5‐C5Me4R)2SiMe2)]2(μ‐η2,η 2‐N2) (223), where a flanked SiMe2 moiety holds the Cp rings rigidly together, could be functionalized with CO2 to generate a trans‐(NCO2)2 bridged bis‐zirconium complex 224. Treatment of 224 with excess electrophiles (e.g. MeOTf or Me3SiI) allowed releasing the corresponding methylated or silylated organic synthons (Scheme 40). The rigid ligand framework was proposed to minimize the N2 loss via lowering its ligand exchange rate favoring the formation of more reactive dimeric ansa‐zirconocene dinitrogen complex. [171]
Scheme 40.
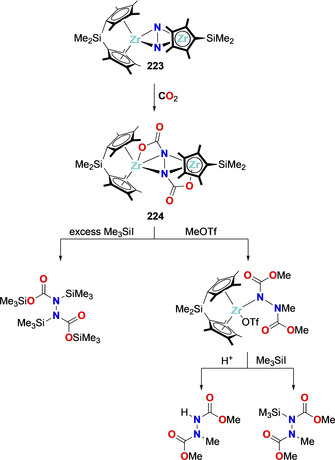
Associative N2 functionalization with CO2 and subsequent methylation/silylation by the ansa‐zirconocene complex 223.
The hafnium analogues of side‐on bound [{Zr(η 5‐C5Me4R)2}2(μ‐η2,η 2‐N2)] were also explored for N−C bond formation using CO2 [172] or electrophiles such as organic isocyanates[ 65 , 172 ] and alkyl halides. [65] Unlike the Zr analogues, reaction of CO2 with the dimeric Hf metallocene N2 complex [{(η5‐C5Me4H)2Hf}2‐(μ2‐η2,η 2‐N2)] (225) revealed the formation of the N2C2O4 2− bridged Hf complex [(η5‐C5Me4H)2Hf]2(NCO2)2 (226), where both CO2 molecules reacted at the same nitrogen atom (Scheme 41 a). [172] The activated N2 ligand in complex 225 could be further functionalized by the stepwise addition of phenyl isocyanate in an alternating manner (Scheme 41 b). Three equivalents of isocyanate were necessary to functionalize both nitrogen atoms, as the second equivalent reacts on the N‐site of the isocyanate itself, to generate a N4C2O2 2− bridging dimeric Hf cluster. The bond distances of these species indicate a substantial imido character for the nitrogen atom bound to the Hf center.
Scheme 41.
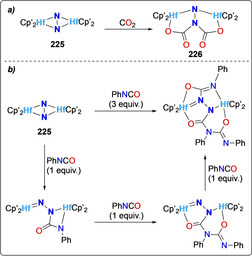
Associative N2 functionalization by hafnocene complex 225 with CO2 (a) and isocyanate (b).
The scope of N2 functionalization was further broadened upon replacing one cyclopentadiene ligand in 225 with a guanidinate ligand (227). In presence of this new ligand set, fast mono alkylation could be obtained using ethyl bromide, while only sluggish reactivity was observed using same reaction conditions with the bis cyclopentadienyl analogue (Scheme 42 a). [65] The stronger electron‐donating properties of the guanidinate ligand likely increased the nucleophilicity of the N2‐moiety and explains its easier functionalization. This effect can be further strengthened upon exchanging hafnium with scandium in such half‐sandwich architecture (complex 228) to generate a further reduced N2 3− moiety. [173] The electropositive nature of scandium assists in furnishing more electron density on the bridging N2 3− site and allowed isolating the bis‐methylated (N2Me2)2−‐bridged discandium complex 229 in a 1:1 ratio together with the N2 2− bound complex 230 via a similar disproportion mechanism than introduced in Section 3.1.1. Complex 229 proved to be a versatile precursor for the preparation of tetrasubstituted hydrazides upon subsequent alkylation (Scheme 42 b).
Scheme 42.
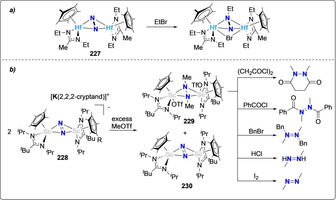
Associative N2 alkylation by hafnium complex 227 (a), and scandium complex 228 (b).
NPN complexes bearing a bridging N2 ligand had also been investigated for alkylation. The [(μ‐η 1:η 2‐N2)Ta2(NPN)2(μ‐H)2] (190) complex reacted with benzyl bromide to afford the corresponding N‐alkylated complex [(NPN)Ta‐(μ‐η 1:η 2‐N2CH2Ph)(μ‐H)2TaBr(NPN)] (231) (Scheme 43 a). [174] Complex 190 proved to be very versatile for N2 functionalization, showing reactivity with carbodiimide, sulfidoimides or CS2 (Scheme 43 b). [175]
Scheme 43.
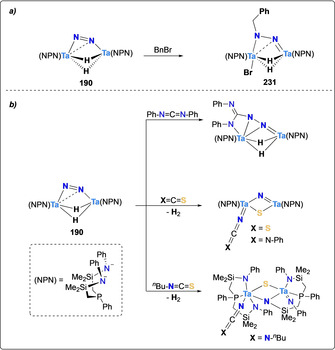
Associative N2 alkylation (a) and functionalization (b) by tantalum complex 190 using various electrophiles,.
Functionalization of side‐on bridged N2 complexes was later demonstrated on the diphosphino‐diamido zirconium dinitrogen complex 159 supported by the macrocyclic ligand scaffold (Scheme 44). [176] Cycloaddition of substituted alkynes across a Zr−N bond resulted in the formation of zircona‐aza‐cyclobutene intermediates 232 that subsequently underwent cleavage of the Zr−C bond by protonation from another terminal alkyne to produce N2‐substituted alkyne bound complexes 233–235. It should be noted that such reactivity with terminal alkynes could not be observed for the Ta complex 190, suggesting an increased basicity of the side‐on bridged N2 moiety in the Zr complex 159. [162]
Scheme 44.
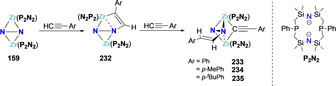
Associative N2 alkylation by zirconium complex 159.
Alkoxide ligands have also demonstrated to be very efficient at increasing the nucleophilicity of metal‐bound N2‐moieties. Kawaguchi and co‐workers have recently reported a TiIII complex supported by a tris‐phenoxide carbide ligand scaffold (O3C)4− that binds an N2 molecule to generate the anionic complex [(O3C)2Ti2(μ‐N2)K3(thf)3][K(thf)6] (236). [177] Reaction of excess CO2 with this end‐on bound dinitrogen ligand resulted in the formation of a dimeric Ti complex 237 bridged by a rare [N2C3O6]4− moiety. Removal of this [N2C3O6]4− unit using Me3SiI afforded N2(SiMe3)(CO2SiMe3)3 that degraded via CO2 loss to convert to 1,1‐ and 1,2‐N2(SiMe3)2(CO2SiMe3)2 (Scheme 45). Treatment of complex 236 with an excess of tert‐butyl isocyanate showed insertion of isocyanate at each nitrogen atom of the N2 ligand to generate a [N2(CONtBu)2] bridging group in complex 238 while reaction with phenylallene afforded two isomers of N‐substituted alkene complexes 239 and 240 via 2+2 cycloaddition reactions on the Ti=N bond.
Scheme 45.
Associative N2 functionalization by titanium complex 236 with allene, isocyanate and CO2.
Finally, the dinuclear UIII complex 147 from Mazzanti and co‐workers introduced above also demonstrated N−C bond formation upon addition of CO onto the N2 4− unit, promoting the full cleavage of the N−N bond. [139] Both the nitride and the hydrazido bridging groups reacted with CO affording the bridging oxo/cyanate UIV/UIV complex 241 and KCN as a byproduct (Scheme 46 a). In case of the analogous oxo‐hydrazido‐complex 242 (Scheme 46 b) obtained from dinitrogen and the corresponding oxo‐complexhighlighted 243, reaction with CO however differed and the formation of complex 244 with a bridging cyanamide ligand was observed. [178]
Scheme 46.
Associative N2 functionalization by uranium siloxide complexes, 147 (a), and 243 (b).
In addition, it should be noted that, beyond direct N2 transformation to another chemical entity, Lewis acid binding to metal‐coordinated N2 is key to further increase the charge delocalization from the metal to the N2 moiety and, hence, enable its catalytic transformation, as recently by Simonneau and Etienne. [21]
3.4. Summary
The cooperativity between the metal center and the functionalizing group in associative pathways enables the efficient polarization and activation of dinitrogen via “push–pull” mechanisms,[ 32 , 179 , 180 ] in which the functionalizing group increases electron density withdrawal at one side of dinitrogen and triggers enhanced π‐back‐donation from the metal center. This facilitated activation explains that associative mechanisms constitute the vast majority of currently reported stoichiometric N2 functionalization routes. It should be noted, however, that incomplete N≡N bond cleavage is often observed in associative reaction pathway, increasing the likelihood to afford a broader distribution of products. This constitutes a key challenge for the development of catalytic reactions with high selectivity.
4. Catalytic systems
As discussed previously, the synthesis of dinitrogen metal complexes and the stoichiometric transformation of coordinated N2 under ambient conditions have been widely investigated. Yet, only a limited number of catalytic systems have been reported to date.[ 12 , 14 , 15 ] The main catalytic systems will be discussed in this section according to the nature of the bond formed (N−H, N−Si, N−C). Finally, electrocatalytic systems will be discussed as a promising path towards sustainable nitrogen fixation. [181] All catalytic systems reported here for N−H, N−Si, and N−C bond formation are overviewed in Scheme 47 and their respective catalytic performances are summarized in Tables S6 and S7.
4.1. Catalytic formation of N−H bonds
In the 1960s, Vol'pin and Shur reported the catalytic reduction of dinitrogen to a mixture of ammonia and hydrazine using metal chlorides and acetylacetonate complexes in the presence of strong alkali reducing agents and hydrogen gas.[ 182 , 183 ] This very early work was followed by reports from Shilov and co‐workers of N2 reduction in protic media, using trivalent group 5–6 hydroxides [184] or a Mo–Mg cluster, Mg[Mg2Mo8O22(OMe)6(MeOH)4] (245) [185] and sodium mercury amalgam. Complex 245 was shown to be selective for hydrazine with TONs of up to 1000 in protic solvents. However, the reaction mechanisms for N2 reduction using these systems has not been investigated and the molecular origin of the active sites was not unambiguously demonstrated.
The first example of a well‐defined mononuclear complex capable of catalytically converting dinitrogen to ammonia was disclosed in 2003 by Schrock and co‐workers. [186] The use of a very bulky hexa‐iso‐propyl‐terphenyl (HIPT) triamidoamine ligand allowed the isolation of the mononuclear dinitrogen complex [MoIII(N2)(HIPTN3N)] (246).
Treatment of complex 246 with excess CrCp* 2 and 2,6‐lutidinium tetraarylborate ([LutH][BArF 4]) afforded ammonia with 66 % yield. In contrast of Cummins’ tris‐amidate MoIII complex 1, [27] the steric bulk of the ligand in the case of 246 prevented the binuclear dinitrogen cleavage to the corresponding nitride. The full catalytic mechanism was determined via a combination of theoretical studies[ 187 , 188 , 189 ] and the spectroscopic [190] and structural characterization of reaction intermediates.[ 191 , 192 , 193 ] The key steps of the reaction could be identified by the stepwise addition of reactant to isolate reaction intermediates. Reaction of 246 with 1 equiv of [LutH][BArF 4] and 2 equiv of cobaltocene (CoCp2) led to quantitative formation of [Mo(N=NH)(HIPTN3N)] (247). Treating 246 with an excess of [LutH][BArF 4] (7 equiv) and CoCp2 (8.2 equiv) led to the isolation of the last intermediate of the reaction cycle, the ammonia complex [Mo(NH3)(HIPTN3N)] (248). The stepwise protonation of complex 247 afforded complex [Mo=N−NH3)(HIPTN3N)]+ (249) which could be further reduced to yield the nitride complex [Mo≡N(HIPTN3N)] (250). These results unambiguously confirmed that catalyst 246 follows an associative distal pathway as presented in Scheme 48 a.
Scheme 48.
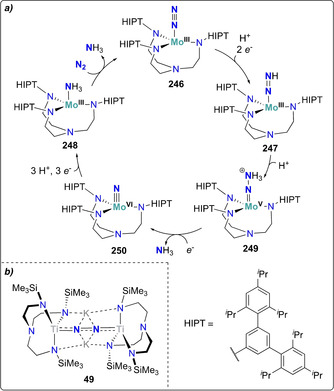
Proposed catalytic cycle for catalyst 246 (a) and structure of complex 49 (b).
