

Abstract
The increasing prevalence of metallo‐β‐lactamase (MBL)‐expressing bacteria presents a worrying trend in antibiotic resistance. MBLs rely on active site zinc ions for their hydrolytic activity and the pursuit of MBL‐inhibitors has therefore involved the investigation of zinc chelators. To ensure that such chelators specifically target MBLs, a series of cephalosporin prodrugs of two potent zinc‐binders: dipicolinic acid (DPA) and 8‐thioquinoline (8‐TQ) was prepared. Although both DPA and 8‐TQ bind free zinc very tightly (K d values in the low nm range), the corresponding cephalosporin conjugates do not. The cephalosporin conjugates are efficiently hydrolyzed by MBLs to release DPA or 8‐TQ, as confirmed by using both NMR and LC‐MS studies. Notably, the cephalosporin prodrugs of DPA and 8‐TQ show potent inhibitory activity against NDM, VIM, and IMP classes of MBLs and display potent synergy with meropenem against MBL‐expressing clinical isolates of K. pneumoniae and E. coli.
Keywords: antibiotic resistance, enzyme inhibition, metallo-β-lactamase, synergy, zinc chelators
Stopping bacterial resistance: Metallo‐β‐lactamases (MBLs) are bacterial resistance enzymes that degrade β‐lactam antibiotics, including last line of defense carbapenems like meropenem. A new series of cephalosporin prodrug inhibitors that exploit the hydrolytic action of the target MBL itself to activate the inhibitor is reported. The prodrugs display potent MBL inhibition and effectively synergize with meropenem against highly resistant MBL‐expressing isolates.
Introduction
Antimicrobial resistance presents a growing challenge to global health in the treatment of bacterial infections and carries with it a significant economic burden.[ 1 , 2 , 3 ] Among the numerous antibiotic resistance mechanisms known to be of clinical relevance, the activity of β‐lactamase enzymes is of increasing concern. These enzymes can hydrolyze the β‐lactam ring of penicillins, cephalosporins, and carbapenems, rendering them inactive. The family of β‐lactamase enzymes can be divided into two main classes based on their mechanism of action: serine β‐lactamases (SBLs), which utilize an active site serine for the hydrolysis of the β‐lactam ring, and metallo‐β‐lactamases (MBLs), which rely on active site zinc ions to activate a water molecule for the hydrolysis.[ 4 , 5 ] A variety of clinically approved SBL inhibitors are available, which, when combined with a conventional β‐lactam antibiotic, can be used to effectively treat infections owing to SBL‐producing strains. [6] In contrast, there are currently no MBL inhibitors approved for use in humans, making it very difficult for physicians to treat patients infected with MBL‐producing pathogens.[ 7 , 8 , 9 ]
The common feature shared by all MBLs is the presence of essential zinc ions in their active site, which play a key role in the hydrolytic mechanism. The chelation or stripping of zinc from the active site has been demonstrated to be an effective approach for deactivating MBLs. Ethylene‐diaminetetraacetic acid (EDTA), dipicolinic acid (DPA), aspergillomarasmine A (AMA), and a variety of aminocarboxylic acids effectively bind zinc and have been shown to inhibit MBLs, including the clinically relevant New Delhi metallo‐β‐lactamase (NDM‐1).[ 10 , 11 , 12 ] Although such compounds are effective at inhibiting MBLs under optimized in vitro assay conditions, their potent metal binding properties would be expected to result in severe off‐target effects in more complex biological settings. Metalloproteins are estimated to comprise about a third of the human proteome and, more specifically, zinc‐dependent enzymes account for almost 10 % of the human proteome.[ 13 , 14 ] For this reason, the direct use of strong zinc binding MBL inhibitors is likely to be of limited therapeutic application. In addition to the chelators described above, a number of thiol‐based compounds have also been investigated as MBL inhibitors, including captopril, bisthiazolidines, and various thiol carboxylates.[ 15 , 16 , 17 ] Recent reports describing 8‐thioquinoline (8‐TQ) as a potent zinc binding motif also prompted us to investigate its capacity to inhibit MBLs.[ 18 , 19 , 20 ] Although we were able to show that 8‐TQ is a potent zinc binder with the capacity to inhibit MBLs, we also found that it has the tendency to rapidly oxidize to the corresponding disulfide, eliminating its zinc binding and MBL inhibiting properties. This problem is common to many thiol‐based MBL inhibitors and thus limits their potential for therapeutic use. [17]
