

Abstract
T-cell receptor mimic (TCRm) antibodies combine the capacity of a T cell to target intracellular antigens with other capacities unique to antibodies. Neoantigens are abnormal proteins that arise as a consequence of somatic mutations. Technological advances promote the development of neoantigen-targeting therapies including TCRm antibody therapies. This review summarizes key characteristics of TCRm antibodies, in particular those targeting neoantigens, and further introduces discussion of obstacles that must be overcome to advance TCRm therapeutics.
Keywords: neoantigen, TCR receptor mimic antibody, bispecific antibody T cell engager, CAR-T cell therapy, cancer testis antigen (CTA)
Statement of Significance
The field of immunotherapy is evolving rapidly and new tools, including TCRm antibodies, are being introduced and optimized. This review highlights the promise of cancer neoantigens as targets for TCRm antibodies and presents a perspective on risks and benefits associated with TCRm therapies being developed.
INTRODUCTION
Hippocrates, generally recognized as the father of modern medicine, believed the major goal of medicine should be to build the patient’s own strength. However, in facing cancer, today’s therapies often resort to methods whose effects can challenge a patient even more than the illness itself. Personalized immunotherapy that builds the strength of the patient’s own immune system offers potentially enduring, highly specific, and safe treatments. Many efforts in immunotherapy research aim to enhance T cell responses. Another approach is to enhance antibody responses. Monoclonal antibodies (mAbs) show highest efficiency in treating cancer. Current mAb therapies rely on specific cell surface molecules or soluble molecules as targets. However, cell surface proteins account for only a small portion of tumor cell proteins. Currently, the FDA has approved fewer than 20 cell surface or extracellular proteins as anti-cancer mAb targets [1] (Table 1). Making mAbs to target intracellular proteins could greatly extend the influence of mAb therapy and benefit cancer patients.
Table 1.
Extracellular proteins as anti-cancer mAb targets validated in the clinic
| Target | Therapeutic indication(s) | References |
|---|---|---|
| CD19 | Precursor cell lymphoblastic leukemia-lymphoma | [87] |
| CD20 | Relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin lymphoma (NHL) | [88] |
| CD22 | B-cell acute lymphoblastic leukemia | [89] |
| CD30 | Hodgkin lymphoma (HL), systemic anaplastic large cell lymphoma | [90] |
| CD33 | AML | [91] |
| CD38 | Multiple myeloma | [92] |
| CD52 | B-cell chronic lymphocytic leukemia | [93] |
| CTLA-4 | Melanoma | [94] |
| EGFR | Carcinoma, non-small-cell lung | [95] |
| EpCAM | Head and neck cancer | [96] |
| GD2 | Neuroblastoma | [97] |
| HER2 | Breast cancer | [98] |
| PD-1 | Carcinoma; non-small-cell lung carcinoma; renal cell Hodgkin disease melanoma | [99], [100] |
| PDGFR-α | Sarcoma | [101] |
| PD-L1 | Metastatic Merkel cell carcinoma | [102], [103] |
| SLAMF7 | Multiple myeloma | [104] |
| VEGF | Stomach neoplasms | [95], [105], [106] |
Trenevska et al. [2] reviewed approaches to target intracellular antigens with therapeutic antibodies. One major strategy is to deliver mAbs into cells. Two ways in which this may be performed are as follows: (1) a “gene therapy” approach using vectors to express mAbs in the cytoplasm; and (2) using vehicles such as nanoparticles, liposomes, or fusion to cell-penetrating peptides to deliver mAbs into the cytoplasm. A second major strategy is to target externalized intracellular proteins. In rare cases, intracellular proteins in tumor cells could translocalize to the cell surface or extracellular environment, making them become mAb targets. PRL3 and gp75 are two examples of normally intracellular proteins externalized by cancer cells and targeted with antibody therapy [3, 4].
More often, intracellular proteins are degraded by the proteasome to form short peptides, some of which are presented at the cell surface by the major histocompatibility class I (MHC-I) complex. At the cell surface, peptide-MHC-I (pMHC) assemblies can be recognized by T-cell receptors (TCRs) on CD8+ T cells. T-cell receptor mimic (TCRm) or TCR-like antibodies can also recognize epitopes comprising the MHC-I molecule and a short peptide derived from intracellular proteins presented by the major histocompatibility complex (MHC).
Scientists are taking advantage of understanding the process of antigen presentation by MHC and developing novel therapeutic strategies against cancer, including therapies based on TCRm antibodies, T-cell receptor modified T cells (TCR-T), and chimeric antigen receptor engineered T cells (CAR-T) [5–7]. Information on TCRm antibodies is well summarized in the 2017 review by Trenevska et al. describing therapeutic antibodies against intracellular tumor antigens [2]. Other reviews present further overviews of targets, methods, challenges, and opportunities associated with TCRm antibodies [8, 9].
The current review presents an updated analysis of the anti-cancer strategy using TCRm antibodies targeting intracellular proteins with an emphasis on a recent rising star target: the neoantigen. We discuss classification of pMHC complexes, identification of tumor-associated antigens (TAAs), methods of generating TCRm antibodies, and the most recent progress in therapeutic applications of these antibodies. Finally, we present a perspective on how this discussion aids understanding of TCRm advantages and how key obstacles to the development to TCRm cancer therapeutics may best be overcome.
CLASSIFICATION OF pMHC COMPLEX
TAAs are mostly proteins produced by tumors that could be recognized by the immune system, such as antibodies. pMHC complexes are the tumor antigens for TCRm antibodies. A basic pathway of intracellular antigen processing is shown in Figure 1. Exogenous and endogenous intracellular proteins are processed by proteasomes. Resulting short peptides are transported into the endoplasmic reticulum to form assemblies with MHC-I molecules, which pass through the Golgi apparatus to be presented at the cell surface. Based on the origin of their short peptide component, pMHC complexes can be classified into three types: tumor-associated viral antigens, tumor-associated self-antigens, and neoantigens.
Figure 1.
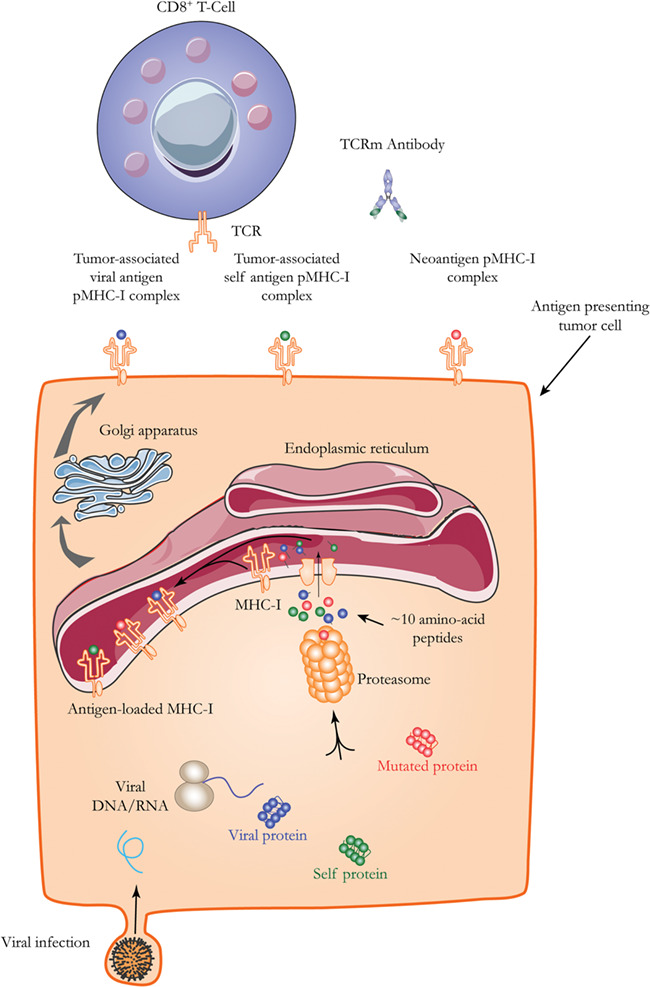
A basic pathway of antigen processing for cell surface presentation. A proteasome processes cytosolic proteins, including foreign proteins from viral infections and phagocytosis. The process generates short peptides, which are transported into the endoplasmic reticulum. There, they bind MHC-I molecules. The pMHC assembly is transported through the Golgi apparatus to the cell surface. There, the assembly can engage the TCR of a CD8+ cell.
In some virus-related cancers, such as hepatitis B virus (HBV)-related liver cancer and human papilloma virus-related cervical cancer, viral proteins that localize to the cytoplasm can be degraded and presented to the cell surface [6]. These are considered tumor-associated viral antigens. Tumor-associated self-antigens are normal proteins that are abnormally expressed in tumor cells. Some examples are MAGE [10], NY-ESO-1 [11] and WT1 [12], Her2 [13], PSA [14], and MUC1 [15].
Neoantigens, on the other hand, are generated as a result of gene mutations that happen in tumor cells, which are considered by the host immune system as “non-self” [16]. Thus, neoantigens are sometimes also called tumor-specific antigens (TSAs). As covered in several previous reviews [17–25], there is increasing interest in studies of neoantigens, their roles in cancer immunity, and the potential for their application in personalized cancer immunotherapy. The number of neoantigens was reported to be a critical parameter in evaluating the outcome of immunotherapy for several types of cancers. In triple-negative breast cancers, a high immune cell infiltration cohort associated with good prognosis also showed significantly lower mutation and neoantigen counts than the low immune cell infiltration cohort [26]. In lung adenocarcinomas, a high diversity of mutation-driven neoantigen expression in tumor cells is associated with higher tumor-infiltrating T-cell intratumor heterogeneity (ITH). This result suggests that spatial differences in the T-cell repertoire may be driven by distinct neoantigens in different tumor regions. More importantly, the authors found that higher degree of TCR ITH correlates with higher risk of relapse and shorter disease free survival [27]. In melanoma patients treated with nivolumab, those who responded well showed mutation and neoantigen load lower than baseline (before treatment). Correspondingly, clonal heterogeneity was significantly lower after nivolumab treatment in responding patients. This result suggests that nivolumab activates cytotoxic T cells to eliminate those tumor cells with high immunogenic neoantigens [28]. Luksza et al. [29] reported that the response to checkpoint blockade immunotherapy in a melanoma model could be predicted based on the likelihood of neoantigen presentation by the MHC and subsequent recognition by T cells. The apparent contradiction of these reports indicates that neoantigen load itself is not a determinant of patient outcome. Rather, the host immune response to the neoantigens is the key factor. In the case of immune activation, more tumor-infiltrating T cells means more killing of neoantigen bearing tumor cells, which in turn leads to lower neoantigen counts and better prognosis. In the case of immune suppression, tumor-infiltrating T cells are not active. When immune checkpoint inhibitors are given, the T cells switch to active status and the killing effect is positively associated with higher neoantigen load.
To address the question of how neoantigens are generated in cancer cells, Giovanni et al. [30] inactivated MLH1, which is critical for DNA repair, in colorectal, breast, and pancreatic mouse cancer cells. They found this inactivation increased neoantigen generation and led to poor tumor growth in immune competent mice. Reviews have covered current research on cancer neoantigens and their potential in immunotherapy for the management of human cancers including leukemia [31], head and neck cancer [25]. Current strategies for developing therapeutics against neoantigens, however, mainly focus on cancer vaccines and T cell-based therapies. TCRm antibodies are less studied due to the highly individual-specific characteristic of neoantigens.
