

Summary
Cannabis (Cannabis sativa L.) is one of the oldest cultivated plants purported to have unique medicinal properties. However, scientific research of cannabis has been restricted by the Single Convention on Narcotic Drugs of 1961, an international treaty that prohibits the production and supply of narcotic drugs except under license. Legislation governing cannabis cultivation for research, medicinal and even recreational purposes has been relaxed recently in certain jurisdictions. As a result, there is now potential to accelerate cultivar development of this multi‐use and potentially medically useful plant species by application of modern genomics technologies. Whilst genomics has been pivotal to our understanding of the basic biology and molecular mechanisms controlling key traits in several crop species, much work is needed for cannabis. In this review we provide a comprehensive summary of key cannabis genomics resources and their applications. We also discuss prospective applications of existing and emerging genomics technologies for accelerating the genetic improvement of cannabis.
Keywords: breeding, cannabinoids, cannabis, crop improvement, genome assembly, genomics
Introduction
Cannabis sativa L. (cannabis), a member of the Cannabaceae family, is one of the world’s oldest domesticated crops (Bradshaw et al., 1981; Long et al., 2017). It is believed to have originated in Central Asia, from where its cultivation rapidly spread throughout Asia and Europe. Nowadays legal and illegal cannabis cultivation occurs globally (Van Bakel et al., 2011).
The exact number of species comprising the Cannabis genus is controversial. Some claim the genus consists of three species that display distinct phenotypic differences; namely C. sativa L., C. indica Lam (Lamarck) and C. ruderalis (Sawler et al., 2015; Clarke & Merlin, 2016; Henry et al., 2020). The alternative, and perhaps most accepted, viewpoint is that Cannabis is a monotypic genus consisting of a single species, Cannabis sativa L. (referred to as cannabis hereafter) (Small & Cronquist, 1976). Cannabis has a diploid genome (2n = 20) consisting of nine autosomes and a pair of sex chromosomes (X and Y) (Braich et al., 2019; McKernan et al., 2020). It is predominantly dioecious, meaning a plant is either a male or a female, with estimated haploid genome sizes of 843 Mb and 818 Mb for male and female plants, respectively (Van Bakel et al., 2011). Despite the presence of defined sex chromosomes, environmental factors such as reduced photoperiod and low temperature, and foliar applications of chemicals such as silver nitrate and the ethylene hormone inhibitor silver thiosulfate induce pollen production in female flowers, leading to the production of ‘feminised seeds’ (Ram & Sett, 1982; Kaushal, 2012; Lubell & Brand, 2018). This technique has been exploited as a useful tool in cannabis breeding (for example, selfing or crossing female plants) and in generating populations for dissection of the genetic bases of important traits.
Cannabis can be classified as fibre‐type (hemp or industrial hemp) and drug‐type (medicinal cannabis or marijuana) based on usage and cannabinoid content; fibre‐type plants contain < 0.3% ∆9‐tetrahydrocannabinol (THC) whereas drug‐type plants contain > 0.3% THC. Both have been exploited by humans for various applications since 8000 bce (Srinivasababu, 2014). For instance, the stalk of hemp is an important fibre source whilst oil extracted from its seeds is used in several food and nonfood applications (Clarke & Merlin, 2016). More recently, there have been applications of hemp in construction, geotextiles, cosmetics, as a food product and as a therapeutic agent (Piluzza et al., 2013). Historical medicinal and recreational usage of cannabis has been reported, particularly the use of marijuana for its mood‐altering narcotic properties. Consequently, marijuana is the most cultivated, trafficked and abused illicit drug in the world (Sawler et al., 2015). Its prolonged usage has been associated with detrimental health outcomes, such as impaired cognitive development and psychomotor performance, leading to chronic health conditions (Andre et al., 2016). Hence, there is an urgent need to conduct evidence‐based research to safeguard purity and quality of products, and to better understand the mode of action of cannabinoids for therapeutic applications.
Cannabis plants grown for medicinal and recreational end‐uses are generally shorter, have thinner stems, more branches and a higher density of floral tissues than industrial hemp plants. Cultivars also can be discriminated by their cannabinoid profile, also termed chemotype (Piluzza et al., 2013; Clarke & Merlin, 2016). Cannabinoids are secondary metabolites produced in capitate stalked glandular trichomes (Fig. 1), more than 120 of which have been identified (Braich et al., 2019; Kovalchuk et al., 2020). Two of these, THC and CBD (cannabidiol), are highly sought after by cannabis breeders and pharmaceutical industries (Adams et al., 1940; Weiblen et al., 2015; Andre et al., 2016). Precursor synthesis of these cannabinoids occurs from two distinct metabolic pathways; the polyketide pathway and the methylerythritol phosphate (MEP) pathway (Fig. 1) (Kovalchuk et al., 2020). These produce alkylresorcinolic acids, including olivetolic acid (OA) that is specific to cannabis, and geranyl diphosphate (GPP), respectively. CBGA (cannabigerolic acid) is then synthesized from OA and GPP to produce the acidic precursors of THC (tetrahydrocannabinolic acid; THCA) and CBD (cannabidiolic acid; CBDA) (Weiblen et al., 2015). THC is the main psychoactive/intoxicant in cannabis. It induces sensations of euphoria, anxiety, paranoia and cognitive deficits, and is associated primarily with the narcotic status of cannabis (Boggs et al., 2018). However, THC also has therapeutic benefits as it confers relief from nausea caused by certain anti‐cancer treatments and acts as an anti‐inflammatory agent (Andre et al., 2016). CBD, which is an isomer of THC, has an analgesic effect, and also is purported to have neuroprotective, anti‐cancer and anti‐diabetic properties (Andre et al., 2016). Epidiolex, the first CBD‐based product approved by the US Food and Drug Administration, also has been shown to reduce seizures in children with Dravet syndromes (O’Connell et al., 2017; Chen et al., 2019). Industrial hemp and recreational drug chemotypes differ in their THC : CBD ratios. Cannabis plants are frequently classified into three main chemotypes based on this ratio; chemotype I plants (drug‐type) exhibit a THC : CBD ratio well beyond 1.0, chemotype II plants have an intermediate ratio of 0.5–2.0 and chemotype III plants (fibre‐type) have a ratio well below 1.0 (Aizpurua‐Olaizola et al., 2016). Additionally, the DW of THC in mature female inflorescences is used to demarcate cultivars for industrial hemp end‐uses such as seed or fibre production (Piluzza et al., 2013).
Fig. 1.
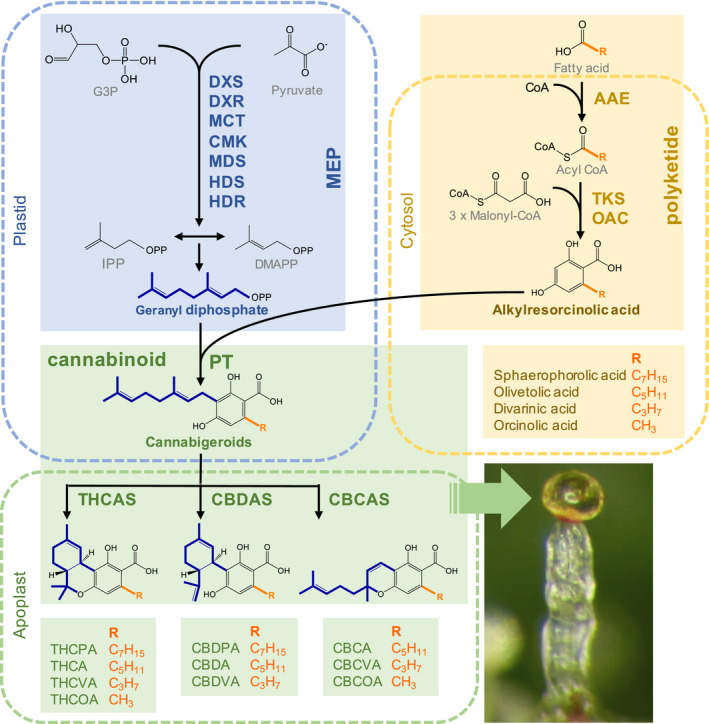
Schematic diagram of cannabinoid biosynthesis including polyketide and isoprenoid precursor pathways. Precursor pathways are merged by a plastid‐localized aromatic prenyltransferase, with alkylresorcinolic acids and geranyl diphosphate intermediates forming cannabigeroids with a linear isoprenyl residue (Gülck & Møller, 2020). Cannabinoid synthesis concludes in the apoplastic storage cavity of glandular trichomes. Here, cannabigeroids are converted to tri‐ and di‐cyclic cannabinoids such as ∆9‐tetrahydrocannabinolic acid (THCA) and cannabidiolic acid (CBDA) via stereoselective oxidative cyclisation of the isoprenyl moiety. This occurs enzymatically by the cannabinoid synthases THCAS, CBDAS and CBCAS. The green arrow indicates location of the extracellular storage cavity of a Cannabis stalked glandular trichome; bar, 100 µm. Subcellular locations of cannabinoid and precursor pathway enzymes were predicted with the subcellular location software TargetP‐2.0 (http://www.cbs.dtu.dk/services/TargetP/). AAE, acyl‐activating enzyme; CBCA, cannabichromenic acid; CBCAS, cannabichromenic acid synthase; CBCOA, cannabiorcichromenic acid; CBCVA, cannabichromevarinic acid; CBDA, cannabidiolic acid; CBDAS, cannabidiolic acid synthase; CBDPA, cannabidiphorolic acid; CBDVA, cannabidivarinic acid; CMK, 4‐(cytidine 50‐diphospho)‐2‐C‐methyl‐d‐erythritol kinase; DXR, 1‐deoxy‐D‐xylulose‐5‐phosphate reductoisomerase; DXS, 1‐deoxy‐d‐xylulose 5 phosphate synthase; HDR, 1‐hydroxy‐2‐methyl‐2‐butenyl 4‐diphosphate reductase; HDS, 1‐hydroxy‐2‐methyl‐2‐butenyl 4‐diphosphate synthase; MCT, 2‐C‐methyl‐d‐erythritol 4‐phosphate cytidylyltransferase; MDS, 2‐C‐methyl‐d‐erythritol 2,4‐cyclodiphosphate synthase; MEP, 2‐C‐methyl‐d‐erythritol‐4‐phosphate; OAC, olivetolic acid cyclase; PT, prenyltransferase (e.g. geranylpyrophosphate:olivetolate geranyltransferase (GOT)); THCA, tetrahydrocannabinolic acid; THCAS, tetrahydrocannabinolic acid synthase; THCOA, tetrahydrocannabiorcolic acid; THCPA, tetrahydrocannabipgorolic acid; THCVA, tetrahydrocannabivarinic acid; TKS, tetraketide synthase.
The prospects of using contemporary breeding technologies to improve cannabis traits for medicinal applications are promising. However, progress in this area is hampered by several issues. First, the genetics of cannabis is poorly understood and causes incorrect classification of cultivars/strains, with implications for researchers, growers, cannabis users and regulators (Vergara et al., 2016; Welling et al., 2016; Schwabe & McGlaughlin, 2019). Secondly, intensive clandestine breeding practices since the early 1970s have led to a genetic bottleneck and reduction in allelic diversity in marijuana plants (Clarke & Merlin, 2016). Thirdly, restrictions such as the international narcotics conventions and associated legislation have hampered the exchange of cannabis genetic resources and research materials (Welling et al., 2016).