To address the aforementioned shortcomings of zinc binding amino‐carboxylates and small molecule thiols as MBL inhibitors, we here describe a cephalosporin prodrug strategy that allows for the active inhibitor to be released only in the presence of the target MBL. Specifically, this prodrug strategy is based on the mechanism of the β‐lactamase‐mediated hydrolysis of cephalosporins (Figure 1 A). It is well established that for cephalosporins containing a suitable leaving group at the 3‐position, hydrolysis of the β‐lactam ring leads to the subsequent release of the leaving group. This unique reactivity has been previously exploited in the design of cephalosporin‐based probes for β‐lactamase activity [21] as well as in the development of other prodrugs capable of delivering toxic payloads to cells expressing β‐lactamases.[ 22 , 23 ] In our design, the incorporation of the strong zinc binding compound is achieved by covalently linking either the carboxylate or thiol moiety involved in zinc binding to the cephalosporin 3‐position. In this way, zinc binding is temporarily shut off and, in the case of thiols, disulfide formation is also blocked. Only upon hydrolysis of the β‐lactam ring are the strong zinc binders released from the 3‐position in their active form. To investigate this prodrug strategy as an approach to developing MBL inhibitors, DPA and 8‐TQ were selected as representative zinc binders and linked to a 7‐phenacyl‐cephalosporin core (Figure 1 B). The zinc binding properties of both the free chelators and the corresponding cephalosporin prodrugs were assessed by using isothermal titration calorimetry (ITC). In addition, the mechanism of MBL‐mediated prodrug cleavage was confirmed by using both NMR and LC‐MS techniques. Furthermore, the inhibitory activity of these prodrugs was evaluated against different members of the MBL family and synergy assays used to measure their capacity to resensitize MBL‐expressing clinical isolates to the carbapenem antibiotic meropenem.
Figure 1.
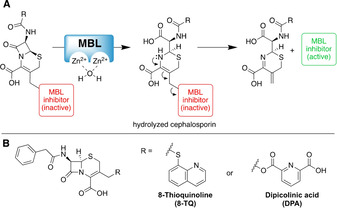
(A) Proposed mechanism of MBL‐mediated hydrolysis of the β‐lactam ring in cephalosporin prodrugs with subsequent release of the zinc binding MBL inhibitor. (B) Structures of the cephalosporin prodrug conjugates of 8‐thioquinoline (8‐TQ) and dipicolinic acid (DPA).
Results and Discussion
The cephalosporin prodrug conjugates of 8‐TQ and DPA were prepared by linking both compounds to the 7‐phenacyl‐cephalosporin scaffold as indicated in Scheme 1. For the preparation of the 8‐TQ conjugates, 8‐TQ was freshly reduced with tris(2‐carboxyethyl)phosphine (TCEP) by using either resin‐bound TCEP or a solution of TCEP in 0.1 m HCl followed by extraction in chloroform. Next, commercially available 4‐methoxybenzyl 3‐(chloromethyl)‐8‐oxo‐7‐[(phenylacetyl)amino]‐5‐thia‐1‐azabicyclo[4.2.0]oct‐2‐ene‐2‐carboxylate (GCLE) was converted into the corresponding iodide in situ by treatment with sodium iodide followed by addition of freshly reduced 8‐TQ⋅HCl in the presence of sodium bicarbonate. Deprotection of the p‐methoxybenzyl (PMB) protecting group by using a mixture of TFA and anisole (5:1) followed by HPLC purification provided compound 6 in excellent yield. For the synthesis of the corresponding DPA conjugate 13, one of the carboxylate moieties of DPA was first PMB‐protected by treatment of DPA with the PMB‐isourea (generated from diisopropylcarbodiimide and p‐methoxybenzyl alcohol) and 4‐dimethylaminopyridine (DMAP) in DMF, affording acid 9. GCLE was again converted to its corresponding iodide by using sodium iodide in situ, followed by coupling of the mono‐PMB‐protected DPA 9 to yield protected conjugate 10 in good yield. Attempted deprotection of the ester linked conjugate with the same conditions used in the preparation of compound 6, however, led to the cleavage of the ester bond between DPA and the cephalosporin core. Gratifyingly, milder conditions (TFA and anisole (2.5:1) in CH2Cl2 at 0 °C) allowed for PMB group removal without cleavage of the ester. Subsequent purification by preparative HPLC yielded compound 13 in good yield. Notably, in the synthesis of both 6 and 13 no indication of Δ2–Δ3 double bond isomerization, which is known to occur in cephalosporins under basic coupling conditions, was observed. To further expand upon conjugates 6 and 13, the corresponding sulfoxides and sulfones were also synthesized to investigate the impact of the oxidation state of the cephalosporin sulfur atom on the activity and stability of the conjugates. Oxidation was achieved by using 1 or 2.5 equivalents of meta‐chloroperoxybenzoic acid (mCPBA) to provide the sulfoxide or sulfone, respectively. For 8‐TQ conjugates 7 and 8, mCPBA oxidation was first performed on GCLE to avoid oxidation of the thioether linkage present in the 8‐TQ conjugates. In generating the DPA conjugates 14 and 15, oxidation was performed on the PMB‐protected intermediate 10.
