

Abstract
The humanization of antibodies for therapeutics is a critical process that can determine the success of antibody drug development. However, the science underpinning this process remains elusive with different laboratories having very different methods. Well-funded laboratories can afford automated high-throughput screening methods to derive their best binder utilizing a very expensive initial set of equipment affordable only to a few. Often within these high-throughput processes, only standard key parameters, such as production, binding and aggregation are analyzed. Given the lack of suitable animal models, it is only at clinical trials that immunogenicity and allergy adverse effects are detected through anti-human antibodies as per FDA guidelines. While some occurrences that slip through can be mitigated by additional desensitization protocols, such adverse reactions to grafted humanized antibodies can be prevented at the humanization step. Considerations such as better antibody localization, avoidance of unspecific interactions to superantigens and the tailoring of antibody dependent triggering of immune responses, the antibody persistence on cells, can all be preemptively considered through a holistic sagacious approach, allowing for better outcomes in therapy and for research and diagnostic purposes.
Keywords: therapeutics antibodies, VH/VL families, constant isotype, super antigen binding, antibody humanization
Statement of Significance
Recent investigations of antibody elements have revealed effects that when combined with already known functions of elements such as isotypes, can open a new frontier in antibody humanization to reduce adverse effects in sagacious design.
INTRODUCTION
The therapeutic antibody market is steadily increasing over the past few years [1]. Already an expensive developmental process, drug candidates can fail from adverse effects during clinical trials that include allergic reactions from immunogenicity [2]. While this is often difficult to predict during the early stages of drug development, recent insights into antibody functions and activity may allow for early mitigation of such adverse reactions at the humanization step. By taking a holistic and rational approach, preventing adverse reactions can be sagaciously taken, and this review discusses the parameters with respect to the various antibody elements.
Many of the current antibody therapeutics were obtained through the immunization of animals, e.g. (mice), harvesting of lymphocytes or spleen and generating hybridoma cells [3 and 4]. When purposed for therapy, rodent antibodies are typically humanized. This humanization process can occur to varying extents, involving the displacement of antibody elements, such as the constant region (mouse-human chimera akin to class isotype switching [5]), or grafting the rodent complementarity determining regions (CDRs) to a human antibody scaffold. While the early generation of therapeutic antibodies are typically chimeras of constant region humanization (e.g. rituximab, infliximab and cetuximab), there remains arguments that the reduction of immunogenicity is very subjective to the particular antibody, and the humanization process may not necessarily correlate to reducing immune reactions [6], thus, it is still necessary to test for anti-human antibodies during clinical trials. Regardless of the nuances, the humanization process has allowed for the addition to desired functions to antibody therapeutics that include antibody recycling [7], prevention of IgG4 light chain swapping [8], better localization [9] and even cross-isotype engineering to activate multiple IgFcRs [10–12]. In this review, we discuss the recent findings on antibody elements i.e. the variable (V-) and constant (C-) regions of both the heavy and light chains, as well as their role in humanization for therapeutics (Fig. 1), and how they can be leveraged upon to make safer therapeutics as well as research and diagnostic antibodies.
Figure 1.
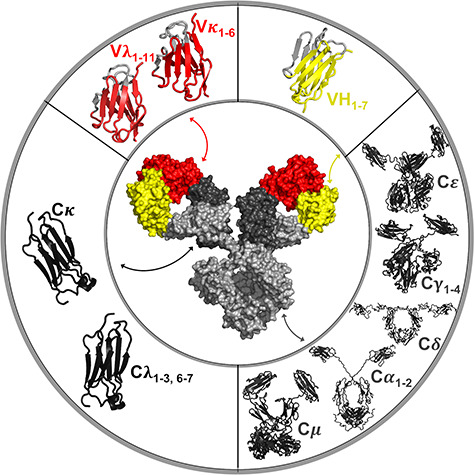
A schematic of antibody elements that can be humanized. Top left represents the variable light chain (VL, red) consisting of Vκ1–6 and Vλ1–11 families. Top right represents the variable heavy chain (VH, yellow) consisting of VH1–7 families. Bottom right represents the constant heavy chain (CH, light gray) consisting of γ1–4, α1–2, μ, δ, ε. Bottom left represents the constant light chain (CL, dark gray) consisting of Cκ and Cλ1–3, 6–7. Augmented reality for this figure can be seen using APD AR Holistic review App [13].
Heavy chain isotype
The earliest attempts of humanization as rodent-human chimeras were proposed to reduce immunogenicity, increasing the safety of such therapeutics. While recent Fc engineering methods include detailed edits in the antibody constant region to allow for the formation of bispecific antibodies [14] amongst above already mentioned functions, the constant region has innate features that can allow tailoring for more effective functions for therapeutics, and even for diagnostics and research use. In this, we highlight the known human isotypes and how they can be considered for various purposes.
Immunoglobulin G (IgG) 1–4
IgG is the most abundant isotype found in blood at 73% (calculated from study population results in [15]). It has four subtypes: IgG1, IgG2, IgG3 and IgG4 at 60%, 32%, 4% and 4%, respectively, of total IgG [16], each highly similar but with their own unique functions and effects. All current Food and Drug Administration (FDA) approved antibody therapeutics are of the IgG isotype, with the majority as IgG1 subtype. IgG1 can activate antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and has a long half-life of 33.2 days [17]. Alternatively, IgG2 weakly activates ADCC and CDC, and is seldom used for therapeutic purposes especially since natural polymorphisms in FcγR2A (H131R), a receptor for IgGs, can result in weak interactions particular to IgG2 [18]. Nonetheless, IgG2 therapeutics do exist, and is often chosen for inhibiting receptor-ligand interactions [19], being more resistant to proteolysis at the hinge [20]. IgG3, on the other hand, has the shortest half-life of 25.9 days [17] among the IgGs and it can activate ADCC and CDC very efficiently. It is the largest of the IgGs with an extended hinge and the only IgG that contains O-link glycosylation with effects discussed later.
IgG4 is similar to IgG2 in being a weak activator of ADCC and CDC. Yet, it is also unique in that it occasionally swaps its light chain, posing a challenge in antigen recognition. While there are methods to prevents this by mutations in the CH1 region [8,21], IgG4 is classified to be bispecific and functionally monovalent [22].
Within these IgG subtypes, there are little differences between their transient recombinant production rates [9] and their antigen binding properties [9,23–25] as shown in our prior comparison using Trastuzumab and Pertuzumab models across all known human constant isotypes and subtypes on both heavy (constant heavy (CH)) and light chains (CL) [9].