Genomic analyses have facilitated a paradigm shift in the improvement of cultivars of major crop species over the last two decades (Morrell et al., 2012; Yuan et al., 2017). This has been driven by high‐throughput sequencing and single nucleotide polymorphism (SNP) marker‐based genotyping platforms, as well as the development of high‐quality reference genome and transcriptome assemblies. These technologies have increased our understanding of gene content, genomic variation and the genetic basis of complex agronomic traits in multiple plant species (Xie et al., 2015; Xu et al., 2017; Appels et al., 2018; Thomas et al., 2019). The status of cannabis as an emerging, high‐value and clinically efficacious crop and the potential of genomics‐assisted breeding, coupled with the relaxation of regulations and restrictions, warrant expanded research efforts. In this review we summarize available cannabis genomics resources and report on the application of these tools. We also discuss future applications and emerging genomics technologies relevant to the genetic improvement of cannabis.
Cannabis sativa genomics resources and the discoveries they have enabled
Genome assemblies
De novo assembly of plant genomes remains challenging and the cannabis genome is no exception (Schatz et al., 2012). The cannabis genome is highly heterozygous (estimated at 12.5–40.5%) and contains large amounts of repetitive elements (estimated at 70%) (Van Bakel et al., 2011; Pisupati et al., 2018; Gao et al., 2020; Kovalchuk et al., 2020). Several attempts have been made to assemble this complex genome, as illustrated by publicly available genome and transcriptome assemblies of varying sizes and completeness for 12 different cannabis cultivars (Table 1a,b). Initial efforts relied upon the use of short‐read sequencing technology, but these proved computationally challenging. Application of third‐generation long‐read sequencing technologies such as Single‐Molecule Real‐Time (SMRT) sequencing (PacBio) and MinION (Oxford Nanopore Technologies) have greatly improved the contiguity of cannabis reference sequences, as has the anchoring of scaffolds using genetic linkage maps coupled with Hi‐C data (Grassa et al., 2018; Laverty et al., 2019; Gao et al., 2020; McKernan et al., 2020). This has resulted in the creation of four chromosome‐level assemblies for Purple Kush (PK; drug‐type), Finola (FN; hemp), JL (wild accession) and CBDRx (cs10; high‐CBD) (Grassa et al., 2018; Laverty et al., 2019; Gao et al., 2020). The cs10 assembly is the most complete and contiguous chromosome‐level assembly, comprising 25 302 protein‐coding genes (Fig. 2; Maoz, 2020). The current version of this assembly (v.2.0; GenBank acc. no. GCA_900626175.2) has recently been updated with the chromosomes renumbered according to an agreed community standard (Table 2; https://www.ncbi.nlm.nih.gov/assembly/GCF_900626175.2#/st; Maoz, 2020). Earlier this year, the International Cannabis Genomics Research Consortium (ICGRC) proposed that cs10 be used as the reference for cannabis genomics (Maoz, 2020). Cannabis genome assemblies other than cs10 also have much to offer. For instance, the PK, FN and the contig‐level Jamaican Lion trio assemblies have been key to confirming findings from earlier, lower‐resolution studies as well as uncovering important biology of the Cannabis genus (Van Bakel et al., 2011; Laverty et al., 2019; Prentout et al., 2019; Vergara et al., 2019; Booth et al., 2020; McKernan et al., 2020). Further improvement of these assemblies will assist in fully realising their potential.
Table 1.
Statistics for the latest Cannabis sativa reference genome and transcriptome assemblies.
| Genome assemblies | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cultivar (GenBank acc. no.) | Sex | Total sequence length (bp) | Total ungapped length (bp) | Number of scaffolds | Scaffold N50 (bp) | Number of contigs | Contig N50 (bp) | GC content (%) | Number of chromosomes and plasmids | Genome coverage | Sequencing technology | Number of protein‐coding genes/transcripts | Reference |
| cs10/CBDRx a (GCA_900626175.2) | Female | 876 147 649 | 736 579 359 | 221 | 91 913 889 | 1052 | 1959 202 | 34 | 10 | 100× | Oxford Nanopore Technology | 25 302 protein‐coding genes | Grassa et al. (2018) |
| Purple Kush b (GCA_000230575.5) | Female | 891 964 663 | 891 346 362 | 6653 | 60 968 100 | 12 836 | 133 904 | 34 | 10 | 79× | PacBio | 30 074 transcripts | Laverty et al. (2019) |
| Finola (GCA_003417725.2) | Male | 1009 674 739 | 1009 380 638 | 2362 | 77 135 887 | 5303 | 370 471 | 34 | 10 | 97.64× | PacBio | 37 689 transcripts | Lavertyet al. (2019) |
| Pineapple Banana Bubba Kush (GCA_002090435.1) | Male | 512 174 223 | 512 174 223 | 18 355 | 51 819 | 18 355 | 51 819 | 34 | – | 72× | PacBio | – | – |
| LA Confidential (GCA_001510005.1) | Female | 595 358 288 | 595 357 797 | 311 039 | 2649 | 311 039 | 2649 | 35 | – | 50× | 454 sequencing | – | – |
| Chemdog 91 (GCA_001509995.1) | Female | 285 932 793 | 285 527 436 | 175 088 | 2250 | 190 122 | 2189 | 33 | – | 50× | Illumina GAII | – | – |
| Cannatonic (GCA_001865755.1) | Female | 585 823 666 | 585 823 666 | 11 110 | 128 718 | 11 110 | 128 718 | 34 | – | 10× | PacBio | – | – |
| Jamaican Lion (female parent)c (GCA_012923425.1) | Female | 876 735 611 | 876 735 611 | – | – | 1599 | 3283 100 | 34 | – | 125× | PacBio Sequel | 27 664 genes | McKernan et al. (2020) |
| Jamaican Lion (male parent)c (GCA_013030025.1) | Male | 1009 156 132 | 1009 156 132 | – | – | 1264 | 1668 042 | 34 | – | 125× | PacBio Sequel | 31 591 genes | McKernan et al. (2020) |
| Jamaican Lion (F1) c (JAATIR000000000.1) | Female | 999 122 115 | 999 122 115 | – | – | 658 | 1668 042 | 34 | – | – | PacBio Sequel | – | McKernan et al. (2020) |
| JL d (GCA_013030365.1) | Female | 812 525 420 | 811 830 406 | 483 | 82 998 198 | 2978 | 509 999 | 34 | 10 | 153× | PacBio Sequel | 38 382 protein‐coding genes | Gao et al. (2020) |
| Transcriptome assemblies | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cultivar | Sex | Total sequence length (bp) | Total ungapped length (bp) | Total Number of transcripts | N50 length (bp) | Shortest transcript length (bp) | Longest transcript length (bp) | Reference | Source |
| Purple Kush | Female | 33 200 961 | 33 200 740 | 30 074 | 1906 | 90 | 12 107 | van Bakel et al. (2011) | http://genome.ccbr.utoronto.ca/ |
| Finola | Male | 25 682 508 | 25 682 508 | 37 689 | 1280 | 88 | 7210 | – | http://genome.ccbr.utoronto.ca/ |
| Cannbio e | Male & Female | 55 924 982 | 55 677 217 | 64 413 | 1796 | 201 | 70 089 | Braich et al. (2019) | NCBI (GenBank acc. no. : GIFP00000000.1) |
Grassa et al. (2018) refer to an earlier version of this assembly in their paper (GenBank acc. no. GCA_900626175.1).
Laverty et al. (2019) refer to an earlier version of this assembly in their paper (GenBank acc. no. GCA_000230575.4).
McKernan et al. (2020) refer to earlier versions of these assemblies in their paper (Jamaican Lion, female parent = CoGe ID 55184; Jamaican Lion, male parent = CoGe ID 55360 and Jamaican Lion, F1 = CoGe ID 55567).
Genome annotation not included with this assembly.
The Cannbio assembly was generated using RNA‐Seq data from the female and male cannabis cultivars, Cannbio‐2 and Cannbio‐male, respectively.
Fig. 2.
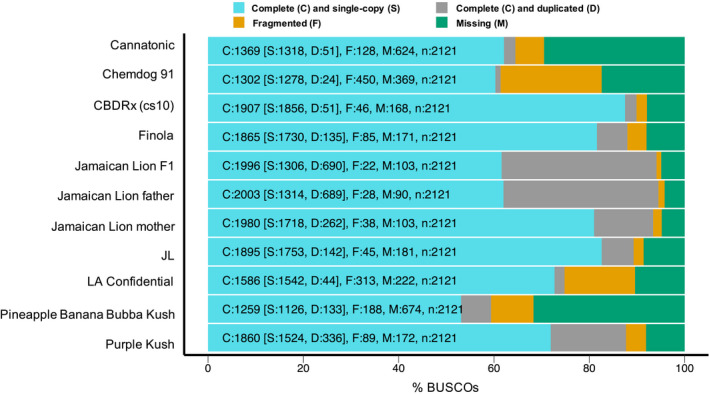
Benchmarking Universal Single‐Copy Ortholog (BUSCO) assessment of the cannabis genome assemblies shown in Table 1(a). The percentages of complete (single‐copy and duplicated), fragmented and missing universal single‐copy orthologue genes were identified using busco v.4.02 (Simão et al., 2015). The Jamaican Lion assemblies (female parent, male parent, F1) have more complete BUSCOs on average, but they also harbour a larger number of duplicated BUSCOs, which reflects the fragmented nature of these assemblies.
Table 2.
Chromosomal nomenclatures of Cannabis sativa genome assemblies highlighting the discrepancies in chromosome numbering among the current assemblies (https://www.ncbi.nlm.nih.gov/assembly/GCF_900626175.2#/st; Maoz, 2020).
| cs10 v.2.0 (GenBank acc. no. GCA_900626175.2) | cs10 v.1.0 (GenBank acc. no. GCA_900626175.1) | Finola v.2.0 (latest; GenBank acc. no. GCA_003417725.2) | Purple Kush v.5.0 (latest; GenBank acc. no. GCA_000230575.5) | ||||
|---|---|---|---|---|---|---|---|
| Chromosome number | GenBank sequence | Chromosome number | GenBank sequence | Chromosome number | GenBank sequence | Chromosome number | GenBank sequence |
| 1 | NC_044371.1 | 2 | NC_044371.1 | 5 | CM011609.1 | 5 | CM010796.2 |
| 2 | NC_044375.1 | 6 | NC_044375.1 | 3 | CM011607.1 | 3 | CM010793.2 |
| 3 | NC_044372.1 | 3 | NC_044372.1 | 4 | CM011608.1 | 4 | CM010794.2 |
| 4 | NC_044373.1 | 4 | NC_044373.1 | 7 | CM011611.1 | 7 | CM010799.2 |
| 5 | NC_044374.1 | 5 | NC_044374.1 | 1 | CM011605.1 | 1 | CM010790.2 |
| 6 | NC_044377.1 | 8 | NC_044377.1 | 2 | CM011606.1 | 2 | CM010792.2 |
| 7 | NC_044378.1 | 9 | NC_044378.1 | 6 | CM011610.1 | 6 | CM010797.2 |
| 8 | NC_044379.1 | 10 | NC_044379.1 | 9 | CM011613.1 | 9 | CM010798.2 |
| 9 | NC_044376.1 | 7 | NC_044376.1 | 8 | CM011612.1 | 8 | CM010795.2 |
| 10 | NC_044370.1 | 1 | NC_044370.1 | 10 | CM011614.1 | 10 | CM010791.2 |
Chromosome 10 (chromosome 1 in cs10 v1.0) corresponds to the sex chromosome in cs10 v.2.0, Purple Kush v.5.0 and Finola v.2.0.