Scheme 1.
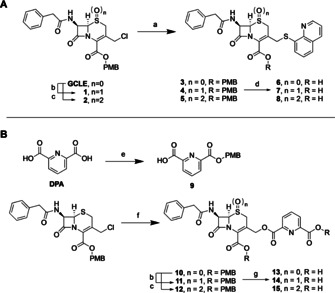
Synthesis of (A) 8‐thioquinoline conjugates 6–8 and (B) dipicolinic acid conjugates 13–15. Reagents and conditions: a) 1) NaI, DMF, rt, 1 h, 2) 8‐thioquinoline hydrochloride, NaHCO3, DMF, rt, 4 h; b) mCPBA (1 equiv), CH2Cl2, 0 °C, 3 h; c) m‐CPBA (2.5 equiv), CH2Cl2, 0 °C to rt, overnight; d) TFA/anisole (5:1), 0 °C to rt, 1 h; e) O‐PMB‐N,N′‐diisopropylisourea, DMAP, DMF, CH2Cl2, rt, overnight; f) 9, NaHCO3, DMF, rt, overnight, g) TFA/anisole (2.5:1), CH2Cl2, 0 °C, 2 h.
To quantify the zinc binding properties of DPA and 8‐TQ, and evaluate the masking efficiency of the prodrug approach, dissociation constants (K d values) for Zn2+ were determined by using ITC. Titration of zinc sulfate into DPA revealed a K d value of 389±45 nm with a 2:1 stoichiometry of DPA to Zn2+. This K d value is somewhat different (ca. 6‐fold lower) than that previously reported by our group, [24] a difference likely attributable to the optimized lower concentration of TRIS buffer used in the current study (20 mm versus 100 mm). Prior to its use in the ITC experiment, 8‐TQ was freshly reduced by using resin‐bound TCEP followed by titration with zinc sulfate. This revealed a very strong zinc‐binding interaction with a low nanomolar K d value close to the ITC's limit of detection. To fully quantify the 8‐TQ/Zn2+ interaction, a competition experiment was established wherein freshly reduced 8‐TQ was titrated into a premixed solution of DPA and zinc sulfate. The results of this analysis confirmed 8‐TQ to be a highly potent zinc chelator with a K d value of 1.87±0.15 nm and a 2:1 stoichiometry of 8‐TQ to Zn2+. Next, the zinc binding capacities of the cephalosporin prodrugs of DPA and 8‐TQ were evaluated to assess how effectively zinc binding is masked. All of the 8‐TQ conjugates were found to be comparably stable and among the DPA conjugates sulfoxide analog 14 demonstrated the best stability in the buffer conditions used for the zinc binding assay and was therefore used in the ITC experiment. No appreciable zinc binding was observed for compounds 6 and 14, confirming that the zinc binding properties of 8‐TQ and DPA are completely blocked upon conjugation to the cephalosporin (thermograms provided in Figure S4 in the Supporting Information).
