

Abstract
Primary cilia are small microtubule‐based organelles capable of transducing signals from growth factor receptors embedded in the cilia membrane. Developmentally, oligodendrocyte progenitor cells (OPCs) express genes associated with primary cilia assembly, disassembly, and signaling, however, the importance of primary cilia for adult myelination has not been explored. We show that OPCs are ciliated in vitro and in vivo, and that they disassemble their primary cilia as they progress through the cell cycle. OPC primary cilia are also disassembled as OPCs differentiate into oligodendrocytes. When kinesin family member 3a (Kif3a), a gene critical for primary cilium assembly, was conditionally deleted from adult OPCs in vivo (Pdgfrα‐CreER™:: Kif3a fl/fl transgenic mice), OPCs failed to assemble primary cilia. Kif3a‐deletion was also associated with reduced OPC proliferation and oligodendrogenesis in the corpus callosum and motor cortex and a progressive impairment of fine motor coordination.
Keywords: Kif3a, myelin, oligodendrocyte, oligodendrocyte progenitor cell, primary cilia
Main Points
OPCs disassemble primary cilia upon cell cycle re‐entry.
Deletion of Kif3a from OPCs prevents primary cilia assembly and reduces OPC proliferation.
Deletion of Kif3a from adult OPCs in vivo reduces oligodendrogenesis and impairs motor function.
1. INTRODUCTION
Primary cilia are small organelles, ~1–5 μm in length, that protrude from the surface of many proliferative and non‐proliferative cell types, including neural stem cells, astrocytes and neurons in the central nervous system (CNS) (reviewed by Sterpka & Chen, 2018; Wheway, Nazlamova, & Hancock, 2018; Youn & Han, 2018). During primary cilia assembly, the basal body differentiates from the centrosome and associates with membrane vesicles en route to the cell surface, where it anchors and the vesicles fuse with the plasma membrane to form the ciliary membrane compartment (Sorokin, 1968; Wu, Chen, & Tang, 2018). The basal body then nucleates α‐ and β‐tubulin heterodimers and the axonemal microtubules elongate, protruding from the cell surface (Rosenbaum & Child, 1967; Xu et al., 2016). As protein synthesis cannot occur inside the cilium, assembly and elongation is facilitated by intraflagellar transport (IFT) motor proteins, which allow the cilium to remain highly dynamic, with new tubulin being laid down constantly and old tubulin being removed (Marshall & Rosenbaum, 2001). Primary cilium disassembly involves destabilization and depolymerization of the microtubules of the axoneme (reviewed by Sanchez & Dynlacht, 2016), and in proliferating cells, cilia disassembly occurs in two stages of the cell cycle, during G1 and prior to mitosis; while reassembly of the primary cilia occurs at the end of mitosis (Doobin, Kemal, Dantas, & Vallee, 2016; Gupta, Tsuchiya, Ohta, Shiratsuchi, & Kitagawa, 2017; Pugacheva, Jablonski, Hartman, Henske, & Golemis, 2007; Sanchez & Dynlacht, 2016; Wang et al., 2013).
Primary cilia are distinct calcium signaling compartments (DeCaen, Delling, Vien, & Clapham, 2013; Delling, DeCaen, Doerner, Febvay, & Clapham, 2013) that transduce growth factor and morphogen signaling from receptors in the ciliary membrane. For example, primary cilia are required for sonic hedgehog (shh) signaling (Corbit et al., 2005; Rohatgi, Milenkovic, & Scott, 2007; Sterpka & Chen, 2018), regulate Wnt signaling (Balmer et al., 2015; Haycraft et al., 2005), and can be a site of bone morphogenic protein (Monnich et al., 2018; Vion et al., 2018) and platelet‐derived growth factor receptor (PDGFR) α (Schneider et al., 2005; Schneider et al., 2009; Umberger & Caspary, 2015) signaling. Within the CNS, signaling at the primary cilium can impact cellular polarity (Higginbotham et al., 2013), neural patterning or cell fate specification (Cortellino et al., 2009; Gazea et al., 2016; Gorivodsky et al., 2009; Willaredt et al., 2008), proliferation (Amador‐Arjona et al., 2011; Lepanto, Davison, Casanova, Badano, & Zolessi, 2016; Spassky et al., 2008; Tong et al., 2014; Wilson, Wilson, Wang, Wang, & McConnell, 2012), cell death (Marszalek et al., 2000) and neuronal synaptic integration (Guo et al., 2017; Kumamoto et al., 2012). Consequently, dysfunction of the primary cilia or cilia‐associated proteins can result in diseases, including some cancers (Alvarez‐Satta & Matheu, 2018; Barakat, Humke, & Scott, 2013; Eguether & Hahne, 2018; Han et al., 2009; Jackson, 2018; Jenks et al., 2018; Lee et al., 2018; Zingg et al., 2018), that have varied phenotypes but are collectively known as ciliopathies (reviewed by Schock & Brugmann, 2017; Valente, Rosti, Gibbs, & Gleeson, 2014).
Primary cilia may also regulate developmental and adult myelination. In vitro, desert hedgehog signaling at primary cilia on immature Schwann cells increases the number of myelin segments produced in co‐culture with dorsal root ganglion neurons (Yoshimura & Takeda, 2012), suggesting that primary cilia promote myelination in the peripheral nervous system. However, Schwann cells disassemble their primary cilia as they mature (Yoshimura & Takeda, 2012), indicating that primary cilia are not required to maintain peripheral myelin. Microarray (Cahoy et al., 2008) and RNA sequencing (Zhang et al., 2014) studies indicate that oligodendrocyte progenitor cells (OPCs) from the developing mouse cortex express Kinesin family member 3a (Kif3a) and Intraflagellar transport 81 (Ift81), genes critical for primary cilia assembly (Bhogaraju et al., 2013; Kubo et al., 2016; Lin et al., 2003; Marszalek, Ruiz‐Lozano, Roberts, Chien, & Goldstein, 1999) and the Hedgehog signaling receptors Patched 2 (Ptchd2) and Smoothened (Smo) which, in other cell types, signal at the primary cilium (Caspary, Larkins, & Anderson, 2007; Rohatgi et al., 2007; Spassky et al., 2008). Additionally, OPCs cultured from the P1 rat cortex have γ‐tubulin+ puncta, consistent with having centrioles or basal bodies, and discrete regions of ADP‐ribosylation factor‐like (Arl)13b staining, consistent with labeling of primary cilia axonemes (Falcon‐Urrutia, Carrasco, Lois, Palma, & Roth, 2015), suggesting that signaling at the primary cilia may also impact central myelination.
Herein, we report that OPCs have assembled primary cilia that are disassembled during cell division and disassembled during oligodendrocyte differentiation. Furthermore, we show that Kif3a is a critical regulator of OPC proliferation and oligodendrogenesis in vivo, and that its function in OPCs ultimately impacts fine motor coordination.
2. MATERIALS AND METHODS
2.1. Transgenic mice
Pdgfrα‐CreER™ (Kang, Fukaya, Yang, Rothstein, & Bergles, 2010); Pdgfrα‐H2BGFP knock‐in mice (Hamilton, Klinghoffer, Corrin, & Soriano, 2003), referred to as Pdgfrα‐histGFP; Plp‐CreER (Doerflinger, Macklin, & Popko, 2003), and Rosa26‐YFP (Srinivas et al., 2001) transgenic mouse lines were obtained from Jackson Laboratories. Kif3a fl/fl (Marszalek et al., 1999) transgenic mice were obtained from the Mutant Mouse Resource and Research Centers. Sox10‐GFP‐DTA transgenic mice (Kessaris et al., 2006), referred to as Sox10‐GFP mice, were a kind gift from Professor William D Richardson (University College London, UK). All mice were maintained on a C57BL/6 background and, except for pups used for primary glial cultures, were weaned after P35 and cohoused with gender‐matched littermates to ensure normal myelin development (Makinodan, Rosen, Ito, & Corfas, 2012). Mice were housed in individually ventilated Optimice™ cages (Animal Care Systems), on a 12‐hr light–dark cycle, with food and water available ad libitum. Male and female mice were used for this study and all experiments were approved by the University of Tasmania Animal Ethics Committee (A0016151 and A0018606) and carried out in accordance with the Australian code for the care and use of animals for scientific purposes.
2.2. Genomic DNA extraction and PCR genotyping
Pdgfrα‐histGFP and Sox10‐GFP mice were genotyped for GFP expression at P0 by exposure to a BlueStar flashlight (Nightsea, Lexington). The presence of other transgenes was evaluated by performing PCR on 50–100 ng of genomic DNA as previously described (O'Rourke et al., 2016). The following primers were used to detect the Rosa26‐YFP transgene: Rosa26 wildtype 5′ AAAGT CGCTC TGAGT TGTTAT; Rosa26 wildtype 3′ GGAGC GGGAG AAATG GATATG and Rosa26 YFP 5′ GCGAAGAGTTTGTCCTCAACC (94°C for 4′; 37 cycles of 94°C for 30″, 60°C for 45″ and 72°C for 60″, and a final 72°C for 10 min); or Kif3a floxed and wildtype alleles: Kif3a_1 5′ AGGGC AGACG GAAGG GTGG, Kif3a_2 5′ TCTGT GAGTT TGTGA CCAGCC and Kif3a_3 5′ TGGCA GGTCA ATGGA CGCAG (94°C for 4′, 34 cycles of 94°C for 30″, 62°C for 45″ and 72°C for 60″, and a final 72°C for 10 min), with an ~350 bp product corresponding to the wildtype Kif3a gene and an ~480 bp product corresponding to the Kif3a fl/fl allele (Marszalek et al., 1999). Pdgfrα‐CreER™ and Plp‐CreER mice were genotyped using primers that amplify a portion of the Cre transgene: Cre 5′ CAGGT CTCAG GAGCT ATGTC CAATT TACTG ACCGTA Cre 3′ GGTGT TATAA GCAAT CCCCA GAA (94°C for 4′, 34 cycles of 94°C for 30″, 62°C for 45″ and 72°C for 60″, and a final 10 min at 72°C).
2.3. Tamoxifen administration and tissue processing
Tamoxifen (Tx; Sigma) was reconstituted to 40 mg/ml in corn oil and placed in a sonicating water bath for ≥1 hr until dissolved. For conditional gene deletion and lineage tracing, postnatal day 57 (P57) mice received 300 mg/kg Tx by oral gavage, daily for four consecutive days. Mice were terminally anaesthetized with a 30 mg/kg intraperitoneal (i.p.) injection of sodium pentobarbitone (Ilium) and perfusion fixed with 4% paraformaldehyde (PFA) (wt/vol) (Sigma) in phosphate buffered saline (PBS) at a rate of ~9 ml/min. Brains were removed and sliced into 2 mm thick coronal slices using a 1 mm brain matrix (Kent Scientific), before being immersion fixed in 4% PFA/PBS for 90 min at ~21°C. Tissue was cryoprotected by immersion in 20% sucrose (Sigma) (wt/vol) in PBS overnight at 4°C prior to embedding in optimal cutting temperature cryomatrix (Thermo Scientific) and storage at −80°C. Mice that received Tx from P57 and were perfusion fixed 7 days later are referred to as P57 + 7.
2.4. Mixed glial culture and OPC isolation
Six well tissue culture plates were pretreated with 0.05 mg/ml >30,000 MW poly‐D‐lysine hydrobromide (PDL; Sigma) for a minimum of 1 hr and washed twice in sterile MilliQ before being left to dry prior to use. Brains were dissected from P0 to P5 Pdgfrα‐histGFP and Pdgfrα‐histGFP:: Kif3a fl/fl transgenic mice and the cortices isolated into Earle's balanced salt solution (EBSS, Invitrogen) and digested in 0.25% (wt/vol) trypsin (Sigma, T4799) in EBSS at 37°C for 5–10 min (Pringle et al., 1996; Young et al., 2013). DNAse I (Sigma) and fetal calf serum (FCS; Invitrogen) were added to a final concentration of 131 μg/ml and 10% (vol/vol), respectively. The cells were gently triturated, passed through a 40 μm cell sieve (BD, 352340), pelleted by centrifugation and gently resuspended in OPC medium (10 ng/ml PDGF‐AA, 10 ng/ml bFGF, 10 ng/ml CNTF, 5 μg/ml NAC, 1 ng/ml NT3, 1 ng/ml Biotin, 10 μM forskolin, 10x penicillin/streptomycin, 2% B27, 5 μg/ml insulin, 60 ng/ml progesterone, 100 μg/ml transferrin, 100 μg/ml bovine serum albumin, 40 ng/ml sodium selenite, 16 μg/ml putrescine in Dulbecco's Modified Eagle's Medium). Each cell preparation was plated across three wells of a six well plate and incubated at 37°C and 5% CO2.
After 7 days in vitro (DIV) primary mixed glial cultures were rinsed twice in EBSS and incubated in 50% TrypLE (Thermofisher) (vol/vol) in EBSS at 37°C and 5% CO2 for 5 min or until cells had detached. Trypsin was inactivated with 200 μl of FCS, the cells pelleted by centrifugation, resuspended in 3 ml of 50% FCS (vol/vol) in EBSS and passed through a 40 μm cell sieve prior to fluorescence‐activated cell sorting of the GFP+ OPCs into OPC medium using a Beckman Coulter MoFlo Astrios cell sorter (Beckman Coulter, CA). For immunocytochemistry or protein lysate generation, GFP+ OPCs were pelleted by centrifugation, resuspended in OPC medium and plated onto PDL‐coated 13 mm #1 glass coverslips (Lomb) at a density of 30,000 cells/well in complete OPC medium. For live imaging, GFP+ OPCs were plated on PDL‐coated 35 mm glass bottom culture dishes with a 14 mm glass diameter (In Vitro Scientific) at a density of 20,000 cells per dish in 2 ml of OPC medium. Cultures were incubated at 37°C and 5% CO2.
2.5. Tat‐Cre treatment and transfection of cultured cells
Tat‐Cre (Excellgen, EG‐1001; 4 μM in OPC medium) was added to 8–10 DIV OPC cultures, diluting the existing medium 1:1, such that the final concentration of tat‐Cre was 2 μM. Cultures were incubated at 37°C/5% CO2 for 90 min, before the medium was exchanged for OPC medium and the cultures incubated for a further 72 hr. Culture medium was gently removed and replaced with ice‐cold 4% PFA (wt/vol) in PBS and incubated on ice for 5 min, before being replaced with ice‐cold methanol and incubated for a further 5 min. Cells were washed thrice with PBS and stored in PBS at 4°C for immunohistochemistry. Where stated, OPCs were transfected using a NeuroMag magnetofection kit (OZ Biosciences, MN50200) according to manufacturer's instructions, using 0.5–1 μg of plasmid DNA and 0.5–1 μl of NeuroMag solution per transfection. The culture medium was replaced 1–2 hr after transfection.
2.6. EdU administration
For EdU labeling in vitro, filter sterilized stocks of 2.5 mg/ml 5‐ethynyl‐2′‐deoxyuridine (EdU; Life Technologies) in EBSS were stored at −20°C and added to OPC culture medium, to achieve a final concentration of 2.5 μg/ml. To label proliferating cells over a period of days to weeks in vivo, EdU was administered to adult mice via the drinking water at a concentration of 0.2 mg/ml as previously described (Clarke et al., 2012). EdU is light sensitive and was administered via opaque water bottles (Optimice) and replenished every 48 hr. To acutely label proliferating cells in vivo, adult mice received 20 μg of EdU (in PBS) i.p at 0, 2, and 4 hr and mice were perfusion fixed at 6 hr (as per Wang et al., 2016). EdU labeling was visualized using the AlexaFluor‐647 Click‐IT EdU kit (Invitrogen). Floating cryosections were incubated for 15 min in 0.5% triton x‐100 (vol/vol) in PBS at 21°C, transferred into the EdU developing cocktail, and incubated in the dark for 45 min before being washed in PBS to commence immunohistochemistry. EdU‐treated OPC cultures were fixed with ice‐cold 4% PFA (wt/vol) in PBS for 5 min, washed thrice with PBS and incubated with 50 μl of the EdU developing cocktail in the dark for 30 min. The coverslips were washed in PBS to commence immunohistochemistry.
2.7. Analyzing protein expression by Western blot
Cultured cells were rinsed twice with ice‐cold PBS before being lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate and protease inhibitors). Cell lysates were centrifuged and frozen at −80°C. Western blot analyses were performed as previously described (Auderset et al., 2016). Protein expression was assessed using rabbit anti‐Kif3a (1:2000, Abcam ab11259) and mouse anti‐β‐actin (1:15000, Sigma A2228) detected with anti‐rabbit HRP (1:5000, Dako P0448) and anti‐mouse HRP (15,000, Dako P0447), respectively.
2.8. DNA plasmids
The pEGFP‐Arl13b plasmid was generously donated by Judith Paridaen and Weiland Huttner (Max Planck Institute, Dresden, Germany; (Paridaen, Wilsch‐Brauninger, & Huttner, 2013) and the N‐terminal mCherry fusion vector purchased from Clontech (632523; Mountain View, CA). For transformations, DSHΔ chemically competent Escherichia coli were incubated with 500 ng of plasmid DNA in ice‐cold KCM (10 mM KCl, 30 mM CaCl2, 50 mM MgCl2) at 42°C for 60 s, before being rapidly transferred to ice for a further 5 min. 1 ml of antibiotic‐free luria broth was added, and after 45 min at 37°C, the bacteria were transferred to agar plates containing 100 μg/ml kanamycin (Thermo Fisher) and incubated at 37°C overnight. Colonies were picked for mini‐prep amplification of the DNA and the purified DNA used to generate the CMV‐Arl13b‐mCherry plasmid (see Figure S1).