Interestingly, the catalytic activity appeared specific to Mo in this exact ligand framework: Cr, W and V complexes bearing HIPTN ligands were prepared[ 194 , 195 , 196 ] along with Mo complexes supported by tripodal amide ligands with different substituents, [197] but none presented a significant catalytic activity towards N2 reduction. Using a less sterically encumbered triamidoamine ligand scaffold, Liddle and co‐workers reported recently that the titanium dimer [{(TrenTMS)Ti}2(μ‐η1:η1:η2:η2‐N2K2)] (49) was catalytically active for N2 reduction, achieving up to 18 equiv of ammonia using potassium graphite and [Cy3PH]I as a reductant/acid source (Scheme 48 b). [56] Control experiments suggested that N2H4 was formed and then converted into NH3 in the presence of acid and reductant, which is in agreement with an alternating associative mechanism.
A key example for the catalytic conversion of dinitrogen to ammonia was provided by Nishibayashi and co‐workers in 2011, using the PNP Mo complex [MoCl3(PNP)] (251). [43] Reduction of 251 under an N2 atmosphere allowed the isolation of the terminal/bridging dinitrogen complex [{Mo(N2)2(PNP)}2(μ‐N2)] (22) Treating complex 22 with excess cobaltocene and lutidinium trifluoromethanesulfonate ([LutH]OTf) allowed for the generation of up to 23 equiv of ammonia, yet with significant amounts of dihydrogen as a byproduct (44 equiv). The mechanism was postulated as depicted in Scheme 49 a, following an associative distal pathway. Despite the high number of coordinated N2 moieties in 22, only one of the terminal dinitrogen ligands is involved in the catalytic cycle. The formation of the dimeric species was, however, shown to be key to catalytic activity: the activation of N2 occurs at one Mo center while the other Mo center acts as an electron storage site, donating electrons through the bridging dinitrogen ligand (Scheme 49 a). Involvement of a terminal nitride intermediate 252 was suggested based on the catalytic activity of the independently synthetized molybdenum nitride complex [Mo≡N(PNP)Cl]+ (253) for N2 reduction to NH3. [44]
Scheme 49.
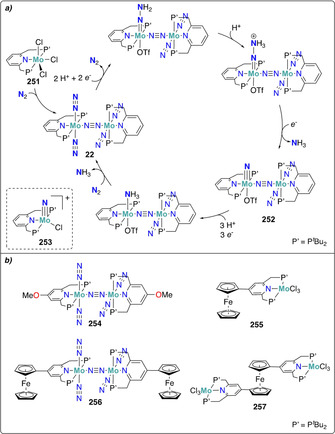
Proposed catalytic cycle for catalyst 22 (a) and structure of molybdenum complexes bearing PNP ligands 254–257 (b).
This study triggered a significant number of studies aimed at modulating the PNP ligand framework to improve catalytic activity. The effect of the para‐substituent on the PNP scaffold was investigated, and an increase in activity was observed in the case of an electron‐donating methoxy group (complex 254), [198] as well as in the case of a ferrocene moiety with complexes 255 and 256 (Scheme 49 b). [199] The activity did not further increase when ferrocene was used as a bridging moiety (complex 257). [200] It is worth mentioning that H2 evolution was similarly observed in all these systems due to the easy formation of Mo‐hydride species upon reaction of the reduced complex with protons. Interestingly, varying the halide ligand in the starting complex had a tremendous influence on catalytic activity. As mentioned in Section 2.2, at the difference of its chloride analogue forming a bridging N2 complex that remains stable during reducing conditions, reduction of [Mo(PNP)I3] (20) under N2 allowed for the isolation of the terminal molybdenum nitride complex 21. Higher activity and selectivity vs. H2 evolution were observed for the molybdenum iodide complex 21 suggesting that it operates via a different catalytic cycle than the binuclear complex 22. The reaction was proposed to proceed via the dissociative cleavage of a bridging N2 to form a terminal Mo nitride. Favoring this reaction path allowed increasing activity while decreasing the competitive hydrogen evolution by destabilizing potential Mo‐hydride intermediates (Scheme 50 a). [41] Recently, the introduction of an anionic pyrrole‐based PNP′ ligand allowed the isolation of the complex [Mo(PNP′)I2] (258), which reacted with N2 in the presence of metallic lithium at −50 °C to afford the dimeric MoIV nitride complex [{Li(PhMe)Mo≡N(PNP′)I}2] (259) (Scheme 50 b). [201] Complex 259 catalyzes the formation of NH3 (12 equiv) in the presence of SmI2 and ethylene glycol, via a similar mechanism than the one proposed for complex 20. It is interesting to note that contrary to the previously reported PNP complexes, the coordination and cleavage of N2 is reversible in this case. The lower stability of the formed nitride was proposed to constitute a key factor to explain the higher turnover numbers observed with this complex.
Scheme 50.
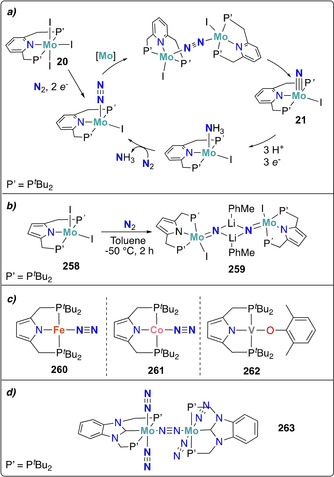
Proposed catalytic cycle for complex 20 (a). Structure of PNP‐pyrrole molybdenum 258 (b), iron 260, cobalt 261 and vanadium 262 (c) catalysts and of NHC‐PCP molybdenum catalyst 263.
The same anionic pyrrole‐based PNP ligand also allowed for the isolation of the iron‐based complex 260, [202] along with the first well‐defined cobalt [203] and vanadium [204] complexes 261 and 262 acting as catalysts for nitrogen reduction (Scheme 50 c). The Fe complex 260 was able to catalyze the formation of NH3 and N2H4 with TONs of up to 14.3 and 2.4 respectively, while the Co and V analogues 261 and 262 catalyzed the formation of NH3 with TONs of 4.2 and 12, respectively. Interestingly, the ratio of NH3 and N2H4 produced using complex 260 was shown to depend on the nature of the solvent used with stronger coordinating solvents, such as THF, favoring N2H4 formation. An associative distal mechanism was proposed according to DFT calculations.
Variation of the ligand scaffold and use of PPP‐pincer analogues permitted the synthesis of the terminal Mo nitride complex 26 (Scheme 6 d). As highlighted in Section 2.2, the lower Brønsted acidity of the PPP‐pincer ligands with respect to their PNP counterparts increases their stability towards protonation, which helps preventing the competitive hydrogen evolution arising from the reduction of the protonated ligand. This strategy allowed significantly decreasing H2 formation and accordingly increasing ammonia selectivity of catalyst 26, to reach TONs of up to 63 for ammonia, compared to the 23 TONs of the PNP‐based complex 22. [47]
The highest catalytic activity reported to date was disclosed by Nishibayashi and co‐workers using the molybdenum catalyst 263 supported by a NHC‐PCP type ligand (Scheme 50 d) via a proposed associative distal pathway. Similar to complex 22,[ 42 , 205 ] TONs as high as 58 per Mo for NH3 could be obtained using CrCp*2 as a reducing agent. [205] A benchmark TON of 2175 was obtained using SmI2 as a reducing agent in the presence of mild proton sources such as alcohols or water. [42] By analogy with the reduction of enamines and carbonyls with SmI2 in the presence of water, a PCET mechanism was proposed here to explain the increased activity.[ 206 , 207 ]
Mechanistic investigations of N2 reduction at the nitrogenase co‐factor and identification of the role of iron sites in N2 activation[ 8 , 11 , 33 ] have been the driving force for the intense development of iron‐based catalysts for dinitrogen reduction. In 2013, Peters et al. reported that the P3 B Fe complex 264 (Scheme 51 a) catalyzed the formation of ammonia at −78 °C in the presence of Brookhart's acid (7 equiv). [208] The flexibility of the coordination sphere of the iron center was demonstrated to be of prime importance for the activity of the complex. Structural, computational and spectroscopic studies allowed for the identification of the formation of a terminal nitride intermediate and an associative distal reaction path was therefore proposed, as highlighted in Scheme 51 b.[ 171 , 209 , 210 , 211 ] The ligand scaffold showed high versatility and allowed for the isolation of a Co‐based complex which proved to be also active for N2 reduction to NH3. [212]
Scheme 51.
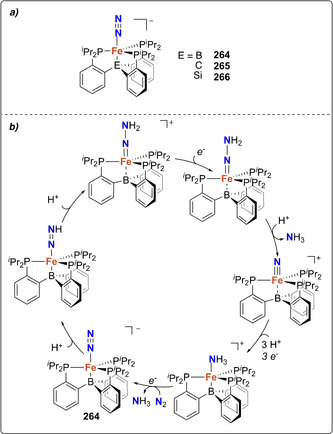
Structure of complexes 264–266 (a) and proposed catalytic cycle for 264 (b).
The existence of a central carbide moiety in the Fe–Mo cofactor motivated the investigation of complexes containing Fe−C bonds. Peters and co‐workers hence isolated an Fe tris(phoshine) complex (265) with a carbon anchor provided by a P3 C ligand (P3 C=tri(phosphine)methyl) (Scheme 51 a). Complex 265 is an active catalyst for the reduction of N2 to ammonia, albeit with lower efficiency than complex 264 (4.6 equiv). [213] However, yield for both systems could be increased by raising the loading of electron and proton sources to form up to 64 equiv of ammonia for 264 and 47 equiv for 265. The effectiveness of 264 as a catalyst could be further increased by using milder reductants and acids. Using potassium graphite (E 0<−3.0 V vs. Fc+/0) and [H(OEt2)2[BArF 4] (pK a<0 in THF) as a reducing agent and proton source resulted in lower ammonia yields than using the milder decamethylcobaltocene (E 0=−1.96 V vs. Fc+/0) and [Ph2NH2][OTf] (pK a=3.2 in THF) (7 equiv of NH3 vs. 12.8 equiv, respectively). [214] This enhanced activity was rationalized by DFT calculations, proving that the protonated metallocene could act as an effective PCET mediator.
Surprisingly, the P3 Si analogue 266 bearing a ligand with a silicon anchor showed only minor activity for NH3 generation (Scheme 51 a). This low reactivity enabled the isolation and structural characterization of a [Fe=NNH2(P3 Si)]+ intermediate and the spectroscopic identification of [Fe=NH2NH2(P3 Si)]+ providing evidence of a so‐called hybrid associative mechanism, where the distal nitrogen is being doubly protonated before the proximal nitrogen is also doubly protonated. This modulation of reactivity can be linked to the contrasting associative distal mechanism proposed for complex 264.[ 170 , 215 ] The P3 Si ligand scaffold also allowed for the preparation of the first ruthenium and osmium‐based catalysts for dinitrogen reduction, and TONs of up to 120 were observed for the osmium system. [216] The multielectron reduction of N2 was rendered possible by the redox properties of the ligand scaffold that can share the reducing equivalents of the metal center, similar to metalloclusters in biological systems.
A more flexible and electron rich PPP‐pincer scaffold was used by Peters and co‐workers to afford the unusual iron dihydride dinitrogen complex 267, showing enhanced catalytic activity under photolytic conditions (Scheme 52). Combining complex 267 with excess [HOEt2][BArF 4] and potassium graphite at −78 °C led to the formation of 66.7 equiv of NH3 upon irradiation with a mercury lamp, vs. 24.5 equiv in the absence of light. [217] While mechanistic studies are ongoing, this observation was attributed to the known tendency of iron dihydride species to undergo H2 elimination under photolytic conditions, allowing for in situ complex reactivation and faster turnover. Mézailles and co‐workers reported another PPP iron complex, [(PPhP2 Cy)Fe(N2)(H)2] (268) (Scheme 52), which yields 2.7 equiv of NH3 when treated with 200 equiv of potassium graphite and HBAr4 F at −80 °C. [218] The weak activity for both complexes can be attributed to the propensity of the iron center to be competitively protonated and generate stable iron hydride complexes.
Scheme 52.