We next set out to confirm the mechanism of release of 8‐TQ and DPA from the corresponding cephalosporin conjugates. To investigate the MBL‐mediated release of 8‐TQ and DPA by hydrolysis of the β‐lactam ring, we devised an in situ NMR assay wherein compounds 6 and 13 were incubated with NDM‐1 (Figure 2). Briefly, NDM‐1 was added at a final concentration of 374 nm to an NMR tube containing cephalosporin conjugate 6 or 13 in 20 mm HEPES with 500 μm zinc sulfate in D2O. The 1H NMR data presented in Figure 2 clearly show the appearance of the vinylic cephalosporin byproduct formed upon elimination of 8‐TQ (compound 6) and DPA (compound 13). As can also be seen, the hydrolysis of DPA conjugate 13 proceeds more rapidly than hydrolysis of 8‐TQ conjugate 6. Notably, no indication of hydrolysis of either 6 or 13 was found in control experiments where no enzyme was present. These results support the proposed mechanism of action wherein the zinc binding 8‐TQ or DPA are released in an MBL‐dependent manner. It is also worth noting that to achieve complete hydrolysis of the cephalosporin conjugates a relatively high 374 nm concentration of NDM‐1 was required. When working with lower concentrations of enzyme, the hydrolysis of 6 and 13 would proceed only partially before stalling, presumably owing to inhibition of the enzyme by the newly released 8‐TQ or DPA (data not shown). The formation of the vinylic byproduct and release of 8‐TQ and DPA was further confirmed by using LC‐MS by incubation of compounds 6 and 13 with NDM‐1 (see Figures S6 and S7 in the Supporting Information).
Figure 2.
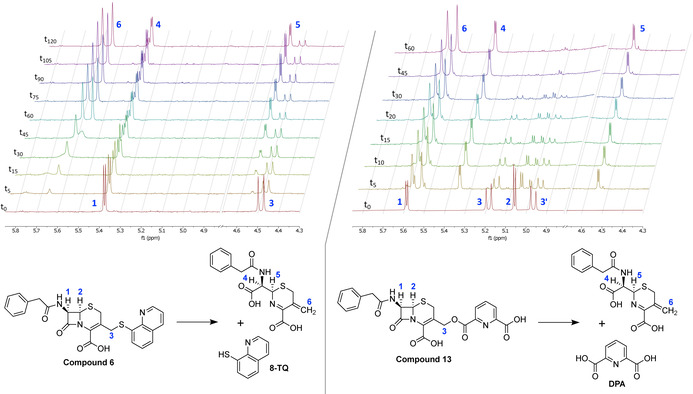
1H NMR spectra showing the MBL‐mediated hydrolysis of compounds 6 (left panel) and 13 (right panel) over time. For both compounds, the hydrolysis by NDM‐1 leads to the clean formation of the vinylic product and release of the zinc binding compound. For clarity, the region of the spectra including the suppressed water signal has been removed. In the case of compound 6, the signals for the proton at position 2 and one of the protons at position 3 overlap with the suppressed water signal and are therefore not visible (full NMR spectra provided in the Supporting Information).
The MBL inhibitory activity of 8‐TQ and DPA and their corresponding cephalosporin prodrugs was next assessed against a panel of purified MBLs, including members of the clinically most relevant NDM, VIM, and IMP families (Table 1). Enzyme inhibition assays were performed as previously described by using a fluorescent cephalosporin substrate for assessing MBL activity, added following a 10 min pre‐incubation of the enzyme with the inhibitor. [21] Effective inhibition was observed for 8‐TQ as well as its conjugates 6–8 against all MBLs tested with IC50 values in the low micromolar range. Interestingly, the conjugates were generally more potent inhibitors of NDM‐1 and VIM‐2 than against the IMP enzymes tested. By comparison, 8‐TQ showed similar activity against all MBLs evaluated suggesting that cephalosporin conjugates 6–8 may be better substrates for NDM‐1 and VIM‐2 than for IMP‐type MBLs. Similar inhibitory potency was measured for DPA and prodrug conjugates 13–15; however, in this case, the oxidation state of the cephalosporin sulfur atom had a more pronounced effect on activity. Although DPA and conjugate 13 showed similar inhibition against all enzymes tested with IC50 values in the low micromolar range, sulfoxide 14 was nearly 10‐fold more active with an IC50 value of 0.299 μm against NDM‐1 and 0.756 μm against VIM‐2. Interestingly, the increased potency of sulfoxide 14 did not extend to the IMP‐type enzymes tested. By comparison, sulfone analog 15 was found to be significantly less active with an IC50 of 18.78 μm for NDM‐1 and 10.83 μm for VIM‐2. Against IMP‐1 and IMP‐28, however, both sulfoxide 14 and sulfone 15 showed a similarly reduced activity relative to DPA or conjugate 13.
Table 1.