2.9. RNA probe synthesis and in situ hybridization
Stab cultures of Escherichia coli, containing plasmids coding for Pdgfrα, Ift81, Kif3a, Ptchd2 and Smo, were purchased from Source Biosciences (Nottingham, UK). Each clone was streaked onto a 0.5% (wt/vol) agar plate containing the appropriate antibiotic and incubated at 37°C overnight. DNA was amplified, purified, and digested using a restriction enzyme that would linearize the DNA, cutting at the 5′ end of the coding sequence. The linearized DNA template was purified using a QIAquick PCR purification kit (QIAGEN) according to the manufacturer's instructions. The quality of the DNA template was evaluated by running 5 μl on a 1% (wt/vol) agarose gel. RNA probes were synthesized from the linearized DNA template using either T7, T3, or SP6 RNA polymerase (Promega) and the digoxigenin (DIG) labeling mix (Roche), according to the manufacturer's instructions and were stored at −80°C until required.
Each RNA probe was diluted 1:500 with preheated hybridization solution (50% formamide, 0.2 M NaCl, 5 mM EDTA, 10 mM Tris–HCL (pH 7.5), 5 mM NaH2PO4, 5 mM Na2HPO4, 0.1 mg/ml yeast tRNA, 10% dextran sulphate and 1 x Denhardts) and incubated at 70°C for 5 min before being applied to coverslips containing 7–10 DIV OPCs on 13 mm glass coverslips, that had been fixed with 4% PFA in PBS and transferred into 12 well plates. Then, 1 ml of PBS was added to each empty well and the plate was sealed and incubated overnight at 65°C in a hybridization oven. The coverslips were washed thrice with 1 ml of preheated wash solution (50% formamide, 1 x SSC, 0.1% tween‐20) at 65°C (30 min per wash) and twice with maleic acid buffer‐Tween 20 (MABT) at ~21°C (10 min per wash). Coverslips were incubated with in situ blocking solution (2% [vol/vol] blocking reagent [Sigma, 11096176001] and 10% [vol/vol] heat inactivated sheep serum in MABT) at ~21°C for 1 hr before being exchanged for anti‐DIG‐AP (Roche; 1:1500) in blocking solution overnight at 4°C. The coverslips were washed thrice in MABT at ~21°C and twice in pre‐staining buffer (0.2 M NaCl, 0.1 M MgCl2, 0.2 M Tris pH 9.5, 0.2% tween 20 in milliQ water) before the addition of staining buffer (prestaining buffer containing 0.1% nitro blue tetrazolium chloride, 0.1% 5‐bromo‐4‐chloro‐3‐indolyl phosphate and 5% polyvinyl alcohol). The plate was incubated in the dark at 37°C for 2 hr or until a blue color was visible. The reaction was stopped by replacing the staining buffer with milliQ water. Coverslips were ethanol dehydrated, dipped in xylene, and mounted using DPX (Sigma).
2.10. Immunohistochemistry
Immunohistochemistry was performed as previously described (O'Rourke et al., 2016). Primary and secondary antibodies were diluted in PBS blocking solution (0.1% [vol/vol] Triton X‐100 and 10% FCS in PBS) and applied to cryosections overnight at 4°C, unless staining involved the use of mouse anti‐CC1 (1:100, Calbiochem), in which case antibodies were diluted in Tris buffered saline (TBS) blocking solution (0.1% [vol/vol] Triton X‐100 and 10% fetal calf serum in TBS). Other primary antibodies included goat anti‐PDGFRα (1:100; GeneTex, CA), rabbit anti‐OLIG2 (1:400 Millipore), rat anti‐GFP (1:2000; Nacalai Tesque, Kyoto, Japan), mouse anti‐γ‐tubulin (1:500; Sigma), and rabbit anti‐Arl13b (1:200 Proteintech). Secondary antibodies were conjugated to AlexaFluor‐488, ‐568, or ‐647 (Invitrogen) and included: donkey anti‐goat (1:1000), donkey anti‐rabbit (1:1000), donkey anti‐mouse (1:1000), and donkey anti‐rat (1:500). Nuclei were labeled using Hoechst 33342 (1:1000; Invitrogen).
2.11. Light microscopy
Following in situ hybridization, OPC cultures were imaged using ×40 and ×60 air objectives on an Axio Lab.A1 microscope with an Axiocam ICc5 camera and Axiocam software (Zeiss, Germany). Confocal fluorescent images of cultures and tissue sections were collected using an UltraView spinning disk confocal microscope with Volocity Software (Perkin Elmer, MA) with standard excitation and emission filters for DAPI (Hoechst 33342), FITC (Alexa Fluor‐488), TRITC (Alexa Fluor‐568) and far red (Alexa Fluor‐647). Hoechst 33342 was used to consistently define regions of interest within each tissue section. For low magnification images for cell number quantification, confocal stacks were collected at 2 μm Z‐intervals using a ×20 or ×40 objective and were stitched using Volocity software. For high magnification images for quantification of assembled primary cilia, confocal stacks were collected at 0.5 μm Z‐intervals using a 100x oil objective and stitched using Volocity software. Cell counts were performed manually from exported images, using ImageJ (NIH, Washington DC) and Adobe Photoshop CS6 by an experimenter blind to genotype, timepoint or treatment. Images were collected from ≥3 mice per genotype for in vivo experiments and ≥ 3 independent biological replicates per treatment condition for in vitro experiments.
2.12. Live time‐lapse imaging
Live imaging was performed using a Nikon Eclipse TI microscope (Tokyo, Japan) and the live imaging protocol adapted from Paridaen et al. (2013). Live videos were recorded for 8–12 hr using differential interference contrast (DIC), 488 and 568 emission filters and a ×60 water immersion lens. Images were acquired every 10 min and at 1 μm z‐plane slices with an Andor Zyla camera (Andor Technology, Belfast, Northern Ireland). Cells were maintained at 37°C and 5% CO2 during the imaging period. Videos were analyzed using NIS Elements software (Nikon, Tokyo, Japan).
2.13. Transmission electron microscopy
Control and Kif3a‐deleted mice (P57+150) were perfused with Karnovsky's fixative (2.5% glutaraldehyde, 2% PFA, 0.25 mM CaCl2, 0.5 mM MgCl2 in 0.1 M sodium cacodylate buffer). Brains were cut into 1 mm‐thick coronal slices using a 1 mm brain matrix (Kent Scientific) and postfixed in Karnovsky's fixative for 2 hr at 21°C. The tissue blocks were rinsed and stored in 0.1 M sodium cacodylate buffer overnight. The medial part of the CC (~Bregma +0.5 to −0.5) was dissected and incubated in 1% osmium tetroxide/1.5% potassium ferricyanide [OsO4/K3Fe(III)(CN)6] in 0.1 M sodium cacodylate buffer in the dark for 2 hr at 4°C, before being dehydrated in ethanol and propylene oxide, and embedded in Epon812 resin. Ultrathin 70 nm sections were cut using a Leica Ultra‐cut UCT7 and stained with uranyl acetate and lead citrate. High resolution electron microscopy imaging at 80 kV was performed using a Hitachi HT7700 transmission electron microscope.
Image analysis was carried out using Adobe Photoshop (CS6) by an experimenter blind to genotype. The g‐ratio of myelinated axons was determined by measuring axon diameter with and without including the ensheathing myelin and calculating (axon diameter/[axon + myelin diameter]). The number of myelin wraps was quantified by counting major dense lines for a minimum of 40 transected axons per mouse.
2.14. Mouse motor coordination testing
Motor coordination testing of control and Kif3a‐deleted male and female mice was performed prior to Tx administration (P57‐1) to establish baseline performance and up to P57+150. Each testing session was carried out at the same time of day (14:00–17:00), during the light phase of the 12‐hr light/dark cycle, in low light conditions (~20 lx). Mice were habituated to the test room for at least an hour prior the start of testing.
Gait analysis
Running gait was assessed using the DigiGait™ Imaging System (Mouse Specifics, Inc., MA). Briefly, each individual mouse was placed within the brightly lit, enclosed plexiglass chamber on a transparent motorized treadmill belt for 5 min to habituate to the chamber. The treadmill was then turned on at a speed of 5 cm/s for 5 s before the belt speed was slowly increased to 22 cm/s over a 10‐s period. A 10 s video of the mouse running at 22 cm/s was recorded at 164 frames/s using a Basler scA640‐74fc digital camera mounted underneath the treadmill. Then, 3–4 s video segments (~10 steps) were analyzed using DigiGait™ Analyzer software version 15 (Mouse Specifics, Inc., MA). Each video was analyzed to quantify forelimb and hindlimb gait parameters, particularly stride length (the average length of a complete step), stride frequency (the average number of steps per second), swing time (the portion of the stride where the paw is moving forward but is not in contact with the treadmill) and stride time (the time taken to complete one step). Data for the left and right limbs were pooled, but forelimbs and hindlimbs were analyzed separately.
Beam walk
Mice were trained to cross a 100 cm length of wooden dowel (10 mm diameter) that was fixed horizontally and elevated 50 cm above the surface of the bench. Individual trials were video recorded at 60 frames/s using a hand‐held Samsung FHD video camera. The videos were then scored by an experimenter blind to genotype to determine the total number of foot slips for a single crossing. A foot slip was defined as any paw falling below the level of the beam.
Grid walk
Mice were placed onto a horizontal, 2 cm wire mesh grid elevated 50 cm above the ground and left to explore for 5‐min. Each mouse was video recorded from underneath the grid using a Samsung FHD video camera (60 frames/s) and the videos scored by an experimenter blind to genotype. The total number of missteps that occurred within the first 100 steps, or over the 5 min if 100 steps were not taken, was counted and analyzed as a percentage. A misstep was defined as the stepping foot slipping through the grid.
2.15. Statistical analyses
To determine the proportion (%) of OPCs that have assembled primary cilia, each OPC was examined for the presence of a γ‐tubulin+ basal body immediately adjacent to an Arl13b+ axoneme. When quantifying OPC proliferation, each PDGFRα+ OPC was examined for the presence or absence of EdU in the nucleus of the cell, and the data presented as the proportion (%) of OPCs that had incorporated EdU (EdU+ PDGFRα+/PDGFRα+ × 100) (as per Clarke et al., 2012; Young et al., 2013). The recombination efficiency of the Rosa26‐YFP transgene was quantified as the proportion (%) of PDGFRα+ OPC that were YFP‐labeled (YFP+ PDGFRα+/PDGFRα+ × 100). The total number of newly differentiated oligodendrocytes (YFP+ OLIG2+ PDGFRα‐neg) is expressed as a proportion (%) of all YFP+ OLIG2+ cells in the region (as per Rivers et al., 2008 and Young et al., 2013). As the vast majority of YFP‐labeled cells are OPCs, this approach allows the rate of oligodendrogenesis to be standardized to the fraction of YFP‐labeled OPCs within a region and negates any effect of recombination efficiency. When the number of labeled cells within a region is expressed as a density, the total number of cells counted within the region of interest was divided by the size of the area (defined in x‐y coordinates only, as the z‐depth was consistently 30 μm) and expressed as cells per mm2.
All statistical analyses were performed using GraphPad Prism 6 or8 (GraphPad Software). Data were first assessed using the Shapiro–Wilk normality test where n > 5, or the Kolmogorov–Smirnov normality test where n ≤ 5 and were further analyzed by parametric or nonparametric tests as appropriate. Data sets with n = 3 in any group were analyzed using parametric tests, as the nonparametric equivalents rely on ranking and are unreliable for small sample sizes (GraphPad Prism 8.0). For culture experiments, data are presented as mean ± SD for a minimum of three independent cultures per condition. The number of cultures analyzed in each group (n) is indicated in the corresponding figure legend. For each region analyzed histologically, the data are presented per mouse and expressed as mean ± SD. Behavioral data are presented as mean ± SEM. The number of animals analyzed in each group (n) is indicated in the corresponding figure legend. Normally distributed data with two independent factors (e.g., genotype and time post‐Tx) were analyzed by 2‐way ANOVA with a Bonferroni posttest. Statistical significance was defined as p < .05. ANOVA main effects are given in each figure legend or, where data are not graphically represented, in text. The data that support the findings of this study are available from the corresponding author upon reasonable request.
3. RESULTS
3.1. OPCs express genes associated with primary cilium structure and function
OPCs purified from the early postnatal mouse cortex express a number of genes associated with primary cilia assembly or disassembly, maintenance and function (Cahoy et al., 2008; Zhang et al., 2014). To determine whether cultured mouse OPCs express these genes, we generated mixed primary glial cultures from the cortex of P0‐P5 Pdgfrα‐histGFP transgenic mice, in which the Pdgfrα promoter drives expression of a histone‐targeted form of GFP in the nucleus of PDGFRα+ OPCs. When GFP+ cells were purified from 7DIV cultures by flow cytometry (Figure 1a,b), and cultured until 10 DIV, 90 ± 7% of the cells were GFP+ PDGFRα+ OPCs (mean of n = 8 independent cultures ± SD; Figure 1c). All cells that lacked GFP and PDGFRα expression had the morphological characteristics of astrocytes (not shown). By performing in situ hybridization using an anti‐sense Pdgfrα RNA probe, we confirmed that 92 ± 1% of the cells expressed Pdgfrα mRNA (mean ± SD of n = 3 cultures; haemotoxylin (Htx) counterstain, pink; Pdgfrα mRNA, blue; Figure 1d,i). Ift81 mRNA, which codes for a protein that maintains the cilium cytoskeleton (Bhogaraju et al., 2013), was expressed throughout the cytoplasm of 96 ± 1% of the cultured cells (mean ± SD of n = 6 cultures; Figure 1e,i), and the Kif3a gene, which is involved in primary cilium assembly (Lin et al., 2003; Marszalek et al., 1999), was expressed at a low level in 95 ± 1% of the cells (mean ± SD of n = 7 cultures; Figure 1f,i). The expression of Smoothened (Smo) and Patched 2 (Ptchd2) mRNAs by 97 ± 1% and 96 ± 0.6% of cells, respectively (Figure 1g–i), further supported OPCs having assembled primary cilia in vitro, as Hedgehog receptors signal at the primary cilium.
FIGURE 1.
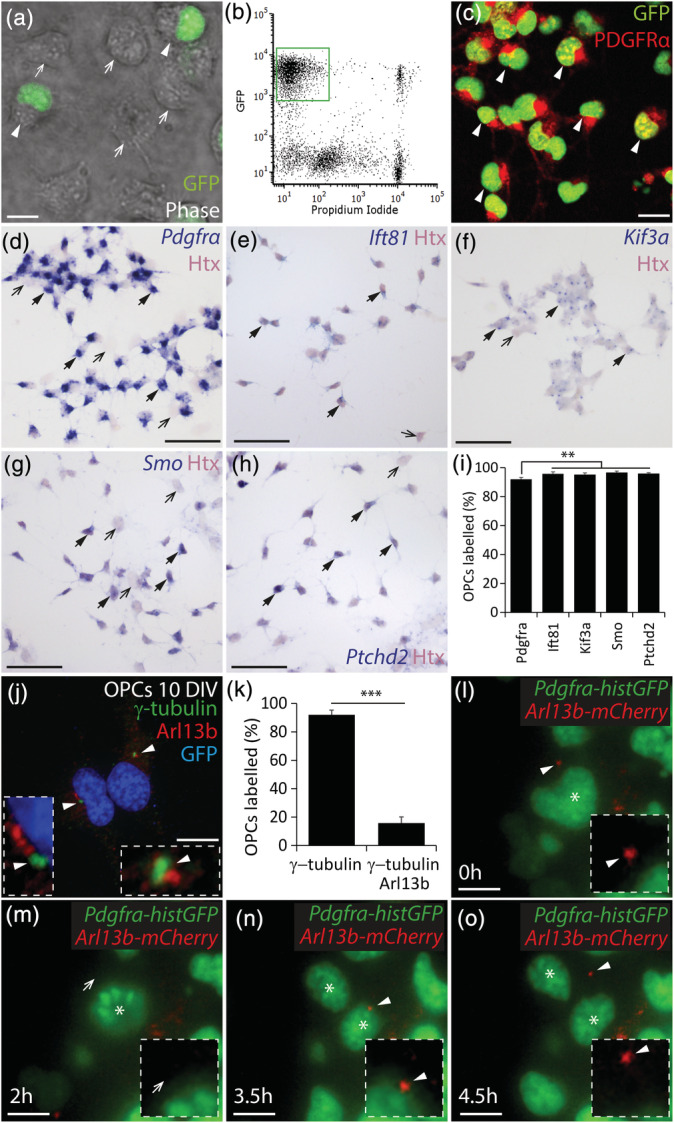
OPCs assemble primary cilia in vitro. (a) Photomicrograph of 5 DIV mixed glial cultures (phase, greyscale), derived from Pdgfrα‐histGFP mice, showing the presence of GFP+ (green; white arrow heads) and GFP‐negative cells (white arrows). (b) Representative fluorescence activated cell sorting profile for 7 DIV mixed glial cultures derived from Pdgfrα‐histGFP mice. The green box denotes the GFP+ propidium iodide‐negative presumptive OPCs population to be isolated. (c) Representative image of 10 DIV GFP+ cells that were purified from 7DIV mixed glial cultures and immunostained to detect GFP (green) and PDGFRα (red). White arrow heads denote GFP+ PDGFRα+ OPCs. (d–h) 10 DIV OPC cultures that were stained with hematoxylin (Htx, pink) following in situ hybridization (blue) to detect Pdgfrα (d), Ift81 (e), Kif3a (f), Smo (g), and Ptched2 (h). Black arrows with solid arrow heads denote example Htx+ cells that co‐label for the target RNA. Black arrows denote example Htx+ cells that do not express the target RNA. (i) Graphical representation of the proportion of Htx+ cells that expressed each target RNA (%). N = 3–7 cultures were exposed to each probe and the data are expressed as mean ± SD. One‐way ANOVA F (4, 8) = 8.10, p = .006. Bonferroni post hoc analysis ** p < .003. (j) 10 DIV OPC culture immunostained to detect Arl13b (red), γ‐tubulin (green) and Hoescht 33,342 (HST, blue). White arrowheads denote primary cilia (Arl13b+ puncta adjacent to γ‐tubulin+ puncta). Insets show the primary cilia at a higher magnification. (k) Graphical representation of the proportion of GFP+ OPCs within each culture that have γ‐tubulin+ puncta and the proportion of GFP+ OPCs that elaborate γ‐tubulin+ Arl13b+ primary cilia (mean ± SD, n = 4–5 cultures). Unpaired t test (t = 28.7, df = 7), ***p < .0001. (l–o) Representative images from a time‐lapse movie of a dividing Pdgfrα‐histGFP OPC transfected with CMV‐Arl13b‐mCherry and imaged to visualize GFP+ nuclei (green) and mCherry+ primary cilia (red) before (l), during (m,n) and after cell division (o). A white arrowhead indicates a mCherry+ primary cilium. A white arrow head indicates the cell surface lacking the primary cilium. An asterisk denotes the mother and daughter cells. Insets show the cell surface at higher magnification, highlighting the presence or absence of a primary cilium on the mother cell. Scale bars represent 5 μm (a), 10 μm (c), 50 μm (d–h) and 12 μm (j, l–o) [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Only a subset of OPCs have assembled primary cilia in vitro
To visualize primary cilia on 10 DIV Pdgfrα‐histGFP OPC cultures, we performed immunohistochemistry to detect GFP (blue) and the primary cilia markers Arl13b (red) and γ‐tubulin (green) (Figure 1j). γ‐tubulin is expressed at the basal body, a structure associated with primary cilia assembly, that also forms the centrioles during cell division, and Arl13b is present in the primary cilium axoneme. We found that 92 ± 3% of cultured OPCs had a γ‐tubulin+ puncta, often two distinct puncta, suggesting two conjoined centrioles. This configuration has been reported in other cells and includes the mother centriole, which forms the basal body of the primary cilium, and the daughter centriole (reviewed in Winey & O'Toole, 2014). A low level of Arl13b staining was detected throughout the cytoplasm of some OPCs, but an intense Arl13b+ puncta (red) was associated with a γ‐tubulin+ basal body, indicative of an assembled primary cilium, on 16 ± 4.5% of cultured GFP+ OPCs (Figure 1j,k).
