

Abstract
Objective:
Traumatic brain injury is a major risk factor for acquired epilepsies and understanding the mechanisms underlying the early pathophysiology could yield viable therapeutic targets. Growing evidence indicates a role for inflammatory signaling in modifying neuronal excitability and promoting epileptogenesis. Here we examined the effect of innate immune receptor, toll-like receptor 4 (TLR4) on excitability of the hippocampal dentate gyrus and epileptogenesis after brain injury.
Methods:
Slice and in vivo electrophysiology and western blots were conducted in rats subject to fluid percussion brain injury or sham injury.
Results:
The studies identify that TLR4 signaling in neurons, augments dentate granule cell calcium-permeable AMPA receptor (CP-AMPAR) currents after brain injury. Blocking TLR4 signaling in vivo shortly after brain injury reduced dentate network excitability and seizure susceptibility. When blocking of TLR4 signaling after injury was delayed, however, this treatment failed to reduce post-injury seizure susceptibility. Furthermore, blocking TLR4 signal was less efficacious in limiting seizure susceptibility when AMPAR currents, downstream targets of TLR4 signaling, were transiently enhanced. Paradoxically, blocking TLR4 signaling augmented both network excitability and seizure susceptibility in uninjured controls. Despite the differential effect on seizure susceptibility, TLR4 antagonism suppressed cellular inflammatory responses after injury without impacting sham controls.
Interpretation:
These findings demonstrate that independently of glia, the immune receptor TLR4 directly regulates post-traumatic neuronal excitability. Moreover, the TLR4-dependent early increase in dentate excitability is causally associated with epileptogenesis. Identification and selective targeting of the mechanisms underlying the aberrant TLR4-mediated increase in CP-AMPAR signaling after injury may prevent epileptogenesis after brain trauma.
Keywords: innate immune receptor, glutamate, AMPA, excitotoxicity, epilepsy
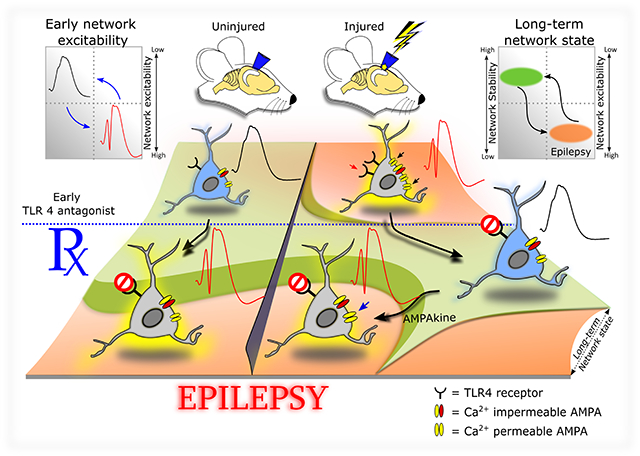
Graphical Abstract
Summary of interactions between TLR4 signaling and brain injury on network excitability and epileptogenesis.
Graphic illustration of the effect of injury and early TLR4 antagonist treatment on early network excitability and the long-term network state. The schematic neurons include TLR4 and AMPAR subunit expression profiles in the acute phase of sham or brain injury. The corresponding early effects on network excitability are depicted by schematic population response traces (inset on upper left). Note the increase in excitability of the uninjured neuron after TLR4 antagonism without changes in AMPAR expression. Note also the increase in TLR4, calcium permeable AMPARs and population excitability after injury and its reduction by TLR4 antagonist treatment. Ampakine enhancement of excitability during TLR4 antagonism is illustrated. The early phase responses and manipulations (including injury, treatments, and molecular responses) are superimposed on a two-tone color-coded network state topology where green indicates low-normal network excitability, ensuring network stability and low risk for epilepsy (Inset on upper right). Note the correspondence between early excitability state (population response profile) and long-term seizure susceptibility and the effects of pharmacological manipulations.
Introduction
Acquired epilepsies that develop after brain insults such as trauma are particularly refractory to treatments yet are potentially preventable if the underlying mechanisms are identified and appropriately targeted (1–3). A wealth of preclinical and clinical studies predict that neuronal excitability and plasticity act alongside inflammatory processes and contribute to epileptogenesis (3–9). However, the interaction between neurophysiological and inflammatory responses to injury, and the underlying mechanisms are not fully understood (10).
Activation of the innate immune receptor toll-like receptor 4 (TLR4) by its ligand HMGB1, which is released during neuronal damage, is thought to play a critical role in the inflammatory responses to seizures and brain injury (6, 11). TLR4 is expressed in both neurons and glia (11, 12). Glial TLR4 signaling has been proposed to underlie increases in neuronal excitability and excitotoxicity by activating glial cytokines, which enhance neuronal NMDA receptor (NMDAR) currents (11, 13, 14). In contrast, we recently identified TLR4 expression and enhancement in hippocampal dentate neurons after experimental brain injury, but not in astrocytes or microglia (12). Curiously, acute ex vivo antagonism of TLR4 signaling modulated AMPA receptor (AMPAR), but not NMDAR currents, in dentate neurons in slices one week after brain injury. These data raise the possibility that TLR4 signaling after brain injury does not engage the classical glial pathways (15), but instead acts through neuronal effectors which are yet to be identified (16, 17). The ability of TLR4 antagonists to reduce early network excitability in slices (12) raises the untested, clinically relevant, possibility that TLR4 signaling contributes to and could be harnessed to limit epileptogenesis after brain injury. Moreover, it is imperative to identify whether the paradoxical increase in network excitability in slices from uninjured rats treated with TLR4 antagonists (12) has both immediate and lasting consequences on dentate networks in vivo in order to evaluate the feasibility of targeting TLR4 for therapeutics. The divergent effect of TLR4 on network excitability underscores the need to identify the effectors and mechanisms underlying these disparate responses. Simultaneously, differential TLR4-mediated effects present an unparalleled opportunity to address the long-standing question concerning the role for altered network excitability in post-traumatic epileptogenesis (4, 5) and to discriminate between the roles for neuronal and classical inflammatory effectors in injury-induced epileptogenesis.
Here we use a rodent fluid percussion injury (FPI) model of concussive brain injury and post-traumatic epileptogenesis to identify the cellular, synaptic and receptor mechanisms underlying TLR4 enhancement of hippocampal dentate excitability within a week after brain injury. Focusing on cellular and molecular processes in the injured brain, we demonstrate that TLR4 signaling in neurons rapidly enhances calcium-permeable AMPAR (CP-AMPAR) currents. Our data show that early changes in hippocampal dentate excitability predict risk for epileptogenesis, regardless of the presence or absence of cellular inflammation. We identify that transient and early inhibition of the TLR4-AMPAR signaling in vivo after brain injury has the potential to prevent post-traumatic epileptogenesis.
Materials and Methods
Animals:
All procedures were approved by the Institutional Animal Care and Use Committee of the Rutgers New Jersey Medical School, Newark, New Jersey.
Fluid percussion injury:
Juvenile male Wistar rats (25–27 days old) were subject to the moderate (2.0–2.2 atm) lateral fluid percussion injury or sham-injury using standard methods (12, 18, 19). Only rats with >10 sec acute apnea and startle/seizures acutely after injury were included in the injury group and only the ipsilateral hemisphere was examined.
Drug administration and electrode implantation:
One day (20–24 hrs.) after injury, a randomly assigned cohort of FPI and sham rats received either vehicle (saline) or a synthetic TLR4 antagonist, CLI-095 (0.5mg/kg, s.c.) for 3 days. A second group underwent stereotaxic injection (Hamilton syringe-26 G) of 5μl of saline or LPS-RS Ultrapure (LPS-RSU, 2mg/ml) to the ipsilateral hippocampus. Injections were delivered through the implanted syringe hub, which was used to deliver FPI, one day after injury at a rate of 1μl/5 mins. Some animals received Ampakine (CX546, 300μM) along with LPS-RSU. A third group of animals underwent stereotaxic placement of a cannula electrode in the ipsilateral hippocampus (18). Briefly, after injury, sham and FPI rats underwent stereotaxic placement of a cannula electrode through the implanted syringe hub. Drugs were then injected through the cannula directly to the hippocampus. Some animals underwent stereotaxic implantation of an ipsilateral hippocampal depth or surface electrode 25 days or 85 days after FPI or sham injury. Drug dosing was based on published studies in vivo (20–23).
Electrophysiology:
Six to eight days after FPI or sham-injury, rats were anesthetized with isoflurane and decapitated. Horizontal brain slices (400 μm for field recordings and 350 μm for whole cell recordings) were prepared as described previously (12, 24). Field recording were conducted in an interface field recording chamber (BSC2, Automate Scientific, Berkeley, CA), perfused, and recorded in ACSF at 32–33°C making note of the electrode positions. Field recordings of dentate population responses were obtained using patch pipettes containing ACSF (12, 25). Slices were then incubated in CLI-095 or ACSF for 45 min and transferred back to the recording chamber for recordings. For whole cell patch clamp recordings of afferent evoked AMPARs, currents were recorded and analyzed as detailed previously (12). CP-AMPAR current was recorded at −60 mV and +40 mV holding potential in the presence of SR95531 (10μM) and D-APV (50 μM). ClampFit 10 and SigmaPlot 12.3 were used to analyze the data.
Cell surface protein extraction and Western blotting:
The cell surface proteins of hippocampus were isolated using Pierce cell surface protein isolation kit (Pierce, Thermo Scientific, MA). Biotinylated proteins bound to NeutrAvidin beads (bound fraction) were isolated from the nonbiotinylated (unbound) fraction by centrifugation (3000 RPM, 1 min). The bound proteins were then released by incubating with Laemmli sample buffer containing 50mM of dithiothereitol. Cell surface proteins were isolated from experimental groups and the extracted proteins as well as total proteins were used for western blotting analysis. Western blots were performed as described previously (12). Membranes were then washed and incubated overnight at 4°C with the corresponding primary and secondary antibodies. Chemiluminescent detection was performed using FluoroChem 8800, and images were quantified using Image-J (NIH). Surface protein expression was normalized to corresponding total protein levels and total protein levels were normalized and normalized to corresponding β-actin density. While surface proteins did include a band for β-actin, this was much lower than the total β-actin levels in the total protein and likely reflect proteins associated with intracellular membranes. All antibodies used in the study are in Supplementary Table 3.
Neuronal Cultures:
Primary hippocampal neurons were obtained from the hippocampi of Embryonic day 17–18 mice (WT or TLR4−/−) embryos using standard methods (26, 27). Hippocampal tissue was centrifuged at 1000 rpm for 1 min with Hanks balanced salt solution (HBSS), digested DNAse (200 Units/mg) in 25% trypsin solution washed with and transferred to Neurobasal media-A (NB-A) and mechanically dissociated. The cells were pelleted by centrifugation in NB-A and 0.5–1.0 × 106 cells per well were plated on Poly-D-Lysine (PDL) coated 15mm coverslips in NB-A medium containing B27 and L-glutamine and cultured at 37°C in 5% C02/95% air. The medium was changed every 3–4 days. Ara-C was added to media on day 3 for complete removal of glia. Cultured coverslips were stained for MAP2, GFAP and IBA-1 to identify cell types. 11 to 13 DIV cultures were used for patch clamp recordings described above.