Inhibitory activity of 8‐TQ, DPA, and conjugates 6–8 and 13–15 against selected MBLs.
|
|
NDM‐1 IC50 [μm][a] |
VIM‐2 IC50 [μm] |
IMP‐1 IC50 [μm] |
IMP‐28 IC50 [μm] |
|---|---|---|---|---|
|
8‐TQ |
2.99±0.58 |
4.20±1.00 |
4.42±0.14 |
4.20±0.11 |
|
6 |
2.31±0.25 |
3.64±0.27 |
12.21±0.25 |
10.32±0.22 |
|
7 |
2.18±0.14 |
6.35±0.56 |
33.57±1.72 |
71.87±3.05 |
|
8 |
8.47±1.02 |
4.17±0.65 |
28.92±1.29 |
46.24±3.13 |
|
DPA |
3.13±0.12 |
4.34±0.67 |
15.16±0.17 |
9.65±0.62 |
|
13 |
2.13±0.07 |
1.87±0.10 |
5.22±0.25 |
7.39±0.10 |
|
14 |
0.299±0.005 |
0.756±0.056 |
15.09±1.09 |
46.70±2.76 |
|
15 |
18.78±1.22 |
10.83±0.60 |
18.71±2.43 |
61.87±1.82 |
[a] Half‐maximal inhibitory concentration of the compounds tested against NDM‐1, VIM‐2, IMP‐1, and IMP‐28. Inhibitors pre‐incubated with the MBL for 10 min followed by addition of the FC5 substrate (full assay details provided in the Supporting Information). Values reported based on triplicate experiments.
As the activation of these prodrugs is governed by hydrolysis of the cephalosporin β‐lactam ring, the time‐dependent activity of inhibitors 6 and 14 was next evaluated. The compounds were incubated with NDM‐1 for 0, 10, 20, 30, 45, or 60 min prior to the addition of the fluorescent enzyme substrate. For compound 6, no significant change in IC50 was observed with longer incubation times. However, for compound 14, a clear decrease in IC50 from 560 nm to 120 nm was observed with longer incubation times (see Figure S2 in the Supporting Information). The observation that a decrease in IC50 is only observed for DPA and not for 8‐TQ might be explained by the parallel formation of the inactive disulfide balancing out any decrease in IC50. To further investigate whether these inhibitors work via stripping zinc from the MBL active site or through formation of a ternary complex with zinc still bound in the MBL active site, the zinc dependence of the inhibition observed for 8‐TQ, DPA, and conjugates 6 and 14 was next evaluated. The compounds were tested for their inhibitory activity against NDM‐1 in the presence of 0.1, 1.0, and 10 μm zinc sulfate. Interestingly, although the activity of 8‐TQ and DPA showed a strong dependence on the concentration of zinc in the media, this effect was not observed for conjugates 6 and 14 (see Figure S3 in the Supporting Information). An explanation for this observation may be that although 8‐TQ and DPA function as simple chelators in solution, in prodrug form they are only released in the active site of an MBL where they may then be capable of functioning in a more localized manner. This localized release of 8‐TQ or DPA may allow for formation of a ternary complex with the zinc bound in the MBL active site, in a manner independent of the concentration of free zinc in solution.
The ability of the 8‐TQ and DPA cephalosporin prodrugs to resensitize MBL‐expressing bacteria to a clinically used β‐lactam antibiotic was next investigated. The stability of the compounds in the assay buffers was first evaluated to ensure that any observed activity could be fairly attributed to MBL‐mediated hydrolysis of the conjugates rather than degradation or auto‐hydrolysis under the assay conditions used. In the HEPES buffer used for the NMR hydrolysis experiments and the enzyme inhibition assays, the conjugates showed good stability over the time course of the experiment (no significant hydrolysis after 6 h at room temperature). However, for bacterial growth assays, standard conditions involve the use of Mueller–Hinton Broth (MHB) at 37 °C. The stability of the 8‐TQ and DPA conjugates under these conditions revealed that the DPA conjugates 13–15 showed appreciable degradation over time with stability depending on the oxidation state of the cephalosporin sulfur. Specifically, DPA conjugate 13 (sulfide) showed 85 % degradation after 2 h, whereas sulfone 15 showed a half‐life of about 5 h and sulfoxide 14 showed a half‐life of about 14 h. By comparison, 8‐TQ conjugates were found to be much more stable. Compounds 6 and 7 showed very little sign of degradation with >90 % remaining intact after 24 h. Sulfone 8 showed some degradation over time, with an estimated half‐life of about 24 h (see Figure S5 in the Supporting Information). These stability data indicate that the ester linkage in the DPA‐cephalosporin conjugates is not sufficiently stable under the conditions used for the bacterial growth inhibition experiments. Therefore, for