

Abstract
The interplay between life sciences and advancing technology drives a continuous cycle of chemical data growth; these data are most often stored in open or partially open databases. In parallel, many different types of algorithms are being developed to manipulate these chemical objects and associated bioactivity data. Virtual screening methods are among the most popular computational approaches in pharmaceutical research. Today, user-friendly web-based tools are available to help scientists perform virtual screening experiments. This article provides an overview of internet resources enabling and supporting chemical biology and early drug discovery with a main emphasis on web servers dedicated to virtual ligand screening and small-molecule docking. This survey first introduces some key concepts and then presents recent and easily accessible virtual screening and related target-fishing tools as well as briefly discusses case studies enabled by some of these web services. Notwithstanding further improvements, already available web-based tools not only contribute to the design of bioactive molecules and assist drug repositioning but also help to generate new ideas and explore different hypotheses in a timely fashion while contributing to teaching in the field of drug development.
Keywords: virtual screening, in silico drug design, chemical biology
Introduction
The development of new drugs is characterized by high cost, long development cycles and low success rate [1–3]. The field faces numerous challenges but at the same time new opportunities are appearing. Numerous diseases tend to be much more complex than originally thought while the potential of genomic medicine to revolutionize health care will take much more time than anticipated [4, 5]. Despite important progress, there are still many medical conditions for which treatments are inadequate or missing. Fortunately, experimental high-throughput technologies generate a significant amount of data that facilitate, in some cases, the understanding of molecular mechanisms involved in the health and disease states and the development of novel drugs. However, with big data comes also novel challenges that will need to be addressed so as to fully benefit from advances in artificial intelligence, virtual screening and machine learning approaches [6–18].
There are different types of therapeutic molecules (e.g. small chemical compounds and biologics) and here we will focus on small molecules although many web services or online tools are also available to assist the development of biologics such as therapeutic antibodies and peptides. The traditional drug discovery process, in simple terms, encompasses several stages, starting with identification of targets or molecular pathways likely involved in the investigated disease and the search for hit compounds that modulate the putative target(s) and pathway(s) [19] (NB: the steps are different when the studies start with phenotypic screening [20, 21]. Then, various properties of the initial hit compounds have to be optimized (e.g. affinity, solubility, toxicity at the required dose…, usually performed during the so-called hit to lead step and beyond) up to the identification of clinical candidates. Different experimental approaches/skills are required to identify and optimize these early stage compounds (screening, biophysical methods, biochemistry, medicinal chemistry…). Considering only the initial discovery and preclinical studies (and thus not mentioning the clinical phases), the first steps of the process are known to be time consuming (5–6 years) and expensive (~$430 millions) [2, 22, 23]. To assist this highly challenging process, various types of in silico approaches are being used, depending on the available data and the stage of the discovery process.
In silico approaches contribute to drug discovery [24–30] but many algorithms, often not yet integrated in commercial packages, could be very valuable for a given project but can be difficult to install, redevelop and/or use. To improve the usability of such computational resources, new in silico approaches are often implemented online. These web applications and/or databases help not only wet-lab researchers but also computational experts to quickly integrate many different types of data and advanced drug design tools in their everyday research tasks. Some online tools allow users to create an account and manage their data. On most systems, data are automatically deleted within a few days. It should also be born in mind that uploading highly confidential data online can be risky and as such users may have to check with the web developers for potential security issues. Indeed, these recent years, one observes that new databases and algorithms that attempt to solve new questions and better address known or emerging drug discovery challenges are reported almost every week [31–44]. Of importance, these resources can also assist teaching, for example chemistry databases, online tools and freely available software packages are being used in different universities for that purpose [44–47].
In the present communication, among the different computational approaches that can be used in drug discovery, we essentially focus on recently reported and easily accessible virtual screening web servers. The first part of the review introduces the field of virtual screening and some related computational approaches that might be required prior to or after virtual screening experiments. We then discuss ligand-based virtual screening (LBVS) and structure-based virtual screening (SBVS) tools. Along the presentation, we mention, when documented (i.e. some tools are very new and have not yet been used by wet-lab scientists), some experimental case studies that made use of these web applications. The tools that we report are essentially peer-reviewed published methods with URLs tested in February 2020 (i.e. tools with broken links after several trials during 2–4 weeks are not described) and tools that we have identified by internet search. The list of tools might not be exhaustive yet the searches have been performed twice a week in PubMed and in several scientific journals during these last 10 years. Citations of the tools were investigated via Google Scholar. The URLs of the various tools are reported in three tables but are also stored online, updated on a regular basis and made available on the shortlist page at www.vls3d.com [39, 44].
Virtual ligand screening: key concepts and related tools
Virtual screening or virtual ligand screening, first coined in the literature in 1997 [48], is a computational technique that is used, in general, in the early stages of the drug discovery process, to search libraries of small molecules in order to identify chemical compounds that are likely to bind to one or several drug targets [49–59]. This type of computation can be conceived as a sort of experimental biochemical high-throughput screening (HTS) performed in silico [60, 61]. In general, virtual screening computations associated with interactive analysis of the data will generate a list of about top 30–500 compounds (or more) that will then need to be validated experimentally. The in silico methods are of course not without pitfalls [62, 63] and are generally used prior to or in parallel to experimental screening. As compound collections for experimental screening usually contain from about 50 000 (academic lab) to 5 million (big pharma) compounds, the time and cost required for a project can be significantly reduced with in silico screening, not only in terms of purchasing biological and chemical materials but also in terms of analysis of the HTS results (i.e. it can take several months to identify false-positives…) [64]. Further, as the chemical space is almost infinite [65, 66], virtual screening can explore novel regions of the chemical space and even molecules that are not yet synthesized but yet ‘medchem friendly’ [67, 68]. Key computational methods to identify hits and/or perform the first rounds of compound optimization can be subdivided in two broad categories, LBVS and SBVS approaches. In addition, several other computational approaches (a.k.a. in this review named ‘related tools’) can be used prior to or after in silico screening (e.g. investigation of absorption, distribution, metabolism, excretion and toxicity—ADME-Tox—properties, predictions of binding pocket, evaluation of flexibility via molecular simulations…) (Fig. 1).
Figure 1.
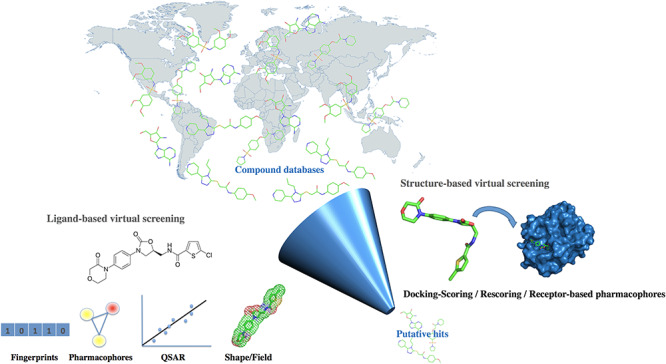
Virtual screening online. Several research laboratories worldwide provide online virtual screening services. In the present survey, the identified main countries offering online virtual screening services (active URLs in February 2020) are (by alphabetic order): Austria, Australia, Brazil, Canada, China, Czech Republic, Denmark, France, Germany, India, Israel, Italy, Japan, Poland, Republic of Ecuador, Republic of Korea, Saudi Arabia, Spain, Switzerland, The Netherlands, Taïwan, Turkey, the UK and the USA. The map chart was created with Mapchart.net. The main LBVS methods are 2D similarity search, pharmacophores, QSAR models and shape/field alignments while for SBVS, the approaches involve docking, scoring and rescoring with various methods including consensus scoring, or different flavors of free energy calculations.
For the first category (LBVS), 2D or 3D chemical structures or molecular descriptors of the known actives are used to retrieve other (‘similar’) compounds of interest in a database using different types of similarity measures or by seeking a common substructure or pharmacophore between the query molecule and the scanned libraries. The methods rely on the knowledge of small molecules (the queries) that bind to the target(s) of interest (for example obtained after mining annotated compound collections or after a first round of experimental screening). The main tools that are used in ligand-based screening are 2D molecular similarity approaches, 3D similarity searches (pharmacophore, molecular shapes colored or not by physicochemical properties, energy fields around the molecules such as electrostatic properties) and 2D/3D QSAR (quantitative structure–activity relationship) modeling. In 2D molecular similarity methods, the molecular fingerprint of known ligands that bind to a target is used to find molecules with similar fingerprints in the electronic libraries [69–73]. Various types of similarity or distance metrics can be used among which the Tanimoto coefficient values between 0 and 1. Here, the higher the threshold, the closer the compounds in the database are to the input query [74]. In ligand-based pharmacophore modeling (a pharmacophore is a molecular framework that defines the essential features responsible for the biological activity of a compound), usually performed in 3D, common and key structural features of the ligands that bind to a target are identified and used to do develop a model and to screen a compound collection with that model such as to identify compounds that match the model requirements [75–77]. Other 3D similarity methods involve shape similarity. Here again, the algorithms can be used for virtual screening but also in molecular target prediction, drug repositioning and scaffold hopping. A wide range of methods have been developed to describe the molecular shape and to determine the similarity between shapes (including electrostatic similarity or ‘force field’ colored shape similarity) [78–83]. Machine learning and QSAR modeling are computational methods that model the relationship between a set of selected features (e.g. molecular descriptors like fingerprint and/or various physicochemical properties) of the ligands that bind to a target and the corresponding biological activity effect. In contrast to traditional similarity searches, here the knowledge of true active and true inactive compounds is, in general, needed to develop statistical predictive models, although in some cases artificial decoys can be used to represent inactive molecules [13, 84–96]. Predictive machine learning models (e.g. classification, regression and clustering) can thus provide a list of compounds that is different or that complements the one obtained with, for instance, 2D or 3D similarity search [17, 97]. Of importance, different types of metrics can be used to evaluate statistical models and explanations about the applicability domain have to be provided to users [13, 86, 96]. As described in this section, most of these approaches are available online and in some cases, the same web servers can indeed offer different types of computations.
For the second category (SBVS), compounds (or fragments) from a database are docked into a binding site (or over the entire surface or in several binding sites or taking into account distance constraints such as in covalent docking) and are ranked using generally one (e.g. force field based, empirical or knowledge-based) scoring function [98–108].
The targets are in general proteins, but other macromolecules can also be investigated such as nucleic acids. Different types of postprocessing steps can be applied such as consensus scoring or consensus docking and scoring, rescoring with more rigorous binding free energy calculations or with machine learning scoring functions. Other types of rescoring approaches involve various types of molecular interaction fingerprints as the different ligands of a given target often share key molecular interaction patterns. In SBVS, no prior knowledge on known ligands hitting the investigated target is required, although, of course, knowledge about binders (e.g. known ligands with some affinity for the target of interest or cocrystallized compounds) helps to calibrate (or select) the best available computational method(s). De novo ligand design can be performed in 2D [109] but structure-based approaches tend to be used more and more as the amount of structural data increases. Thus in the structure-based category of tools, approaches such as fragment-based de novo ligand design are being used [110–113]. Receptor-based pharmacophore screening fits also in this category of structure-based screening (i.e. in this situation knowledge of the binding pocket is used in generating pharmacophore models) [75–77]. Other computations that are not SBVS per se can be used and combined with SBVS. For example, ligand may not need to be docked into a binding pocket but can be transposed (pocket matching algorithms) as some binding sites are very similar despite the fact that the 3D structures of the targets can be very different [35, 114–117]. Along the same line of reasoning, databases of comparative structure models of drug–target interactions can be built [118].
The structure-based methods are applicable when the 3D structure of the target has been determined with biophysical methods such as X-ray, cryo-electron microscopy and nuclear magnetic resonance, but homology models can also be used [119–125]. Molecular dynamics (or related approaches) are frequently used in structure-based screening to explore the likely poses of a compound, to rescore molecules, to investigate the importance of water molecules, the pathways of interaction (e.g. kinetics of binding, unbinding events) and to investigate target flexibility, explore binding pockets and/or discover cryptic pockets and to rationalize allosteric events [126–131]. In some conditions, ligand (including machine learning models) and SBVS can be combined such as, for instance, in the case of hierarchical screening with various types of ligand-based filtering steps followed by docking and rescoring [132–135].
Ligand-based and SBVS approaches are most often used to search libraries of compounds for molecules that are most likely to bind to a specific target. However, these approaches can also be used, with some modifications, to identify the most likely targets of a query molecule. One goal here is to try to predict the bioactivity or mechanism of action of compound, to, for instance, detect drug polypharmacology, predict potential adverse effects, help rationally design multitarget drugs or assist drug repositioning endeavors [25, 30, 136–141]. This is often referred in the literature to as target-fishing or in silico profiling. The popular approaches to perform target-fishing involve ligand-based or structure-based approaches. The simplest methods for target prediction are based on chemical similarity and make use of the known bioactivity of millions of small molecules stored in databases. In this case, the putative targets of a molecule can be predicted by identifying proteins with known ligands that are highly similar to the query compound. 2D similarity search, 3D search and machine learning approaches are available. It is also possible to dock a query compound into a small or large database of prepared receptor 3D structures. This process of target-fishing relies on inverse or reverse docking methods.
A first step prior to virtual screening computations usually involves gathering information about targets and compounds. Numerous databases are freely available and can be mined (e.g. target already drugged, novel targets, search for approved drugs, commercially available compounds, medicinal chemistry aware virtual compounds, natural products, small macrocycles, short peptides, toxic compounds, compounds reported in patents…). Three very recent reviews on chemistry databases [42, 142] and Omics and target databases [143] have just been reported and cover all the major databases that most users will need to embark in a virtual screening project.
Further, as mentioned above, for most receptor-based screening experiments, it is important to define (or refine) the likely binding pocket for the putative ligands. Often, this zone is known from the literature (e.g. a catalytic site) but, if not or in case the researchers need to screen outside these well-known binding cavities, different types of computational methods have been developed. Of importance, pocket with a point mutation identified in patients could be of major importance for drug discovery endeavors. The tools to predict, prepare, compare and explore putative binding pockets have been reviewed [115, 144–151]. Some recently (since 2017) reported web services dedicated to pockets or allowing the exploration of target plasticity are reported in Supplementary Table S1.
Most projects at some point will require the investigation of the ADME-Tox properties of the small molecules. Undesirable pharmacokinetic properties or potential toxicity or the presence of assay interfering chemical (sub)structures can be evaluated in silico. Recent reviews about some available in silico tools and the way these approaches are being used have been reported [34, 42, 152–167]. Different concepts are explored with these computational approaches. A first level of in silico prediction involves various types of physicochemical property filtering together with addition of flags or even removing molecules that contain some ‘risky’ structural alerts (i.e. either toxicity alerts or unwanted promiscuous binders and frequent hitters). Other approaches predict more specific toxicity endpoints. Further, once some initial hit compounds are obtained, optimization is usually required (e.g. increasing the affinity to the target while improving solubility and reducing potential binding to antitargets) (see Supplementary Table S1). Of course, ligand-based and structure-based tools can be used, but more specific engines can be valuable to assist the process, for instance methods that enable fast and user friendly searches for bioisoteric replacements or tools that attempt to optimize several ADME-Tox properties at the same time (multiparameter optimization) (see Supplementary Table S1).
LBVS and target-fishing web servers
The different tools are reported in Table 1.
Table 1.