Genome assemblies have made enormous contributions to our understanding of the cannabinoid biosynthetic pathways through the underlying synthase genes. In particular, the assemblies have shed light on the inheritance of these genes (Grassa et al., 2018; Laverty et al., 2019; McKernan et al., 2020). Before the availability of genome sequence data, de Meijer et al. proposed a Mendelian inheritance model of chemotype that involved a single locus, B, with two co‐dominant alleles, BT and BD, encoding for THCAS and CBDAS, respectively (De Meijer et al., 2003). Homozygosity at the B locus led to the production of either THC (drug‐type; BT/BT) or CBD (fibre‐type; BD/BD), whilst heterozygous individuals (BT/BD) had a mixed THC‐CBD chemotype. However, the creation of high‐quality genome assemblies supports an alternative, multilocus model whereby THCAS and CBDAS are two different genes located in close proximity in a retrotransposon‐rich region of the cannabis genome (Grassa et al., 2018; Laverty et al., 2019; McKernan et al., 2020). This finding supports other earlier nongenomics studies (De Meijer et al., 2003; Kojoma et al., 2006; Weiblen et al., 2015).
The identification of the less‐studied CBCA synthase (cannabichromenic acid synthase; CBCAS) gene (also known as inactive THCAS) is another notable discovery originating from genomics datasets (Grassa et al., 2018; Laverty et al., 2019; McKernan et al., 2020). This gene catalyses the synthesis of cannabichromene (CBC), which is an emerging target for medicinal cannabis breeding as it is nonintoxicating, can reduce pain sensations and act as an anti‐inflammatory agent (Laverty et al., 2019). The functional, chemotype‐determining forms of CBCAS, THCAS and CBDAS are highly similar at the nucleotide and amino acid levels (Fig. 3a,b). The fact that long‐read sequencing enabled the assembly of such highly similar loci reflects the importance of this technology in resolving complex, repetitive regions in the cannabis genome (Schatz et al., 2012; Michael & VanBuren, 2020). Our survey of the contig‐level PacBio genome assemblies of Pineapple Banana Bubba Kush (PBBK) and Cannatonic also supports these findings. We identified a larger number of THCAS and CBCAS gene loci in these assemblies compared to Chemdog91 and LA Confidential, which were generated using short‐read sequencing (Fig. 4; Supporting information Tables S1, S2). It is likely that underestimation of THCAS, CBDAS and CBCAS loci has occurred in these collapsed and relatively smaller assemblies (<595 Mb). However, the presence of true biological variation among these cultivars cannot be ruled out. For instance, structural variants (SVs) in the form of copy number variations (CNVs) and presence/absence variations (PAVs) are known to affect the gene content of many plant species (Saxena et al., 2014).
Fig. 3.
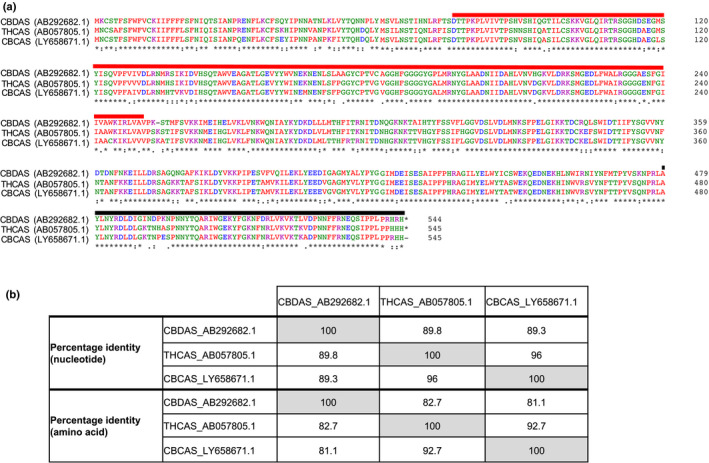
Sequence similarity between the Cannabis sativa cannabinoid synthase genes tetrahydrocannabinolic acid synthase (THCAS; GenBank acc. no. AB057805.1), cannabidiolic acid synthase (CBDAS: GenBank acc. no. AB292682.1) and cannabichromenic acid synthase (CBCAS; GenBank acc. no. LY658671.1). (a) Protein sequence alignments of THCAS, CBDAS and CBCAS were performed using Clustal Omega and protein domains were annotated using interproscan v.5.41‐78.0 (Sievers et al., 2011; Jones et al., 2014). The p‐cresol methylhydroxylase (PCMH)‐type flavin adenine dinucleotide (FAD)‐binding domain (residues 77–251, PrositeProfiles: PS51387, InterPro:IPR016166) and berberine and berberine‐like domain (residues 480–538 for THCAS and CBCAS, residues 479–537 for CBDAS, Pfam: PF08031, InterPro: IPR012951) are highlighted in red and black, respectively. The FAD‐binding domain (residues 81–214, Pfam: PF01565, IPR006094) is not shown. (b) THCAS is more similar to CBCAS than CBDAS at the nucleotide and amino acid levels. It is possible that the presence of CBCAS may lead to the production of THCA as a by‐product (McKernan et al., 2020).
Fig. 4.
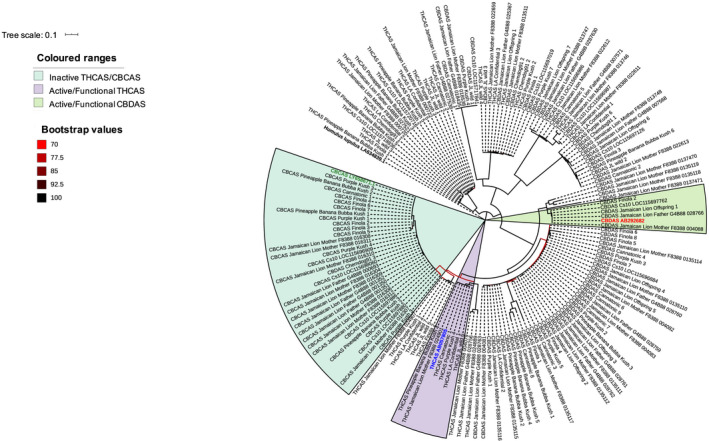
Maximum‐likelihood phylogenetic tree depicting the relationship among the Cannabis sativa cannabinoid synthase genes tetrahydrocannabinolic acid synthase (THCAS), cannabidiolic acid synthase (CBDAS) and cannabichromenic acid synthase (CBCAS). The published nucleotide sequences of the active/functional forms of THCAS (GenBank acc. no. AB057805.1), CBDAS (GenBank acc. no. AB292682.1), CBCAS (GenBank acc. no. LY658671.1) and the paralogues of these genes as annotated in the cs10 v.2.0 and Jamaican Lion (female parent and male parent) assemblies (Supporting Information Tables S1, S2) were aligned against the latest C. sativa reference genome assemblies (Table 1) using Blast+/2.2.29 (Altschul et al., 1990). Best hits corresponding to a percentage identity > 98.5%, query coverage > 75% and alignment length = query length ± 100 bp were retained (Tables S1, S2). The nucleotide sequences of these best hits were extracted from each assembly (where applicable) using bedtools v.2.26.0 (Quinlan & Hall, 2010). transdecoder v.3.0 was used to predict the longest open reading frame from the extracted regions (https://transdecoder.github.io/). The predicted proteins along with amino acid sequences (complete CDS) of AB057805.1 (gene ID in blue), AB292682.1 (gene ID in red), LY658671.1 (gene ID in green) and the other cannabinoid synthase gene copies annotated in the cs10 v.2.0 and Jamaican Lion (female parent and male parent) genome assemblies were used for multiple sequence alignment using clustal Omega (Sievers et al., 2011). The phylogenetic tree was reconstructed from these alignments using raxml v.8.12.12. with 500 bootstrap replicates under the JTT model of amino acid substitution and visualized using Interactive Tree Of Life (iTOL) (Letunic & Bork, 2007; Stamatakis et al., 2008). The tree was rooted with the Humulus lupulus THCAS homolog (GenBank acc. no. LA634839.1). Only bootstrap values of > 70% are shown. It is worth noting that all CBCAS genes cluster with some the THCAS genes reflecting the high sequence similarity between these two cannabinoid synthase genes (Fig. 3).
The identification of sex chromosomes in the cannabis genome is another notable genomics‐driven achievement (Prentout et al., 2019; McKernan et al., 2020). Of the 565 sex‐linked genes identified in the PK transcriptome, 363 were mapped to cs10 v.1.0 chromosome 1 (cs10 v.2.0 chromosome 10), indicating that this chromosome pair constitutes the sex chromosomes (Prentout et al., 2019). This enabled the identification of sex‐specific molecular markers to aid cannabis breeding. THCA and CBDA are produced at much higher concentrations in the inflorescences of female cannabis plants compared with males, and hence female plants are economically more valuable (Braich et al., 2019; Prentout et al., 2019; McKernan et al., 2020). Having the capacity to identify male and female plants at an early stage enables yield improvement and better management of cannabis crops. Approximately 3500 sex‐biased genes have been identified, which are differentially expressed between female and male cannabis plants, with a subset being expressed in the flower buds (Prentout et al., 2019). These genes are not restricted to the sex chromosomes: some are located on the Y‐chromosome of male plants and are involved in trichome development, sex determination, hermaphroditism and photoperiod‐independence (McKernan et al., 2020).
Consistent chromosome nomenclature is important when working with multiple genome assemblies of the same species. However, discrepancies exist between the cs10, PK and FN assemblies. Chromosomal mappings performed by NRGene determined that cs10 v.2.0 chromosome 7 (cs10 v.1.0 chromosome 9) corresponds to chromosome 6 of PK and FN (Table 2; Maoz, 2020). Whilst our synteny analyses broadly agree with the findings from NRGene, they also show the lack of synteny between these genomes in some regions as illustrated by breaks in the chromosomal alignments (Fig. 5). These could have occurred due to SVs which would indicate true biological variation between these unrelated chemotypes. Another possibility relates to the more fragmented and less contiguous nature of the PK and FN assemblies relative to the cs10 v.2.0 assembly. Such lack of assembly contiguity can cause an underestimation of the syntenic relationship, causing genomic regions to erroneously appear as absent in one assembly relative to another (Liu et al., 2018).
Fig. 5.