Histology:
For immunostaining, rats were perfused with 4% paraformaldehyde 24hr after FPI or sham-injury and hippocampal sections (50 μm) were obtained. Immunostaining was performed as detailed previously using antibodies listed in supplementary table 3. Nissl staining was performed on coronal sections (50 μm) from experimental rats perfused with 4% paraformaldehyde 1 week after injury. Sections were mounted on gelatinized slides, air dried, stained in Cresyl violet and differentiated in alcohol and xylene prior to mounting. Cell counts were performed using the optical fractionator of Stereo Investigator V.10.02 (MBF Bioscience, Williston, VT) on an Olympus BX51 microscope with a 40X objective. In each section, the hilus was outlined by a contour traced using a 10X objective. Sampling parameters were set at 40X: counting frame = 50 μm by 50 μm, dissector height = 30 μm, and top guard zone = 5 μm. Approximately 25 sites per contour were selected using randomized systematic sampling protocols in Stereo Investigator(28).
Video-EEG monitoring:
One and three months after injury, rats were implanted with a hippocampal depth electrode as described above and seizure susceptibility to Kainic acid (KA, 5 mg/kg i.p.) was examined using a video EEG recording system (18, 29). Three to four months after brain injury a group of experimental rats were implanted with subdural screw electrodes and monitored for development of spontaneous seizures using video EEG recording system as described previously (24). Animals were monitored for 8 continuous hours overnight for 3–5 days (minimum of 5 days if seizures were not detected). Following EEG monitoring, rats were transcardially perfused with saline followed by 4% paraformaldehyde under surgical anesthesia.
EEG data analysis:
Analysis was performed by investigators blinded to treatment groups. Electrographic seizures were defined as rhythmic activity exceeding a threshold of mean baseline amplitude + 2.5 S.D. for >5 seconds. Simultaneous video recordings were used to exclude movement artifacts. For kainic acid evoked seizures, latency of first onset electrographic and behavioral seizure, and seizure severity were analyzed for 3 hours post injection. Seizure severity was assessed off-line by ranking the behavior according to a modified Racine’s scale (30): 0 = no behavioral change (subclinical), 1 = facial movements (twitching of vibrissae, sniffing, eye blinking or jaw automatisms), 2 = head nodding, stiff tail, 3 = forelimb clonus, chewing, 4 = rearing with myoclonus and tonic immobility and 5= Rearing and falling with myoclonus (tonic-clonic seizures). For spontaneous seizures, only electrical seizures associated with stage 3 or higher behavioral seizures and lasting at least 30 seconds were included in the analysis.
Flow Cytometry:
Three days after injury, animals were anesthetized, perfused with cold saline and the hippocampus from the injured hemisphere and spleen (positive staining control) extracted, dissociated and processed as detailed previously (31). Following LIVE/DEAD staining, cells were resuspended in FACS buffer and probed for CD45, CD3, CD4, GR-1, and OX42. Appropriate isotype controls were included to reduce non-specific signal (Supplementary Table 3). Samples (1,000,000 events/sample) were acquired using BD LSRII flow cytometer and data analyzed using FlowJo (V 10.0.8).
Statistical analysis:
Statistical analyses were performed using SigmaPlot 12.3. Analysis was performed once, midway during experiments to determine sample size. Data are shown as mean ± s.e.m. in Supplementary Table 1. Statistical results are presented in Supplementary Table 2.
Results
TLR4 signaling enhances CP-AMPAR current in dentate granule cells after brain injury
Immunostaining for TLR4 in the hippocampal dentate gyrus neurons was confirmed by co-localization of TLR4 with the neuronal marker NeuN (Fig. 1a) (12). Consistent with the effects of LPS-RSU (12), CLI-095 (or TAK-242/Resatorvid, 10ng/ml), an antagonist at the intracellular domain of TLR4 (32), reversed increases in afferent-evoked dentate population spike amplitude and field EPSP slope in slices one week after brain injury (Fig. 1b–e, Supplementary tables). However, CLI-095 increased excitability in slices from sham operated controls (Fig. 1c–e) demonstrating an opposite effect in the uninjured brain. Previously, we identified a selective increase in granule cell AMPAR currents one week after brain injury, with no changes in NMDAR currents (12, 33). TLR4 antagonists abolished post-injury increases in AMPAR current amplitude without altering AMPAR or NMDAR currents in controls (12). Granule cells express heteromeric AMPARs containing both GluA1 and GluA2 subunits, with the presence of GluA2 rendering the AMPAR currents calcium impermeable and non-rectifying (34, 35). In controls, pharmacologically isolated, perforant-path evoked granule cell AMPAR currents recorded from holding potentials of +40 and −60 mV were symmetrical (non-rectifying) indicating a calcium-impermeable phenotype (Fig. 1f–g). The selective TLR4 antagonist LPS-RSU (1μg/ml) failed to alter AMPAR current rectification in controls. In contrast, granule cell AMPAR currents were inwardly-rectifying one week after brain injury (Fig. 1f–g), indicating an increase in CP-AMPAR currents. Incubation in LPS-RSU reduced rectification (Fig. 1f–g), demonstrating that TLR4 signaling underlies increases in CP-AMPAR currents after injury. The molecular underpinnings of the post-injury increase in CP-AMPAR currents were examined in hippocampal slices from control and injured rats treated with saline or LPS-RSU (1μg/ml for 1hr). Western blots of surface biotinylated AMPAR subunits identified an increase in surface expression of GluA1 subunit after brain injury, which was reversed by a brief (1hr) incubation in LPS-RSU (Fig. 1h–i). LPS-RSU failed to alter GluA1 surface expression in controls, and GluA2 surface expression was unchanged after injury or with LPS-RSU treatment (Fig. 1i–j). Unlike surface GluA1 expression, total GluA1 and GluA2 expression were unchanged after FPI and with drug treatments (Fig 1i, k–l). These data demonstrate that TLR4 signaling selectively and reversibly augments membrane surface expression of the GluA1 subunit early after brain injury. The increase in GluA1 in the absence of corresponding increase in GluA2 would increase the fraction of GluA2 lacking AMPARs and contribute to the post-injury increase in CP-AMPAR currents.
Fig. 1. TLR4 signaling augments CP-AMPAR currents and GluA1 surface expression after brain injury.
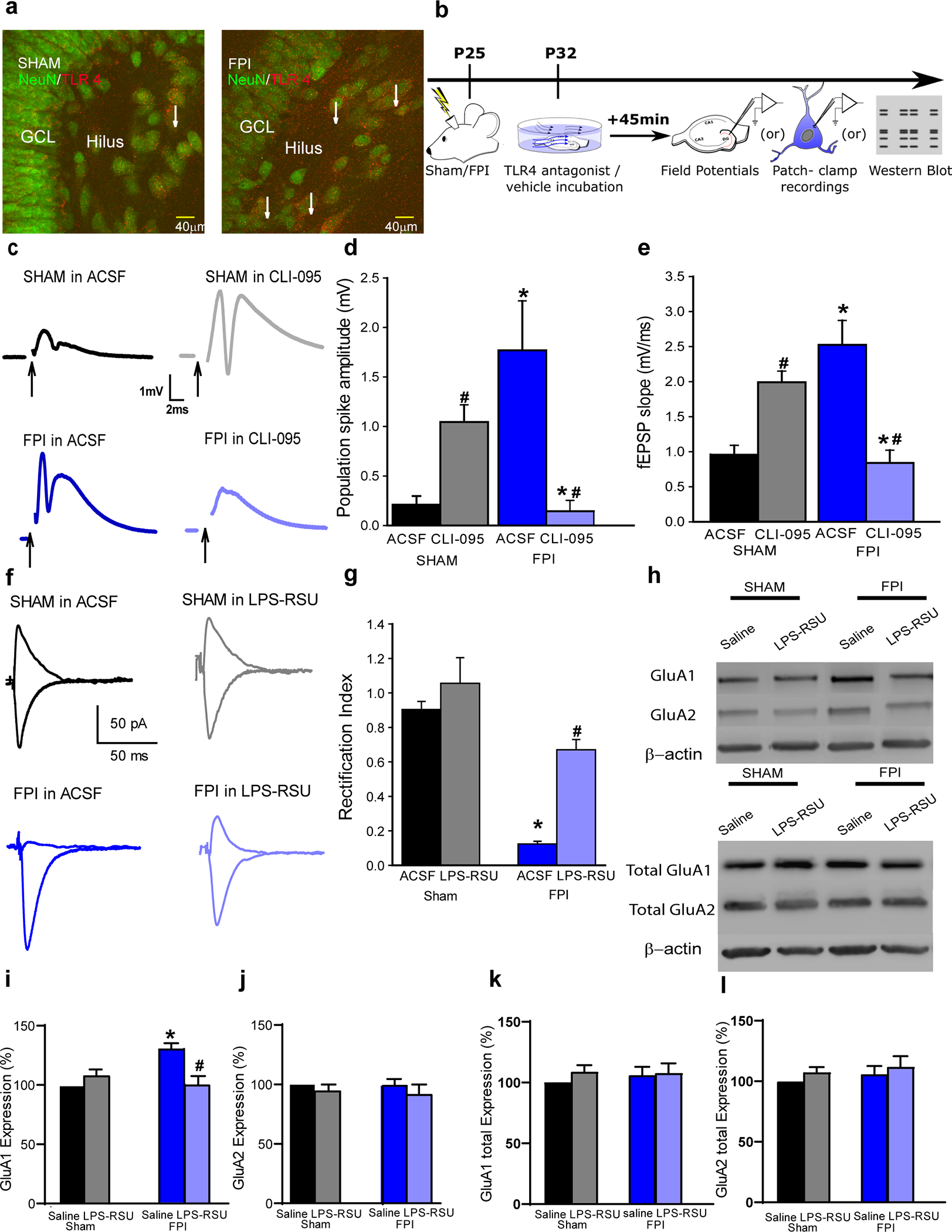
a. Representative images of dentate granule and hilar cells in rats sacrificed 24 hours after sham or FPI illustrate co-localization of TLR4 with NeuN (arrows). b. Schematic of experimental design for panels c-j. Following sham or FPI at postnatal day (P) 25, hippocampal tissue was isolated at P32 for experiments. c. Granule cell population responses to perforant path stimulation (4mA) in a slice from a sham (upper panels) and FPI rats (lower panels) incubated in ACSF (left) and CLI-095 (10 ng/ml for 1 h, right). Arrows indicate truncated stimulus artifact. d-e. Summary histogram of population spike amplitude (d) and field EPSP slope (e) in slices from rats 1-week post-injury. (* indicates p<0.05 compared to without drug; # indicates p<0.05 compared to sham by TW-RM-ANOVA followed by post-hoc Tukey’s test). f. Perforant path-evoked granule cell AMPAR current traces recorded at +40 and −60 mV, in the presence of GABAAR and NMDAR blockers, show lack of rectification in sham (above left) and in LPS-RSU (above right). One week after FPI, AMPAR currents show rectification (below left) which was reversed in LPS-RSU (below right). g. Summary of rectification index measured as the ratio of peak AMPAR current at +40 mV and −60 mV. (* indicates p<0.05 by TW-ANOVA). h. Western blots for GluA1 and GluA2 and β-actin expression in surface fractions extracted following biotinylation assay on hippocampal sections from sham and FPI rats incubated in ACSF or LPS-RSU. Western blots for total protein expression of GluA1 and GluA2 and β-actin in corresponding samples are illustrated below. Note that surface fractions were extracted from 250 μg of sample while total protein was from 50 μg and the image exposure for surface blots was three fold longer than that of blots for total protein in order to reliably visualize proteins expressed in low levels in surface fractions. i-l. Summary plots quantify the relative expression of surface biotinylated GluA1 (i) and GluA2 (j) expression normalized to total GluA1 and GluA2 respectively and total GluA1 (k) and GluA2 (l) normalized to β-actin and presented as % of control. *p< 0.05 from sham and # indicates p<0.05 compared to saline within injury type by TW ANOVA followed by pairwise comparison.