LBVS and target-fishing web tools
| Tools | URL | Aims/main algorithms (2D, 3D and/or machine learning.) | Input/output open and/or registration | Data policy/privacy | References |
|---|---|---|---|---|---|
| Well-known databases with 2D similarity search utilities | |||||
| ChEMBL | https://www.ebi.ac.uk/chembl | Annotated compound database | Main input: chemical or text query Main output: similar compounds Open |
Data privacy documented. Queries are not stored nor analyzed | [168] |
| DrugBank | https://www.drugbank.ca/ | Drug database | Main input: chemical or text query Main output: similar compounds Open for noncommercial use, if not users have to contact the authors |
Data privacy documented. Different situations in case of simple search or in case the user is registered | [171] |
| hmdb | http://www.hmdb.ca | The Human Metabolome database | Main input: chemical or text query Main output: similar compounds Open for noncommercial use, if not users have to contact the authors |
Data privacy documented. Some minimum data are collected about users | [171] |
| PubChem | https://pubchem.ncbi.nlm.nih.gov | Annotated compound database | Main input: chemical or text query Main output: similar compounds Open |
Data privacy documented. Some minimum data are collected about users | [170] |
| SuperDRUG 2 | http://cheminfo.charite.de/superdrug2 | Drug database | Main input: chemical or text query Main output: similar compounds Open for noncommercial use if not users have to contact the authors |
n/a | [172] |
| SureCheMBL | https://www.surechembl.org | Patented molecules | Main input: chemical or text query Main output: similar compoundsOpen |
Data privacy documented. Some minimum data are collected about users | [169] |
| ZINC | http://zinc15.docking.org | Compounds from various chemical vendors and other utilities | Main input: chemical query Main output: similar compoundsOpen |
Data privacy documented | [173] |
| 2D or 3D similarity search, structural diversity and computation of molecular descriptors | |||||
| BRUSELAS | http://bio-hpc.eu/software/Bruselas/ | Database with 3D shape and pharmacophore ligand similarity search utilities | Main input: compound in SMILES, MOL2, SDF) Main output: similar compounds (if 3D search, aligned ligands can be downloaded as PyMOL session) Open |
n/a | [180] |
| ChemDes | http://www.scbdd.com/chemdes/ | Compute molecular descriptors and fingerprints | Main input: compounds in SMILES or SDF format Main output: Computed descriptors Requires registration |
n/a | [184] |
| ChemMine Tools | http://chemmine.ucr.edu/ | Small-molecule similarity calculation and clustering and physchem property calculation | Main input: Compounds in SMILES/SDF format (can be drawn) Output: similar compounds Open |
All data provided by the user is stored in a secure space on our server and only used to perform the intended analyses requested by the user. Once an analysis session by a user is completed, all its records and intermediate files are deleted within 30 days | [185] |
| MXFP | http://similaritysearch.gdb.tools/ | Non-Lipinski ligand similarity search utility | Main input: Compound in SMILES or can be drawn Main output: Compound positioned on the selected chemical space Open but possible to register and save data | Data privacy documented | [188] |
| pepMMsMIMIC | http://mms.dsfarm.unipd.it/pepMMsMIMIC/ | Database with pharmacophore and shape peptidomimetic ligand similarity search utility | Main input: peptide in 3D PDB format Main output: top 200 chemical compounds that mimic the peptide. Results can be downloaded as SDF file Open | n/a | [189] |
| PharmaGist | https://bioinfo3d.cs.tau.ac.il/PharmaGist/ | Pharmacophore generator | Main input: up to 32 ligands in MOL2 format Main output: tables with computed pharmacophores and aligned molecules with a score Open | n/a | [191] |
| Pharmit | http://pharmit.csb.pitt.edu | Database with pharmacophore and molecular shape ligand similarity search utility | Main input: a ligand in 3D PDB format Main output: similar aligned on the query. Results can be downloaded as SDF file Open and registration is possible | Collect information only when registration but data remains secure | [193] |
| Rchempp | http://shiny.bioinf.jku.at/Analoging/ | Ligand similarity search utility in ChEMBL, Drugbank and the Connectivity Map databases | Main input: Compounds in MOL/SDF format. Main output: similar compounds identified. Open | n/a | [195] |
| SwissSimilarity | http://www.swisssimilarity.ch/ | Compound database with 2D and 3D ligand similarity search utility | Main input: Compounds in SMILES format (can be drawn). Output: compounds with similarity scores. Open | The institute does not look at user data | [182] |
| USR-VS | http://usr.marseille.inserm.fr/ | Database with molecular shape ligand similarity search utility | Main input: a query molecule in 3D SDF format. Main output: aligned molecules. Open | n/a | [181] |
| ZINCPharmer | http://zincpharmer.csb.pitt.edu/ | Database with pharmacophore ligand similarity search utility | Main input: a ligand in 3D, a PDB file with a protein and the ligand, a protein–protein PDB file, a pharmacophore generated by another software Main output: aligned compounds (can also be downloaded) Open | Data privacy documented. Some minimum data are collected about users | [199] |
| Machine learning models and LBVS | |||||
| ChemSAR | http://chemsar.scbdd.com/ | SAR model building using machine learning | Main input: compounds in SMILES or SDF, data sets in CSV format Main output: compounds with predicted affinity to the target that fit the statistical model. SDF file can be downloaded Open and users can create their own working space | n/a | [200] |
| DPubChem | https://www.cbrc.kaust.edu.sa/dpubchem/ | Machine learning/QSAR | Main input: PubChem bioassay Main output: statistical model that can be used to screen compounds Open and users can upload a saved statistical model |
n/a | [201] |
| DeepScreening | http://deepscreening.xielab.net/ | Deep learning model construction for virtual screening. Find if compounds could bind to a selected target | Main input: training data set in SDF format or select data from ChEMBL Main output: predictive models with diverse evaluation metrics and possibilities to perform predictions Open |
n/a | [202] |
| MLViS | http://www.biosoft.hacettepe.edu.tr/MLViS/ | Machine learning/QSAR | Main input: PubChem Compound ID numbers Main output: predictive models Open |
n/a | [203] |
| OCHEM | http://www.ochem.eu | Machine learning/QSAR | Main input: training sets or molecules to pass in a previously developed model Main output: predictive models or filtered compounds Open or possibility to register and have working space. License agreement has to be accepted |
n/a The situation is different for guest users and registered users |
[205] |
| Ligand-based target-fishing or disease/target specific screening | |||||
| Anglerfish | http://anglerfish.urv.cat/anglerfish/ | Target-fishing using one or more fingerprints | Main input: paste compounds in SMILES or InChi or upload SDF file Main output: List of possible targets and expected affinity, can be exported as CSV file Open |
n/a | Drs. Garcia-Vallve and Pujadas team, EURECAT Technology Centre of Catalonia |
| ChemProt-3 | http://potentia.cbs.dtu.dk/ChemProt/ | Target-fishing using 2D similarity search or QSAR models | Main input: paste compounds in SMILES or compound name or target name Main output: similar ligands found by similarity search or statistical prediction together with target names. The list can be downloaded Open |
n/a | [213] |
| DIA-DB | http://bio-hpc.eu/software/dia-db/ | Shape similarity search against antidiabetic drugs | Main input: paste compounds in SMILES or draw the molecule. Main output: email with a link to the results, aligned molecules Open but users have to provide an email address |
n/a | [215] |
| HitPickV2 | http://mips.helmholtz-muenchen.de/HitPickV2/target_prediction.jsp | Predict targets of chemical compounds using 2D similarity search | Main input: paste compounds in SMILES Main output: top 10 predicted targets. CSV file can be downloaded Open, the server follows EU regulations for data storage. It is mentioned that the server is free for academic users | Data privacy and data management documented | [216] |
| MolTarPred | http://moltarpred.marseille.inserm.fr/ | Target profiling using 2D similarity search | Main input: a compound in SMILES Main output: predicted targets and similar compounds. Data can be downloaded as TSV files. Open | n/a | [217] |
| MuSSel | http://mussel.uniba.it:5000/ | Target-fishing using 2D similarity search | Main input: draw a compound online. Main output: similar compounds, predicted targets. Data can be downloaded as CSV files Open | n/a | [218] |
| PPB2 | http://gdbtools.unibe.ch:8080/PPB/ | Polypharmacology browser, target prediction combining nearest neighbors with machine learning | Main input: draw a compound online Main output: similar compounds, predicted targets. Data can be downloaded as TXT files Open | n/a | [221] |
| RFQSAR | http://rfqsar.kaist.ac.kr | Ligand-based screening and machine learning | Main input: compounds in SMILES Main output: predicted targets Open | n/a | [222] |
| SEA | http://sea.bkslab.org/ | Target-fishing by 2D similarity search with scores corrected by a statistical model | Main input: a compound in SMILES Main output: a predicted target list that can be downloaded. Compounds are also linked to ZINC Open | n/a | [214] |
| SuperPred | http://prediction.charite.de/index.php | Target-fishing by 2D similarity search or ATC code prediction | Main input: a PubChem name or a SMILES or by drawing the molecule Main output: a predicted target list with probability scores or the ATC code Open | n/a | [223] |
| SwissTarget Prediction | http://www.swisstargetprediction.ch/ | Target-fishing with a combined 2D-3D similarity score | Main input: a compound in SMILES Main output: a predicted target list that can be downloaded in CSV format or others Open | The institute does not look at user data | [224, 225] |
| TargetNet | http://targetnet.scbdd.com/calcnet/index/ | Target-fishing using QSAR models | Main input: a compound in SMILES, can be sketched or file with compounds in SMILES can be uploaded Main output: a predicted target list Open | n/a | [227] |
| WDL-RF | https://zhanglab.ccmb.med.umich.edu/WDL-RF/ | Machine learning approach to predict bioactivities against G protein-coupled receptors | Main input: paste compounds in SMILES or upload a file with compounds in SMILES. Select the GPCR for activity prediction Main output: predicted activity Open but results (link) are sent via email to the user | Some minimum data are collected about users | [228] |
2D similarity search utilities on some selected well-known databases
As mentioned above, we will not cover compound databases in the present review. We here highlight some well-known services where it is possible to carry out ligand-based similarity search. Users can usually upload or draw a compound of interest, for example known to be active on a given target and search for similar compounds present in the investigated database (e.g. ChEMBL [168], SureCheMBL [169], PubChem [170], DrugBank [171], hmdb [171], SuperDRUG [172], ZINC [173] etc.). Most often, one or several structural fingerprints are available to carry out the search [174, 175]. Fingerprints in general encode the presence of substructural fragments. A similarity measure can be obtained by determining the numbers of chemical substructures in common between the query compound and the molecules in the database. These so-called similar compounds are expected to have similar activities following the Similar Property Principle (see for instance [176] and comments in [175]). It is also important to note that the experimental data available in these databases can be very valuable to assist compound validation and optimization while giving some hints about the putative targets of the investigated compound.
2D or 3D similarity search, structural diversity and computation of molecular descriptors
Balanced rapid and unrestricted server for extensive ligand-aimed screening
It is a web tool for 3D shape (e.g. computed with Screen3D [177], WEGA [178]) and pharmacophore (computed with SHAFTS [179]) searches in libraries extracted for instance from ChEMBL, KEGG or DrugBank [180]. A wide panel of shape and pharmacophore similarity algorithms is combined. Balanced rapid and unrestricted server for extensive ligand-aimed screening (BRUSELAS) was tested against related servers (e.g. USR-VS [181], SwissSimilarity [182] and ChemMapper [183]) to search for potential antidiabetic drugs.
ChemDes
In many projects, it will be necessary to compute molecular descriptors and fingerprints (e.g. for machine learning, similarity search, etc.). Most of the tools needed to compute these properties are distributed as standalone software or packages that require in some cases complex installation or programming efforts. Second, many of the tools can only calculate a subset of molecular descriptors, and the results from multiple tools need to be manually merged to generate a comprehensive set of descriptors. Third, some packages only provide application programming interfaces and are implemented in different computer languages, which pose additional challenges to the integration of these tools. ChemDes is a free web-based platform for the calculation of 2D or 3D molecular descriptors (Chemopy, CDK, RDKit, Pybel, BlueDesc and PaDEL descriptors) and fingerprints; currently, it computes 3679 molecular descriptors and 59 types of molecular fingerprints such as topological fingerprints, electro-topological state (E-state) fingerprints, MACCS keys, FP4 keys, atom pairs fingerprints, topological torsion fingerprints and Morgan/circular fingerprints among others [184].
ChemMine tools
This is an online service for small molecule data analysis [185]. The primary functionalities of this service fall into five major application areas: data visualization, structure comparisons, similarity searching, compound clustering and prediction of chemical properties. Users can upload compound data sets and use utilities such as compound viewing, structure drawing and format interconversion. Pairwise structural similarities among compounds can be quantified. Interfaces to ultrafast structure similarity search algorithms are available to mine the chemical space in the public domain databases (PubChem or ChEMBL). The service also includes a clustering toolbox to enable systematic structure- and activity-based analyses. Further, physicochemical property descriptors of compound sets can be calculated and, for instance, use for QSAR studies. The tools were, among others, used to analyze molecules inhibiting Chikungunya virus-induced cell death [186] or to plot a dendogram showing structural similarity of hit compounds acting on Dengue viruses [187].
Macromolecule extended atom-pair fingerprint
Seven million of the currently 94 million entries in the PubChem database break at least one of the four Lipinski constraints for oral bioavailability while 183 185 of which are also found in the ChEMBL database. These non-Lipinski PubChem (NLP) and ChEMBL (NLC) subsets can be of interest in many projects. The macromolecule extended atom-pair fingerprint (MXFP) web-based application has been developed to explore larger molecules and to position users’ compounds in this chemical space [188]. MXFP is a 217-D fingerprint tailored to analyze large molecules in terms of molecular shape and pharmacophores. The approach can perform MXFP nearest neighbor searches in the NLP and NLC space.
pepMMsMIMIC
This tool is a web-oriented peptidomimetic compound virtual screening tool based on a multiconformers 3D similarity search strategy [189]. Key to the development of pepMMsMIMIC has been the creation of a library of 17 million conformers calculated from 3.9 million commercially available chemicals. Using as input the 3D structure of a peptide bound to a protein, pepMMsMIMIC suggests which chemical structures are able to mimic the peptide using both pharmacophore and shape similarity techniques. The tool was for instance used for the development of Mdm2 small molecule modulators [190].
PharmaGist
This is a tool for ligand-based pharmacophore detection and does not require the 3D structure of the target [191]. The input is a set of structures of drug-like molecules that are known to bind to the receptor of interest. The output consists of candidate pharmacophores that are computed by multiple flexible alignment of the input ligands. The method handles the flexibility of the input ligands explicitly and in deterministic manner within the alignment process. This tool was, for example, used to study pregnane X receptor ligands, a xenobiotic sensor [192].
Pharmit
This tool provides an online, interactive environment for the virtual screening of large compound databases (over 200 million compounds in total) using pharmacophores, molecular shape and energy minimization [193]. Users can import, create and edit virtual screening queries in an interactive browser-based interface. Queries are specified in terms of a pharmacophore, a spatial arrangement of the essential features of an interaction, and molecular shape. Search results can be further ranked and filtered using energy minimization. In addition to nine prebuilt databases of popular compound libraries (e.g. ChEMBL, molecules from chemical vendors…), users may submit their own compound libraries for screening. The Pharmit web service was, for example, used to search for novel cholinesterase inhibitors, molecules that could be used in the treatment of Alzheimer’s disease [194].
Rchempp
Rchempp is a web service that identifies structurally similar compounds (structural analogs) in large compound databases [195]. The service allows compounds to be queried in the widely used ChEMBL (version 18) and DrugBank (version 4.0) databases mentioned above and the Connectivity Map database [196]. Rchemcpp utilizes similarity functions, i.e. molecule kernels, as measures for structural similarity. By exploiting information contained in public databases, the web service facilitates many applications crucial for the drug development process, such as prioritizing compounds after screening or reducing adverse side effects during late phases.
SwissSimilarity
This is a web tool for rapid LBVS of small to large libraries of small molecules [182]. Screenable compounds include drugs, bioactive (databases used: ChEMBL, Chemical Entities of Biological Interest (ChEBI), G protein-coupled receptor (GPCR) ligands, hmdb and ligands from the protein data bank (PDB)) and commercial molecules (databases used: Zinc, Asinex, Aldrich, ChemBridge, ChemDiv, Enamine…), as well as over 280 million virtual compounds (Sigma Aldrich library) readily synthesizable from commercially available synthetic reagents. Predictions can be carried out on-the-fly using six different screening approaches, including 2D molecular fingerprints (e.g. FP2 fingerprints) as well as 3D similarity methodologies (e.g. Shape-IT, Align-IT). SwissSimilarity was for instance used to find analogs of novel inhibitors of the membrane-associated inhibitory kinase PKMYT1 [197] or to generate a screening library to find histamine H3 receptor ligands [198].
Ultrafast Shape Recognition-Virtual Screening (USR-VS)
It is a web server that uses two validated ligand-based 3D methods (Ultrafast Shape Recognition (USR) or its pharmacophoric extension (USRCAT)) for large-scale prospective virtual screening [181]. Total 93.9 million 3D conformers, expanded from 23.1 million purchasable molecules, are screened and the 100 most similar molecules to the user 3D query input compound in terms of 3D shape and pharmacophoric properties are shown. USR-VS also provides interactive visualization of the similarity of the query molecule against the hit molecules as well as vendor information to purchase selected hits.