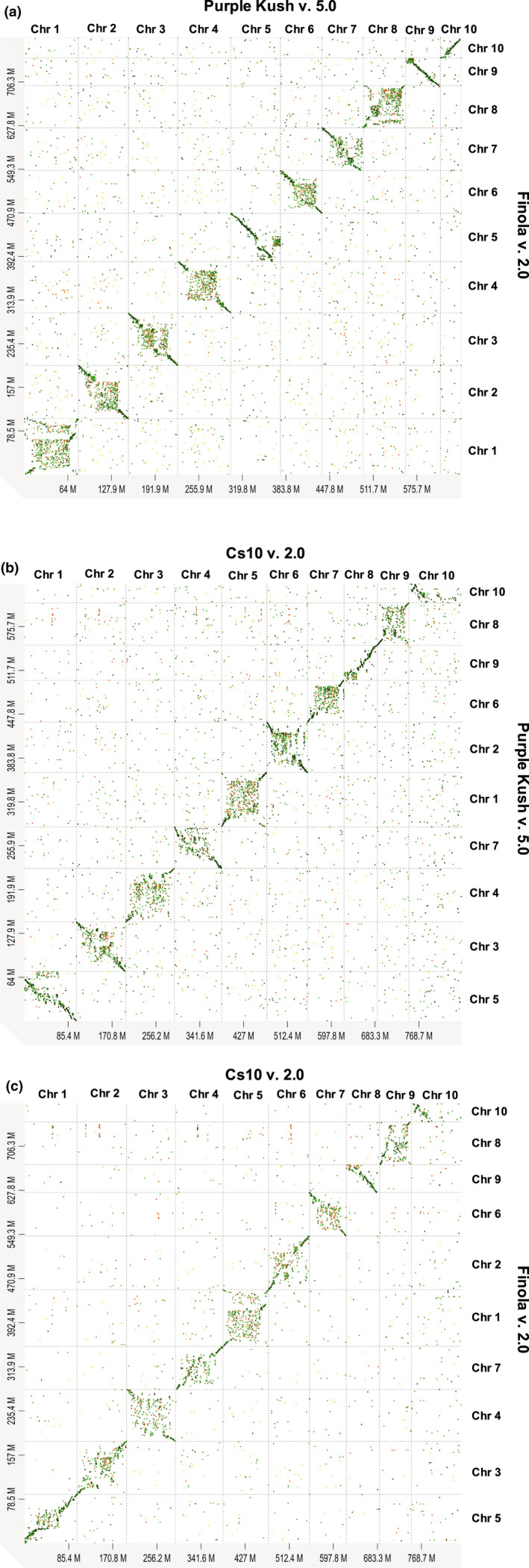
Dotplots showing the syntenic relationship between genomes of three Cannabis sativa cultivars. Pairwise genome alignments for (a) PK v.5.0 (GenBank acc. no. GCA_000230575.5) and FN v.2.0 (GenBank acc. no. GCA_003417725.2), (b) cs10 v.2.0 (GenBank acc. no. GCA_900626175.2) and PK v.5.0 and (c) cs10 v.2.0 and FN v.2.0 were performed using Minimap2 and the alignments were visualized using d‐genies (Cabanettes & Klopp, 2018; Li, 2018) (Supporting Information Table S3). Breaks in the alignment could be due to the presence of structural variants or the less contiguous nature of the PK and FN assemblies. The difference in chromosome orientation between the assemblies also can be seen. Only chromosome‐level alignments are shown. PK, Purple Kush; FN, Finola.
Our synteny analyses also revealed the presence of multiple CBDAS, CBDAS‐like and inactive THCAS (CBCAS) genes on chromosome 6 and unplaced scaffolds of the PK and FN assemblies (Tables S1, S3). We found that the scaffolds harbouring the CBDAS gene copies could be best aligned (start to end) against the CBDAS gene cluster region (29.63–30.93 Mbp) of cs10 v.2.0 chromosome 7 (Table S3). Likewise, the scaffolds containing the CBCAS genes could be best aligned against the inactive THCAS gene cluster region (25.82–26.05 Mbp) of cs10 v.2.0 chromosome 7. These findings suggest that the unplaced scaffolds belong to chromosome 6 of PK and FN and that the regions surrounding the candidate genes on these scaffolds share synteny with cs10 v.2.0 chromosome 7. Although these results highlight the syntenic relationship that exists between these various cannabis chemotypes, they also illustrate the difficulty in inferring such a relationship when presented with fragmented assemblies. The continuous improvement of current cannabis assemblies will therefore be required for more accurate comparative genomics analyses within and between species.
Gene expression
Large‐scale gene expression studies on cannabis have been instrumental in elucidating cannabinoid metabolism, but application of these approaches to other important traits is limited (Guerriero et al., 2017; Spitzer‐Rimon et al., 2019; McKernan et al., 2020). Many enzymes required for the conversion of primary metabolic precursors through to the synthesis of THCA and CBDA were identified by early comparisons between expressed sequence tags from trichome and leaf tissue (Marks et al., 2009; Van Bakel et al., 2011; Stout et al., 2012; Braich et al., 2019; Livingston et al., 2019; Zager et al., 2019). RNA‐Seq analysis identified 15‐fold increases in cannabinoid pathway genes in flowers of PK compared with FN, although this preliminary investigation lacked a sufficient number of biological replicates for robust statistical analyses (Van Bakel et al., 2011). Tissue and organ comparisons were recently improved, with female and male plants compared as well as various trichome morphotypes (Braich et al., 2019; Livingston et al., 2019). Gene co‐expression network analysis identified functionally relevant cannabis terpene synthases (CsTPS) involved in mono‐ and sesquiterpene accumulation (Zager et al., 2019). The differential expression of CsTPS genes among cultivars also has been linked to variation in terpene profiles of these cultivars (Booth et al., 2020). However, the genes underlying the synthesis of many minor cannabinoids and associated expression patterns are less well‐defined (Pollastro et al., 2011; Citti et al., 2019; Welling et al., 2019; Basas‐Jaumandreu & de las Heras, 2020).
Substantial CNV exists among cannabinoid synthase loci
The CNV of cannabinoid synthases has been reported in the cannabis genome and may have resulted from either natural or artificial selection (Grassa et al., 2018; Vergara et al., 2019; McKernan et al., 2020). Overall, it appears that CBDAS exists in significantly larger copy numbers compared to THCAS and CBCAS (Fig. 4; Tables S1, S2, S4). We also identified a larger number of CBDAS gene clusters relative to the other cannabinoid synthases, suggesting the presence of higher sequence variation among CBDAS gene copies. A previous report also suggested a more recent evolution of THCAS and CBCAS genes, which originated from the ancestral gene, CBDAS, as a result of gene duplication (Onofri et al., 2015). Additionally, we found the functional copies of CBDAS to be highly similar (>99% nucleotide identity) among accessions (Table S1). This also was the case for the functional THCAS and CBCAS loci, suggesting that intensive breeding practices have been performed to select these desirable cannabinoid synthase loci, which have become less polymorphic as a result. Beyond their similarity at the nucleotide and amino acid sequence levels, studies on the evolution of these genes would shed more light on their origin and functional divergence.
The relationship between cannabinoid synthase CNVs and cannabinoid content
The association between cannabinoid synthases, CNVs and overall cannabinoid yield remains unclear and may, in fact, not be significant (Grassa et al., 2018). Of the five quantitative trait loci (QTL) for total cannabinoid content recently identified, none belong to the cannabinoid synthase gene cluster on cs10 v.2.0 chromosome 7 (cs10 v.1.0 chromosome 9) (Grassa et al., 2018), suggesting that other non‐cannabinoid synthase‐related loci contribute to cannabinoid yield. Regardless, the functional forms of THCAS and CBDAS rather than their other copies are essential for the synthesis of THCA and CBDA, respectively. This is supported by our survey of the genome assemblies of the high‐THC producing cultivars PBBK and PK. Whilst both assemblies contained similar numbers of THCAS and CBDAS copies, they also contain a functional THCAS copy that bears > 99% nucleotide identity with the THCA‐producing gene locus identified by Sirikantaramas and colleagues (GenBank acc. no. AB057805.1; Fig. 4; Table S1) (Sirikantaramas et al., 2004). It is likely that nonfunctional CBDAS loci, inferred by the presence of premature stop codons and frameshift mutations, also are present in these assemblies (Weiblen et al., 2015). This is probable because CBDAS is a stronger competitor for CBGA than THCAS (Weiblen et al., 2015). However, when the nonfunctional form of CBDAS and the functional form of THCAS are both present, THCA is produced instead of CBDA (Weiblen et al., 2015). Indeed, we identified two loci in PBBK and PK that share > 96% nucleotide identity with the published nonfunctional CBDAS homologues from Skunk #1 (Table S5) (Weiblen et al., 2015). We also identified these loci in Cannatonic and JL, and found that these cultivars also contain one locus which is highly similar (>99% nucleotide identity) to AB057805.1 (Table S1).
It is important to validate hypotheses generated in silico to explain chemotypic properties of cannabis cultivars by conducting either in vitro or in vivo studies. This can be achieved using genetic approaches such as co‐segregation of known chemotypes and enzyme genotypes (Weiblen et al., 2015). For example, the association between functional forms of either THCAS or CBDAS and chemotype was demonstrated by analysis of an F2 population derived from crossing hemp and marijuana chemotype plants. The presence of functional THCAS or CBDAS in F2 individuals was determined by genotype analysis, then related to the plants' cannabinoid contents (Weiblen et al., 2015).
Short read sequencing creates challenges during CNV and expression analyses of the highly similar cannabinoid synthase loci
The predominant use of short read methods to analyze cannabis transcriptomes may create misleading artefacts resulting from the very high sequence similarity of cannabinoid synthase gene loci. Short reads that originate from paralogous and pseudogenic regions of a genome frequently cannot be distinguished (Ju et al., 2017; Dougherty et al., 2018). These challenges are illustrated in our analysis of trichome transcriptome data from nine medicinal cannabis cultivars generated by Zager et al. (2019) (Fig. 6a–c). Using cs10 v.1.0 as the reference, we identified two highly similar inactive THCAS loci (>99% nucleotide identity), LOC115697880 and LOC115697886, which were expressed much more highly than the remaining THCAS‐like and inactive THCAS‐like copies across all cultivars (Table S6). These loci are more similar to CBCAS (>99% nucleotide identity) than to the functional THCAS gene, AB057805.1 (>94% nucleotide identity) (Fig. 4; Table S1). It was reported that THCA (>13 % DW on average) was present in these cultivars (Zager et al., 2019). Therefore, we suspect that although the high‐THCA cultivars each harbour at least one functional THCAS gene copy, the reads that originated from this locus were forced to map to LOC115697880 and LOC115697886 owing to the higher similarity between the reads and these two loci, and the fact that cs10 lacks a functional THCAS gene. Whilst our findings agree with our observations above that CNV does not impact the synthesis and accumulation of THCA, they reflect the inadequacy of short reads to differentiate between highly similar loci and highlight the challenges of using an unrelated reference assembly for expression analyses.
Fig. 6.