IDENTIFICATION OF TAAs
The key first step in the development of strategies targeting pMHC complexes is the identification of TAAs. By the early 1970s, TAAs such as carcinoembryonic antigen [32] and α-fetoprotein [33] had been defined by analysis of heteroimmune sera. The association of Epstein–Barr virus (EBV) with Burkitt’s lymphoma and nasopharyngeal carcinoma was demonstrated [34]. In the early 1990s, the first human tumor antigen, melanoma antigen-1 (MAGE-1), was successfully cloned [10]. Subsequent studies demonstrated that MAGE-1 (also known as MAGE-A1) is frequently expressed in cancers. However, it is not expressed in normal tissues except testis and placenta [35]. The approach used to identify MAGE-A1 contains two steps. The first step is to expand cytotoxic T lymphocytes (CTLs) from the peripheral blood of cancer patients and stimulate the CTL with autologous tumor cells. The second step is to re-stimulate the CTL clones with cells transfected with cDNA libraries constructed from autologous tumor cells. Using this two-step approach, genes that encode the relevant antigens are identified [10]. Studies that used this strategy, termed T-cell epitope cloning, successfully identified several other tumor antigens [36–38]. Later, Pfreundschuh et al. [11] developed a serological approach, which uses antibodies from patients instead of T cells for the immunoscreening of tumor cDNA expression libraries. This technology, termed serological analysis of cDNA expression libraries, has enabled the discovery of several novel immunogenic TAAs and the highly immunogenic tumor antigen New York esophageal squamous cell carcinoma 1 (NY-ESO-1) [11]. Old and Chen [11] used the term cancer testis antigen to describe products of genes that are expressed in malignancies of various histotypes, but not in normal tissue except testis and placenta. TAAs can be identified using several other techniques in addition to immunological methods. First, reverse immunology, using dedicated software sometimes supported by proteasome-cleavage programs, predicts motifs for binding of the human leukocyte antigen (HLA) complex, the human version of MHC [39]. Second, biochemical methods using elution and fractionation of TAA peptides followed by analysis by chromatography and mass spectrometry can identify TAAs [40]. Third, TAAs can be identified by DNA microarray technology that compares gene expression profiles in tumor tissues and normal counterparts [7].
Before 2005, neoantigens did not gain much attention because they were so difficult to identify and are highly individually specific. However, by 2008, cancer genome sequencing showed that somatic mutations happened frequently in all cancer types [41]. This evidence supported the hypothesis that neoantigens are not rare in cancers. Computational predictions showed that mutations in breast cancer and colon cancer lead to the generation of HLA-binding peptides that can stimulate CD8+ T cell response [42]. In 2012, using a combination of next generation sequencing, in silico epitope prediction, and immunological approaches, two laboratories independently identified and validated distinct TSAs in murine melanoma tumor cells and in sarcoma cells [43, 44]. In 2014, Gubin et al. [45] used genomics and bioinformatics approaches and found that tumor-specific mutant proteins are a major class of T-cell antigens inducing tumor rejection following checkpoint blockade therapy targeting PD-1 or CTLA-4 in mouse sarcomas. In the same issue of Nature journal, Yadav et al. [46] reported an approach that combines whole-exome and transcriptome sequencing analysis with mass spectrometry to identify neo-epitopes in two widely used murine tumor models. Further study showed that, in human melanoma and non-small cell lung cancer, neoantigen load was significantly associated with clinical benefit in immune checkpoint blockade therapy [47–51]. Mutant MHC-II epitopes were also reported to drive therapeutic immune responses to cancer [52].
METHODS OF GENERATING TCRm ANTIBODIES
After identifying targets, producing TCRm antibodies is an important next step in the development of TCRm antibody therapeutics. Immunization of an animal with pMHC complex followed by hybridoma generation is the traditional way to obtain a TCRm antibody. Either antigen presenting cells that express the pMHC on their surface or the purified pMHC complex can serve as antigen to immunize the mice. Dadaglio et al. [53] reported that 1 out of 1000 hybridoma clones showed specific binding to the target pMHC following immunization of mice with whole cells. Another study showed 4 out of 1000 hybridoma clones with specific binding to the target pMHC [54]. Many attempts failed to generate any specific antibodies against the target pMHC. These cases illustrate that peptide-specific, MHC-restricted antibodies are quite rare even under optimal conditions when using whole cells to immunize animals. More recently, several groups have been successful in using recombinant MHC/peptide complexes for the immunization and high throughput screening of a few thousand clones for the isolation of rare TCRm antibodies [55–59]. These attempts showed much higher efficiency than the whole cell immunization method [55].
In 1996, Engberg et al. [60] demonstrated an immunization and phage display approach for rapid and efficient isolation of antibodies with unique specificity. The approach starts with cDNA produced from total RNA isolated from the spleen of an immunized mouse. Amplified variable regions of the immunoglobulin gene are cloned into a pFab5c vector for construction of phage library displaying the antibody Fab (antigen binding fragment). These phage particles are purified by repeated rounds of panning on cells expressing a specific antigen. Usually, a de-selection process using a control antigen is involved in the panning process. Thus, the phage display procedure shows a much higher positive rate of production of that peptide-specific, MHC-restricted mAbs than the hybridroma approach. Notably, the immunization step may be omitted; a human naïve peripheral blood mononuclear cell (PBMC) phage library may be used to screen for TCRm antibodies that target a specific pMHC complex [61–63]. Traditional immunization and hybridoma methods take several months and generate few positive clones. In contrast, the naïve phage library method generates positive clones in a few weeks. Another advantage over the immunization and hybridoma method is the capability to generate fully human antibodies from phage display libraries, which eliminates the need for a humanized mouse or antibody humanization steps otherwise necessary to produce antibodies that can be used in human patients.
However, due to the lack of an in vivo affinity maturation step, TCRm antibodies obtained from naïve phage libraries usually show lower binding affinity (in the range of hundreds of nanomolar) than do antibodies generated through the immunization and hybridoma method (in the lower nanomolar range) [55, 56]. Thus, many of the TCRm antibodies generated by the naïve phage library method require in vitro affinity maturation. In vitro affinity maturation can be performed using the random mutation method, which randomly mutates the amino acids in the target peptide, or a structurally directed mutation method such as that reported by Stewart-Jones et al. [64] in 2009. In this method, researchers first obtained high-resolution crystal structure of two Fabs in complex with NY-ESO-1157-165/HLA-A*0201. Then, they compared them with the structure of a TCR that recognized the same pMHC complex. Based on the comparison, they evaluated the contributions of individual amino acids of the Fab and the NY-ESO-1 peptide to binding affinity and specificity. They mutated the amino acids at positions where side chains could be optimized for direct interactions with the peptide but not the HLA molecule. In this way, they improved the affinity of two Fabs by 20 fold to 2 nm without changing the binding specificity. By contrast, the random mutation method often leads to increased affinity but lower specificity [20, 65].