These features of the various IgG subtypes could be chosen for their mode of action (ADCC/blocker) or target-specific (receptor/bacteria/virus) effects. Depending on desired properties, the interaction between IgG and superantigens, i.e. protein G and protein A (except non-binding IgG3) can be utilized for purification or for avoidance of unwanted activation by superantigen binding in the physiological sites of activity.
IgM
IgM is a penta/hexamer connected by the joining chain (J-chain). Given its unique interconnectedness, it has a higher valency of 10 or 12 that is multiple folds higher than the other isotypes, contributing to its avidity effects [9]. As the primary response antibody [26] accounting for 10% of total Ig in blood [15], IgM can activate the complement system [27] and is a potent CDC inducer [27] and [28]. IgM can oligomerize in culture conditions without the J-chain [9, 29] which encourages the formation of the hexameric form that it is more effective than the pentameric form for complement system activation [29]. Unlike IgG, common superantigens, e.g. proteins G, A and L do not bind to the IgM constant region. While one IgM therapeutic (Centoxin) failed to reach the clinics due to production problems and unpromising clinical data [30], there are many learning points for IgM antibody therapeutics. At the point of writing, IgM Bioscience Inc., has developed IgM therapeutic candidates that showed promise in clinical trials with more following in the preclinical pipeline [31]. While IgM has clear superior avidity and agglutination effects over IgG, it is not a blanket rule that IgM can be leveraged upon to create better antigen binders. In one example, we found that the location of the epitope on the antigen to severely limit full IgM valency through steric hindrances where Pertuzumab-IgM is shown to be better Pertuzumab-IgG1, but not for Trastuzumab-IgM and Trastuzumab-IgG1 [32] due to epitope location in the same HER2 antigen. However, in this particular case, the large size of IgM was also advantageous for steric inhibition of HER2 dimerization, providing an advantage for IgM in such mechanistic inhibition.
IgE
The use of IgE therapeutics for cancer immunotherapy to mount exaggerated allergic responses to treat cancer was proposed and termed—“AllergoOncology” [33]. As the activation of mast cells can lead to type-I hypersensitivity inflammatory response that is deemed more effective than IgG ADCC in tissues, research in AllergroOncology is increasingly popular [34] and [35]. IgE plays a central role in this type-I hypersensitivity [36 and 37], and despite being one of the least abundant isotypes in blood (60 000 times < IgG) [38], it is often found on mast cells lining mucosal areas that are also colonized by microorganisms. Thus, the binding of bacterial superantigens, e.g. A or L [39] can potentially cross-link and activate these IgE sensitized mast cells [40] leading to allergic symptoms. While the biotechnological production of potential IgE therapeutics can avoid these superantigens by using the high affinity receptor FcεRIα for affinity purification, it is not possible to do so for other isotypes given the low equilibrium dissociation constant rates with their respective receptors. This limits the affinity purification of other isotypes to rely on superantigens, i.e. protein A/G/L. Considering that many drugs fail clinical trials due to adverse effects including anaphylaxis, it is important to ensure that such IgE biologics do not aggravate or cause such effects. At the time of writing, there are patents on using IgE as a cancer therapeutic drug (e.g. [41]), with companies working on IgE therapeutics (e.g. IGEM Therapeutics Ltd), and candidates in clinical trials (e.g. NCT02546921, www.clinicaltrials.gov).
IgAs 1 and 2
The two human IgA subtypes: IgA1 and IgA2 are the dominant Ig in mucosal areas where it mounts the defence against bacterial and virus [42] infections as 17% of total antibodies in blood [15]. IgA1 has a longer hinge, and the differences in hinges were found to modulate allosteric signaling between the antigen (Fab) and receptor binding regions or Fc [43]. IgA1 differs from IgA2 in having O-linked glycans in this region [44], allowing IgA1, but not IgA2, to be recognized by certain T-lymphocyte receptors [45 and 46]. The main immune effects of IgA are ADCC, Antibody-Dependent Cellular Phagocytosis (ADCP) and the triggering of cytokines release via FcαRI or CD89 on effector cells [47]. In human isotype comparison experiments using Trastuzumab and Pertuzumab, transient recombinant IgA production was the highest of the isotypes and subtypes [9]. Experiments to increase its short half-life of 3–6 days [47] by removing the N-glycosylation sites (N166 and N337), stabilizing linkage of heavy chain to light chain and removing free cysteines (C311 and C472) are promising [48]. Despite great interest in considering IgA for therapeutics [49], there is none in clinical trials. While the short half-life may be a factor, this may be an advantage when designing a fast acting biologic that can remain active in the gastrointestinal environment with potential for oral administration (passive immunisation of IgA via human milk and colostrum) [50]. IgA can dimerize [50] to its predominant form for secretion [51], and future exploitations may involve possible combinations of IgA1–A2 dimers for oral administration, or monomeric forms for intravenous administration to decrease off-site side effects and increase localization to mucosal areas [50]. Given that there are no known functional differences other than avidity between monomeric and dimeric IgA [52], further investigations with regards to immune efficacy are warranted. IgA does not or binds weakly to common superantigens Protein G, A(weakly) and L (VL), making it a possibly safer isotype than IgE for therapy in mucosal areas colonized by microflora.
IgD
IgD is similar to IgA1 and IgG3 in possessing O-linked glycans with a long hinge between CH2 and CH3 domains. IgD is typically secreted in the upper respiratory tract and bone marrow, and binds to certain T-lymphocyte receptors via the O-linked glycan [45 and 46]. Together with IgM, it is often the B cell receptor [53] and is rarely found in secreted forms, making up to about 0.25% of total Ig in serum [54]. IgD levels are associated with viral infections such as HIV [55] providing a hint to a possible role in infectious diseases although much remains to be investigated. It has a short half-life of 2.8 days [54], poor cell culture production [9] and showed diminished antigen binding effect of the V-regions in limited experiments [9], making it a poor choice for a therapeutic. At the point of writing, there is no known interest in IgD therapeutics.
The CH region plays an important role in localization, half-lives and even immune effector functions. While the latter is not a consideration for diagnostic and research use, the right isotype may be a matter of convenience in laboratories to the secondary antibodies that various labs already possess for their immunological assays or when designing research antibodies with long shelf lives. On the diagnostic front, CH may be of interest for their avidity effects from multimeric formation of IgM in hemagglutination-based assays, and naturally, the half-life and stability of the antibodies for the shelf-life of diagnostic kits [56].