ZINCPharmer
This is an online server for searching the purchasable compounds of the ZINC database using the Pharmer pharmacophore search technology [199]. The commercial collection MolPort can also be screened. A pharmacophore describes the spatial arrangement of the essential features of an interaction. Compounds that match a well-defined pharmacophore serve as potential lead compounds for drug discovery. ZINCPharmer provides tools for constructing and refining pharmacophore hypotheses directly from an input molecular structure. A search of 176 million conformers of 18.3 million compounds is performed. The results can be analyzed interactively, or the aligned structures can be downloaded for offline analysis.
Machine learning models and LBVS
ChemSAR
Predictive models based on machine learning techniques have proven to be effective in drug discovery. However, to develop such statistical models, researchers usually have to use multiple tools and the process requires many different steps (e.g. RDKit or ChemoPy package for molecular descriptor calculation, ChemAxon Standardizer for structure preprocessing, scikit-learn package for model building and statistical analysis and ggplot2 package for data visualization, etc.). Strong programming skills are needed to develop such models. ChemSAR is a web-based pipelining platform for generating SAR classification models (random forest, support vector machine, naive bayes, K-nearest neighbors and decision tree) of small molecules [200]. The capabilities of ChemSAR include the validation and standardization of chemical structure representation, the computation of 783 1D/2D molecular descriptors and 10 types of widely used fingerprints for small molecules, the filtering methods for feature selection, the generation of predictive models via a step-by-step job submission process, model interpretation in terms of feature importance and tree visualization, as well as a helpful report generation system.
DPubChem
This is a web tool for deriving QSAR models that implement the state-of-the-art machine learning techniques (classification models with random forest, AdaBoost, support vector machine, naive Bayes, K-nearest neighbors and decision tree) to enhance the precision of the models and enable efficient analyses of experiments from PubChem BioAssay database [201]. DPubChem has a simple interface that provides various options to users. Users select a PubChem BioAssay, compute chemical features (e.g. various fingerprints and molecular descriptors are available) and correction for class imbalance can be turned on. Once the statistical model is built, users can screen molecules. DPubChem predicted active compounds for 300 datasets with an average geometric mean and F1 score of 76.68% and 76.53%, respectively. Furthermore, DPubChem builds interaction networks that highlight novel predicted links between chemical compounds and biological assays. Using such a network, the tool successfully suggested a novel drug for the Niemann-Pick type C disease.
DeepScreen
This is a web server with integration of the state-of-art deep learning algorithm, which utilizes either annotated databases such as ChEMBL or user-provided datasets and performs virtual screening to propose chemical probes or drugs for a specific target [202]. With DeepScreening, users can construct a deep learning model to generate target-focused compound libraries. The constructed classification or regression models can then be subsequently used for virtual screening against chemical vendor collections or other libraries (e.g. Specs, Enamine, ion channels, epigenetics, DrugBank or GPCR libraries) or a de novo compound library can be generated via the de novo module.
Machine learning-based virtual screening tool
As discussed above, virtual screening can be used in the early-phase of drug discovery. Because there are thousands of bioactive compounds, it might be possible to partially distinguish drug-like and nondrug-like molecules. Statistical machine learning methods can be used for classification purpose. Machine learning-based virtual screening tool (MLViS) is a tool that attempts to classify molecules as drug-like and nondrug-like based on various machine learning methods, including discriminant, tree-based, kernel-based, ensemble and other algorithms [203]. The application can also create heat map and dendrogram for visual inspection of the molecules through hierarchical cluster analysis. Moreover, users can connect the PubChem database to download molecular information and to build 2D structures of the selected compounds.
Online Chemical Modeling Environment
The Online Chemical Modeling Environment (OCHEM) is a web-based platform that aims to automate and simplify the typical steps required for QSAR modeling [204, 205]. The platform consists of two major subsystems: the database of experimental measurements and the modeling framework. A user-contributed database contains a set of tools for easy input, search and modification of the records. The OCHEM database is based on the wiki principle and focuses primarily on the quality and verifiability of the data. The database is tightly integrated with the modeling framework, which supports all the steps required to create a predictive model: data search, calculation and selection of a vast variety of molecular descriptors, application of machine learning methods (e.g. neural networks, K-nearest neighbors, support vector machine, multiple linear regression, partial least square, decision tree and random forest), validation, analysis of the model and assessment of the applicability domain. The system was for instance use to model human ether-a-go-go related gene (hERG) K+ channel blockage, an important protein that can be involved in cardiotoxicity [206].
Ligand-based target-fishing or disease/target specific ligand-based screening
Anglerfish
The tool performs similarity search combining several different molecular fingerprints (which can be chosen by the users such as the ones computed by RDKit, OpenBabel FP3, MACCS166…) and by searching in-house prepared ChEMBL activity data to predict potential new targets for the query molecules. (Drs Garcia-Vallve and Pujadas team, EURECAT Technology Centre of Catalonia).
ChemProt-3
The tool compiles multiple chemical–protein annotation resources integrated with diseases and clinical outcomes information. The data sources include ChEMBL [168], BindingDB [207], DrugBank [171], STITCH [208]… information from the Anatomical Therapeutic Classification System and side effect data from Sider [209]. Biological data were for instance obtained from KEGG [210], Reactome [211], GeneCards [212]… All the compound bioactivity data were stored in the ChemProt database following an internal curation procedure [213]. Compounds can be compared with the Daylight-like fingerprints as implemented in RDKit and calculation of the Tanimoto coefficient. The Similarity Ensemble Approach [214] is also implemented. Naïve Bayes classifier models have also been developed for about 850 proteins (i.e. for proteins with sufficient compound bioactivity data) and a visual interface that enables navigation of the pharmacological space for the identified small molecules is also available. In the last version of the service, the authors take the example of caffeine as a query compound and show that the molecule is similar to 105 compounds present in the database and could possibly be associated to 449 proteins. The visual heatmap display shows weak-to-strong association with the different proteins identified [213].
DIA-DB
This server aims at identifying novel antidiabetic drugs [215]. A database of approved and experimental has been collected. A query compound can then be compared with existing molecules via shape similarity search (performed with WEGA [178] or SHAFT [179]). Aligned molecules are returned to users of the service.
HitPickV2
This is a ligand-based approach for the prediction of 2739 human druggable protein targets (e.g. identified in STITCH [208]) for compounds provided by a user [216]. For each query compound, the server predicts up to 10 distinct targets. The Functional-Class Fingerprints (FCFP)-like circular Morgan fingerprints as implemented in RDKit are used. The tool places the query compounds into its surrounding chemical space of annotated compound–target interactions using k-nearest neighbor (k-NN) chemical similarity search. Then HitPick scores these 10 targets based on three parameters: the computed Tanimoto coefficient (Tc) between the query and the most similar compound interacting with the target, a target rank that considers Tc and Laplacian-modified naïve Bayesian target models scores and a parameter that considers the number of compounds interacting with each target.
MolTarPred
The service predicts potential targets for an input query compound [217]. Tanimoto similarities between the Morgan fingerprints of the query molecule and that of each of the 607 659 molecules extracted from the ChEMBL database are computed (these compounds act on 4553 targets). A list of putative targets is returned with an estimated confidence score. Molecules can be visualized online with the percent of similarity.
Multifingerprint Similarity Search aLgorithm (MuSSel)
MuSSel is a predictive tool to find putative protein drug targets for a query compound. Predictions are automatically made by screening a large collection of 611 333 small molecules having high-quality experimental bioactivity data covering 3357 protein drug targets selected from the ChEMBL database [218, 219]. Eighteen different fingerprints are calculated, different similarity threshold values were investigated and the notion of activity cliffs was also explored and the approach was then implemented online.
PPB2
The first version of the tool proposes putative protein targets for a query compound [220]. It searches through a database containing the ligands of 4613 targets extracted from the ChEMBL database. PPB performs target prediction using 10 different fingerprints separately and returns the predefined number (by default 20) of the top predicted targets using each of them. The result is provided as a consolidated table of annotated targets and the similarity values for each selected fingerprints are shown. Links to the ChEMBL compounds and targets are available and results can be downloaded. Another version of PPB is available and is named PPB2. In this version, 1720 targets were extracted from the ChEMBL database. Ligand similarities are computed with three main types of fingerprints and machine learning approaches are also used (e.g. nearest neighbor, Naïve Bayes) [221]. The tools were used to study the polypharmacology profile of a potent inhibitor of TRPV6 transmembrane calcium channel. Twenty-four targets were selected based on the prediction and several targets could be validated experimentally.
RFQSAR
This web server applies LBVS model comprising 1121 target structure–activity relationships models built using a random forest algorithm to predict the activity of ligands toward each target and ranking candidate targets for a query ligand using a unified scoring scheme [222]. This approach is thus at the frontier between ligand-based screening, machine learning and target-fishing. The user interface is user friendly and intuitive, offering many useful information and cross references.
SEA
The tool takes a query molecule as input and performs predictions for about 4160 targets using annotated chemical data extracted from the ChEMBL database [214]. A list of predicted targets annotated with P-values and Tanimoto coefficients of the most similar ligand of each of the predicted targets of the query molecule are provided. SEA only returns predicted targets for which P-values are significantly low. SEA uses the concept of raw similarity scores coupled with a statistical model to compare query molecules to another set of ligands. Pairwise extended-connectivity fingerprints (ECFP4) Tanimoto coefficient similarity is initially computed (using the RDKit toolbox). The significance of the score is then further assessed by using a statistical model built on a random distribution of the raw scores. At the end, raw scores are converted in z-scores and P-values (i.e. the P-value for a target indicates the probability of chance similarity between ligands of a target and a query compound). SEA was, for example, used to identify new targets for known drugs [214]. Out of 30 novel drug–target interactions suggested by prediction, 23 were confirmed experimentally.
SuperPred
The web server comprises two methods, one for drug classification based on approved drugs classified by the World Health Organization (WHO) and one for target prediction based on available compound–target interaction data [223]. The drug classification method takes into account 2D and fragment similarity and a method for 3D superposition of the small molecules. The consensus of these methods is taken into account. If at least two methods predict the same ATC class, that class is considered as final prediction. If three different ATC classes are predicted, a threshold for every method is used to decide for the most probable ATC class. The method for target prediction uses the similarity distribution among ligands for estimating the targets’ individual thresholds and probabilities to avoid false positive predictions. The ECFP (that belongs to the class of radial fingerprints) was found to exhibit the best performance and the calculated fingerprints are then subsequently compared by computing Tanimoto similarity scores. Data were extracted from the ChEMBL database (~341 000 compounds, ~1800 targets and ~665 000 compound–target interactions). Structural information about drug–target are also provided when available.
SwissTargetPrediction
The tool performs predictions by searching for similar molecules, in 2D and 3D, within a collection of 376 342 compounds known to be experimentally active on an extended set of 3068 macromolecular targets (extracted from ChEMBL). The quantification of similarity is 2-fold. In both cases, it consists in computing a pairwise comparison of 1D vectors describing molecular structures: the 2D measure uses the Tanimoto index between path-based binary fingerprints (FP2), whereas the 3D measure is based on a Manhattan distance similarity quantity between Electroshape 5D (ES5D) float vectors. For both 2D and 3D similarity measures, the principle is that two similar molecules are represented by analogous vectors, which exhibit a quantified similarity close to 1. The SwissTargetPrediction model was trained by fitting a multiple logistic regression on various ligand size-related subsets of known actives to weight 2D and 3D similarity parameters in a so-called combined score [224, 225]. A list of putative targets (maximum 100) is reported with various graphical views and links to UniProt and GeneCard. The data can be downloaded in various formats and so are the list of SMILES and list of interactions used by the service. Moreover, targets tagged ‘by homology’ are predictions based on similar molecules active on proteins showing sufficient level of homology. Users can also select a species for the target predictions: Homo sapiens, Mus musculus and Rattus norvegicus. SwissTargetPrediction ranks the targets based on these combined scores, which are converted to probabilities that give an estimate of the likelihood of correct predictions. The tool was for instance used to find putative targets for cudraflavone C, a naturally occurring flavonol with reported antiproliferative activities, that were validated experimentally [226].
TargetNet
Users here can submit query compounds and the server predicts the activity of the user’s molecules across 623 human proteins using 623 QSAR models related to these proteins. Seven types of fingerprints can be selected. Data were obtained from BindingDB. After curation, the final database used contained 623 protein targets, 228 415 compounds and 359 353 bioactivity data [227].
Weighted deep learning and random forest (WDL-RF)
The server uses weighted deep learning and random forest, to model the bioactivity of GPCR-associated ligand molecules [228]. The protocol consists of two consecutive stages: (i) molecular fingerprint generation through a new weighted deep learning method and (ii) bioactivity calculations with a random forest model. GPCRs data were taken from the UniProt database and from the GPCR–Ligand Association (GLASS) database, which included over 519 000 unique GPCR–ligand interaction entries. Compounds in SMILES format can be uploaded and the user selects the GPCR name/type for activity predictions.
SBVS, compound docking and target-fishing web servers
The different tools are reported in Table 2.
Table 2.