Cannabinoid synthase gene expression in relation to cannabinoid content and composition in nine high cannabinoid yielding cannabis cultivars (data taken from Zager et al., 2019). (a) Tetrahydrocannabinolic acid : cannabidiolic acid (THCA : CBDA) ratio and (b) cannabinoid contents of the cultivars. The lower panel in (b) shows a zoomed‐in view of cannabinoid content (% DW) in the range 0–0.5%. (c) Trichome‐specific expression patterns of 13 cannabinoid synthase genes from cs10 v.1.0 genome assembly (GenBank acc. no. GCA_900626175.1) in these cultivars. Positions on chromosomes represent one or more cannabinoid synthase locus. The reference CBDAS (LOC115697762) and inactive THCAS (LOC115697880) loci are underlined. LOC115697762 bears 100% nucleotide identity with the functional CBDAS identified by Taura et al. (2007) (GenBank acc. no. AB292682.1), whereas LOC115697880 is 99% identical to CBCAS (GenBank acc. no. LY658671.1) at the nucleotide level (Taura et al., 2007). Of the 13 loci, two (LOC115698060 and LOC115697886) are pseudogenic inactive THCAS copies containing in‐frame stop codons, whereas the remaining 11 genes produce full‐length CDS. Trichome enriched RNA‐seq reads previously reported by Zager et al. (2019) were accessed from the NCBI Sequence Read Archive (SRA project no. PRJNA498707; Zager et al., 2019). The reads were mapped to the Cannabis sativa cs10 v.1.0 genome assembly (using Hisat2 v.2.1.0 and sorted by genomic location using samtools v.1.9 Li et al., 2009; Kim et al., 2019). stringtie v.1.3.5 was used to assemble RNA‐Seq alignments into potential transcripts and to calculate gene abundances (TPM) (Supporting Information Table S6; Pertea et al., 2015). Chromosome numbers have been changed to community standard nomenclature in accordance with cs10 v.2.0. (GenBank acc. no. GCA_900626174.2.). Cannabis sativa var. cs10 is associated with a high CBD chemotype. BB, Black Berry Kush; BL, Black Lime; CC, Cherry Chem; CT, Canna Tsu; MT, Mama Thai; SD, Sour Diesel; TP, Terple; TPM, Transcripts per million; VF, Valley Fire; WC, White Cookies. Error bars represent ± 1 SD of the mean metabolite content of each cultivar (n = 3).
Likewise, the higher expression of the CBDAS gene, LOC115697762, and higher CBDA content in Canna Tsu (7.76 ± 0.3% DW) relative to the other high‐THCA cultivars suggests that only one copy of the CBDAS gene is predominantly responsible for CBDA synthesis (Table S6). LOC115697762 bears 100% nucleotide identity with the functional CBDAS identified by Taura et al. (GenBank acc. no. AB292682.1; Fig. 4; Table S1) (Taura et al., 2007). Further, the moderate expression of another highly similar CBDAS‐like gene copy (LOC115696884, 88% nucleotide identity to LOC115697762) in all cultivars could explain the low concentrations (<0.45% DW) of CBDA detected in the high‐THCA cultivars (Zager et al., 2019).
Overall, our findings suggest that CNV does not affect cannabinoid content. Recent studies using long‐read approaches from PacBio (SMRT isoform sequencing, Iso‐Seq) and Oxford Nanopore support this (McKernan et al., 2020; Michael, 2020). Both concluded that only single copies of the functional THCAS and CBDAS genes were expressed in the cannabis genome and contribute to the production of THCA and CBDA, respectively (McKernan et al., 2020; Michael, 2020). This finding highlights the strength of long‐read sequencing at more accurately identifying and quantifying the expression of paralogous genes (Dougherty et al., 2018).
Terpene synthases
Cannabis is a prolific producer of terpenes and these compounds are thought to contribute to the therapeutic efficacy of cannabis preparations via the ‘entourage effect’, by acting in combination with cannabinoids (Ben‐Shabat et al., 1998; LaVigne et al., 2020). However, evidence for the existence of the entourage effect is largely anecdotal and lacks mechanistic explanation, for example whether the effect is additive or multiplicative. Despite their therapeutic potential and similar biosynthetic origin, genetic prediction of terpene composition is challenging. Genomic analysis of 55 CsTPS genes suggests a complex genetic background characterized by CsTPS nonhomologous gene clusters and tandem duplication events (Allen et al., 2019; Booth et al., 2020; McKernan et al., 2020). Further, the presence of extensive CNVs within the CsTPS‐a (sesquiterpene synthase) and CsTPS‐b (monoterpene synthase) gene subfamilies point to their highly diverse nature (Booth et al., 2020). It appears that some members of the CsTPS‐b gene subfamily can produce sesquiterpene in both cannabis and sandalwood. This indicates that similar selective pressures have occurred on the species‐specific monoterpene synthase ancestors to produce these loci (Gao et al., 2012). The diversity of the CsTPS genes complicates studies of gene regulation at various levels. Terpene composition can vary between cultivars, tissue types, trichome morphotypes and across development, whilst nonenzymatic modifications such as the oxidation of β‐myrcene cause variation independent of genomic and transcriptomic regulation (Marchini et al., 2014; Aizpurua‐Olaizola et al., 2016; Allen et al., 2019; Livingston et al., 2019; Booth et al., 2020). Consequently, future transcriptional studies would need to consider gene‐environment interactions, as well as organ, tissue and cell‐type specificity.
SNP studies
High‐throughput SNP studies have contributed substantially to our understanding of cannabis evolutionary history. Key findings include: the genome‐wide distinction between drug and hemp types that resulted from selective breeding; the association between chemotypic identity and variation of loci encoding cannabinoid synthases; and errors in cultivar classification and ancestry by breeders (Van Bakel et al., 2011; Sawler et al., 2015; Lynch et al., 2016; Soorni et al., 2017). These findings are likely to facilitate the development of more accurate diagnostic systems for cannabis germplasm to assist with product compliance, traceability, provenance and consumer education (Henry et al., 2020). Domestication and intensive breeding have narrowed the genetic and allelic diversity of cannabis gene pools (Sawler et al., 2015; Soorni et al., 2017). However, these can be expanded by a comprehensive, genomics‐based assessment of cannabis germplasm that defines the diversity available. This would in turn help with the identification of heterotic groups for use in crosses to achieve hybrid vigour and hence assist with the creation of elite cultivars (Huang et al., 2015).
Genomics approaches that could be applied in the near‐term to improve cannabis traits
Our knowledge of how cannabis traits relate to genotype is currently limited. Many next generation sequencing (NGS)‐based tools exist that can rapidly identify genetic variation underlying traits of interest. The available genetic and genomic resources provide an opportunity to apply NGS tools for trait discovery and molecular breeding in cannabis, as has been achieved in other crops (Varshney et al., 2014; Kang et al., 2016; Crossa et al., 2017).
QTL mapping and gene discovery
Low‐density molecular markers such as amplified fragment length polymorphisms (AFLPs) and simple sequence repeats (SSRs) have been employed in cannabis to identify QTLs associated with sex determination and cannabinoid composition (Weiblen et al., 2015; Faux et al., 2016). High‐density genetic maps recently developed for cannabis should improve the accuracy of QTL mapping (Grassa et al., 2018; Laverty et al., 2019). Several NGS‐based methods are available for high‐resolution mapping and interval detection in plants (Schneeberger et al., 2009; Abe et al., 2012; Takagi et al., 2013). A common feature of these methods is that they combine NGS with bulked‐segregant analysis to study mutated or natural populations. The methods have been deployed mainly in self‐pollinating species with homozygous genomes (Jaganathan et al., 2020). However, recent protocol improvements have allowed their application to heterozygous, outcrossing species, for example to identify loci involved in sex determination, flowering, trichome formation, anthocyanin accumulation and leaf shape in Dioscorea rotundata (Guinea yam), Brassica rapa and Vitis vinifera (grapevine) (Tamiru et al., 2017; Demmings et al., 2019; Itoh et al., 2019; Zhang et al., 2020). Similar approaches could be used in cannabis.
Ethyl methanesulfonate (EMS) mutagenesis has been applied successfully to hemp and a protocol exists for cannabis cell culture (Bielecka et al., 2014; Hari, 2020). However, large‐scale generation and screening of mutant lines can be a logistical challenge in cannabis owing to its size and the requirement in many jurisdictions to grow it in secure and licensed facilities. Consequently, the exploitation of the cannabis natural diversity provides a better option for mining important genes in the short term (Vergara et al., 2016; Welling et al., 2016). Therefore, the available gene/QTL mapping tools should go beyond simple genetic variations such as SNPs and small insertions/deletions (Indels) and consider SVs including large Indels, rearrangements and CNVs, all of which have been shown to affect trait diversity in several crops including cannabis (Wang et al., 2016; Chakraborty et al., 2019; McKernan et al., 2020).
Genome‐wide association studies (GWAS)
There are only two published cannabis GWAS studies, one of which is ongoing and the other reporting marker‐trait association using a limited number of SNPs (B. J. Campbell et al., 2019; Henry et al., 2020; http://multihemp.eu/). The paucity of studies is likely a consequence of the previous limited accessibility of cannabis genetic diversity and insufficient marker density. GWAS relies on linkage disequilibrium (LD) for detecting common genetic variants associated with a trait in natural and experimental populations (Brachi et al., 2011). Consequently, it has been widely deployed in self‐crossing species that generally have more extensive LD. Nevertheless, GWAS has been used successfully for genotype–trait association in outcrossing and vegetatively propagated species including date palm, sweet potato and hop (Humulus lupulus), the species most closely related to cannabis (Henning et al., 2015; Hazzouri et al., 2019; Okada et al., 2019). Application of efficient and high‐throughput phenotyping systems to cannabis will help GWAS studies owing to the species’ high phenotypic plasticity; applicable commercial and open‐source solutions already exist (L. G. Campbell et al., 2019). A variant of GWAS termed mGWAS (metabolite‐based GWAS) that combines genotyping and metabolic profiling has proved powerful for dissecting the genetic bases of metabolic diversity in plants (Fang & Luo, 2019; Chen et al., 2020). A similar approach could be applied to the cannabinoid and terpene biosynthesis pathways of cannabis.
Genomic selection
Genomic selection utilizes genome‐wide marker information to predict the breeding value of genotypes, which is an estimate of the value of a genotype as a parent. It integrates genotypic and phenotypic data from a reference population and uses statistical models to determine the genomic‐estimated breeding values (GEBVs) of other individuals for which only genotype information is available. Elite lines with the highest GEBVs are then selected for use as parents in breeding programs. Genomic selection is considered particularly promising for genetic improvement of complex traits controlled by many genes with minor effects (Heslot et al., 2015; Spindel & McCouch, 2016). The approach has been successfully implemented in breeding programmes for outcrossing heterozygous species such as maize and cassava (Crossa et al., 2017; Elias et al., 2018). It might consequently make a notable contribution to the genetic improvement of complex cannabis traits once marker density, population size, statistical models and accuracy of phenotyping improve.
Emerging genomics technologies with high potential in cannabis research
Phased genome assemblies
The assembly of heterozygous plant genomes remains challenging despite the use of long‐read sequencing technology (Michael & VanBuren, 2020). Genome assembly of outcrossing species such as cannabis is particularly challenging because haplotypes consist of various repeating units, short Indels and SVs (Chin et al., 2016; VanBuren et al., 2018). As a result, the majority of near‐complete haploid cannabis assemblies are fully unphased, meaning they are a synthetic patchwork of collapsed segments of homologous chromosomes that do not fully capture genome composition (Chin et al., 2016; Grassa et al., 2018; Laverty et al., 2019; Gao et al., 2020). There have been recent efforts to produce partially phased genome assemblies for cannabis (two draft assemblies are now available for the maternal Jamaican Lion cultivar) and hop (var. Cascade) (Padgitt‐Cobb et al., 2019; Medicinal Genomics, 2020b). In addition, NRGene recently announced the creation of fully‐phased genome assemblies for two proprietary elite cultivars using cs10 as the basis for scaffold ordering (Weisshaus, 2020). These assemblies are likely to improve our understanding of haplotype structure and heterozygosity, which is essential for allele‐specific analyses of quantitative traits (Chin et al., 2016).
Cannabis pangenomics
Plant pangenomics studies conducted over the last five years have demonstrated the inadequacy of a single reference genome in representing the entire genetic diversity of a species (Golicz et al., 2016; Hurgobin et al., 2018). The creation of a cannabis pangenome promises to shed more light on the extent of gene content variation, as well as forming the basis for cannabis breeding programmes. For instance, the inclusion of diverse genotypes and wild cannabis populations (sometime referred to as C. ruderalis), would facilitate the identification of elite marker genes in the dispensable/variable genome of cannabis and drive the process of heterosis to create resilient and high‐yielding cultivars (Tao et al., 2019). Additionally, combining gene PAV and pangenome‐wide SNPs would lead to a more accurate identification of causal variants associated with traits of interest (Hurgobin & Edwards, 2017).
Independent cannabis pangenome initiatives are being led by NRGene and Medicinal Genomics (NRGene, 2018; Medicinal Genomics, 2020a). Allelic variations and additional genes involved in cannabinoid biosynthesis were identified by comparing the recent fully‐phased assemblies from NRGene with existing chromosome‐level, unphased cannabis assemblies (Weisshaus, 2020). Conserved genomic regions, as well as variable regions harbouring SVs such as CNVs, PAVs and large rearrangements, which may be implicated in cannabis and hemp breeding, were also identified. The increased accessibility of long‐read sequencing will likely encourage the construction of graph‐based pangenomes, which are considered to be the future of plant pangenomics studies (Bayer et al., 2020). A graph‐based pangenome consists of a single, nonredundant reference that contains SVs from multiple individuals/accessions. It can be visualized as a sequence graph with branches representing accession‐specific sequences (Garrison et al., 2018). The first graph‐based pangenome of soybean was recently produced using 29 wild and cultivated accessions (Liu et al., 2020). Using this resource and the SVs that they had identified among the accessions, the authors performed a GWAS and identified a 10‐kbp PAV, which was associated with seed lustre variation. This type of analysis would be helpful for cannabis as it could be used to determine the SV landscape of this important crop and its association with traits of interest.
Single cell genomics
Transcriptomics has been revolutionized by single cell RNA‐Sequencing (scRNA‐Seq) because it enables researchers to investigate the complex interplay of molecular regulators and identify active cellular processes at true cellular resolution (Rich‐Griffin et al., 2020). Whilst only a handful of scRNA‐Seq studies have been conducted in plants to date, they demonstrate that this technology may have great potential in cannabis breeding (Rich‐Griffin et al., 2020). For instance, given the specificity of the cannabinoid biosynthetic pathway to the capitate stalked trichomes, a detailed view of gene expression in the cell‐types involved in this pathway would assist with the selection of marker genes. An understanding of cell‐types which respond most strongly to environmental cues such as biotic or abiotic stresses also would be valuable. Combining this information with single‐cell assay for transposable accessible chromatin (scATAC) sequencing data would enable the identification of regulatory regions of the genome that drive cell‐type specific expression (Rich‐Griffin et al., 2020). Integration of scRNA‐Seq with CRISPR/Cas9‐based genetic screens would enable more effective selection of cell types for targeted gene manipulation whilst reducing the impact of pleiotropy (Yuan et al., 2018; Shahan, 2019; Marand et al., 2020; Rich‐Griffin et al., 2020).
Concluding remarks and future perspectives
Cannabis biology remains poorly understood but the relaxation of legislation, together with the application of existing and emerging genomics technologies, is likely to fuel more scientific research on this fascinating plant. The rapid generation of sequencing data and the creation of additional, fully phased, chromosome‐level genome assemblies will enable a more comprehensive assessment of the genetic architecture of important traits and aid in marker discovery. Whilst genomics‐assisted breeding will have a pivotal role in increasing the efficiency and precision of cannabis crop improvement, the utility of integrating the other ‘omics technologies cannot be overlooked. In line with the overarching goal of the ICGRC, the integration of individual ‘omics approaches such as transcriptomics, phenomics, metabolomics and proteomics, among others, and the ongoing development of computational methods will be useful for understanding gene function, biological and metabolic pathways, and regulatory networks underlying traits of interest. Complementing these ‘omics approaches with traditional breeding practices will provide a multifaceted strategy for the improvement of this emerging crop species.
Author contributions
BH and MGL conceived the manuscript; BH and MW conducted genome sequence analyses; BH, MTO and MW wrote the manuscript; BH, MTO, MTW, MGL, MSD, AB and JW revised the manuscript. All authors read and approved the final version.
Supporting information
Table S1 Summary of best Blast hits of the functional cannabinoid biosynthesis genes against the latest C. sativa reference genome assemblies.
Table S2 Summary of best Blast hits of other THCAS‐ and CBDAS‐like gene copies against the latest C. sativa reference genome assemblies.
Table S3 Alignment of Purple Kush v.5.0 (GenBank acc. no. GCA_000230575.5) and Finola v.2.0 (GenBank acc. no. GCA_003417725.2) unplaced scaffolds against the cs10 v.2.0 genome assembly (GenBank acc. no. GCA_900626175.2).
Table S4 Cannabinoid synthase genes from cs10 v.2.0 (GenBank acc. no. GCA_900626175.2) and Jamaican Lion (female parent: GenBank acc. no. GCA_012923435.1; male parent: GenBank acc. no. GCA_013030025.1) assemblies.
Table S5 Summary of best Blast hits of nonfunctional CBDAS homologs from Skunk #1 against the latest C. sativa reference genome assemblies.
Table S6 Transcript per million (TPM) counts of the 13 cannabinoid synthase genes from the cs10 v.1.0 genome assembly (GenBank acc. no. GCA_900626175.1) in nine medicinal cannabis cultivars from Zager et al. (2019).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was funded by the Australian Research Council Industrial Transformation Hub in Medicinal Agriculture (IH180100006), institutional and industry partners. Cann Group Limited are partners within the Australian Research Council Industrial Transformation Hub in Medicinal Agriculture.
References
- Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M et al. 2012. Genome sequencing reveals agronomically important loci in rice using MutMap. Nature Biotechnology 30: 174–178. [DOI] [PubMed] [Google Scholar]
- Adams R, Hunt M, Clark J. 1940. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. I. Journal of the American Chemical Society 62: 196–200. [Google Scholar]
- Aizpurua‐Olaizola O, Soydaner U, Öztürk E, Schibano D, Simsir Y, Navarro P, Etxebarria N, Usobiaga A. 2016. Evolution of the cannabinoid and terpene content during the growth of Cannabis sativa plants from different chemotypes. Journal of Natural Products 79: 324–331. [DOI] [PubMed] [Google Scholar]
- Allen KD, McKernan K, Pauli C, Roe J, Torres A, Gaudino R. 2019. Genomic characterization of the complete terpene synthase gene family from Cannabis sativa . PLoS ONE 14: e0222363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Andre CM, Hausman J‐F, Guerriero G. 2016. Cannabis sativa: the plant of the thousand and one molecules. Frontiers in Plant Science 4: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ, Choulet F, Distelfeld A, Poland J. 2018. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361: eaar7191. [DOI] [PubMed] [Google Scholar]
- Basas‐Jaumandreu J, De las Heras FXC. 2020. GC‐MS metabolite profile and identification of unusual homologous cannabinoids in high potency Cannabis sativa . Planta Medica 86: 338–347. [DOI] [PubMed] [Google Scholar]
- Bayer PE, Golicz AA, Scheben A, Batley J, Edwards D. 2020. Plant pan‐genomes are the new reference. Nature Plants 6: 914–920. [DOI] [PubMed] [Google Scholar]
- Ben‐Shabat S, Fride E, Sheskin T, Tamiri T, Rhee MH, Vogel Z, Bisogno T, De Petrocellis L, Di Marzo V, Mechoulam R. 1998. An entourage effect: inactive endogenous fatty acid glycerol esters enhance 2‐arachidonoyl‐glycerol cannabinoid activity. European Journal of Pharmacology 353: 23–31. [DOI] [PubMed] [Google Scholar]
- Bielecka M, Kaminski F, Adams I, Poulson H, Sloan R, Li Y, Larson TR, Winzer T, Graham IA. 2014. Targeted mutation of Δ12 and Δ15 desaturase genes in hemp produce major alterations in seed fatty acid composition including a high oleic hemp oil. Plant Biotechnology Journal 12: 613–623. [DOI] [PubMed] [Google Scholar]
- Boggs DL, Nguyen JD, Morgenson D, Taffe MA, Ranganathan M. 2018. Clinical and preclinical evidence for functional interactions of cannabidiol and Δ 9‐tetrahydrocannabinol. Neuropsychopharmacology 43: 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth JK, Yuen MM, Jancsik S, Madilao L, Page J, Bohlmann J. 2020. Terpene synthases and terpene variation in Cannabis sativa . Plant Physiology 184: 130–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachi B, Morris GP, Borevitz JO. 2011. Genome‐wide association studies in plants: the missing heritability is in the field. Genome Biology 12: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw RHW, Coxon P, Greig JRA, Hall AR. 1981. New fossil evidence for the past cultivation and processing of hemp (Cannabis sativa L.) in Eastern England. New Phytologist 89: 503–510. [Google Scholar]
- Braich S, Baillie RC, Jewell LS, Spangenberg GC, Cogan NOI. 2019. Generation of a comprehensive transcriptome atlas and transcriptome dynamics in medicinal cannabis. Scientific Reports 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanettes F, Klopp C. 2018. D‐GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6: e4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BJ, Berrada AF, Hudalla C, Amaducci S, McKay JK. 2019. Genotype × environment interactions of industrial hemp cultivars highlight diverse responses to environmental factors. Agrosystems, Geosciences & Environment 2: 1–11. [Google Scholar]
- Campbell LG, Naraine SG, Dusfresne J. 2019. Phenotypic plasticity influences the success of clonal propagation in industrial pharmaceutical Cannabis sativa . PLoS ONE 14: e0213434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Emerson J, Macdonald SJ, Long AD. 2019. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nature Communications 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hu X, Shi T, Yin H, Sun D, Hao Y, Xia X, Luo J, Fernie AR, He Z et al. 2020. Metabolite‐based genome‐wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnology Journal 18: 1722–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Borgelt LM, Blackmer AB. 2019. Cannabidiol: a new hope for patients with Dravet or Lennox‐Gastaut syndromes. Annals of Pharmacotherapy 53: 603–611. [DOI] [PubMed] [Google Scholar]
- Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa‐Balderas R, Morales‐Cruz A. 2016. Phased diploid genome assembly with single‐molecule real‐time sequencing. Nature Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citti C, Linciano P, Panseri S, Vezzalini F, Forni F, Vandelli MA, Cannazza G. 2019. Cannabinoid profiling of hemp seed oil by liquid chromatography coupled to high‐resolution mass spectrometry. Frontiers in Plant Science 10: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RC, Merlin MD. 2016. Cannabis domestication, breeding history, present‐day genetic diversity, and future prospects. Critical Reviews in Plant Sciences 35: 293–327. [Google Scholar]
- Crossa J, Pérez‐Rodríguez P, Cuevas J, Montesinos‐López O, Jarquín D, DelosCampos G, Burgueño J, González‐Camacho JM, Pérez‐Elizalde S, Beyene Y. 2017. Genomic selection in plant breeding: methods, models, and perspectives. Trends in Plant Science 22: 961–975. [DOI] [PubMed] [Google Scholar]
- De Meijer EPM, Bagatta M, Carboni A, Crucitti P, Moliterni VMC, Ranalli P, Mandolino G. 2003. The inheritance of chemical phenotype in Cannabis sativa L. Genetics 163: 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmings EM, Williams BR, Lee CR, Barba P, Yang S, Hwang CF, Reisch BI, Chitwood DH, Londo JP. 2019. Quantitative Trait Locus analysis of leaf morphology indicates conserved shape loci in grapevine. Frontiers in Plant Science 10: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty ML, Underwood JG, Nelson BJ, Tseng E, Munson KM, Penn O, Nowakowski TJ, Pollen AA, Eichler EE. 2018. Transcriptional fates of human‐specific segmental duplications in brain. Genome Research 28: 1566–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias AA, Rabbi I, Kulakow P, Jannink JL. 2018. Improving genomic prediction in cassava field experiments using spatial analysis. G3: Genes, Genomes, Genetics 8: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Luo J. 2019. Metabolic GWAS‐based dissection of genetic bases underlying the diversity of plant metabolism. The Plant Journal 97: 91–100. [DOI] [PubMed] [Google Scholar]
- Faux AM, Draye X, Flamand MC, Occre A, Bertin P. 2016. Identification of QTLs for sex expression in dioecious and monoecious hemp (Cannabis sativa L.). Euphytica 209: 357–376. [Google Scholar]
- Gao S, Wang B, Xie S, Xu X, Zhang J, Pei L, Yu Y, Yang W, Zhang Y. 2020. A high‐quality reference genome of wild Cannabis sativa . Horticulture Research 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Honzatko RB, Peters RJ. 2012. Terpenoid synthase structures: a so far incomplete view of complex catalysis. Natural Product Reports 29: 1153–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Sirén J, Novak AM, Hickey G, Eizenga JM, Dawson ET, Jones W, Garg S, Markello C, Lin MF. 2018. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nature Biotechnology 36: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz AA, Bayer PE, Barker GC, Edger PP, Kim H, Martinez PA, Chan CKK, Severn‐Ellis A, McCombie WR, Parkin IA. 2016. The pangenome of an agronomically important crop plant Brassica oleracea . Nature Communications 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassa CJ, Wenger JP, Dabney C, Poplawski SG, Motley ST, Michael TP, Schwartz C, Weiblen GD. 2018. A complete Cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv: doi: 10.1101/458083 [DOI] [Google Scholar]
- Guerriero G, Mangeot‐Peter L, Legay S, Behr M, Lutts S, Siddiqui KS, Hausman JF. 2017. Identification of fasciclin‐like arabinogalactan proteins in textile hemp (Cannabis sativa L.): in silico analyses and gene expression patterns in different tissues. BMC Genomics 18: 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gülck T, Møller BL. 2020. Phytocannabinoids: origins and biosynthesis. Trends in Plant Science 25: 985–1004. [DOI] [PubMed] [Google Scholar]
- Hari V. 2020. Generation of new varieties of cannabis by chemical mutagenesis of cannabis cell suspensions. United States Patent Application 16/594,733, filed April 9, 2020.
- Hazzouri KM, Gros‐Balthazard M, Flowers JM, Copetti D, Lemansour A, Lebrun M, Masmoudi K, Ferrand S, Dhar MI, Fresquez ZA. 2019. Genome‐wide association mapping of date palm fruit traits. Nature Communications 10: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning J, Coggins J, Hill S, Hendrix D, Townsend S. 2015. Genome‐wide association study on ten traits of economic importance in hop (Humulus lupulus L.). IV International Humulus Symposium 1236: 93–104. [Google Scholar]
- Henry P, Khatodia S, Kapoor K, Gonzales B, Middleton A, Hong K, Hilyard A, Johnson S, Allen D, Chester Z. 2020. A Single Nucleotide Polymorphism assay sheds light on the extent and distribution of genetic diversity, population structure and functional basis of key traits in cultivated North American cannabis. bioRxiv: doi: 10.1101/2020.02.16.951459. [DOI] [PMC free article] [PubMed]
- Heslot N, Jannink JL, Sorrells ME. 2015. Perspectives for genomic selection applications and research in plants. Crop Science 55: 1–12. [Google Scholar]
- Huang X, Yang S, Gong J, Zhao Y, Feng Q, Gong H, Li W, Zhan Q, Cheng B, Xia J et al. 2015. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nature Communications 6: 6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurgobin B, Edwards D. 2017. SNP discovery using a pangenome: has the single reference approach become obsolete? Biology 6: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurgobin B, Golicz AA, Bayer PE, Chan CKK, Tirnaz S, Dolatabadian A, Schiessl SV, Samans B, Montenegro JD, Parkin IA. 2018. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus . Plant Biotechnology Journal 16: 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Segawa T, Tamiru M, Abe A, Sakamoto S, Uemura A, Oikawa K, Kutsuzawa H, Koga H, Imamura T et al. 2019. Next‐generation sequencing‐based bulked segregant analysis for QTL mapping in the heterozygous species Brassica rapa . Theoretical and Applied Genetics 132: 2913–2925. [DOI] [PubMed] [Google Scholar]
- Jaganathan D, Bohra A, Thudi M, Varshney RK. 2020. Fine mapping and gene cloning in the post‐NGS era: advances and prospects. Theoretical and Applied Genetics 133: 1791–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G. 2014. InterProScan 5: genome‐scale protein function classification. Bioinformatics 30: 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju CJT, Zhao Z, Wang W. 2017. Efficient approach to correct read alignment for pseudogene Abundance Estimates. IEEE/ACM Transactions on Computational Biology and Bioinformatics 14: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Lee T, Lee J, Shim S, Jeong H, Satyawan D, Kim MY, Lee SH. 2016. Translational genomics for plant breeding with the genome sequence explosion. Plant Biotechnology Journal 14: 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal S. 2012. Impact of physical and chemical mutagens on sex expression in Cannabis sativa . Indian Journal of Fundamental and Applied Life Sciences 2: 97–103. [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nature Biotechnology 37: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojoma M, Seki H, Yoshida S, Muranaka T. 2006. DNA polymorphisms in the tetrahydrocannabinolic acid (THCA) synthase gene in “drug‐type” and “fiber‐type” Cannabis sativa L. Forensic Science International 159: 132–140. [DOI] [PubMed] [Google Scholar]
- Kovalchuk I, Pellino M, Rigault P, van Velzen R, Ebersbach J, Ashnest JR, Mau M, Schranz M, Alcorn J, Laprairie R. 2020. The genomics of cannabis and its close relatives. Annual Review of Plant Biology 71: 713–739. [DOI] [PubMed] [Google Scholar]
- Laverty KU, Stout JM, Sullivan MJ, Shah H, Gill N, Holbrook L, Deikus G, Sebra R, Hughes TR, Page JE. 2019. A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC/CBD acid synthase loci. Genome Research 29: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVigne J, Hecksel R, Streicher JM. 2020. In defense of the “Entourage Effect”: Terpenes found in Cannabis sativa activate the Cannabinoid Receptor 1 in vivo . FASEB Journal 34: 1. [Google Scholar]
- Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128. [DOI] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Hunt M, Tsai IJ. 2018. Inferring synteny between genome assemblies: a systematic evaluation. BMC Bioinformatics 19: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Du H, Li P, Shen Y, Peng H, Liu S, Zhou G‐A, Zhang H, Liu Z, Shi M. 2020. Pan‐genome of wild and cultivated soybeans. Cell 182: 162–176. e113. [DOI] [PubMed] [Google Scholar]
- Livingston S, Quilichini T, Booth J, Wong D, Rensing K, Laflamme‐Yonkman J, Castellarin S, Bohlmann J, Page J, Samuels AL. 2019. Cannabis glandular trichomes alter morphology and metabolite content during flower maturation. The Plant Journal 101: 37–56. [DOI] [PubMed] [Google Scholar]
- Long T, Wagner M, Demske D, Leipe C, Tarasov PE. 2017. Cannabis in Eurasia: origin of human use and Bronze Age trans‐continental connections. Vegetation History and Archaeobotany 26: 245–258. [Google Scholar]
- Lubell JD, Brand MH. 2018. Foliar sprays of silver thiosulfate produce male flowers on female hemp plants. Horttechnology 28: 743–747. [Google Scholar]
- Lynch RC, Vergara D, Tittes S, White K, Schwartz C, Gibbs MJ, Ruthenburg TC, deCesare K, Land DP, Kane NC. 2016. Genomic and chemical diversity in cannabis. Critical Reviews in Plant Sciences 35: 349–363. [Google Scholar]
- Maoz TY. 2020. Making Cannabis History in 2020. [WWW document] URL https://www.nrgene.com/blog/making‐cannabis‐history‐in‐2020/
- Marand AP, Chen Z, Gallavotti A, Schmitz RJ. 2020. A cis‐regulatory atlas in maize at single‐cell resolution. bioRxiv: doi: 2020.2009.2027.315499 [DOI] [PubMed]
- Marchini M, Charvoz C, Dujourdy L, Baldovini N, Filippi JJ. 2014. Multidimensional analysis of cannabis volatile constituents: identification of 5, 5‐dimethyl‐1‐vinylbicyclo [2.1. 1] hexane as a volatile marker of hashish, the resin of Cannabis sativa L. Journal of Chromatography A 1370: 200–215. [DOI] [PubMed] [Google Scholar]
- Marks MD, Tian L, Wenger JP, Omburo SN, Soto‐Fuentes W, He J, Gang DR, Weiblen GD, Dixon RA. 2009. Identification of candidate genes affecting Δ9‐tetrahydrocannabinol biosynthesis in Cannabis sativa . Journal of Experimental Botany 60: 3715–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan KJ, Helbert Y, Kane LT, Ebling H, Zhang L, Liu B, Eaton Z, McLaughlin S, Kingan S, Baybayan P. 2020. Sequence and annotation of 42 cannabis genomes reveals extensive copy number variation in cannabinoid synthesis and pathogen resistance genes. bioRxiv: doi: 10.1101/2020.01.03.894428. [DOI]
- Medicinal Genomics . 2020a. The cannabis pan‐genome project: advancing cannabis breeding . [WWW document] URL https://www.medicinalgenomics.com/cannabis‐pan‐genome‐project‐advancing‐cannabis‐breeding/ [accessed 1 September 2020].
- Medicinal Genomics . 2020b. Jamaican Lion data release . [WWW document] URL https://www.medicinalgenomics.com/jamaican‐lion‐data‐release/ [accessed 1 September 2020].
- Michael TP. 2020. Long‐read nanopore cDNA sequencing and direct DNA methylation detection resolves copy number debate in cannabis . London Calling 2020 Online. [Webinar] URL https://londoncallingconf.co.uk/resource‐centre/long‐read‐nanopore‐cdna‐sequencing‐and‐direct‐dna‐methylation‐detection‐resolves‐0 [accessed 20 August 2020].
- Michael TP, VanBuren R. 2020. Building near‐complete plant genomes. Current Opinion in Plant Biology 54: 26–33. [DOI] [PubMed] [Google Scholar]
- Morrell PL, Buckler ES, Ross‐Ibarra J. 2012. Crop genomics: advances and applications. Nature Reviews Genetics 13: 85–96. [DOI] [PubMed] [Google Scholar]
- NRGene . 2018. Creating the first cannabis pangenome . [WWW document] URL https://www.analyticalcannabis.com/news/creating‐the‐first‐cannabis‐pangenome‐308027 [accessed 1 September 2020].
- O'Connell BK, Gloss D, Devinsky O. 2017. Cannabinoids in treatment‐resistant epilepsy: a review. Epilepsy & Behavior 70: 341–348. [DOI] [PubMed] [Google Scholar]
- Okada Y, Monden Y, Nokihara K, Shirasawa K, Isobe S, Tahara M. 2019. Genome‐wide association studies (GWAS) for yield and weevil resistance in sweet potato (Ipomoea batatas (L.) Lam). Plant Cell Reports 38: 1383–1392. [DOI] [PubMed] [Google Scholar]
- Onofri C, de Meijer EP, Mandolino G. 2015. Sequence heterogeneity of cannabidiolic and tetrahydrocannabinolic acid‐synthase in Cannabis sativa L. and its relationship with chemical phenotype. Phytochemistry 116: 57–68. [DOI] [PubMed] [Google Scholar]
- Padgitt‐Cobb LK, Kingan SB, Wells J, Elser J, Kronmiller B, Moore D, Concepcion G, Peluso P, Rank D, Jaiswal P et al. 2019. A phased, diploid assembly of the Cascade hop (Humulus lupulus) genome reveals patterns of selection and haplotype variation. bioRxiv: doi: 10.1101/786145 [DOI] [PubMed]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piluzza G, Delogu G, Cabras A, Marceddu S, Bullitta S. 2013. Differentiation between fiber and drug types of hemp (Cannabis sativa L.) from a collection of wild and domesticated accessions. Genetic Resources and Crop Evolution 60: 2331–2342. [Google Scholar]
- Pisupati R, Vergara D, Kane NC. 2018. Diversity and evolution of the repetitive genomic content in Cannabis sativa . BMC Genomics 19: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollastro F, Taglialatela‐Scafati O, Allara M, Munoz E, Di Marzo V, De Petrocellis L, Appendino G. 2011. Bioactive prenylogous cannabinoid from fiber hemp (Cannabis sativa). Journal of Natural Products 74: 2019–2022. [DOI] [PubMed] [Google Scholar]
- Prentout D, Razumova O, Rhoné B, Badouin H, Henri H, Feng C, Käfer J, Karlov G, Marais GA. 2019. A high‐throughput segregation analysis identifies the sex chromosomes of Cannabis sativa . bioRxiv: doi: 10.1101/721324 [DOI] [PMC free article] [PubMed]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram HM, Sett R. 1982. Induction of fertile male flowers in genetically female Cannabis sativa plants by silver nitrate and silver thiosulphate anionic complex. Theoretical and Applied Genetics 62: 369–375. [DOI] [PubMed] [Google Scholar]
- Rich‐Griffin C, Stechemesser A, Finch J, Lucas E, Ott S, Schäfer P. 2020. Single‐cell transcriptomics: a high‐resolution avenue for plant functional genomics. Trends in Plant Science 25: 186–197. [DOI] [PubMed] [Google Scholar]
- Sawler J, Stout JM, Gardner KM, Hudson D, Vidmar J, Butler L, Page JE, Myles S. 2015. The genetic structure of marijuana and hemp. PLoS ONE 10: e0133292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena RK, Edwards D, Varshney RK. 2014. Structural variations in plant genomes. Briefings in Functional Genomics 13: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz MC, Witkowski J, McCombie WR. 2012. Current challenges in de novo plant genome sequencing and assembly. Genome Biology 13: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL, Jørgensen JE, Weigel D, Andersen SU. 2009. SHOREmap: simultaneous mapping and mutation identification by deep sequencing. Nature Methods 6: 550–551. [DOI] [PubMed] [Google Scholar]
- Schwabe AL, McGlaughlin ME. 2019. Genetic tools weed out misconceptions of strain reliability in Cannabis sativa: implications for a budding industry. Journal of Cannabis Research 1: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahan R. 2019. The future is now: gene expression dynamics at single cell resolution. The Plant Cell 31: 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J. 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirikantaramas S, Morimoto S, Shoyama Y, Ishikawa Y, Wada Y, Shoyama Y, Taura F. 2004. The gene controlling marijuana psychoactivity molecular cloning and heterologous expression of Δ1‐tetrahydrocannabinolic acid synthase from Cannabis sativa L. Journal of Biological Chemistry 279: 39767–39774. [DOI] [PubMed] [Google Scholar]
- Soorni A, Fatahi R, Haak DC, Salami SA, Bombarely A. 2017. Assessment of genetic diversity and population structure in Iranian cannabis germplasm. Scientific Reports 7: 15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindel JE, McCouch SR. 2016. When more is better: how data sharing would accelerate genomic selection of crop plants. New Phytologist 212: 814–826. [DOI] [PubMed] [Google Scholar]
- Spitzer‐Rimon B, Duchin S, Bernstein N, Kamenetsky R. 2019. Architecture and florogenesis in female Cannabis sativa plants. Frontiers in Plant Science 10: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasababu N. 2014. Assessing the mechanical performance Cannabis sativa composites – reinforced with long time dried fibre. Procedia Engineering 97: 986–993. [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology 57: 758–771. [DOI] [PubMed] [Google Scholar]
- Stout JM, Boubakir Z, Ambrose SJ, Purves RW, Page JE. 2012. The hexanoyl‐CoA precursor for cannabinoid biosynthesis is formed by an acyl‐activating enzyme in Cannabis sativa trichomes. The Plant Journal 71: 353–365. [DOI] [PubMed] [Google Scholar]
- Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S et al. 2013. QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The Plant Journal 74: 174–183. [DOI] [PubMed] [Google Scholar]
- Tamiru M, Natsume S, Takagi H, White B, Yaegashi H, Shimizu M, Yoshida K, Uemura A, Oikawa K, Abe A et al. 2017. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biology 15: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Zhao X, Mace E, Henry R, Jordan D. 2019. Exploring and exploiting pan‐genomics for crop improvement. Molecular Plant 12: 156–169. [DOI] [PubMed] [Google Scholar]
- Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y, Morimoto S. 2007. Cannabidiolic‐acid synthase, the chemotype‐determining enzyme in the fiber‐type Cannabis sativa . FEBS Letters 581: 2929–2934. [DOI] [PubMed] [Google Scholar]
- Thomas J, Kim HR, Rahmatallah Y, Wiggins G, Yang Q, Singh R, Glazko G, Mukherjee A. 2019. RNA‐seq reveals differentially expressed genes in rice (Oryza sativa) roots during interactions with plant‐growth promoting bacteria, Azospirillum brasilense . PLoS ONE 14: e0217309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bakel H, Stout J, Cote A, Tallon C, Sharpe A, Hughes T, Page J. 2011. The draft genome and transcriptome of Cannabis sativa . Genome Biology 12: R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBuren R, Wai CM, Ou S, Pardo J, Bryant D, Jiang N, Mockler TC, Edger P, Michael TP. 2018. Extreme haplotype variation in the desiccation‐tolerant clubmoss Selaginella lepidophylla . Nature Communications 9: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Terauchi R, McCouch SR. 2014. Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biology 12: e1001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara D, Baker H, Clancy K, Keepers KG, Mendieta JP, Pauli CS, Tittes SB, White KH, Kane NC. 2016. Genetic and genomic tools for Cannabis sativa . Critical Reviews in Plant Sciences 35: 364–377. [Google Scholar]
- Vergara D, Huscher EL, Keepers KG, Givens RM, Cizek CG, Torres A, Gaudino R, Kane NCJB. 2019. Gene copy number is associated with phytochemistry in Cannabis sativa . AoB Plants 11: plz074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F. 2016. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nature Genetics 48: 1233–1241. [DOI] [PubMed] [Google Scholar]
- Weiblen GD, Wenger JP, Craft KJ, ElSohly MA, Mehmedic Z, Treiber EL, Marks MD. 2015. Gene duplication and divergence affecting drug content in Cannabis sativa . New Phytologist 208: 1241–1250. [DOI] [PubMed] [Google Scholar]
- Weisshaus O. 2020. Cannabis gene variation – comparison of multiple genome assemblies . Webinar on Cannabis Genome. [WWW document] URL https://www.omicsonline.org/open‐access/cannabis‐gene‐variation‐‐comparison‐of‐multiple‐genome‐assemblies.pdf [accessed 25 August 2020].
- Welling MT, Liu L, Raymond CA, Kretzschmar T, Ansari O, King GJ. 2019. Complex patterns of cannabinoid alkyl side‐chain inheritance in cannabis. Scientific Reports 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling MT, Shapter T, Rose TJ, Liu L, Stanger R, King GJ. 2016. A belated green revolution for cannabis: virtual genetic resources to fast‐track cultivar development. Frontiers in Plant Science 7: 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Wang G, Yuan M, Yao W, Lyu K, Zhao H, Yang M, Li P, Zhang X, Yuan J. 2015. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proceedings of the National Academy of Sciences, USA 112: E5411–E5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Xu C, Xu S. 2017. Prediction and association mapping of agronomic traits in maize using multiple omic data. Heredity 119: 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Bayer PE, Batley J, Edwards D. 2017. Improvements in genomic technologies: application to crop genomics. Trends in Biotechnology 35: 547–558. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Lee H, Hu H, Scheben A, Edwards D. 2018. Single‐cell genomic analysis in plants. Genes 9: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager JJ, Lange I, Srividya N, Smith A, Lange BM. 2019. Gene networks underlying cannabinoid and terpenoid accumulation in cannabis. Plant Physiology 180: 1877–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang K, Wu J, Guo N, Liang J, Wang X, Cheng F. 2020. QTL‐Seq and sequence assembly rapidly mapped the gene BrMYBL2.1 for the purple trait in Brassica rapa . Scientific Reports 10: 2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Summary of best Blast hits of the functional cannabinoid biosynthesis genes against the latest C. sativa reference genome assemblies.
Table S2 Summary of best Blast hits of other THCAS‐ and CBDAS‐like gene copies against the latest C. sativa reference genome assemblies.
Table S3 Alignment of Purple Kush v.5.0 (GenBank acc. no. GCA_000230575.5) and Finola v.2.0 (GenBank acc. no. GCA_003417725.2) unplaced scaffolds against the cs10 v.2.0 genome assembly (GenBank acc. no. GCA_900626175.2).
Table S4 Cannabinoid synthase genes from cs10 v.2.0 (GenBank acc. no. GCA_900626175.2) and Jamaican Lion (female parent: GenBank acc. no. GCA_012923435.1; male parent: GenBank acc. no. GCA_013030025.1) assemblies.
Table S5 Summary of best Blast hits of nonfunctional CBDAS homologs from Skunk #1 against the latest C. sativa reference genome assemblies.
Table S6 Transcript per million (TPM) counts of the 13 cannabinoid synthase genes from the cs10 v.1.0 genome assembly (GenBank acc. no. GCA_900626175.1) in nine medicinal cannabis cultivars from Zager et al. (2019).
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.