the purpose of the subsequent antibacterial synergy assays, we focused on the 8‐TQ conjugates 6–8. The compounds were first tested for inherent activity against a series of MBL‐expressing clinical isolates of E. coli, K. pneumoniae, and P. aeruginosa (see Table S3 in the Supporting Information). Standard minimum inhibitory concentration (MIC) assays revealed that none of the compounds were active on their own (MIC values >256 μg mL−1). Next, the capacity for 8‐TQ and prodrugs 6–8 to synergize with meropenem against MBL‐expressing strains was evaluated. In an initial screen, two different NDM‐1 expressing K. pneumoniae strains were used to assess the synergy of the compounds at 25 % of the maximum concentration tested in the previous MIC assays (64 μg mL−1) to exclude additive effects. The results of the synergy studies conducted with 8‐TQ itself were clouded by formation of the insoluble 8‐TQ disulfide (the disulfide species precipitates and impairs the visual readout of the bacterial growth assay). In the case of prodrugs 6–8, however, a clear and significant level of synergy with meropenem was observed free of disulfide precipitation. These findings suggest that the prodrug strategy enables effective delivery of otherwise reactive thiols as MBL inhibitors. In a preliminary screen with two highly resistant NDM‐1 expressing isolates, compounds 6–8 all showed effective synergy. Notably, the most stable 8‐TQ‐cephalosporin conjugate 6 was also found to be the most effective synergist, lowering the MIC of meropenem from 16–32 μg mL−1 (resistant) to 1–4 μg mL−1 (sensitive) against the two strains tested. Compound 6 was therefore selected for further evaluation against an expanded panel of MBL‐producing clinical isolates of E. coli, K. pneumoniae, and P. aeruginosa (Table 2 and Figures S8–S9 in the Supporting Information). Potent synergy with meropenem was consistently observed against all K. pneumoniae and E. coli strains tested expressing the main MBL classes (NDM, VIM, IMP) with measured enhancements ranging from 8‐ to 128‐fold. It is it notable that compound 6 exhibited the most effective synergy when applied at 64 μg mL−1 (corresponding to 130 μm), a concentration approximately 60 times higher than the IC50 value measured for NDM‐1 in the inhibition assays with purified enzyme (Table 1). The higher concentration of compound 6 required to elicit a notable effect in the bacterial assays might be explained by the need for the compound to pass the outer membrane to engage the target MBL. From the synergy data obtained, the fractional inhibitory concentration (FIC) values were also determined with values below 0.5, indicating a synergistic effect between meropenem and the MBL inhibitor. Notably, significantly less synergy was found against the P. aeruginosa strains tested, an observation in keeping with the known low‐permeability of the P. aeruginosa outer membrane to antibiotics and other antiseptic agents. [25]
Table 2.
Bacterial growth inhibition by meropenem in the presence of cephalosporin prodrug 6 against clinical isolates of MBL‐producing Gram‐negative bacteria.
|
Strain |
MBL |
MIC [μg mL−1][a] |
FIC[b] |
|
|---|---|---|---|---|
|
|
|
meropenem |
+6 |
|
|
K. pneumoniae (strain JS022) |
NDM‐1 |
16 |
1 (16) |
≤0.188 |
|
K. pneumoniae (strain N11‐2218) |
NDM‐1 |
32 |
4 (8) |
≤0.250 |
|
K. pneumoniae (strain RC0048) |
VIM‐1 |
64 |
4 (16) |
≤0.188 |
|
K. pneumoniae (strain RC0021) |
VIM‐1 |
16 |
0.5 (32) |
≤0.156 |
|
K. pneumoniae (strain NRZ‐293) |
VIM‐2 |
4 |
0.13 (32) |
≤0.156 |
|
K. pneumoniae (strain JS265) |
IMP‐28 |
4 |
0.13 (32) |
≤0.156 |
|
E. coli (strain RC0089) |
NDM‐1 |
32 |
1 (32) |
≤0.156 |
|
E. coli (strain 2018–2015) |
NDM‐1 |
8 |
0.5 (16) |
≤0.188 |
|
E. coli (strain 1322) |
VIM‐2 |
4 |
0.25 (16) |
≤0.188 |
|
E. coli (strain 2018–014) |
IMP‐4 |
8 |
0.063 (128) |
≤0.133 |
|
P. aeruginosa (strain NRZ‐8418) |
NDM‐1 |
64 |
32 (2) |
NS[c] |
|
P. aeruginosa (strain 581) |
VIM‐2 |
16 |
8 (2) |
NS[c] |
|
P. aeruginosa (strain 2018–009) |
IMP‐13 |
32 |
32 (1) |
NS[c] |
[a] Minimum inhibitory concentration (MIC) values given in μg mL−1 with the fold improvement in brackets. Compound 6 administered at 64 μg mL−1. Values are the mean of at least three experiments. [b] FIC=(MIC of meropenem in combination/MIC of meropenem alone)+(MIC of inhibitor in combination/MIC of inhibitor alone). FIC values below 0.5 indicate a synergistic effect. [c] NS=no synergy.