SBVS and target-fishing web tools
| Tools | URL | Method | Input/output open and/or registration | Data policy | References |
|---|---|---|---|---|---|
| Screening and receptor-based pharmacophore screening | |||||
| ACFIS | http://chemyang.ccnu.edu.cn/ccb/server/ACFIS/ | Fragment-based drug discovery | Major input: the protein structure file is uploaded in pdb format and the user can choose the appropriate fragment database for screening Major output: the results can be visualized online and outputs can be downloaded Open | n/a | [229] |
| CaverWeb | https://loschmidt.chemi.muni.cz/caverweb/ | Docking in protein tunnels and channels using Vina | Main input: upload target PDB file or input PDB code. Users can provide multiple ligands either as files with coordinates, ZINC database accession codes or by drawing the structure in a sketcher Main output: docked compounds can be interactively analyzed and visualized. Also, the results can be generated in a PDF document Open | Data privacy and data management documented | [231, 232] |
| DOCK Blaster | http://blaster.docking.org/ | Docking with DOCK 3.6 | Main input: Target protein structure in PDB or MOL2 format; Users have an option to specify up to 50 actives and up to 50 inactives as SMILES or ZINC IDs. Docking of larger databases (e.g. lead-like, fragment-like) is allowed after initial control calculations are completed successfully Main output: results can be visualized online or downloaded Open | n/a | [235] |
| DOCKovalent | http://covalent.docking.org/ | Covalent docking | Main input: supply the protein structure file in PDB or MOL2 format and provide information about the binding site and the attachment point. The user has to select the covalent database for docking Main output: docked compounds can be visualized online and downloaded Open | Data privacy and data management documented | [238] |
| DockThor | https://dockthor.lncc.br/v2/ | Virtual screening, score with the MMFF94S force field. The version 2 can also deal with short peptides | Main input: The target structure is uploaded in PDB format. The ligand file (1 or more ligands) should be supplied in MOL2, PDB or SDF format Main output: the results (different binding modes) can be interactively visualized online) are downloadable Open It is possible to create an account and register | n/a | [240] |
| EasyVS | https://easyvs.unifei.edu.br/ | Virtual screening with AutoDock Vina and rescoring with NNScore | Main input: upload target PDB file or supply PDB code; upload a compound collection in 3D sdf format or select libraries of interest from the prepared molecule databases Main output: molecules docked into the target can be downloaded or visualized online Open | n/a | Developed by Wandré N. de P. Veloso, Pâmela M. Rezende, David Ascher, Carlos H. da Silveira and Douglas E. V. Pires |
| e-LEA3D | https://chemoinfo.ipmc.cnrs.fr/LEA3D/index.html | De novo design, docking with PLANTS | Main input: upload a library of molecules or selects the de novo drug design option. The choice for selecting the FDA-approved drug structure data set is also provided; Upload a protein structure file in PDB or MOL2 format Main output: results from docking or de novo design can be visualized online or downloaded Open | Data privacy and data management documented | [243] |
| ezCADD | http://dxulab.org/software | Docking with Vina or Smina, virtual screening. Protein–ligand interactions can be visualized with ezLigPlot. Other tools are available in beta versions such as pocket search | Main input: the receptor can be provided as PDB file or PDB ID; the ligand file is uploaded in MOL, MOL2, sdf, or PDB format. Also, ligand details can be provided as SMILES, InChI key or by drawing using the PubChem Sketcher Main output: the results from docking/screening can be visualized online The server is free for academic use | n/a | [246] |
| iScreen | http://iscreen.cmu.edu.tw/basic.php# | Docking with PLANTs | Major input: Protein structure is uploaded in pdb or mol2 format without any compound/ligand/inhibitor in the binding site; the ligand file is provided in mol2 or sdf format Major output: A notice is sent to the user when the job is finished. The results can be visualized online Open | n/a | [249] |
| MTiOpenScreen | https://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/ | Virtual screening with Autodock (blind docking of up to 10 molecules) or screen via Autodock Vina | Main input: upload or paste a compound collection in 3D sdf or MOL2, or use prepared in-house collections including approved drugs; paste or upload a protein 3D PDB file Main output: compounds docked into the target. Predicted structures can be downloaded or visualized online Open | Data privacy and data management documented. Different situations for guest users and registered users | [250, 252] |
| Pharmit | http://pharmit.csb.pitt.edu | Database with receptor-based pharmacophore search utility | Main input: ligand with its receptor in 3D PDB format Main output: similar compounds positioned in the binding pocket. Results can be downloaded as SDF file Open and registration is possible | Collect information only when registration but data remains secure | [193] |
| Sanjeevini | http://www.scfbio-iitd.res.in/sanjeevini/sanjeevini.jsp | Virtual screening | Major input: upload a (protein) target and a candidate drug in PDB format. The user can also select a relevant chemical library for screening Major output: docked protein–ligand complexes along with their predicted binding affinities are emailed to the user. Additionally, a link is provided to download the docked structure files Open | n/a | [254] |
| Dock/undock or rescoring tools | |||||
| AMMOS2 | http://drugmod.rpbs.univ-paris-diderot.fr/ammosHome.php | Post-docking energy optimization and rescoring | Main input: upload docked ligands in mol2 format; upload a protein 3D PDB file. Main output: The optimized ligand poses are ranked by the predicted binding energy. The results can be downloaded or visualized online with for instance PLIP to see noncovalent interactions. Open | Data privacy and data management documented. Different situations for guest users and registered users | [255] |
| CompScore | http://bioquimio.udla.edu.ec/compscore/ | Rescoring | Major input: upload a. TXT file containing compound ID, classification of the compound, weighting criterion (number of heavy atoms) and docking scores Major output: Links for downloading the results are sent to the e-mail address of the user Open | n/a | [257] |
| CSM–Lig | http://biosig.unimelb.edu.au/csm_lig/ | Predict the binding affinity of a protein–small-molecule complex based on structural signatures | Main input: upload a single PDB structure of the protein–small-molecule complex or a compressed file with multiple structures. The molecule information is provided as a tab-separated file containing HET ID and SMILES string for each uploaded PDB Main output: results can be visualized online or downloaded as a Pymol session file for visualizing the ligand interactions Open | n/a | [299] |
| dockNmine | http://www.ufip.univ-nantes.fr/tools/docknmine/ | Assemble and analyze virtual and experimental interaction data | Main input: The target protein is added by entering the UniProt ID and then selecting the relevant structure from the enumerated entries; Ligand information is added via PDB ID, PubChem ID, or ChEMBL ID; or multiple ligands can be added by uploading an sdf file Main output: The results from docking can be visualized online and graphs can be downloaded Registration is required before job submission | n/a Different situations if users are registered or not | [259] |
| MedusaDock | https://dokhlab.med.psu.edu/medusadock/ | Flexible docking with constraints | Main input: upload the protein structure in PDB or MOL2 format or input the PDB ID of the receptor; the ligand file is uploaded in mol2 format or ZINC ID of the ligand can be entered Main output: The results from docking can be visualized online Open | n/a | [260] |
| MoMA-LigPath | http://moma.laas.fr | Ligand unbinding | Main input: the protein–ligand complex is uploaded in PDB format Main output: results are downloadable and the ligand unbinding pathway can be visualized online Open | Different situations if users are registered or not | [234] |
| nAPOLI | http://bioinfo.dcc.ufmg.br/napoli/ | Analysis of protein–ligand interactions | Main input: submit PDB files or provide a list of pdb ids, its chains and ligands to be analyzed Main output: The protein–ligand interaction can be visualized online; figures and statistical data can be downloaded Open | n/a | [262] |
| PlayMolecule | https://www.playmolecule.org/ | Prepare proteins, rescoring, machine learning, pocket search, predict interaction of a ligand with a pathway | Major input: different modules have their own specifications for uploading input files Major output: rescored compounds can be visualized online and downloaded Partially open. Users can use a guest account or register | Different situations if users are registered or not | [263–265] |
| PRODIGY-LIG | http://milou.science.uu.nl/services/PRODIGY-LIG | Binding affinity | Main input: provide PDB ID of the protein–small-molecule complex or can upload a file in PDB or mmCIF format. An archive of multiple PDB/mmCIF files (.tar, .tgz, .zip, .bz2, or .tar.gz) can also be uploaded Major output: results can be visualized online and archive file of all the outputs (.tgz) can be downloaded Open | Different situations if users are registered or not | [266] |
| Screening Explorer | http://stats.drugdesign.fr | Analysis of screening results, consensus scoring | Major input: supply a csv file with compounds identifiers, docking scores and Activity tags Major output: Interactive charts & screening metrics can be visualized online and downloaded Open | n/a | [267] |
| SwissDock | http://www.swissdock.ch | Protein–ligand docking and advanced binding affinity prediction | Main input: provide either PDB code, protein name, sequence, URL or upload PDB file or a ZIP file containing the target protein in the CHARMM format; Ligand information can be supplied as ZINC accession number, ligand name, URL or by uploading ligand file in the mol2 format or as a ZIP file containing files in the CHARMM format Main output: docking poses can be visualized online and downloaded This server is free for academic use | The institute does not look at user data | [268] |
| Webina | https://durrantlab.pitt.edu/webina/ | Docking with AutoDock Vina | Main input: upload a receptor and ligand file in the pdbqt format Main output: compound docked into the receptor, this can be visualized online. Output files can also be downloaded Open | n/a | Durrant’s lab (https://www.biorxiv.org/content/10.1101/2019.12.18.881789v1) |
| Designed to investigate 3D protein-protein interfaces | |||||
| AnchorQuery | http://anchorquery.csb.pitt.edu | Receptor-based pharmacophore screening | Major input: The protein–ligand complex is uploaded in PDB format. The ligand file or library is uploaded for pharmacophore-based screening Major output: The results can be visualized online and downloaded Open | n/a | [274] |
| farPPI | http://cadd.zju.edu.cn/farppi/ | Rescoring PPI inhibitors with MM/PB(GB)SA | Main input: docked pose uploaded in sdf or mol2 format; Receptor file uploaded in PDB format; Selection of the appropriate force field and rescoring procedure Main output: Results file can be downloaded in CSV and PDF format Open | n/a | [275] |
| Receptor-based target-fishing or docking into specific protein families | |||||
| ACID | http://chemyang.ccnu.edu.cn/ccb/server/ACID/ | Consensus inverse docking strategy. Docking with for instance AutoDock Vina, LEDOCK, PLANTS and PSOVina | Major input: select a protein set of interest; ligand information is fetched by drawing a molecule or by uploading a file in sdf, pdb or mol2 format Major output: results can be visualized online Open | n/a | [276] |
| CRDS | http://pbil.kaist.ac.kr/CRDS | Consensus inverse docking | Major input: PubChem or ZINC ID or a ligand file can be uploaded in mol2 or sdf format (3D structure). Compounds are docked to a selected set of targets Major output: the results from docking and the predicted binding pose can be visualized online and the structure files can be downloaded. Information about pathways are also reported. Target UniProt ID and PDB id are provided Open, results are sent by email | n/a | [283] |
| DIA-DB | http://bio-hpc.eu/software/dia-db/ | Inverse docking with Vina | Main input: paste compounds in SMILES or draw the molecule Main output: email with a link to the results, docked molecules Open, results are sent by email | n/a | [215] |
| EDMON v3 | http://atome.cbs.cnrs.fr/ATOME_V3/SERVER/EDMon_v3.html | Combine machine learning on the docking outputs of @TOME and PLANTS and ligand similarity measurements. Inverse screening | Main input: upload a compound collection (maximum ten ligands) in MOL2 format; Select models or structures of the nuclear receptors of interest on which docking will be performed Main output: The results from inverse screening and poses can be visualized online Open | n/a | [284] |
| GOMoDo | http://gomodo.grs.kfa-juelich.de/php/begin.php | Docking with AutoDock Vina or HADDOCK | Main input: provide protein sequence or select sequence from the human GPCRs database and upload a compound structure in pdb or sdf format or select a ligand from the ‘odor ligand’ tab Main output: compound docked into the predicted structure can be downloaded Open | n/a | [286] |
| GUT-DOCK | http://gut-dock.miningmembrane.com/ | Docking with AutoDock Vina | Main input: ligand file can be uploaded in .sdf, .pdbqt, .pdbq, .mol, .mol2, .pdb, or .smi format. The compounds are then docked to gut hormone GPCRs Main output: the docking poses of the ligands can be downloaded as PDB files and visualized online with Ligplot Open | Data privacy and data management documented | [289] |
| idTarget | http://idtarget.rcas.sinica.edu.tw | Inverse docking with MEDock | Major input: upload a ligand file in cif, pdb, mol2, or pdbqt format. Compounds are cross docked to representative pockets Major output: the can be interactively visualized online Open | n/a | [290] |
Screening and receptor-based pharmacophore screening
Auto Core Fragment in silico Screening
Fragment-based drug design is an effective approach for lead discovery. In silico it is however not easy to discriminate binders as fragments are small and can bind in many different regions with similar predicted affinity. Auto Core Fragment in silico Screening (ACFIS) is a web-server for fragment-based drug discovery [229]. It offers three modules to perform fragment-based drug design. ACFIS can, for instance, generate core fragment structure from the active molecule using fragment deconstruction analysis and perform in silico screening by growing fragments to the junction of core fragment structure. Fragments are linked using a modified version of the AutoGrow algorithm [230]. An integrated advanced energy calculation rapidly identifies which fragments fit the binding site. The interface enables users to view top-ranking molecules in 2D and the binding mode in 3D for further exploration. The tool was assessed on many protein crystal structures and was very accurate. It was recently used to dock a small fragment in the binding site of VEGFR-2 kinase (31677447).
CaverWeb
Protein tunnels and channels are key transport pathways that allow ligands to pass between proteins’ external and internal environments. CaverWeb (combine Caver and CaverDock implemented online) is a new tool for analyzing the ligand passage through the biomolecules [231, 232]. The method uses a modified version of the AutoDock Vina docking algorithm for ligand placement docking and implements a parallel heuristic algorithm to search the space of possible trajectories. It can assess many ligands in contrast to many other tools that typically involve the use of molecular dynamics. The tool was found more robust than two related packages SLITHER [233] and MoMA-LigPath [234] on the evaluated datasets. In its present form, CaverDock cannot robustly address the conformational dynamics of the protein structure.
DOCK blaster
The service allows users to dock ligands obtained from the ZINC database or from the Directory of Useful Decoys (DUD) Decoy maker service in the protein of interest using the program DOCK [235]. The tool was benchmarked and found to give reasonable results and half the time, the known bioactive ligands ranked among the top 5% of 100 physically matched decoys chosen on the fly. The tool was for instance used to find opioid analgesics [236] or allosteric modulators of the M2 muscarinic acetylcholine receptor [237].
DOCKovalent
The server allows for the screening large virtual libraries of electrophilic small molecules [238]. This is an adaptation of DOCK 3.6 [239] allowing for covalent docking and targeting proteins’ nucleophiles such as cysteine residues. Given a pregenerated set of ligand conformation and a covalent attachment point in the target protein, it exhaustively samples ligand conformations around the covalent bond and selects the lowest energy pose using a physics-based energy function. It was applied to discover reversible covalent fragments that target distinct protein nucleophiles, including the catalytic serine of AmpC β-lactamase and noncatalytic cysteines in RSK2, MSK1 and JAK3 kinases.
DockThor
This web server is dedicated to protein–ligand docking simulation [240]. The DockThor program uses a grid-based method that employs a steady-state genetic algorithm as a search engine and the MMFF94S force field as the scoring function for pose evaluation. The webserver provides the major steps of ligand and protein preparation, it is possible to change the protonation states of the target amino acid and to define the degree of flexibility of the ligand. At present, it is possible to perform virtual screening experiments with a maximum of 100 compounds as a guest user or 1000 compounds as a registered user. Several optimizations have been introduced in the tool and the last version, DockThor 2, has been shown very efficient to dock flexible ligands such as peptides [241].
EasyVS
It is a web-based platform built to simplify molecule library selection and virtual screening. With an intuitive interface, the tool allows users to go from selecting a protein target with a known structure and tailoring a purchasable molecule library to performing and visualizing docking in a few clicks. The docking is performed with AutoDock Vina. After docking, NNScore 2.01 [242] is used as an external scoring function to estimate Kd values between the target and ligands. This tool has been developed by Wandré N. de P. Veloso, Pâmela M. Rezende, David Ascher, Carlos H. da Silveira and Douglas E. V. Pires.
e-LEA3D
The web server integrates several complementary tools to perform computer-aided drug design based on molecular fragments [243]. It can be considered as 2D or 3D depending on the algorithm selected by the user. In drug discovery projects, there is a considerable interest in identifying novel and diverse molecular scaffolds. The de novo drug design module based on LEA3D [244] is used to invent new ligands to optimize a user-specified scoring function. The composite scoring function includes both structure- and ligand-based evaluations. A heuristic based on a genetic algorithm rapidly finds which fragments or combination of fragments fit a QSAR model or the binding site of a protein. The approach is well-suited for scaffold hopping and the module also allows a scan for possible substituents to a user-specified scaffold. The second module offers SBVS computations (docking with PLANTS [245]) and the filtering of an uploaded library of compounds. The third module addresses the combinatorial library design that is based on a user-drawn scaffold and reactants coming, for example, from a chemical supplier.
ezCADD
This is a web-based modeling environment that allows users to perform different types of computations [246]. The service was also noted to be an effective tool for promoting science, technology, engineering and mathematics education. The different services include ezSMDock (Small-Molecule Docking), ezPPDock (Protein–Protein Docking), ezPocket (Binding Site Detection), ezLigPlot (2D/3D Visualization of Protein–Ligand Interactions), ezHTVS (High-Throughput Virtual Screening), ezGrow (de Novo Lead Optimization), ezTargetSearch (An Integrative Cross-Database Molecule Search Engine for Drug Discovery, Drug Repurposing and Drug Safety Research), ezPocketSearch (Drug Target and Polypharmacology Identification) and ezFAERS (Drug Repurposing using the Food and Drug Administration (FDA) Adverse Event Reporting System). In brief, the docking application ezSMDock requires the uploading of a Protein Data Bank file or entering a PDB file ID. The ligand structures can be provided in different formats. Docking is performed via AutoDock Vina [247] or Smina [248].
iScreen
This server is dedicated to virtual screening, it uses the traditional Chinese medicine (TCM) database [249] for the compound collection and PLANTS [245] for docking. The service offers a protein preparation tool that both extracts protein of interest from a raw input file and estimates the size of the ligand bind site. For customized docking, several services are available including standard, in-water, pH environment and flexible docking modes. TCM de novo drug design can also be carried out via iScreen.
MTiOpenScreen
This tool performs docking and virtual screening of small molecules, offering the possibility to screen in one run up to 5000 molecules uploaded by the user or up to 10 000 molecules taken from different compound collections provided by the system including approved drugs, food compounds, natural products or putative inhibitors of protein–protein interactions (PPI) [250–252]. Two services, MTiAutoDock and MTiOpenScreen, are available. MTiAutoDock, based on AutoDock [247], performs docking into a binding pocket defined by the user or blind docking over the entire protein surface. MTiOpenScreen based on AutoDock Vina [247] (19499576) docking performs automated virtual ligand screening. MTiOpenScreen was for instance used to search for proteasome inhibitors [253].