These Arl13b+ primary cilia varied in length, with some being quite short and circular and others linear, perhaps reflecting different states of assembly and disassembly. To determine whether primary cilia are dynamically assembled and disassembled on the surface of OPCs as they proliferate, we transfected 8DIV Pdgfrα‐histGFP OPC cultures with CMV‐Arl13b‐mCherry. After 24 hr, OPCs with mCherry+ primary cilia were selected for live imaging at high magnification (images collected every 10 min for 4.5 hr; Figure 1l–o). Of the 53 transfected mCherry+ OPCs imaged, only 5 started and completed a division during the imaging period. In each instance, the assembled primary cilium was disassembled during cell division (compare Figure 1l and m). This was detected as a complete disappearance of the Arl13b‐mCherry fluorescent signal or, in one case, a visible shortening of the mCherry+ primary cilium and the retention of a feint, diffuse red fluorescent signal. In all cases, a mCherry+ primary cilium was reassembled by one daughter OPC, as soon as cytokinesis was complete (Figure 1n,o). In other cell types, the primary cilium is disassembled during cell division and the basal body forms the centriole, which is preferentially inherited by one daughter cell following division (Paridaen et al., 2013). That daughter cell can use the basal body to immediately reassemble a primary cilium, while the other daughter cell must synthesize a basal body before it can assemble a primary cilium (Paridaen et al., 2013). These data show that primary cilia are disassembled by OPCs as they enter the cell cycle in vitro and reassembled once they exit.
3.3. Kif3a deletion reduces OPC primary cilium assembly and proliferation in vitro
To determine whether primary cilia are critical regulators of OPC division in vitro, we used a Cre‐lox approach to delete the Kif3a gene from OPCs and prevent primary cilia assembly. GFP+ OPCs were isolated from primary glial cultures generated from P0 to P5 Pdgfrα‐histGFP (control) and Pdgfrα‐histGFP:: Kif3a fl/fl mice, in which both copies of the Kif3a gene are flanked by loxP sites, rendering them susceptible to Cre‐mediated gene deletion. Pdgfrα‐histGFP control OPCs were treated with 2 μM tat‐Cre and Pdgfrα‐histGFP:: Kif3a fl/fl OPCs treated with 0 μM (diluent only) or 2 μM tat‐Cre for 90 min. OPCs were cultured for a further 3 days before protein lysates were generated to examine Kif3a expression by Western blot (Figure 2a). Kif3a expression was equivalent in Pdgfrα‐histGFP control OPCs cultures that received 2 μM tat‐Cre and Pdgfrα‐histGFP:: Kif3a fl/fl OPCs treated with diluent only, however, Pdgfrα‐histGFP:: Kif3a fl/fl OPCs treated with 2 μM tat‐Cre expressed significantly less Kif3a, consistent with Kif3a deletion in vitro (Figure 2b). Pdgfrα‐histGFP:: Kif3a fl/fl OPCs treated with 0 and 2 μM tat‐Cre were also analyzed by immunocytochemistry to evaluate their ability to assemble γ‐tubulin+ (green) and Arl13b+ (red) primary cilia (Figure 2c–e). In 0 μM (diluent‐treated) cultures, 42 ± 3% of the GFP+ OPCs had assembled primary cilia (Figure 2c,d), compared with only 4 ± 0.7% of GFP+ OPCs in the 2 μM tat‐Cre treated cultures (Figure 2c,e). These data suggest that Kif3a deletion prevents primary cilia assembly by OPCs, however a similar phenotype could be observed if Kif3a deletion instead increased the rate of OPC proliferation, such that more cells enter the cell cycle and have disassembled primary cilia.
FIGURE 2.
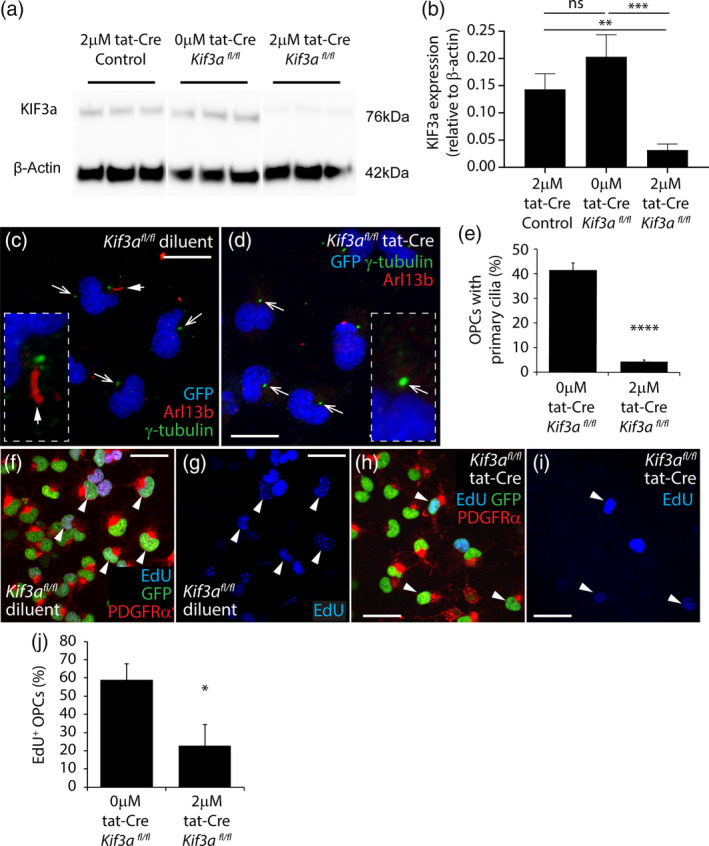
Deleting Kif3a from cultured OPCs reduces their proliferation. (a) Image of a Western blot detecting Kif3A and β‐actin protein expression in lysates generated from Pdgfrα‐histGFP or Pdgfrα‐histGFP:: Kif3a fl/fl OPCs 3 days after exposure to 0 or 2 μM tat‐Cre for 90 min. (b) Quantification of Kif3a expression (relative to β‐actin protein expression) in lysates generated from Pdgfrα‐histGFP or Pdgfrα‐histGFP:: Kif3a fl/fl OPCs 3 days after exposure to 0 or 2 μM tat‐Cre for 90 min. Data are expressed as mean ± SD for n = 3 independent cultures per treatment. One way ANOVA (F = 26.34, p = .001) with a Bonferroni post‐test **p < .01, ***p < .001. (c, d) Confocal image of 0 μM (c) and 2 μM (d) tat‐Cre‐treated Pdgfrα‐histGFP:: Kif3a fl/fl OPC cultures stained to detect γ‐tubulin (green), Arl13b (red) and GFP (blue). White arrow with solid arrow head identifies a γ‐tubulin+ Arl13b+ primary cilium. White arrows indicate γ‐tubulin+ puncta without assembled primary cilia. Insets show an example primary cilia (c) and γ‐tubulin+ puncta (d) at higher magnification. (e) Quantification of the proportion of GFP+ OPCs that have a γ‐tubulin+ basal body associated with an assembled Arl13b+ primary cilium in 0 and 2 μM tat‐Cre‐treated Pdgfrα‐histGFP:: Kif3a fl/fl OPC cultures. Data is expressed as mean ± SD for n = 4 independent cultures per treatment. Unpaired two‐tailed t test (t = 25.7, df = 6), ***p < .0001. (f–i) Confocal images (single z‐planes) of 0 μM (f, g) or 2 μM (h, i) tat‐Cre‐treated Pdgfrα‐histGFP:: Kif3a fl/fl OPCs that were exposed to EdU for 12 hr and immunostained to detect GFP (green), PDGFRα (red) and EdU (blue). White arrowheads indicate example GFP+ PDGFRα+ EdU+ cells. (j) Quantification of the proportion of OPCs that were EdU‐labeled in 0 and 2 μM tat‐Cre‐treated Pdgfrα‐histGFP:: Kif3a fl/fl OPC cultures. Data is expressed as mean ± SD for n = 3 independent cultures. Unpaired two‐tailed t test (t = 4.3, df = 4), *p = .01. Scale bars represent 25 μm (c, d) and 40 μm (f–i) [Color figure can be viewed at wileyonlinelibrary.com]
To examine this possibility, 8–9 DIV Pdgfrα‐histGFP:: Kif3a fl/fl OPC cultures treated with 0 μM (diluent alone; Figure 2f,g) or 2 μM (Figure 2h,i) tat‐Cre were grown for 5 days before EdU was added to the culture medium for 12 hr. EdU is a thymidine analog, which is taken up by cells in S‐phase of the cell cycle, effectively labeling all OPCs that divide during the labeling period. Immunocytochemistry was performed to detect GFP (green), PDGFRα (red) and EdU (blue), and 59 ± 9% of PDGFRα+ GFP+ OPCs were EdU‐labeled in diluent‐treated cultures, compared with only 23 ± 12% of PDGFRα+ GFP+ OPCs in the tat‐Cre treated cultures (Figure 2j). These data indicate that the deletion of Kif3a effectively prevented primary cilia assembly by OPCs and reduced their proliferation.
3.4. OPCs, but not oligodendrocytes, assemble primary cilia in the adult mouse brain
To determine whether cells of the oligodendrocyte lineage assemble primary cilia in vivo, 30 μm cryosections were collected from the brains of young adult Sox10‐GFP mice, in which all cells of the oligodendrocyte lineage are GFP‐labeled (Kessaris et al., 2006). Immunohistochemistry was performed to detect GFP and the primary cilia markers Arl13b and γ‐tubulin (green) within the corpus callosum (CC). We found that only 7 ± 0.1% of GFP+ cells had assembled primary cilia (Figure 3a), a proportion very similar to the fraction of SOX10+ cells previously reported to be OPCs (Rivers et al., 2008). Indeed, by performing immunohistochemistry to detect OPCs (PDGFRα, blue) and primary cilia (Arl13b, red; γ‐tubulin, green), we determined that 69 ± 10% of PDGFRα+ adult OPCs were ciliated (Figure 3a,b), suggesting that OPCs account for the majority of ciliated cells of the oligodendrocyte lineage. This was confirmed by staining cryosections from P57+7 Plp‐CreER:: Rosa26‐YFP transgenic mice, in which immature and mature oligodendrocytes, but not OPCs, are YFP‐labeled (Doerflinger et al., 2003). The vast‐majority of YFP+ callosal oligodendrocytes did not have Arl13b+ γ‐tubulin+ primary cilia assembled on their surface (Figure 3a,c). As OPCs can directly differentiate into oligodendrocytes in the adult mouse brain (Hughes, Kang, Fukaya, & Bergles, 2013), this process must include disassembly of the primary cilium during or at the conclusion of differentiation. It is possible that the 2% ± 0.5% of YFP+ cells that had an assembled primary cilium included oligodendrocytes that had recently differentiated, such as premyelinating (ENPP6+) oligodendrocytes (Xiao et al., 2016).
FIGURE 3.
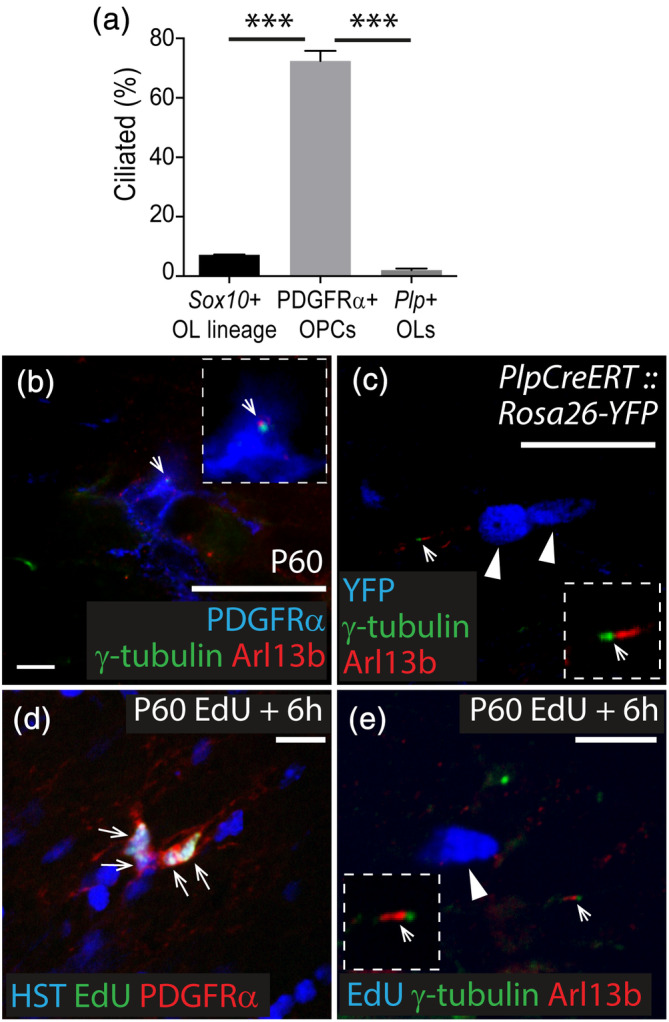
Primary cilia are disassembled as OPCs enter the cell cycle and are not assembled by oligodendrocytes. (a) Quantification of the proportion of Sox10 + cells of the oligodendrocyte lineage, PDGFRα+ OPCs and Plp + oligodendrocytes that have assembled primary cilia. Data are expressed as mean ± SD for n = 3–4 mice per group. One‐way ANOVA F (2, 7) = 148.4, p < .0001. Bonferroni post hoc analysis ***p < .0001. (b) Representative confocal image from the CC of a P60 C57BL/6 mouse immunostained to detect PDGFRα (blue), Arl13b (red), and γ‐tubulin (green). White arrow indicates an Arl13b+ γ‐tubulin+ primary cilium on a PDGFRα+ OPC. Inset shows primary cilium at a higher magnification. (c) Representative confocal image from the CC of a P57+7 Plp‐CreERT:: Rosa26‐YFP mouse immunostained to detect YFP (blue), Arl13b (red) and γ‐tubulin (green). White arrow indicates Arl13b+ γ‐tubulin+ primary cilia assembled on a YFP‐negative cell (higher magnification inset). White arrow heads indicate YFP+ oligodendrocytes that do not express Arl13b or γ‐tubulin. (d) Representative confocal image from the CC proper of a P60 C57BL/6 mouse that received EdU for 6 hr and was immunostained to detect EdU (green), PDGFRα (red) and HST (blue). White arrows indicate EdU+ PDGFRα+ OPCs. (e) Representative confocal image from the CC proper of a P60 C57BL/6 mouse that received EdU for 6 hr and was immunostained to detect EdU (blue), Arl13b (red) and γ‐tubulin (green). White arrow indicates an assembled primary cilia on an EdU‐negative cell (inset at higher magnification). White arrow head indicates an EdU+ cell without primary cilia. Scale bars represent 30 μm (b, c) or 15 μm (d, e) [Color figure can be viewed at wileyonlinelibrary.com]
In vivo, OPCs proliferate asynchronously, such that only a subset of OPCs are in S‐ and M‐phases of the cell cycle at any given time (Young et al., 2013). To determine whether actively dividing OPCs account for the OPCs that lack assembled primary cilia in vivo, we administered EdU to P60 C57Bl6 mice for 6 hr, to label cells entering or transitioning through S‐phase. OPCs in the CC divide approximately once a week and have an S‐phase of 19 ± 7 hr (Young et al., 2013) and an M‐phase of 1.2 ± 0.2 hr (Psachoulia, Jamen, Young, & Richardson, 2009), such that very few OPCs labeled in S‐phase will successfully complete their cell cycle within the 6‐hr labeling period. Immunohistochemistry to detect EdU (green) and PDGFRα (red) confirmed that the small number of EdU+ cells within the CC proper were almost exclusively PDGFRα+ OPCs (94 of 95 EdU+ cells examined across n = 3 mice; Figure 3e). Furthermore, essentially all EdU+ cells within the CC proper lacked Arl13b+ (red), γ‐tubulin+ (green) primary cilia (80 of the 81 EdU+ cells examined across n = 3 mice; Figure 3f), indicating that OPCs disassemble their primary cilia to enter the cell cycle in vivo.