Neuronal signaling underlies TLR4-modulation of AMPAR currents
While dentate neurons express TLR4 (12), direct regulation of CNS neuronal excitability by TLR4 remains to be established. To isolate the effects of neuronal TLR4, neuronal cultures at day in vitro (DIV) 12 were established from embryonic mouse hippocampi and verified for neuronal purity by positive immunolabeling of the neuronally enriched microtubule associate protein 2 (MAP2), and lack of immunolabeling for GFAP and Iba1 (Fig. 2a–b), markers for astrocytes and microglia, respectively. In neuronal cultures from wild-type (WT) mice, neuropil stimulation evoked synaptic AMPAR currents with inward rectification, indicating calcium-permeable currents (Fig. 2c–d), similar to currents seen in granule cells after brain injury. This is consistent with reports of increased CP-AMPAR currents during development in vitro (36, 37). However, AMPAR currents in neuronal cultures from mice lacking TLR4 (TLR4−/−) were non-rectifying (Fig. 2c–d) demonstrating that neuronal TLR4 is necessary for expression of the inwardly rectifying CP-AMPAR currents. In neurons cultured from WT mice, the TLR4 agonist HMGB1 (10ng/ml) failed to increase AMPAR rectification, while LPS-RSU (1 μg/ml) reversed AMPAR current rectification (Fig. 2e–f). Together, the data from cultured WT and TLR4−/− neurons demonstrate that neuronal TLR4 is contributes to the increase in CP-AMPAR currents in vitro and is sufficient to modulate CP-AMPAR currents.
Fig. 2. TLR4 modulation of calcium-permeable AMPAR currents in hippocampal neuron-only cultures in vitro.
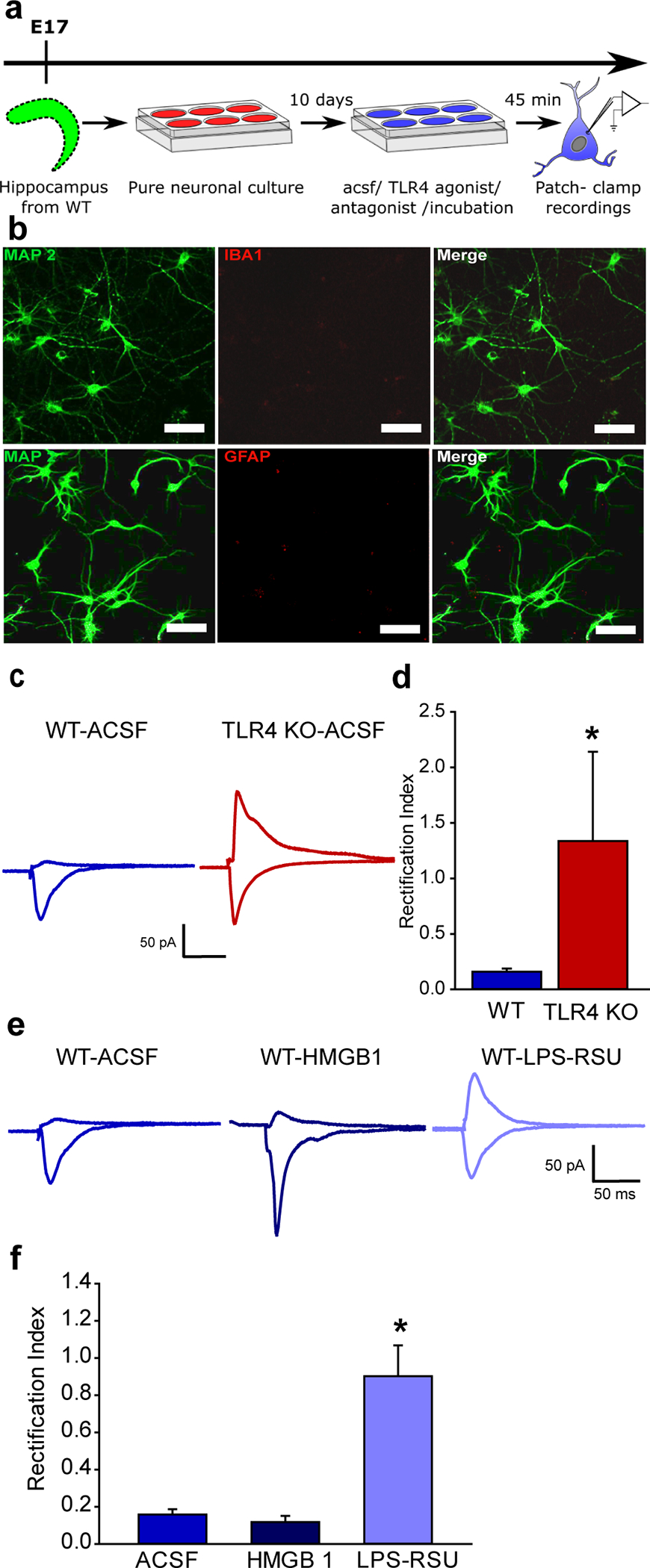
a. Schematic of the experimental paradigm illustrates the timeline for isolation of the hippocampus and maintenance in vitro followed by electrophysiology. b. Representative maximum intensity projection of confocal image stacks from a neuronal culture plated at embryonic day 17 and stained for MAP2 at DIV 12 to reveal neurites. c. Representative traces of AMPAR current recordings in neurons cultured from WT (left) and TLR4−/− mice (right) in response to a 1 mA stimulus to the neuropil. Recordings were obtained from holding potentials of −60 mV and +40 mV as in Fig. 1f. Note the AMPAR current rectification in neurons from WT and symmetric currents in TLR4-/−. d. Summary plot of rectification index measured as the ratio of peak AMPAR current at +40 mV to the peak current at −60 mV. (* indicates p<0.05 by Mann-Whitney test). e. Sample AMPAR current traces recorded in response to neuropil stimulation in WT neuronal cultures. Neurons were held at +40mV and −60 mV to obtain recordings in ACSF (left), TLR4 agonist HMGB1 (middle) and TLR4 antagonist LPS-RSU (right). Note the AMPAR current rectification in the absence of drugs and symmetric currents in LPS-RSU. f. Summary plot of rectification index. * indicates p<0.05 by One Way ANOVA on ranks followed by post-hoc Tukey’s test.
Systemic TLR4 antagonism in vivo has opposing effects on early dentate excitability and epileptogenicity in control and injured brains
Next, we focused on the impact of the early post-injury TLR4 signaling on subsequent epileptogenesis which has been shown to occur after fluid percussion injury (38, 39). Latency to seizures evoked by low-dose kainic acid (KA, 5 mg/kg i.p.) challenge was used to assess epileptogenicity (29). While only a few sham animals injected with low-dose KA developed electrographic and behavioral seizures (Fig. 3a–b), rats one month after brain injury reliably developed seizures within 30 minutes of KA injection. Latency to KA-induced seizures was significantly reduced in rats one month after brain injury (Fig. 3c). Additionally, sham injured rats reached a maximum Racine score of 2 or 3 and brain injured rats reached maximum score of 5 (Fig. 3d). Overall, compared to sham-injured controls, brain-injured rats develop more-severe seizures with shorter latency following low-dose KA injection demonstrating post-traumatic enhancement of seizure susceptibility. In a small cohort, we monitored animals for spontaneous seizures after brain injury. In 30–40-hour simultaneous video- and surface EEG recordings from rats 12–15 weeks after sham or FPI, none of the sham rats (n=9) showed electrographic seizures, while spontaneous electrographic and behavioral seizures were observed in 8 of 13 FPI rats (~62%, Fig. 4a–d). In rats with spontaneous seizures the average seizure severity was Racine score 3.37±0.18 (range of 3–4) and average seizure duration was 97.88±10.20 sec (range of 50–140 sec) confirming the risk for post-traumatic epileptogenesis in the experimental model. It is possible that strain differences, use of younger juvenile rats which are more susceptible to adverse effects of brain injury (40), implementation of injury on the day after surgery to minimize neuroprotection due to surgical anesthesia and inclusion criteria based on apnea duration contributed to greater proportion of rats developing spontaneous seizures compared to earlier studies (41–43).
Figure 3. Enhanced susceptibility to chemically evoked seizures after brain injury.
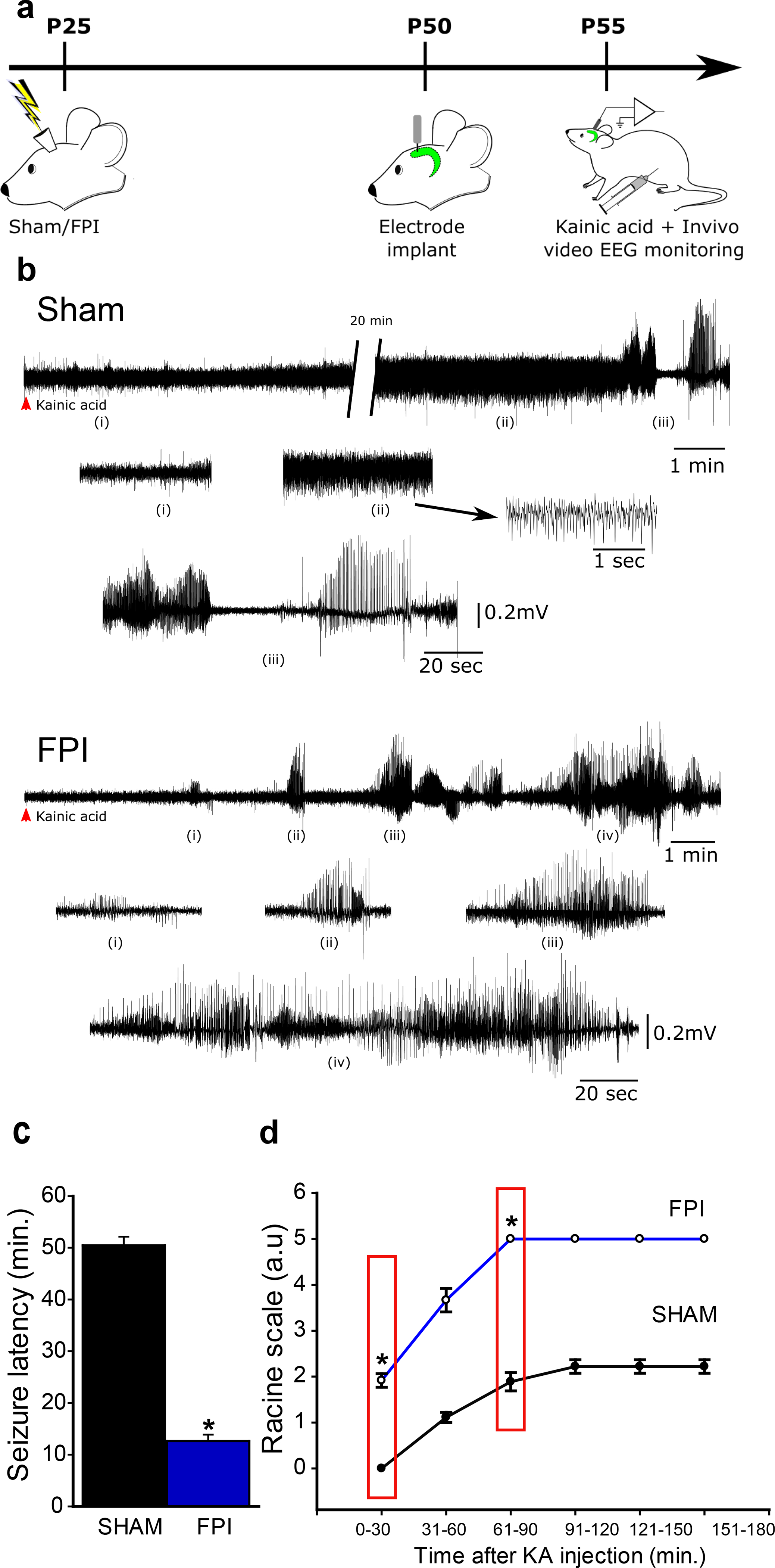
a. Schematic of timeline for in vivo injury followed by electrode implantation and low-does Kainic Acid challenge b. Sample hippocampal depth electrode recordings show evolution of electrographic activity following kainic acid injection in rats one month after sham (above) and FPI. Note that FPI animals develop electrographic seizures early which quickly develops into convulsive seizures (FPI, i-iv). c. Summary of latency to kainic acid induced seizures in rats 1-month after sham or brain injury. d. Summary of progression of seizure severity by Racine’s scale over time in sham (n=9) and FPI (n=12) rats. Boxed areas indicate time points at which statistical comparisons are reported. * indicates p<0.05 by Student’s t-test.
Fig. 4. TLR4 antagonism alters delayed occurrence of spontaneous epileptic seizures.
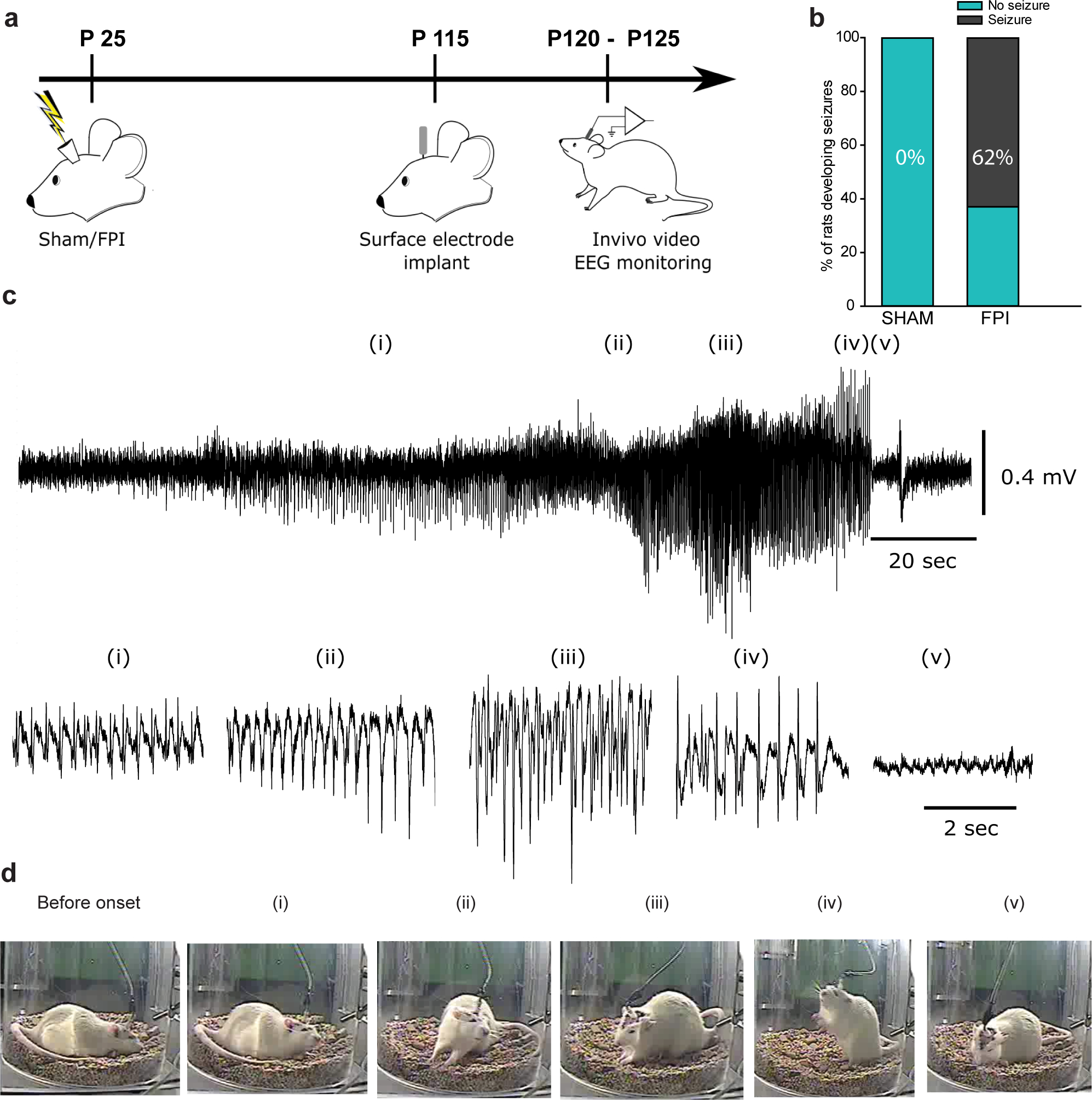
a. Timeline for assessment of spontaneous seizures following sham or FPI in rat. b. Summary plot illustrates percentage of rats developing spontaneous seizures. c. Sample subdural electrode recordings in brain injured rat obtained 12–15 weeks after injury. Panels below show expanded segments of EEG data from regions i-v. d. Video snapshots corresponding to segments i-v are illustrated. The time of recoding first ictal activity is noted. * indicates p<0.05 by z-test based on 10 sham and 13 FPI rats.
To transition our findings on TLR4 modulation of excitability in ex vivo slices to in vivo, we examined whether systemic TLR4 antagonism could modify dentate excitability one week after injury. Sham and FPI rats were treated with CLI-095 (0.5mg/kg, s.c. for 3 days) starting 20–24 hours after injury and dentate excitability was examined in hippocampal slices 6–8 days later. As with acute incubations, CLI-095 treatment in vivo increased dentate excitability in sham-injured rats, while effectively suppressing excitability after FPI (Fig. 5a–c). These data justify the use of in vivo treatments in evaluating the effect of TLR4 signaling on early dentate excitability and epileptogenesis after brain injury.
Fig. 5. Systemic TLR4 antagonism in vivo modulates dentate network excitability in both sham- and brain-injured rats.
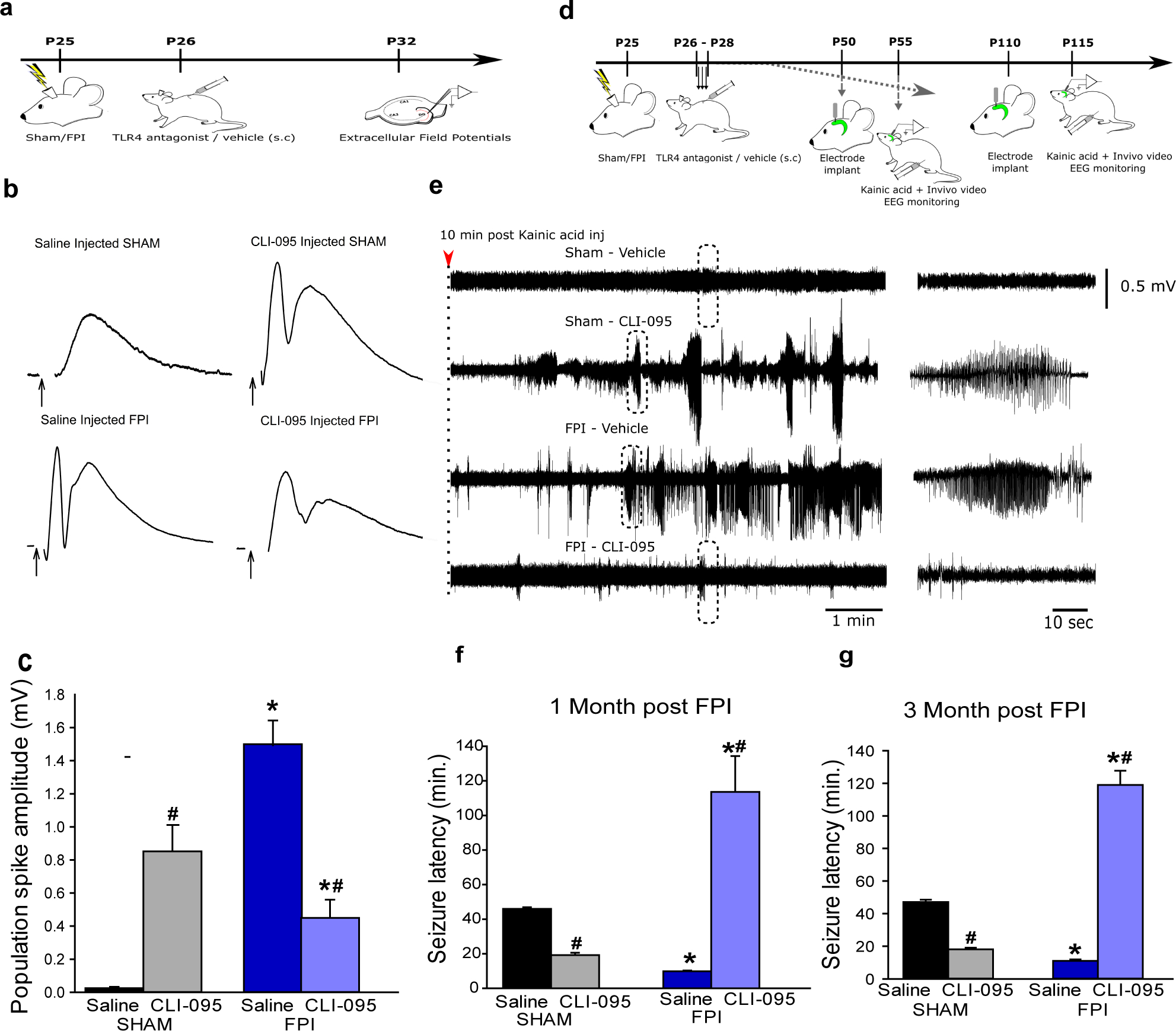
a. Illustration of timeline for in vivo treatments and slice physiology in sham- and brain-injured rats. b. Dentate population responses evoked by a 4-mA stimulus to the perforant path in slices from sham (above) and brain injured (below) rats treated in vivo with saline (left) and CLI-095 (0.5mg/kg, right). Arrows indicate truncated stimulus artifact. c. Summary data demonstrate the effect of CLI-095 on perforant path-evoked granule cell population spike amplitude in slices from sham (6 slices each from 3 rats) and FPI rats (6 slices each from 3 rats). Error bars indicate s.e.m. * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type, by TW-ANOVA followed by Tukey’s post hoc test. d. Illustration of timeline for surgical procedures, in vivo treatments and EEG recordings in sham and brain injured rats. e. Sample hippocampal depth electrode recordings in sham and brain injured rats treated with vehicle or CLI-095 (0.5mg/kg). Recordings show EEG activity 10 minutes after KA injection (right panel). Expanded traces of the activity in boxed areas illustrated in the left panels show lack of seizure activity in sham-vehicle and FPI-drug conditions and development of seizure activity in FPI-vehicle and sham-drug rats in the 10–20-minute period after KA injection. f-g. Summary of latency to KA-induced seizures in rats 1-month (f) and 3-months (g) after sham or brain injury. * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type, by TW-ANOVA followed by Tukey’s post hoc test.
Although the post-injury increase in dentate excitability is postulated to underlie a long-term increased risk for epilepsy (4, 5, 44), this association remains untested. Since blocking TLR4 reduces excitability after injury and increases excitability in uninjured rats, TLR4 antagonism provides an opportunity to test the link between early excitability and subsequent risk for seizures. Rats were treated with systemic CLI-095 (0.5mg/kg) or saline once daily for 3 days starting 24 hours after sham or brain injury, when hippocampal TLR4 levels were found to be maximal (12, 45). As expected, saline-treated injured rats had a shorter latency to KA-induced seizures compared to saline-treated sham rats when examined 1 and 3 months after injury (Fig. 5d–f). CLI-095 treatment decreased latency to KA-evoked seizures in sham injured rats while prolonging seizure latency in FPI rats at one-month and three months post-FPI (Fig. 5f–g). Mechanistically, the data reveal that the risk for seizures is driven by early changes in dentate excitability. Translationally, these results show that transiently blocking TLR4 early after brain injury has the potential to prevent post-traumatic epileptogenesis.
Early focal TLR4 antagonism is sufficient reduce post-traumatic increases in seizure susceptibility while delayed treatment is ineffective.
To probe the association between hippocampal TLR4 signaling and epileptogenesis, we adopted a focal TLR4 antagonist treatment. Ipsilateral bolus hippocampal injection of LPS-RSU (2mg/ml) 24 hours after FPI/sham led to decreased latency to KA-evoked seizures in sham rats and eliminated seizures altogether in FPI rats when examined one and three months after brain injury (Fig. 6a, b, d). Hippocampal LPS-RSU treatment enhanced seizure severity in controls and reduced seizure severity in FPI rats at one and three months after FPI/sham. (Fig. 6c, e). Thus, local suppression of TLR4 in the injured hippocampus and its modification of early dentate excitability can predict long-term seizure susceptibility.
Fig. 6. Early but not delayed focal hippocampal TLR4 antagonism eliminates post-traumatic increase in seizure susceptibility.
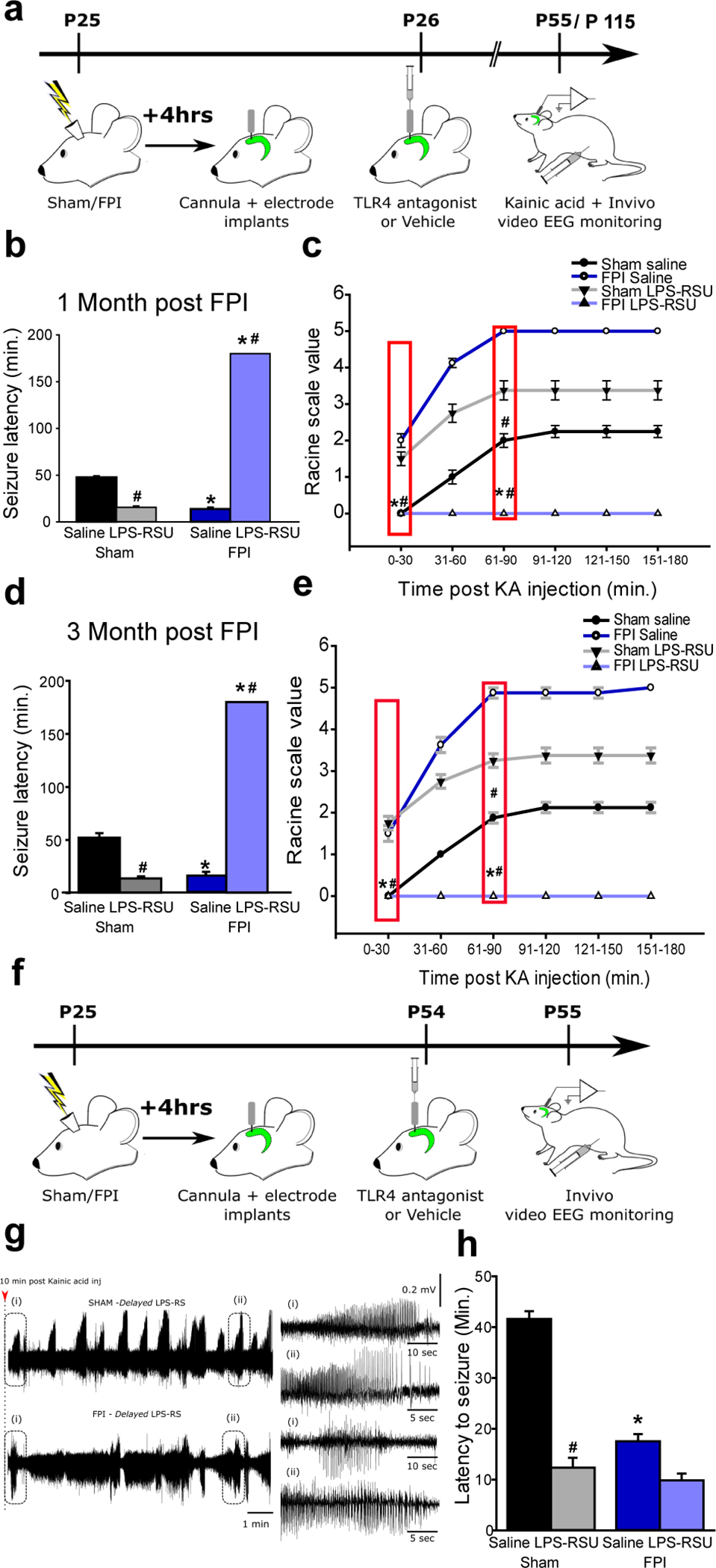
a. Schematic of treatments and surgical procedures relevant to panels b-e for experimental rats. b and d. Summary of latency to KA (5 mg/kg) induced seizures in sham and FPI rats 1-month (b) and 3-months (d) post injury obtained using hippocampal depth electrodes. Rats were injected with saline or LPS-RSU (2mg/ml, single bolus injection) unilaterally on the side of the FPI implant 24 hours after injury. c and e. Progression of seizure severity and maximum seizure score by Racine’s scale reached in saline or drug treated sham and FPI rats 1-month (c) and 3-months (e) post injury. * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type. N=8 rats per group. f. Illustration of timeline for delayed treatment. g. Sample hippocampal depth electrode recordings in sham and brain injured rats treated with LPS-RSU (2mg/ml) one-month post FPI. EEG recordings show rapid development of seizures within 10–15 minutes after KA injection. Panels to the left show expanded traces from the boxed areas. h. Summary plot of latency to kainic acid induced seizures. * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type, by TW-ANOVA followed by Tukey’s post hoc test.
Interestingly, analysis of mortality among experimental animals revealed that sham-injured rats treated with TLR4 antagonists had increased mortality while post-traumatic rats treated with TLR4 antagonists showed lower mortality (% mortality between one week after treatment and experimental end point: sham-saline: 0%, 0/20; Sham-LPS-RSU: 26.9%, 7/26; FPI-saline: 44.4%, 16/36; FPI-LPS-RSU: 13.3%, 4/30). Moreover, evaluation of the spontaneous seizures in TLR4 antagonist-treated sham and FPI animals revealed that one LPS-RSU-treated sham rat (of 6 examined; 16.7%) and none of the saline-treated sham rats developed spontaneous seizures. However, three (of 6 tested; 50%) saline-treated FPI rats exhibited spontaneous seizures and none of the 6 LPS-RSU-treated FPI rats showed electrographic or behavioral seizures in the same duration of testing.
The salience of the early period of excitability to seizure susceptibility, was tested by delivering focal LPS-RSU or saline treatments 1 month after injury followed by evaluation of KA-induced seizures on the following day. Delayed LPS-RSU treatment reduced the latency to seizures in uninjured rats confirming that blocking TLR4 can modify network excitability in controls (Fig. 6f–h). Conversely, delayed LPS-RSU treatment failed to reduce seizure susceptibility after FPI (Fig. 6f–h), highlighting the importance of the early post-injury increases in dentate network excitability in long-term seizure risk.
TLR4 antagonism reduces cellular inflammation following FPI without altering inflammatory milieu in controls.
Since TLR4 signaling leads to cellular inflammation, prior studies have attributed beneficial effects of blocking TLR4 after brain trauma to reducing inflammatory responses (46). To test whether inflammatory responses could mediate the divergent effects of TLR4 antagonists on seizure susceptibility, we examined the inflammatory responses in hippocampal tissue obtained from saline/CLI-095-treated rats three days after sham/FPI. In western blots, CLI-095 significantly reduced hippocampal expression of TLR4, GFAP and Iba1 in the injured brain without enhancing expression in sham animals (Fig. 7a–e). A cohort of animals were examined one month after FPI and saline/CLI-095 treatment. Consistent with our previous findings demonstrating that TLR4 returns to control levels by one-month (12), hippocampal expression of TLR4 expression in saline treated FPI rats was not different from sham-saline rats when examined one month after treatment (Sham-saline: 1.00±0.0; FPI-Saline: 1.10±0.28, p=0.92 by Tukey’s post hoc test, n= 3 each). TLR4 expression in FPI rats treated with CLI-095 one day after injury was not different from sham-saline or FPI-saline groups (TLR4 expression normalized to sham-saline: FPI-CLI 0.82±0.18, p=0.58 by One-Way ANOVA, Tukey’s post hoc test, n= 3 each). Curiously, GFAP protein levels were highly variable after FPI, increasing in some and decreasing in others and was not significantly different between groups (FPI-saline normalized to sham Saline, Sham-saline: 1.00±0.0, n=3; FPI-Saline: 1.74±0.67, p=0.35, n=4). However, GFAP protein levels in FPI-CLI group trended to decrease in FPI-saline group one-month post-injury (GFAP expression in FPI-CLI normalized to sham-saline, FPI-saline: 1.74±0.67, n=4; FPI-CLI: 0.17±0.06, p=0.063, n=4 by Tukey’s post hoc test). Finally, we examined whether FPI and early CLI-095 treatment altered GluA1 expression one-month after injury. Surface expression of GluA1 was not different between saline treated sham and FPI groups (surface/total GluA1 ratio normalized to sham-saline: sham-saline: 1.00±0.0; FPI-Saline: 1.12±0.29, p=0.94, n=3 each). Additionally, early CLI-095 treatment did not alter surface-expression of GluA1 tested one month after FPI (surface/total GluA1 ratio normalized to sham-saline: FPI-CLI 1.23±0.43, p=0.96, n=3 each). However, total GluA1 levels showed a significant increase one month after FPI (total GluA1 normalized to sham-saline, sham-saline: 1.00±0.0; FPI-Saline: 1.39±0.15, p=0.03, n=4 each) and reduced significantly following early CLI treatment (total GluA1 normalized to sham-Saline: FPI-Saline: 1.39±0.15, FPI-CLI: 0.75±0.05, p=0.002, n=4 each, by One-Way ANOVA, Tukey’s post hoc test). These changes in total GluA1 expression are consistent with persistent post-FPI increase in hippocampal excitability after FPI (47) and demonstrates that early treatment with CLI-095 reduces a potential molecular substrate for persistent posttraumatic increase in excitability.
Fig. 7. TLR4 antagonism suppresses cellular inflammation and loss after brain injury without perturbing inflammatory response in uninjured sham rats.
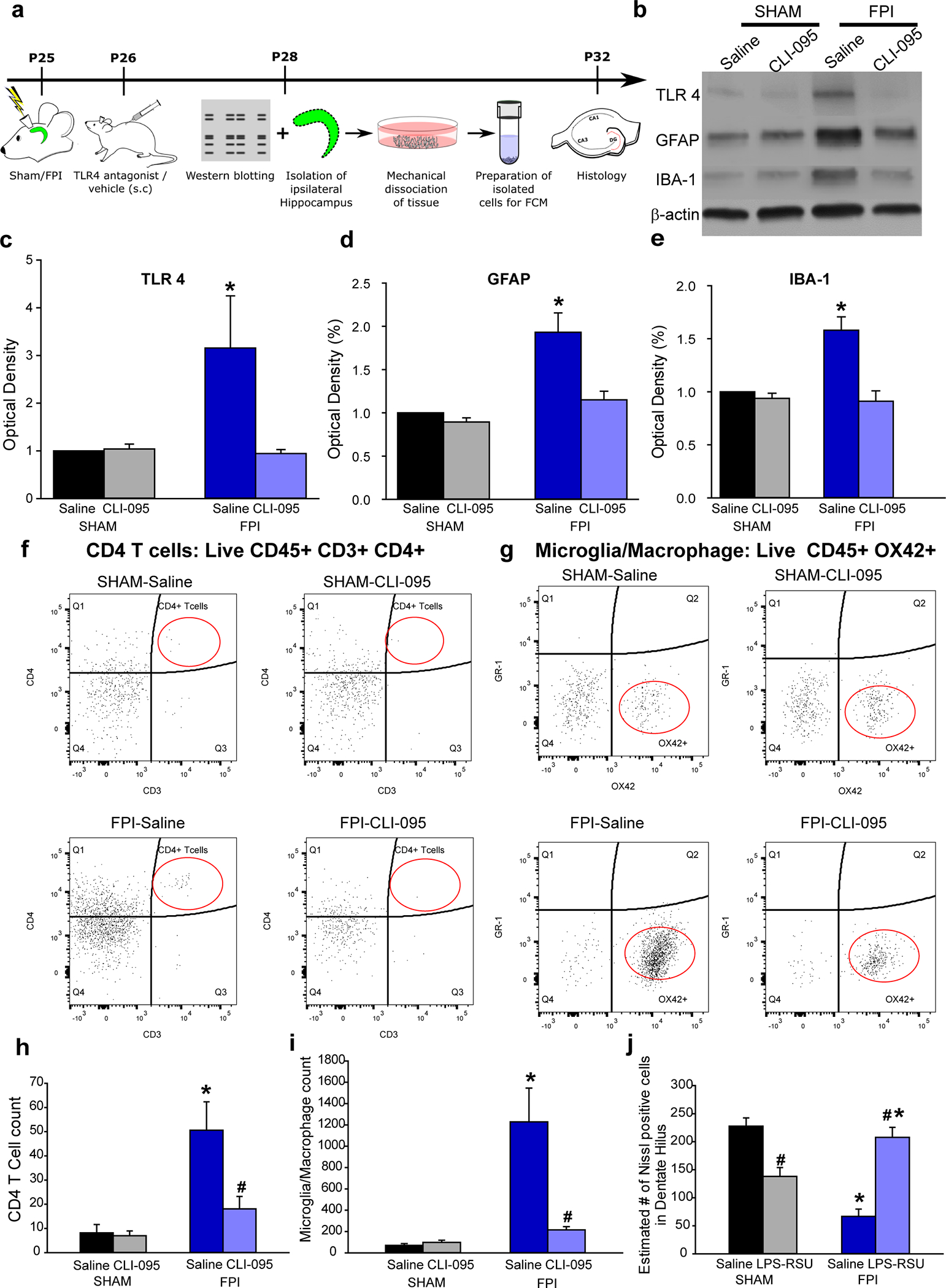
a. Experimental timeline for western blot, flow cytometry and histological studies. b. Representative western blots for TLR4, GFAP and IBA-1 in hippocampal samples from the injured side obtained 3 days after saline/drug treatment. Treatments began 24 hours after injury. Corresponding β-actin bands are illustrated. c-e. Summary histograms of expression of TLR4 (c), GFAP (d) and IBA-1 (e) as a % of the expression levels in sham-saline treated controls. f. Representative CD4/CD3 scatter plots from the hippocampus on the injured side of saline- and (left) and CLI-095 treated (right) sham (above) and brain injured (below) rats. CD4/CD3 scatter plots were gated on live CD45+ cells. The population of interest is noted by the red oval. g. Representative GR-1/Ox42 scatter plots from the hippocampus on the injured side of saline- and (left) and CLI-095 treated (right) sham (above) and brain injured (below) rats. GR-1/OX42 scatter plots were gated on live CD45+ cells. h-i. Quantification of total CD45+CD3+CD4+ T cells (i) and CD45+OX42+ microglia in the experimental groups. Data are presented as mean ± s.e.m., n = 12 animals/treatment (4/group with 3 replicates) * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type by Kruskal-Wallis One Way ANOVA on ranks followed by post-hoc pairwise multiple comparison by Student-Newman-Keuls Method. j. Estimate of Nissl stained cells/ section in the ipsilateral dentate hilus in the experimental groups. n = 11 slices in 3 rats in sham-saline, 11 slices in 3 rats for sham treatment 12 slices in 4 rats from FPI-saline and 11 slices from 4 rats in FPI-treatment. * indicates p<0.05 from sham and # indicates p<0.05 compared to saline within injury type by TW-ANOVA followed by post-hoc Tukey’s test.
Flow cytometry to quantify the cellular inflammation in the hippocampus was performed three days after sham/FPI followed by treatments. The data revealed a significant increase in CD45+CD3+CD4+ T-cells and CD45+Ox42+ microglia/macrophages in saline-treated rats compared to uninjured controls consistent with timeline for post-injury inflammatory responses (48). CLI-095 treatment reduced T-cell and microglial/macrophage count in the hippocampus of brain injured rats (Fig. 7f–i). Contrary to its effects on excitability, CLI-095 failed to increase T-cells or microglia/macrophage counts in sham rats (Fig. 7f–i). To directly assess the effect of treatment on histopathology, we examined the dentate hilus for neuronal loss one week after injury and treatments. Nissl staining for surviving hilar neurons demonstrated that TLR4 antagonists (ipsilateral intrahippocampal LPS-RSU) prevented hilar cell loss observed after brain injury and reduced hilar cell counts in uninjured rats (Fig. 7j) demonstrating that early cell loss tracks changes in excitability regardless of the inflammatory milieu in controls.
Driving excitability limits ability of TLR4 antagonists to suppress seizure susceptibility after brain injury
To test whether the ability of TLR4 antagonists to suppress excitability can influence post-injury seizure susceptibility independently of its ability to reduce inflammation we tested whether enhancing AMPAR currents during TLR4-antagonism would increase excitability downstream of TLR4. Ampakines, positive allosteric modulators of AMPARs, have been used in vivo to enhance AMPAR currents (49, 50). Since TLR4 modulates AMPAR currents only after brain injury, we first validated the ability of an Ampakine to augment AMPAR currents in the presence of TLR4 antagonists in slices from brain-injured rats. Afferent-evoked granule cell AMPAR current charge transfer was enhanced one week after brain injury (Fig.8a–c) and reduced by incubation in LPS-RSU (Fig. 8d–e). Incubation in LPS-RSU (1μg/ml) together with an Ampakine (CX546; 300μM) enhanced charge transfer confirming that this Ampakine can enhance AMPAR currents in the presence of LPS-RSU (Fig. 8d–e). Following ex vivo validation, rats one day after FPI were treated with LPS-RSU followed by an Ampakine (CX546; 300μM) delivered through an implanted hippocampal cannula electrode (Fig. 8f). Although local treatment with LPS-RSU early after injury eliminated seizures during the KA-challenge, simultaneous administration of an Ampakine resulted in development of seizures and an apparent reduction in seizure latency demonstrating a role for TLR4-mediated excitability in seizure susceptibility. However, the latency to seizures in injured animals receiving combined LPS-RSU and Ampakine treatment was longer than those treated with saline (Fig. 8f–h). These results identify TLR4-mediated enhancement of glutamatergic currents after brain injury as critical contributor to post-traumatic epileptogenesis.
Fig. 8. Augmenting AMPAR currents increases seizure susceptibility in TLR4 antagonist-treated brain-injured rats.
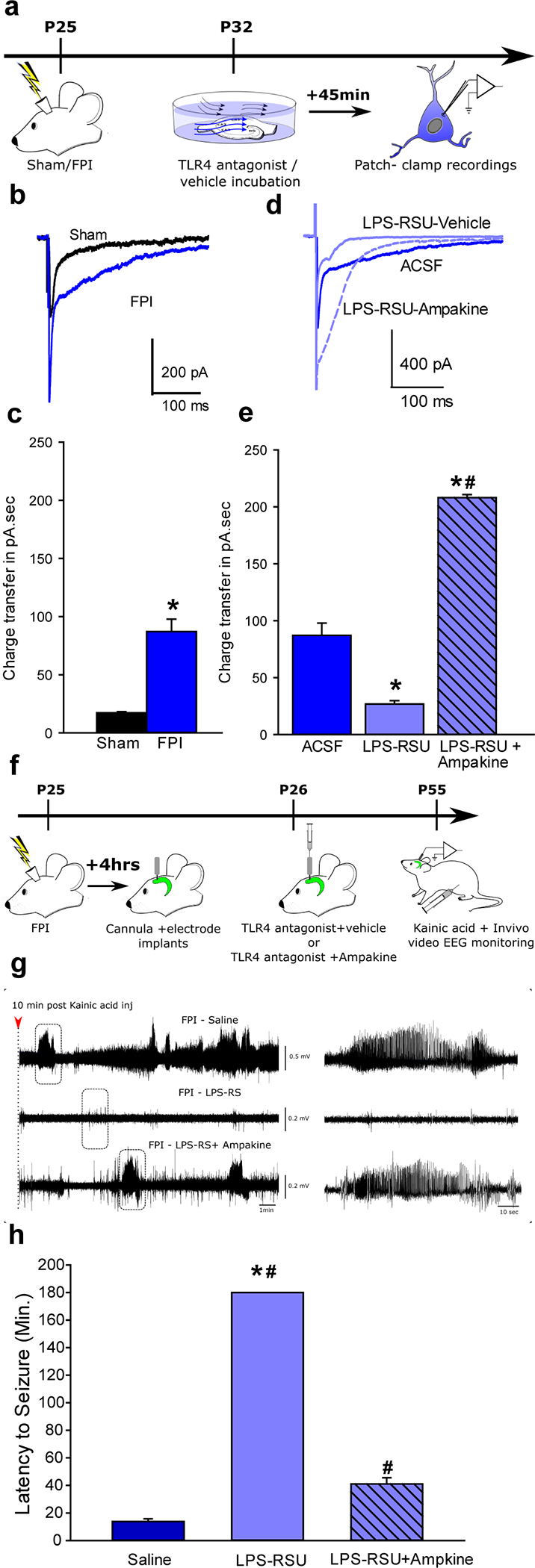
a. Schematic for experiments testing the efficacy of Ampakine to enhance perforant path-evoked AMPAR currents in ex vivo preparations. b. Overlay of afferent evoked AMPAR currents in slices from sham (black) and FPI (blue traces) rats. c. Summary histograms of AMPAR charge transfer in experimental groups. *indicate p<0.05 by Mann Whitney test. d. Overlay of afferent-evoked AMPAR currents in slices from FPI rats incubated in ACSF/saline (dark blue), LPS-RSU and saline (light blue solid line) and LPS-RSU and CX546 (light blue dashed line). e. Summary histograms of AMPAR charge transfer in experimental groups. *indicate p<0.05 by One Way ANOVA on ranks followed by post-hoc Tukey’s test for differences between slices from FPI following different drug incubations. The same data set for FPI-ACSF is used in panels c and e. f. Illustration of treatments and timeline for testing the ability of CX546 to alter seizure susceptibility. g. Sample hippocampal depth electrode recordings in brain injured rats treated with saline or LPS-RSU (2mg/ml) or LPS-RSU (2mg/ml) + CX546 (300μM). EEG recordings obtained 10 minutes after KA (5 mg/kg, i.p) injection shows the early development of seizures in saline treated FPI rats (above), lack of seizure activity in FPI rats treated with LPS-RSU 24 hours after injury (middle) and development of seizure activity in FPI rats treated with CX546and LPS-RSU (lower). Expanded traces in the boxed areas are shown (right panels). h. Summary plot of latency to KA induced seizures. Saline and LPS-RSU data in h were presented in 5b. * indicates p<0.05 from saline and # indicates p<0.05 compared to LPS-RSU by one-way ANOVA followed by Tukey’s post hoc test.
Discussion:
This study reveals a Janus head-like dichotomy in the effect of TLR4 signaling in vivo on neuronal excitability in the uninjured versus injured hippocampal dentate gyrus, which impacts network excitability and seizure susceptibility. Our results highlight the complex interactions between immune signaling, brain injury, and epileptogenesis at distinct functional levels and temporal scales. At a time scale of minutes to days, TLR4 signaling enhanced GluA1 surface expression and CP-AMPAR currents in the injured brain which led to enhanced network excitability. TLR4 antagonist treatment, ex vivo and in vivo, reversed post-traumatic increases in TLR4 and GluA1 surface expression and network excitability to control levels (Figs. 1 and 5). Notably, TLR4 modulation of CP-AMPAR currents was observed in isolated neuronal cultures, revealing a direct effect of neuronal TLR4 in regulating excitability, independent of glial contribution. It is attractive to speculate that TLR4 recruits glia-independent signals to promote GluA1 trafficking and surface expression in the injured brain. These results constitute the first demonstration of a functional role for central neuronal TLR4 in modulating excitability and AMPAR currents.
In contrast to its effects after injury, TLR4 antagonists enhanced network excitability in controls (12). in contrast to effects on excitability, TLR4 antagonists suppressed the post-traumatic cellular inflammatory responses without inducing inflammation in controls. Although the mechanisms by which TLR4 signaling increases dentate excitability in controls is currently unknown, our findings demonstrate that enhanced inflammatory responses do not underlie these effects. Moreover, consistent with our prior study (12), TLR4 does not modulate glutamate currents in controls. Of note, our treatments included two mechanistically distinct antagonists, LPS-RSU delivered locally and CLI-095 administered systemically, to demonstrate the divergent effects of TLR4 signaling on excitability and seizure susceptibility in controls and after injury. These findings suggest that a distinct TLR4 signaling pathway is utilized after injury. Overall, analysis of the short-term effects of TLR4 signaling revealed constitutive suppression of excitability under basal conditions and enhanced excitability mediated by neuronal CP-AMPAR currents after injury.
Our data demonstrating that transient perturbations in hippocampal TLR4 signaling have long-term implications for network excitability and stability constitutes a significant advance in understanding the relevance of TLR4 signaling for post-traumatic neuropathology since our earlier acute studies in vitro (12). Brain injury and TLR4 antagonism both affect TLR4 signaling, augment network excitability, and lead to increased susceptibility to seizures long-term. Blocking TLR4 signaling in vivo early after brain injury reduced network excitability and cellular inflammatory responses, hippocampal GluA1 expression and diminished seizure susceptibility in the long-term. However, transiently increasing AMPAR currents in the injured hippocampus reduced the ability of LPS-RSU to prevent chemically evoked seizures. These data show that, independent of the effect on inflammation, TLR regulation of excitability impacts seizure susceptibility. Moreover, transiently antagonizing TLR4 enhanced dentate excitability and promoted seizures without inducing cellular inflammation in controls. Therefore, regardless of effects on the inflammatory milieu, manipulations that increase in hippocampal dentate excitability drive long-term increased risk for epilepsy. Importantly, the aberrant neuronal TLR4-mediated increase in CP-AMPAR may be targeted after brain trauma to prevent epileptogenesis.
There is growing evidence for interactions between inflammatory signaling and neuronal physiology in the normal brain and in disease (6, 51–53). Brain injury and seizures lead to increases in endogenous damage associated molecular patterns (DAMPs), such as HMGB1, which can activate pattern-recognition receptors, including TLR4 (11, 45, 54, 55). While mice lacking TLR4 show improved neurological outcomes after brain injury, the beneficial effects have been attributed to reduced inflammation (56). TLR4 antagonists have been shown to reduce chemoconvulsive seizures by modulating NMDAR currents, suggesting that TLR4 modifies neuronal excitability (11, 13). Although TLR4 is expressed in neurons (11, 12), TLR4 modulation of NMDAR-dependent calcium entry was found to involve astroglial purinergic signaling or generation of the pro-inflammatory cytokine Tumor Necrosis Factor α (TNFα) (11, 13, 53, 57, 58). Accordingly, glial signaling has been proposed to underlie the neurophysiological effects of TLR4 (6, 53, 59). Our demonstration that TLR4 activity modulates neuronal CP-AMPAR currents, independent of glia, identifies a novel mechanism by which TLR4 promotes excitotoxic damage and epileptogenesis independent of inflammatory signaling. Our findings uncover a unique neuronal axis of TLR4 signaling similar to reports in Major Histocompatibility class I molecules (60). These data raise the possibility that neurons may repurpose the complexity of immune receptors to modulate neuronal excitability. Like enhanced excitability, cellular inflammatory response is a hallmark of traumatic brain injury (3, 61). While TLR4 blockers limited hippocampal cellular inflammation in brain injured animals and failed to induce inflammation in controls, they had an opposite effect on excitability and epileptogenicity depending on the injury profile. Augmenting AMPAR currents with an Ampakine alongside LPS-RSU, 24 hours after brain injury, decreased latency to seizures compared to LPS-RSU administered alone demonstrating that TLR4 enhancement of AMPAR currents contributes to epileptogenesis. However, latency to seizure in injured rats receiving an Ampakine and LPS-RSU was longer than following saline treatment, which may underscore a parallel beneficial effect of reducing inflammation. Taken together, our findings identify a neuronal axis for TLR4 signaling that enhances excitability after brain injury and demonstrate that suppression of both excitability and inflammatory responses contribute to anti-epileptogenic effects of TLR4 antagonists.
The contribution of glial mediators to TLR4 regulation of neuronal excitability has been identified in earlier studies (10, 13, 53). Our findings in neuronal cultures demonstrate that glial signals are not engaged in the TLR4 enhancement of CP-AMPAR currents, revealing neuronal TLR4 signaling in the CNS. Future studies are needed to address the molecular underpinnings of TLR4 signaling in CNS neurons as has been examined in peripheral neurons (16, 62, 63). We previously reported a ~40% increase in granule cell peak AMPAR currents in slices from brain injured rats, which was abolished by incubation in TLR4 blockers (12). Our finding that blocking TLR4 in slices selectively decreases GluA1 surface expression after brain injury without altering GluA2 expression indicates that an increase in GluA2 lacking AMPAR with calcium permeable GluA1 containing receptors underlies the post-injury increase in AMPAR currents. This could occur from exocytosis and surface expression of a reserve pool of GluA1, similar to the processes mediated by glia-derived TNFα in culture systems (64). Increases in granule cell CP-AMPARs have been shown to contribute to excitotoxic damage after experimental stroke (34), suggesting that an increase in CP-AMPARs may drive excitotoxic neuronal loss and epileptogenesis after brain injury. Similarly, TLR4 also augments AMPAR currents in mossy cells after FPI (12) and enhanced mossy cell activity drives an increase in granule cell AMPAR charge transfer after brain injury (33). Thus, the rapid TLR4 regulation of AMPAR currents likely plays an important role in increasing excitability shortly after brain injury. In contrast to the lack of increase in total GluA1 levels one week after FPI, hippocampal total GluA1 expression appears increased one month after FPI suggesting that the mechanisms underlying enhanced excitability evolve with disease progression. However, CLI-095 treatment early after FPI significantly lowered GluA1 expression one month after injury demonstrating that the early reduction in excitability limits long term increases in GluA1 expression and seizure susceptibility.
We find that blocking TLR4 signaling in uninjured rats promotes neuronal loss in the dentate hilus and raises the concern that systemic TLR4 antagonists may contribute to cell loss in uninjured brain regions and compromise circuit function. These findings underscore the need to resolve the distinct mechanisms underlying TLR4 regulation of neuronal excitability in the injured and uninjured brain to enable selective targeting of the pathological mechanisms. Curiously, AMPAR current rectification in cultured neurons from WT mice resembled the injured brain while those from TLR4−/− mice were similar to the uninjured brain. This demonstration that TLR4 is necessary for AMPAR current rectification in cultured neurons provides a mechanism for the previously reported developmental increase in CP-AMPARs in neuronal cultures (36). The parallels between TLR4 effects on AMPAR currents in cultured neurons and the injured brain suggest that the process of generating cultures may impart an “injured” AMPAR phenotype.
Although early increases in dentate excitability after brain injury have been proposed to contribute to epileptogenesis (4–6), a convincing demonstration of a causal association has been lacking. We recently showed that suppressing post-traumatic increases in neurogenesis reduced dentate excitability and epileptogenesis in parallel, supporting the association between excitability and epileptogenesis (18). By identifying that early excitability, regardless of injury phenotype or inflammatory milieu, predicts epileptogenesis, our study supports the causal association between enhanced dentate excitability and remote development of epilepsy. Indeed, our experiments using two TLR4 antagonists, LPS-RSU, an extracellular blocker, and CLI-095, a blocker of the intracellular signaling domain (32), demonstrated the same paradoxical effects on excitability in control and brain-injured animals and eliminate the possibility of nonspecific effects. Moreover, CLI-095 was administered systemically while LPS-RSU was administered by focal injection, demonstrating that both systemic and focal TLR4 antagonism could enhance epileptogenesis in controls and reduce epileptogenesis after brain injury. Interestingly, a recent study found that blocking HMGB1 prior to pediatric brain injury in mice failed to reduce seizure susceptibility (65). It is possible that suppression of HMGB1 prior to injury rather than after injury, and recruitment of TLR4 signaling by alternative injury-induced DAMPs may have contributed to the inability of HMGB1 blockers to limit seizures. While early TLR4 antagonist treatment after injury limits seizure susceptibility, delayed treatment is ineffective in reducing seizure susceptibility (Fig. 6d–f), validating the critical role for early network excitability and a potential “therapeutic window” in post-traumatic epileptogenesis.
Traumatic brain injury is a major risk factor for epilepsy among young adults. Consistent with earlier studies (39, 66), 50–60% of rats develop spontaneous seizures 12–15 weeks after moderate FPI. The ability of TLR4 antagonists to reduce seizure susceptibility when administered 24 hours after brain injury presents an opportunity to target TLR4 to prevent post-traumatic epilepsy. Indeed, CLI-095 (Resatorvid) crosses the blood-brain-barrier, has a half-life of 6 hours (20), and is FDA approved for clinical trials (67). However, its proepileptogenic effect and potential for inducing neuronal loss in uninjured animals poses a major impediment to systemic use of TLR4 antagonists. Since we identify that TLR4 regulation of CP-AMPARs is unique to the injured neurons, elucidating the specific molecular pathways underlying TLR4 enhancement of AMPAR currents after injury would enable targeted suppression of the pathological mechanisms.
Supplementary Material
Acknowledgements
We thank Drs. Rizie Kumar and Amy Davidow for support with statistical analysis of flow cytometry data, Dr. Roman Shirakov for help with culture studies and Dr. Kelly Hamilton for helpful comments and discussions. This research was supported by CURE Foundation CF 259051, NJCBIR CBIR14RG024, NIH/NINDS R01 NS069861 and R01NS097750 to V.S. and NJCBIR CBIR15FEL011 to A.K.
Footnotes
Potential Conflicts of Interest
Nothing to report
References
- 1.Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia 2009;50 Suppl 2:4–9. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt D, and Sillanpaa M. Prevention of Epilepsy: Issues and Innovations. Curr Neurol Neurosci Rep 2016;16(11):95. [DOI] [PubMed] [Google Scholar]
- 3.Saletti PG, Ali I, Casillas-Espinosa PM, Semple BD, Lisgaras CP, Moshe SL, et al. In search of antiepileptogenic treatments for post-traumatic epilepsy. Neurobiol Dis 2019;123:86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neuberger EJ, Gupta A, Subramanian D, Korgaonkar AA, and Santhakumar V. Converging early responses to brain injury pave the road to epileptogenesis. J Neurosci Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt RF, Boychuk JA, and Smith BN. Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci 2013;7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein P, Dingledine R, Aronica E, Bernard C, Blumcke I, Boison D, et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018;59(1):37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vezzani A, French J, Bartfai T, and Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dedeurwaerdere S, Friedman A, Fabene PF, Mazarati A, Murashima YL, Vezzani A, et al. Finding a better drug for epilepsy: antiinflammatory targets. Epilepsia 2012;53(7):1113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West PJ, Saunders GW, Remigio GJ, Wilcox KS, and White HS. Antiseizure drugs differentially modulate theta-burst induced long-term potentiation in C57BL/6 mice. Epilepsia 2014;55(2):214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frigerio F, Flynn C, Han Y, Lyman K, Lugo JN, Ravizza T, et al. Neuroinflammation Alters Integrative Properties of Rat Hippocampal Pyramidal Cells. Mol Neurobiol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. NatMed 2010;16(4):413–9. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Korgaonkar AA, Swietek B, Wang J, Elgammal FS, Elkabes S, et al. Toll-like receptor 4 enhancement of non-NMDA synaptic currents increases dentate excitability after brain injury. Neurobiol Dis 2015;74:240–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balosso S, Liu J, Bianchi ME, and Vezzani A. Disulfide-Containing High Mobility Group Box-1 Promotes N-Methyl-d-Aspartate Receptor Function and Excitotoxicity by Activating Toll-Like Receptor 4-Dependent Signaling in Hippocampal Neurons. Antioxidants & redox signaling. 2014. [DOI] [PubMed] [Google Scholar]
- 14.Vezzani A, Lang B, and Aronica E. Immunity and Inflammation in Epilepsy. Cold Spring Harb Perspect Med 2015;6(2):a022699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, et al. Toll-like receptors modulate adult hippocampal neurogenesis. NatCell Biol 2007;9(9):1081–8. [DOI] [PubMed] [Google Scholar]
- 16.Okun E, Griffioen KJ, and Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci 2011;34(5):269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanke ML, and Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond). 2011;121(9):367–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neuberger EJ, Swietek B, Corrubia L, Prasanna A, and Santhakumar V. Enhanced Dentate Neurogenesis after Brain Injury Undermines Long-Term Neurogenic Potential and Promotes Seizure Susceptibility. Stem Cell Reports. 2017;9(3):972–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta A, Elgammal FS, Proddutur A, Shah S, and Santhakumar V. Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J Neurosci 2012;32(7):2523–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hua F, Tang H, Wang J, Prunty MC, Hua X, Sayeed I, et al. TAK-242, an antagonist for Toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab 2015;35(4):536–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garate I, Garcia-Bueno B, Madrigal JL, Caso JR, Alou L, Gomez-Lus ML, et al. Toll-like 4 receptor inhibitor TAK-242 decreases neuroinflammation in rat brain frontal cortex after stress. J Neuroinflammation 2014;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xing F, Zhang W, Wen J, Bai L, Gu H, Li Z, et al. TLR4/NF-kappaB signaling activation in plantar tissue and dorsal root ganglion involves in the development of postoperative pain. Mol Pain 2018;14:1744806918807050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karasawa J, Kotani M, Kambe D, and Chaki S. AMPA receptor mediates mGlu 2/3 receptor antagonist-induced dopamine release in the rat nucleus accumbens shell. Neurochem Int 2010;57(5):615–9. [DOI] [PubMed] [Google Scholar]
- 24.Yu J, Proddutur A, Swietek B, Elgammal FS, and Santhakumar V. Functional Reduction in Cannabinoid-Sensitive Heterotypic Inhibition of Dentate Basket Cells in Epilepsy: Impact on Network Rhythms. Cereb Cortex 2016;26(11):4229–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuberger EJ, Abdul-Wahab R, Jayakumar A, Pfister BJ, and Santhakumar V. Distinct effect of impact rise times on immediate and early neuropathology after brain injury in juvenile rats. Journal of Neuroscience Research. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilgenberg LG, and Smith MA. Preparation of dissociated mouse cortical neuron cultures. J Vis Exp 2007(10):562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fath T, Ke YD, Gunning P, Gotz J, and Ittner LM. Primary support cultures of hippocampal and substantia nigra neurons. Nat Protoc 2009;4(1):78–85. [DOI] [PubMed] [Google Scholar]
- 28.West MJ, Slomianka L, and Gundersen HJG. Unbiased Stereological Estimation of the Total Number of Neurons in the Subdivisions of the Rat Hippocampus Using the Optical Fractionator. Anatomical Record 1991;231(4):482–97. [DOI] [PubMed] [Google Scholar]
- 29.Echegoyen J, Armstrong C, Morgan RJ, and Soltesz I. Single application of a CB1 receptor antagonist rapidly following head injury prevents long-term hyperexcitability in a rat model. Epilepsy Res 2009;85(1):123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972;32(3):281–94. [DOI] [PubMed] [Google Scholar]
- 31.Wilson NM, Titus DJ, Oliva AA Jr., Furones C, and Atkins CM. Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus. Front Syst Neurosci 2016;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, and Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. European journal of pharmacology. 2008;584(1):40–8. [DOI] [PubMed] [Google Scholar]
- 33.Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z, et al. Granule cell hyperexcitability in the early post-traumatic rat dentate gyrus: the ‘irritable mossy cell’ hypothesis. J Physiol 2000;524 Pt 1:117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Lau L, Wei J, Zhu D, Zou S, Sun HS, et al. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 2004;43(1):43–55. [DOI] [PubMed] [Google Scholar]
- 35.Kamboj SK, Swanson GT, and Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol 1995;486 ( Pt 2):297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen JB, Schousboe A, and Pickering DS. Development of calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors in cultured neocortical neurons visualized by cobalt staining. J Neurosci Res 1998;54(2):273–81. [DOI] [PubMed] [Google Scholar]
- 37.Jensen JB, Lund TM, Timmermann DB, Schousboe A, and Pickering DS. Role of GluR2 expression in AMPA-induced toxicity in cultured murine cerebral cortical neurons. J Neurosci Res 2001;65(3):267–77. [DOI] [PubMed] [Google Scholar]
- 38.Kharatishvili I, and Pitkanen A. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res 2010;90(1–2):47–59. [DOI] [PubMed] [Google Scholar]
- 39.D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, and Miller JW. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127(Pt 2):304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nasr IW, Chun Y, and Kannan S. Neuroimmune responses in the developing brain following traumatic brain injury. Exp Neurol 2019;320:112957. [DOI] [PubMed] [Google Scholar]
- 41.Casillas-Espinosa PM, Andrade P, Santana-Gomez C, Paananen T, Smith G, Ali I, et al. Harmonization of the pipeline for seizure detection to phenotype post-traumatic epilepsy in a preclinical multicenter study on post-traumatic epileptogenesis. Epilepsy Res 2019;156:106131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reid AY, Bragin A, Giza CC, Staba RJ, and Engel J Jr. The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia 2016;57(10):1558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brady RD, Casillas-Espinosa PM, Agoston DV, Bertram EH, Kamnaksh A, Semple BD, et al. Modelling traumatic brain injury and posttraumatic epilepsy in rodents. Neurobiol Dis 2019;123:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garga N, and Lowenstein DH. Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr 2006;6(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao SS, Hua R, Zhao XP, Qin X, Sun ZQ, Zhang Y, et al. Exogenous administration of PACAP alleviates traumatic brain injury in rats through a mechanism involving the TLR4/MyD88/NF-kappaB pathway. Journal of neurotrauma 2012;29(10):1941–59. [DOI] [PubMed] [Google Scholar]
- 46.Dong XQ, Yu WH, Hu YY, Zhang ZY, and Huang M. Oxymatrine reduces neuronal cell apoptosis by inhibiting Toll-like receptor 4/nuclear factor kappa-B-dependent inflammatory responses in traumatic rat brain injury. Inflammation research : official journal of the European Histamine Research Society [et al]. 2011;60(6):533–9. [DOI] [PubMed] [Google Scholar]
- 47.Santhakumar V, Ratzliff AD, Jeng J, Toth K, and Soltesz I. Long-term hyperexcitability in the hippocampus after experimental head trauma. AnnNeurol 2001;50(6):708–17. [DOI] [PubMed] [Google Scholar]
- 48.Holmin S, Mathiesen T, Shetye J, and Biberfeld P. Intracerebral inflammatory response to experimental brain contusion. Acta Neurochir (Wien). 1995;132(1–3):110–9. [DOI] [PubMed] [Google Scholar]
- 49.Arai AC, Kessler M, Rogers G, and Lynch G. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol Pharmacol 2000;58(4):802–13. [DOI] [PubMed] [Google Scholar]
- 50.Baudry M, Kramar E, Xu X, Zadran H, Moreno S, Lynch G, et al. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol Dis 2012;47(2):210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terrone G, Salamone A, and Vezzani A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr Pharm Des 2017;23(37):5569–76. [DOI] [PubMed] [Google Scholar]
- 52.Pribiag H, and Stellwagen D. Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology. 2014;78:13–22. [DOI] [PubMed] [Google Scholar]
- 53.Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, and Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132(Pt 9):2478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu XJ, Xu J, Ma BY, Chen G, Gu PY, Wei D, et al. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chinese journal of traumatology = Zhonghua chuang shang za zhi / Chinese Medical Association. 2014;17(1):1–7. [PubMed] [Google Scholar]
- 55.Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, and Cuzzocrea S. Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS One. 2013;8(3):e57208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reis WL, Yi CX, Gao Y, Tschop MH, and Stern JE. Brain Innate Immunity Regulates Hypothalamic Arcuate Neuronal Activity and Feeding Behavior. Endocrinology. 2015:en20141849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pascual O, Ben Achour S, Rostaing P, Triller A, and Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 2012;109(4):E197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, and Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Annals of neurology. 2011;70(4):646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fourgeaud L, Davenport CM, Tyler CM, Cheng TT, Spencer MB, and Boulanger LM. MHC class I modulates NMDA receptor function and AMPA receptor trafficking. Proc Natl Acad Sci U S A. 2010;107(51):22278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raghupathi R, and Huh JW. Age-at-injury effects of microglial activation following traumatic brain injury: implications for treatment strategies. Neural Regen Res 2017;12(5):741–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu T, Gao YJ, and Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull 2012;28(2):131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Diogenes A, Ferraz CC, Akopian AN, Henry MA, and Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res 2011;90(6):759–64. [DOI] [PubMed] [Google Scholar]
- 64.Stellwagen D, Beattie EC, Seo JY, and Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 2005;25(12):3219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Webster KM, Shultz SR, Ozturk E, Dill LK, Sun M, Casillas-Espinosa P, et al. Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: Chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp Neurol 2019;320:112979. [DOI] [PubMed] [Google Scholar]
- 66.Kharatishvili I, Nissinen JP, McIntosh TK, and Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140(2):685–97. [DOI] [PubMed] [Google Scholar]
- 67.Wittebole X, Castanares-Zapatero D, and Laterre PF. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediators of inflammation. 2010;2010:568396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.