The light chain constant (constant light (CL))
While a lot of functions and effects are attributed to the CH, the light chain constant region or CL can have effects on overall antibody stability. In humans, the CL consists of two isotypes: kappa (Cκ) and lambda (Cλ), where the latter has another five known functional subtypes of Cλ1–3 and Cλ6–7 (Cλ 4 and 5 are classified as pseudo genes) [57 and 58]. The human ratio of usage of κ to λ is close to 1, but in mice, the ratio is around 20:1 [59]. As a result, humanized antibodies of rodent origins following isotype lineages are predominantly κ, and only about 6 out of more than 50 approved antibodies utilize λ-chains: Avelumab, Belimumab, Evolocumab, Raxibacumab, Guselkumab and Erenumab.
There are little differences between the recombinant production of Cκ and Cλ [9], although our previous experiments showed Cλ7 to be produced in slightly lower levels in a hybrid form fused to a variable kappa (Vκ) [9]. In general, λ-chains are associated with more rigid stable interdomain interactions [60], longer hydrophobic CDR3s [61] and shorter half-lives [62]. For unknown reasons, it is also the dominant light chain produced in response to some viral immunisations [59]. As a therapeutic, λ-chains can be considered if it is more energetically favorable for pairing with the heavy chain for stability in diagnostic kits or for faster clearance [62].
Variable (V-) region
The three categories of V-regions in human follow the division of variable heavy (VH) 1–7 for the heavy chain, and variable lambda (Vλ) 1–11 and Vκ1–7 for the respective light chains [57 and 58]. For more complete humanization, the CDRs of animal antibodies are grafted onto the human scaffold, and in this, the V-region framework regions (FWRs) that were traditionally thought to primarily hold up the CDRs, are also attributed to have the main role of forming the paratope to bind the epitopes. While the FWRs were assumed to have a small role in antigen recognition, recent work have demonstrated the importance of FWRs via loop structure alteration [63–71], VH–variable light (VL) interaction affecting CDRs orientation [72–77], binding to superantigen protein L [78–81], allosteric communication to Fc receptors (FcR) [82] and antibody production [82 and 83] amongst many others.
These new data confound an already challenging CDR grafting process that can affect the success of the therapeutic, where many factors are in play, e.g. presence of microbiome and their proteins.
Within the V-regions, the segregation of CDRs and FWRs have debatable boundaries. Given that CDRs can have varying lengths, there are at least six boundaries that are error prone as denoted by the dashes in FRW1-CDR1-FRW2-CDR2-FRW3-CDR3-*FRW4*. While both heavy and light chains start with FRW1, opinion is divided on the end of the light chain where some determined it to end at CDR3 without a FWR4 (denoted by *) due to the absence of the diversity (D) gene in V(D)J rearrangement in the light chains. This difference in opinion adds to the nonstandardized grafting methods across labs to yield varying results.
Pre-humanization considerations
FWRs and CDRs
There are currently six numbering systems for the V-regions in attempts to numerically determine the FWR–CDR boundaries, each with their pros and cons summarized from [84], as shown in Table 1.
Table 1.
Summary of the various antibody numbering schemes used for framework and complementary determining region identification
| Numbering Scheme | Pros | Cons |
|---|---|---|
| Kabat [85–91] | Based on sequence alignment. Includes v-region of antibodies and T cell receptor. Basis of KABATMAN database. Considered standard |
Based on limited numbers of sequences. May be biased as it is based on most common length sequence. Unconventional insertion or deletion in FWRs not included. Do not match well with 3D structure |
| Chothia [92–94] | Based on crystal structure alignment. Better CDR definition reflected in structural loop. Can be optimized by defining new insertion points |
Possible confusion when used with other schemes. Based on most common CDR length. Ignores sequence with unconventional length. Insertion points are not consistent |
| Martin [95] | Uses larger Abysis database. Based on structural alignment. Accept unconventional length sequences. Uses ABnum software based on Chothia. Considered to be the upgraded version of Chothia scheme. Incorporate database from IMGT, Kabat and Chothia |
May incorporate all the weakness from various database |
| Gelfand [96–100] | Features “two span bridge.” Precise comparison of secondary structures in aligned sequences |
Complex nomenclature. Does not include gaps and deletions. Definition of CDR loops is different from other schemes |
| IMGT [101–108] | Based on Ig superfamily from different species. Based on germ-line V gene alignment. Includes variable region of antibodies and T cell receptor. Uses continuous numbering system. Used by WHO-IUIS |
Due to continuous numbering system, it is difficult to visualize insertions. Less flexible. Difficulty adapting to sequences with new insertions |
| Honneger (AHo’s) [109] | Based on structural alignment. Pre-defined C23, W43, C106 and G140 as conversed residues. Takes into account of “two span bridge” conformation in CDR1. Matches well to antibody structures |
Conserved residues positions are only from 28 structures. Possibility of number skipping. Less flexible on insertions |
Each numbering system comes with its own set of assumptions with very little cross-validation agreement. Pure reliance on these methods alone may not result in successful grafting requiring additional checks that may involve structural of further sequence alignment. In fact, the challenges analysis and accurately establishing the artificial CDR/FWR boundaries contributed to the need of back-mutations in many CDR-grafted antibodies to restore binding affinities [110 and 111].
While the FWRs are clearly involved in antigen engagement, CDRs play the major role as evident from the conferment of antigen binding properties in grafting alone. While different CDRs have different weightages in antigen binding (with the highest weightage typically assigned to VH CDR3), slight changes in CDRs can affect antigen recognition. The difficulty in determining this CDR–FWR region is especially pronounced in sequence analysis methods that investigates only one chain at a time, ignoring the combination of both heavy and light chains. In addition, hypervariability and the varying lengths of CDRs further complicates the attempts to determine specificity determining residues (SDRs) that requires the in-depth analysis of the structural fit of the six CDRs (three from each chain) to form the paratope. Current methods to manage this include surface reshaping [112–118]. Thus, despite some attempts to utilize SDR grafting to minimize immunogenicity in the CDRs [119 and 120], this methodology is far less used (>16 000 articles in Google Scholar for “CDR grafting” as opposed to > 2000 articles for “SDR grafting” at the point of writing), leaving CDR grafting as the more commonly used method.
CONSIDERATIONS OF THE PROCESSES POST- HUMANIZATION
Production
Poorly grafted antibodies will lead to poor production or non-binding antibodies. Even single amino acid deletions on the “less important” light chain FWRs can have significant effects [63]. In such cases, the pairing of heavy and light chains can mitigate such effects to an extent where poor producers can yield functional antibodies [82] when paired with the right partner chains. The factors involved in the pairing is still elusive, preventing efficient in silico surface reshaping, often requiring trials and errors. It is only through large scale rational approaches that some clues where certain VH and VLs would be better producers were ascertained, e.g. Vκ3 & 4 for the light chain, and VH1, 3, 5 and 7 over VH2, 4 and 6 [82].