3.5. OPCs lacking Kif3a do not assemble primary cilia and have impaired proliferation
To determine whether Kif3a and primary cilia assembly are important for OPC function in vivo, we administered Tx to P57 Pdgfrα‐CreER™:: Rosa26‐YFP (control) and Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl (Kif3a‐deleted) mice, which enabled the fluorescent labeling and lineage tracing of OPCs with intact and deleted Kif3a, respectively. In the motor cortex of control and Kif3a‐deleted mice, an equivalent proportion of OPCs (>90%) was YFP‐labeled at P57+14 (control, 95 ± 2%; Kif3a‐deleted, 97 ± 1%), P57+30 (control, 93 ± 3%; Kif3a‐deleted, 96 ± 2%) and P57+45 (control, 94 ± 3%; Kif3a‐deleted, 93 ± 2%), indicating that the recombination efficiency of the Rosa26‐YFP transgene was equivalent between genotypes (n = 3–4 mice per group, mean ± SD, two‐way ANOVA: time F [2, 13] = 1.347, p = .3; genotype F [1, 13] = 1.667, p = .2; interaction F [2, 13] = 1.223, p = .3). The proportion of OPCs that was YFP‐labeled in the CC of control and Kif3a‐deleted mice also remained stable throughout the labeling period, from P57+14 (control, 88% ± 6; Kif3a‐deleted, 92 ± 3%), to P57+30 (control, 85 ± 5%; Kif3a‐deleted, 89 ± 4%) and P57+45 (control, 88 ± 2%; Kif3a‐deleted, 90 ± 2%) (n = 3–4 mice per group, mean ± SD, two‐way ANOVA: time F [2, 13] = 1.191, p = .3; genotype F [1, 13] = 4.108, p = .08; interaction F [2, 13] = 0.113, p = .8).
As a proxy for Kif3a deletion, we performed high magnification imaging of the CC, to quantify the proportion of YFP+ cells with an assembled primary cilium (Arl13b, red and γ‐tubulin, green). In control mice, >70% of YFP+ cells had an assembled primary cilium at P57+7 and P57+14 (Figure 4a,b,i). Perhaps unsurprisingly, fewer YFP+ cells (<50%) were ciliated in control mice by P57+30 and P57+60 (Figure 4c,i; control P57+7 and P57+14 versus control P57+30 and P57+60, p < .0001). This decline can be readily explained by the growing fraction of YFP+ cells that differentiate into oligodendrocytes over time (Rivers et al., 2008) and disassemble their primary cilia. By contrast, in Kif3a‐deleted mice only 17.07 ± 1.5% of YFP+ cells were ciliated at P57+7 (mean ± SD, Figure 4e,i) and the proportion of YFP+ cells that were ciliated was not influenced by time, remaining stable at P57+14, P57+30, and P57+60 (Figure 4f–i; p > .4). These data confirmed that Kif3a was deleted from OPCs in vivo and prevented their assembly of primary cilia, but also suggested that the deletion of Kif3a from OPCs stabilized the YFP+ population, mostly likely by preventing oligodendrogenesis.
FIGURE 4.
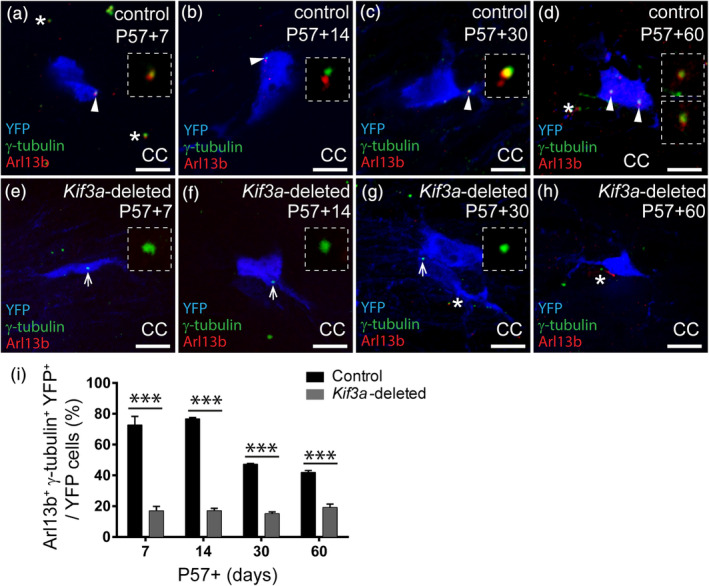
Deleting Kif3a from OPCs prevents cilia assembly in vivo. (a–d) Representative confocal images from the CC of control (Pdgfrα‐CreER™:: Rosa26‐YFP) mice at P57+7 (a), P57+14 (b), P57+30 (c) and P57+60 (d), immunostained to detect YFP (blue), Arl13b (red) and γ‐tubulin (green). White arrowheads indicate primary cilia on YFP‐labeled cells (inset at higher magnification). White asterisks indicate primary cilia on YFP‐negative cells. Scale bars represent 8 μm. (e–h) Representative confocal images from the CC of Kif3a‐deleted (Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice at P57+7 (e), P57+14 (f), P57+30 (g) and P57+60 (h), immunostained to detect YFP (blue), Arl13b (red) and γ‐tubulin (green). White arrows show γ‐tubulin puncta only on YFP‐labeled cells in the CC (inset at higher magnification). White asterisks indicate primary cilia on YFP‐negative cells. Scale bars represent 8 μm. (i) The proportion of YFP+ cells in the CC of control and Kif3a‐deleted mice with assembled primary cilia. Data are presented as mean ± SD for n = 3 mice per genotype per timepoint. Two‐way ANOVA: Time F (3, 16) = 76, p < .0001; genotype F (1, 16) = 1800, p < .0001; interaction F (3, 16) = 80.5, p < .0001. Bonferroni post hoc ***p < .0001 [Color figure can be viewed at wileyonlinelibrary.com]
To determine whether OPCs lacking Kif3a were capable of dividing, P57+25 control and Kif3a‐deleted mice received EdU via the drinking water for 5 days. Brain cryosections were immunolabeled to detect PDGFRα (green) and EdU+ (red), and the fraction of OPCs that had undergone division (EdU+ PDGFRα+/total PDGFRα+) was quantified in the CC and motor cortex. Consistent with previous reports (Psachoulia et al., 2009; Young et al., 2013) in control mice, the fraction of OPCs incorporating EdU over the 5 day labeling period was higher in the CC (~65% of PDGFRα+ OPCs were EdU‐labeled; Figure 5a,e) than the motor cortex (~11%; Figure 5b,e; p < .0001). This was also true for the Kif3a‐deleted OPCs, with ~45% of callosal PDGFRα+ OPCs incorporating EdU (Figure 5c,e) compared with only ~6% in the motor cortex (Figure 5d,e; p < .0001). However, in the CC the fraction of EdU‐labeled OPCs was significantly reduced in Kif3a‐deleted mice relative to controls (Figure 5e), suggesting that deleting Kif3a from OPCs did not prevent all OPCs from dividing, but significantly reduced their rate of proliferation. Kif3a‐deletion did not significantly alter the number of cortical OPCs that incorporated EdU over a 5‐day labeling period, however, this is likely just a reflection of the small number of cortical OPCs that divided in the labeling period (even in control mice) rather than a regional difference in Kif3a function.
FIGURE 5.
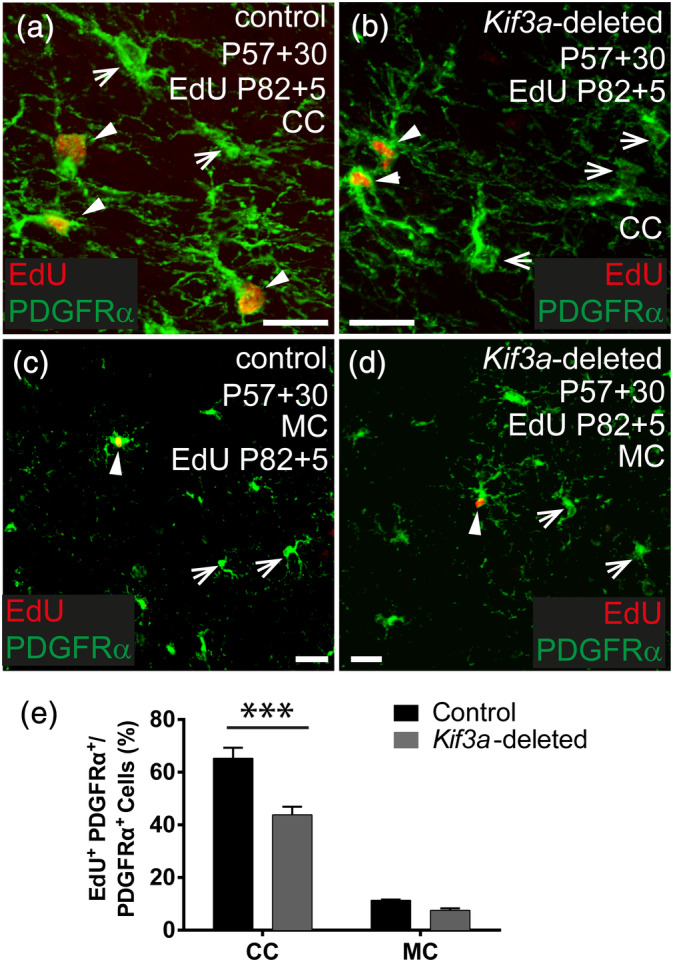
Deleting Kif3a from OPCs reduces proliferation in vivo. (a, b) Representative confocal images from the CC of a Kif3a fl/fl (control) (a) and Pdgfrα‐CreER™:: Kif3a fl/fl (Kif3a‐deleted) (b) mouse that received EdU via the drinking water between P57+25 and P57+30. Brain cryosections were immunostained to detect PDGFRα (green) and EdU (red). (c, d) Representative confocal images from the motor cortex (MC) of a Kif3a fl/fl (control) (c) and Pdgfrα‐CreER™:: Kif3a fl/fl (Kif3a‐deleted) (d) mouse that received EdU via their drinking water from P57+25 to P5+30. Brain cryosections were immunostained to detect PDGFRα (green) and EdU (red). (e) The proportion of PDGFRα+ OPCs that divided and incorporated EdU in the CC and MC of control and Kif3a‐deleted mice. Data are presented as mean ± SD for n = 3–4 mice per genotype at each timepoint. Two‐way ANOVA: brain region F (1, 8) = 932, p < .0001; genotype F (1, 8) = 73.5, p < .0001; interaction F (1, 8) = 35.7, p = .0003. Bonferroni post‐test ***p < .0001. Solid white arrowheads show EdU+ PDGFRα+ OPCs. White arrows show PDGFRα+ OPCs that were EdU‐negative. Scale bars represent 30 μm [Color figure can be viewed at wileyonlinelibrary.com]
3.6. Kif3a‐deficient adult OPCs generate fewer oligodendrocytes than control OPCs
As OPC proliferation was reduced in Kif3a‐deleted mice, we predicted that the OPCs would produce fewer oligodendrocytes. To quantify the rate of oligodendrocyte production, coronal brain cryosections from P57+7, P57+14, P57+30, and P57+45 control and Kif3a‐deleted mice were processed to detect YFP (green), as well as the OPC marker PDGFRα (red), the oligodendrocyte lineage marker OLIG2, and the nuclear stain Hoescht 33342 (blue) (Figure 6). At P57+7, control and Kif3a‐deleted mice had a similar number of new oligodendrocytes (YFP+, PDGFRα‐negative, OLIG2+ cells) in the CC (control 24 ± 6%; Kif3a‐deleted 16 ± 1%, mean ± SD; Figure 6a,d,m) or motor cortex (control 11 ± 0.5%; Kif3a‐deleted 7 ± 2%, mean ± SD; Figure 6g,j,n). However, by P57+14, 32% ± 8% of YFP+ cells were PDGFRα‐negative OLIG2+ cells in the CC of control mice, compared with only 15 ± 2% in the same region of Kif3a‐deleted mice (Figure 6b.e,m). This equated to a ~60% reduction in the density of new oligodendrocytes (Figure S2: control 45 ± 4 new oligodendrocytes/mm2; Kif3a‐deleted 18 ± 2 new oligodendrocytes/mm2, mean ± SD) and suggested that Kif3a deletion significantly impaired adult oligodendrogenesis. Similarly, in the motor cortex, 15 ± 0.8% of the YFP+ cells were new oligodendrocytes in control mice, compared with only 8 ± 1% in Kif3a‐deleted mice (Figure 6h,k,n), which equated to a ~50% decrease in the density of new oligodendrocytes (Figure S2: control 23 ± 2 new oligodendrocytes/mm2; Kif3a‐deleted 12 ± 3 new oligodendrocytes /mm2, mean ± SD). This relative reduction in oligodendrogenesis was also maintained at P57+30 and P57+45 in the CC (Figure 6c,f,m; Figure S2) and motor cortex (Figure 6i,l,n; Figure S2).
FIGURE 6.
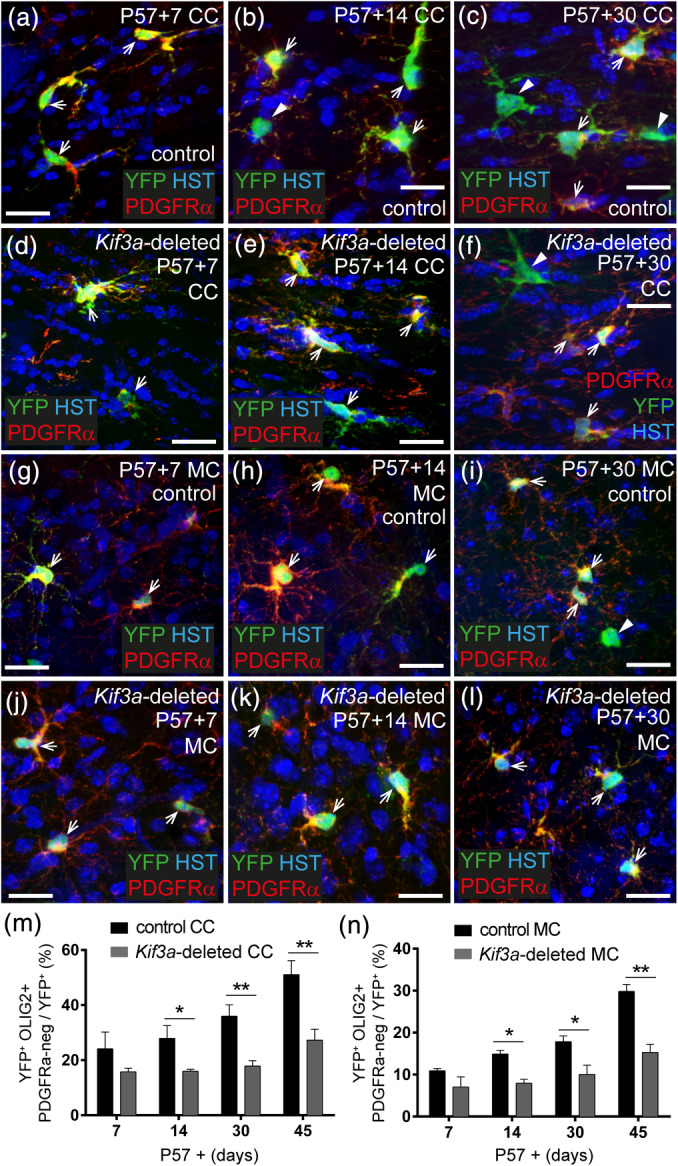
Deleting Kif3a from OPCs reduces oligodendrogenesis in the brain. (a–c) Representative confocal images from the CC of control (Pdgfrα‐CreER™:: Rosa26‐YFP) mice at P57+7 (a), P57+14 (b), and P57+30 (c), immunostained to detect YFP (green), PDGFRα (red) and HST (blue). (d–f) Representative confocal images from the CC of Kif3a‐deleted (Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice at P57+7 (d), P57+14 (e), and P57+30 (f), immunostained to detect YFP (green), PDGFRα (red) and HST (blue). (g–i) Representative confocal images from the motor cortex of control mice at P57+7 (g), P57+14 (h), and P57+30 (i), immunostained to detect YFP (green), PDGFRα (red) and HST (blue). (j–l) Representative confocal images from the motor cortex of Kif3a‐deleted mice at P57+7 (j), P57+14 (k) and P57+30 (l), immunostained to detect YFP (green), PDGFRα (red) and HST (blue). (m, n) The proportion of YFP+ cells in the CC (m) and MC (n) that have differentiated into oligodendrocytes. Data are presented as mean ± SD for n = 3–4 mice per genotype at each timepoint. Two‐way ANOVA for the CC: time F (3, 17) = 30.24, p < .0001; genotype F (1, 17) = 93.10, p < .0001; interaction F (3, 17) = 4.663, p = .01. Two‐way ANOVA for the MC: time F (3, 16) = 37, p < .0001; genotype F (1, 16) = 92, p < .0001; interaction F (3, 16) = 8.5, p = .001. Bonferroni post‐tests *p < .01 and **p < .001. White arrowheads indicate newborn oligodendrocytes (YFP+ PDGFRα‐negative cells). White arrows indicate YFP‐labeled adult OPCs (YFP+ PDGFRα+ cells). Scale bars represent 20 μm (a–f) and 100 μm (g–l) [Color figure can be viewed at wileyonlinelibrary.com]
When additional P57+45 brain cryosections were processed to detect YFP (green) and the oligodendrocyte marker CC1 (red), we similarly found that 53 ± 1% of YFP+ cells in the CC co‐expressed CC1 in control mice (Figure S2), compared with 32 ± 3% in Kif3a‐deleted mice. We detected a similar reduction in new oligodendrocyte addition to the motor cortex, as 26 ± 2% of YFP+ cells in the motor cortex of control mice co‐labeled with CC1, compared with only 15 ± 2% in the motor cortex of Kif3a‐deleted mice (Figure S2; mean ± SD). These data suggest that mice in which OPCs cannot assemble primary cilia, have a reduced rate of OPC proliferation that ultimately leads to fewer new oligodendrocytes being added to the brain.
3.7. Deleting Kif3a from adult OPCs reduces the fraction of myelinated axons in the corpus callosum by ~7 months of age
As Kif3a‐deletion reduces adult oligodendrogenesis, we predicted that it would also impair myelination over time. To explore this possibility, Tx was delivered to control (Kif3a fl/fl) and Kif3a‐deleted (Pdgfrα‐CreER™:: Kif3a fl/fl) mice at P57 and sagittal sections transecting the CC analyzed by transmission electron microscopy at P57+150 (~7 months of age). The CC of control and Kif3a‐deleted mice contained a mixture of myelinated and unmyelinated axons (Figure 7a–d). We first analyzed the myelinated axon population and determined that within the Kif3a‐deleted CC, axonal g‐ratio was shifted toward axons having a larger g‐ratio (axon diameter/[axon + myelin diameter]; Figure 7e). Indeed, the Kif3a‐deleted mice had a larger average g‐ratio for myelinated callosal axons than equivalently treated control mice (Figure 7f). A larger g‐ratio is often attributed to thinner myelin but an increase in axon diameter due to, for example, axon swelling can also increase this measurement. We examined both possibilities and found that the average diameter of myelinated callosal axons was larger in Kif3a‐deleted mice than control mice (Figure 7g), while the distribution and average number of myelin wraps per axon was unchanged by genotype (Figure 7h,i), which indicated that the elevated g‐ratio in Kif3a‐deleted mice resulted from a change in axon diameter rather than thinner myelin. These data may suggest that adult oligodendrogenesis normally leads to the myelination of smaller callosal axons over time in control mice, and that this is not occurring in the Kif3a‐deleted mice or, alternatively, that the reduction in adult myelination or the loss of cilia signaling by adult OPCs results in myelinated axons swelling slightly in the Kif3a‐deleted mice. However, we also found that the proportion of callosal axons that were myelinated was higher in P57+150 control mice compared with Kif3a‐deleted mice (Figure 7j), which confirmed that the deletion of Kif3a from adult OPCs impaired adult myelination.
FIGURE 7.
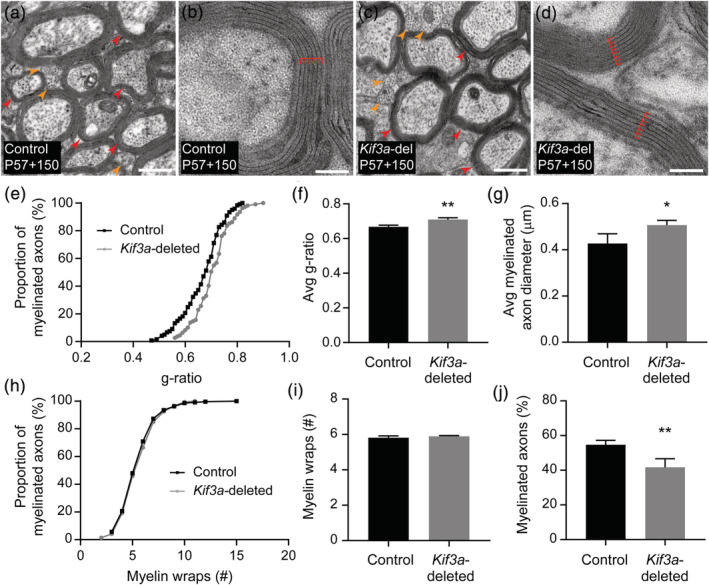
Kif3a‐deletion is associated with an increase in g‐ratio attributed to increased axon diameter rather than a change in the number of myelin lamellae. (a–d) Transmission electron micrographs of transected axons in the CC of P57+150 Kif3a fl/fl (control; a, b) and Pdgfrα‐CreER™:: Kif3a fl/fl (Kif3a‐deleted; c, d) mice. The CC contains a mixture of myelinated and unmyelinated axons, and examples of these are indicated by the red and orange arrows, respectively. At high magnification, it is possible to identify the major dense lines of the myelin sheath (indicated by red lines) to count the number of myelin wraps (lamellae). Scale bars represent 0.5 μm (a, c) or 100 nm (b, d). (e) The g‐ratio was measured from transmission electron micrographs of the CC of n = 4 P57+150 control mice (n = 131 axons from n = 4 mice) and Kif3a‐deleted mice (n = 110 axons from n = 3 mice). This graph shows the cumulative proportion of axons across each g‐ratio for control and Kif3a‐deleted mice. K‐S test (K‐S D = 0.19), p = .02. (f) The average g‐ratio for myelinated axons in the CC of P57+150 Kif3a fl/fl (n = 4 control) and Pdgfrα‐CreER™:: Kif3a fl/fl (n = 3 Kif3a‐deleted) mice. Unpaired t test (t = 5.70, df = 5), p = .002. (g) The average diameter of myelinated axons in the CC of P57+150 Kif3a fl/fl (n = 4 control) and Pdgfrα‐CreER™:: Kif3a fl/fl (n = 3 Kif3a‐deleted) mice. Unpaired t test (t = 2.95, df = 5), p = .031. (h) The number of myelin wraps surrounding myelinated axons in the CC of P57+150 control mice (n = 213 axons from n = 4 mice) and Kif3a‐deleted mice (n = 146 axons across n = 3 mice). This graph shows the cumulative proportion of axons with a given number of myelin wraps. K‐S test (K‐S D = 0.04), p = .995. (i) The average number of myelin wraps surrounding myelinated axons in the CC of P57+150 Kif3a fl/fl (n = 4 control) and Pdgfrα‐CreER™:: Kif3a fl/fl (n = 3 Kif3a‐deleted) mice. Unpaired t test (t = 1.28, df = 5), p = .255. (j) The average proportion (%) of callosal axons that are myelinated in P57+150 control (n = 4) and Kif3a‐deleted (n = 3 ) mice. Unpaired t test (t = 4.61, df = 5), p = .005 [Color figure can be viewed at wileyonlinelibrary.com]
3.8. Deleting Kif3a from adult OPCs impairs fine motor function
Adult‐born oligodendrocytes play a role in complex motor learning (McKenzie et al., 2014), but adult OPCs and/or newborn oligodendrocytes can also play a role in maintaining motor function in the mature mouse CNS (Pepper, Pitman, Cullen, & Young, 2018; S. Schneider et al., 2016). To determine whether conditionally deleting Kif3a from adult OPCs was sufficient to impact motor function, we analyzed the running gait, balance and coordination of adult control and Kif3a‐deleted mice (Figures 8 and S3). To examine gait prior to Tx administration, P57‐1 mice were placed on a DigiGait™ treadmill to run for 10 s at a belt speed of 22 cm/s. Stride length (Forelimbs: Figure 8a, p> .99; Hindlimbs: Figure S3a, p> .99), stride frequency (Forelimbs: Figure 8b, p > 0.99; Hindlimbs: Figure S3b, p >.99), swing time (Forelimbs: Figure S3c, p = .59; Hindlimbs: Figure S3d, p> .99) and stride time (Forelimbs: Figure S3e, p > .99; Hindlimbs: Figure S3f, p > .99) were equivalent for control and Kif3a‐deleted mice, indicating that they initially had a similar running gait. However, following Tx delivery, the Kif3a‐deleted mice experienced a progressive reduction in forelimb stride length (Figure 8a; control P57‐1 vs. P57+150, p = .13; Kif3a‐deleted P57‐1 vs. P57+150, p = .01) and a corresponding increase in stride frequency (Figure 8b; control P57‐1 vs. P57+150, p > .99; Kif3a‐deleted P57‐1 vs. P57+150, p = .0003) that was not observed in the control mice. Consequently, by P57+150, Kif3a‐deleted mice had a significantly shorter stride length (Figure 8a) and greater stride frequency (Figure 8b) than control mice. Within the 150‐day tracing period, Kif3a‐deletion from OPCs only detectably affected forelimb gait, as hindlimb gait parameters remained normal (Figure S3).
FIGURE 8.
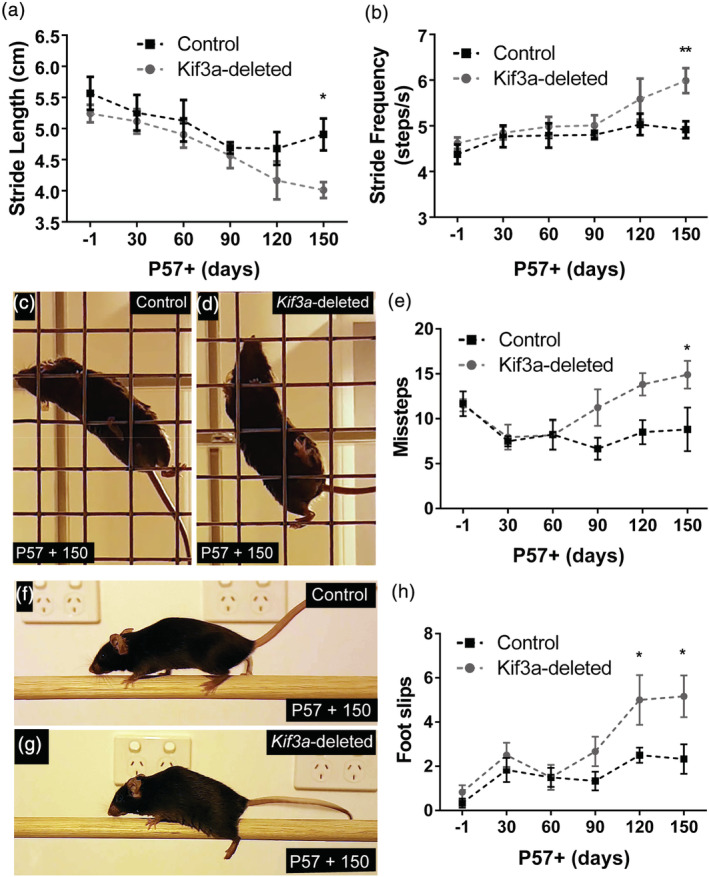
The conditional deletion of Kif3a from adult OPCs leads to a progressive decline in fine motor balance and coordination. (a) Graphical representation of forelimb stride length for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data are expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.15, p = .002; genotype F (5, 85) = 5.57, p = .02; interaction F (5, 85) = 0.37, p = .86. Bonferroni post hoc *p < .05. (b) Graphical representation of forelimb stride frequency for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data are expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.87, p = .0006; genotype F (5, 85) = 8.05, p = .0057; interaction F (5, 85) = 1.48, p = .21. Bonferroni post hoc **p < .01. (c, d) Representative video snapshot of a control (c) and Kif3a‐deleted (d) mouse performing the grid walk task at P57+150. (e) Graphical representation of the number of missteps made by control and Kif3a‐deleted mice during the grid walk task. Data expressed as mean ± SEM for n = 6 mice per genotype at each timepoint. Two‐way ANOVA time F (5, 60) = 2.96, p = .018; genotype F (5, 60) = 9.65, p = .0029; interaction F (5, 60) = 1.88, p = .11. Bonferroni post hoc *p < .05. (f, g) Representative video snapshot of a control (f) and Kif3a‐deleted (g) mouse performing the beam walk task P57+150. (h) Graphical representation of the number of foot slips made by control and Kif3a‐deleted mice during a single beam crossing. Data expressed as mean ± SEM for n = 6 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 60) = 8.25, p < .0001; genotype F (5, 60) = 13.36, p = .005; interaction F (5, 60) = 1.70, p = .14. Bonferroni post hoc *p < .05 [Color figure can be viewed at wileyonlinelibrary.com]
To examine the impact of Kif3a‐deletion on fine motor balance and coordination, we compared the performance of a subset of control and Kif3a‐deleted mice in the grid (Figure 8c–e) and beam (Figure 8f–h) walk tasks. At P57‐1, control and Kif3a‐deleted mice made an equivalent number of missteps (Figure 8e; p > .99) and foot slips (Figure 8h; p > .99) in the grid and beam walk tasks, respectively. However from P57+60, Kif3a‐deleted mice made an increasing number of missteps in the grid walk task, while the performance of control mice was unchanged (Figure 8e; control P57+60 vs. P57+150, p > .99; Kif3a‐deleted P57+60 vs. P57+150, p = .03). By P57+150, Kif3a‐deleted mice made significantly more missteps than their control counterparts (Figure 8e). The number of foot slips made in a single beam crossing also increased from P57+60 in Kif3a‐deleted mice, while performance remained stable for control mice (control P57+60 vs. P57+150, p > .99; Kif3a‐deleted P57+60 vs. P57+150, p = .001), such that Kif3a‐deleted mice experienced more foot slips than controls by P57+120 (Figure 8h). Therefore, the conditional deletion of Kif3a from adult OPCs in vivo, which impaired primary cilia assembly, oligodendrogenesis and myelination, also had a negative impact on fine motor coordination within 5 months.
4. DISCUSSION
We report that OPCs cultured from the early postnatal mouse cortex express genes associated with primary cilia assembly and signaling, and that OPCs assemble and disassemble their primary cilia as they transition through the cell cycle (Figure 1). The deletion of Kif3a from OPCs in vitro impaired primary cilia assembly by these cells and reduced the rate of OPC proliferation (Figure 2). In the adult mouse brain, OPCs had assembled primary cilia while not actively dividing, but disassembled their cilia as they transitioned through the cell cycle, and as they differentiated into oligodendrocytes (Figure 3). The deletion of Kif3a from adult OPCs in vivo reduced primary cilia assembly (Figure 4) and OPC proliferation (Figure 5), which led to a significant reduction in oligodendrogenesis and myelination in the mouse brain (Figures 6 and 7 ), and ultimately impaired fine motor coordination (Figure 8). These data indicate that Kif3a is critical to primary cilia assembly by OPCs and acts as a key regulator of central myelination and motor function.
4.1. How does Kif3a deletion influence OPC proliferation and differentiation?
OPCs, other than those that are progressing through the cell cycle, have assembled primary cilia on their surface. This conclusion is supported by a number of observations: (a) ~7% of Sox10‐GFP + cells in the CC of adult mice have an assembled Arl13b+ primary cilia extending from a γ‐tubulin+ basal body (Figure 3), a proportion similar to the fraction of Sox10‐GFP + cells that would be PDGFRα+ OPCs (Rivers et al., 2008); (b) ~75% of PDGFRα+ OPCs in the CC of adult mice have assembled primary cilia (Figure 3), consistent with the large fraction of adult OPCs that reside in G0 of the cell cycle at any one time (Psachoulia et al., 2009), and (c) OPCs acutely labeled with EdU in vivo, identifying them as cells that have entered or have very recently exited s‐phase of the cell cycle, do not have assembled primary cilia (Figure 3). These data suggest that the fraction of OPCs that have assembled primary cilia is also a proxy for the fraction of OPCs not actively dividing. As the rate of OPC proliferation slows over time in OPC primary cultures, prior to the onset of differentiation (Barres, Lazar, & Raff, 1994; Noble et al., 1990; Raff, Abney, & Fok‐Seang, 1985), this idea is also supported by our observation that ~18% of 10DIV OPC cultures have assembled primary cilia compared with ~40% in 13 DIV OPCs cultures (compare Figures 1k and 2e ).
Kif3a is an essential component of Kinesin‐2, which is required for ciliogenesis (Marszalek et al., 1999), and the conditional deletion of Kif3a from OPCs revealed that primary cilia are not only assembled and disassembled as OPCs progress through the cell cycle, but are able to influence the rate of OPC proliferation (Figures 2, 5) and differentiation (Figure 6). Our data may, in part, explain a previous report of impaired OPC proliferation and survival in early postnatal Bbs1 M390R/M390R mice, which model the human ciliopathy Bardet–Biedl syndrome (Carter et al., 2012). In other cell types, primary cilia can regulate proliferation by facilitating Shh signaling (Han et al., 2009; Spassky et al., 2008; Tong et al., 2014), which is restricted to the primary cilium in vertebrates (reviewed in Goetz & Anderson, 2010). Shh is important for OPC generation during embryonic development (Bian, Zheng, Li, Luo, & Ding, 2016; Merchan et al., 2007; Shin, Xue, Mattson, & Rao, 2007; Tekki‐Kessaris et al., 2001), but can also directly regulate OPC proliferation in vitro (Falcon‐Urrutia et al., 2015; Ortega, Radonjic, & Zecevic, 2013) and in vivo (Ferent, Zimmer, Durbec, Ruat, & Traiffort, 2013). Blocking Shh signaling can reduce OPC proliferation (Falcon‐Urrutia et al., 2015), making it possible that Kif3a‐deleted OPCs experience impaired proliferation due to lost Shh signaling.
Excessive Wnt/β‐catenin signaling could also reduce oligodendrogenesis (Fancy et al., 2009; Tsai et al., 2016) following Kif3a‐deletion. Kif3a knockout mice have elevated Wnt signaling by embryonic day 9 (Corbit et al., 2008), and the conditional deletion of Kif3a from renal tubular epithelial cells (Cdh16‐Cre:: Kif3a fl/fl; Lin et al., 2003) or maturing adult‐born neurons (Kumamoto et al., 2012) also increases canonical Wnt signaling. This phenotype is not specific to Kif3a‐deletion, but is more broadly relevant to primary cilia loss, as mouse embryonic fibroblasts that are hypomorphic for intraflagellar transport 88 (Ift88 orpk/orpk), a key component of the intraflagellar transport that supports ciliogenesis; or lack oral facial digital type 1 (Ofd1), which is required for a basal body formation and ciliogenesis (Ferrante et al., 2006), also show excessive canonical Wnt signaling following exposure to Wnt3a (Corbit et al., 2008). In the context of oligodendrogenesis, β‐catenin acts on neural progenitor cells to suppress OPC production, but acts on cells of the oligodendrocyte lineage to promote oligodendrocyte maturation (Dai et al., 2014). β‐catenin achieves this by competing with HDAC1 and HDAC2 for binding to the transcription factor TCF7L2/TCF4 (Ye et al., 2009). Mice lacking Hdac1/2 or expressing a stable and active form of β‐catenin lack OPCs and oligodendrocytes, while mice lacking tcf7l2 produce OPCs but lack oligodendrocytes (Ye et al., 2009), suggesting that the Wnt/β‐catenin effector TCF7L2 is not required for OPC production but is required for oligodendrocyte maturation. Once bound to TCF7L2, β‐catenin can activate the oligodendrocyte inhibitors Id2 and Id4 and reduce mbp transcription (Ye et al., 2009). If preventing primary cilia assembly caused excessive β‐catenin signaling within OPCs, this could conceivably suppress oligodendrogenesis.
4.2. How are primary cilia disassembled during oligodendrocyte differentiation?
Unlike OPCs, mature oligodendrocytes do not have assembled primary cilia on their surface. While the vast majority of Sox10‐GFP + cells are mature oligodendrocytes, we found that only 7% of Sox10‐GFP + cells had assembled primary cilia. Furthermore, ~98% of all YFP‐labeled oligodendrocytes in the CC of Plp‐CreER :: Rosa26‐YFP transgenic mice were unciliated. The idea that OPCs disassemble their primary cilia as they differentiate into oligodendrocytes is also supported by microarray (Cahoy et al., 2008) and RNA sequencing data showing that the major cilium cytoskeleton‐associated genes, including Kif3a, the IFT proteins Ift81, Ift57, Ift172, and Ift88, the basal body associated proteins Cep290 and Cep97, and the cilium membrane associated protein Arl13b, are more highly expressed by OPCs than oligodendrocytes (Zhang et al., 2014). It is unclear why a very small number of YFP+ oligodendrocytes have primary cilia, but it is possible that these YFP+ ciliated cells are newly differentiated premyelinating oligodendrocytes (Trapp, Nishiyama, Cheng, & Macklin, 1997; Xiao et al., 2016) that expressed plp mRNA at the time of Tx administration and had not fully disassembled their primary cilia at the time of analysis. Nevertheless, adultborn oligodendrocytes do not appear to retain their primary cilia for a long time after differentiation is initiated, as our lineage tracing experiment, that followed the fate of adult OPCs using Pdgfrα‐CreER™:: Rosa26‐YFP transgenic mice, showed that the accumulation of YFP+ adult‐born oligodendrocytes was closely accompanied by an increase in the proportion of unciliated YFP+ cells.
It is not uncommon for cells to lose their capacity to assemble primary cilia as they differentiate from a proliferative progenitor to a post‐mitotic cell type, and many post mitotic cells lose their basal bodies after differentiation (Cunha‐Ferreira, Bento, & Bettencourt‐Dias, 2009; Debec, Sullivan, & Bettencourt‐Dias, 2010). Some differentiated cell types, including neurons and astrocytes, have assembled primary cilia (Arellano, Guadiana, Breunig, Rakic, & Sarkisian, 2012; Kasahara, Miyoshi, Murakami, Miyazaki, & Asanuma, 2014), however, Schwann cells, the myelinating cells of the peripheral nervous system, do not. In mouse dorsal root ganglion neuron and Schwann cell co‐cultures, a high proportion of the proliferative pre‐myelinating Schwann cells have assembled primary cilia, but these structures are rarely detected on the mature myelinating Schwann cells (Yoshimura & Takeda, 2012). Schwann cells and oligodendrocytes are both post‐mitotic, myelinating cell types, and their common disassembly of primary cilia suggests that this type of signaling is not required for myelin maintenance.
Primary cilia disassembly may be regulated by microRNAs (miRNAs) that orchestrate oligodendrocyte differentiation in parallel. The overexpression of miR‐219 and miR‐338 can induce precocious oligodendrocyte differentiation, while deleting these miRNAs impairs oligodendrocyte differentiation in the developing zebrafish and mouse spinal cord (Dugas et al., 2010; Wang et al., 2017; Zhao et al., 2010). Similarly, in the developing mouse nervous system, conditionally deleting the enzyme required for miRNA synthesis, Dicer1, from cells of the oligodendrocyte lineage (Olig1‐Cre:: Dicer1 fl/fl), prevents the expression of miR‐219 and miR‐338 during oligodendrocyte differentiation (Dugas et al., 2010; Zhao et al., 2010), which increases OPC proliferation and impairs central nervous system myelination (Zhao et al., 2010). miR‐219 promotes oligodendrocyte differentiation by binding to the 3′ UTRs of FoxJ3, Sox6, Pdgfrα, ZFP238 and Hes5 genes to repress gene translation (Dugas et al., 2010; Zhao et al., 2010), but it is intriguing to consider the possibility that miR‐219 also promotes primary cilia disassembly. In neural precursor cells, miR‐219 reduces expression of the apical Par polarity proteins, partitioning‐defective (Pard) 3 and protein kinase C iota (Prkci) (Hudish et al., 2016; Hudish, Blasky, & Appel, 2013), which complex at the apical contact point (Afonso & Henrique, 2006; Chen & Zhang, 2013) to promote ciliogenesis (Fan et al., 2004; Krock & Perkins, 2014; Sfakianos et al., 2007). In zebrafish injected with miR‐219 antisense morpholino oligonucleotides, neural precursor cells have long cilia and generate an abnormally large number of Sox2+ cells (Hudish et al., 2016). While this phenotype is largely normalized by Shh antagonism, it is recapitulated by specifically preventing miR‐219 binding at the pard3 3'UTR (Hudish et al., 2016). As OPCs in the early postnatal mouse cortex express pard3 and prkci mRNA (Zhang et al., 2014), and miR‐219 is upregulated with oligodendrocyte differentiation (Dugas et al., 2010), it seems likely that miR‐219 suppresses expression of these genes to underpin the primary cilia disassembly that occurs during oligodendrocyte differentiation.
4.3. How does Kif3a‐deletion impair motor function?
OPCs proliferate and differentiate into oligodendrocytes throughout life (Dimou, Simon, Kirchhoff, Takebayashi, & Gotz, 2008; Hill, Li, & Grutzendler, 2018; Hughes, Orthmann‐Murphy, Langseth, & Bergles, 2018; Kang et al., 2010; Rivers et al., 2008; Young et al., 2013; Zhu et al., 2011). A reduction in oligodendrogenesis was observed in the CC and motor cortex of Kif3a‐deleted mice from P57+14 (Figure 6), and by P57+150 significantly fewer axons were myelinated in the CC of Kif3a‐deleted mice relative to control mice (Figure 7). The loss of adult myelination and the accompanying metabolic support it would provide (Hirrlinger & Nave, 2014; Lee et al., 2012; Meyer et al., 2018), may account for the increase in average axon diameter that we detected, that was not accompanied by any change in myelin thickness. However, it is unclear whether this reduction in adult myelination can also account for the impaired motor performance of Kif3a‐deleted mice (Figure 8). Kif3a‐deletion impacts the proliferative adult OPCs, and it is interesting to draw parallels between the results of this study and a previous study that deleted the cell cycle regulator, Esco2, from all cells of the oligodendrocyte lineage (Sox10‐CreER:: Esco2 fl/fl). The deletion of Esco2 induced the death of proliferating adult OPCs and reduced oligodendrogenesis, but also resulted in impaired balance in the beam and grid walk tests within 4–6 weeks (Schneider et al., 2016). A similar phenotype, that took longer to manifest (~8 months), was also detected when Esco2 was conditionally deleted from adult OPCs (NG2‐CreER:: Esco2 fl/fl; Schneider et al., 2016). However, as adult OPCs perform a number of regulatory functions in the central nervous system [reviewed by (Pepper et al., 2018)], it is difficult to discount the possibility that disrupted adult oligodendrogenesis only partially accounts for the progressive motor phenotype detected in Kif3a‐deleted mice, that develops over time (Figure 8).
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Kaylene M. Young, Carlie L. Cullen, Megan O'Rourke, and Robert Gasperini developed the project and wrote the manuscript. Carlie L. Cullen, Megan O'Rourke, Shannon J. Beasley, Loic Auderset, Robert Gasperini, Yilan Zhen, Renee E. Pepper, and Kaylene M. Young carried out the experiments. Kaylene M. Young, Carlie L. Cullen, and Robert Gasperini obtained the funding. Carlie L. Cullen, Megan O'Rourke, Shannon J. Beasley, and Kaylene M. Young performed the statistical analyses and generated the figures. Kaylene M. Young, Carlie L. Cullen, and Robert Gasperini provided supervision.
Supporting information
Figure S1 Generation of a CMV‐Arl13b‐mCherry plasmid to label primary cilia with a red fluorescent protein
(a) A schematic of the cloning strategy used to generate a CMV‐Arl13b‐mCherry plasmid to mCherry‐label primary cilia. The Arl13b fragment was excised from the CMV‐eGFP‐Arl13b plasmid by performing a restriction digest with the enzymes Pst1 and Kpn1, followed by gel purification of the ~1,300 bp band. CMV‐mCherry‐C1 vector (Clontech, 632,524), which has a multiple closing site (MCS) at the C‐terminal end of the mCherry sequence was linearized by performing a restriction digest with Pst1 and Kpn1. Linearized DNA was treated with Antarctic Phosphatase (New England Biolabs, Ipswich, MA) as per the manufacturer's instructions. The Arl13b DNA fragment was ligated into the linearized CMV‐mCherry‐C1 plasmid and the resulting CMV‐Arl13b‐mCherry plasmid transformed into E. coli.
(b) Image showing cytoplasmic expression of mCherry in HEK293 cells transfected with CMV‐C1‐mCherry.
(c) HEK293 cells transfected with CMV‐Arl13b‐mCherry have a low level of cytoplasmic mCherry expression. A subset of transfected cells has a single point of intense fluorescence, where the Arl13‐b‐mCherry fusion protein is expressed at the primary cilium (arrow heads).
Figure S2 Deleting Kif3a from adult OPCs reduces the addition of CC1 + oligodendrocytes to the mouse brain
(a‐b) The density of YFP+ OLIG2+ PDGFRα‐neg new oligodendrocytes in the corpus callosum (CC; a) and motor cortex (MC; b) of control (Pdgfrα‐CreER™:: Rosa26‐YFP) and Kif3a‐deleted (Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice from P57 + 7 to P57 + 45. Data are presented as mean ± SD for n = 3–4 mice per genotype at each timepoint. Two‐way ANOVA for the CC: time F (3, 17) = 655.5, p < 0.0001; genotype F (1, 17) = 382.7, p < 0.0001; interaction F (3, 17) = 76.6, p < 0.0001. Two‐way ANOVA for the MC: time F (3, 17) = 158.0, p < 0.0001; genotype F (1, 17) = 191.2, p < 0.0001; interaction F (3, 17) = 29.06, p < 0.0001. Bonferroni post‐test *** p < 0.001 and **** p < 0.0001.
(c‐d) Compressed confocal image of YFP (green), CC1 (red) and HST (blue) labeling in the CC of P57 + 45 control (c) and Kif3a‐deleted (d; Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice.
(e‐f) Compressed confocal image of YFP (green), CC1 (red) and HST (blue) labeling in the MC of P57 + 45 control (e) and Kif3a‐deleted (f; Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice.
(g) The proportion (%) of YFP+ cells in the CC and MC that have differentiated into CC1+ oligodendrocytes by P57 + 45. Data are presented as mean ± SD for n = 3 mice per genotype. Two‐way ANOVA: region F (1, 8) = 258.2, p < 0.0001; genotype F (1, 8) = 132.6, p < 0.0001; interaction F (1, 8) = 11.64, p = 0.009. Bonferroni post‐test *** p < 0.001 and **** p < 0.0001.
Solid arrowheads denote newborn oligodendrocytes (YFP+ CC1+ cells). Arrows denote YFP+ CC1‐neg cells (presumptive OPCs). Scale bars represent 20 μm.
Figure S3 Kif3a‐deletion does not impact hind limb gait parameters
(a) Graphical representation of hindlimb stride length for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.3, p = 0.002; genotype F (1, 85) = 0.4, p = 0.5; interaction F (5, 85) = 0.22, p = 0.9.
(b) Graphical representation of hindlimb stride frequency for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 5.4, p < 0.001; genotype F (1, 85) = 0.56, p = 0.5; interaction F (5, 85) = 0.27, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 control, p < 0.05, and P57‐1 v P57 + 150 or P57 + 60 v P57 + 150 Kif3a‐deleted, p = 0.02.
(c) Graphical representation of forelimb swing time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 6.308, p < 0.0001; genotype F (1, 85) = 8.970, p = 0.004; interaction F (5, 85) = 0.7138, p = 0.6. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.03 and P57‐1 v P57 + 150 Kif3a‐deleted p = 0.003.
(d) Graphical representation of hindlimb swing time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 3.4, p = 0.007; genotype F (1, 85) = 0.14, p = 0.7; interaction F (5, 85) = 0.3, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.02.
(e) Graphical representation of forelimb stride time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.150, p = 0.002; genotype F (1, 85) = 5.579, p = 0.02; interaction F (5, 85) = 0.3746, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 Kif3a‐deleted p = 0.01.
(f) Graphical representation of hindlimb stride time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 5.0, p < 0.001; genotype F (1, 85) = 1.0, p = 0.3; interaction F (5, 85) = 0.48, p = 0.8. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.02, P57‐1 v P57 + 150 Kif3a‐deleted p = 0.05, and P57 + 60 v P57 + 150 p = 0.04.
ACKNOWLEDGMENTS
We thank Judith Paridaen and Wieland Huttner (Max‐Planck Institute of Molecular Cell Biology and Genetics, Germany) for providing the Arl13b‐GFP plasmid. We thank Prof. William Richardson (University College London) for the kind gift of Sox10‐GFP‐DTA transgenic mice. We thank our colleagues at the University of Tasmania for their assistance with animal care and genotyping, as well as constructive feedback and suggestions for improvement of this manuscript.
Cullen CL, O'Rourke M, Beasley SJ, et al. Kif3a deletion prevents primary cilia assembly on oligodendrocyte progenitor cells, reduces oligodendrogenesis and impairs fine motor function. Glia. 2021;69:1184–1203. 10.1002/glia.23957
Carlie L. Cullen, Megan O'Rourke, and Shannon J. Beasley contributed equally to this manuscript.
Funding information Australian Research Council, Grant/Award Number: DP180101494; Medical Research Future Fund, Grant/Award Number: EPCD0000008; Multiple Sclerosis Research Australia, Grant/Award Numbers: 11014, 15054, 16105, 17007; National Health and Medical Research Council, Grant/Award Numbers: 1030939, 1045240, 1077792, 1139041
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Afonso, C. , & Henrique, D. (2006). PAR3 acts as a molecular organizer to define the apical domain of chick neuroepithelial cells. Journal of Cell Science, 119(Pt 20), 4293–4304. 10.1242/jcs.03170 [DOI] [PubMed] [Google Scholar]
- Alvarez‐Satta, M. , & Matheu, A. (2018). Primary cilium and glioblastoma. Therapeutic Advances in Medical Oncology, 10, 1758835918801169. 10.1177/1758835918801169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador‐Arjona, A. , Elliott, J. , Miller, A. , Ginbey, A. , Pazour, G. J. , Enikolopov, G. , … Terskikh, A. V. (2011). Primary cilia regulate proliferation of amplifying progenitors in adult hippocampus: Implications for learning and memory. The Journal of Neuroscience, 31(27), 9933–9944. 10.1523/JNEUROSCI.1062-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arellano, J. I. , Guadiana, S. M. , Breunig, J. J. , Rakic, P. , & Sarkisian, M. R. (2012). Development and distribution of neuronal cilia in mouse neocortex. The Journal of Comparative Neurology, 520(4), 848–873. 10.1002/cne.22793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auderset, L. , Cullen, C. L. , & Young, K. M. (2016). Low density lipoprotein‐receptor related protein 1 is differentially expressed by neuronal and glial populations in the developing and mature mouse central nervous system. PLoS One, 11(6), e0155878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer, S. , Dussert, A. , Collu, G. M. , Benitez, E. , Iomini, C. , & Mlodzik, M. (2015). Components of Intraflagellar transport complex a function independently of the cilium to regulate canonical Wnt signaling in drosophila. Developmental Cell, 34(6), 705–718. 10.1016/j.devcel.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat, M. T. , Humke, E. W. , & Scott, M. P. (2013). Kif3a is necessary for initiation and maintenance of medulloblastoma. Carcinogenesis, 34(6), 1382–1392. 10.1093/carcin/bgt041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres, B. A. , Lazar, M. A. , & Raff, M. C. (1994). A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development, 120(5), 1097–1108. [DOI] [PubMed] [Google Scholar]
- Bhogaraju, S. , Cajanek, L. , Fort, C. , Blisnick, T. , Weber, K. , Taschner, M. , … Lorentzen, E. (2013). Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science, 341(6149), 1009–1012. 10.1126/science.1240985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian, J. , Zheng, J. , Li, S. , Luo, L. , & Ding, F. (2016). Sequential differentiation of embryonic stem cells into neural epithelial‐like stem cells and Oligodendrocyte progenitor cells. PLoS One, 11(5), e0155227. 10.1371/journal.pone.0155227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy, J. D. , Emery, B. , Kaushal, A. , Foo, L. C. , Zamanian, J. L. , Christopherson, K. S. , … Barres, B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. The Journal of Neuroscience, 28(1), 264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, C. S. , Vogel, T. W. , Zhang, Q. , Seo, S. , Swiderski, R. E. , Moninger, T. O. , … Sheffield, V. C. (2012). Abnormal development of NG2+PDGFR‐alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nature Medicine, 18(12), 1797–1804. 10.1038/nm.2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary, T. , Larkins, C. E. , & Anderson, K. V. (2007). The graded response to sonic hedgehog depends on cilia architecture. Developmental Cell, 12(5), 767–778. 10.1016/j.devcel.2007.03.004 [DOI] [PubMed] [Google Scholar]
- Chen, J. , & Zhang, M. (2013). The Par3/Par6/aPKC complex and epithelial cell polarity. Experimental Cell Research, 319(10), 1357–1364. 10.1016/j.yexcr.2013.03.021 [DOI] [PubMed] [Google Scholar]
- Clarke, L. E. , Young, K. M. , Hamilton, N. B. , Li, H. , Richardson, W. D. , & Attwell, D. (2012). Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse. The Journal of Neuroscience, 32(24), 8173–8185. 10.1523/JNEUROSCI.0928-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit, K. C. , Aanstad, P. , Singla, V. , Norman, A. R. , Stainier, D. Y. , & Reiter, J. F. (2005). Vertebrate smoothened functions at the primary cilium. Nature, 437(7061), 1018–1021. 10.1038/nature04117 [DOI] [PubMed] [Google Scholar]
- Corbit, K. C. , Shyer, A. E. , Dowdle, W. E. , Gaulden, J. , Singla, V. , Chen, M. H. , … Reiter, J. F. (2008). Kif3a constrains beta‐catenin‐dependent Wnt signalling through dual ciliary and non‐ciliary mechanisms. Nature Cell Biology, 10(1), 70–76. 10.1038/ncb1670 [DOI] [PubMed] [Google Scholar]
- Cortellino, S. , Wang, C. , Wang, B. , Bassi, M. R. , Caretti, E. , Champeval, D. , … Bellacosa, A. (2009). Defective ciliogenesis, embryonic lethality and severe impairment of the sonic hedgehog pathway caused by inactivation of the mouse complex a intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Developmental Biology, 325(1), 225–237. 10.1016/j.ydbio.2008.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha‐Ferreira, I. , Bento, I. , & Bettencourt‐Dias, M. (2009). From zero to many: Control of centriole number in development and disease. Traffic, 10(5), 482–498. 10.1111/j.1600-0854.2009.00905.x [DOI] [PubMed] [Google Scholar]
- Dai, Z. M. , Sun, S. , Wang, C. , Huang, H. , Hu, X. , Zhang, Z. , … Qiu, M. (2014). Stage‐specific regulation of oligodendrocyte development by Wnt/beta‐catenin signaling. The Journal of Neuroscience, 34(25), 8467–8473. 10.1523/JNEUROSCI.0311-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debec, A. , Sullivan, W. , & Bettencourt‐Dias, M. (2010). Centrioles: Active players or passengers during mitosis? Cellular and Molecular Life Sciences, 67(13), 2173–2194. 10.1007/s00018-010-0323-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen, P. G. , Delling, M. , Vien, T. N. , & Clapham, D. E. (2013). Direct recording and molecular identification of the calcium channel of primary cilia. Nature, 504(7479), 315–318. 10.1038/nature12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling, M. , DeCaen, P. G. , Doerner, J. F. , Febvay, S. , & Clapham, D. E. (2013). Primary cilia are specialized calcium signalling organelles. Nature, 504(7479), 311–314. 10.1038/nature12833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou, L. , Simon, C. , Kirchhoff, F. , Takebayashi, H. , & Gotz, M. (2008). Progeny of Olig2‐expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. The Journal of Neuroscience, 28(41), 10434–10442. 10.1523/JNEUROSCI.2831-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger, N. H. , Macklin, W. B. , & Popko, B. (2003). Inducible site‐specific recombination in myelinating cells. Genesis, 35(1), 63–72. 10.1002/gene.10154 [DOI] [PubMed] [Google Scholar]
- Doobin, D. J. , Kemal, S. , Dantas, T. J. , & Vallee, R. B. (2016). Severe NDE1‐mediated microcephaly results from neural progenitor cell cycle arrests at multiple specific stages. Nature Communications, 7, 12551. 10.1038/ncomms12551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas, J. C. , Cuellar, T. L. , Scholze, A. , Ason, B. , Ibrahim, A. , Emery, B. , … Barres, B. A. (2010). Dicer1 and miR‐219 are required for normal oligodendrocyte differentiation and myelination. Neuron, 65(5), 597–611. 10.1016/j.neuron.2010.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguether, T. , & Hahne, M. (2018). Mixed signals from the cell's antennae: Primary cilia in cancer. EMBO Reports, 19(11), e46589. 10.15252/embr.201846589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon‐Urrutia, P. , Carrasco, C. M. , Lois, P. , Palma, V. , & Roth, A. D. (2015). Shh signaling through the primary cilium modulates rat Oligodendrocyte differentiation. PLoS One, 10(7), e0133567. 10.1371/journal.pone.0133567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, S. , Hurd, T. W. , Liu, C. J. , Straight, S. W. , Weimbs, T. , Hurd, E. A. , … Margolis, B. (2004). Polarity proteins control ciliogenesis via kinesin motor interactions. Current Biology, 14(16), 1451–1461. 10.1016/j.cub.2004.08.025 [DOI] [PubMed] [Google Scholar]
- Fancy, S. P. , Baranzini, S. E. , Zhao, C. , Yuk, D. I. , Irvine, K. A. , Kaing, S. , … Rowitch, D. H. (2009). Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes & Development, 23(13), 1571–1585. 10.1101/gad.1806309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferent, J. , Zimmer, C. , Durbec, P. , Ruat, M. , & Traiffort, E. (2013). Sonic hedgehog signaling is a positive oligodendrocyte regulator during demyelination. The Journal of Neuroscience, 33(5), 1759–1772. 10.1523/JNEUROSCI.3334-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante, M. I. , Zullo, A. , Barra, A. , Bimonte, S. , Messaddeq, N. , Studer, M. , … Franco, B. (2006). Oral‐facial‐digital type I protein is required for primary cilia formation and left‐right axis specification. Nature Genetics, 38(1), 112–117. 10.1038/ng1684 [DOI] [PubMed] [Google Scholar]
- Gazea, M. , Tasouri, E. , Tolve, M. , Bosch, V. , Kabanova, A. , Gojak, C. , … Blaess, S. (2016). Primary cilia are critical for sonic hedgehog‐mediated dopaminergic neurogenesis in the embryonic midbrain. Developmental Biology, 409(1), 55–71. 10.1016/j.ydbio.2015.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz, S. C. , & Anderson, K. V. (2010). The primary cilium: A signalling Centre during vertebrate development. Nature Reviews. Genetics, 11(5), 331–344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorivodsky, M. , Mukhopadhyay, M. , Wilsch‐Braeuninger, M. , Phillips, M. , Teufel, A. , Kim, C. , … Westphal, H. (2009). Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Developmental Biology, 325(1), 24–32. 10.1016/j.ydbio.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J. , Otis, J. M. , Higginbotham, H. , Monckton, C. , Cheng, J. , Asokan, A. , … Anton, E. S. (2017). Primary cilia signaling shapes the development of Interneuronal connectivity. Developmental Cell, 42(3), 286–300 e284. 10.1016/j.devcel.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A. , Tsuchiya, Y. , Ohta, M. , Shiratsuchi, G. , & Kitagawa, D. (2017). NEK7 is required for G1 progression and procentriole formation. Molecular Biology of the Cell, 28(15), 2123–2134. 10.1091/mbc.E16-09-0643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, T. G. , Klinghoffer, R. A. , Corrin, P. D. , & Soriano, P. (2003). Evolutionary divergence of platelet‐derived growth factor alpha receptor signaling mechanisms. Molecular and Cellular Biology, 23(11), 4013–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, Y. G. , Kim, H. J. , Dlugosz, A. A. , Ellison, D. W. , Gilbertson, R. J. , & Alvarez‐Buylla, A. (2009). Dual and opposing roles of primary cilia in medulloblastoma development. Nature Medicine, 15(9), 1062–1065. 10.1038/nm.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft, C. J. , Banizs, B. , Aydin‐Son, Y. , Zhang, Q. , Michaud, E. J. , & Yoder, B. K. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genetics, 1(4), e53. 10.1371/journal.pgen.0010053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham, H. , Guo, J. , Yokota, Y. , Umberger, N. L. , Su, C. Y. , Li, J. , … Anton, E. S. (2013). Arl13b‐regulated cilia activities are essential for polarized radial glial scaffold formation. Nature Neuroscience, 16(8), 1000–1007. 10.1038/nn.3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R. A. , Li, A. M. , & Grutzendler, J. (2018). Lifelong cortical myelin plasticity and age‐related degeneration in the live mammalian brain. Nature Neuroscience, 21(5), 683–695. 10.1038/s41593-018-0120-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirrlinger, J. , & Nave, K. A. (2014). Adapting brain metabolism to myelination and long‐range signal transduction. Glia, 62(11), 1749–1761. 10.1002/glia.22737 [DOI] [PubMed] [Google Scholar]
- Hudish, L. I. , Blasky, A. J. , & Appel, B. (2013). miR‐219 regulates neural precursor differentiation by direct inhibition of apical par polarity proteins. Developmental Cell, 27(4), 387–398. 10.1016/j.devcel.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudish, L. I. , Galati, D. F. , Ravanelli, A. M. , Pearson, C. G. , Huang, P. , & Appel, B. (2016). miR‐219 regulates neural progenitors by dampening apical par protein‐dependent hedgehog signaling. Development, 143(13), 2292–2304. 10.1242/dev.137844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, E. G. , Kang, S. H. , Fukaya, M. , & Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self‐repulsion to achieve homeostasis in the adult brain. Nature Neuroscience, 16(6), 668–676. 10.1038/nn.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, E. G. , Orthmann‐Murphy, J. L. , Langseth, A. J. , & Bergles, D. E. (2018). Myelin remodeling through experience‐dependent oligodendrogenesis in the adult somatosensory cortex. Nature Neuroscience, 21(5), 696–706. 10.1038/s41593-018-0121-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, P. K. (2018). EZH2 inactivates primary cilia to activate Wnt and drive melanoma. Cancer Cell, 34(1), 3–5. 10.1016/j.ccell.2018.06.011 [DOI] [PubMed] [Google Scholar]
- Jenks, A. D. , Vyse, S. , Wong, J. P. , Kostaras, E. , Keller, D. , Burgoyne, T. , … Tanos, B. E. (2018). Primary cilia mediate diverse kinase inhibitor resistance mechanisms in cancer. Cell Reports, 23(10), 3042–3055. 10.1016/j.celrep.2018.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S. H. , Fukaya, M. , Yang, J. K. , Rothstein, J. D. , & Bergles, D. E. (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron, 68(4), 668–681. 10.1016/j.neuron.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara, K. , Miyoshi, K. , Murakami, S. , Miyazaki, I. , & Asanuma, M. (2014). Visualization of astrocytic primary cilia in the mouse brain by immunofluorescent analysis using the cilia marker Arl13b. Acta Medica Okayama, 68(6), 317–322. 10.18926/AMO/53020 [DOI] [PubMed] [Google Scholar]
- Kessaris, N. , Fogarty, M. , Iannarelli, P. , Grist, M. , Wegner, M. , & Richardson, W. D. (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nature Neuroscience, 9(2), 173–179. 10.1038/nn1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock, B. L. , & Perkins, B. D. (2014). The par‐PrkC polarity complex is required for cilia growth in zebrafish photoreceptors. PLoS One, 9(8), e104661. 10.1371/journal.pone.0104661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, T. , Brown, J. M. , Bellve, K. , Craige, B. , Craft, J. M. , Fogarty, K. , … Witman, G. B. (2016). Together, the IFT81 and IFT74 N‐termini form the main module for intraflagellar transport of tubulin. Journal of Cell Science, 129(10), 2106–2119. 10.1242/jcs.187120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto, N. , Gu, Y. , Wang, J. , Janoschka, S. , Takemaru, K. , Levine, J. , & Ge, S. (2012). A role for primary cilia in glutamatergic synaptic integration of adult‐born neurons. Nature Neuroscience, 15(3), 399–405, S391. 10.1038/nn.3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Yi, S. , Won, M. , Song, Y. S. , Yi, H. S. , Park, Y. J. , … Shong, M. (2018). Loss‐of‐function of IFT88 determines metabolic phenotypes in thyroid cancer. Oncogene, 37(32), 4455–4474. 10.1038/s41388-018-0211-6 [DOI] [PubMed] [Google Scholar]
- Lee, Y. , Morrison, B. M. , Li, Y. , Lengacher, S. , Farah, M. H. , Hoffman, P. N. , … Rothstein, J. D. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature, 487(7408), 443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepanto, P. , Davison, C. , Casanova, G. , Badano, J. L. , & Zolessi, F. R. (2016). Characterization of primary cilia during the differentiation of retinal ganglion cells in the zebrafish. Neural Development, 11, 10. 10.1186/s13064-016-0064-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. , Hiesberger, T. , Cordes, K. , Sinclair, A. M. , Goldstein, L. S. , Somlo, S. , & Igarashi, P. (2003). Kidney‐specific inactivation of the KIF3A subunit of kinesin‐II inhibits renal ciliogenesis and produces polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America, 100(9), 5286–5291. 10.1073/pnas.0836980100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan, M. , Rosen, K. M. , Ito, S. , & Corfas, G. (2012). A critical period for social experience‐dependent oligodendrocyte maturation and myelination. Science, 337(6100), 1357–1360. 10.1126/science.1220845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, W. F. , & Rosenbaum, J. L. (2001). Intraflagellar transport balances continuous turnover of outer doublet microtubules: Implications for flagellar length control. The Journal of Cell Biology, 155(3), 405–414. 10.1083/jcb.200106141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek, J. R. , Liu, X. , Roberts, E. A. , Chui, D. , Marth, J. D. , Williams, D. S. , & Goldstein, L. S. (2000). Genetic evidence for selective transport of opsin and arrestin by kinesin‐II in mammalian photoreceptors. Cell, 102(2), 175–187. [DOI] [PubMed] [Google Scholar]
- Marszalek, J. R. , Ruiz‐Lozano, P. , Roberts, E. , Chien, K. R. , & Goldstein, L. S. (1999). Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin‐II. Proceedings of the National Academy of Sciences of the United States of America, 96(9), 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie, I. A. , Ohayon, D. , Li, H. , de Faria, J. P. , Emery, B. , Tohyama, K. , & Richardson, W. D. (2014). Motor skill learning requires active central myelination. Science, 346(6207), 318–322. 10.1126/science.1254960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchan, P. , Bribian, A. , Sanchez‐Camacho, C. , Lezameta, M. , Bovolenta, P. , & de Castro, F. (2007). Sonic hedgehog promotes the migration and proliferation of optic nerve oligodendrocyte precursors. Molecular and Cellular Neurosciences, 36(3), 355–368. 10.1016/j.mcn.2007.07.012 [DOI] [PubMed] [Google Scholar]
- Meyer, N. , Richter, N. , Fan, Z. , Siemonsmeier, G. , Pivneva, T. , Jordan, P. , … Kettenmann, H. (2018). Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Reports, 22(9), 2383–2394. 10.1016/j.celrep.2018.02.022 [DOI] [PubMed] [Google Scholar]
- Monnich, M. , Borgeskov, L. , Breslin, L. , Jakobsen, L. , Rogowski, M. , Doganli, C. , … Pedersen, L. B. (2018). CEP128 localizes to the subdistal appendages of the mother centriole and regulates TGF‐beta/BMP signaling at the primary cilium. Cell Reports, 22(10), 2584–2592. 10.1016/j.celrep.2018.02.043 [DOI] [PubMed] [Google Scholar]
- Noble, M. , Barnett, S. C. , Bogler, O. , Land, H. , Wolswijk, G. , & Wren, D. (1990). Control of division and differentiation in oligodendrocyte‐type‐2 astrocyte progenitor cells. CIBA Foundation Symposium, 150, 227–243; discussion 244‐229. 10.1002/9780470513927.ch14 [DOI] [PubMed] [Google Scholar]
- O'Rourke, M. , Cullen, C. L. , Auderset, L. , Pitman, K. A. , Achatz, D. , Gasperini, R. , & Young, K. M. (2016). Evaluating tissue‐specific recombination in a Pdgfrα‐CreERT2 transgenic mouse line. PLoS One, 11(9), e0162858. 10.1371/journal.pone.0162858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega, J. A. , Radonjic, N. V. , & Zecevic, N. (2013). Sonic hedgehog promotes generation and maintenance of human forebrain Olig2 progenitors. Frontiers in Cellular Neuroscience, 7, 254. 10.3389/fncel.2013.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paridaen, J. T. , Wilsch‐Brauninger, M. , & Huttner, W. B. (2013). Asymmetric inheritance of centrosome‐associated primary cilium membrane directs ciliogenesis after cell division. Cell, 155(2), 333–344. 10.1016/j.cell.2013.08.060 [DOI] [PubMed] [Google Scholar]
- Pepper, R. E. , Pitman, K. A. , Cullen, C. L. , & Young, K. M. (2018). How do cells of the Oligodendrocyte lineage affect neuronal circuits to influence motor function, memory and mood? Frontiers in Cellular Neuroscience, 12, 399. 10.3389/fncel.2018.00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle, N. P. , Yu, W. P. , Guthrie, S. , Roelink, H. , Lumsden, A. , Peterson, A. C. , & Richardson, W. D. (1996). Determination of neuroepithelial cell fate: Induction of the oligodendrocyte lineage by ventral midline cells and sonic hedgehog. Developmental Biology, 177(1), 30–42. 10.1006/dbio.1996.0142 [DOI] [PubMed] [Google Scholar]
- Psachoulia, K. , Jamen, F. , Young, K. M. , & Richardson, W. D. (2009). Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biology, 5(3–4), 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva, E. N. , Jablonski, S. A. , Hartman, T. R. , Henske, E. P. , & Golemis, E. A. (2007). HEF1‐dependent aurora A activation induces disassembly of the primary cilium. Cell, 129(7), 1351–1363. 10.1016/j.cell.2007.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff, M. C. , Abney, E. R. , & Fok‐Seang, J. (1985). Reconstitution of a developmental clock in vitro: A critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell, 42(1), 61–69. 10.1016/s0092-8674(85)80101-x [DOI] [PubMed] [Google Scholar]
- Rivers, L. E. , Young, K. M. , Rizzi, M. , Jamen, F. , Psachoulia, K. , Wade, A. , … Richardson, W. D. (2008). PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nature Neuroscience, 11(12), 1392–1401. 10.1038/nn.2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi, R. , Milenkovic, L. , & Scott, M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science, 317(5836), 372–376. 10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- Rosenbaum, J. L. , & Child, F. M. (1967). Flagellar regeneration in protozoan flagellates. The Journal of Cell Biology, 34(1), 345–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, I. , & Dynlacht, B. D. (2016). Cilium assembly and disassembly. Nature Cell Biology, 18(7), 711–717. 10.1038/ncb3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, L. , Clement, C. A. , Teilmann, S. C. , Pazour, G. J. , Hoffmann, E. K. , Satir, P. , & Christensen, S. T. (2005). PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Current Biology, 15(20), 1861–1866. 10.1016/j.cub.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Schneider, L. , Stock, C. M. , Dieterich, P. , Jensen, B. H. , Pedersen, L. B. , Satir, P. , … Pedersen, S. F. (2009). The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR‐alpha in the primary cilium. The Journal of Cell Biology, 185(1), 163–176. 10.1083/jcb.200806019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, S. , Gruart, A. , Grade, S. , Zhang, Y. , Kroger, S. , Kirchoff, F. , … Dimou, L. (2016). Decrease in newly generated oligodendrocytes leads to motor dysfunctions and changed myelin structures that can be rescued by transplanted cells. Glia, 64, 2201–2218. [DOI] [PubMed] [Google Scholar]
- Schock, E. N. , & Brugmann, S. A. (2017). Discovery, diagnosis, and etiology of craniofacial Ciliopathies. Cold Spring Harbor Perspectives in Biology, 9(9), a028258. 10.1101/cshperspect.a028258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos, J. , Togawa, A. , Maday, S. , Hull, M. , Pypaert, M. , Cantley, L. , … Mellman, I. (2007). Par3 functions in the biogenesis of the primary cilium in polarized epithelial cells. The Journal of Cell Biology, 179(6), 1133–1140. 10.1083/jcb.200709111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, S. , Xue, H. , Mattson, M. P. , & Rao, M. S. (2007). Stage‐dependent Olig2 expression in motor neurons and oligodendrocytes differentiated from embryonic stem cells. Stem Cells and Development, 16(1), 131–141. 10.1089/scd.2006.0023 [DOI] [PubMed] [Google Scholar]
- Sorokin, S. P. (1968). Centriole formation and ciliogenesis. Aspen Emphysema Conference, 11, 213–216. [PubMed] [Google Scholar]
- Spassky, N. , Han, Y. G. , Aguilar, A. , Strehl, L. , Besse, L. , Laclef, C. , … Alvarez‐Buylla, A. (2008). Primary cilia are required for cerebellar development and Shh‐dependent expansion of progenitor pool. Developmental Biology, 317(1), 246–259. 10.1016/j.ydbio.2008.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas, S. , Watanabe, T. , Lin, C. S. , William, C. M. , Tanabe, Y. , Jessell, T. M. , & Costantini, F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology, 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterpka, A. , & Chen, X. (2018). Neuronal and astrocytic primary cilia in the mature brain. Pharmacological Research, 137, 114–121. 10.1016/j.phrs.2018.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekki‐Kessaris, N. , Woodruff, R. , Hall, A. C. , Gaffield, W. , Kimura, S. , Stiles, C. D. , … Richardson, W. D. (2001). Hedgehog‐dependent oligodendrocyte lineage specification in the telencephalon. Development, 128(13), 2545–2554. [DOI] [PubMed] [Google Scholar]
- Tong, C. K. , Han, Y. G. , Shah, J. K. , Obernier, K. , Guinto, C. D. , & Alvarez‐Buylla, A. (2014). Primary cilia are required in a unique subpopulation of neural progenitors. Proceedings of the National Academy of Sciences of the United States of America, 111(34), 12438–12443. 10.1073/pnas.1321425111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, B. D. , Nishiyama, A. , Cheng, D. , & Macklin, W. B. (1997). Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. Journal of Cell Biology, 137(2), 459–468. 10.1083/jcb.137.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, H. H. , Niu, J. , Munji, R. , Davalos, D. , Chang, J. , Zhang, H. , … Fancy, S. P. (2016). Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science, 351(6271), 379–384. 10.1126/science.aad3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umberger, N. L. , & Caspary, T. (2015). Ciliary transport regulates PDGF‐AA/alphaalpha signaling via elevated mammalian target of rapamycin signaling and diminished PP2A activity. Molecular Biology of the Cell, 26(2), 350–358. 10.1091/mbc.E14-05-0952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente, E. M. , Rosti, R. O. , Gibbs, E. , & Gleeson, J. G. (2014). Primary cilia in neurodevelopmental disorders. Nature Reviews. Neurology, 10(1), 27–36. 10.1038/nrneurol.2013.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vion, A. C. , Alt, S. , Klaus‐Bergmann, A. , Szymborska, A. , Zheng, T. , Perovic, T. , … Gerhardt, H. (2018). Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. The Journal of Cell Biology, 217(5), 1651–1665. 10.1083/jcb.201706151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G. , Chen, Q. , Zhang, X. , Zhang, B. , Zhuo, X. , Liu, J. , … Zhang, C. (2013). PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. Journal of Cell Science, 126(Pt 6), 1355–1365. 10.1242/jcs.114918 [DOI] [PubMed] [Google Scholar]
- Wang, H. , Moyano, A. L. , Ma, Z. , Deng, Y. , Lin, Y. , Zhao, C. , … Lu, Q. R. (2017). miR‐219 cooperates with miR‐338 in myelination and promotes myelin repair in the CNS. Developmental Cell, 40(6), 566–582 e565. 10.1016/j.devcel.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Bolos, M. , Clark, R. , Cullen, C. L. , Southam, K. A. , Foa, L. , … Young, K. M. (2016). Amyloid beta precursor protein regulates neuron survival and maturation in the adult mouse brain. Molecular and Cellular Neurosciences, 77, 21–33. 10.1016/j.mcn.2016.09.002 [DOI] [PubMed] [Google Scholar]
- Wheway, G. , Nazlamova, L. , & Hancock, J. T. (2018). Signaling through the primary cilium. Frontiers in Cell and Development Biology, 6, 8. 10.3389/fcell.2018.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willaredt, M. A. , Hasenpusch‐Theil, K. , Gardner, H. A. , Kitanovic, I. , Hirschfeld‐Warneken, V. C. , Gojak, C. P. , … Tucker, K. L. (2008). A crucial role for primary cilia in cortical morphogenesis. The Journal of Neuroscience, 28(48), 12887–12900. 10.1523/JNEUROSCI.2084-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, S. L. , Wilson, J. P. , Wang, C. , Wang, B. , & McConnell, S. K. (2012). Primary cilia and Gli3 activity regulate cerebral cortical size. Developmental Neurobiology, 72(9), 1196–1212. 10.1002/dneu.20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey, M. , & O'Toole, E. (2014). Centriole structure. Philosophical transactions of the Royal Society of London. Series B, Biological Sciences, 369(1650), 20130457. 10.1098/rstb.2013.0457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. T. , Chen, H. Y. , & Tang, T. K. (2018). Myosin‐Va is required for preciliary vesicle transportation to the mother centriole during ciliogenesis. Nature Cell Biology, 20(2), 175–185. 10.1038/s41556-017-0018-7 [DOI] [PubMed] [Google Scholar]
- Xiao, L. , Ohayon, D. , McKenzie, I. A. , Sinclair‐Wilson, A. , Wright, J. L. , Fudge, A. D. , … Richardson, W. D. (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor‐skill learning. Nature Neuroscience, 19(9), 1210–1217. 10.1038/nn.4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Zhang, Y. , Wei, Q. , Huang, Y. , Hu, J. , & Ling, K. (2016). Phosphatidylinositol phosphate kinase PIPKIgamma and phosphatase INPP5E coordinate initiation of ciliogenesis. Nature Communications, 7, 10777. 10.1038/ncomms10777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, F. , Chen, Y. , Hoang, T. , Montgomery, R. L. , Zhao, X. H. , Bu, H. , … Lu, Q. R. (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta‐catenin‐TCF interaction. Nature Neuroscience, 12(7), 829–838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, K. , & Takeda, S. (2012). Hedgehog signaling regulates myelination in the peripheral nervous system through primary cilia. Differentiation, 83(2), S78–S85. 10.1016/j.diff.2011.10.006 [DOI] [PubMed] [Google Scholar]
- Youn, Y. H. , & Han, Y. G. (2018). Primary cilia in brain development and diseases. The American Journal of Pathology, 188(1), 11–22. 10.1016/j.ajpath.2017.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, K. M. , Psachoulia, K. , Tripathi, R. B. , Dunn, S.‐J. , Cossell, L. , Attwell, D. , … Richardson, W. D. (2013). Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron, 77(5), 873–885. 10.1016/j.neuron.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Chen, K. , Sloan, S. A. , Bennett, M. L. , Scholze, A. R. , O'Keeffe, S. , … Wu, J. Q. (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience, 34(36), 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , He, X. , Han, X. , Yu, Y. , Ye, F. , Chen, Y. , … Lu, Q. R. (2010). MicroRNA‐mediated control of oligodendrocyte differentiation. Neuron, 65(5), 612–626. 10.1016/j.neuron.2010.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Hill, R. A. , Dietrich, D. , Komitova, M. , Suzuki, R. , & Nishiyama, A. (2011). Age‐dependent fate and lineage restriction of single NG2 cells. Development, 138(4), 745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg, D. , Debbache, J. , Pena‐Hernandez, R. , Antunes, A. T. , Schaefer, S. M. , Cheng, P. F. , … Sommer, L. (2018). EZH2‐mediated primary cilium deconstruction drives metastatic melanoma formation. Cancer Cell, 34(1), 69–84 e14. 10.1016/j.ccell.2018.06.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Generation of a CMV‐Arl13b‐mCherry plasmid to label primary cilia with a red fluorescent protein
(a) A schematic of the cloning strategy used to generate a CMV‐Arl13b‐mCherry plasmid to mCherry‐label primary cilia. The Arl13b fragment was excised from the CMV‐eGFP‐Arl13b plasmid by performing a restriction digest with the enzymes Pst1 and Kpn1, followed by gel purification of the ~1,300 bp band. CMV‐mCherry‐C1 vector (Clontech, 632,524), which has a multiple closing site (MCS) at the C‐terminal end of the mCherry sequence was linearized by performing a restriction digest with Pst1 and Kpn1. Linearized DNA was treated with Antarctic Phosphatase (New England Biolabs, Ipswich, MA) as per the manufacturer's instructions. The Arl13b DNA fragment was ligated into the linearized CMV‐mCherry‐C1 plasmid and the resulting CMV‐Arl13b‐mCherry plasmid transformed into E. coli.
(b) Image showing cytoplasmic expression of mCherry in HEK293 cells transfected with CMV‐C1‐mCherry.
(c) HEK293 cells transfected with CMV‐Arl13b‐mCherry have a low level of cytoplasmic mCherry expression. A subset of transfected cells has a single point of intense fluorescence, where the Arl13‐b‐mCherry fusion protein is expressed at the primary cilium (arrow heads).
Figure S2 Deleting Kif3a from adult OPCs reduces the addition of CC1 + oligodendrocytes to the mouse brain
(a‐b) The density of YFP+ OLIG2+ PDGFRα‐neg new oligodendrocytes in the corpus callosum (CC; a) and motor cortex (MC; b) of control (Pdgfrα‐CreER™:: Rosa26‐YFP) and Kif3a‐deleted (Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice from P57 + 7 to P57 + 45. Data are presented as mean ± SD for n = 3–4 mice per genotype at each timepoint. Two‐way ANOVA for the CC: time F (3, 17) = 655.5, p < 0.0001; genotype F (1, 17) = 382.7, p < 0.0001; interaction F (3, 17) = 76.6, p < 0.0001. Two‐way ANOVA for the MC: time F (3, 17) = 158.0, p < 0.0001; genotype F (1, 17) = 191.2, p < 0.0001; interaction F (3, 17) = 29.06, p < 0.0001. Bonferroni post‐test *** p < 0.001 and **** p < 0.0001.
(c‐d) Compressed confocal image of YFP (green), CC1 (red) and HST (blue) labeling in the CC of P57 + 45 control (c) and Kif3a‐deleted (d; Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice.
(e‐f) Compressed confocal image of YFP (green), CC1 (red) and HST (blue) labeling in the MC of P57 + 45 control (e) and Kif3a‐deleted (f; Pdgfrα‐CreER™:: Rosa26‐YFP:: Kif3a fl/fl) mice.
(g) The proportion (%) of YFP+ cells in the CC and MC that have differentiated into CC1+ oligodendrocytes by P57 + 45. Data are presented as mean ± SD for n = 3 mice per genotype. Two‐way ANOVA: region F (1, 8) = 258.2, p < 0.0001; genotype F (1, 8) = 132.6, p < 0.0001; interaction F (1, 8) = 11.64, p = 0.009. Bonferroni post‐test *** p < 0.001 and **** p < 0.0001.
Solid arrowheads denote newborn oligodendrocytes (YFP+ CC1+ cells). Arrows denote YFP+ CC1‐neg cells (presumptive OPCs). Scale bars represent 20 μm.
Figure S3 Kif3a‐deletion does not impact hind limb gait parameters
(a) Graphical representation of hindlimb stride length for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.3, p = 0.002; genotype F (1, 85) = 0.4, p = 0.5; interaction F (5, 85) = 0.22, p = 0.9.
(b) Graphical representation of hindlimb stride frequency for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 5.4, p < 0.001; genotype F (1, 85) = 0.56, p = 0.5; interaction F (5, 85) = 0.27, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 control, p < 0.05, and P57‐1 v P57 + 150 or P57 + 60 v P57 + 150 Kif3a‐deleted, p = 0.02.
(c) Graphical representation of forelimb swing time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 6.308, p < 0.0001; genotype F (1, 85) = 8.970, p = 0.004; interaction F (5, 85) = 0.7138, p = 0.6. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.03 and P57‐1 v P57 + 150 Kif3a‐deleted p = 0.003.
(d) Graphical representation of hindlimb swing time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 3.4, p = 0.007; genotype F (1, 85) = 0.14, p = 0.7; interaction F (5, 85) = 0.3, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.02.
(e) Graphical representation of forelimb stride time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 4.150, p = 0.002; genotype F (1, 85) = 5.579, p = 0.02; interaction F (5, 85) = 0.3746, p = 0.9. Bonferroni post‐test: P57‐1 v P57 + 150 Kif3a‐deleted p = 0.01.
(f) Graphical representation of hindlimb stride time for control and Kif3a‐deleted mice running on the DigiGait™ treadmill at 22 cm/s. Data is expressed as mean ± SEM for n = 6–10 mice per genotype at each timepoint. Two‐way ANOVA: time F (5, 85) = 5.0, p < 0.001; genotype F (1, 85) = 1.0, p = 0.3; interaction F (5, 85) = 0.48, p = 0.8. Bonferroni post‐test: P57‐1 v P57 + 150 control p = 0.02, P57‐1 v P57 + 150 Kif3a‐deleted p = 0.05, and P57 + 60 v P57 + 150 p = 0.04.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.