Generating TCRm antibodies against neoantigens is more complicated. The identification of neoantigens is a rate-limiting step. Due to the rapid progress in next generation sequencing method and bioinformatics algorithms, the workflow of generating TCRm antibodies against highly personalized neoantigens can now be completed in a more timely manner [44, 46], as illustrated in Figure 2.
Figure 2.
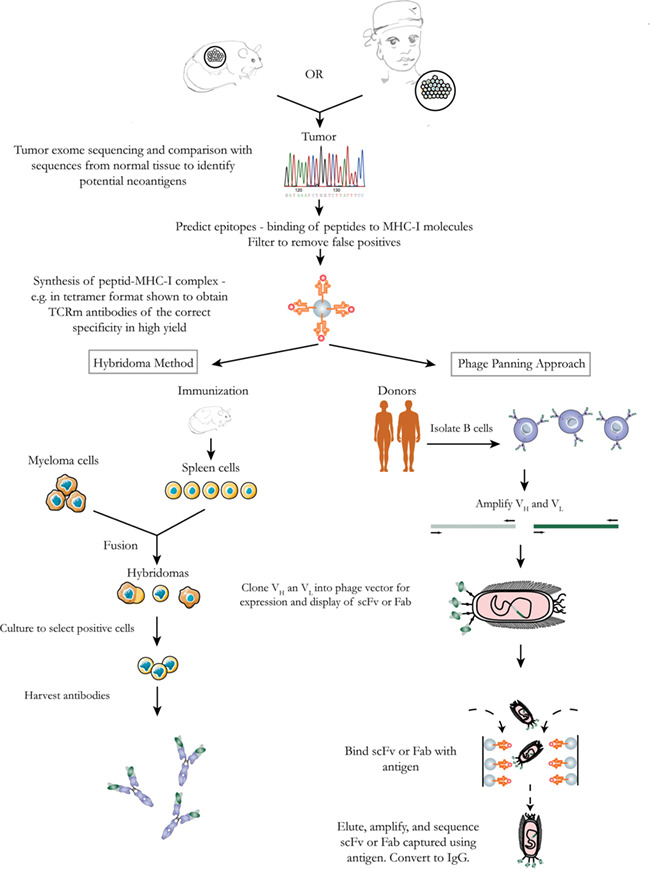
Strategies for production of TCRm antibodies against neoantigens. Analysis of tumor tissues from a human patient or animal model identifies candidate antigens. Further analysis identifies a subset of potential antigens that might be presented by MHC-I. Then, pMHC complexes are synthesized and used to generate antibodies either through screening of a phage-display library or through a traditional hybridoma method.
THERAPEUTIC APPLICATION OF TCRm ANTIBODIES
Since their introduction, TCRm antibodies have shown increasing promise as advances are made in the target identification and antibody production processes. In laboratory and preclinical studies, researchers are testing TCRm antibodies in the development of cancer therapeutics. These antibodies target two types of pMHC complexes: tumor-associated viral antigens and tumor-associated self-antigens. Although targeting neoantigens represents a major advance due to high tumor specificity, the extreme rareness of specific neoantigens greatly limits their application in TCRm antibody therapy.
Tumor-associated virus antigens, foreign proteins specifically expressed in virus-affected cells, are being validated as targets. EBV is a human gamma herpes virus found in more than 90% of the human population. EBV is associated with a number of human cancers such as Burkitt’s lymphoma, Hodgkin’s lymphoma, and nasopharyngeal carcinoma. A group from the National University of Singapore reported the generation of three novel TCRm mAbs against EBV proteins. These proteins, EBNA1562–570, LMP1125–133, and LMP2A426–434, are presented by (HLA)-A*0201 to cell surface [66]. The group used these TCRm antibodies to treat B lymphoblastoid cell lines (BLCLs). Results showed that BLCLs are inhibited in engrafted immunodeficient mice. Mice treated with TCRm antibodies showed significantly improved survival [67]. Liver cancer is another human malignancy associated with viral infection [68]. Currently, there is no antibody target therapy for liver cancer, although some potential targets, such as glypican-3 [69, 70], show promise. A TCRm antibody was reported to be an effective way to treat HBV positive liver cancer [71]. However, since HBV antigens are expressed not only by cancer cells but also by infected hepatocytes, the risk of inducing severe liver damage with this TCRm antibody should not be ignored [71].
Tumor-associated self-antigens are another group of potential targets for TCRm antibodies for cancer treatment. As mentioned earlier, an immunization and hybridoma method is used to generate TCRm antibodies of mouse origin. These were demonstrated to inhibit cancer progression: a TCRm mouse mAb specific for the PR1/HLA-A0201 inhibited acute myeloid leukemia (AML) progenitor cell growth in vitro [55]; a mouse mAb specific for hCGβ (human chorionic gonadotropin-beta) bound to HLA-A02 was shown to slow tumor growth in orthotopic xenograft models of breast cancer [72]. Later, fully human antibodies were generated by human naïve PBMC phage library and tested in vitro and in vivo for their ability to treat variety of cancers. The WT1 (Wilms tumor 1) oncoprotein is an intracellular transcriptional factor that is overexpressed in a wide range of cancers and was ranked as the top cancer target for immunotherapy. A 9-mer peptide WT1126–134, RMFPNAPYL (RMF), is processed and presented by HLA-A0201 [12]. Scheinberg et al. [73] generated a fully human IgG1 mAb named “ESK1” that binds the WT1 RMF/HLA-A0201 complex. The antibody was shown to bind fresh AML CD34+/CD33+ leukemia cells in an HLA-A2 and WT1 restricted manner. However, ESK1 did not bind normal CD33+ PBMCs[74]. Finally, Dao et al. [74] demonstrated that ESK1 induces a dramatic antitumor effect in established human xenografts in NSG mice. Other studies from the same group extended the research based on ESK1 antibody to bispecific T-cell engager (BiTE) antibody [73] and chimeric antigen receptor (CAR) T cells [75]. Both showed promising therapeutic effect against leukemia.
Early clinical safety studies of TCRm antibodies may focus on previously characterized tumor antigens and may work together with TCR-based therapies such as CAR-T and TCR-T. For example, ESK1 TCRm against WT1 holds promise for clinical trials. Similar to TCR-based therapies, TCRm antibodies face limitations of animal models in which the MHC or tumor peptide epitope may not be conserved.
Target identification and development of TCRm antibodies are active fields of research. For instance, Ahmed et al. [76] have developed bispecific TCRm antibodies targeting LMP2A for treatment of EBV malignancies. Transmembrane localization makes the target difficult to recognize by conventional antibody therapeutics. Phage display was employed to isolate TCRm specific for a portion of the target that could be presented on cell surfaces by HLA class I. Another example of progress is the work of a team at The Cancer Research UK Oxford Antibody Therapeutics Programme. They are developing TCRm antibodies against HLA-A*0201 peptide derived from the cancer target p53 (http://commercial.cancerresearchuk.org/sites/default/files/CB_TCR%20Mimic_November%202016.pdf). This work addresses a great need, as the tumor protein p53 is mutated or deregulated in a majority of human cancers. One example of a state-of-the-art target identification platform is the Immatics XPRESIDENT system [77]. XPRESIDENT enables identification of large numbers of tumor-associated proteins, facilitating development of T-cell receptor therapeutics.
PERSPECTIVE
TCRm antibodies hold the promise of targeting intracellular proteins for cancer therapy. A variety of potential mechanisms are available for therapies based on TCRm antibodies, as illustrated in Figure 3. However, the application of TCRm antibodies in treating human patients still faces major challenges. Since the MHC component is invariant for a given haplotype, the specificity of a TCRm antibody is determined by the embedded peptide [78]. Off-target effects are one major concern that impairs the therapeutic application of TCRm antibodies. Off-target effects may arise from two sources. First, the TAAs, which are targets of TCRm antibodies, may not be expressed exclusively on tumor cells. For instance, Her2 protein is pathologically overexpressed in certain cancers but also expressed at physiological levels on the surfaces of normal cells. Second, only a few key amino acids in the short peptide are required for the binding of TCRm antibodies to their target pMHC complex [55, 74]. Thus, the likelihood is high that other off-target epitopes share the key amino acids essential for binding. Of course, potential off-target peptide sequences may not be correctly processed and presented at the cell surface by MHC. Considering all factors, many tests are warranted to guard against potential off-target effects before advancing TCRm antibodies to human trials.
Figure 3.
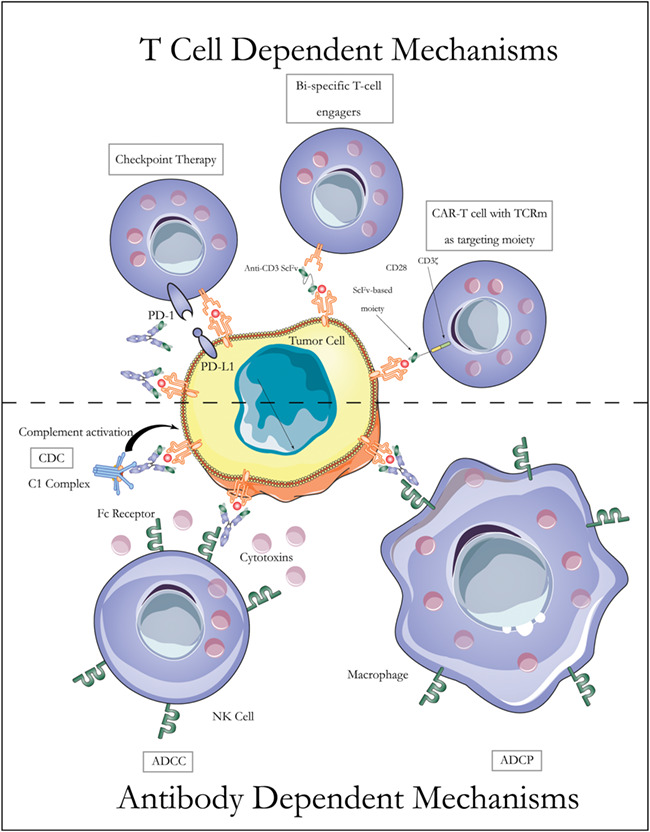
Mechanisms of TCRm based cancer therapies. Like conventional therapeutic antibodies against cancer, TCRm antibodies can be effective through antibody-dependent mechanisms. For example, they can initiate ADCC, ADCP, and CDC. TCRm antibodies can also be used in combination with checkpoint therapy, BiTE antibodies, and CAR-T cell therapy.
Another factor to consider is the density of the target pMHC on the cell surface. Presentation of a peptide by MHC is a complicated process, which makes it challenging to predict the degree of cell-surface expression for a given epitope [6]. Two major factors contribute to pMHC presentation: the level of protein expression and the rate of protein degradation [79]. Better understanding of the antigen processing mechanism and subsequent pharmacological modulation of these processes will greatly support rational development of TCRm antibody therapies. On the other hand, down-regulation or loss of MHC-I complex expression is one of the major mechanisms by which tumor cells escape immune surveillance [80]. There is an urgent need to recover MHC class I in cancers to enhance immunotherapy, including TCRm antibody therapy. Some efforts have been devoted to deal with this issue, including enhance HLA gene expression, gene therapy for tumors with HLA or beta-2-microglobulin, and a strategy employing NK cells to clear MHC-I negative cells [81].
One solution to the problem of low cell-surface density of epitope is to enhance the potency of TCRm antibody. Effector functions of IgG include antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytolysis (CDC). ADCC and ADCP are dependent on immune cells, specifically NK and macrophage. CDC does not require the presence of immune cells. Enhancing ADCC and ADCP has been widely used to improve the therapeutic efficacy of many mAbs. Veomtt et al. [82] reported an Fc-enhanced TCRm antibody against WT1-derived peptide that was engineered to yield an afucosylated chain at Asn297 within the Fc region. Fc enhancement of the TCRm antibody led to enhanced ADCC and therapeutic efficacy in both in vitro and animal studies.
Increased potency could also be achieved by direct fusion of the antibody with a drug or by combination therapy using a TCRm antibody with another therapy. Antibody-drug-conjugation (ADC) has been shown to be highly efficacious in treatment of certain types of cancers. One report showed that fusing TCRm Fab antibodies specific for melanoma antigens MART-1 26–35/A2 or gp100 280–288/A2 to a truncated form of Pseudomonas endotoxin dramatically inhibited melanoma xenograft progression in vivo [83]. However, due to the low rate of MHC complex internalization, ADC might not work as well for the majority of TCRm antibodies. Combination therapy with other drugs could also improve therapeutic efficacy. When anti-WT1 TCRm antibody ESKM was combined with a variety of tyrosine kinase inhibitors, greater inhibition of chronic myelogenous leukemia was observed in mouse models than was observed with either treatment alone [84].
Another way to increase the potency of TCRm antibody is to engineer bispecific or bispecific T-cell engager antibodies. Bispecific TCRm antibodies target two pMHC molecules at the same time, which could increase the chance of a therapeutic effect. The powerful T-cell cytotoxicity of BiTEs antibodies has been shown to be an effective strategy in mouse tumor models. As mentioned earlier, a BiTE TCRm antibody, ESK-BiTE, was generated to selectively bind WT1/HLA-A*0201 positive human tumor cells. This bispecific antibody showed potent therapeutic activity against multiple human cancers in vitro and in vivo [73]. Engineered T cells with CARs based on TCRm antibodies could enhance therapeutic effects, further extending the potential applications of TCRm antibody. Several CARs have been generated in laboratories to target self-antigens including WT1 and GP100 [75, 85]. Another study showed that a neoantigen, H3.3K27M, led to activation of CD8+ T cells and could be used as a target for T cell-based therapy to treat patients with diffuse intrinsic pontine glioma [86].
Different methods for the production of pMHC complexes is an area open for further optimization. The pMHC complex proteins used in antibody discovery may or may not match their conformations as presented on cell surfaces. Results of studies to assess and validate the conformations of peptide-pMHC complexes will be helpful for the ongoing development of therapeutic TCRm antibodies.
A final issue to consider is the antigen density required for effectiveness of the various TCRm mechanisms of action illustrated in Figure 3. The antigen densities necessary for TCRm mechanisms is another topic warranting further study.
In conclusion, although hurdles have impeded their progress to clinical application, nevertheless TCRm antibodies hold significant potential for development as cancer therapeutics due to their unique combination of high specificity and high affinity.
FUNDING
This study was supported in part by Welch foundation [AU-0042-20030616] and Cancer Prevention and Research Institute of Texas (CPRIT) [RP150551].
Conflict of interest statement. The authors declare no potential conflicts of interest.
REFERENCES
- 1. Strohl, WR. Current progress in innovative engineered antibodies. Protein Cell 2018; 9: 86–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trenevska, I, Li, D, Banham, AH. Therapeutic antibodies against intracellular tumor antigens. Front Immunol 2017; 8: 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takechi, Y, Hara, I, Naftzger, C et al. A melanosmal membrane protein is a cell surface target for melanoma therapy. Clin Cancer Res 1996; 2: 1837–1842. [PubMed] [Google Scholar]
- 4. Thura, M, Al-Aidaroos, AQO, Yong, WP et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight 2016; 1: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caron, E, Kowalewski, DJ, Chiek Koh, C et al. Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry. Mol Cell Proteomics 2015; 14: 3105–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blum, JS, Wearsch, PA, Cresswell, P. Pathways of antigen processing. Annu Rev Immunol 2013; 31: 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Novellino, L, Castelli, C, Parmiani, G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother 2005; 54: 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen, M, Reiter, Y. T-cell receptor-like antibodies: targeting the intracellular proteome therapeutic potential and clinical applications. Antibodies 2013; 2: 517–534. [Google Scholar]
- 9. Dubrovsky, L, Dao, T, Gejman, RS et al. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Bruggen P, Traversari C, Chomez P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254: 1643–7. [DOI] [PubMed] [Google Scholar]
- 11. Chen, YT, Scanlan, MJ, Sahin, U et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A 1997; 94: 1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinilla-Ibarz, J, May, RJ, Korontsvit, T et al. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia 2006; 20: 2025–2033. [DOI] [PubMed] [Google Scholar]
- 13. Disis, ML, Cheever, MA. HER-2/neu protein: a target for antigen-specific immunotherapy of human cancer. Adv Cancer Res 1997; 71: 343–371. [DOI] [PubMed] [Google Scholar]
- 14. Horig, H, Lee, CS, Kaufman, HL. Prostate-specific antigen vaccines for prostate cancer. Expert Opin Biol Ther 2002; 2: 395–408. [DOI] [PubMed] [Google Scholar]
- 15. Vlad, AM, Kettel, JC, Alajez, NM et al. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol 2004; 82: 249–293. [DOI] [PubMed] [Google Scholar]
- 16. Gubin, MM, Artyomov, MN, Mardis, ER et al. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015; 125: 3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capietto, AH, Jhunjhunwala, S, Delamarre, L. Characterizing neoantigens for personalized cancer immunotherapy. Curr Opin Immunol 2017; 46: 58–65. [DOI] [PubMed] [Google Scholar]
- 18. Desrichard, A, Snyder, A, Chan, TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res 2016; 22: 807–812. [DOI] [PubMed] [Google Scholar]
- 19. Efremova, M, Finotello, F, Rieder, D et al. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol 2017; 8: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li, Y, Moysey, R, Molloy, PE et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 2005; 23: 349–354. [DOI] [PubMed] [Google Scholar]
- 21. Schumacher, TN, Schreiber, RD. Realising the promise: neoantigens in cancer immunotherapy. Science 2015; 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 22. Wang, RF, Wang, HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res 2017; 27:11–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer, Avances in immunology. Vol. 130. Academic Press. 2016.25–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wirth, TC, Kühnel, F. Neoantigen targeting—dawn of a new era in cancer immunotherapy? Front Immunol 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zolkind, P, Dunn, GP, Lin, T et al. Neoantigens in immunotherapy and personalized vaccines: implications for head and neck squamous cell carcinoma. Oral Oncol 2017; 71: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karn, T, Jiang, T, Hatzis, C et al. Association between genomic metrics and immune infiltration in triple-negative breast cancer. JAMA Oncol 2017; 3: 1707–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reuben, A, Gittelman, R, Gao, J et al. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov 2017; 7: 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riaz, N, Havel, JJ, Makarov, V et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017; 171: 934–949 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luksza, M, Riaz, N, Makarov, V et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017; 551: 517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Germano, G, Lamba, S, Rospo, G et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017; 552: 116–120. [DOI] [PubMed] [Google Scholar]
- 31. Yang, L, Han, Y, Saurez Saiz, F et al. A tumor suppressor and oncogene: the WT1 story. Leukemia 2007; 21: 868–876. [DOI] [PubMed] [Google Scholar]
- 32. Gold, P, Freedman, SO. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J Exp Med 1965; 121: 439–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Houstek, J, Masopust, J, Kithier, K et al. Hepatocellular carcinoma in association with a specific fetal alpha-1-globulin, fetoprotein. J Pediatr 1968; 72: 186–193. [DOI] [PubMed] [Google Scholar]
- 34. Klein, G, Pearson, G, Nadkarni, JS et al. Relation between Epstein–Barr viral and cell membrane immunofluorescence of Burkitt tumor cells. I. Dependence of cell membrane immunofluorescence on presence of EB virus. J Exp Med 1968; 128: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Plaen, E, Arden, K, Traversari, C et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics 1994; 40: 360–369. [DOI] [PubMed] [Google Scholar]
- 36. Boel, P, Wildmann, C, Sensi, ML et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995; 2: 167–175. [DOI] [PubMed] [Google Scholar]
- 37. De Backer, O, Arden, KC, Boretti, M et al. Characterization of the GAGE genes that are expressed in various human cancers and in normal testis. Cancer Res 1999; 59: 3157–3165. [PubMed] [Google Scholar]
- 38. Gaugler B, Van den Eynde B, van der Bruggen P et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med 1994; 179:921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kessler, JH, Melief, CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia 2007; 21: 1859–1874. [DOI] [PubMed] [Google Scholar]
- 40. Admon, A, Barnea, E, Ziv, T. Tumor antigens and proteomics from the point of view of the major histocompatibility complex peptides. Mol Cell Proteomics 2003; 2: 388–398. [DOI] [PubMed] [Google Scholar]
- 41. Cancer Genome Atlas Research . N: comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Segal, NH, Parsons, DW, Peggs, KS et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008; 68: 889–892. [DOI] [PubMed] [Google Scholar]
- 43. Castle, JC, Kreiter, S, Diekmann, J et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72: 1081–1091. [DOI] [PubMed] [Google Scholar]
- 44. Matsushita, H, Vesely, MD, Koboldt, DC et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gubin, MM, Zhang, X, Schuster, H et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515: 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yadav, M, Jhunjhunwala, S, Phung, QT et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515: 572–576. [DOI] [PubMed] [Google Scholar]
- 47. Van Allen, EM, Miao, D, Schilling, B et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sacco, E, Cortes, M, Josseaume, N et al. Mutation landscape of acquired cross-resistance to glycopeptide and beta-lactam antibiotics in Enterococcus faecium. Antimicrob Agents Chemother 2015; 59: 5306–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McGranahan, N, Furness, AJ, Rosenthal, R et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351: 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stronen, E, Toebes, M, Kelderman, S et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016; 352: 1337–1341. [DOI] [PubMed] [Google Scholar]
- 51. Anagnostou, V, Smith, KN, Forde, PM et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 2017; 7: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kreiter S, Vormehr M, van de Roemer N et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dadaglio, G, Nelson, CA, Deck, MB et al. Characterization and quantitation of peptide-MHC complexes produced from hen egg lysozyme using a monoclonal antibody. Immunity 1997; 6: 727–738. [DOI] [PubMed] [Google Scholar]
- 54. Porgador, A, Yewdell, JW, Deng, Y et al. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 1997; 6: 715–726. [DOI] [PubMed] [Google Scholar]
- 55. Sergeeva, A, Alatrash, G, He, H et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 2011; 117: 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bernardeau, K, Gouard, S, David, G et al. Assessment of CD8 involvement in T cell clone avidity by direct measurement of HLA-A2/Mage3 complex density using a high-affinity TCR like monoclonal antibody. Eur J Immunol 2005; 35: 2864–2875. [DOI] [PubMed] [Google Scholar]
- 57. Verma, B, Hawkins, OE, Neethling, FA et al. Direct discovery and validation of a peptide/MHC epitope expressed in primary human breast cancer cells using a TCRm monoclonal antibody with profound antitumor properties. Cancer Immunol Immunother 2010; 59: 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hawkins, O, Verma, B, Lightfoot, S et al. An HLA-presented fragment of macrophage migration inhibitory factor is a therapeutic target for invasive breast cancer. J Immunol 2011; 186: 6607–6616. [DOI] [PubMed] [Google Scholar]
- 59. Wittman, VP, Woodburn, D, Nguyen, T et al. Antibody targeting to a class I MHC-peptide epitope promotes tumor cell death. J Immunol 2006; 177: 4187–4195. [DOI] [PubMed] [Google Scholar]
- 60. Engberg, J, Andersen, S, Nielsen, K et al. Phage-display libraries of murine and human antibody fab fragments. Mol Biotechnol 1996; 6:287–310. [DOI] [PubMed] [Google Scholar]
- 61. Chames, P, Hufton, SE, Coulie, PG et al. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1-MAGE-A1 from a nonimmunized phage-Fab library. Proc Natl Acad Sci U S A 2000; 97: 7969–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Biddison, WE, Turner, RV, Gagnon, SJ et al. Tax and M1 peptide/HLA-A2-specific Fabs and T cell receptors recognize nonidentical structural features on peptide/HLA-A2 complexes. J Immunol 2003; 171: 3064–3074. [DOI] [PubMed] [Google Scholar]
- 63. Cohen, CJ, Hoffmann, N, Farago, M et al. Direct detection and quantitation of a distinct T-cell epitope derived from tumor-specific epithelial cell-associated mucin using human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells. Cancer Res 2002; 62: 5835–5844. [PubMed] [Google Scholar]
- 64. Stewart-Jones, G, Wadle, A, Hombach, A et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci U S A 2009; 106: 5784–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao, Y, Bennett, AD, Zheng, Z et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol 2007; 179: 5845–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sim, AC, Too, CT, Oo, MZ et al. Defining the expression hierarchy of latent T-cell epitopes in Epstein–Barr virus infection with TCR-like antibodies. Sci Rep 2013; 3: 3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lai, J, Tan, WJ, Too, CT et al. Targeting Epstein–Barr virus-transformed B lymphoblastoid cells using antibodies with T-cell receptor-like specificities. Blood 2016; 128: 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Matsushita, H, Vesely, MD, Koboldt, DC et al. Cancer exome analysis reveals a T cell dependent mechanism of cancer immunoediting. Nature 2013; 482: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Feng, M, Gao, W, Wang, R et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A 2013; 110: E1083–E1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gao, W, Kim, H, Feng, M et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology 2014; 60: 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bertoletti, A, Brunetto, M, Maini, MK et al. T cell receptor-therapy in HBV-related hepatocellularcarcinoma. Oncoimmunology 2015; 4: e1008354 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Verma, B, Neethling, FA, Caseltine, S et al. TCR mimic monoclonal antibody targets a specific peptide/HLA class I complex and significantly impedes tumor growth in vivo using breast cancer models. J Immunol 2010; 184: 2156–2165. [DOI] [PubMed] [Google Scholar]
- 73. Dao, T, Pankov, D, Scott, A et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 2015; 33: 1079–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dao, T, Yan, S, Veomett, N et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med 2013; 5: 176ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rafiq, S, Purdon, TJ, Daniyan, AF et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 2017; 31: 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ahmed, M, Lopez-Albaitero, A, Pankov, D et al. TCR-mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weinschenk T, Rajkovic E, Reusch U et al. Abstract 3753: identification of antibodies against a novel tumor-associated MHC/peptide-target and generation of highly specific and potent HLA-A*02 MMP1-003 /CD3 TandAbs. Cancer Res 2017; 77(13 Suppl): Abstract nr 3753. [Google Scholar]
- 78. Simons, AL, Mattson, DM, Dornfeld, K et al. Opportunities and challenges for TCR mimic antibodies in cancer therapy. J Cancer Res Ther 2009; 5: 1–7.19293480 [Google Scholar]
- 79. Bassani-Sternberg, M, Pletscher-Frankild, S, Jensen, LJ et al. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics 2015; 14: 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bubenik, J. MHC class I down-regulation: tumour escape from immune surveillance? (review). Int J Oncol 2004; 25: 487–491. [PubMed] [Google Scholar]
- 81. Garrido, F, Aptsiauri, N, Doorduijn, EM et al. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 2016; 39: 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Veomett, N, Dao, T, Liu, H et al. Therapeutic efficacy of an Fc-Enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin Cancer Res 2015; 20: 4036–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Klechevsky, E, Gallegos, M, Denkberg, G et al. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res 2008; 68: 6360–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dubrovsky, L, Pankov, D, Brea, EJ et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 2014; 123: 3296–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang, G, Wang, L, Cui, H et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep 2014; 4: 3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chheda, ZS, Kohanbash, G, Okada, K et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med 2018; 215: 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Przepiorka, D, Ko, CW, Deisseroth, A et al. FDA approval: Blinatumomab. Clin Cancer Res 2015; 21: 4035–4039. [DOI] [PubMed] [Google Scholar]
- 88. Oflazoglu, E, Audoly, LP. Evolution of anti-CD20 monoclonal antibody therapeutics in oncology. MAbs 2010; 2: 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Haso, W, Lee, DW, Shah, NN et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013; 121: 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Moskowitz, CH, Nademanee, A, Masszi, T et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015; 385: 1853–1862. [DOI] [PubMed] [Google Scholar]
- 91. Castaigne, S, Pautas, C, Terre, C et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012; 379: 1508–1516. [DOI] [PubMed] [Google Scholar]
- 92. Lokhorst, HM, Plesner, T, Laubach, JP et al. Targeting CD38 with Daratumumab monotherapy in multiple myeloma. N Engl J Med 2015; 373: 1207–1219. [DOI] [PubMed] [Google Scholar]
- 93. Ishizawa, K, Fukuhara, N, Nakaseko, C et al. Safety, efficacy and pharmacokinetics of humanized anti-CD52 monoclonal antibody alemtuzumab in Japanese patients with relapsed or refractory B-cell chronic lymphocytic leukemia. Jpn J Clin Oncol 2017; 47: 54–60. [DOI] [PubMed] [Google Scholar]
- 94. Hodi, FS, O'Day, SJ, McDermott, DF et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wainberg, Z, Hecht, JR. A phase III randomized, open-label, controlled trial of chemotherapy and bevacizumab with or without panitumumab in the first-line treatment of patients with metastatic colorectal cancer. Clin Colorectal Cancer 2006; 5: 363–367. [DOI] [PubMed] [Google Scholar]
- 96. Sebastian, M, Kuemmel, A, Schmidt, M et al. Catumaxomab: a bispecific trifunctional antibody. Drugs Today (Barc) 2009; 45: 589–597. [DOI] [PubMed] [Google Scholar]
- 97. Dhillon, S. Dinutuximab: first global approval. Drugs 2015; 75: 923–927. [DOI] [PubMed] [Google Scholar]
- 98. Baselga, J, Norton, L, Albanell, J et al. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res 1998; 58: 2825–2831. [PubMed] [Google Scholar]
- 99. Garon, EB, Rizvi, NA, Hui, R et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015; 372: 2018–2028. [DOI] [PubMed] [Google Scholar]
- 100. Wolchok, JD, Kluger, H, Callahan, MK et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shirley, M. Olaratumab: first global approval. Drugs 2017; 77: 107–112. [DOI] [PubMed] [Google Scholar]
- 102. Kaufman, HL, Russell, J, Hamid, O et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 2016; 17: 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Fehrenbacher, L, Spira, A, Ballinger, M et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016; 387: 1837–1846. [DOI] [PubMed] [Google Scholar]
- 104. Lonial, S, Dimopoulos, M, Palumbo, A et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373: 621–631. [DOI] [PubMed] [Google Scholar]
- 105. Rosenfeld, PJ, Brown, DM, Heier, JS et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355: 1419–1431. [DOI] [PubMed] [Google Scholar]
- 106. McCarthy, M. Antiangiogenesis drug promising for metastatic colorectal cancer. Lancet 2003; 361: 1959. [DOI] [PubMed] [Google Scholar]