Pharmit
This tool provides an online, interactive environment for the virtual screening of large compound databases [193]. As seen above, Pharmit can be used as a ligand-based pharmacophore screening tool, but it can also perform receptor-based pharmacophore screening when the 3D structure of a ligand–protein complex is available. Pharmit uses the Volumetric Aligned Molecular Shapes method to search shapes. The shape queries may be provided by the user or extracted directly from the Protein Data Bank code of the protein–ligand complex.
Sanjeevini
The server allows users to dock Lipinski compliant ligands present in several in-built databases or to draw and dock specific ligands [254]. Sanjeevini performs a series of computational steps such as preparation of the protein and the ligand from the files uploaded, docks the candidate molecule at the binding site via a Monte Carlo algorithm (for protein or nucleic acid targets), minimizes and scores the docked complex (three scoring functions are available, one for protein–ligand complexes, one for Zn containing metalloproteinase and one for nucleic acid targets) in an automated mode. The binding pocket can be specified by the users or predicted.
Dock/undock or rescoring tools
Automatic Molecular Mechanics Optimization for in silico Screening (AMMOS2) is a web server for protein–ligand–water complexes refinement via molecular mechanics [255]. The protocol employs atomic-level energy minimization of experimental ligand–protein complexes or of ligands docked for instance via SBVS computations. The web server is based on the standalone software Automatic Molecular Mechanics Optimization for in silico Screening (AMMOS). AMMOS utilizes the physics-based force field AMMP sp4 and performs optimization of protein–ligand interactions at five levels of flexibility of the protein receptor. In AMMOS2, the users can also include explicit water molecules and individual metal ions during the minimization. The 3D structure of the complexes can be visualized and specific protein–ligand interactions can be seen using Protein–Ligand Interaction Profiler (PLIP) computations [256].
CompScore
This web service implements an algorithm that searches for the combination of docking scoring functions components that maximizes any of the BEDROC or Enrichment Factor virtual screening metrics through a genetic algorithm search [257]. It is also possible to rescore a dataset using the CompScore method.
CSM–Lig
Accurately predicting binding affinities is a challenging and difficult task. This a web server tailored to predict the binding affinity of protein–small-molecule complex based on structural signatures. CSM–Lig was first built and evaluated on PDBbind data [258]. Over the PDBbind core set, a blind test of 195 diverse complexes with binding affinities ranging from millimolar to picomolar indicates that the models outperformed many well established scoring functions and predictors. CSM–Lig should there be valuable to helping assess docking poses, the effects of multiple mutations, including insertions, deletions and alternative splicing events, in protein–small-molecule affinity, unraveling important aspects that drive protein–compound recognition.
dockNmine
This portal aims at gathering public and private data into a unique service [259]. Automated queries on protein targets and ligand definitions are performed to UniProt, PubChem and ChEMBL to enhance the results of precomputed docking experiments. When available, public data are automatically added to the docking results to produce protein–ligand binding analysis such as receiver operating characteristic (ROC) curves or enrichment analysis. Users can also upload their own private data. The docking results are categorized into three classes (e.g. good, AutoDock or Vina scores, kcal/mol, are ≤−10; intermediate, −10 > energy ≤ −6.5; and bad, higher scores).
MedusaDock
Existing flexible docking approaches model the ligand and receptor flexibility either separately or in a loosely coupled manner, which usually captures the conformational changes inefficiently. MedusaDock allows for flexible docking and models both ligand and receptor flexibility simultaneously with sets of discrete rotamers [260]. The ligand rotamer library is built on-the-fly during docking simulations. Initially, coarse docking computations are performed with representative ligand conformations, at this stage the ligand is kept rigid. Then the top 10% lowest energy poses are selected for a round of more extensive docking computations. Constraints can be incorporated. The authors tested the approach on several targets (e.g. cyclin-dependent kinase 2, vascular endothelial growth factor receptor 2, HIV reverse transcriptase, and HIV protease) and found significant improvements in virtual screening enrichments when compared to rigid-receptor methods.
MoMA-LigPath
Protein–ligand interactions taking place far away from the active site, during ligand binding or release, may determine molecular specificity and activity. However, obtaining information about these interactions with experimental or computational methods remains difficult. The computational tool MoMA-LigPath is based on a mechanistic representation of the molecular system, considering partial flexibility (in the current version, flexibility is considered for the ligand and all the protein side chains), and on the application of a robotics-inspired algorithm to explore the conformational space [234]. Such a purely geometric approach, together with the efficiency of the exploration algorithm, enables the simulation of ligand unbinding within short computing time. Ligand unbinding pathways generated by MoMA-LigPath represent a first approximation that can be explored further with other molecular modeling approaches. Thus, for example, starting from the model of a protein–ligand complex, MoMA-LigPath computes the ligand exit path from the active site to the protein surface. As such, the tool proposes residues that could be important for binding despite being far away from the binding pocket. The tool was, for instance, used to investigate the unbinding of compound inhibiting phosphatidylinositol 3-kinase-gamma (PI3Kγ) an important target in oncology [261].
Analysis of PrOtein–Ligand Interactions (nAPOLI)
Protein–ligand recognition is mostly driven by specific noncovalent interactions. The large-scale data sets of protein–ligand complexes are available to study recognition. nAPOLI combines large-scale analysis of conserved interactions in protein–ligand complexes at the atomic level, interactive visual representations and comprehensive reports of the interacting residues/atoms to detect and explore conserved noncovalent interactions [262]. The tool was, for example, used to study kinases and human nuclear receptor proteins.
PlayMolecule
This is a platform that offers different applications for preparing a target, search for cryptic and druggable cavities, run molecular dynamics, machine learning and rescoring. For instance, DeepSite is a protein-binding site predictor tool that uses 3D-convolutional neural networks [263], KDEEP allows to protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks [264] and BindScope, a web application for large-scale active–inactive classification of compounds based on deep convolutional neural networks [265] (30169549).
PRODIGY-LIG
PROtein binDIng enerGY (PRODIGY prediction) is a collection of web services focused on the prediction of binding affinity in biological complexes as well as the identification of biological interfaces from crystallographic one. PRODIGY-LIG aimed at the prediction of affinity in protein-small ligand complexes [266]. The original predictive method was readapted for small ligand by making use of atomic instead of amino acid contacts. It was successfully applied for the blind prediction of 102 protein–ligand complexes during the D3R Grand Challenge 2.
Screening explorer
This is a web-based application that allows for an intuitive evaluation of the results of screening experiments using complementary metrics [267]. The service facilitates screening results by linking different metrics interactively in an interactive usable web-based application. Charts representing predictiveness, ROC, enrichment curves, scores and active compounds distributions can be obtained. Simple consensus scoring methods based on scores normalization, standardization (z-scores), and compounds ranking to evaluate the enrichments that can be expected through methods combination are also available.
SwissDock
This is a web server dedicated to ligand docking [268], either on a known binding site or on the entire surface. It is based on the protein–ligand docking program EADock dihedral space sampling (DSS) [269]. Ligand and protein files can be prepared using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/) and the results visualized online or downloaded. As the tool is based on the accurate physics-based estimation of binding modes and free energies using the Chemistry at HARvard Macromolecular Mechanics (CHARMM) force field [270], it cannot handle many ligands but should be used after a fast screening engine (e.g. AutoDock Vina) to further validate a small list of preselected compounds. The tool was, for instance, used to dock molecules into the muscarinic acetylcholine receptors [271], to investigate potential antiprion compounds [272] or inhibitors of Dub3, a protein playing a role in cancer [273].
Webina is an open-source library and web app that runs AutoDock Vina in a web browser (Durrant’s lab). The service allows users to upload a prepared receptor structure and a ligand. Several parameters can be selected, for instance users can upload a reference experimental ligand structure and decide about the docking box where the search will be performed among others. The tool outputs the docked ligand poses and offers efficient interactive visualization. The service has been tested on several ligands and proteins including La-related protein 1 and poly-(ADP-ribose)-polymerase 1.
Designed to investigate 3D protein–protein interfaces
AnchorQuery
This is a web application for the rational structure-based design of PPI inhibitors [274]. A specialized variant of pharmacophore search is used to rapidly screen libraries consisting of more than 31 million synthesizable compounds biased by design to preferentially target PPIs. AnchorQuery provides all the tools necessary for users to perform online interactive virtual screens of millions of compounds, including pharmacophore elucidation and search, and enrichment analysis.
farPPI
Designing PPI inhibitors is difficult as often, the binding pockets tend to be more flat and larger than regular binding sites such as those found in enzymes or in membrane receptors. The Fast Amber Rescoring webserver offers a freely available service for rescoring the docking poses of PPI inhibitors by using the MM/PB(GB)SA methods [275].
Receptor-based target-fishing or docking into specific protein families
Auto in silico Consensus Inverse Docking (ACID)
The ACID server combines the results of four docking methods into a consensus inverse docking scheme [276]. The selected docking tools involve AutoDock Vina [277], LEDOCK (http://www.lephar.com), PLANTS [245], and PSOVina [278]. The reasons for this choice mentioned by the authors is that the docking methods use different conformational search algorithms and different scoring functions and that in general, combining different tools has been shown beneficial in term of hit finding. In addition, the server applies a Molecular Mechanics/Poisson-Boltzmann Surface Area [279] protocol and X-SCORE [280, 281] for the final binding energy calculations. Users can upload a compound or draw a molecule and dock it in selected target families.
Consensus Reverse Docking System
The CRDS docks a compound into 5254 protein X-ray structures collected from the sc-PDB database [282]. Gold, Vina and LeDock binding scores are reported and the targets are ranked by consensus scoring. The first target seen in the output table has thus the highest consensus score. Another output shows which pathways the resulting top 50 targets are associated with [283]. The top 10 pathways are displayed on a pie chart.
DIA-DB
This server aims at helping users identifying potential antidiabetic drugs [215]. As seen above, it can be used via ligand-based shape similarity search but inverse virtual screening can also be carried out with Autodock Vina. About 20 protein targets known to play a role in diabetes are stored in DIA-DB. A docking score for a novel input query compound against the different targets is returned as well as the structure of the predicted complexes and various graphical views of the binding pockets.
Endocrine Disruptor MONitoring (EDMON v3)
The approach [284] takes advantage of a new interface between the virtual screening standalone package PLANTS [245] and the web server @TOME [285] to screen multiple conformations in parallel. It allows users to systematically deduce a shape restraint and binding site boundaries based on the geometry of the original ligand from each crystal structure in a fully automatic manner. Subsequent postprocessing is performed using various chemoinformatics tools including several scoring functions to predict protein–ligand affinity and select an optimal pose. The approach evaluates the applicability of machine learning on the docking outputs of @TOME and PLANTS and ligand similarity measurements. Users have to supply a ligand that is inversely docked into several nuclear hormone receptors, an important class of targets.
GPCR Online MOdeling and DOcking server (GOMoDo)
This tool can automatically perform template choice, homology modeling and either blind or information-driven docking [286], with, for instance, AutoDock Vina [247, 277] or high ambiguity-driven protein–protein DOCKing (HADDOCK) [287]. This server was for example used to investigate muscarinic acetylcholine receptors (M1 to M5) expressed in murine brain microvascular endothelium and suggested that drug development should focus on the allosteric sites of the M1 and M3 receptors [288].
GUT-DOCK
The prolonged use of many currently available drugs results in the severe side effect of the disruption of glucose metabolism leading to type 2 diabetes mellitus. Gut hormone receptors including glucagon receptor (GCGR) and the incretin hormone receptors: glucagon-like peptide 1 receptor (GLP1R) and gastric inhibitory polypeptide receptor (GIPR) are important drug targets for the treatment of diabetes, as they play roles in the regulation of glucose and insulin levels and of food intake. GUT-DOCK allows users to compute the binding affinities between a small molecule and various class B GPCRs, gut hormone receptors, vasoactive intestinal polypeptide receptor 1 (VIPR1) and pituitary adenylate cyclase-activating polypeptide receptor (PAC1R) [289]. Several protein structures were modeled and simulated using molecular dynamics. The docking procedure is carried out with AutoDock Vina. Users have to provide the small-molecule ligands. Short peptides can also be docked.
idTarget
This web service predicts possible binding targets of small chemical molecules using the maximum entropy-based docking (MEDock) program, which generates initial docking poses of the ligands based on the divide-and-conquer docking approach. The docking poses are rescored according to the modified AutoDock4 scoring function that was developed on robust regression analysis and quantum chemical charge models [290, 291]. Affinity profiles of the protein targets are used to provide confidence levels of prediction. The program was efficaciously used to identify many known off-targets of drugs or drug-like compounds.
Discussion
The identification of bioactive compounds acting on thousands of putative therapeutic targets via the experimental screening of large compound collections is time-consuming and costly. Virtual screening assists the process by providing a small list of molecules to assess experimentally instead of a collection of thousands or millions of compounds. Performing virtual screening computations using standalone approaches is still a challenging task. However, many online tools are available today to facilitate the search of bioactive compounds. This review provides a list of web servers that should assist wet-lab researchers identifying tools that could be beneficial for their screening projects. We discussed different types of LBVS engines and then reported various approaches that perform receptor-based or SBVS computations. Experimental teams have now not only access to different types of virtual screening web servers but also to tools that evaluate some ADME-Tox properties or that generate ideas for compound optimization (see Supplementary Table S1). These approaches should help identify new hits but it is also important to note that several in silico screening approaches can also be used, after some tuning, for drug repositioning, target-fishing or profiling, for studying drugs’ adverse effects and for polypharmacology prediction. As seen above, different types of virtual screening computations can also be valuable for rational multitarget drug design [292–298]. To further assist users in the selection of virtual screening methods or in combining applications, we provide some examples of workflows that could be used, depending on the study questions and the available input data (Supplementary Figure S1). Taken together, we are convinced that novel hit compounds will be discovered by combining online calculations and experimental studies, and possibly, some will become drug candidates and/or will help to explore complex disease conditions. In the era of open and big data, the web servers developed within academic or public–private collaborative settings should definitively play a major role in the coming years to advance the success of drug discovery and chemical biology projects.
Key Points
We report ligand-based and SBVS/docking web servers.
Based on the description of the tools, experimentalists should be able to identify the screening services that best suit their needs.
This survey can also be valuable for computer scientists who aim at developing novel approaches or novel interfaces.
Supplementary Material
N. Singh has been working the field of in silico drug design for several years and presently investigates different types of rescoring protocols.
L. Chaput has been working in the field of in silico drug design for several years and presently works on ligand-based pharmacological profiling methods.
B. Villoutreix has been working at the interface between molecular medicine, structural bioinformatics and chemoinformatics for over 25 years. He is director of research at the Inserm Institute (the French National Institute of Health & Medical Research, a leading academic biomedical research institution).
Funding
The Inserm Institute, Lille I-Site and Lille Region.
References
- 1. Hammel B, Michel MC. Why are new drugs expensive and how can they stay affordable? Handb Exp Pharmacol 2019;260:453–66. [DOI] [PubMed] [Google Scholar]
- 2. Khanna I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov Today 2012;17:1088–102. [DOI] [PubMed] [Google Scholar]
- 3. Rubin EH, Gilliland DG. Drug development and clinical trials--the path to an approved cancer drug. Nat Rev Clin Oncol 2012;9:215–22. [DOI] [PubMed] [Google Scholar]
- 4. Joyner MJ, Paneth N. Promises, promises, and precision medicine. J Clin Invest 2019;129:946–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shendure J, Findlay GM, Snyder MW. Genomic medicine-progress, pitfalls, and promise. Cell 2019;177:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen H, Kogej T, Engkvist O. Cheminformatics in drug discovery, an industrial perspective. Mol Inform 2018;37:e1800041. [DOI] [PubMed] [Google Scholar]
- 7. Ekins S, Puhl AC, Zorn KM, et al. Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 2019;18:435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyer JG, Liu S, Miller IJ, et al. Learning drug functions from chemical structures with convolutional neural networks and random forests. J Chem Inf Model 2019;59:4438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paricharak S, Mendez-Lucio O, Chavan Ravindranath A, et al. Data-driven approaches used for compound library design, hit triage and bioactivity modeling in high-throughput screening. Brief Bioinform 2018;19:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richter L, Ecker GF. Medicinal chemistry in the era of big data. Drug Discov Today Technol 2015;14:37–41. [DOI] [PubMed] [Google Scholar]
- 11. Sieg J, Flachsenberg F, Rarey M. In need of bias control: evaluating chemical data for machine learning in structure-based virtual screening. J Chem Inf Model 2019;59:947–61. [DOI] [PubMed] [Google Scholar]
- 12. Wallach I, Heifets A. Most ligand-based classification benchmarks reward memorization rather than generalization. J Chem Inf Model 2018;58:916–32. [DOI] [PubMed] [Google Scholar]
- 13. Yang X, Wang Y, Byrne R, et al. Concepts of artificial intelligence for computer-assisted drug discovery. Chem Rev 2019;119:10520–94. [DOI] [PubMed] [Google Scholar]
- 14. Fourches D, Muratov E, Tropsha A. Trust, but Verify II: a practical guide to Chemogenomics data curation. J Chem Inf Model 2016;56:1243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Varnek A, Baskin II. Chemoinformatics as a theoretical chemistry discipline. Mol Inform 2011;30:20–32. [DOI] [PubMed] [Google Scholar]
- 16. Koromina M, Pandi MT, Patrinos GP. Rethinking drug repositioning and development with artificial intelligence, machine learning, and omics. OMICS 2019;23:539–48. [DOI] [PubMed] [Google Scholar]
- 17. Lo YC, Rensi SE, Torng W, et al. Machine learning in chemoinformatics and drug discovery. Drug Discov Today 2018;23:1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu H. Big data and artificial intelligence Modeling for drug discovery. Annu Rev Pharmacol Toxicol 2020;60:573–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hughes JP, Rees S, Kalindjian SB, et al. Principles of early drug discovery. Br J Pharmacol 2011;162:1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aulner N, Danckaert A, Ihm J, et al. Next-generation phenotypic screening in early drug discovery for infectious diseases. Trends Parasitol 2019;35:559–70. [DOI] [PubMed] [Google Scholar]
- 21. Brown DG, Wobst HJ. Opportunities and challenges in phenotypic screening for neurodegenerative disease research. J Med Chem 2019. doi: 10.1021/acs.jmedchem.9b00797. [DOI] [PubMed] [Google Scholar]
- 22. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 2016;47:20–33. [DOI] [PubMed] [Google Scholar]
- 23. Rawlins MD. Cutting the cost of drug development? Nat Rev Drug Discov 2004;3:360–4. [DOI] [PubMed] [Google Scholar]
- 24. Hillisch A, Heinrich N, Wild H. Computational chemistry in the pharmaceutical industry: from childhood to adolescence. ChemMedChem 2015;10:1958–62. [DOI] [PubMed] [Google Scholar]
- 25. Rognan D. The impact of in silico screening in the discovery of novel and safer drug candidates. Pharmacol Ther 2017;175:47–66. [DOI] [PubMed] [Google Scholar]
- 26. Taboureau O, Baell JB, Fernandez-Recio J, et al. Established and emerging trends in computational drug discovery in the structural genomics era. Chem Biol 2012;19:29–41. [DOI] [PubMed] [Google Scholar]
- 27. Clark DE. What has virtual screening ever done for drug discovery? Expert Opin Drug Discov 2008;3:841–51. [DOI] [PubMed] [Google Scholar]
- 28. Koppen H. Virtual screening—what does it give us? Curr Opin Drug Discov Devel 2009;12:397–407. [PubMed] [Google Scholar]
- 29. Rester U. From virtuality to reality—virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective. Curr Opin Drug Discov Devel 2008;11:559–68. [PubMed] [Google Scholar]
- 30. Westermaier Y, Barril X, Scapozza L. Virtual screening: an in silico tool for interlacing the chemical universe with the proteome. Methods 2015;71:44–57. [DOI] [PubMed] [Google Scholar]
- 31. Agamah FE, Mazandu GK, Hassan R, et al. Computational/in silico methods in drug target and lead prediction. Brief Bioinform 2019. doi: 10.1093/bib/bbz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Banegas-Luna AJ, Ceron-Carrasco JP, Perez-Sanchez H. A review of ligand-based virtual screening web tools and screening algorithms in large molecular databases in the age of big data. Future Med Chem 2018;10:2641–58. [DOI] [PubMed] [Google Scholar]
- 33. Ekins S, Williams AJ. Precompetitive preclinical ADME/Tox data: set it free on the web to facilitate computational model building and assist drug development. Lab Chip 2010;10:13–22. [DOI] [PubMed] [Google Scholar]
- 34. Jia CY, Li JY, Hao GF, et al. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today 2020;25:248–58. [DOI] [PubMed] [Google Scholar]
- 35. Naderi M, Lemoine JM, Govindaraj RG, et al. Binding site matching in rational drug design: algorithms and applications. Brief Bioinform 2019;20:2167–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ertl P, Jelfs S. Designing drugs on the internet? Free web tools and services supporting medicinal chemistry. Curr Top Med Chem 2007;7:1491–501. [DOI] [PubMed] [Google Scholar]
- 37. Nicola G, Liu T, Gilson MK. Public domain databases for medicinal chemistry. J Med Chem 2012;55:6987–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tetko IV, Maran U, Tropsha A. Public (Q)SAR services, integrated Modeling environments, and model repositories on the web: state of the art and perspectives for future development. Mol Inform 2017;36. doi: 10.1002/minf.201600082. [DOI] [PubMed] [Google Scholar]
- 39. Villoutreix BO, Renault N, Lagorce D, et al. Free resources to assist structure-based virtual ligand screening experiments. Curr Protein Pept Sci 2007;8:381–411. [DOI] [PubMed] [Google Scholar]
- 40. Yuriev E, Ramsland PA. Carbohydrates in cyberspace. Front Immunol 2015;6:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hohman M, Gregory K, Chibale K, et al. Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discov Today 2009;14:261–70. [DOI] [PubMed] [Google Scholar]
- 42. Pawar G, Madden JC, Ebbrell D, et al. In silico toxicology data resources to support read-across and (Q)SAR. Front Pharmacol 2019;10:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singla D, Dhanda SK, Chauhan JS, et al. Open source software and web services for designing therapeutic molecules. Curr Top Med Chem 2013;13:1172–91. [DOI] [PubMed] [Google Scholar]
- 44. Villoutreix BO, Lagorce D, Labbe CM, et al. One hundred thousand mouse clicks down the road: selected online resources supporting drug discovery collected over a decade. Drug Discov Today 2013;18:1081–9. [DOI] [PubMed] [Google Scholar]
- 45. Daina A, Blatter MC, Gerritsen VB, et al. Educational tools to introduce computer-aided drug design to students and to the public at large. Chimia (Aarau) 2018;72:55–61. [DOI] [PubMed] [Google Scholar]
- 46. Martinez X, Krone M, Alharbi N, et al. Molecular graphics: bridging structural biologists and computer scientists. Structure 2019;27:1617–23. [DOI] [PubMed] [Google Scholar]
- 47. Sydow D, Morger A, Driller M, et al. TeachOpenCADD: a teaching platform for computer-aided drug design using open source packages and data. J Chem 2019;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horvath D. A virtual screening approach applied to the search for trypanothione reductase inhibitors. J Med Chem 1997;40:2412–23. [DOI] [PubMed] [Google Scholar]
- 49. Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol 2001;5:375–82. [DOI] [PubMed] [Google Scholar]
- 50. Cavasotto CN, Adler NS, Aucar MG. Quantum chemical approaches in structure-based virtual screening and lead optimization. Front Chem 2018;6:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shoichet BK. Virtual screening of chemical libraries. Nature 2004;432:862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phatak SS, Stephan CC, Cavasotto CN. High-throughput and in silico screenings in drug discovery. Expert Opin Drug Discov 2009;4:947–59. [DOI] [PubMed] [Google Scholar]
- 53. Heikamp K, Bajorath J. The future of virtual compound screening. Chem Biol Drug Des 2013;81:33–40. [DOI] [PubMed] [Google Scholar]
- 54. Slater O, Kontoyianni M. The compromise of virtual screening and its impact on drug discovery. Expert Opin Drug Discov 2019;14:619–37. [DOI] [PubMed] [Google Scholar]
- 55. Villoutreix BO, Kuenemann MA, Poyet JL, et al. Drug-like protein-protein interaction modulators: challenges and opportunities for drug discovery and chemical biology. Mol Inform 2014;33:414–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Forli S. Charting a path to success in virtual screening. Molecules 2015;20:18732–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hutter MC. The current limits in virtual screening and property prediction. Future Med Chem 2018;10:1623–35. [DOI] [PubMed] [Google Scholar]
- 58. Scior T, Bender A, Tresadern G, et al. Recognizing pitfalls in virtual screening: a critical review. J Chem Inf Model 2012;52:867–81. [DOI] [PubMed] [Google Scholar]
- 59. Yuriev E, Holien J, Ramsland PA. Improvements, trends, and new ideas in molecular docking: 2012-2013 in review. J Mol Recognit 2015;28:581–604. [DOI] [PubMed] [Google Scholar]
- 60. Polgar T, Keseru GM. Integration of virtual and high throughput screening in lead discovery settings. Comb Chem High Throughput Screen 2011;14:889–97. [DOI] [PubMed] [Google Scholar]
- 61. Tanrikulu Y, Kruger B, Proschak E. The holistic integration of virtual screening in drug discovery. Drug Discov Today 2013;18:358–64. [DOI] [PubMed] [Google Scholar]
- 62. Martin EJ, Jansen JM. Biased diversity for effective virtual screening. J Chem Inf Model 2020. doi: 10.1021/acs.jcim.9b01155. [DOI] [PubMed] [Google Scholar]
- 63. Stumpfe D, Bajorath J. Current trends, overlooked issues, and unmet challenges in virtual screening. J Chem Inf Model 2020. doi: 10.1021/acs.jcim.9b01101. [DOI] [PubMed] [Google Scholar]
- 64. Davies JW, Glick M, Jenkins JL. Streamlining lead discovery by aligning in silico and high-throughput screening. Curr Opin Chem Biol 2006;10:343–51. [DOI] [PubMed] [Google Scholar]
- 65. Polishchuk PG, Madzhidov TI, Varnek A. Estimation of the size of drug-like chemical space based on GDB-17 data. J Comput Aided Mol Des 2013;27:675–9. [DOI] [PubMed] [Google Scholar]
- 66. Reymond JL, Awale M. Exploring chemical space for drug discovery using the chemical universe database. ACS Chem Nerosci 2012;3:649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grebner C, Malmerberg E, Shewmaker A, et al. Virtual screening in the cloud: how big is big enough? J Chem Inf Model 2019. doi: 10.1021/acs.jcim.9b00779. [DOI] [PubMed] [Google Scholar]
- 68. Lyu J, Wang S, Balius TE, et al. Ultra-large library docking for discovering new chemotypes. Nature 2019;566:224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov 2016;11:137–48. [DOI] [PubMed] [Google Scholar]
- 70. Stumpfe D, Hu H, Bajorath J. Evolving concept of activity cliffs. ACS Omega 2019;4:14360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stumpfe D, Ripphausen P, Bajorath J. Virtual compound screening in drug discovery. Future Med Chem 2012;4:593–602. [DOI] [PubMed] [Google Scholar]
- 72. Vogt M. Progress with modeling activity landscapes in drug discovery. Expert Opin Drug Discov 2018;13:605–15. [DOI] [PubMed] [Google Scholar]
- 73. Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discov Today 2006;11:1046–53. [DOI] [PubMed] [Google Scholar]
- 74. Bajusz D, Racz A, Heberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Chem 2015;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lu X, Yang H, Chen Y, et al. The development of pharmacophore Modeling: generation and recent applications in drug discovery. Curr Pharm Des 2018;24:3424–39. [DOI] [PubMed] [Google Scholar]
- 76. Seidel T, Schuetz DA, Garon A, et al. The pharmacophore concept and its applications in computer-aided drug design. Prog Chem Org Nat Prod 2019;110:99–141. [DOI] [PubMed] [Google Scholar]
- 77. Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 2010;15:444–50. [DOI] [PubMed] [Google Scholar]
- 78. Ballester PJ. Ultrafast shape recognition: method and applications. Future Med Chem 2011;3:65–78. [DOI] [PubMed] [Google Scholar]
- 79. Diller DJ, Connell ND, Welsh WJ. Avalanche for shape and feature-based virtual screening with 3D alignment. J Comput Aided Mol Des 2015;29:1015–24. [DOI] [PubMed] [Google Scholar]
- 80. Hawkins PC, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J Med Chem 2007;50:74–82. [DOI] [PubMed] [Google Scholar]
- 81. Kumar A, Zhang KYJ. Advances in the development of shape similarity methods and their application in drug discovery. Front Chem 2018;6:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nicholls A, McGaughey GB, Sheridan RP, et al. Molecular shape and medicinal chemistry: a perspective. J Med Chem 2010;53:3862–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shin WH, Zhu X, Bures MG, et al. Three-dimensional compound comparison methods and their application in drug discovery. Molecules 2015;20:12841–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Carpenter KA, Huang X. Machine learning-based virtual screening and its applications to Alzheimer's drug discovery: a review. Curr Pharm Des 2018;24:3347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jain AN, Cleves AE. Does your model weigh the same as a duck? J Comput Aided Mol Des 2012;26:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Baskin II, Winkler D, Tetko IV. A renaissance of neural networks in drug discovery. Expert Opin Drug Discov 2016;11:785–95. [DOI] [PubMed] [Google Scholar]
- 87. Cherkasov A, Muratov EN, Fourches D, et al. QSAR modeling: where have you been? Where are you going to? J Med Chem 2014;57:4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Scior T, Medina-Franco JL, Do QT, et al. How to recognize and workaround pitfalls in QSAR studies: a critical review. Curr Med Chem 2009;16:4297–313. [DOI] [PubMed] [Google Scholar]
- 89. Sosnin S, Vashurina M, Withnall M, et al. A survey of multi-task learning methods in Chemoinformatics. Mol Inform 2019;38:e1800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen H, Engkvist O, Wang Y, et al. The rise of deep learning in drug discovery. Drug Discov Today 2018;23:1241–50. [DOI] [PubMed] [Google Scholar]
- 91. Gertrudes JC, Maltarollo VG, Silva RA, et al. Machine learning techniques and drug design. Curr Med Chem 2012;19:4289–97. [DOI] [PubMed] [Google Scholar]
- 92. Mater AC, Coote ML. Deep learning in chemistry. J Chem Inf Model 2019;59:2545–59. [DOI] [PubMed] [Google Scholar]
- 93. Neves BJ, Braga RC, Melo-Filho CC, et al. QSAR-based virtual screening: advances and applications in drug discovery. Front Pharmacol 2018;9:1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 2019;18:463–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhang L, Tan J, Han D, et al. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov Today 2017;22:1680–5. [DOI] [PubMed] [Google Scholar]
- 96. Robinson MC, Glen RC, Lee AA. Validating the validation: reanalyzing a large-scale comparison of deep learning and machine learning models for bioactivity prediction. J Comput Aided Mol Des 2020. doi: 10.1007/s10822-019-00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Plewczynski D, Spieser SA, Koch U. Performance of machine learning methods for ligand-based virtual screening. Comb Chem High Throughput Screen 2009;12:358–68. [DOI] [PubMed] [Google Scholar]
- 98. Batool M, Ahmad B, Choi S. A structure-based drug discovery paradigm. Int J Mol Sci 2019;20. doi: 10.3390/ijms20112783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bjij I, Olotu FA, Agoni C, et al. Covalent inhibition in drug discovery: filling the void in literature. Curr Top Med Chem 2018;18:1135–45. [DOI] [PubMed] [Google Scholar]
- 100. De Cesco S, Kurian J, Dufresne C, et al. Covalent inhibitors design and discovery. Eur J Med Chem 2017;138:96–114. [DOI] [PubMed] [Google Scholar]
- 101. Guedes IA, Pereira FSS, Dardenne LE. Empirical scoring functions for structure-based virtual screening: applications, critical aspects, and challenges. Front Pharmacol 2018;9:1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li J, Fu A, Zhang L. An overview of scoring functions used for protein-ligand interactions in molecular docking. Interdiscip Sci 2019;11:320–8. [DOI] [PubMed] [Google Scholar]
- 103. Luo J, Wei W, Waldispuhl J, et al. Challenges and current status of computational methods for docking small molecules to nucleic acids. Eur J Med Chem 2019;168:414–25. [DOI] [PubMed] [Google Scholar]
- 104. Pason LP, Sotriffer CA. Empirical scoring functions for affinity prediction of protein-ligand complexes. Mol Inform 2016;35:541–8. [DOI] [PubMed] [Google Scholar]
- 105. Bian Y, Xie XS. Computational fragment-based drug design: current trends, strategies, and applications. AAPS J 2018;20:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Campagna-Slater V, Therrien E, Weill N, et al. Methods for docking small molecules to macromolecules: a user's perspective. 2. Applications. Curr Pharm Des 2014;20:3360–72. [DOI] [PubMed] [Google Scholar]
- 107. Weill N, Therrien E, Campagna-Slater V, et al. Methods for docking small molecules to macromolecules: a user's perspective. 1. The theory. Curr Pharm Des 2014;20:3338–59. [DOI] [PubMed] [Google Scholar]
- 108. Abramyan TM, An Y, Kireev D. Off-pocket activity cliffs: a puzzling facet of molecular recognition. J Chem Inf Model 2020;60:152–61. [DOI] [PubMed] [Google Scholar]
- 109. Segall M. Advances in multiparameter optimization methods for de novo drug design. Expert Opin Drug Discov 2014;9:803–17. [DOI] [PubMed] [Google Scholar]
- 110. Fischer T, Gazzola S, Riedl R. Approaching target selectivity by De novo drug design. Expert Opin Drug Discov 2019;14:791–803. [DOI] [PubMed] [Google Scholar]
- 111. Schneider G. De novo design - hop(p)ing against hope. Drug Discov Today Technol 2013;10:e453–60. [DOI] [PubMed] [Google Scholar]
- 112. Schneider G, Clark DE. Automated De novo drug design: are we nearly there yet? Angew Chem Int Ed Engl 2019;58:10792–803. [DOI] [PubMed] [Google Scholar]
- 113. Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov 2005;4:649–63. [DOI] [PubMed] [Google Scholar]
- 114. Bietz S, Fahrrolfes R, Rarey M. The art of compiling protein binding site ensembles. Mol Inform 2016;35:593–8. [DOI] [PubMed] [Google Scholar]
- 115. Ehrt C, Brinkjost T, Koch O. Binding site characterization - similarity, promiscuity, and druggability. Medchemcomm 2019;10:1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Haupt VJ, Daminelli S, Schroeder M. Drug promiscuity in PDB: protein binding site similarity is key. PLoS One 2013;8:e65894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pu L, Govindaraj RG, Lemoine JM, et al. DeepDrug3D: classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput Biol 2019;15:e1006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Naderi M, Govindaraj RG, Brylinski M. eModel-BDB: a database of comparative structure models of drug-target interactions from the binding database. Gigascience 2018;7. doi: 10.1093/gigascience/giy091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cavasotto CN. Homology models in docking and high-throughput docking. Curr Top Med Chem 2011;11:1528–34. [DOI] [PubMed] [Google Scholar]
- 120. Davis AM, Teague SJ, Kleywegt GJ. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew Chem Int Ed Engl 2003;42:2718–36. [DOI] [PubMed] [Google Scholar]
- 121. Muhammed MT, Aki-Yalcin E. Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem Biol Drug Des 2019;93:12–20. [DOI] [PubMed] [Google Scholar]
- 122. Wlodawer A, Minor W, Dauter Z, et al. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J 2008;275:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Carugo O. How large B-factors can be in protein crystal structures. BMC Bioinformatics 2018;19:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Deller MC, Rupp B. Models of protein-ligand crystal structures: trust, but verify. J Comput Aided Mol Des 2015;29:817–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lange J, Baakman C, Pistorius A, et al. Facilities that make the PDB data collection more powerful. Protein Sci 2020;29:330–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Amaro RE, Baudry J, Chodera J, et al. Ensemble docking in drug discovery. Biophys J 2018;114:2271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Aminpour M, Montemagno C, Tuszynski JA. An overview of molecular Modeling for drug discovery with specific illustrative examples of applications. Molecules 2019;24. doi: 10.3390/molecules24091693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Gioia D, Bertazzo M, Recanatini M, et al. Dynamic docking: a paradigm shift in computational drug discovery. Molecules 2017;22. doi: 10.3390/molecules22112029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bera I, Payghan PV. Use of molecular dynamics simulations in structure-based drug discovery. Curr Pharm Des 2019;25:3339–49. [DOI] [PubMed] [Google Scholar]
- 130. Sledz P, Caflisch A. Protein structure-based drug design: from docking to molecular dynamics. Curr Opin Struct Biol 2018;48:93–102. [DOI] [PubMed] [Google Scholar]
- 131. Vajda S, Beglov D, Wakefield AE, et al. Cryptic binding sites on proteins: definition, detection, and druggability. Curr Opin Chem Biol 2018;44:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Drwal MN, Griffith R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov Today Technol 2013;10:e395–401. [DOI] [PubMed] [Google Scholar]
- 133. Kumar A, Zhang KY. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015;71:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Li B, Kang X, Zhao D, et al. Machine learning models combined with virtual screening and molecular docking to predict human topoisomerase I inhibitors. Molecules 2019;24. doi: 10.3390/molecules24112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem 2005;5:739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Awale M, Reymond JL. Web-based tools for Polypharmacology prediction. Methods Mol Biol 1888;2019:255–72. [DOI] [PubMed] [Google Scholar]
- 137. Cereto-Massague A, Ojeda MJ, Valls C, et al. Tools for in silico target fishing. Methods 2015;71:98–103. [DOI] [PubMed] [Google Scholar]
- 138. Pinzi L, Rastelli G. Identification of target associations for Polypharmacology from analysis of crystallographic ligands of the protein data Bank. J Chem Inf Model 2020;60:372–90. [DOI] [PubMed] [Google Scholar]
- 139. Trosset JY, Cave C. In silico drug-target profiling. Methods Mol Biol 1953;2019:89–103. [DOI] [PubMed] [Google Scholar]
- 140. Luo Q, Zhao L, Hu J, et al. The scoring bias in reverse docking and the score normalization strategy to improve success rate of target fishing. PLoS One 2017;12:e0171433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Xu X, Huang M, Zou X. Docking-based inverse virtual screening: methods, applications, and challenges. Biophys Rep 2018;4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tanoli Z, Seemab U, Scherer A, et al. Exploration of databases and methods supporting drug repurposing: a comprehensive survey. Brief Bioinform 2020. doi: 10.1093/bib/bbaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Paananen J, Fortino V. An omics perspective on drug target discovery platforms. Brief Bioinform 2019. doi: 10.1093/bib/bbz122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Trosset JY, Cave C. In silico target Druggability assessment: from structural to systemic approaches. Methods Mol Biol 1953;2019:63–88. [DOI] [PubMed] [Google Scholar]
- 145. Vukovic S, Huggins DJ. Quantitative metrics for drug-target ligandability. Drug Discov Today 2018;23:1258–66. [DOI] [PubMed] [Google Scholar]
- 146. Ehrt C, Brinkjost T, Koch O. Impact of binding site comparisons on medicinal chemistry and rational molecular design. J Med Chem 2016;59:4121–51. [DOI] [PubMed] [Google Scholar]
- 147. Kufareva I, Chen YC, Ilatovskiy AV, et al. Compound activity prediction using models of binding pockets or ligand properties in 3D. Curr Top Med Chem 2012;12:1869–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Perot S, Sperandio O, Miteva MA, et al. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today 2010;15:656–67. [DOI] [PubMed] [Google Scholar]
- 149. Skolnick J, Gao M, Roy A, et al. Implications of the small number of distinct ligand binding pockets in proteins for drug discovery, evolution and biochemical function. Bioorg Med Chem Lett 2015;25:1163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Stank A, Kokh DB, Fuller JC, et al. Protein binding pocket dynamics. Acc Chem Res 2016;49:809–15. [DOI] [PubMed] [Google Scholar]
- 151. Ferreira de Freitas R, Schapira M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 2017;8:1970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dmitriev AV, Lagunin AA, Karasev Dcapital AC, et al. Prediction of drug-drug interactions related to inhibition or induction of drug-metabolizing enzymes. Curr Top Med Chem 2019;19:319–36. [DOI] [PubMed] [Google Scholar]
- 153. Idakwo G, Luttrell J, Chen M, et al. A review on machine learning methods for in silico toxicity prediction. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2018;36:169–91. [DOI] [PubMed] [Google Scholar]
- 154. Lagorce D, Douguet D, Miteva MA, et al. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci Rep 2017;7:46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Liu H, Dong K, Zhang W, et al. Prediction of brain:blood unbound concentration ratios in CNS drug discovery employing in silico and in vitro model systems. Drug Discov Today 2018;23:1357–72. [DOI] [PubMed] [Google Scholar]
- 156. Petito ES, Foster DJR, Ward MB, et al. Molecular Modeling approaches for the prediction of selected pharmacokinetic properties. Curr Top Med Chem 2018;18:2230–8. [DOI] [PubMed] [Google Scholar]
- 157. Raies AB, Bajic VB. In silico toxicology: comprehensive benchmarking of multi-label classification methods applied to chemical toxicity data. Wiley Interdiscip Rev Comput Mol Sci 2018;8:e1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Saini N, Bakshi S, Sharma S. In-silico approach for drug induced liver injury prediction: recent advances. Toxicol Lett 2018;295:288–95. [DOI] [PubMed] [Google Scholar]
- 159. Ferreira LLG, Andricopulo AD. ADMET modeling approaches in drug discovery. Drug Discov Today 2019;24:1157–65. [DOI] [PubMed] [Google Scholar]
- 160. Montanari F, Ecker GF. Prediction of drug-ABC-transporter interaction--recent advances and future challenges. Adv Drug Deliv Rev 2015;86:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Moroy G, Martiny VY, Vayer P, et al. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today 2012;17:44–55. [DOI] [PubMed] [Google Scholar]
- 162. Stark C, Steger-Hartmann T. Nonclinical safety and toxicology. Handb Exp Pharmacol 2016;232:261–83. [DOI] [PubMed] [Google Scholar]
- 163. Tao L, Zhang P, Qin C, et al. Recent progresses in the exploration of machine learning methods as in-silico ADME prediction tools. Adv Drug Deliv Rev 2015;86:83–100. [DOI] [PubMed] [Google Scholar]
- 164. Villoutreix BO, Taboureau O. Computational investigations of hERG channel blockers: new insights and current predictive models. Adv Drug Deliv Rev 2015;86:72–82. [DOI] [PubMed] [Google Scholar]
- 165. Wang Y, Xing J, Xu Y, et al. In silico ADME/T modelling for rational drug design. Q Rev Biophys 2015;48:488–515. [DOI] [PubMed] [Google Scholar]
- 166. Stoll F, Goller AH, Hillisch A. Utility of protein structures in overcoming ADMET-related issues of drug-like compounds. Drug Discov Today 2011;16:530–8. [DOI] [PubMed] [Google Scholar]
- 167. Cavalluzzi MM, Imbrici P, Gualdani R, et al. Human ether-a-go-go-related potassium channel: exploring SAR to improve drug design. Drug Discov Today 2019. doi: 10.1016/j.drudis.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 168. Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019;47:D930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Papadatos G, Davies M, Dedman N, et al. SureChEMBL: a large-scale, chemically annotated patent document database. Nucleic Acids Res 2016;44:D1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47:D1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Siramshetty VB, Eckert OA, Gohlke BO, et al. SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res 2018;46:D1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sterling T, Irwin JJ. ZINC 15--ligand discovery for everyone. J Chem Inf Model 2015;55:2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Eckert H, Bajorath J. Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches. Drug Discov Today 2007;12:225–33. [DOI] [PubMed] [Google Scholar]
- 175. Willett P. Similarity searching using 2D structural fingerprints. Methods Mol Biol 2011;672:133–58. [DOI] [PubMed] [Google Scholar]
- 176. Johnson M, Lajiness M, Maggiora G. Molecular similarity: a basis for designing drug screening programs. Prog Clin Biol Res 1989;291:167–71. [PubMed] [Google Scholar]
- 177. Kalaszi A, Szisz D, Imre G, et al. Screen3D: a novel fully flexible high-throughput shape-similarity search method. J Chem Inf Model 2014;54:1036–49. [DOI] [PubMed] [Google Scholar]
- 178. Yan X, Li J, Liu Z, et al. Enhancing molecular shape comparison by weighted Gaussian functions. J Chem Inf Model 2013;53:1967–78. [DOI] [PubMed] [Google Scholar]
- 179. Liu X, Jiang H, Li H. SHAFTS: a hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J Chem Inf Model 2011;51:2372–85. [DOI] [PubMed] [Google Scholar]
- 180. Banegas-Luna AJ, Ceron-Carrasco JP, Puertas-Martin S, et al. BRUSELAS: HPC generic and customizable software architecture for 3D ligand-based virtual screening of large molecular databases. J Chem Inf Model 2019;59:2805–17. [DOI] [PubMed] [Google Scholar]
- 181. Li H, Leung KS, Wong MH, et al. USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res 2016;44:W436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Zoete V, Daina A, Bovigny C, et al. SwissSimilarity: a web tool for low to ultra high throughput ligand-based virtual screening. J Chem Inf Model 2016;56:1399–404. [DOI] [PubMed] [Google Scholar]
- 183. Gong J, Cai C, Liu X, et al. ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics 2013;29:1827–9. [DOI] [PubMed] [Google Scholar]
- 184. Dong J, Cao DS, Miao HY, et al. ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J Chem 2015;7:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Backman TW, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res 2011;39:W486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Cruz DJ, Bonotto RM, Gomes RG, et al. Identification of novel compounds inhibiting chikungunya virus-induced cell death by high throughput screening of a kinase inhibitor library. PLoS Negl Trop Dis 2013;7:e2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Cruz DJ, Koishi AC, Taniguchi JB, et al. High content screening of a kinase-focused library reveals compounds broadly-active against dengue viruses. PLoS Negl Trop Dis 2013;7:e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Capecchi A, Awale M, Probst D, et al. PubChem and ChEMBL beyond Lipinski. Mol Inform 2019;38:e1900016. [DOI] [PubMed] [Google Scholar]
- 189. Floris M, Masciocchi J, Fanton M, et al. Swimming into peptidomimetic chemical space using pepMMsMIMIC. Nucleic Acids Res 2011;39:W261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Niazi S, Purohit M, Niazi JH. Role of p53 circuitry in tumorigenesis: a brief review. Eur J Med Chem 2018;158:7–24. [DOI] [PubMed] [Google Scholar]
- 191. Schneidman-Duhovny D, Dror O, Inbar Y, et al. PharmaGist: a webserver for ligand-based pharmacophore detection. Nucleic Acids Res 2008;36:W223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Lille-Langoy R, Goldstone JV, Rusten M, et al. Environmental contaminants activate human and polar bear (Ursus maritimus) pregnane X receptors (PXR, NR1I2) differently. Toxicol Appl Pharmacol 2015;284:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sunseri J, Koes DR. Pharmit: interactive exploration of chemical space. Nucleic Acids Res 2016;44:W442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Williams A, Zhou S, Zhan CG. Discovery of potent and selective butyrylcholinesterase inhibitors through the use of pharmacophore-based screening. Bioorg Med Chem Lett 2019;29:126754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Klambauer G, Wischenbart M, Mahr M, et al. Rchemcpp: a web service for structural analoging in ChEMBL, Drugbank and the connectivity map. Bioinformatics 2015;31:3392–4. [DOI] [PubMed] [Google Scholar]
- 196. Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929–35. [DOI] [PubMed] [Google Scholar]
- 197. Najjar A, Platzer C, Luft A, et al. Computer-aided design, synthesis and biological characterization of novel inhibitors for PKMYT1. Eur J Med Chem 2019;161:479–92. [DOI] [PubMed] [Google Scholar]
- 198. Ghamari N, Zarei O, Reiner D, et al. Histamine H3 receptor ligands by hybrid virtual screening, docking, molecular dynamics simulations, and investigation of their biological effects. Chem Biol Drug Des 2019;93:832–43. [DOI] [PubMed] [Google Scholar]
- 199. Koes DR, Camacho CJ. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res 2012;40:W409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Dong J, Yao ZJ, Zhu MF, et al. ChemSAR: an online pipelining platform for molecular SAR modeling. J Chem 2017;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Soufan O, Ba-Alawi W, Magana-Mora A, et al. DPubChem: a web tool for QSAR modeling and high-throughput virtual screening. Sci Rep 2018;8:9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Liu Z, Du J, Fang J, et al. DeepScreening: a deep learning-based screening web server for accelerating drug discovery. Database (Oxford) 2019;2019. doi: 10.1093/database/baz104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Korkmaz S, Zararsiz G, Goksuluk D. MLViS: a web tool for machine learning-based virtual screening in early-phase of drug discovery and development. PLoS One 2015;10:e0124600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Oprisiu I, Novotarskyi S, Tetko IV. Modeling of non-additive mixture properties using the online CHEmical database and Modeling environment (OCHEM). J Chem 2013;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Sushko I, Novotarskyi S, Korner R, et al. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des 2011;25:533–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Li X, Zhang Y, Li H, et al. Modeling of the hERG K+ channel blockage using online chemical database and Modeling environment (OCHEM). Mol Inform 2017;36. doi: 10.1002/minf.201700074. [DOI] [PubMed] [Google Scholar]
- 207. Gilson MK, Liu T, Baitaluk M, et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res 2016;44:D1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Szklarczyk D, Santos A, Mering C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res 2016;44:D380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Kuhn M, Campillos M, Letunic I, et al. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 2010;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Croft D, O'Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 2011;39:D691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Stelzer G, Dalah I, Stein TI, et al. In-silico human genomics with GeneCards. Hum Genomics 2011;5:709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kringelum J, Kjaerulff SK, Brunak S, et al. ChemProt-3.0: a global chemical biology diseases mapping. Database (Oxford) 2016;2016. doi: 10.1093/database/bav123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Keiser MJ, Setola V, Irwin JJ, et al. Predicting new molecular targets for known drugs. Nature 2009;462:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Pereira ASP, Haan H, Pena-Garcia J, et al. Exploring African medicinal Plants for potential anti-diabetic compounds with the DIA-DB inverse virtual screening web server. Molecules 2019;24. doi: 10.3390/molecules24102002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hamad S, Adornetto G, Naveja JJ, et al. HitPickV2: a web server to predict targets of chemical compounds. Bioinformatics 2019;35:1239–40. [DOI] [PubMed] [Google Scholar]
- 217. Peon A, Li H, Ghislat G, et al. MolTarPred: a web tool for comprehensive target prediction with reliability estimation. Chem Biol Drug Des 2019;94:1390–401. [DOI] [PubMed] [Google Scholar]
- 218. Alberga D, Trisciuzzi D, Montaruli M, et al. A new approach for drug target and bioactivity prediction: the multifingerprint similarity search algorithm (MuSSeL). J Chem Inf Model 2019;59:586–96. [DOI] [PubMed] [Google Scholar]
- 219. Montaruli M, Alberga D, Ciriaco F, et al. Accelerating drug discovery by early protein drug target prediction based on a multi-fingerprint similarity search. Molecules 2019;24. doi: 10.3390/molecules24122233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Awale M, Reymond JL. The polypharmacology browser: a web-based multi-fingerprint target prediction tool using ChEMBL bioactivity data. J Chem 2017;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Awale M, Reymond JL. Polypharmacology browser PPB2: target prediction combining nearest Neighbors with machine learning. J Chem Inf Model 2019;59:10–7. [DOI] [PubMed] [Google Scholar]
- 222. Lee K, Lee M, Kim D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target-fishing server. BMC Bioinformatics 2017;18:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Nickel J, Gohlke BO, Erehman J, et al. SuperPred: update on drug classification and target prediction. Nucleic Acids Res 2014;42:W26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Gfeller D, Grosdidier A, Wirth M, et al. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res 2014;42:W32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 2019;47:W357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Soo HC, Chung FF, Lim KH, et al. Cudraflavone C induces tumor-specific apoptosis in colorectal cancer cells through inhibition of the phosphoinositide 3-kinase (PI3K)-AKT pathway. PLoS One 2017;12:e0170551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Yao ZJ, Dong J, Che YJ, et al. TargetNet: a web service for predicting potential drug-target interaction profiling via multi-target SAR models. J Comput Aided Mol Des 2016;30:413–24. [DOI] [PubMed] [Google Scholar]
- 228. Wu J, Zhang Q, Wu W, et al. WDL-RF: predicting bioactivities of ligand molecules acting with G protein-coupled receptors by combining weighted deep learning and random forest. Bioinformatics 2018;34:2271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Hao GF, Jiang W, Ye YN, et al. ACFIS: a web server for fragment-based drug discovery. Nucleic Acids Res 2016;44:W550–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Durrant JD, Amaro RE, McCammon JA. AutoGrow: a novel algorithm for protein inhibitor design. Chem Biol Drug Des 2009;73:168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Pinto GP, Vavra O, Filipovic J, et al. Fast screening of inhibitor binding/unbinding using novel software tool CaverDock. Front Chem 2019;7:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Vavra O, Filipovic J, Plhak J, et al. CaverDock: a molecular docking-based tool to analyse ligand transport through protein tunnels and channels. Bioinformatics 2019;35:4986–93. [DOI] [PubMed] [Google Scholar]
- 233. Lee PH, Kuo KL, Chu PY, et al. SLITHER: a web server for generating contiguous conformations of substrate molecules entering into deep active sites of proteins or migrating through channels in membrane transporters. Nucleic Acids Res 2009;37:W559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Devaurs D, Bouard L, Vaisset M, et al. MoMA-LigPath: a web server to simulate protein-ligand unbinding. Nucleic Acids Res 2013;41:W297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Irwin JJ, Shoichet BK, Mysinger MM, et al. Automated docking screens: a feasibility study. J Med Chem 2009;52:5712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Manglik A, Lin H, Aryal DK, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016;537:185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Korczynska M, Clark MJ, Valant C, et al. Structure-based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc Natl Acad Sci U S A 2018;115:E2419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. London N, Miller RM, Krishnan S, et al. Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Biol 2014;10:1066–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model 2010;50:1561–73. [DOI] [PubMed] [Google Scholar]
- 240. Silveira NJF, Pereira FSS, Elias TC, et al. Web Services for Molecular Docking Simulations. Methods Mol Biol 2019;2053:221–9. [DOI] [PubMed] [Google Scholar]
- 241. Santos KB, Guedes IA, Karl ALM, et al. Highly flexible ligand docking: benchmarking of the DockThor program on the LEADS-PEP protein-peptide data set. J Chem Inf Model 2020. doi: 10.1021/acs.jcim.9b00905. [DOI] [PubMed] [Google Scholar]
- 242. Durrant JD, McCammon JA. NNScore 2.0: a neural-network receptor-ligand scoring function. J Chem Inf Model 2011;51:2897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Douguet D. E-LEA3D: a computational-aided drug design web server. Nucleic Acids Res 2010;38:W615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Douguet D, Munier-Lehmann H, Labesse G, et al. LEA3D: a computer-aided ligand design for structure-based drug design. J Med Chem 2005;48:2457–68. [DOI] [PubMed] [Google Scholar]
- 245. Korb O, Stutzle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model 2009;49:84–96. [DOI] [PubMed] [Google Scholar]
- 246. Tao A, Huang Y, Shinohara Y, et al. ezCADD: a rapid 2D/3D visualization-enabled web Modeling environment for democratizing computer-aided drug design. J Chem Inf Model 2019;59:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Forli S, Huey R, Pique ME, et al. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 2016;11:905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model 2013;53:1893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Tsai TY, Chang KW, Chen CY. iScreen: world's first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J Comput Aided Mol Des 2011;25:525–31. [DOI] [PubMed] [Google Scholar]
- 250. Labbe CM, Rey J, Lagorce D, et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res 2015;43:W448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Lagarde N, Goldwaser E, Pencheva T, et al. A free web-based protocol to assist structure-based virtual screening experiments. Int J Mol Sci 2019;20. doi: 10.3390/ijms20184648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Lagarde N, Rey J, Gyulkhandanyan A, et al. Online structure-based screening of purchasable approved drugs and natural compounds: retrospective examples of drug repositioning on cancer targets. Oncotarget 2018;9:32346–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Villoutreix BO, Khatib AM, Cheng Y, et al. Blockade of the malignant phenotype by beta-subunit selective noncovalent inhibition of immuno- and constitutive proteasomes. Oncotarget 2017;8:10437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Jayaram B, Singh T, Mukherjee G, et al. Sanjeevini: a freely accessible web-server for target directed lead molecule discovery. BMC Bioinformatics 2012;13:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Labbe CM, Pencheva T, Jereva D, et al. AMMOS2: a web server for protein-ligand-water complexes refinement via molecular mechanics. Nucleic Acids Res 2017;45:W350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Salentin S, Schreiber S, Haupt VJ, et al. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res 2015;43:W443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Perez-Castillo Y, Sotomayor-Burneo S, Jimenes-Vargas K, et al. CompScore: boosting structure-based virtual screening performance by incorporating docking scoring function components into consensus scoring. J Chem Inf Model 2019;59:3655–66. [DOI] [PubMed] [Google Scholar]
- 258. Wang R, Fang X, Lu Y, et al. The PDBbind database: methodologies and updates. J Med Chem 2005;48:4111–9. [DOI] [PubMed] [Google Scholar]
- 259. Gheyouche E, Launay R, Lethiec J, et al. DockNmine, a web portal to assemble and analyse virtual and experimental interaction data. Int J Mol Sci 2019;20. doi: 10.3390/ijms20205062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Wang J, Dokholyan NV. MedusaDock 2.0: efficient and accurate protein-ligand docking with constraints. J Chem Inf Model 2019;59:2509–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Jamal MS, Parveen S, Beg MA, et al. Anticancer compound plumbagin and its molecular targets: a structural insight into the inhibitory mechanisms using computational approaches. PLoS One 2014;9:e87309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Fassio AV, Santos LH, Silveira SA, et al. nAPOLI: a graph-based strategy to detect and visualize conserved protein-ligand interactions in large-scale. IEEE/ACM Trans Comput Biol Bioinform 2019. doi: 10.1109/TCBB.2019.2892099. [DOI] [PubMed] [Google Scholar]
- 263. Jimenez J, Doerr S, Martinez-Rosell G, et al. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017;33:3036–42. [DOI] [PubMed] [Google Scholar]
- 264. Jimenez J, Skalic M, Martinez-Rosell G, et al. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model 2018;58:287–96. [DOI] [PubMed] [Google Scholar]
- 265. Skalic M, Martinez-Rosell G, Jimenez J, et al. PlayMolecule BindScope: large scale CNN-based virtual screening on the web. Bioinformatics 2019;35:1237–8. [DOI] [PubMed] [Google Scholar]
- 266. Vangone A, Schaarschmidt J, Koukos P, et al. Large-scale prediction of binding affinity in protein-small ligand complexes: the PRODIGY-LIG web server. Bioinformatics 2019;35:1585–7. [DOI] [PubMed] [Google Scholar]
- 267. Empereur-Mot C, Zagury JF, Montes M. Screening explorer-An interactive tool for the analysis of screening results. J Chem Inf Model 2016;56:2281–6. [DOI] [PubMed] [Google Scholar]
- 268. Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res 2011;39:W270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Grosdidier A, Zoete V, Michielin O. EADock: docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007;67:1010–25. [DOI] [PubMed] [Google Scholar]
- 270. MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 1998;102:3586–616. [DOI] [PubMed] [Google Scholar]
- 271. Thal DM, Sun B, Feng D, et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016;531:335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Ferreira NC, Marques IA, Conceicao WA, et al. Anti-prion activity of a panel of aromatic chemical compounds: in vitro and in silico approaches. PLoS One 2014;9:e84531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Wu Y, Wang Y, Lin Y, et al. Dub3 inhibition suppresses breast cancer invasion and metastasis by promoting Snail1 degradation. Nat Commun 2017;8:14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Koes DR, Domling A, Camacho CJ. AnchorQuery: rapid online virtual screening for small-molecule protein-protein interaction inhibitors. Protein Sci 2018;27:229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Wang Z, Wang X, Li Y, et al. farPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics 2019;35:1777–9. [DOI] [PubMed] [Google Scholar]
- 276. Wang F, Wu FX, Li C-Z, et al. ACID: a free tool for drug repurposing using consensus inverse docking strategy. J Chem 2019;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Ng MC, Fong S, Siu SW. PSOVina: the hybrid particle swarm optimization algorithm for protein-ligand docking. J Bioinform Comput Biol 2015;13:1541007. [DOI] [PubMed] [Google Scholar]
- 279. Sun H, Li Y, Tian S, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys 2014;16:16719–29. [DOI] [PubMed] [Google Scholar]
- 280. Obiol-Pardo C, Rubio-Martinez J. Comparative evaluation of MMPBSA and XSCORE to compute binding free energy in XIAP-peptide complexes. J Chem Inf Model 2007;47:134–42. [DOI] [PubMed] [Google Scholar]
- 281. Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des 2002;16:11–26. [DOI] [PubMed] [Google Scholar]
- 282. Meslamani J, Rognan D, Kellenberger E. Sc-PDB: a database for identifying variations and multiplicity of `druggable' binding sites in proteins. Bioinformatics 2011;27:1324–6. [DOI] [PubMed] [Google Scholar]
- 283. Lee A, Kim D. CRDS: consensus reverse docking system for target fishing. Bioinformatics 2019. doi: 10.1093/bioinformatics/btz656. [DOI] [PubMed] [Google Scholar]
- 284. Schneider M, Pons JL, Bourguet W, et al. Towards accurate high-throughput ligand affinity prediction by exploiting structural ensembles, docking metrics and ligand similarity. Bioinformatics 2020;36:160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Pons JL, Labesse G. @TOME-2: a new pipeline for comparative modeling of protein-ligand complexes. Nucleic Acids Res 2009;37:W485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Sandal M, Duy TP, Cona M, et al. GOMoDo: a GPCRs online modeling and docking webserver. PLoS One 2013;8:e74092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Zundert GCP, Rodrigues J, Trellet M, et al. The HADDOCK2.2 web server: user-friendly integrative Modeling of biomolecular complexes. J Mol Biol 2016;428:720–5. [DOI] [PubMed] [Google Scholar]
- 288. Radu BM, Osculati AMM, Suku E, et al. All muscarinic acetylcholine receptors (M1-M5) are expressed in murine brain microvascular endothelium. Sci Rep 2017;7:5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Pasznik P, Rutkowska E, Niewieczerzal S, et al. Potential off-target effects of beta-blockers on GUT hormone receptors: in silico study including GUT-DOCK-A web service for small-molecule docking. PLoS One 2019;14:e0210705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Wang JC, Chu PY, Chen CM, et al. idTarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res 2012;40:W393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Chang DT, Oyang YJ, Lin JH. MEDock: a web server for efficient prediction of ligand binding sites based on a novel optimization algorithm. Nucleic Acids Res 2005;33:W233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Cheng T, Hao M, Takeda T, et al. Large-scale prediction of drug-target interaction: a data-centric review. AAPS J 2017;19:1264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Fourches D, Ash J. 4D- quantitative structure-activity relationship modeling: making a comeback. Expert Opin Drug Discov 2019;14:1227–35. [DOI] [PubMed] [Google Scholar]
- 294. Sydow D, Burggraaff L, Szengel A, et al. Advances and challenges in computational target prediction. J Chem Inf Model 2019;59:1728–42. [DOI] [PubMed] [Google Scholar]
- 295. Chen C, Wu M, Cen S, et al. MTLD, a database of multiple target ligands, the updated version. Molecules 2017;22. doi: 10.3390/molecules22091375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Hu Y, Gupta-Ostermann D, Bajorath J. Exploring compound promiscuity patterns and multi-target activity spaces. Comput Struct Biotechnol J 2014;9:e201401003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Ramsay RR, Popovic-Nikolic MR, Nikolic K, et al. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med 2018;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Zhang W, Bai Y, Wang Y, et al. Polypharmacology in drug discovery: a review from systems pharmacology perspective. Curr Pharm Des 2016;22:3171–81. [DOI] [PubMed] [Google Scholar]
- 299. Pires DEV, Ascher DB. CSM-lig: a web server for assessing and comparing protein–small molecule affinities Nucleic Acids Res. 2016; 44: W557–W561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.