Aggregation
Apart from reducing the useable fraction of therapeutics (and thereby manufacturing costs and final treatment costs), aggregation has also been found to affect the immunogenicity of therapeutic antibodies [121]. The aggregation of antibodies, particularly IgE [122], can also lead to unspecific activation of mast cells in the absence of the known antigen, exhibiting “cytokinergic” effects. While cytokinergic activity is not reported in other Igs since IgE is unique in its modus operandi by activation of sensitized cells, such aggregative effects can lead to other diseases even when on other isotypes, e.g. IgA nephropathy [123]. In addition to mitigating extrinsic factors by controlling buffer and temperature conditions, intrinsic interventions via protein engineering methods to reduce glycosylation, adding tetra-peptide extensions, and mutagenesis of residues in specific parts of antibodies have been incorporated to reduce antibody aggregation [121]. In our previous work [9], we found the IgG versions of our model antibodies to be more consistent in exhibiting lower levels of unwanted aggregations compared to the other isotypes (with exception of the expected oligomerization of IgM). These effects can be mitigated by VH-VL pairing (see Supplementary material of [82]), thus requiring a rational and holistic approach to antibody humanization considering the desired properties of the therapeutic antibody. It is conceivable that by combining methods to control extrinsic and intrinsic factors in aggregation, immunogenicity can be reduced without compromising desired biological effects conferred by specific antibody elements. Certainly, further optimization of these factors in a combinatorial manner is required, and taking consideration of the varying processes adopted in different labs.
Binding affinity
While the primary desired effect of CDR grafting in humanization is to ensure retention of antigen binding, recent studies have demonstrated the impact of CDRs and FWRs on FcR engagement (Fig. 2) [39, 82]. Even with the best scaffold for production and retention of antigen binding, immune effector cell engagement is a key function of many therapeutics. Apart from production, the CH and the VH–VL pairings have slight but notable effects. When trying to leverage on the unique functions of the various CHs (e.g. IgE sensitization on mast cells as a form of passive immunization for immediate reaction upon antigen challenge), consideration of VH and VH–VL effects are also to be taken when tweaking the antibody drug for persistence on receptors [39]. In such attempts, there is a need to also consider the innate ability of FWR effects to bind superantigens that may trigger or inactivate the antibodies, e.g. choosing Vκ2, 5 and 6 to avoid superantigen protein L binding if the therapeutic target is not deployed in otherwise sterile environments such as blood, but in areas with microflora. All at the same time, balancing the other desired parameters of the final product in localization production, aggregation, half-life, etc.
Figure 2.
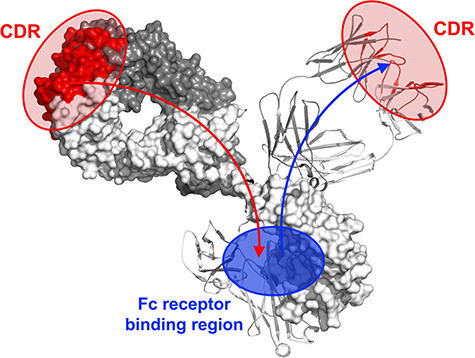
Illustration of allosteric communication found between CDRs/FWRs and Fc engagement as shown from our previous work [39, 43, 82].
Added optional safeguards and considerations
As mentioned earlier, the upcoming trend of designing tandem or cross-isotype Fc antibodies [10–12] to combine desired effects of more than one CH is interesting and promising. Yet, with these artificial challenges, the risks of immunogenicity are also increased. While there are in silico methods to perform T- and B-cell epitope predictions (see review [124]), there should be significant care in the interpretation of results, in particularly, the prediction of allergenicity. For example, in the case of in silico predictions of food allergens [125], the presence of an epitope (e.g. on peanut) does not necessary equate to Type 1 hypersensitivities if the eventual response is not IgE (not everyone mounted IgE responses to peanut). In fact, promising treatments for allergy include oral (OIT)/sublingual (SIT) immunotherapies [126 and 127] to elicit IgG4 antibodies over IgE. Furthermore, in silico T- and B-cell epitope predictions are severely limited in being unable to consider the intrinsic immunological factors of the human leukocyte antigen system fully, which has the crucial role in the development of anti-drug antibodies [128]. Thus, despite the numerous assays [129–131] developed as added checks before the dive to clinical trials, sagacity is limited here given that immunogenicity and allergy adverse effects can only truly be detected during clinical trials as per FDA guidelines.
CONCLUSION—HOLISTIC VIEW IN ANTIBODY HUMANIZATION
The location of the target, the immunological mechanisms to be deployed against the target, the purpose of the drug, and many others, are among the numerous considerations in antibody drug design and development beyond obvious biotechnological considerations, such as production cost. The complex nature of the various elements of the antibodies are shown by recent work to be more interconnected than previously reduced. While a simple CH swap for chimerization may meet the needs of designing antibodies toward research use or diagnostics, there is an increasing requirement for sagacity in antibody therapeutic design beyond brute force methods and the incorporation of desensitization interventions as rescue measures. On top of increasing the success of the antibody therapeutics, the cost savings may translate to more affordable drugs where a holistic view [132] toward meeting the key features of the final drug product could make the difference between failure and success.
ACKNOWLEDGEMENTS
This work was supported by A*STAR core funds. We thank Mr Chan K.F. for creating the augmented reality.
ABBREVIATIONS
- VH
Variable heavy
- CH
Constant heavy
- VL
Variable light
- CL
Constant light
- Vκ
Variable kappa
- Cκ
Constant kappa
- Vλ
Variable lambda
- Cλ
Constant lambda
- FWR
Framework regions
- CDR
Complementarity determining regions
- ADCP
Antibody-dependent cellular phagocytosis
- SDR
Specificity determining residue
- V-region
Variable -region
- C-region
Constant -region
- ADCC
Antibody dependent cellular cytotoxicity
- CDC
Complement-dependent cytotoxicity
- J-chain
Joining chain
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial interests.
References
- 1). Grilo, AL, Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol 2019; 37: 9–16. [DOI] [PubMed] [Google Scholar]
- 2). Roa-Medellin, D, Garcia-Gutierrez, I, Lillo, MCet al. Adverse reaction to humanised monoclonal antibodies. J Allergy Clin Immunol 2017; 139: AB37. [Google Scholar]
- 3). Kim, HY, Stojadinovic, A, Izadjoo, MJ. Immunization, hybridoma generation, and Selection FOR monoclonal antibody production. In: Ossipow, V, Fischer, N (eds). Monoclonal Antibodies. Methods in Molecular Biology (Methods and Protocols) vol. 1131. Totowa, NJ: Humana Press, 2014, pp. 33–45 [DOI] [PubMed] [Google Scholar]
- 4). Zhang, C. Hybridoma technology for the generation of monoclonal antibodies. In: Proetzel, G, Ebersbach, H (eds). Antibody Methods and Protocols. Methods in Molecular Biology (Methods and Protocols), vol. 901. Totowa, NJ: Humana Press, 2012, pp. 117–35 [DOI] [PubMed] [Google Scholar]
- 5). Stavnezer, J, Schrader, CE. IgH chain class switch recombination: mechanism and regulation. J Immunol 2014; 193: 5370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Getts, DR, Getts, MT, McCarthy, DPet al. Have we overestimated the benefit of human(ized) antibodies? MAbs 2010; 2: 682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Sampei, Z, Haraya, K, Tachibana, Tet al. Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. Plos One 2018; 13: e0209509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Labrijn, AF, Buijsse, AO, van denBremer, ETJet al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009; 27: 767–71. [DOI] [PubMed] [Google Scholar]
- 9). Lua, WH, Ling, WL, Yeo, JYet al. The effects of antibody engineering CH and CL in Trastuzumab and Pertuzumab recombinant models: impact on antibody production and antigen-binding. Sci Rep 2018; 8: 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Chintalacharuvu, KR, Vuong, LU, Loi, LAet al. Hybrid IgA2/IgG1 antibodies with tailor-made effector functions. Clin Immunol 2001; 101: 21–31. [DOI] [PubMed] [Google Scholar]
- 11). Kelton, W, Mehta, N, Charab, Wet al. IgGA: “a cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol 2014; 21: 1603–9. [DOI] [PubMed] [Google Scholar]
- 12). Borrok, MJ, Luheshi, NM, Beyaz, Net al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcRI (CD89) binding. MAbs 2015; 7: 743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Poh, JJ, Phua, SX, Chan, KFet al. Commentary: augmented reality scientific phone apps–making the APD AR holistic review app and using existing AR apps for scientific publications. Sci Phone Apps Mobile Device 2018; 4: 4. [Google Scholar]
- 14). Ha, JH, Kim, JE, Kim, YS. Immunoglobulin Fc heterodimer platform technology: from design to applications in therapeutic antibodies and proteins. Front Immunol 2016; 7: 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Gonzalez-Quintela, A, Alende, R, Gude, Fet al. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol 2007; 151: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Vidarsson, G, Dekkers, G, Rispens, T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014; 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Wasserman, RL, Church, JA, Peter, HHet al. Pharmacokinetics of a new 10% intravenous immunoglobulin in patients receiving replacement therapy for primary immunodeficiency. Eur J Pharm Sci 2009; 37: 272–8. [DOI] [PubMed] [Google Scholar]
- 18). Warmerdam, PA, van deWinkel, JG, Vlug, Aet al. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol 1991; 147: 1338–43. [PubMed] [Google Scholar]
- 19). Kreschmer, A, Schwanbeck, R, Valerius, Tet al. Antibody isotypes for tumor immunotherapy. Transfus Med Hemother 2017; 44: 320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Berzski, RJ, Oberholtzer, A, Strake, Bet al. The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs 2011; 3: 558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). Salfeld, JG. Isotype selection in antibody engineering. Nat Biotechnol 2007; 25: 1369–72. [DOI] [PubMed] [Google Scholar]
- 22). Schuurman, J, vanRee, R, Perdok, GJet al. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology 1999; 97: 693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Torres, M, Casadevall, A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol 2008; 29: 91–7. [DOI] [PubMed] [Google Scholar]
- 24). Beenhouwer, DO, Yoo, EM, Lai, CWet al. Human immunoglobulin G2 (IgG2) and IgG4, but not IgG1 or IgG3, protect mice against Cryptococcus neoformans infection. Infect Immun 2007; 75: 1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Cooper, LJ, Robertson, D, Granzow, Ret al. Variable domain-identical antibodies exhibit IgG subclass-related differences in affinity and kinetic constant as determined by surface plasmon resonance. Mol Immunol 1994; 31: 577–84. [DOI] [PubMed] [Google Scholar]
- 26). Carsetti, R, Rosado, MM, Wardmann, H. Peripheral development of B cells in mouse and man. Immunol Rev 2004; 197: 179–91. [DOI] [PubMed] [Google Scholar]
- 27). Hurst, MM, Volanakis, JE, Stroud, RMet al. C1 fixation and classical complement pathway activation by a fragment of the Cmu4 domain of IgM. J Exp Med 1975; 142: 1322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Devarapu, SK, Mamidi, S, Ploger, Fet al. Cytotoxic activity against human neuroblastoma and melanoma cells mediated by IgM antibodies derived from peripheral blood of healthy donors. Int J Cancer 2016; 138: 2963–73. [DOI] [PubMed] [Google Scholar]
- 29). Wiersma, EJ, Collins, C, Fazel, Set al. Structural and functional analysis of J chain-deficient IgM. J Immunol 1998; 160: 5979–89. [PubMed] [Google Scholar]
- 30). Marks, L. The birth pangs of monoclonal antibody therapeutics: the failure and legacy of Centoxin. MAbs 2012; 4: 403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Baliga, R, Li, K, Manlusoc, Met al. High avidity IgM-based CD20xCD3 bispecific antibody (IGM-2323) for enhanced T-cell dependent killing with minimal cytokine release. Blood 2019; 134: 1574. [Google Scholar]
- 32). Samsudin, F, Yeo, JY, Gan, SKEet al. Not all therapeutic antibody isotypes are equal: the case of IgM versus IgG in Pertuzumab and Trastuzumab. Chem Sci 2020. doi: 10.1039/C9SC04722K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Jensen-Jarolim, E, Achatz, G, Turner, MCet al. AllergoOncology: the role of IgE-mediated allergy in cancer. Allergy 2008; 63: 1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Karagiannis, SN, Josephs, DH, Bax, HJet al. Therapeutic IgE antibodies: harnessing a macrophage-mediated immune surveillance mechanism against cancer. Cancer Res 2017; 77: 2779–83. [DOI] [PubMed] [Google Scholar]
- 35). Karagiannis, P, Singer, J, Hunt, Jet al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol Immunother 2009; 58: 915–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Gould, HJ, Sutton, BJ. IgE in allergy and asthma today. Nat Rev Immunol 2008; 8: 205–17. [DOI] [PubMed] [Google Scholar]
- 37). Akin, C. Mast cell activation syndromes. J Allergy Clin Immunol 2017; 140: 349–55. [DOI] [PubMed] [Google Scholar]
- 38). Gould, HJ, Sutton, BJ, Beavil, AJet al. The biology of IgE and the basis of allergic disease. Annu Rev Immunol 2003; 21: 579–628. [DOI] [PubMed] [Google Scholar]
- 39). Lua, WH, Su, CTT, Yeo, JYet al. Role of the IgE variable heavy chain in FceRIa and superantigen binding in allergy and immunotherapy. J Allergy Chin Immunol 2019; 144: 514–23. [DOI] [PubMed] [Google Scholar]
- 40). Genovese, A, Bouvet, JP, Florio, Get al. Bacterial immunoglobulin superantigen proteins a and L activate human heart mast cells by interacting with immunoglobulin E. Infect Immun 2000; 68: 5517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Penichet, ML, Schultes, BC, Nicodemus, CFet al. Patent US-8697079B2. 2014.
- 42). Gould, VMW, Francis, JN, Anderson, KJet al. Nasal IgA provides protection against human influenza challenge in volunteers with low serum influenza antibody titre. Front Microbiol 2017; 8: 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Su, CTT, Lua, WH, Ling, WLet al. Allosteric effects between the antibody constant and variable regions: a study of IgA Fc mutations on antigen binding. Antibodies 2018; 7: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Woof, JM, Russell, MW. Structure and function relationship in IgA. Mucosal Immunol 2011; 4: 590–7. [DOI] [PubMed] [Google Scholar]
- 45). Rudd, PM, Fortune, F, Patel, Tet al. A human T-cell receptor recognises ‘O’-linked sugars from the hinge region of human IgA1 and IgD. Immunology 1994; 83: 99–106. [PMC free article] [PubMed] [Google Scholar]
- 46). Swenson, CD, Patel, T, Parekh, RBet al. Human T cell IgD receptors react with O-glycans on both human IgD and IgA1. Eur J Immunol 1998; 28: 2366–72. [DOI] [PubMed] [Google Scholar]
- 47). Monteiro, RC, van deWinkel, JG. IgA Fc receptors. Annu Rev Immunol 2003; 21: 177–204. [DOI] [PubMed] [Google Scholar]
- 48). Lohse, S, Meyer, S, Meulenbroek, LAPMet al. An anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res 2016; 76: 1–15. [DOI] [PubMed] [Google Scholar]
- 49). Leusen, JHW. IgA as therapeutic antibody. Mol Immunol 2015; 68: 35–9. [DOI] [PubMed] [Google Scholar]
- 50). Woof, JM, Kerr, MA. The function of immunoglobulin A in immunity. J Pathol 2006; 208: 270–82. [DOI] [PubMed] [Google Scholar]
- 51). Woof, JM, Kerr, MA. IgA function—variations on a theme. Immunology 2004; 113: 175–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Lombana, TN, Rajan, S, Zorn, JAet al. Production, characterization, and in vivo half-life extension of polymeric IgA molecules in mice. MAbs 2019; 11: 1122–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53). Preud’homme, JL, Petit, I, Barra, Aet al. Structural and functional properties of membrane and secreted IgD. Mol Immunol 2000; 37: 871–87. [DOI] [PubMed] [Google Scholar]
- 54). Rogentine, GNJ, Rowe, DS, Bradley, Jet al. Metabolism of human immunoglobulin D (IgD). J Clin Investig 1966; 45: 1467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55). Moskophidis, D, Moskophidis, M, Lohler, J. Virus-specific IgD in acute viral infection of mice. J Immunol 1997; 158: 1254–61. [PubMed] [Google Scholar]
- 56). Johnson, M. Antibody storage and antibody shelf life. Mater Methods 2012; 2: 120. [Google Scholar]
- 57). Lefranc, MP, Lefranc, G. The Immunoglobulin FactsBook. London, UK: Academic Press, 2001 [Google Scholar]
- 58). Giudicelli, V, Chaume, D, Lefranc, MP. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 2005; 33: D256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59). Smith, K, Shah, H, Muther, JJet al. Antigen nature and complexity influence human antibody light chain usage and specificity. Vaccine 2016; 34: 2813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60). Ponomarenko, N, Chatziefthimiou, SD, Kurkova, Iet al. Role of κ-λ light chain constant-domain switch in the structure and functionality of A17 reactibody. Acta Crystallogr D Biol Crystallogr 2014; 70: 708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61). Townsend, CL, Laffy, JMJ, Wu, YCBet al. Significant differences in physicochemical properties of human immunoglobulin kappa and lambda CDR3 regions. Front Immunol 2016; 7: 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62). Montano, RF, Morrison, SL. Influence of the isotype of the light chain on the properties of IgG. J Immunol 2002; 168: 224–31. [DOI] [PubMed] [Google Scholar]
- 63). Su, CTT, Ling, WL, Lua, WHet al. The role of antibody Vκ framework 3 region towards antigen binding: effects on recombinant production and protein L binding. Sci Rep 2017; 7: 3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Haidar, JN, Yuan, QA, Zeng, Let al. A universal combinatorial design of antibody framework to graft distinct CDR sequences: a bioinformatics approach. Proteins 2012; 80: 896–912. [DOI] [PubMed] [Google Scholar]
- 65). Sedrak, P, Hsu, K, Mohan, C. Molecular signatures of anti-nuclear antibodies—contribution of heavy chain framework residues. Mol Immunol 2003; 40: 491–9. [DOI] [PubMed] [Google Scholar]
- 66). Xiang, J, Sha, Y, Jia, Zet al. Framework residue-71 and residue-93 of the chimeric B72.3 antibody are major determinants of the conformation of heavy-chain hypervariable loops. J Mol Biol 1995; 253: 385–90. [DOI] [PubMed] [Google Scholar]
- 67). Rodríguez-Rodríguez, ER, Ledezma-Candanoza, LM, Contreras-Ferrat, LGet al. A single mutation in framework 2 of the heavy variable domain improves the properties of a diabody and a related single-chain antibody. J Mol Biol 2012; 423: 337–50. [DOI] [PubMed] [Google Scholar]
- 68). Foote, J, Winter, G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol 1992; 224: 487–99. [DOI] [PubMed] [Google Scholar]
- 69). Tramontano, A, Chothia, C, Lesk, A. Framework residue-71 is a major determinant of the position and conformation of the 2nd hypervariable region in the VH domains of immunoglobulins. J Mol Biol 1990; 215: 175–82. [DOI] [PubMed] [Google Scholar]
- 70). Holmes, M, Buss, T, Foote, J. Structural effects of framework mutations on a humanized anti-lysozyme antibody. J Immunol 2001; 167: 296–301. [DOI] [PubMed] [Google Scholar]
- 71). Kettleborough, CA, Saldanha, J, Heath, VJet al. Humanization of a mouse monoclonal antibody by CDR–grafting: the importance of framework residues on loop conformation. Protein Eng Des Sel 1991; 4: 773–83. [DOI] [PubMed] [Google Scholar]
- 72). Banfield, M, King, D, Mountain, Aet al. V-L:V-H domain rotations in engineered antibodies: crystal structures of the Fab fragments from two murine antitumor antibodies and their engineered human constructs. Proteins 1997; 29: 161–71. [DOI] [PubMed] [Google Scholar]
- 73). Nakanishi, T, Tsumoto, K, Yokota, Aet al. Critical contribution of VH-VL interaction to reshaping of an antibody: the case of humanization of anti-lysozyme antibody, HyHEL-10. Protein Sci 2008; 17: 261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74). Stanfield, R, Takimotokamimura, M, Rini, Jet al. Major antigen-induced domain rearrangements in an antibody. Structure 1993; 1: 83–93. [DOI] [PubMed] [Google Scholar]
- 75). Tan, P, Sandmaier, B, Stayton, P. Contributions of a highly conserved VHNL hydrogen bonding interaction to scFv folding stability and refolding efficiency. Biophys J 1998; 75: 1473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76). Essen, L, Skerra, A. The de-novo design of an antibody combining site—crystallographic analysis of the V-L domain confirms the structural model. J Mol Biol 1994; 238: 226–44. [DOI] [PubMed] [Google Scholar]
- 77). Masuda, K, Sakamoto, K, Kojima, Met al. The role of interface framework residues in determining antibody V-H/V-L interaction strength and antigen-binding affinity. FEBS J 2006; 273: 2184–94. [DOI] [PubMed] [Google Scholar]
- 78). Björck, L. Protein L, A novel bacterial cell wall protein with affinity for Ig L chains. J Immunol 1988; 140: 1194–7. [PubMed] [Google Scholar]
- 79). Graille, M, Stura, EA, Housden, NGet al. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure 2001; 9: 679–87. [DOI] [PubMed] [Google Scholar]
- 80). Kastern, W, Sjöbring, U, Björck, L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J Biol Chem 1992; 267: 12820–5. [PubMed] [Google Scholar]
- 81). Nilson, BH, Solomon, A, Björck, Let al. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J Biol Chem 1992; 267: 2234–9. [PubMed] [Google Scholar]
- 82). Ling, WL, Lua, WH, Poh, JJet al. Effect of VH–VL families in Pertuzumab and Trastuzumab recombinant production, Her2 and FcγIIA binding. Front Immunol 2018; 9: 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83). Corti, A, Barbanti, E, Tempest, PRet al. Idiotope determining regions of a mouse monoclonal antibody and its humanized versions: identification of framework residues that affect idiotype expression. J Mol Biol 1994; 235: 53–60. [DOI] [PubMed] [Google Scholar]
- 84). Dondelinger, M, Filée, P, Sauvage, Eet al. Understanding the significance and implications of antibody numbering and antigen-binding surface/residue definition. Front Immunol 2018; 9: 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85). Wu, TT, Kabat, EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 1970; 132: 211–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86). Kabat, EA, Wu, TT. Attempts to locate complementary-determining residues in the variable positions of light and heavy chains. Ann N Y Acad Sci 1971; 190: 382–93. [DOI] [PubMed] [Google Scholar]
- 87). Capra, JD, Kehoe, JM. Variable region sequences of five human immunoglobulin heavy chains of the VH3 subgroup: definitive identification of four heavy chain hypervariable regions. Proc Natl Acad Sci USA 1974; 71: 845–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88). Kabat, EA, Wu, TT, Bilofsky, H. Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J Biol Chem 1977; 252: 6609–16. [PubMed] [Google Scholar]
- 89). Kabat, EA, Wu, TT, Bilofsky, H. Sequences of immunoglobulin chains: tabulation analysis of amino acid sequences of precursors, V-regions, C-regions, J-chain BP-microglobulins. Department of Health, Education, Welfare, Public Health Service, National Institutes of Health, Bethesda, MD, 1979.
- 90). Kabat, EA, Wu, TT, Foeller, Cet al. Sequences of Proteins of Immunological Interest. Collingdale, PA: Diane Publishing Company, 1992 [Google Scholar]
- 91). Martin, AC. Accessing the Kabat antibody sequence database by computer. Proteins 1996; 25: 130–3. [DOI] [PubMed] [Google Scholar]
- 92). Chothia, C, Lesk, AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196: 901–17. [DOI] [PubMed] [Google Scholar]
- 93). Al-Lazikani, B, Lesk, AM, Chothia, C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273: 927–48. [DOI] [PubMed] [Google Scholar]
- 94). Chothia, C, Lesk, AM, Tramontano, Aet al. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342: 877–83. [DOI] [PubMed] [Google Scholar]
- 95). Abhinandan, KR, Martin, ACR. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol 2008; 45: 3832–9. [DOI] [PubMed] [Google Scholar]
- 96). Gelfand, IM, Kister, AE. Analysis of the relation between the sequence and secondary and three-dimensional structures of immunoglobulin molecules. Proc Natl Acad Sci USA 1995; 92: 10884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97). Gelfand, I, Kister, A, Kulikowski, Cet al. Geometric invariant core for the V(L) and V(H) domains of immunoglobulin molecules. Protein Eng 1998; 11: 1015–25. [DOI] [PubMed] [Google Scholar]
- 98). Gelfand, IM, Kister, AE, Leshchiner, D. The invariant system of coordinates of antibody molecules: prediction of the “standard” C alpha framework of VL and VH domains. Proc Natl Acad Sci USA 1996; 93: 3675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99). Gelfand, I, Kister, A, Kulikowski, Cet al. Algorithmic determination of core positions in the VL and VH domains of immunoglobulin molecules. J Comput Biol 1998; 5: 467–77. [DOI] [PubMed] [Google Scholar]
- 100). Tramontano, A, Chothia, C, Lesk, AM. Structural determinants of the conformations of medium-sized loops in proteins. Proteins 1989; 6: 382–94. [DOI] [PubMed] [Google Scholar]
- 101). Giudicelli, V, Chaume, D, Bodmer, Jet al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 1997; 25: 206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102). Lefranc, MP. Unique database numbering system for immunogenetic analysis. Immunol Today 1997; 18: 509. [DOI] [PubMed] [Google Scholar]
- 103). Lefranc, MP, Pommié, C, Ruiz, Met al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol 2003; 27: 55–77. [DOI] [PubMed] [Google Scholar]
- 104). Lefranc, MP, Giudicelli, V, Ginestoux, Cet al. IMGT-ONTOLOGY for immunogenetics and immunoinformatics. In Silico Biol 2004; 4: 17–29. [PubMed] [Google Scholar]
- 105). Ruiz, M, Giudicelli, V, Ginestoux, Cet al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 2000; 28: 219–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106). Ehrenmann, F, Kaas, Q, Lefranc, MP. IMGT/3dstructure-DB and IMGT/domaingapalign: a database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MHcSF. Nucleic Acids Res 2009; 38: 301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107). Lefranc, MP. IMGT collier de perles for the variable (V), constant (C), and groove (G) domains of IG, TR, MH, IgSF, and MhSF. Cold Spring Harb Protoc 2011; 6: 643–51. [DOI] [PubMed] [Google Scholar]
- 108). Brochet, X, Lefranc, MP, Giudicelli, V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 2008; 36: W503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109). Honegger, A, Plückthun, A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol 2001; 309: 657–70. [DOI] [PubMed] [Google Scholar]
- 110). Sun, F, Wang, T, Jiang, Jet al. Engineering a high-affinity humanized anti-CD24 antibody to target hepatocellular carcinoma by a novel CDR grafting design. Oncotarget 2017; 8: 51238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111). Zhang, D, Chen, CF, Zhao, BBet al. A novel antibody humanization method based on epitopes scanning and molecular dynamics simulation. Plos One 2013; 8: e80636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112). Pedersen, JT, Henry, AH, Searle, SJet al. Comparison of surface accessible residues in human and murine immunoglobulin Fv domains: implication for humanization of murine antibodies. J Mol Biol 1994; 235: 959–73. [DOI] [PubMed] [Google Scholar]
- 113). Hurle, MR, Gross, M. Protein engineering techniques for antibody humanization. Curr Opin Biotechnol 1994; 5: 428–33. [DOI] [PubMed] [Google Scholar]
- 114). Winter, GP. Antibody engineering. Phil Trans R Soc Lond B 1989; 324: 537–47. [DOI] [PubMed] [Google Scholar]
- 115). Morea, V, Lesk, AM, Tramontano, A. Antibody modeling: implications for engineering and design. Methods 2000; 20: 267–79. [DOI] [PubMed] [Google Scholar]
- 116). Jones, P, Dear, P, Foote, Jet al. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986; 321: 522–5. [DOI] [PubMed] [Google Scholar]
- 117). Riechmann, L, Clark, M, Waldmann, Het al. Reshaping human antibodies for therapy. Nature 1988; 332: 323–7. [DOI] [PubMed] [Google Scholar]
- 118). Verhoeyen, M, Milstein, C, Winter, G. Reshaping human antibodies: grafting an antilysozyme activity. Science 1988; 239: 1534–6. [DOI] [PubMed] [Google Scholar]
- 119). Kashmiri, SV, De Pascalis, R, Gonzales, NRet al. SDR grafting—a new approach to antibody humanization. Methods 2005; 36: 25–34. [DOI] [PubMed] [Google Scholar]
- 120). Kim, J.H. and Hong, H.J. (2012) Humanization by CDR grafting and specific-determining residue grafting. In: Chames, P. (eds) Antibody Engineering. Methods in Molecular Biology (Methods and Protocols), vol. 907, Totowa, NJ: Humana Press, pp. 237–245. [DOI] [PubMed] [Google Scholar]
- 121). Ratanji, KD, Derrick, JP, Dearman, RJet al. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol 2014; 11: 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122). Bax, HJ, Bowen, H, Beavil, RLet al. IgE trimers drive SPE-7 cytokinergic activity. Sci Rep 2017; 7: 8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123). Barratt, J, Feehally, J. IgA nephropathy. J Am Soc Nephrol 2005; 16: 2088–97. [DOI] [PubMed] [Google Scholar]
- 124). Sanchez-Trincado, JL, Comez-Perosanz, M, Reche, PA. Fundamentals and methods for T-and B- cell epitope prediction. J Immunol Res 2017; 2017: 2680160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125). Maurer-Stroh, S, Krutz, NL, Kern, PSet al. AllerCatPro-prediction of protein allergenicity potential from the protein sequence. Bioinformatics 2019; 35: 3020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126). Wood, RA. Food allergen immunotherapy: current status and prospects for the future. J Allergy Chin Immunol 2016; 137: 973–82. [DOI] [PubMed] [Google Scholar]
- 127). Kim, EH, Yang, L, Ye, Pet al. Long-term sublingual immunotherapy for peanut allergy in children: clinical and immunologic evidence of desensitization. J Allergy Chin Immunol 2019; 144: 1320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128). Sazonovs, A, Kennedy, NA, Moutsianas, Let al. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and Adalimumab in patients with Crohn’s disease. Gastroenterology 2020; 158: 189–99. [DOI] [PubMed] [Google Scholar]
- 129). Swanson, SJ, Bussiere, J. Immunogenicity assessment in non-clinical studies. Curr Opin Microbiol 2012; 15: 337–47. [DOI] [PubMed] [Google Scholar]
- 130). Groell, F, Jordan, O, Borchard, G. In vitro models for immunogenicity prediction of therapeutic protein. Eur J Pharm Biopharm 2018; 130: 128–42. [DOI] [PubMed] [Google Scholar]
- 131). Tuordot, S, Hickling, TP. Nonclinical immunogenicity risk assessment of therapeutic proteins. Bioanalysis 2019; 11: 1631–43. [DOI] [PubMed] [Google Scholar]
- 132). Phua, SX, Chan, KF, Su, CTet al. Perspective: the promises of a holistic view of proteins-impact on antibody engineering and drug discovery. Biosci Rep 2019; 39: BSR20181958. [DOI] [PMC free article] [PubMed] [Google Scholar]