Conclusion
The emergence of MBL‐driven antibiotic resistance and a lack of clinically approved MBL inhibitors pose a serious threat to global health. We here describe a new series of cephalosporin prodrugs designed to deliver zinc binding MBL inhibitors in a targeted manner. Recent reports have demonstrated that such strategies can be effective for delivering antibacterial metal chelators such as pyrithione.[ 23 , 26 ] In the present study, we focused on new cephalosporin prodrugs of 8‐thioquinoline (8‐TQ) and dipicolinic acid (DPA). This approach allows for the controlled activation of the zinc‐binding properties of the inhibitor only in the presence of the target MBL and, in the case of 8‐TQ, also protects the active species from oxidative deactivation prior to target engagement. The 8‐TQ and DPA prodrugs here studied effectively inhibit purified NDM‐, VIM‐, and IMP‐type enzymes by an activation mechanism shown to be dependent on MBL‐mediated β‐lactam ring hydrolysis. Notably, although elevated concentrations of free zinc were found to diminish the inhibitory potency of 8‐TQ and DPA alone, the same effect was not observed for the corresponding cephalosporin conjugates. These findings point to a proximity effect for the prodrugs, wherein the zinc‐binding MBL inhibitor is released within the MBL active site itself, serving to enhance its selectivity. In bacterial growth assays, the 8‐TQ conjugates demonstrated potent synergy with meropenem against a number of clinical isolates of E. coli and K. pneumoniae expressing NDM‐, VIM‐, and IMP‐ type MBLs. Taken together, these results indicate that such prodrug strategies can effectively deliver zinc‐binding MBL inhibitors to resistant bacteria in a selective manner. In the present study, the conjugates prepared contained the phenacyl substituent at the cephalosporin C‐7 position. Among the 3rd and 4th generation cephalosporins, a range of optimized substituents are found at this position imparting increased activity and stability. Ongoing efforts are therefore aimed at similarly enhancing the activity of the MBL inhibitor‐cephalosporin prodrugs here described.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the European Research Council (ERC consolidator grant to NIM, grant agreement no. 725523).
M. J. van Haren, K. H. M. E. Tehrani, I. Kotsogianni, N. Wade, N. C. Brüchle, V. Mashayekhi, N. I. Martin, Chem. Eur. J. 2021, 27, 3806.
References
- 1. United States Centers for Disease Control and Prevention: Report on Antibiotic/Antimicrobial Resistance (https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf), 2019.
- 2. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, from http://www.jpiamr.eu/wp-content/uploads/2014/12/AMR-Review-Paper-Tackling-a-crisis-for-the-health-and-wealth-of-nations 1-2.pdf, J. O'Neill, 2014.
- 3. Årdal C., Balasegaram M., Laxminarayan R., McAdams D., Outterson K., Rex J. H., Sumpradit N., Nat. Rev. Microbiol. 2020, 18, 267–274. [DOI] [PubMed] [Google Scholar]
- 4. Crowder M. W., Aitha M., Bonomo R. A., Lisa M.-N., Moreno D. M., Vila A. J., Llarrull L. I., Palacios A. R., Tierney D. L., González M. M., Spencer J., Nat. Commun. 2017, 8, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun Z., Hu L., Sankaran B., Prasad B. V. V., Palzkill T., Nat. Commun. 2018, 9, 4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tehrani K. H. M. E., Martin N. I., MedChemComm 2018, 9, 1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ju L. C., Cheng Z., Fast W., Bonomo R. A., Crowder M. W., Trends Pharmacol. Sci. 2018, 39, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rotondo C. M., Wright G. D., Curr. Opin. Microbiol. 2017, 39, 96–105. [DOI] [PubMed] [Google Scholar]
- 9. Somboro A. M., Osei Sekyere J., Amoako D. G., Essack S. Y., Bester L. A., Appl. Environ. Microbiol. 2018, 84, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tehrani K. H. M. E., Fu H., Brüchle N. C., Mashayekhi V., Prats Luján A., van Haren M. J., Poelarends G. J., Martin N. I., Chem. Commun. 2020, 56, 3047–3049. [DOI] [PubMed] [Google Scholar]
- 11. King A. M., Reid-Yu S. A., Wang W., King D. T., De Pascale G., Strynadka N. C., Walsh T. R., Coombes B. K., Wright G. D., Nature 2014, 510, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen A. Y., Thomas P. W., Stewart A. C., Bergstrom A., Cheng Z., Miller C., Bethel C. R., Marshall S. H., Credille C. V., Riley C. L., Page R. C., Bonomo R. A., Crowder M. W., Tierney D. L., Fast W., Cohen S. M., J. Med. Chem. 2017, 60, 7267–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lothian A., Hare D. J., Grimm R., Ryan T. M., Masters C. L., Roberts B. R., Front. Aging Neurosci. 2013, 5, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andreini C., Banci L., Bertini I., Rosato A., J. Proteome Res. 2006, 5, 3173–3178. [DOI] [PubMed] [Google Scholar]
- 15. Büttner D., Kramer J. S., Klingler F. M., Wittmann S. K., Hartmann M. R., Kurz C. G., Kohnhäuser D., Weizel L., Brüggerhoff A., Frank D., Steinhilber D., Wichelhaus T. A., Pogoryelov D., Proschak E., ACS Infect. Dis. 2018, 4, 360–372. [DOI] [PubMed] [Google Scholar]
- 16. González M. M., Kosmopoulou M., Mojica M. F., Castillo V., Hinchliffe P., Pettinati I., Brem J., Schofield C. J., Mahler G., Bonomo R. A., Llarrull L. I., Spencer J., Vila A. J., ACS Infect. Dis. 2015, 1, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tehrani K. H. M. E., Martin N. I., ACS Infect. Dis. 2017, 3, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez C., Li J., Parlati F., Rouffet M., Yuyong M., Mackinnon A. L., Chou T. F., Deshaies R. J., Cohen S. M., J. Med. Chem. 2017, 60, 1343–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Yakushi T., Parlati F., Mackinnon A. L., Perez C., Ma Y., Carter K. P., Colayco S., Magnuson G., Brown B., Nguyen K., Vasile S., Suyama E., Smith L. H., Sergienko E., Pinkerton A. B., Chung T. D. Y., Palmer A. E., Pass I., Hess S., Cohen S. M., Deshaies R. J., Nat. Chem. Biol. 2017, 13, 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hameed D. S., Sapmaz A., Burggraaff L., Amore A., Slingerland C. J., van Westen G. J. P., Ovaa H., Angew. Chem. Int. Ed. 2019, 58, 14477–14482; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14619–14624. [Google Scholar]
- 21. Cain R., Schofield C. J., Brem J., Fishwick C. W. G., van Berkel S. S., Owens R. J., Spencer J., Rydzik A. M., Salimraj R., Verma A., J. Med. Chem. 2013, 56, 6945–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Evans L. E., Krishna A., Ma Y., Webb T. E., Marshall D. C., Tooke C. L., Spencer J., Clarke T. B., Armstrong A., Edwards A., J. Med. Chem. 2019, 62, 4411–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jackson A. C., Zaengle-Barone J. M., Puccio E. A., Franz K. J., ACS Infect. Dis. 2020, 6, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tehrani K. H. M. E., Brüchle N. C., Wade N., Mashayekhi V., Pesce D., van Haren M. J., Martin N. I., ACS Infect. Dis. 2020, 6, 1366–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chevalier S., Bouffartigues E., Bodilis J., Maillot O., Lesouhaitier O., Feuilloley M. G. J., Orange N., Dufour A., Cornelis P., FEMS Microbiol. Rev. 2017, 41, 698–722. [DOI] [PubMed] [Google Scholar]
- 26. Zaengle-Barone J. M., Jackson A. C., Besse D. M., Becken B., Arshad M., Seed P. C., Franz K. J., ACS Infect. Dis. 2018, 4, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary