

Abstract
Kidney tubular dysfunction contributes to acute kidney injury and to the transition to chronic kidney disease. Although tubular mitochondria have been implicated in the pathophysiology of kidney failure, the mechanisms are not yet clear. Here, we demonstrated that ischemia-reperfusion injury induced acute translocation and activation of mitochondrial protein kinase B (also known as AKT1) in the kidney tubules. We hypothesized that mitochondrial AKT1 signaling protects against the development of acute kidney injury and subsequent chronic kidney disease. To test this prediction, we generated two novel kidney tubule-specific transgenic mouse strains with inducible expression of mitochondria-targeted dominant negative AKT1 or constitutively active AKT1, using a Cre-Lox strategy. Inhibition of mitochondrial AKT1 in mitochondria-targeted dominant negative AKT1 mice aggravated azotemia, tubular injuries, kidney fibrosis, glomerulosclerosis, and negatively impacted survival after ischemia-reperfusion injury. Conversely, enhancing tubular mitochondrial AKT1 signaling in mitochondria-targeted constitutively active AKT1 mice attenuated kidney injuries, protected kidney function, and significantly improved survival after ischemia-reperfusion injury (76.9% vs. 20.8%, respectively). Uncoupled mitochondrial respiration and increased oxidative stress was found in the kidney tubules when mitochondria AKT1 was inhibited, supporting the role of mitochondrial dysfunction in the pathophysiology of kidney failure. Thus, our studies suggest tubular mitochondrial AKT1 signaling could be a novel target to develop new strategies for better prevention and treatment of kidney injury.
Keywords: Mitochondrial Akt1, Ischemia Refusion Injury, Acute Kidney Injury, Chronic Kidney Disease
Graphical Abstract
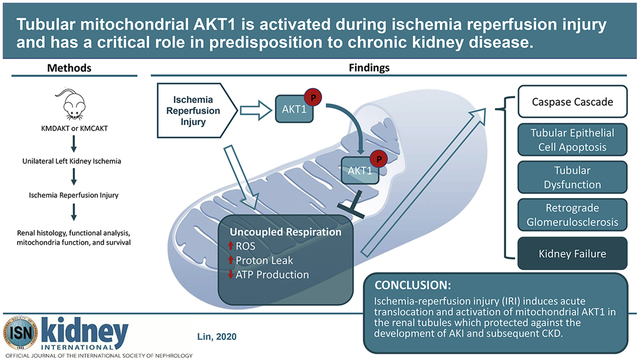
Translational Statement
Acute kidney injury (AKI) and subsequent development of chronic kidney disease (CKD) are significant health issues. We developed transgenic mouse models and defined the role of proximal renal tubule mitochondria AKT activation in AKI and CKD. This rescue mechanism of renal protection may represent a novel target to develop new strategies for better prevention and treatment of AKI and CKD. Future studies should confirm the involvement of mitochondrial AKT signaling in human kidney injury and explore druggable targets in the mitochondrial AKT1 pathway1.
Introduction
Acute kidney injury (AKI) and subsequent development of chronic kidney disease (CKD) are significant health issues. Currently, specific medical therapies for these disease states are limited, in part due to lack of understanding of the molecular mechanisms of renal damage. The kidney is one of the most energy-demanding organs in human body being necessary to facilitate active transport, maintain proper electrochemical gradient, and regulate fluid balance.2 Mitochondria are the main source of cellular ATP production through oxidative phosphorylation. Perturbation of ATP production and mitochondrial dysfunction can lead to increased reactive oxygen species (ROS) production which may, in turn, impair renal cell function and induce cell death. Although mitochondrial damage and dysfunction have been reported in humans with kidney disease and animal models of acute and chronic kidney injury,3,4 knowledge of the pathophysiological roles of mitochondria in kidney injury remains incomplete.
Enhancing mitochondrial function may protect against ischemia reperfusion injury (IRI).5,6 During the ischemic phase of IRI, depletion of oxygen inhibits electron transport in the mitochondrial respiratory chain and decreases production of ATP. The subsequent influx of sodium ions and efflux of protons helps restore normal pH and can promote cell survival by restoring intracellular volume.7 During reperfusion, restoration of blood flow is associated with increased reactive oxygen species (ROS) production, Ca2+ influx into mitochondria, loss of mitochondrial cross-membrane electrochemical gradient, and activation of apoptotic pathways.8 These intracellular changes could be detrimental to cells that survive the initial ischemic insult.
AKI is common among hospitalized patients and may precipitate onset of new CKD, worsen existing CKD, and lead to renal death.9 The mechanisms underlying transition of AKI to CKD are poorly understood. Recent animal studies suggest that renal tubular dysfunction plays an important role in the progression of AKI to CKD, reflecting the AKI to CKD transition in humans.10,11 Understanding the signaling pathways modulating kidney injury will allow us to identify new therapeutic targets that can mitigate renal damage and prevent progression of AKI to CKD.
The pathways linking cytosolic signaling to mitochondrial function during renal injuries are largely unknown. Mitochondrial function is intimately connected with cytosolic signaling, other organelles, and nuclear pathways.12 Our laboratory had identified activation and translocation of AKT1 into mitochondria as a key step in protecting cardiomyocytes against IRI by modulating oxidative phosphorylation complexes, improving oxidative phosphorylation efficiency, and reducing ROS.1,13 However, whether IRI can trigger translocation of activated AKT1 from cytosol to mitochondria in renal tubules is not yet known. If mitochondrial AKT1 can be activated in renal tubules upon kidney injury, it will be important to assess the pathophysiological role of mitochondrial AKT1 in the development of acute kidney injury (AKI) and chronic kidney disease (CKD). Herein, we have used two inducible renal-tubule specific transgenic mouse models to modulate mitochondrial AKT1 signaling in vivo and demonstrate a protective role for the renal tubular mitochondrial AKT1 activation during IRI against AKI and subsequent development of CKD.
Results
Activation of Mitochondrial AKT1 in Renal Tubules upon IRI
We first investigated whether IRI could activate AKT1 in renal mitochondria. To do so, we established a renal IRI model by ligation of the left renal artery for 30 minutes, followed by contralateral nephrectomy (Nx). Ligation of the left renal artery was reversed to end the ischemic phase for reperfusion and the left kidney was harvested at indicated time intervals.
The renal cortex, which is enriched with renal tubules, was isolated and the mitochondria were prepared by sub-fractionation. Upon reperfusion, the abundance of mitochondrial pAKT1 and AKT1 increased, suggesting translocation of activated AKT1 into mitochondria (Fig 1A). This is similar to what we had observed in cardiac muscle.13 Significant increase of mitochondrial AKT1 occurred 30–60 minutes after reperfusion (Fig 1A and B) as compared to the sham-operated control group or nephrectomized control (n=11–15). Mitochondrial pAKT1 was also increased. These results indicate IRI induced activation and translocation of AKT1 to mitochondria in kidney. Phosphorylation of AKT1 in renal tubules after IRI was confirmed with IHC staining (Fig 1C). There was minimal background pAKT1 staining in the renal tubule of the sham-operated control group, whereas pAKT1 was sharply increased in the renal tubules after IRI (Fig 1C and 1D). No pAKT1 was visible in other kidney regions than tubules. To confirm subcellular pAKT1 localization in renal tubules, pAKT1 was co-stained with a mitochondria marker. Upon IRI, distinct co-localization of p-AKT1 and mitochondria was observed in the renal tubular cells (Figure 1E). These results collectively indicated IRI translocation and activation of AKT1 to mitochondria.
Figure 1. Ischemia reperfusion induced acute translocation and activation of AKT1 in renal mitochondria.
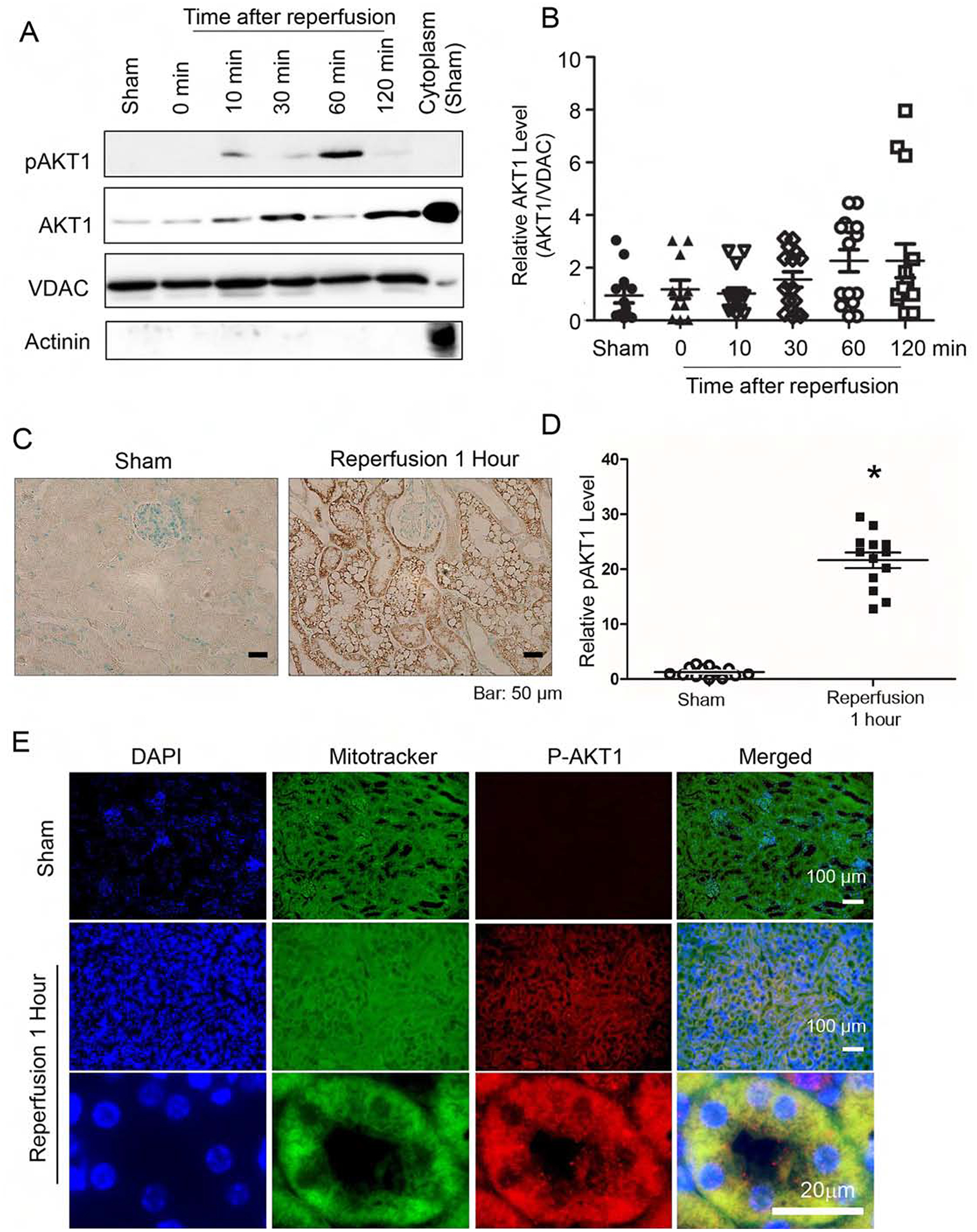
After ischemia, kidneys were harvested at various time points during reperfusion. (A) Increase in AKT1 and pAKT1 in mitochondria following reperfusion. Mitochondrial fractions were isolated and protein lysates were analyzed by western blotting with antibodies for total pAKT1, AKT1, mitochondria marker VDAC and cytosol marker actinin. (B) Mitochondrial AKT1 is increased following reperfusion graph (Kruskall-Wallis test, p<0.05). (C) Renal sections from the sham control or 1 hour after IRI was used to visualize AKT1 phosphorylation in tubules with immunohistochemistry staining. (D) Increased pAKT positive areas (%) 1 hour after IRI (p<0.001, n=13 in each group). (E) Co-localization of pAKT1 with renal tubule mitochondria 60 min post-ischemia. Renal sections were stained with pAKT1 (S473) antibodies and Mitotracker Green.
Generation of transgenic mice with inducible renal tubule-specific expression of a dominant negative AKT in mitochondria
To test the hypothesis that activation of tubular mitochondrial AKT1 during IRI protects the kidney against AKI and subsequent development of CKD, we generated a transgenic mouse model that enables Tamoxifen (TAM)-inducible renal tubule-specific expression of mitochondrial-targeted dominant negative AKT1 (Fig 2A). A dominant negative AKT1 (mdnAKT) and a mitochondria-targeting sequence was added to the N terminus. Tamoxifen-conditional expression of the AKT1 transgene was mediated using pCALNL (see Materials and Methods).14,15 To preclude confounding effects that can be associated with random integration of transgene arrays within the genome, A single copy of the transgene was introduced using targeted transgenesis at the Gt(ROSA)26Sor locus on chromosome 6 via homologous recombination in ES cells. Bigenic mice (KMDAKT) were obtained by crossing TAM-inducible mdnAKT mouse with hemizygous KSP-CreERT2 mice in which TAM-inducible Cre activity is driven by a renal tubular cell specific cadherin 16 promoter. The Cre activity was induced by TAM injections, and mitochondria were isolated and mitochondrial proteins were resolved with SDS-PAGE for immunoblotting. The results showed that TAM successfully induced expression of the mutant AKT1 (Fig 2B). The mutant protein could not be detected in the vehicle (corn oil) injected KMDAKT mice, KSP-CreERT2 mice or wildtype mice.
Figure 2. Bigenic mice (KMDAKT) for renal tubule-specific mitochondria-targeting of a dominant negative AKT1.
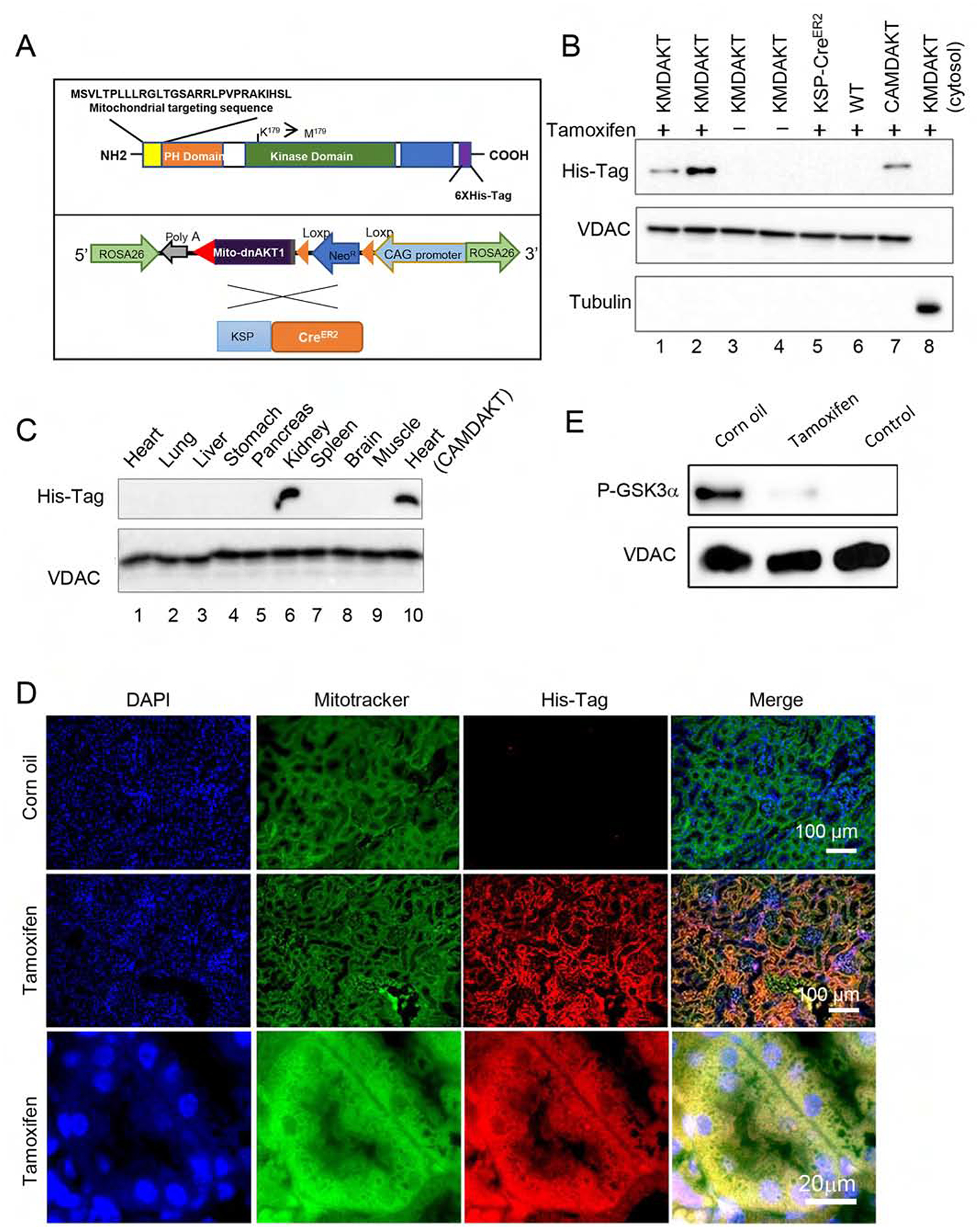
(A) Mitochondrial-targeting dominant negative AKT1 (mdnAKT) was engineered as described in Materials and Methods. A 6X His tag was added to the C terminus of AKT1 cDNA. mdnAKT transgenic mice were crossed with KSP-CreERT2 mice (Cre recombinase in renal tubular epithelial cells) to generate bi-genic mice (KMDAKT) for this series of experiments. (B) Expression of mdnAKT. Eight-week-old KMDAKT mice were injected with tamoxifen (TAM) or corn oil. The mitochondrial fraction was isolated and expression of mdnAKT proteins was analyzed by western blotting. The mdnAKT protein was expressed in the renal mitochondria isolated from Tam-treated KMDAKT mice but not in corn oil-treated KMDAKT mice, TAM-treated KSP-CreERT2 mice or TAM-treated wildtype (WT) mice. No mdnAKT was present in the renal cytosol fraction (last lane). VDAC was used as loading control and mitochondria marker, while tubulin is a cytosolic marker. (C) After TAM injection, the mitochondrial fraction was isolated from different organs and mutant AKT was only expressed in the kidney. The mdnAKT in the cardiac mitochondria of TAM-injected heart-specific mdnAKT mice (CAMDAKT) served as a positive control. (D) Mutant AKT1 co-localized to mitochondria in the renal tubules, immunofluorescence with anti-His-Tag antibody and MitoTracker® Green FM. (E) AKT activity in the mitochondrial protein preps were analyzed by an in vitro kinase assay using recombinant GSK3α as substrate. Tubular mitochondrial AKT was activated by IRI (corn oil injected), but AKT activity was inhibited in the Tam-treated KMDAKT mitochondria after IRI. The supernatant of KMDAKT mitochondria proteins after clearing immunoprecipitation with AKT antibodies served as negative control (lane 3).
To verify renal-specific expression, mitochondrial proteins were extracted from various tissues after TAM injection. His-tagged mutant AKT1 was expressed exclusively in the kidney (Fig 2C). Renal sections were stained with an anti-His-tag antibody and mitochondrial Mitotracker stain for immunofluorescence imaging (Fig 2D). The results showed all mutant AKT1 stained with His-tag antibody localized to mitochondria. No mutant AKT1 was detected in the glomerulus. Enzymatic activities of mitochondria AKT1 were analyzed with recombinant GSK3α to confirm the dominant negative effect in the mitochondria of TAM-treated KMDAKT tubules (Fig 2E). Since renal tubular mitochondrial AKT1 was activated by IRI, we compared the enzymatic activities of AKT1 in the mitochondria isolated from the TAM-treated and corn-oil-treated mice after IRI. The results showed renal mitochondrial AKT1 activities were significantly suppressed in the TAM-treated KMDAKT mice. Therefore this transgenic mouse line is a good model to study the role of tubular mitochondria AKT1.
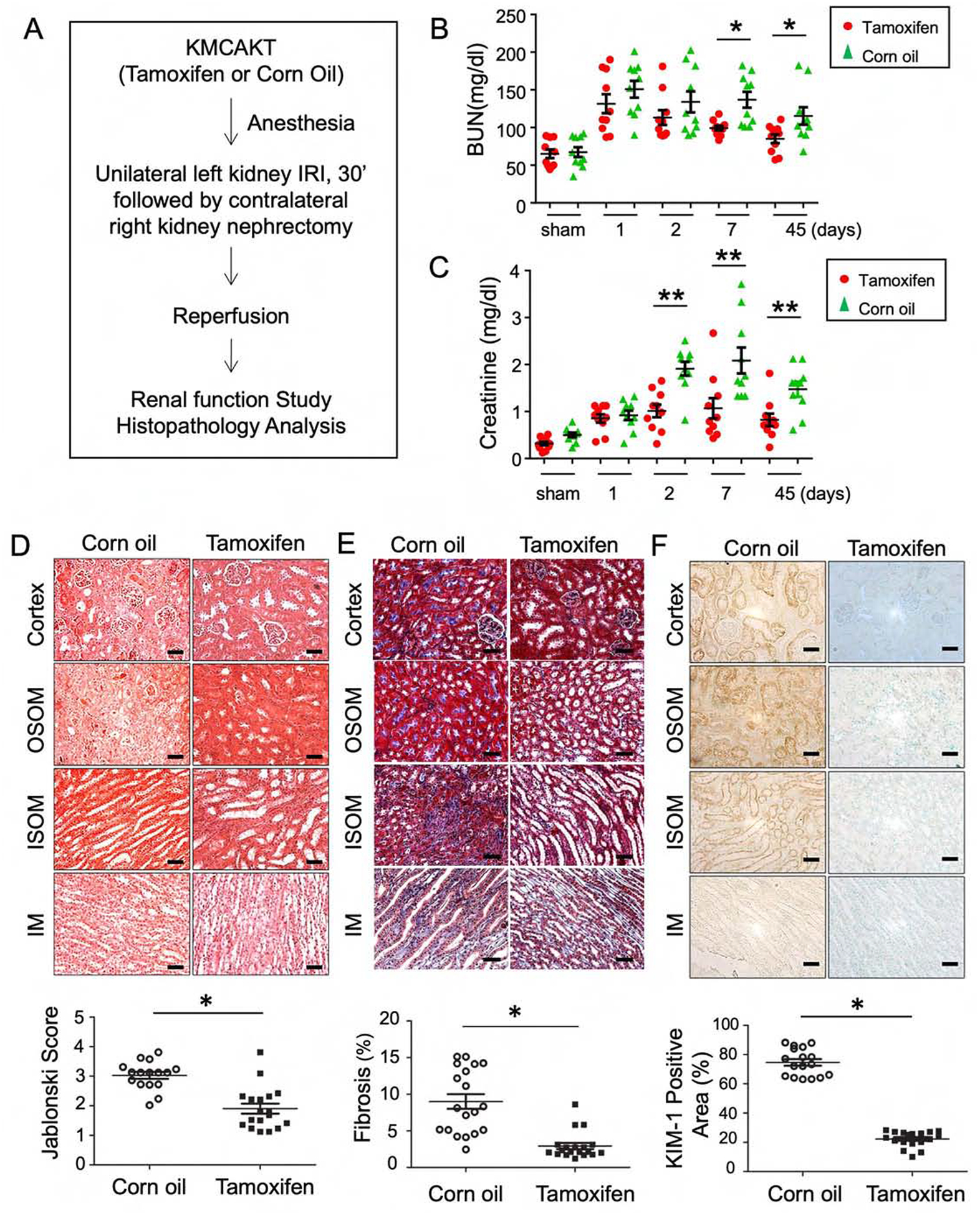
Inhibition of renal tubule mitochondrial AKT aggravated IRI in vivo
IRI was induced with unilateral renal ischemia as outlined (Fig 3A). In the last series of experiments, phosphorylated AKT1 was nearly undetectable in the mitochondria of proximal renal tubules (Fig 1). Renal histology, blood urea nitrogen (BUN), and serum Creatinine (Cr) were indistinguishable in the corn oil-injected KMDAKT mice and the TAM-injected KMDAKT mice (Supplemental Fig 1), suggesting that mitochondrial AKT1 signaling had no overt role in the kidney structure and function at baseline before induction of IRI. To investigate whether mitochondrial AKT1 played a protective role during IRI, AKI was induced with unilateral IRI in the corn oil- or TAM-KMDAKT mice. There was no difference in BUN or Cr levels obtained immediately upon induction of IRI, however, both BUN and Cr were significantly higher 45 days later in the TAM-KMDAKT mice (Fig 3B and 3C). These results indicated that blocking activation of mitochondrial AKT1 in renal tubules during IRI aggravated later development of renal dysfunction. Histological analysis of H&E stained renal sections revealed aggravated renal injuries in the TAM-KMDAKT mice (Fig 3D). Jablonski scores of renal tubular injury were higher with more tubular brush border loss, tubular lysis, and debris in tubular lumen space (p=0.018). To exclude effect of TAM or Cre recombinase, TAM-injected KSP/CreERT2 mice were also analyzed, Jablonski scores were similar in the corn oil-KMDAKT and the TAM-KSP/CreERT2 mice on day 7 after IRI (Fig 3D) (p=0.7613). These data indicate that our findings were not confounded by the effect of TAM or Cre recombinase. Masson’s trichrome stain showed higher fibrosis area (%) in the TAM-KMDAKT mice after IRI (p<0.001), accompanied by disorganized tubule structure with intraluminal casts and debris (Fig 3E). The expression of Col1a was increased, corroborating the results Masson’s trichrome stain (Fig 3F). TGFβ expression was not increased, which suggested that mitochondrial AKT1 did not modulate inflammatory response (Fig 3F). Renal tubule injury marker-KIM-1 expression was increased in TAM-KMDAKT kidney after IRI, which corroborated exacerbation of kidney injury in the TAM-KMDAKT group (p=0.002) (Fig 3G).
Figure 3. Inhibition of tubule mitochondria AKT aggravated kidney failure induced by ischemia reperfusion.
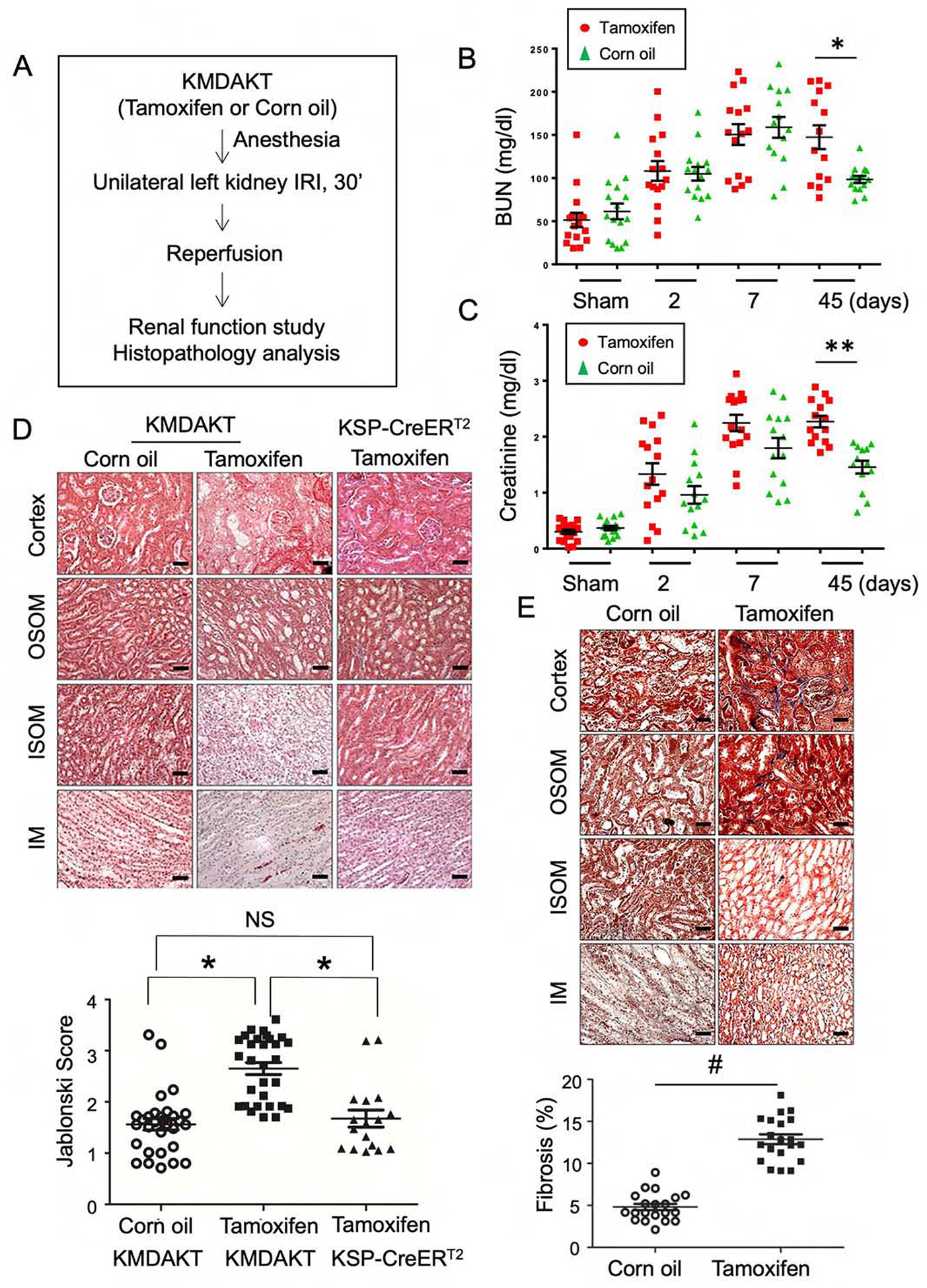
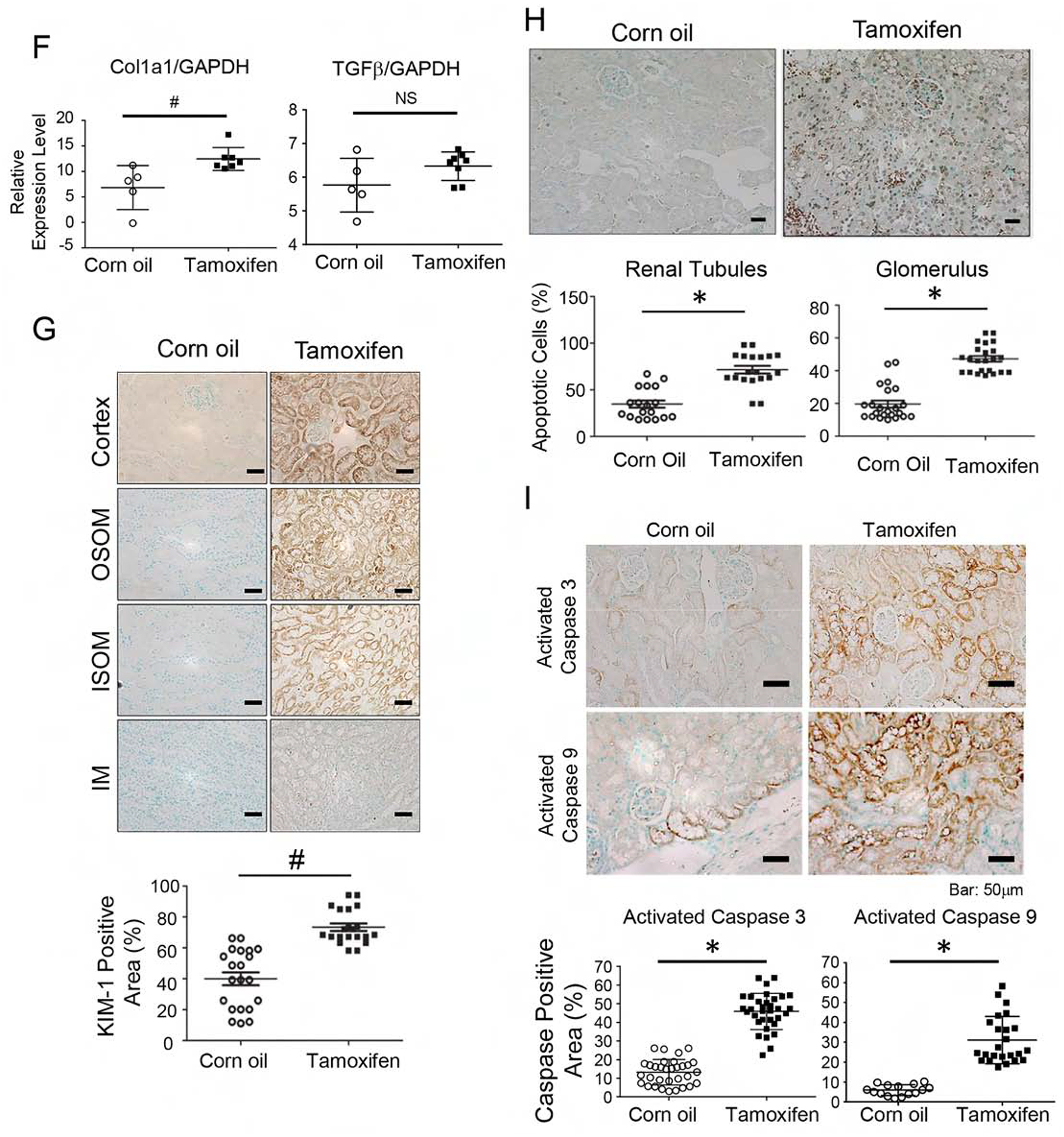
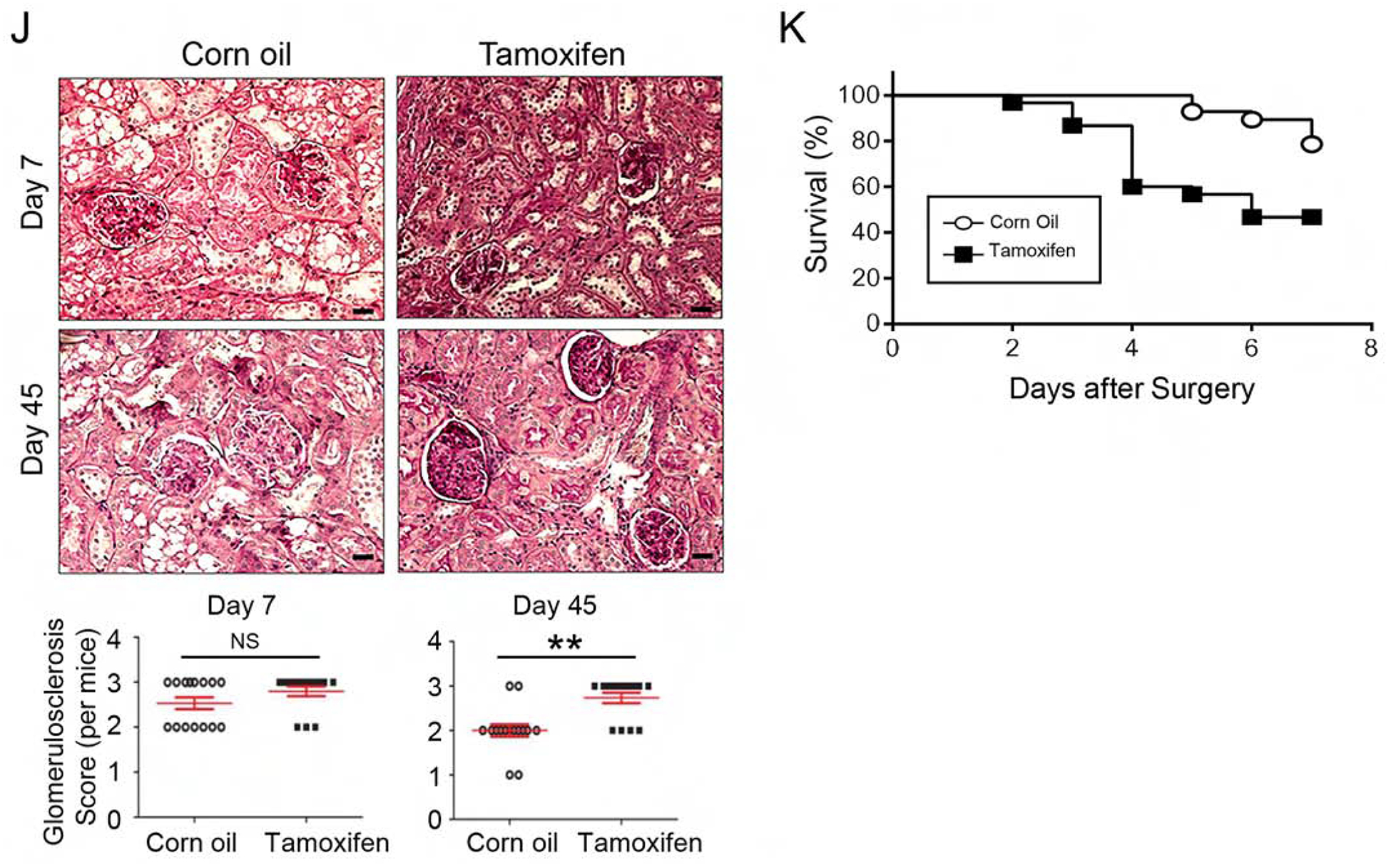
(A) The experimental protocol of ischemia reperfusion injury for this series of study. (B, C) Serial measurements showed rise of BUN and Creatinine (Cr) after IRI. Significantly higher BUN and Cr was observed in the male Tam-treated KMDAKT mice on day 45 (*p<0.002, **p<0.001). (D) Renal tissue histology (7 days after IRI) stained with HE to estimate Jablonski scores. OSOM - outer stripe outer medulla. ISOM - inner stripe outer medulla. IM - inner medulla. Tamoxifen injected KSP-CreERT2 mice served as another control for Tamoxifen-KMDAKT mice. The Jablonski scores were summarized in the bar graph. *p < 0.01. (E) Renal fibrosis was analyzed by Masson’s trichrome staining (7 days after IRI). The bar graph compared the extent of renal fibrosis in the two groups. #p < 0.02. (F) Expression of TGFβ and Col1A in kidney (7 days after IRI). The levels of TGFβ and Col1A mRNA were analyzed and expressed as ratios to GAPDH mRNA. # p < 0.01. (G) KIM-1 expression was examined by immunohistochemistry (7 days after IRI). The bar graph summarized the results of KIM-1 staining in these mice. #p < 0.02. (H) Renal apoptosis was analyzed by TUNEL staining (7 days after IRI). Upper panels show representative microscopic images, while lower panels summarized results of quantification of apoptotic cells from the two groups of mice. (I) Activation of caspase 3 and 9 in kidneys (7 days after IRI). Activated caspase 3 and 9 were analyzed by immunohistochemistry. The bar graph summarized the results of staining in these mice. #p < 0.01. (J) Glomerulosclerosis was analyzed by Periodic acid–Schiff (PAS) staining. The PAS results were graded as follows: 0 (no lesions), 1 (lesions in up to 25% of glomeruli), 2 (lesions in 25 to 50% of glomeruli) or 3 (lesions in > 50% of glomeruli). The representative images were shown in the upper panel. At least 50 glomeruli were scored for each mouse, the results were summarized in the graph (lower panel). **p<0.001. NS - not significant. (K) Survival analysis after IRI was compared in the two groups (TAM n=30, Corn oil n=28, all male). 46.7% TAM-treated KMDAKT mice survived by day 45, as compared to 85.7% control mice. Log rank test, p=0.0013.
TUNEL staining was used to evaluate apoptosis of renal cells (Fig 3H). The number of apoptotic cells was significantly increased in the renal tubules (p=0.0054) and in glomeruli (p<0.001) of TAM-KMDAKT mice compared to the controls. This suggests that inhibiting the tubular mitochondrial AKT1 during IRI could aggravate tubular cell death and escalate glomerular injury. Consistent with this, activation of both caspase 9 (initiator) and caspase 3 (executioner) was substantially increased in the tubular epithelial cells of TAM-KMDAKT kidneys (Fig 3I). To further explore chronic glomerular changes, renal sections were analyzed with Periodic acid-Schiff (PAS) stain to quantitate glomerulosclerosis (Fig 3J). Although no difference in glomerulosclerosis was seen 7 days after IRI (p=0.2634), more glomerulosclerosis was found 45 days after IRI in the TAM-KMDAKT mice (p<0.001). This finding suggests that the renal tubular mitochondria AKT signaling could have modulated glomerular scarring during the development of CKD. Kaplan-Meier survival analysis demonstrated the impact of inhibiting tubular mitochondria AKT on the outcome of IRI (Fig 3K). Compared to the controls, TAM-KMDAKT mice showed reduced survival after IRI (p=0.0013).
Survival of TAM-KMDAKT mice was significantly lower than the controls within 7 days post IRI, but BUN/Cr levels were similar in the TAM- and corn oil-KMDAKT mice during this time period (Fig 3C, 3D and 3K). As BUN/Cr data could not be obtained from the deceased mice, the reported changes of BUN/Cr may have been underestimated in TAM-treated mice. Furthermore, the higher death rate in TAM-KMDAKT mice may have been caused by distant organ dysfunction (lung, heart, brain) beyond changes of BUN/Cr.16
The effects of tubular mitochondrial AKT1 were also evaluated in female KMDAKT mice. Serum BUN/Cr and renal histology were comparably higher in the TAM-KMDAKT female mice 45 days post IRI (Supplementary Fig 2). No obvious gender effect was observed.
Augmentation of renal tubule mitochondrial AKT1 protected against AKI and subsequent CKD
Because inhibiting AKT1 in renal epithelial cells aggravated post-IRI kidney function and animal survival, we next investigated whether enhancing tubular mitochondrial AKT1 signaling could protect the kidney against IRI. To do so, we generated a bigenic mouse line with TAM-inducible tubular cell-specific expression of a mitochondrial-targeted constitutively active AKT1 (mcaAKT) (KMCAKT mice) via the same strategy we developed for KMDAKT mice (Supplemental Fig 3A). Western blots showed kidney-specific expression of constitutively active AKT1 in the mitochondria from TAM-KMCAKT mice, but not the control animals (Supplemental Fig 3B), and was kidney specific (Supplemental Fig 3C). As before, to verify that mutant AKT1 was localized to the mitochondria in tubules, immunofluorescence staining showed co-localization of mutant AKT1 and mitochondria (Supplemental Fig 3D). No mutant AKT1 was detected outside of tubules. To evaluate the activity of mcaAKT, we compared AKT1 enzymatic activities in the mitochondria isolated from the TAM- and corn-oil-treated mice. As expected, renal mitochondrial AKT1 activity were significantly increased in the TAM-KMCAKT mice (Supplemental Fig 3E).
To study whether enhancing tubular mitochondria AKT1 could improve the outcome of IRI, AKI was induced by unilateral IRI with concurrent contralateral Nx in the TAM-KMCAKT and corn-oil-KMCAKT mice (Fig 4A). Corn oil-KMCAKT mice developed higher serum BUN on day 7 and day 45 after IRI compared to TAM-KMCAKT mice (p=0.03) (Fig 4B). Cr was significantly lower in the TAM-KMCAKT mice on days 2, 7, and 45 after IRI (Fig 4C). These results indicated that enhancing mitochondrial AKT in renal tubules attenuated IRI-induced AKI and ameliorated deterioration of renal function and subsequent development of CKD. Histology from these mice showed more severe renal injuries in the corn oil-injected mice (Fig 4D). Jablonski scores for renal tubular injury were higher with more tubular brush border loss, tubular lysis, and debris in tubular lumen than the TAM-KMCAKT kidney (p=0.038). Masson’s trichrome stain showed reduced fibrosis area (%) in the TAM-KMCAKT mice after IRI (p<0.001), (Fig 4E). TAM-KMCAKT kidneys showed less tubular injury and lower KIM-1 expression (Fig 4F). TUNEL assay showed more apoptotic cells in the corn oil-KMCAKT kidney after IRI (Fig 4G). Apoptosis was reduced by 52% in tubules and by 46% in glomeruli in the TAM-KMCAKT kidney (p=0.0064 and p=0.0021). Activation of Caspase 3 and 9 was accordingly suppressed by activation of mitochondrial AKT1 in the tubules (Fig 4H). The severity of glomerulosclerosis was assessed by PAS staining and was reduced by 38% 45 days after IRI in the TAM-KMCAKT mice compared to the corn oil-KMCAKT mice (p=0.001) (Fig 4I). Therefore, enhancing tubular mitochondrial AKT1 protected the kidney against IRI-induced tubular cell apoptosis, prevented accumulation of debris in the renal tubules, and attenuated subsequent glomerular damages that followed tubular injury.
Figure 4. Augmentation of mitochondria AKT in tubules reversed kidney failure induced by ischemia reperfusion.
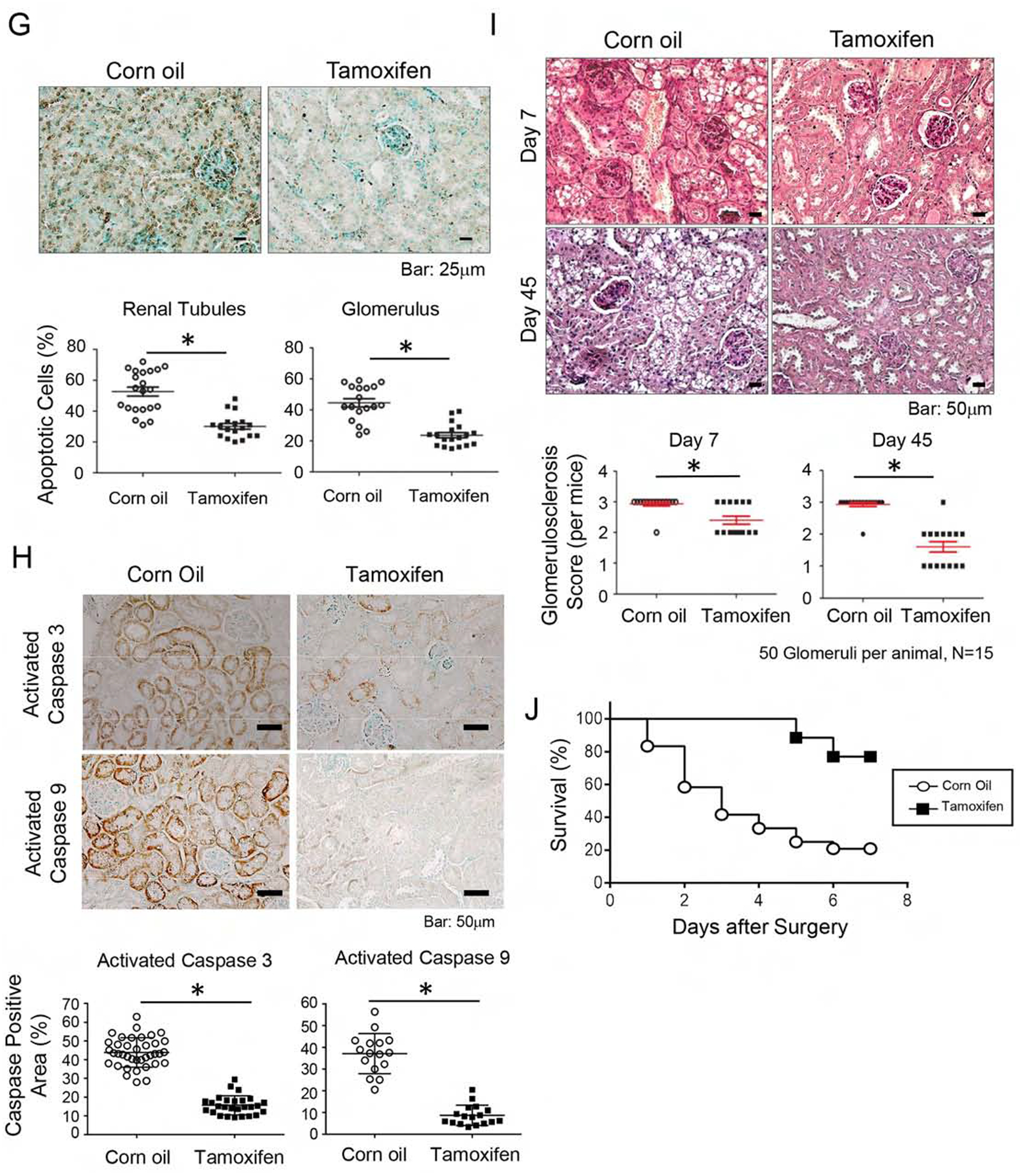
(A) The experimental protocol of ischemia reperfusion injury for this series of study. (B, C) Acute kidney injury was induced by IR in male KMCAKT mice. BUN and Cr were measured after IRI in the TAM-treated KMCAKT and corn oil-treated KMCAKT mice. Tam-KMCAKT mice showed lower BUN and Cr after day 2 (*p<0.032, **p<0.001). (D) Renal histology (7 days after IRI) was HE stained to estimate Jablonski scores. OSOM - outer stripe outer medulla. ISOM - inner stripe outer medulla. IM - inner medulla. Jablonski scores are summarized in the bar graph (*p ≤ 0.001). (E) Renal fibrosis was analyzed by Masson’s trichrome staining (7 days after IRI). The bar graph compared the extent of renal fibrosis in the two groups (p=0.002). (F) KIM-1 expression immunohistochemistry (7 days after IRI). The bar graph summarized the results of KIM-1 staining in these mice (*p<0.002). (G) Renal apoptosis was analyzed by TUNEL staining (7 days after IRI). The upper panel showed representative microscopic images, the lower panel summarized quantification of apoptotic nuclei from the two groups of mice (*p<0.003). (H) Activation of caspase 3 and 9 in kidneys (7 days after IRI). Activated caspase 3 and 9 were analyzed by immunohistochemistry. The bar graph summarized the results of staining in these mice. *p < 0.001. (I) Glomerulosclerosis was analyzed by Periodic acid-Schiff (PAS) staining. The PAS results were graded as follows: 0 (no lesions), 1 (lesions in up to 25% of glomeruli), 2 (lesions in 25 to 50% of glomeruli) or 3 (lesions in > 50% of glomeruli). The representative images were shown in the upper panel. 50 glomeruli were scored for each mouse (15 mice in each group), the results were summarized in the graphs (lower panel, *p<0.003). (J) Survival analysis after IRI was compared in the two groups (TAM n=26, Corn oil n=24) (H). 76.9% of TAM-treated KMCAKT mice survived by day 7, as compared to 20.83% in control mice. Log rank test, p<0.0001.
Kaplan-Meier survival analysis was used to evaluate whether the above histological and functional changes in kidney can be extrapolated into survival (Fig 4J). Indeed, TAM-KMCAKT mice had much better survival than the corn-oil KMCAKT mice after IRI (76.9% vs. 20.8%, p<0.001). This suggests that manipulating tubular mitochondria AKT1 signaling can be considered as a potential target to develop new treatments to improve outcomes.
Mitochondrial AKT1 modulated mitochondria respiration and ATP production in renal tubules
To investigate the direct effect of mitochondrial AKT1 in renal tubule epithelial cells, we isolated primary renal tubular epithelial (RTE) cells from the KMDAKT mice. Analysis of the cell preparations with antibodies to aquaporin1, a proximal tubule epithelial cell marker, showed 97% of the RTE cells expressed this marker (by direct cell count under microscope) (Fig 5A). Seventy-two hours after 4-OH TAM or vehicle treatment, RTE cells were stained with Mitotracker Red before immunostaining with anti-His-Tag antibodies (Green). As shown in Fig 5B, there was no His-Tag signal in the vehicle treated cells, consistent with the expected absence of mdnAKT expression. In contrast, His-Tag was detected in the 4-OH TAM treated cells and was co-localized with Mitotracker Red.
Figure 5. Inhibition of mitochondria AKT uncoupled mitochondria respiration and decreased cellular ATP in renal tubular epithelial cells.
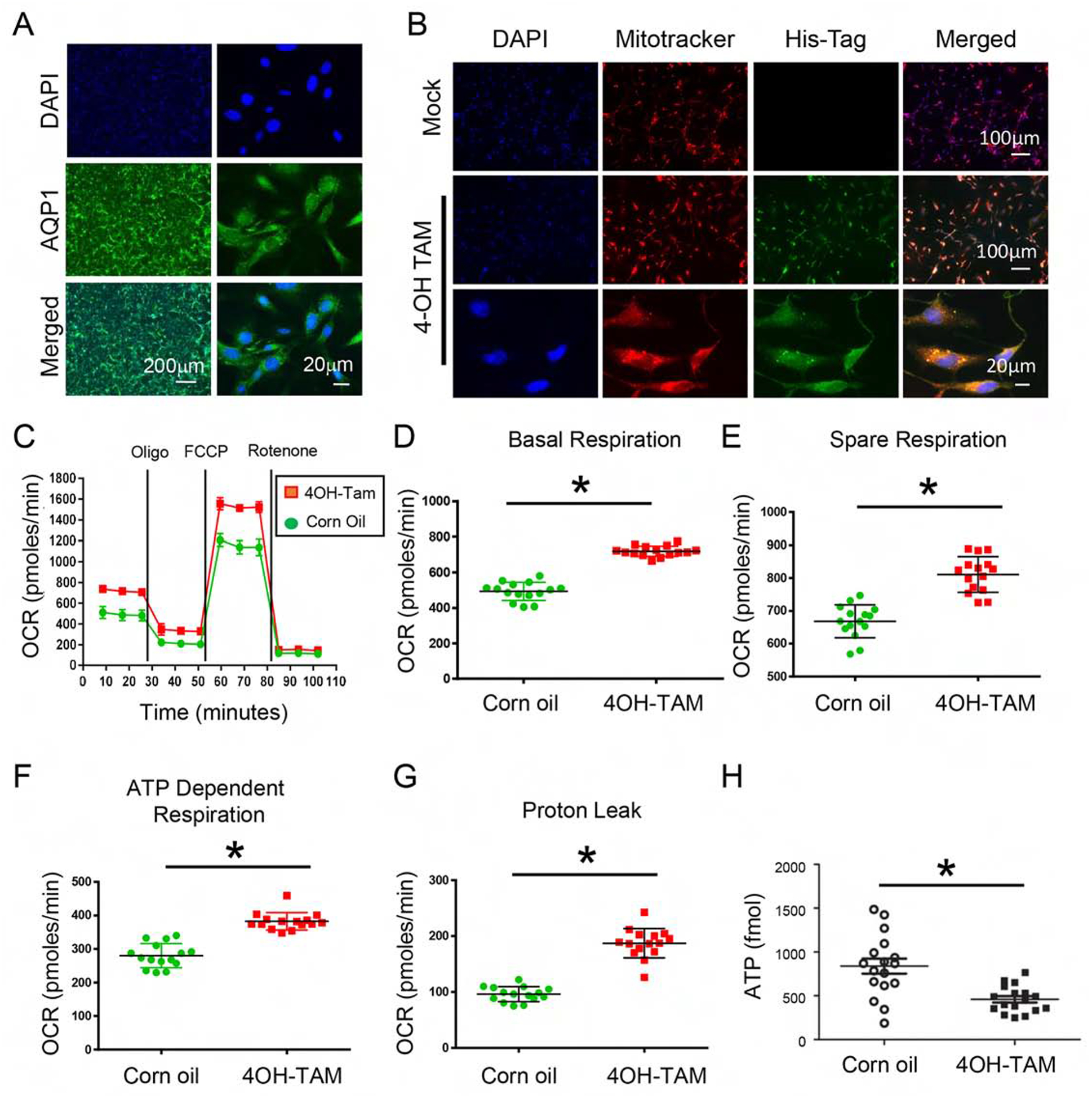
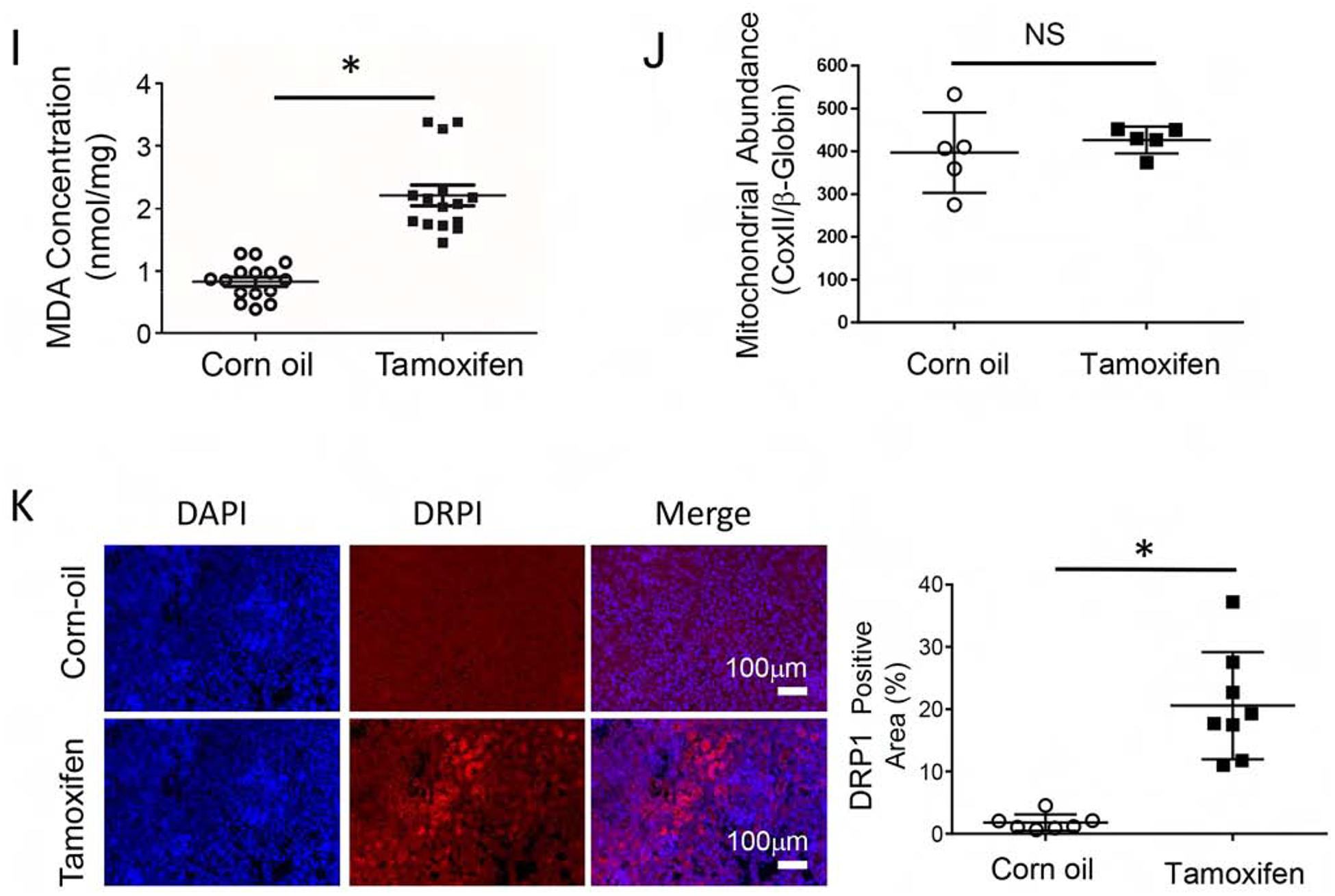
(A) Primary renal tubule epithelial cells (RTE) were isolated from 3-week-old KMDAKT mice and enrichment was quantified using the renal epithelial cell marker AQP1. (B) Primary RTE cells were treated with DMSO or 10ng/ml of tamoxifen (4-OH TAM) for transgene induction. (C) RTE cells were plated for Seahorse XF Analyzer. After basal extracellular respiration rates (OCR) analysis, different inhibitors were injected sequentially to measure different stages of respiration (complex V inhibitor: oligomycin, uncoupler: carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone, and complex I inhibitor: Rotenone) as shown in D–G. The basal respiration, (p<0.001), spare respiration (p=0.00076), ATP dependent respiration (p<0.001) and proton leak (p<0.001) were all significantly higher in the 4-OH TAM treated cells than DMSO treated cells. (H) Sixty hours after 4-OH TAM or DMSO treatment, ATP levels were reduced in KMDAKT cells expressing dnAKT1 (*p<0.04). (I) Lipid peroxidation in KMDAKT kidneys. Lipid peroxidation was measured and the results showed higher lipid peroxidation in the Tam-KMDAKT kidneys. *p < 0.01. (J) Abundance of mitochondria in KMDAKT kidneys. The content of mitochondria DNA was analyzed by real-time PCR and presented as a ratio of mitochondria DNA to nuclear DNA. NS: p value not significant. (K) DRP1 staining in KMDAKT kidneys. Mitochondria fission marker DRP1 was analyzed with immunofluorescence. The bar graph summarized the results from each mice. *p< 0.001.
To analyze how mitochondrial AKT1 signaling modulated RTE cellular bioenergetics, we used a Seahorse extracellular analyzer to assess mitochondrial function. The OCR represents measurements of oxidative respiration (Fig 5C). Overall basal respiration of the 4-OH TAM treated KMDAKT RTE cells was significantly higher than the control (p<0.001) (Fig 5D). The spare respiration profile of the 4-OH TAM incubated KMDAKT RTE cells was also higher than the control (p=0.00076) (Fig 5E). The ATP dependent respiration profile of the 4-OH TAM incubated KMDAKT RTE cells was significantly higher than control (p<0.001) (Fig 5F). The higher proton leak in the 4-OH TAM treated KMDAKT RTE cells indicated uncoupled respiration (p<0.001) (Fig 5G). ATP production was reduced in the RTE cells derived from KMDAKT after 4-OH TAM induction (Fig 5H). Lipid peroxidation was significantly increased in the TAM-induced cells, indicating increased ROS (Fig 5I). The changes of oxidative phosphorylation were not caused by a change in the content of mitochondria in these cells (Fig 5J). However, in the TAM-KMDAKT kidneys we observed higher level of Drp1, a marker for mitochondria fission (Fig 5K), which suggested modulation of mitochondria dynamics. These findings suggest inhibition of mitochondrial AKT1 signaling caused mitochondrial dysfunction with uncoupled respiration, which, combined with reduced ATP production, aggravated renal apoptosis in the KMDAKT kidney under IRI.
We also investigated how activation of mitochondrial AKT1 modulated oxidative phosphorylation in the RTE cells isolated from KMCAKT (Supplemental Fig 4). The results showed opposite effect as compared to the KMDAKT RTE cells. Basal respiration, spare respiration, ATP-dependent respiration, proton leak, and glycolytic potential were lower in the 4OH-TAM incubated cells (Supplemental Fig 4B–4F). These findings indicate the effect of mitochondrial AKT1 activation on improving the efficiency of oxidative phosphorylation and reducing respiration uncoupling. Drp1 analysis showed lower Drp1 staining in the TAM-KMCAKT kidneys (Supplemental Fig 4G), further corroborated the higher Drp1 in the TAM-KMDAKT kidneys and confirmed that mitochondrial AKT1 is involved in the regulation of mitochondria dynamics.
Discussion
This study shows that IRI induced acute activation and translocation of AKT1 in mitochondria in proximal tubular epithelial cells. Activation of mitochondrial AKT1 in renal proximal tubules in response to IRI appears to represent a self-protective mechanism during ischemia reperfusion. This is because inhibition of tubular mitochondria AKT1 aggravated AKI and promoted subsequent development of CKD after IRI. Conversely, enhancing tubular mitochondrial AKT1 signaling protected the kidney against IRI and attenuated development of CKD. These data identify a novel role for tubular mitochondrial AKT1 signaling during evolving kidney failure after IRI.
The Role of Mitochondria in Renal Tubule AKI
The majority of renal mitochondria reside in the proximal tubules. Mitochondria serve as the major cellular organelle to metabolize nutrients for ATP production. Mitochondrial AKT1 regulates cellular oxidative phosphorylation, ROS production, and cell survival.17 As demonstrated in this study, impaired mitochondrial AKT1 signaling produced uncoupled respiration and lowered ATP production. Although renal tubular apoptosis is a common finding in various models of AKI, the signaling pathways upstream from mitochondria remain incompletely understood.18 Renal IRI leads to induction of apoptosis genes and activation of caspases and endonucleases, which contribute to induction of apoptosis.19 Injury and death of tubular cells had been recognized as key factors in the development of AKI. Furthermore, tubule repair and regeneration has been proposed as a major event in the recovery from AKI.20–23 Although sub-lethal injury may be reversible, the death of tubular cells leads to inevitable loss of tubular function.18 Activation of the intrinsic apoptotic pathways in AKI have been characterized in both in vitro and in vivo models, including Bcl-2 protein family and mitochondrial apoptosis machinery.24–27 Loss of mitochondrial cross-membrane electrochemical gradient is considered a pivotal control point that triggers apoptosis.28 Mitochondrial proton leakage, which is modulated by mitochondrial AKT1, combined with reduced ATP synthesis in this energy-demanding organ are likely to contribute to an overall balance in signaling that favors apoptosis.29–31
The Crosstalk between Renal Tubules and Glomerulus
Anatomically, tubulointerstitial injury could cause stenosis of the glomerulo-tubular junction and finally result in atubular glomeruli, i.e. glomeruli without patent connection to the renal tubule.32 Tubular epithelial cell dysfunction, compression and obstruction of adjacent tubules by interstitial matrix, and transition of parietal epithelial cells to fibroblast-like cells are potential mechanisms of atubular glomerulus formation. Proximal RTE, especially at S1 region, are sensitive to ischemic injury.32 In response to renal injury, RTE may undergo incomplete repair, resulting in tubular atrophy and interstitial fibrosis.32
Tubular injury may affect glomerular filtration function through tubulo-glomerular feedback. Tubulo-glomerular feedback is a physiologic cross talk mechanism between tubules and glomeruli.33–35 Little is known about the sequence of events following AKI and the relationship between glomerular filtration rate and tubular alterations. In a clinical study, in patients with severe renal artery stenosis, there were widespread formation of atubular glomeruli,36 suggesting that chronic ischemia might have induced tubular damage and disintegration of renal tubular structure. On the other hand, there is ample evidence indicating that renal tubular dysfunction could lead to loss of tubular cell polarity, loss of gap junction, tubular cell death, subsequent tubular obstruction, and retrograde glomerular damage.37–41
The results of our study provide novel insight into how mitochondria participated in the defense against IRI-induced kidney injury. Inhibiting mitochondrial AKT1 during IRI led to activation of caspases and tubular cell death, which might have triggered retrograde glomerular apoptosis, glomerulosclerosis, and renal fibrosis. Activation of mitochondrial AKT1 likely played an important role during the transition from AKI to CKD, as inhibiting mitochondrial AKT1 during IRI significantly affected BUN/Cr 45 days after the initial IRI.
Transition from AKI to CKD
The exact mechanisms underlying transition of AKI to CKD remain to be defined. Histologically, both AKI and CKD are associated with renal tubule injuries.42 It has been known for years that tubulointerstitial pathology is feature of many types of CKD.43 Perturbation of intracellular signaling in renal tubular cells is believed to play a role during the initiation and progression of AKI to CKD.
In CKD, the transforming growth factor (TGF)-β signaling pathway is activated and contributes to glomerulosclerosis and tubulointerstitial fibrosis by inducing production of profibrotic extracellular matrix proteins.44 However, the role of TGF-β signaling in AKI is inconsistent in different experimental models.45 Our data did not show increased TGF-β expression (Fig 3F). Hypoxia activates hypoxia-inducible factors (HIF) to modulate gene transcription.46 In AKI, global renal expression of HIF exerted renal protective effects in various experimental systems.47 But other studies have failed to confirm the renal protective actions of HIF-1.48
Current evidence suggests the mitochondrion could be a key modulator during the transition of AKI to CKD. Ablation of the proapoptotic BAK and BAX resulted in amelioration of ischemic and cisplatin-induced AKI.49 In ischemic and nephrotoxic models of AKI, inhibition of mitochondrial fragmentation has been shown to attenuate kidney injuries.50 The severity of ischemic AKI in diabetic mice was associated with heightened activation of the mitochondrial apoptosis.51 CKD-associated suppression of mitochondrial function and biogenesis could sensitize kidney cells and tissues to AKI and prevent recovery from AKI.52–54 In CKD, renal mitochondrial dysfunction also occurred during the development and progression of CKD.55 High glucose and albumin overload can induce mitochondria apoptosis signaling in kidney cells in CKD.56 Mitochondrial fragmentation, as a result of pathologic alterations in mitochondrial fusion and fission, was detected in experimental CKD induced by diabetes.57
Identification of the tubulointerstitial histopathology in the pathogenesis of CKD has shifted the glomerulocentric paradigm of kidney injury to the new attention on the pathophysiological role of proximal tubule in AKI and its transition to CKD37–41,58. Our results suggest that mitochondrial AKT1 indirectly protects against the development of glomerulosclerosis, possibly by maintaining the integrity of renal tubular structure and function. The data presented this study help fill an important knowledge gap concerning the role of proximal renal tubule mitochondria in AKI and its subsequent transition to CKD, via activation and translocation of AKT1 into mitochondria. Future studies should be carried out to confirm the role of mitochondrial AKT1 in human kidney injury and explore potential targets in the mitochondrial AKT1 signaling pathway that can be used to improve the outcomes of kidney failure.
Methods
Experimental Animal Model
Constructs to express mitochondrial-targeted dominant negative or constitutively active AKT were generated using strategies similar to those described previously.13,17 inducible overexpression of the mitochondria-targeted dominant-negative mdnAKT or constitutive active mcaAKT in the renal tubule epithelial cells was obtained by crossing ROSA26-CAG-LNL-mdnAKT or ROSA26-CAG-LNL-mcaAKT mice with a well-studied Cre transgenic mouse strain, KSP-CreERT2 mice59. The details of the transgenic mice engineering were presented in the supplementary materials.
Ischemia Reperfusion
Eight-week-old C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) and transgenic mice were used to study the effect of ischemia reperfusion injury. The mice were anesthetized by injecting 2.5% Avertin (Tribromoethanol), 0.01 ml per gram of body weight. Unilateral renal ischemia was induced by ligating the left renal pedicles for 30 minutes under anesthesia and, when indicated, concurrent contralateral nephrectomy. Reperfusion was induced by releasing the ligation. The mice were euthanized at indicated time intervals and kidneys were harvested for further analysis.
Statistics
Data are presented as mean ± SD, unless noted otherwise. Statistical data were analyzed with GraphPad Prism 5 software, with ANOVA when indicated. The region of interest (ROI) of western blot were quantified by ImageJ, normalized with the ROI value of loading control and analyzed with Student’s T-test. The statistical significance level was set at p< 0.05. Survival analysis was calculated with the Kaplan-Meier estimator.
Study Approval
The experimental protocol was approved by the Institutional Animal Care and Use Committee at the University of California at Irvine (AUP-18–113) and the experiments were performed in accordance of federal and local guidelines.
Supplementary Material
Figure S1. Tamoxifen did not change baseline renal histology or renal function in KMDAKT Mice.
Figure S2. Inhibition of tubule mitochondria AKT aggravated renal function induced by IRI in female mice.
Figure S3. Bigenic mice (KMCAKT) for renal tubule-specific mitochondria-targeting of a constitutively active AKT1.
Figure S4. Activation of mitochondrial AKT improved mitochondrial respiration in renal tubular epithelial cells.
Acknowledgments
This study was supported by the National Institutes of Health (R01HL096987), Ko Family Foundation, and Kroc Foundation (to PHW). HL is supported by a fellowship from KMU Alumni Association. The authors acknowledge the support of the Chao Family Comprehensive Cancer Center Transgenic Mouse Facility Shared Resource, supported by the National Cancer Institute of the National Institutes of Health (P30CA062203). The authors would like to thank Dr. Peter Igarashi (UT Southwestern Medical Center) for providing the KSP-CreERT2 mice.
Grant Support
National Institutes of Health, Ko Family Foundation, and Kroc Foundation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional experimental methods are described in Supplementary Methods
Disclosure Statement
The authors have no financial interests to disclose.
References
- 1.Yang JY, Deng W, Chen Y, et al. Impaired translocation and activation of mitochondrial Akt1 mitigated mitochondrial oxidative phosphorylation Complex V activity in diabetic myocardium. J Mol Cell Cardiol. 2013;59:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Ying Z, Bosy-Westphal A, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92(6):1369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li SY, Susztak K. The Role of Peroxisome Proliferator-Activated Receptor gamma Coactivator 1alpha (PGC-1alpha) in Kidney Disease. Semin Nephrol. 2018;38(2):121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Portilla D, Dai G, McClure T, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002;62(4):1208–18. [DOI] [PubMed] [Google Scholar]
- 5.Forbes JM. Mitochondria-Power Players in Kidney Function?. Trends Endocrinol Metab. 2016;27(7):441–4425. [DOI] [PubMed] [Google Scholar]
- 6.Szeto HH, Liu S, Soong Y, et al. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am J Physiol Renal Physiol. 2015;308(1):F11–F21.6. [DOI] [PubMed] [Google Scholar]
- 7.Kalogeris T, Baines CP, Krenz M, et al. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gracia-Sancho J, Casillas-Ramirez A, Peralta C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: a 2015 update. Clin Sci (Lond). 2015;129(4):345–62. [DOI] [PubMed] [Google Scholar]
- 9.See EJ, Jayasinghe K, Glassford N, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019;95(1):160–72. [DOI] [PubMed] [Google Scholar]
- 10.Szeto HH, Liu S, Soong Y, et al. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J Am Soc Nephrol. 2017;28(5):1437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran MT, Zsengeller ZK, Berg AH, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531(7595):528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tait SW, Green DR. Mitochondria and cell signaling. J Cell Sci. 2012;125(Pt 4):807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng W, Leu HB, Chen Y, et al. Protein kinase B (PKB/AKT1) formed signaling complexes with mitochondrial proteins and prevented glycolytic energy dysfunction in cultured cardiomyocytes during ischemia-reperfusion injury. Endocrinology. 2014;155(5):1618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanegae Y, Lee G, Sato Y, et al. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific Cre recombinase. Nucleic Acids Res. 1995;23(19):3816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007. January 16;104(3):1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SA, Cozzi M, Bush EL, et al. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am J Kidney Dis. 2018;72(6):846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su CC, Yang JY, Leu HB, et al. Mitochondrial Akt-regulated mitochondrial apoptosis signaling in cardiac muscle cells. Am J Physiol Heart Circ Physiol. 2012;302(3):H716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ratliff BB, Rabadi MM, Vasko R, et al. Messengers without borders: mediators of systemic inflammatory response in AKI. J Am Soc Nephrol. 2013;24(4):529–36. [DOI] [PubMed] [Google Scholar]
- 19.Basnakian AG, Ueda N, Kaushal GP, et al. DNase I-like endonuclease in rat kidney cortex that is activated during ischemia/reperfusion injury. J Am Soc Nephrol. 2002;13(4):1000–7. [DOI] [PubMed] [Google Scholar]
- 20.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinsey GR, Sharma R, Okusa MD. Regulatory T cells in AKI. J Am Soc Nephrol. 2013;24(11):1720–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. [DOI] [PubMed] [Google Scholar]
- 23.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22(6):999–1006. [DOI] [PubMed] [Google Scholar]
- 24.Saikumar P, Dong Z, Patel Y, et al. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 1998;17(26):3401–15. [DOI] [PubMed] [Google Scholar]
- 25.Wei Q, Dong G, Chen JK, et al. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84(1):138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfs TG, De Vries B, Walter S, et al. Apoptotic cell death is initiated during normothermic ischemia in human kidneys. Am J Transplant. 2005;5(1):68–75. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Wei Q, Wang CY, et al. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279(19):19948–54. [DOI] [PubMed] [Google Scholar]
- 28.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2(1):63–7. [DOI] [PubMed] [Google Scholar]
- 29.Green DR, Ferguson T, Zitvogel L, et al. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9(5):353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. [DOI] [PubMed] [Google Scholar]
- 31.Lan R, Geng H, Singha P, et al. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol. 2016;27(11):3356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–43, 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh P, Okusa MD. The role of tubuloglomerular feedback in the pathogenesis of acute kidney injury. Contrib Nephrol. 2011;174:12–21. [DOI] [PubMed] [Google Scholar]
- 34.Morrell ED, Kellum JA, Hallows KR, et al. Epithelial transport during septic acute kidney injury. Nephrol Dial Transplant. 2014;29(7):1312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weichert W, Paliege A, Provoost AP, et al. Upregulation of juxtaglomerular NOS1 and COX-2 precedes glomerulosclerosis in fawn-hooded hypertensive rats. Am J Physiol Renal Physiol. 2001;280(4):F706–14. [DOI] [PubMed] [Google Scholar]
- 36.Marcussen N Atubular glomeruli in renal artery stenosis. Lab Invest. 1991;65(5):558–65. [PubMed] [Google Scholar]
- 37.Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol. 2016;311(1):F145–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol. 2011;301(1):F110–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galarreta CI, Grantham JJ, Forbes MS, et al. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am J Pathol. 2014;184(7):1957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82(2):172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takaori K, Nakamura J, Yamamoto S, et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J Am Soc Nephrol. 2016;27(8):2393–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linkermann A, Chen G, Dong G, et al. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20(1):1–17. [DOI] [PubMed] [Google Scholar]
- 44.Loeffler I, Wolf G. Transforming growth factor-beta and the progression of renal disease. Nephrol Dial Transplant. 2014;29 Suppl 1(i37–i45). [DOI] [PubMed] [Google Scholar]
- 45.Gewin L, Vadivelu S, Neelisetty S, et al. Deleting the TGF-beta receptor attenuates acute proximal tubule injury. J Am Soc Nephrol. 2012;23(12):2001–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eckardt KU, Rosenberger C, Jurgensen JS, et al. Role of hypoxia in the pathogenesis of renal disease. Blood Purif. 2003;21(3):253–7. [DOI] [PubMed] [Google Scholar]
- 47.Schley G, Klanke B, Schödel J et al. Selective stabilization of HIF-1alpha in renal tubular cells by 2-oxoglutarate analogues. Am J Pathol. 2012;181(5):1595–606. [DOI] [PubMed] [Google Scholar]
- 48.Higgins DF, Kimura K, Bernhardt W, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117(12):3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang M, Pabla N, Murphy R, et al. Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J Biol Chem. 2007;282(4):2636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks C, Wei Q, Cho SG, et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. The J Clin invest. 2009;119(5):1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brooks C, Cho SG, Wang CY, et al. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300(3):C447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith JA, Stallons LJ, Collier JB, et al. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther. 2015;352(2):346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garrett SM, Whitaker RM, Beeson CC, et al. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther. 2014;350(2):257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitaker RM, Wills LP, Stallons LJ, Schnellmann RG. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharmacol Exp Ther. 2013;347(3):626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Granata S, Zaza G, Simone S, et al. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics. 2009;10:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X, Pabla N, Wei Q, et al. PKC-delta promotes renal tubular cell apoptosis associated with proteinuria. J Am Soc Nephrol. 2010;21(7):1115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15(2):186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim BJ, Yang J, Zhong J, et al. Tubulointerstitial fibrosis can sensitize the kidney to subsequent glomerular injury. Kidney Int. 2017;92(6):1395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jablonski P, Howden BO, Rae DA, et al. An experimental model for assessment of renal recovery from warm ischemia. Transplantation. 1983;35:198–204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tamoxifen did not change baseline renal histology or renal function in KMDAKT Mice.
Figure S2. Inhibition of tubule mitochondria AKT aggravated renal function induced by IRI in female mice.
Figure S3. Bigenic mice (KMCAKT) for renal tubule-specific mitochondria-targeting of a constitutively active AKT1.
Figure S4. Activation of mitochondrial AKT improved mitochondrial respiration in renal tubular epithelial cells.