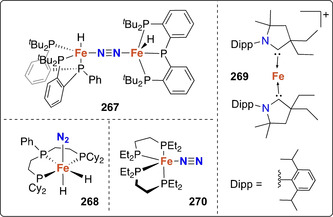
Structure of iron complexes 267–270.
A few types of ligand scaffolds were shown to support effective catalysts for dinitrogen reduction. The carbene complex [Fe(CAAC)2]+ (269) (Scheme 52) was shown by Peters and co‐workers to convert N2 to NH3 at low temperature with excess acid and potassium graphite as a reducing agent, reaching 3.4 equiv of ammonia at −95 °C. [219] Additionally, a rare example of selective catalytic hydrazine formation was reported by Ashley and co‐workers using the previously described complex [Fe(depe)2N2] (270) (Scheme 52) [220] in the presence of excess [LutH][BArF 4] and CoCp*2 (24.5 equiv).[ 105 , 109 ]
The examples above highlight that a broad series of metal complexes (M=Ti, V, Cr, Mo, Fe, Ru, Os, Co) were shown to act as catalysts for dinitrogen reduction to ammonia. Among these, the highest catalytic activities were observed with Mo and Fe complexes bearing multidentate, electron‐rich ligands. Most importantly, the choice of sacrificial reducing agent has evolved throughout the years from strong alkali reductants (sodium mercury amalgam, potassium graphite) to milder reductants that can effectively promote PCET and provide higher activity (CoCp*2, CrCp*2, SmI2). Table S6 aims at providing a comparative view on all examples reported in this section.
4.2. Catalytic formation of N−Si bonds
Historically, dinitrogen silylation by molecular catalysts under mild conditions preceded its conversion to ammonia. Silylation of N2 allows the preparation of value‐added chemicals containing N−Si bonds. In addition, the thus formed silylamines can be converted quantitatively to NH3 upon hydrolysis. Catalytic silylation strategies rely on the in situ generation of silyl radicals, by combining an excess of silylating and alkali reducing agents. Interestingly, such a homolytic N2 functionalization using silyl radicals is somewhat reminiscent of PCET or HAT mechanisms recently proposed for the reduction of N2 to ammonia and discussed above in Section 4.1. Table S7 summarizes the catalytic performances of all systems presented in the text below.
The first molecular catalytic system for N2 silylation was reported by Shiina in 1972. Within a wide range of metal chlorides, CrCl3 was found to be the only one to convert N2 to N(SiMe3)3 in a catalytic fashion (2.7 TONs) in presence of lithium wire as a reducing agent. [221] Two other families of chromium‐based catalysts have been disclosed since then. The macrocyclic Cr complex 130 (Scheme 25) reported by Mock and co‐worker, presented high performances for N2 silylation with TONs up to 17 using metallic sodium as a reducing agent. [125] DFT calculations suggested an associative distal mechanism involving the formation of the hydrazido [Cr=N−N(SiMe3)2](PBn 4NPh 4)] and imido [Cr=NSiMe3)(PBn 4NPh 4)] intermediates. More recently, chromium complexes bearing cyclopentadienyl‐phosphine ligand 271–273 were shown to catalytically convert dinitrogen into tris(trimethylsilyl)amine with a highest the TON of 13 determined for complex 273. The isolation of the chromium hydrazido intermediate 274 by treating the dinitrogen complex 275 with one equivalent of trimethylsilyl chloride is suggested an associative mechanism (Scheme 53). [222]
Scheme 53.
Structure of chromium complexes bearing cyclopentadienyl‐phosphine ligand 271–275.
The second example of a molecular catalyst for N2 silylation was also constituted by group 6 metal complexes. Hidai and co‐workers demonstrated in the late 80s that the molybdenum and tungsten phosphine dinitrogen complexes 94, 95 and 124 (Scheme 20 and 23) converted dinitrogen into tris(trimethylsilyl)amine, the highest TON of 18.3 being obtained using complex 124 and sodium dispersion as a reducing agent. [223] The functionalization of the distal nitrogen of the coordinated N2 by a trimethylsilyl radical was proposed as the first step of the reaction. The ferrocenyldiphosphine analogue, complex 106, was reported by Nishibayashi and co‐workers to convert N2 to tris(trimethylsilyl)amine with TONs about one order of magnitude higher than complex 124. [224] DFT calculations suggested the associative alternating functionalization of N2 with Me3Si. radicals (Scheme 54).
Scheme 54.
Proposed catalytic cycle for N2 silylation mediated by the ferrocenyldiphosphine molybdenum complex 106.
Mo‐mediated catalytic silylation was also reported using bulky tetradentate phosphine ligands by Mézailles and co‐workers. The molybdenum complex [Mo(PPCy 3)Cl]+ (276) (PPCy 3=tris‐[2‐(dicyclohexylphosphino)ethyl]phosphine) could be converted into the dinitrogen complex [Mo(PPCy 3)N2] (277) upon reduction in the presence of N2. Complex 276 catalyzed the silylation of N2 in the presence of trimethylsilyl chloride and potassium with 5.9 TONs. [225] The authors identified and isolated two key intermediates of the reaction: the hydrazido complex [Mo(PPCy 3)=N−N(SiMe3)2] (278) and the imido complex [Mo(PPCy 3)=N(SiMe3)] (279). Both complexes catalyze N2 silylation in the presence of metallic potassium with TONs of 7.5 and 5.9, respectively, suggesting an associative distal mechanism (Scheme 55 a). Reducing the steric bulk of the ligand framework by using the tridentate PPP‐pincer allowed the authors to increase the catalytic performances and to isolate all key intermediates in the catalytic cycle (Scheme 55 b). The molybdenum complex [Mo(PPP)Cl3] (28), previously discussed in Section 2.2.1, also catalyzes the silylation of N2 with up to 38 TONs. In addition to the dinitrogen complex 280, the hydrazido complex 281, the terminal nitride complex 282, and along with the imido complex 283 were also isolated here, allowing to build the full reaction cycle (Scheme 55 b). [226]
Scheme 55.
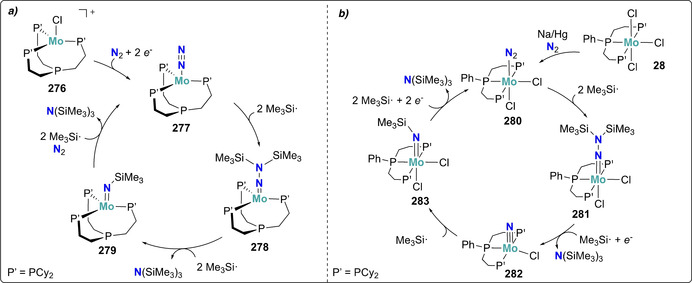
Proposed catalytic cycle for N2 silylation using complexes 276 (a) and 28 (b).
The role of the iron centers in the N2 silylation catalytic activity of the ferrocenyldiphosphine complex 106 was further investigated by Nishibayashi and co‐workers. Ferrocene was shown to be active for N2 silylation in the presence of excess sodium and Me3SiCl, reaching up to 6.5 equiv of N(SiMe3)3 based on Fe. [227] Surprisingly, very similar catalytic activity was obtained using iron pentacarbonyl as an Fe precursor, suggesting that the catalytically active complex was generated in situ and was common to both precursors. In agreement with this hypothesis, DFT calculations allowed proposing the in situ formation of an [FeII(SiMe3)2(THF)] active site (284), which reacts via an associative distal mechanism to afford N(SiMe3)3 under catalytic conditions (Scheme 56 a). This catalytic activity could also be observed using Co analogues, both cobaltocene and dicobalt octacarbonyl mediating N2 silylation. [228] For both precursors, the highest activity (up to 40 TONs for N(SiMe3)3) was observed in presence of 2,2′‐bipyridine (bpy) as an additive. The origin of this increased activity in presence of bpy ligand was suggested by DFT calculations to arise from the stabilization of the reactive Co‐N2 intermediate via the in situ formation of the CoIII complex [Co(SiMe3)3(bpy)(N2)].
Scheme 56.
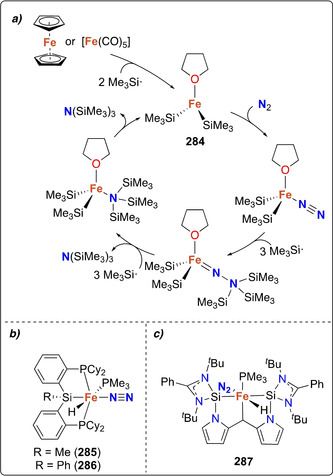
Catalytic N2 silylation using for ferrocene or iron pentacarbonyl (a) and structure of iron‐based catalysts bearing silicon‐containing ligands 285–287 (b).
The identification of complexes with metal−Si bonds as key intermediates in these catalytic cycles motivated the investigation of the iron dinitrogen complexes 285 and 286 as electrocatalysts for N2 silylation (Scheme 56 b). Despite slightly lower TONs than those obtained from the metallocene or carbonyl complexes mentioned above, both PSiP complexes 285 and 286 promote the catalytic silylation of N2 with faster initial kinetics. [229] The higher initial kinetics originate from the immediate availability of the catalytically active species, while the metallocene and carbonyl precursors experienced a long induction period to generate the catalytically active species containing metal−Si bonds. This strategy was further pursued by Li and co‐workers with the recent report of an Fe complex bearing bis(silylene)‐C(sp3) pincer ligand 287 (Scheme 56 c). Complex 287 was shown to be the most efficient Fe‐based catalyst for N2 silylation reaching TONs of 37.5 under ambient conditions. [230]
This series of Fe complexes can be completed by two others: [Fe(CAAC)2]+ (269) [219] and [Fe(depe)2N2] [220] (270) (Scheme 52). Indeed, 269 and 270 selectively reduce dinitrogen to N(SiMe3)3 at room temperature in the presence of Me3SiCl and excess potassium graphite, with TONs of 12.2 [219] and 60.5 respectively. [231] Based on the isolation and characterization of a [Fe=N−N(SiMe3)2(dppe)2] intermediate (288) and DFT calculations, complex 270 was proposed to follow an analogous associative distal mechanism to the one proposed for complexes 257 and 28 (Scheme 57 a). Chelating phosphine ligands were introduced by Mock and co‐workers to provide a stable planar ligand framework able to stabilize axial dinitrogen coordination and to resist the harsh silylation conditions. These guidelines allowed the isolation of the iron complex [Fe(P4N2)N2] (289) (Scheme 57 b) that actively catalyzed the formation of N(SiMe3)3 with TONs up to 37.5 under 100 atm of N2 (TONs of up to 10.5 were determined under 1 atm of N2). The mechanism proposed involved the stepwise addition of ‐SiMe3 groups to the distal N2 site. [232] Chelating phosphine ligands were also used by Mézailles and co‐workers to prepare the iron complexes [(PPhP2 Cy)Fe(N2)2] (290) and [(PtBuP2 Cy)Fe(N2)2] (291) (Scheme 57 c), which catalyze N2 silylation in the presence of Me3SiCl and potassium as a reducing agent to afford N(SiMe3)3 with 32 and 27 TONs, respectively. [218]
Scheme 57.
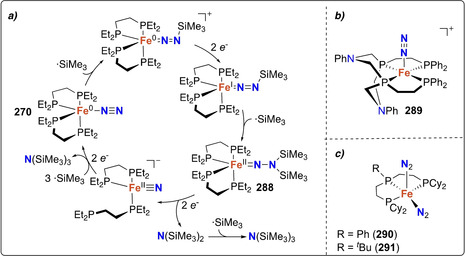
Proposed catalytic cycle for complex 270 (a) and structure of iron‐based catalysts supported by macrocyclic phosphine ligands 289–291 (b) and (c).
Multinuclear Fe complexes were also shown to activate N2 under ambient conditions. The tris‐iron complex 54 supported by a trinuclear tris(β‐diketimine)cyclophane scaffold from Murray et al. and introduced in Section 2.2 (Scheme 11) [233] effectively catalyzed the formation of N(SiMe3)3 from N2 in the presence of excess Me3SiCl and potassium graphite. [234] The same ligand scaffold was reported recently with other first row transition metals (Cr, Mn, Co, Ni), showing that the TON was a function the metal ion employed, and was following the trend Co>Fe>Cr>Ni>Mn, which correlates with the electronegativity of the metal center. [235] High catalytic activities for the silylation of N2 were also reported by Ohki and co‐workers using polynuclear iron hydride clusters. Complexes [Fe4(μ‐H)4(μ3‐H)2{N(SiMe3)2}2(PMe3)4] (292), [Fe6(μ‐H)10(μ3‐H)2(PMe3)10] (293), and (η6‐C7H8)Fe4(μ‐H)2{μ‐N‐ (SiMe3)2}2{N(SiMe3)2}2 (294) exhibited TONs of 80, 91 and 74, respectively, (per catalyst equivalents) for the catalytic silylation of N2 with Me3SiCl and in presence of metallic sodium as reducing agent (Scheme 58 a). [236] As illustrated above with the iron complex 270 (Scheme 57 a), the hemilability of bridging ligands to generate open coordination sites on the metal is a key feature of these polynuclear complexes. This is closely related to the mechanism of the nitrogenase cofactor, as evidence for ligand dissociation to generate active sites was probed recently. [8] The same group took a step further by synthesizing the heteropolymetallic molybdenum‐iron and molybdenum‐manganese hydride clusters 295–298 (Scheme 58 b), which were all shown to catalytically convert N2 to N(SiMe3)3 in the presence of Me3SiCl and metallic sodium as a reducing agent. [237] The iron and manganese clusters 295 and 296 could, respectively afford 69 and 12 equiv of N(SiMe3)3 per equiv of cluster. In light of the high activity of the molybdenum‐iron hydride cluster 295, the authors substituted the trimethylsilyamine ligands by thiolate ligands, resulting in clusters 297 and 298. Both clusters showed high catalytic activity towards dinitrogen silylation with TONs of 65 and 69, respectively.
Scheme 58.
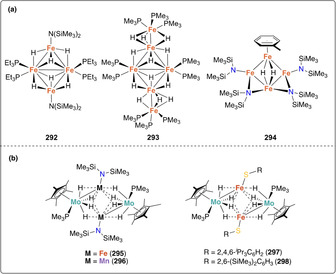
Structure of polynuclear iron hydride clusters 292–294. (a) and heterometallic hydride clusters 295–298 (b).
An original strategy involving multinuclear complexes was investigated by Lu and co‐workers using trisphosphino(triamido)amine ligands (P3N3) to stabilize a dimeric Co complex featuring a Co−Co interaction, [Co2(P3N3)] (299). This complex was revealed to be highly active for N2 silylation, with turnover numbers of ca. 200 being reported. [238] Based on kinetic studies and DFT calculations, the reaction mechanism shown in Scheme 59 a was proposed, which involves a similar associative alternating functionalization of N2 as proposed for complexes 106 and 125. It should be noted that the presence of the second Co center is important to observe high catalytic activity: the Co−Co interaction was shown to present a hemilabile character, being modulated during the catalytic cycle and stabilizing the different steps of the reaction. Such an effect was reduced with an Al−Co analogue, showing a significant drop in catalytic activity. [239]
Scheme 59.
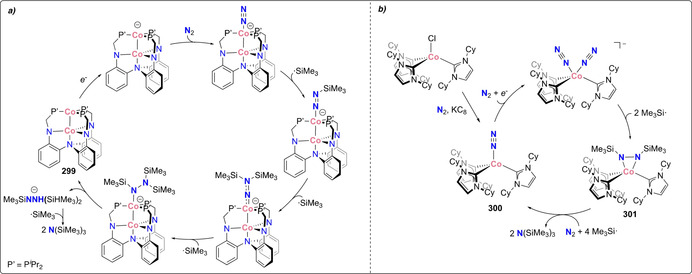
Proposed catalytic cycle for complex 299 (a) and 300 (b).
Another example of a Co‐based catalyst was provided by Deng and co‐workers, who explored the combination of low‐valent CoI and strong σ‐donating NHC ligands to isolate the dinitrogen complex [Co(ICy)3(N2)] (300). Complex 300 catalyzed the silylation of N2 with TONs of 125. [240] Isolation and catalytic activity of the diazene complex 301 pointed towards its role as a potential intermediate in the catalytic cycle (Scheme 59 b), suggesting an alternating associative mechanism. The use of cobalt to mediate N2 silylation was further studied by Fryzuk and Masuda using PNN‐type frameworks. Complexes [Co(NpNP)] (302) [241] and [Co(QuiNacNacP)] (303) [242] (Scheme 60) reduced N2 in the presence of potassium graphite and trimethylsilyl chloride to afford N(SiMe3)3 (TONs of 100 and 19 respectively). Additionally, the cobalt analogue 304 of the above mentioned iron PSiP complex 285 (Scheme 56 b) also promoted the catalytic conversion to N(SiMe3)3 with up to 20.5 TONs. [229]
Scheme 60.
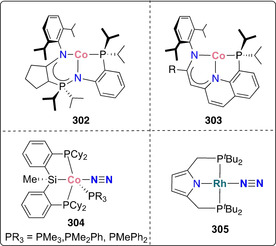
Structure of group 9 complexes 302–305.
Beyond Co, another group 9 catalyst for nitrogen silylation was recently reported by the groups of Yoshizawa and Nishibayashi. Using an anionic pyrrole‐based PNP′‐pincer ligand, the Rh complex [Rh(PNP′)(N2)] (305) (Scheme 60) could be isolated. This constituted the first rhodium complex catalyzing N2 reduction and promoted nitrogen silylation with 11.5 TONs. [243] This activity is noteworthy as contrary to the previously mentioned iron, cobalt and vanadium analogues 260, 261 and 262 (Scheme 50 c) which worked as effective catalysts toward the formation of ammonia and hydrazine, the rhodium‐based N2 complex 305 only favored the production of hydrogen in presence of proton sources.
Other early transition metals such as vanadium and titanium have demonstrated catalytic activity for dinitrogen silylation. Okuda and co‐workers developed a triamidoamine scaffold and prepared the bridging dinitrogen titanium complex K2[{(Xy‐N3N)Ti}2(μ2‐N2)] (306) (Xy=3,5‐dimethylbenzene), which catalyzed N2 silylation to N(SiMe3)3 with a TON of 8.3 (Scheme 61). [244] Polyamine ligands were also recently reported by Nishibayashi and co‐workers as efficient ligands for the preparation of vanadium catalysts for dinitrogen silylation. The dinuclear diamideamine vanadium nitride complexes 38 and 39 previously reported by Cloke et al., [52] were shown to mediate the catalytic silylation of dinitrogen to N(SiMe3)3, achieving up to 12 TONs. [245] Slightly lower TONs of 7.5 and 10.5 were determined under the same conditions as the other vanadium complexes trans‐[VCl2(tmeda)2] (307) (tmeda=N,N,N,N‐tetramethylethylenediamine) and trans‐[Na(thf)][V(N2)2(dppe)2] (308) (Scheme 61). [245]
Scheme 61.
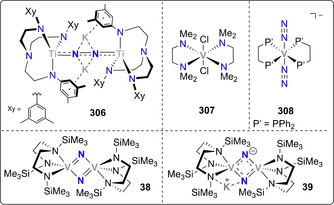
Structures of group 4 and 5 complexes 306–308 and 38–39.
Last, Arnold and co‐workers disclosed very recently the first actinide based catalytic system for N−Si bond formation. The use of the tetradentate meta‐tetraphenol‐arene ligand mTp (mTP=[{2‐(OC6H2‐tBu‐2,Me‐4)2CH}‐C6H4‐1,3]4−) allowed the synthesis of the dinuclear complex [U2(mTP)2] (142), as described in Section 3.1.1 (Scheme 28 b). [18] Upon reduction with potassium graphite, a mild acid and chlorosilane, complex 142 converts catalytically dinitrogen into bis(trimethylsilyl)amine (Scheme 62). Catalytic formation of hexamethyldisilazane (HMDS) (6.4 equiv)—instead of the more common N(SiMe3)3 product—was observed upon treatment of 144 with excess reducing agent, [HNEt3][BPh4] and Me3SiCl.
Scheme 62.
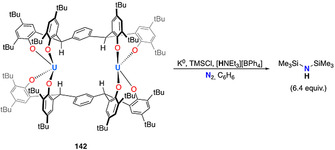
Catalytic N2 silylation using actinide complex 142.
4.3. Catalytic formation of N−C bonds
Surprisingly, only very few catalytic N−C bond formation reactions using N2 as a nitrogen source have been reported, despite being a key strategy to incorporate dinitrogen in value‐added organic molecules.
Mori and co‐workers first showed in 1993 that dinitrogen can be transferred to heterocycles using ketone precursors in the presence of a catalytic amount of TiCl4, excess lithium and Me3SiCl. [246] Hydrolysis or removal of the silyl groups using CsF allowed for the isolation of a wide variety of N‐heterocycles, such as indole, quinoline, pyrrole, pyrrolizine and indolizine, [247] even using dry air instead of pure dinitrogen (Scheme 63).[ 248 , 249 ]
Scheme 63.
Examples of heterocycles formed catalytically from dinitrogen.
The molecular nature of the active catalysts has not been unambiguously determined, but the isolation of the titanium‐nitride complex [TiNMg2Cl2(thf)] upon reaction of TiCl4 or TiCl3 with magnesium under a dinitrogen atmosphere [250] suggested a dissociative mechanism. Nevertheless, the nature of the Ti complex involved in the transfer of the nitrogen synthons to the heterocycles has not been identified so far. [248]
4.4. Electrocatalytic conversion
While all the systems described in Section 4.1 are based on the use of excess proton sources and reducing agents to mimic the PCET‐mediated route to dinitrogen fixation operating in nitrogenases, a more sustainable and scalable approach is to achieve electroreduction of N2. [251] Electroreduction also allows for a better control of the potential applied, avoiding unwanted reductive decomposition of the complex sometimes encountered in the presence of strong reducing agents. The Pourbaix diagram for the N2‐H2O system shows that reduction of N2 to NH3 or NH4 + is thermodynamically possible under mild reducing conditions (Scheme 64). [252] Nevertheless, the electrochemical reduction of N2 utilizing molecular complexes in the presence of proton sources is yet limited by both electrocatalytic overpotential and selectivity over kinetically favored H2 evolution. In that context, Miller and co‐workers have demonstrated that the use of organic solvents for catalytic ammonia reduction further favorizes the thermodynamically favored dinitrogen reduction to ammonium over the competitive hydrogen evolution reaction, a key aspect to develop molecular N2 reduction electrocatalysts. [253]
Scheme 64.
Pourbaix diagram of dinitrogen. Solid lines correspond to N2 reduction to NH4 + or NH3 (red line) and N2 oxidation to NO3 − (blue line). Dotted lines straddle the region of water stability (reduction to H2 for bottom line and oxidation to O2 for top line). [252]
Early work by Pickett and Talarmin paved the way to the electrochemical reduction of dinitrogen to ammonia. By treating trans‐(dppe)2W(N2)2 (94) with tosylic acid in THF, the hydrazido tungsten precursor [W(NNH2)OTs(dppe)2]+ (313) was obtained. Electrolysis of the latter in THF and 0.2 m [NBu4][BF4] saturated with 1 atm N2 at a mercury pool cathode (E=−2,6 V vs. Fc/Fc+) could afford 0.24 equiv of NH3 (Scheme 65 a). [93] In this case, the proton source was not present directly in the media but provided in a stepwise manner after the electrolysis step, forming the hydrazido complex 313.
Scheme 65.
Well‐defined molecular complexes promoting dinitrogen electroreduction 313–315.
To our knowledge, the iron complex [FeN2(P3 B)]+ (314) has been the only molecular complex reported to electrocatalytically convert dinitrogen to ammonia, with Faradic efficiency of up to 28 % (4 equiv of NH3 at −2.1 V vs. Fc+/0) (Scheme 65 b). [254] The role of CoCp*2 as a redox mediator was highlighted as a key feature to enable catalytic activity: DFT calculations suggested that the protonated cobaltocene acts as a PCET donor, essential to promote the first protonation of N2. In that regard, the authors highlighted that the influence of the pK a of the proton source on catalytic activity can be assigned to the ability to generate such protonated decamethylcobaltocene species.
This example was very recently complemented by the report of the first selective molecular electrocatalyst promoting N2 reduction to hydrazine by Dey and co‐workers. The trinuclear nickel complex 315 catalyzed the selective electrochemical N2 reduction to N2H4 using phenol as a proton source at −2.35 V vs. Fc/Fc+. (Scheme 65 c). [255] The author proposed here that the trinuclear arrangement of the Ni centers play a key role for N2 activation.
4.5. Summary
The summary Scheme 47 clearly illustrates that most catalytic systems undergo associative mechanisms. As stressed in Section 3, the cooperativity between functionalizing groups and metal centers is key to facilitate N2 activation and functionalization. Nevertheless, it should be noted that the most efficient molecular system identified so far for dinitrogen reduction to ammonia by Nishibayashi and co‐workers is one of the very few systems proposed to follow a dissociative mechanism. Efficiency of that system however mainly originates from the use of reducing agents and proton sources promoting PCET pathways. We anticipate that the growing numbers of PCET mediated N2 reduction reactions will allow for more comprehensive conclusions to be drawn in the coming years regarding the most favored reaction pathways towards N2 functionalization.
5. Conclusions and Outlook
As the main limitations in N2 functionalization are of kinetic origin, mechanistic consideration of N2 activation are of key importance to enable its transformation under mild conditions. The considerable number of complexes promoting N2 functionalization via dissociative or associative pathways reported in the last 50 years and surveyed here all fulfil this kinetic challenge. As such, they constitute unique s to rationally design new complexes for N2 valorization with improved performances. Nevertheless, among this large number of molecular complexes, only a small portion demonstrates catalytic activity, and significant research efforts are still needed in this area. This Review particularly highlights that among all catalytic reactions reported so far and despite a significant number of studies targeting dissociative pathways, most of the successful catalysts undergo associative mechanisms, as spotlighted in Scheme 47.
The current paradigm is hence shifting from the activation of N2 to the functionalization of N2 to value‐added product in a catalytic way. Besides the simplest silylation of N2, efficient catalytic protonation and N−C bond formation remain mainly elusive, notably since the employed functionalizing agents (protons, carbon‐containing electrophiles) are often incompatible with the reducing agents required for N2 activation and reduction. In addition, the use of complex and expensive sacrificial reducing agents in large excess significantly lowers the applicability and profitability of such functionalization routes.
Therefore, the investigation of more sustainable and tunable catalytic strategies, in particular by utilizing electrical energy sources in electrocatalytic pathways are highly desirable and represent the most exciting development of the field in the coming years.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Fabio Masero completed his Bachelor's and Master's degree in Interdisciplinary Science at ETH Zurich. He carried out his Master thesis at Hokkaido University in the laboratory of Prof. Hajime Ito, working on Cu‐catalyzed borylation chemistry. In 2019, he joined the group of Prof. Victor Mougel as a PhD student with a major focus on studying low‐valent molybdenum complexes for small molecules activation.

Biographical Information
Marie A. Perrin received her Bachelor from Paris‐Saclay University and completed her Master in Molecular Chemistry at Ecole Polytechnique in Paris. She carried out her Master thesis in the group of Prof. Kit Cummins at the Massachusetts Institute of Technology where she worked on metal complexes supported by macrocyclic diphosphines. In 2019, she started her doctoral studies in the group of Prof. Victor Mougel, her work focuses on developing heteropolymetallic assemblies for small molecule activation.

Biographical Information
Subal Dey received his Master's degree in Chemistry from the Indian Institute of Technology Kanpur, India. He pursued his Ph. D. with Prof. Abhishek Dey at IACS, Kolkata. He then moved to Europe as a postdoctoral fellow at College de France, Paris, and he is now a postdoctoral researcher at ETH Zurich in the group of Prof. Victor Mougel. His current area of research is the development of bioinspired catalysis for small molecule transformation.

Biographical Information
Victor Mougel completed his Bachelor's and Master's degree in Chemistry at the ENS of Lyon, and obtained his PhD at the University of Grenoble under the supervision of Prof. Marinella Mazzanti. He then joined ETH Zürich as an ETH/Marie Skłodowska‐Curie Fellow before starting his independent career as a CNRS associate researcher at Collège de France in 2016. Since December 2018, he is a tenure track assistant professor at the Department of Chemistry and Applied Biosciences at ETH Zürich. In 2019, he has been awarded an ERC starting grant to develop molecules and materials for N2 transformation inspired by the nitrogenase enzyme.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. [853064]) and from ETH Zürich via an ETH research grant (ETH‐44 19‐1).
F. Masero, M. A. Perrin, S. Dey, V. Mougel, Chem. Eur. J. 2021, 27, 3892.
References
- 1. Peoples M. B., Herridge D. F., Ladha J. K. in Management of Biological Nitrogen Fixation for the Development of More Productive and Sustainable Agricultural Systems (Eds.: Ladha J. K., Peoples M. B.), Springer Netherlands, Dordrecht, 1995, pp. 3–28. [Google Scholar]
- 2.F. Haber, The Synthesis of Ammonia from its Elements, Nobel Lecture, 1920.
- 3.C. Bosch, The Development of the Chemical High Pressure Method During the Establishment of the New Ammonia Industry, Nobel Lecture, 1932.
- 4. Smil V., Nature 1999, 400, 415. [Google Scholar]
- 5. Smil V., Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, Vol. 79, MIT Press, Cambridge, MA, 2001. [Google Scholar]
- 6. Martín A. J., Shinagawa T., Pérez-Ramírez J., Chem 2019, 5, 263–283. [Google Scholar]
- 7. Zhan C. G., Nichols J. A., Dixon D. A., J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar]
- 8. Sippel D., Rohde M., Netzer J., Trncik C., Gies J., Grunau K., Djurdjevic I., Decamps L., Andrade S. L. A., Einsle O., Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
- 9. Dance I., ChemBioChem 2020, 21, 1671–1709. [DOI] [PubMed] [Google Scholar]
- 10. Kang W., Lee C. C., Jasniewski A. J., Ribbe M. W., Hu Y., Science 2020, 368, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Stappen C., Decamps L., G. E. Cutsail, 3rd , Bjornsson R., Henthorn J. T., Birrell J. A., DeBeer S., Chem. Rev. 2020, 120, 5005–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leigh G. J. in Catalysts for Nitrogen Fixation (Eds.: Smith B. E., Richards R. L., Newton W. E.), Springer Netherlands, Dordrecht, 2004, pp. 33–54. [Google Scholar]
- 13. Ertl G., J. Vac. Sci. Technol. A 1983, 1, 1247–1253. [Google Scholar]
- 14. Foster S. L., Bakovic S. I. P., Duda R. D., Maheshwari S., Milton R. D., Minteer S. D., Janik M. J., Renner J. N., Greenlee L. F., Nat. Catal. 2018, 1, 490–500. [Google Scholar]
- 15. Chalkley M. J., Drover M. W., Peters J. C., Chem. Rev. 2020, 120, 5582–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishibayashi Y., Transition Metal-Dinitrogen Complexes: Preparation and Reactivity, Wiley-VCH, Weinheim, 2019. [Google Scholar]
- 17. Ryan A. J., Balasubramani S. G., Ziller J. W., Furche F., Evans W. J., J. Am. Chem. Soc. 2020, 142, 9302–9313. [DOI] [PubMed] [Google Scholar]
- 18. Arnold P. L., Ochiai T., Lam F. Y. T., Kelly R. P., Seymour M. L., Maron L., Nat. Chem. 2020, 12, 654–659. [DOI] [PubMed] [Google Scholar]
- 19. Kim S., Loose F., Chirik P. J., Chem. Rev. 2020, 120, 5637–5681. [DOI] [PubMed] [Google Scholar]
- 20. Singh D., Buratto W. R., Torres J. F., Murray L. J., Chem. Rev. 2020, 120, 5517–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simonneau A., Etienne M., Chem. Eur. J. 2018, 24, 12458–12463. [DOI] [PubMed] [Google Scholar]
- 22. Himmel H. J., Reiher M., Angew. Chem. Int. Ed. 2006, 45, 6264–6288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6412–6437. [Google Scholar]
- 23. Bercaw J. E., Rosenberg E., Roberts J. D., J. Am. Chem. Soc. 1974, 96, 612–614. [Google Scholar]
- 24. Manriquez J. M., Bercaw J. E., J. Am. Chem. Soc. 1974, 96, 6229–6230. [Google Scholar]
- 25. Gynane M. J. S., Gynane S., Jeffery J., Lappert M. F., J. Chem. Soc. Chem. Commun. 1978, 34–36. [Google Scholar]
- 26. Cusanelli A., Sutton D., Organometallics 1996, 15, 1457–1464. [Google Scholar]
- 27. Laplaza C. E., Cummins C. C., Science 1995, 268, 861–863. [DOI] [PubMed] [Google Scholar]
- 28. Laplaza C. E., Johnson M. J. A., Peters J. C., Odom A. L., Kim E., Cummins C. C., George G. N., Pickering I. J., J. Am. Chem. Soc. 1996, 118, 8623–8638. [Google Scholar]
- 29. Curley J. J., Cook T. R., Reece S. Y., Muller P., Cummins C. C., J. Am. Chem. Soc. 2008, 130, 9394–9405. [DOI] [PubMed] [Google Scholar]
- 30. Pucino M., Allouche F., Gordon C. P., Wrle M., Mougel V., Coperet C., Chem. Sci. 2019, 10, 6362–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shima T., Hu S., Luo G., Kang X., Luo Y., Hou Z., Science 2013, 340, 1549–1552. [DOI] [PubMed] [Google Scholar]
- 32. Hoffman B. M., Lukoyanov D., Yang Z. Y., Dean D. R., Seefeldt L. C., Chem. Rev. 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Einsle O., Rees D. C., Chem. Rev. 2020, 120, 4969–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shima T., Yang J., Luo G., Luo Y., Hou Z., J. Am. Chem. Soc. 2020, 142, 9007–9016. [DOI] [PubMed] [Google Scholar]
- 35. Akagi F., Matsuo T., Kawaguchi H., Angew. Chem. Int. Ed. 2007, 46, 8778–8781; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8934–8937. [Google Scholar]
- 36. Suzuki S., Ishida Y., Kameo H., Sakaki S., Kawaguchi H., Angew. Chem. Int. Ed. 2020, 59, 13444; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13546. [Google Scholar]
- 37. Zanotti-Gerosa A., Solari E., Giannini L., Floriani C., Chiesi-Villa A., Rizzoli C., J. Am. Chem. Soc. 1998, 120, 437–438. [Google Scholar]
- 38. Caselli A., Solari E., Scopelliti R., Floriani C., Re N., Rizzoli C., Chiesi-Villa A., J. Am. Chem. Soc. 2000, 122, 3652–3670. [Google Scholar]
- 39. Connor G. P., Holland P. L., Catal. Today 2017, 286, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hebden T. J., Schrock R. R., Takase M. K., Muller P., Chem. Commun. 2012, 48, 1851–1853. [DOI] [PubMed] [Google Scholar]
- 41. Arashiba K., Eizawa A., Tanaka H., Nakajima K., Yoshizawa K., Nishibayashi Y., Bull. Chem. Soc. Jpn. 2017, 90, 1111–1118. [Google Scholar]
- 42. Ashida Y., Arashiba K., Nakajima K., Nishibayashi Y., Nature 2019, 568, 536–540. [DOI] [PubMed] [Google Scholar]
- 43. Arashiba K., Miyake Y., Nishibayashi Y., Nat. Chem. 2011, 3, 120–125. [DOI] [PubMed] [Google Scholar]
- 44. Tanaka H., Arashiba K., Kuriyama S., Sasada A., Nakajima K., Yoshizawa K., Nishibayashi Y., Nat. Commun. 2014, 5, 3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Silantyev G. A., Forster M., Schluschass B., Abbenseth J., Wurtele C., Volkmann C., Holthausen M. C., Schneider S., Angew. Chem. Int. Ed. 2017, 56, 5872–5876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5966–5970. [Google Scholar]
- 46. Schluschass B., Abbenseth J., Demeshko S., Finger M., Franke A., Herwig C., Wurtele C., Ivanovic-Burmazovic I., Limberg C., Telser J., Schneider S., Chem. Sci. 2019, 10, 10275–10282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arashiba K., Kinoshita E., Kuriyama S., Eizawa A., Nakajima K., Tanaka H., Yoshizawa K., Nishibayashi Y., J. Am. Chem. Soc. 2015, 137, 5666–5669. [DOI] [PubMed] [Google Scholar]
- 48. Liao Q., Cavaille A., Saffon-Merceron N., Mezailles N., Angew. Chem. Int. Ed. 2016, 55, 11212–11216; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11378–11382. [Google Scholar]
- 49. Klopsch I., Finger M., Wurtele C., Milde B., Werz D. B., Schneider S., J. Am. Chem. Soc. 2014, 136, 6881–6883. [DOI] [PubMed] [Google Scholar]
- 50. Lindley B. M., van Alten R. S., Finger M., Schendzielorz F., Wurtele C., Miller A. J. M., Siewert I., Schneider S., J. Am. Chem. Soc. 2018, 140, 7922–7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Alten R. S., Watjen F., Demeshko S., Miller A. J. M., Wurtele C., Siewert I., Schneider S., Eur. J. Inorg. Chem. 2020, 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clentsmith G. K. B., Bates V. M. E., Hitchcock P. B., Cloke F. G. N., J. Am. Chem. Soc. 1999, 121, 10444–10445. [Google Scholar]
- 53. Kawaguchi H., Matsuo T., Angew. Chem. Int. Ed. 2002, 41, 2792–2794; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2916–2918. [Google Scholar]
- 54. Searles K., Carroll P. J., Chen C. H., Pink M., Mindiola D. J., Chem. Commun. 2015, 51, 3526–3528. [DOI] [PubMed] [Google Scholar]
- 55. Ishida Y., Kawaguchi H., J. Am. Chem. Soc. 2014, 136, 16990–16993. [DOI] [PubMed] [Google Scholar]
- 56. Doyle L. R., Wooles A. J., Jenkins L. C., Tuna F., McInnes E. J. L., Liddle S. T., Angew. Chem. Int. Ed. 2018, 57, 6314–6318; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6422–6426. [Google Scholar]
- 57. Doyle L. R., Wooles A. J., Liddle S. T., Angew. Chem. Int. Ed. 2019, 58, 6674–6677; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6746–6749. [Google Scholar]
- 58. Smith J. M., Lachicotte R. J., Pittard K. A., Cundari T. R., Lukat-Rodgers G., Rodgers K. R., Holland P. L., J. Am. Chem. Soc. 2001, 123, 9222–9223. [DOI] [PubMed] [Google Scholar]
- 59. Rodriguez M. M., Bill E., Brennessel W. W., Holland P. L., Science 2011, 334, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dugan T. R., Macleod K. C., Brennessel W. W., Holland P. L., Eur. J. Inorg. Chem. 2013, 3891–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacLeod K. C., McWilliams S. F., Mercado B. Q., Holland P. L., Chem. Sci. 2016, 7, 5736–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grubel K., Brennessel W. W., Mercado B. Q., Holland P. L., J. Am. Chem. Soc. 2014, 136, 16807–16816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee Y., Sloane F. T., Blondin G., Abboud K. A., Garcia-Serres R., Murray L. J., Angew. Chem. Int. Ed. 2015, 54, 1499–1503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1519–1523. [Google Scholar]
- 64. Hirotsu M., Fontaine P. P., Epshteyn A., Zavalij P. Y., Sita L. R., J. Am. Chem. Soc. 2007, 129, 9284–9285. [DOI] [PubMed] [Google Scholar]
- 65. Hirotsu M., Fontaine P. P., Zavalij P. Y., Sita L. R., J. Am. Chem. Soc. 2007, 129, 12690–12692. [DOI] [PubMed] [Google Scholar]
- 66. Fontaine P. P., Yonke B. L., Zavalij P. Y., Sita L. R., J. Am. Chem. Soc. 2010, 132, 12273–12285. [DOI] [PubMed] [Google Scholar]
- 67. Keane A. J., Yonke B. L., Hirotsu M., Zavalij P. Y., Sita L. R., J. Am. Chem. Soc. 2014, 136, 9906–9909. [DOI] [PubMed] [Google Scholar]
- 68. Duman L. M., Farrell W. S., Zavalij P. Y., Sita L. R., J. Am. Chem. Soc. 2016, 138, 14856–14859. [DOI] [PubMed] [Google Scholar]
- 69. Fischler I., von Gustorf E. K., Naturwissenschaften 1975, 62, 63–70. [Google Scholar]
- 70. Rafiq S., Bezdek M. J., Koch M., Chirik P. J., Scholes G. D., J. Am. Chem. Soc. 2018, 140, 6298–6307. [DOI] [PubMed] [Google Scholar]
- 71. Solari E., Da Silva C., Iacono B., Hesschenbrouck J., Rizzoli C., Scopelliti R., Floriani C., Angew. Chem. Int. Ed. 2001, 40, 3907–3909; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 4025–4027. [Google Scholar]
- 72. Huss A. S., Curley J. J., Cummins C. C., Blank D. A., J. Phys. Chem. B 2013, 117, 1429–1436. [DOI] [PubMed] [Google Scholar]
- 73. Bruch Q. J., Connor G. P., Chen C. H., Holland P. L., Mayer J. M., Hasanayn F., Miller A. J. M., J. Am. Chem. Soc. 2019, 141, 20198–20208. [DOI] [PubMed] [Google Scholar]
- 74. Miyazaki T., Tanaka H., Tanabe Y., Yuki M., Nakajima K., Yoshizawa K., Nishibayashi Y., Angew. Chem. Int. Ed. 2014, 53, 11488–11492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11672–11676. [Google Scholar]
- 75. Keane A. J., Farrell W. S., Yonke B. L., Zavalij P. Y., Sita L. R., Angew. Chem. Int. Ed. 2015, 54, 10220–10224; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10358–10362. [Google Scholar]
- 76. Katayama A., Ohta T., Wasada-Tsutsui Y., Inomata T., Ozawa T., Ogura T., Masuda H., Angew. Chem. Int. Ed. 2019, 58, 11279–11284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11401–11406. [Google Scholar]
- 77. Klopsch I., Yuzik-Klimova E. Y., Schneider S. in Nitrogen Fixation. Topics in Organometallic Chemistry (Ed.: Nishibayashi Y.), Springer, Cham, 2017, pp. 71–112. [Google Scholar]
- 78. Sceats E. L., Figueroa J. S., Cummins C. C., Loening N. M., Van der Wel P., Griffin R. G., Polyhedron 2004, 23, 2751–2768. [Google Scholar]
- 79. Figueroa J. S., Piro N. A., Clough C. R., Cummins C. C., J. Am. Chem. Soc. 2006, 128, 940–950. [DOI] [PubMed] [Google Scholar]
- 80. Henderickx H., Kwakkenbos G., Peters A., van der Spoel J., de Vries K., Chem. Commun. 2003, 2050–2051. [DOI] [PubMed] [Google Scholar]
- 81. Curley J. J., Sceats E. L., Cummins C. C., J. Am. Chem. Soc. 2006, 128, 14036–14037. [DOI] [PubMed] [Google Scholar]
- 82. Akagi F., Suzuki S., Ishida Y., Hatanaka T., Matsuo T., Kawaguchi H., Eur. J. Inorg. Chem. 2013, 3930–3936. [Google Scholar]
- 83. Klopsch I., Kinauer M., Finger M., Wurtele C., Schneider S., Angew. Chem. Int. Ed. 2016, 55, 4786–4789; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4864–4867. [Google Scholar]
- 84. MacLeod K. C., Menges F. S., McWilliams S. F., Craig S. M., Mercado B. Q., Johnson M. A., Holland P. L., J. Am. Chem. Soc. 2016, 138, 11185–11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Klopsch I., Schendzielorz F., Volkmann C., Wurtele C., Schneider S., Z. Anorg. Allg. Chem. 2018, 644, 916–919. [Google Scholar]
- 86. Schendzielorz F., Finger M., Abbenseth J., Wurtele C., Krewald V., Schneider S., Angew. Chem. Int. Ed. 2019, 58, 830–834; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 840–844. [Google Scholar]
- 87. Guru M. M., Shima T., Hou Z., Angew. Chem. Int. Ed. 2016, 55, 12316–12320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12504–12508. [Google Scholar]
- 88. Duman L. M., Sita L. R., J. Am. Chem. Soc. 2017, 139, 17241–17244. [DOI] [PubMed] [Google Scholar]
- 89. Chatt J., Wedd A. G., J. Organomet. Chem. 1971, 27, C15–C16. [Google Scholar]
- 90. Hidai M., Uchiday, Tominari K., J. Am. Chem. Soc. 1972, 94, 110–114. [Google Scholar]
- 91. Chatt J., Heath G. A., Richards R. L., J. Chem. Soc. Chem. Commun. 1972, 1010–1011. [Google Scholar]
- 92. Chatt J., Heath G. A., Richards R. L., J. Chem. Soc. Dalton Trans. 1974, 2074–2082. [Google Scholar]
- 93. Pickett C. J., Talarmin J., Nature 1985, 317, 652–653. [Google Scholar]
- 94. Yuki M., Midorikawa T., Miyake Y., Nishibayashi Y., Organometallics 2009, 28, 4741–4746. [Google Scholar]
- 95. Yuki M., Miyake Y., Nishibayashi Y., Wakiji I., Hidai M., Organometallics 2008, 27, 3947–3953. [Google Scholar]
- 96. Yuki M., Miyake Y., Nishibayashi Y., Organometallics 2009, 28, 5821–5827. [Google Scholar]
- 97. Girolami G. S., Salt J. E., Wilkinson G., Thornton-Pett M., Hursthouse M. B., J. Am. Chem. Soc. 1983, 105, 5954–5956. [Google Scholar]
- 98. Mock M. T., Pierpont A. W., Egbert J. D., O'Hagan M., Chen S., Bullock R. M., Dougherty W. G., Kassel W. S., Rousseau R., Inorg. Chem. 2015, 54, 4827–4839. [DOI] [PubMed] [Google Scholar]
- 99. Berben L. A., Kozimor S. A., Inorg. Chem. 2008, 47, 4639–4647. [DOI] [PubMed] [Google Scholar]
- 100. Kendall A. J., Mock M. T., Eur. J. Inorg. Chem. 2020, 1358–1375. [Google Scholar]
- 101. Salt J. E., Wilkinson G., Motevalli M., Hursthouse M. B., J. Chem. Soc. Dalton Trans. 1986, 1141–1154. [Google Scholar]
- 102. Leigh G. J., Jimeneztenorio M., J. Am. Chem. Soc. 1991, 113, 5862–5863. [Google Scholar]
- 103. Hills A., Hughes D. L., Jimenez-Tenorio M., Leigh G. J., Rowley A. T., J. Chem. Soc. Dalton Trans. 1993, 3041–3049. [Google Scholar]
- 104. Hall D. A., Leigh G. J., J. Chem. Soc. Dalton Trans. 1996, 3539–3541. [Google Scholar]
- 105. Hill P. J., Doyle L. R., Crawford A. D., Myers W. K., Ashley A. E., J. Am. Chem. Soc. 2016, 138, 13521–13524. [DOI] [PubMed] [Google Scholar]
- 106. Crossland J. L., Balesdent C. G., Tyler D. R., Inorg. Chem. 2012, 51, 439–445. [DOI] [PubMed] [Google Scholar]
- 107. Yelle R. B., Crossland J. L., Szymczak N. K., Tyler D. R., Inorg. Chem. 2009, 48, 861–871. [DOI] [PubMed] [Google Scholar]
- 108. Gilbertson J. D., Szymczak N. K., Tyler D. R., J. Am. Chem. Soc. 2005, 127, 10184–10185. [DOI] [PubMed] [Google Scholar]
- 109. Doyle L. R., Hill P. J., Wildgoose G. G., Ashley A. E., Dalton Trans. 2016, 45, 7550–7554. [DOI] [PubMed] [Google Scholar]
- 110. George T. A., Rose D. J., Chang Y., Chen Q., Zubieta J., Inorg. Chem. 1995, 34, 1295–1298. [Google Scholar]
- 111. Field L. D., Guest R. W., Vuong K. Q., Dalgarno S. J., Jensen P., Inorg. Chem. 2009, 48, 2246–2253. [DOI] [PubMed] [Google Scholar]
- 112. Chatt J., Dilworth J. R., Richards R. L., Chem. Rev. 1978, 78, 589–625. [Google Scholar]
- 113. Chatt J., Pearman A. J., Richards R. L., Nature 1975, 253, 39–40. [Google Scholar]
- 114. Chatt J., Pearman A. J., Richards R. L., J. Chem. Soc. Dalton Trans. 1977, 1852–1860. [Google Scholar]
- 115. Takahashi T., Mizobe Y., Sato M., Uchida Y., Hidai M., J. Am. Chem. Soc. 1980, 102, 7461–7467. [Google Scholar]
- 116. Hidai M., Mizobe Y., Takahashi T., Uchida Y., Chem. Lett. 1978, 7, 1187–1188. [Google Scholar]
- 117. Chatt J., Pearman A. J., Richards R. L., Nature 1976, 259, 204. [DOI] [PubMed] [Google Scholar]
- 118. Baumann J. A., George T. A., J. Am. Chem. Soc. 1980, 102, 6153–6154. [Google Scholar]
- 119. George T. A., Tisdale R. C., J. Am. Chem. Soc. 1985, 107, 5157–5159. [Google Scholar]
- 120. Khoenkhoen N., de Bruin B., Reek J. N. H., Dzik W. I., Eur. J. Inorg. Chem. 2015, 567–598. [Google Scholar]
- 121. Kilgore U. J., Roberts J. A., Pool D. H., Appel A. M., Stewart M. P., DuBois M. R., Dougherty W. G., Kassel W. S., Bullock R. M., DuBois D. L., J. Am. Chem. Soc. 2011, 133, 5861–5872. [DOI] [PubMed] [Google Scholar]
- 122. Wilson A. D., Shoemaker R. K., Miedaner A., Muckerman J. T., DuBois D. L., DuBois M. R., Proc. Natl. Acad. Sci. USA 2007, 104, 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mock M. T., Chen S., Rousseau R., O'Hagan M. J., Dougherty W. G., Kassel W. S., DuBois D. L., Bullock R. M., Chem. Commun. 2011, 47, 12212–12214. [DOI] [PubMed] [Google Scholar]
- 124. Mock M. T., Chen S., O'Hagan M., Rousseau R., Dougherty W. G., Kassel W. S., Bullock R. M., J. Am. Chem. Soc. 2013, 135, 11493–11496. [DOI] [PubMed] [Google Scholar]
- 125. Kendall A. J., Johnson S. I., Bullock R. M., Mock M. T., J. Am. Chem. Soc. 2018, 140, 2528–2536. [DOI] [PubMed] [Google Scholar]
- 126. Sanner R. D., Manriquez J. M., Marsh R. E., Bercaw J. E., J. Am. Chem. Soc. 1976, 98, 8351–8357. [Google Scholar]
- 127. Manriquez J. M., Mcalister D. R., Rosenberg E., Shiller A. M., Williamson K. L., Chan S. I., Bercaw J. E., J. Am. Chem. Soc. 1978, 100, 3078–3083. [Google Scholar]
- 128. Dilworth J. R., Henderson R. A., Hills A., Hughes D. L., Macdonald C., Stephens A. N., Walton D. R. M., J. Chem. Soc. Dalton Trans. 1990, 1077–1085. [Google Scholar]
- 129. Sanner R. D., Duggan D. M., Mckenzie T. C., Marsh R. E., Bercaw J. E., J. Am. Chem. Soc. 1976, 98, 8358–8365. [Google Scholar]
- 130. Henderson R. A., Morgan S. H., Stephens A. N., J. Chem. Soc. Dalton Trans. 1990, 1101–1106. [Google Scholar]
- 131. Henderson R. A., Morgan S. H., J. Chem. Soc. Dalton Trans. 1990, 1107–1109. [Google Scholar]
- 132. Mizobe Y., Yokobayashi Y., Oshita H., Takahashi T., Hidai M., Organometallics 1994, 13, 3764–3766. [Google Scholar]
- 133. Hillhouse G. L., Bercaw J. E., J. Am. Chem. Soc. 1984, 106, 5472–5478. [Google Scholar]
- 134. Manriquez J. M., Sanner R. D., Marsh R. E., Bercaw J. E., J. Am. Chem. Soc. 1976, 98, 3042–3044. [Google Scholar]
- 135. Shan H., Science 1997, 275, 1460–1462. [Google Scholar]
- 136. Rocklage S. M., Schrock R. R., J. Am. Chem. Soc. 1982, 104, 3077–3081. [Google Scholar]
- 137. Fang M., Lee D. S., Ziller J. W., Doedens R. J., Bates J. E., Furche F., Evans W. J., J. Am. Chem. Soc. 2011, 133, 3784–3787. [DOI] [PubMed] [Google Scholar]
- 138. Korobkov I., Gambarotta S., Yap G. P. A., Angew. Chem. Int. Ed. 2003, 42, 4958–4961; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 5108–5111. [Google Scholar]
- 139. Falcone M., Chatelain L., Scopelliti R., Zivkovic I., Mazzanti M., Nature 2017, 547, 332–335. [DOI] [PubMed] [Google Scholar]
- 140. Yamamoto A., Pu L. S., Kitazume S., Ikeda S., J. Am. Chem. Soc. 1967, 89, 3071. [Google Scholar]
- 141. Hidai M., Takahashi T., Yokotake I., Uchida Y., Chem. Lett. 1980, 9, 645–646. [Google Scholar]
- 142. Nishihara H., Mori T., Tsurita Y., Nakano K., Saito T., Sasaki Y., J. Am. Chem. Soc. 1982, 104, 4367–4372. [Google Scholar]
- 143. Jia G., Morris R. H., Schweitzer C. T., Inorg. Chem. 1991, 30, 593–594. [Google Scholar]
- 144. Nishibayashi Y., Iwai S., Hidai M., Science 1998, 279, 540–542. [DOI] [PubMed] [Google Scholar]
- 145. Nishibayashi Y., Iwai S., Hidai M., J. Am. Chem. Soc. 1998, 120, 10559–10560. [Google Scholar]
- 146. Vidyaratne I., Scott J., Gambarotta S., Budzelaar P. H., Inorg. Chem. 2007, 46, 7040–7049. [DOI] [PubMed] [Google Scholar]
- 147. Fryzuk M. D., Love J. B., Rettig S. J., Young V. G., Science 1997, 275, 1445–1447. [Google Scholar]
- 148. Basch H., Musaev D. G., Morokuma K., Fryzuk M. D., Love J. B., Seidel W. W., Albinati A., Koetzle T. F., Klooster W. T., Mason S. A., Eckert J., J. Am. Chem. Soc. 1999, 121, 523–528. [Google Scholar]
- 149. Pool J. A., Bernskoetter W. H., Chirik P. J., J. Am. Chem. Soc. 2004, 126, 14326–14327. [DOI] [PubMed] [Google Scholar]
- 150. Pool J. A., Lobkovsky E., Chirik P. J., Nature 2004, 427, 527–530. [DOI] [PubMed] [Google Scholar]
- 151. Wang B., Luo G., Nishiura M., Hu S., Shima T., Luo Y., Hou Z., J. Am. Chem. Soc. 2017, 139, 1818–1821. [DOI] [PubMed] [Google Scholar]
- 152. Avenier P., Taoufik M., Lesage A., Solans-Monfort X., Baudouin A., de Mallmann A., Veyre L., Basset J. M., Eisenstein O., Emsley L., Quadrelli E. A., Science 2007, 317, 1056–1060. [DOI] [PubMed] [Google Scholar]
- 153. Solans-Monfort X., Chow C., Goure E., Kaya Y., Basset J. M., Taoufik M., Quadrelli E. A., Eisenstein O., Inorg. Chem. 2012, 51, 7237–7249. [DOI] [PubMed] [Google Scholar]
- 154. Miyazaki T., Tanabe Y., Yuki M., Miyake Y., Nakajima K., Nishibayashi Y., Chem. Eur. J. 2013, 19, 11874–11877. [DOI] [PubMed] [Google Scholar]
- 155. Betley T. A., Peters J. C., J. Am. Chem. Soc. 2003, 125, 10782–10783. [DOI] [PubMed] [Google Scholar]
- 156. Apps S. L., Miller P. W., Long N. J., Chem. Commun. 2019, 55, 6579–6582. [DOI] [PubMed] [Google Scholar]
- 157. Moret M. E., Peters J. C., J. Am. Chem. Soc. 2011, 133, 18118–18121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Suess D. L., Peters J. C., J. Am. Chem. Soc. 2013, 135, 4938–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Rudd P. A., Planas N., Bill E., Gagliardi L., Lu C. C., Eur. J. Inorg. Chem. 2013, 3898–3906. [Google Scholar]
- 160. Fryzuk M. D., MacKay B. A., Patrick B. O., J. Am. Chem. Soc. 2003, 125, 3234–3235. [DOI] [PubMed] [Google Scholar]
- 161. Fryzuk M. D., Acc. Chem. Res. 2009, 42, 127–133. [DOI] [PubMed] [Google Scholar]
- 162. Semproni S. P., Lobkovsky E., Chirik P. J., J. Am. Chem. Soc. 2011, 133, 10406–10409. [DOI] [PubMed] [Google Scholar]
- 163. Chatt J., Heath G. A., Leigh G. J., J. Chem. Soc. Chem. Commun. 1972, 444–445. [Google Scholar]
- 164. Tatsumi T., Hidai M., Uchida Y., Inorg. Chem. 1975, 14, 2530–2534. [Google Scholar]
- 165. Chatt J., Diamantis A. A., Heath G. A., Hooper N. E., Leigh G. J., J. Chem. Soc. Dalton Trans. 1977, 688–697. [Google Scholar]
- 166. Chatt J., Head R. A., Leigh G. J., Pickett C. J., J. Chem. Soc. Chem. Commun. 1977, 299–300. [Google Scholar]
- 167. Chatt J., Hussain W., Leigh G. J., Neukomm H., Pickett C. J., Rankin D. A., J. Chem. Soc. Chem. Commun. 1980, 1024–1025. [Google Scholar]
- 168. Bevan P. C., Chatt J., Leigh G. J., Leelamani E. G., J. Organomet. Chem. 1977, 139, C59–C62. [Google Scholar]
- 169. Rittle J., Peters J. C., J. Am. Chem. Soc. 2016, 138, 4243–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Thompson N. B., Green M. T., Peters J. C., J. Am. Chem. Soc. 2017, 139, 15312–15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Knobloch D. J., Toomey H. E., Chirik P. J., J. Am. Chem. Soc. 2008, 130, 4248–4249. [DOI] [PubMed] [Google Scholar]
- 172. Bernskoetter W. H., Lobkovsky E., Chirik P. J., Angew. Chem. Int. Ed. 2007, 46, 2858–2861; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2916–2919. [Google Scholar]
- 173. Lv Z. J., Huang Z., Zhang W. X., Xi Z., J. Am. Chem. Soc. 2019, 141, 8773–8777. [DOI] [PubMed] [Google Scholar]
- 174. Fryzuk M. D., Johnson S. A., Patrick B. O., Albinati A., Mason S. A., Koetzle T. F., J. Am. Chem. Soc. 2001, 123, 3960–3973. [DOI] [PubMed] [Google Scholar]
- 175. Ballmann J., Yeo A., Patrick B. O., Fryzuk M. D., Angew. Chem. Int. Ed. 2011, 50, 507–510; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 527–530. [Google Scholar]
- 176. Morello L., Love J. B., Patrick B. O., Fryzuk M. D., J. Am. Chem. Soc. 2004, 126, 9480–9481. [DOI] [PubMed] [Google Scholar]
- 177. Nakanishi Y., Ishida Y., Kawaguchi H., Angew. Chem. Int. Ed. 2017, 56, 9193–9197; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9321–9325. [Google Scholar]
- 178. Falcone M., Barluzzi L., Andrez J., Fadaei Tirani F., Zivkovic I., Fabrizio A., Corminboeuf C., Severin K., Mazzanti M., Nat. Chem. 2019, 11, 154–160. [DOI] [PubMed] [Google Scholar]
- 179. Simonneau A., Turrel R., Vendier L., Etienne M., Angew. Chem. Int. Ed. 2017, 56, 12268–12272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12436–12440. [Google Scholar]
- 180. Geri J. B., Shanahan J. P., Szymczak N. K., J. Am. Chem. Soc. 2017, 139, 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Qing G., Ghazfar R., Jackowski S. T., Habibzadeh F., Ashtiani M. M., Chen C.-P., Smith M. R., Hamann T. W., Chem. Rev. 2020, 120, 5437–5516. [DOI] [PubMed] [Google Scholar]
- 182. Vol'pin M. E., Ma I., Larikov E. I., Khidekel M. L., Shvetsov Y. A., Shur V. B., Dokl. Akad. Nauk SSSR 1965, 164, 331–333. [Google Scholar]
- 183. Vol'pin M. E., Shur V. B., Dokl. Akad. Nauk SSSR 1964, 156, 1102–1104. [Google Scholar]
- 184. Bazhenova T. A., Shilov A. E., Coord. Chem. Rev. 1995, 144, 69–145. [Google Scholar]
- 185. Shilov A. E., Russ. Chem. Bull. 2003, 52, 2555–2562. [Google Scholar]
- 186. Yandulov D. V., Schrock R. R., Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- 187. Schenk S., Kirchner B., Reiher M., Chem. Eur. J. 2009, 15, 5073–5082. [DOI] [PubMed] [Google Scholar]
- 188. Schenk S., Le Guennic B., Kirchner B., Reiher M., Inorg. Chem. 2008, 47, 3634–3650. [DOI] [PubMed] [Google Scholar]
- 189. Schrock R. R., Angew. Chem. Int. Ed. 2008, 47, 5512–5522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5594–5605. [Google Scholar]
- 190. Sharma A., Roemelt M., Reithofer M., Schrock R. R., Hoffman B. M., Neese F., Inorg. Chem. 2017, 56, 6906–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Schrock R. R., Acc. Chem. Res. 2005, 38, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Weare W. W., Dai X., Byrnes M. J., Chin J. M., Schrock R. R., Muller P., Proc. Natl. Acad. Sci. USA 2006, 103, 17099–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Yandulov D. V., Schrock R. R., Inorg. Chem. 2005, 44, 1103–1117. [DOI] [PubMed] [Google Scholar]
- 194. Smythe N. C., Schrock R. R., Muller P., Weare W. W., Inorg. Chem. 2006, 45, 7111–7118. [DOI] [PubMed] [Google Scholar]
- 195. Smythe N. C., Schrock R. R., Muller P., Weare W. W., Inorg. Chem. 2006, 45, 9197–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yandulov D. V., Schrock R. R., Can. J. Chem. 2005, 83, 341–357. [Google Scholar]
- 197. Ritleng V., Yandulov D. V., Weare W. W., Schrock R. R., Hock A. S., Davis W. M., J. Am. Chem. Soc. 2004, 126, 6150–6163. [DOI] [PubMed] [Google Scholar]
- 198. Kuriyama S., Arashiba K., Nakajima K., Tanaka H., Yoshizawa K., Nishibayashi Y., Chem. Sci. 2015, 6, 3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Kuriyama S., Arashiba K., Nakajima K., Tanaka H., Kamaru N., Yoshizawa K., Nishibayashi Y., J. Am. Chem. Soc. 2014, 136, 9719–9731. [DOI] [PubMed] [Google Scholar]
- 200. Itabashi T., Arashiba K., Tanaka H., Konomi A., Eizawa A., Nakajima K., Yoshizawa K., Nishibayashi Y., Organometallics 2019, 38, 2863–2872. [Google Scholar]
- 201. Tanabe Y., Sekiguchi Y., Tanaka H., Konomi A., Yoshizawa K., Kuriyama S., Nishibayashi Y., Chem. Commun. 2020, 56, 6933–6936. [DOI] [PubMed] [Google Scholar]
- 202. Kuriyama S., Arashiba K., Nakajima K., Matsuo Y., Tanaka H., Ishii K., Yoshizawa K., Nishibayashi Y., Nat. Commun. 2016, 7, 12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Kuriyama S., Arashiba K., Tanaka H., Matsuo Y., Nakajima K., Yoshizawa K., Nishibayashi Y., Angew. Chem. Int. Ed. 2016, 55, 14291–14295; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14503–14507. [Google Scholar]
- 204. Sekiguchi Y., Arashiba K., Tanaka H., Eizawa A., Nakajima K., Yoshizawa K., Nishibayashi Y., Angew. Chem. Int. Ed. 2018, 57, 9064–9068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9202–9206. [Google Scholar]
- 205. Eizawa A., Arashiba K., Tanaka H., Kuriyama S., Matsuo Y., Nakajima K., Yoshizawa K., Nishibayashi Y., Nat. Commun. 2017, 8, 14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kolmar S. S., Mayer J. M., J. Am. Chem. Soc. 2017, 139, 10687–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Chciuk T. V., W. R. Anderson, Jr. , Flowers, 2nd R. A., J. Am. Chem. Soc. 2018, 140, 15342–15352. [DOI] [PubMed] [Google Scholar]
- 208. Anderson J. S., Rittle J., Peters J. C., Nature 2013, 501, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Anderson J. S., G. E. Cutsail, 3rd , Rittle J., Connor B. A., Gunderson W. A., Zhang L., Hoffman B. M., Peters J. C., J. Am. Chem. Soc. 2015, 137, 7803–7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Del Castillo T. J., Thompson N. B., Peters J. C., J. Am. Chem. Soc. 2016, 138, 5341–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Nesbit M. A., Oyala P. H., Peters J. C., J. Am. Chem. Soc. 2019, 141, 8116–8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Del Castillo T. J., Thompson N. B., Suess D. L., Ung G., Peters J. C., Inorg. Chem. 2015, 54, 9256–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Creutz S. E., Peters J. C., J. Am. Chem. Soc. 2014, 136, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Chalkley M. J., Del Castillo T. J., Matson B. D., Roddy J. P., Peters J. C., ACS Cent. Sci. 2017, 3, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Lee Y., Mankad N. P., Peters J. C., Nat. Chem. 2010, 2, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. J. Fajardo, Jr. , Peters J. C., J. Am. Chem. Soc. 2017, 139, 16105–16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Buscagan T. M., Oyala P. H., Peters J. C., Angew. Chem. Int. Ed. 2017, 56, 6921–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7025–7030. [Google Scholar]
- 218. Cavaillé A., Joyeux B., Saffon-Merceron N., Nebra N., Fustier-Boutignon M., Mezailles N., Chem. Commun. 2018, 54, 11953–11956. [DOI] [PubMed] [Google Scholar]
- 219. Ung G., Peters J. C., Angew. Chem. Int. Ed. 2015, 54, 532–535; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 542–545. [Google Scholar]
- 220. Komiya S., Akita M., Yoza A., Kasuga N., Fukuoka A., Kai Y., J. Chem. Soc. Chem. Commun. 1993, 0, 787–788. [Google Scholar]
- 221. Shiina K., J. Am. Chem. Soc. 1972, 94, 9266–9267. [Google Scholar]
- 222. Yin J., Li J., Wang G. X., Yin Z. B., Zhang W. X., Xi Z., J. Am. Chem. Soc. 2019, 141, 4241–4247. [DOI] [PubMed] [Google Scholar]
- 223. Komori K., Oshita H., Mizobe Y., Hidai M., J. Am. Chem. Soc. 1989, 111, 1939–1940. [Google Scholar]
- 224. Tanaka H., Sasada A., Kouno T., Yuki M., Miyake Y., Nakanishi H., Nishibayashi Y., Yoshizawa K., J. Am. Chem. Soc. 2011, 133, 3498–3506. [DOI] [PubMed] [Google Scholar]
- 225. Liao Q., Saffon-Merceron N., Mezailles N., Angew. Chem. Int. Ed. 2014, 53, 14206–14210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14430–14434. [Google Scholar]
- 226. Liao Q., Saffon-Merceron N., Mézailles N., ACS Catal. 2015, 5, 6902–6906. [Google Scholar]
- 227. Yuki M., Tanaka H., Sasaki K., Miyake Y., Yoshizawa K., Nishibayashi Y., Nat. Commun. 2012, 3, 1254. [DOI] [PubMed] [Google Scholar]
- 228. Imayoshi R., Tanaka H., Matsuo Y., Yuki M., Nakajima K., Yoshizawa K., Nishibayashi Y., Chem. Eur. J. 2015, 21, 8905–8909. [DOI] [PubMed] [Google Scholar]
- 229. Imayoshi R., Nakajima K., Takaya J., Iwasawa N., Nishibayashi Y., Eur. J. Inorg. Chem. 2017, 3769–3778. [Google Scholar]
- 230. Li S., Wang Y., Yang W., Li K., Sun H., Li X., Fuhr O., Fenske D., Organometallics 2020, 39, 757–766. [Google Scholar]
- 231. Piascik A. D., Li R., Wilkinson H. J., Green J. C., Ashley A. E., J. Am. Chem. Soc. 2018, 140, 10691–10694. [DOI] [PubMed] [Google Scholar]
- 232. Prokopchuk D. E., Wiedner E. S., Walter E. D., Popescu C. V., Piro N. A., Kassel W. S., Bullock R. M., Mock M. T., J. Am. Chem. Soc. 2017, 139, 9291–9301. [DOI] [PubMed] [Google Scholar]
- 233. Guillet G. L., Sloane F. T., Ermert D. M., Calkins M. W., Peprah M. K., Knowles E. S., Cizmar E., Abboud K. A., Meisel M. W., Murray L. J., Chem. Commun. 2013, 49, 6635–6637. [DOI] [PubMed] [Google Scholar]
- 234. Ferreira R. B., Cook B. J., Knight B. J., Catalano V. J., Garcia-Serres R., Murray L. J., ACS Catal. 2018, 8, 7208–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Eaton M. C., Knight B. J., Catalano V. J., Murray L. J., Eur. J. Inorg. Chem. 2020, 1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Araake R., Sakadani K., Tada M., Sakai Y., Ohki Y., J. Am. Chem. Soc. 2017, 139, 5596–5606. [DOI] [PubMed] [Google Scholar]
- 237. Ohki Y., Araki Y., Tada M., Sakai Y., Chem. Eur. J. 2017, 23, 13240–13248. [DOI] [PubMed] [Google Scholar]
- 238. Siedschlag R. B., Bernales V., Vogiatzis K. D., Planas N., Clouston L. J., Bill E., Gagliardi L., Lu C. C., J. Am. Chem. Soc. 2015, 137, 4638–4641. [DOI] [PubMed] [Google Scholar]
- 239. Clouston L. J., Bernales V., Carlson R. K., Gagliardi L., Lu C. C., Inorg. Chem. 2015, 54, 9263–9270. [DOI] [PubMed] [Google Scholar]
- 240. Gao Y., Li G., Deng L., J. Am. Chem. Soc. 2018, 140, 2239–2250. [DOI] [PubMed] [Google Scholar]
- 241. Suzuki T., Fujimoto K., Takemoto Y., Wasada-Tsutsui Y., Ozawa T., Inomata T., Fryzuk M. D., Masuda H., ACS Catal. 2018, 8, 3011–3015. [Google Scholar]
- 242. Sanz C. A., Stein C. A. M., Fryzuk M. D., Eur. J. Inorg. Chem. 2020, 1465–1471. [Google Scholar]
- 243. Kawakami R., Kuriyama S., Tanaka H., Arashiba K., Konomi A., Nakajima K., Yoshizawa K., Nishibayashi Y., Chem. Commun. 2019, 55, 14886–14889. [DOI] [PubMed] [Google Scholar]
- 244. Ghana P., van Kruchten F. D., Spaniol T. P., van Leusen J., Kogerler P., Okuda J., Chem. Commun. 2019, 55, 3231–3234. [DOI] [PubMed] [Google Scholar]
- 245. Imayoshi R., Nakajima K., Nishibayashi Y., Chem. Lett. 2017, 46, 466–468. [Google Scholar]
- 246. Kawaguchi M., Hamaoka S., Mori M., Tetrahedron Lett. 1993, 34, 6907–6910. [Google Scholar]
- 247. Mori M., Hori K., Akashi M., Hori M., Sato Y., Nishida M., Angew. Chem. Int. Ed. 1998, 37, 636–637; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 659–661. [Google Scholar]
- 248. Mori M., Akashi M., Hori M., Hori K., Nishida M., Sato Y., Bull. Chem. Soc. Jpn. 2004, 77, 1655–1670. [Google Scholar]
- 249. Mori M., Heterocycles 2009, 78, 281–318. [Google Scholar]
- 250. Yamamoto A., Ookawa M., Ikeda S., J. Chem. Soc. Chem. Commun. 1969, 841–842. [Google Scholar]
- 251. Chen J. G., Crooks R. M., Seefeldt L. C., Bren K. L., Bullock R. M., Darensbourg M. Y., Holland P. L., Hoffman B., Janik M. J., Jones A. K., Kanatzidis M. G., King P., Lancaster K. M., Lymar S. V., Pfromm P., Schneider W. F., Schrock R. R., Science 2018, 360, eaar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Rieger P. H., Electrochemistry , 2nd ed., Chapman & Hall, New York, 1994. [Google Scholar]
- 253. Lindley B. M., Appel A. M., Krogh-Jespersen K., Mayer J. M., Miller A. J. M., ACS Energy Lett. 2016, 1, 698–704. [Google Scholar]
- 254. Chalkley M. J., Del Castillo T. J., Matson B. D., Peters J. C., J. Am. Chem. Soc. 2018, 140, 6122–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.P. Saha, S. Amanullah, A. Dey, ChemRxiv 2020, 10.26434/chemrxiv.12324353.v1. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary