

A membrane protein interacts with PKG and regulates calcium signals at multiple lifecycle stages in malaria parasites.
Abstract
Calcium signaling regulated by the cGMP-dependent protein kinase (PKG) controls key life cycle transitions in the malaria parasite. However, how calcium is mobilized from intracellular stores in the absence of canonical calcium channels in Plasmodium is unknown. Here, we identify a multipass membrane protein, ICM1, with homology to transporters and calcium channels that is tightly associated with PKG in both asexual blood stages and transmission stages. Phosphoproteomic analyses reveal multiple ICM1 phosphorylation events dependent on PKG activity. Stage-specific depletion of Plasmodium berghei ICM1 prevents gametogenesis due to a block in intracellular calcium mobilization, while conditional loss of Plasmodium falciparum ICM1 is detrimental for the parasite resulting in severely reduced calcium mobilization, defective egress, and lack of invasion. Our findings suggest that ICM1 is a key missing link in transducing PKG-dependent signals and provide previously unknown insights into atypical calcium homeostasis in malaria parasites essential for pathology and disease transmission.
INTRODUCTION
Malaria is an important cause of global morbidity and mortality, accounting for around 405,000 deaths in 2018 (1). The disease is caused by parasites of the genus Plasmodium, which have a remarkable life cycle with multiple cellular differentiation events in both humans and mosquitoes. To sense and respond to changes in these very different environments, malaria parasites use an intracellular communication system that relies on intracellular messengers and protein kinases. Two key signaling molecules in these pathways are 3′,5′-guanosine monophosphate (cGMP) and calcium, both of which regulate pivotal processes in most eukaryotes (2, 3).
The sole sensor of cGMP in malaria parasites is the cGMP-dependent protein kinase (PKG), which plays vital roles in most stages of the parasite life cycle, including the asexual blood stages that cause all the manifestations of clinical malaria. These roles include control of cellular events required to trigger release of merozoites from red blood cells (RBCs) (4, 5) and hepatocytes (6), induction of gametogenesis and parasite transmission upon ingestion of gametocytes by a blood-feeding mosquito (7), and sustaining cellular motility necessary for parasite dissemination (8, 9). A major function of PKG in all these processes is the tightly regulated mobilization of calcium from intracellular stores within seconds of activation (8, 10).
In mammalian cells, one of the two PKG isoforms, cGKI (or PKG1), is a well-established regulator of calcium homeostasis. cGKI activation lowers intracellular free calcium upon stimulation by nitric oxide, favoring smooth muscle relaxation or decreasing platelet activation (11). cGKI-dependent phosphorylation is known to regulate levels of cytosolic calcium via distinct mechanisms, including either decreased calcium influx by l-type calcium channels (12) or sequestration of calcium within intracellular stores. Increased calcium sequestration can be mediated by the phosphorylation of (i) the inositol 1,4,5-trisphosphate (IP3) receptor–associated cGMP-kinase substrate (IRAG), which reduces calcium release from intracellular stores by IP3 receptors (13); or (ii) phospholipase-C β3, decreasing IP3 generation and subsequent calcium mobilization (14); or (iii) phospholamban, which enhances calcium sequestration by the sarcoplasmic reticulum calcium/adenosine triphosphatase (ATPase) pump (15). Plasmodium parasites are highly divergent from their mammalian hosts (16), and no genes encoding clear homologs of IRAG, IP3 receptors, phospholamban, or l-type calcium channels have been identified in their genomes (3).
In the absence from Plasmodium of such key signaling molecules, it remains unknown how calcium is mobilized from intracellular stores. We reasoned that the identification of PKG-interacting proteins in Plasmodium would provide valuable new insights into calcium homeostasis. Here, we identify a multipass membrane protein, termed important for calcium mobilization-1 (ICM1; Pf3D7_1231400) that interacts with and is phosphorylated by PKG in two different Plasmodium developmental stages and species. Through stage-specific knockdown and reverse genetic approaches, we show that ICM1 is essential for calcium mobilization in both the clinically relevant asexual blood stages and in gametocytes that mediate transmission to mosquitoes. Our findings highlight this putative transporter or channel as a crucial link between PKG function and calcium signaling.
RESULTS
Plasmodium PKG interacts with ICM1, a multipass membrane protein, in both asexual and sexual stages
To identify potential PKG-interacting partners in Plasmodium falciparum, we used a previously established transgenic parasite line expressing PKG fused at its C terminus to a triple hemagglutinin (HA) tag (PfPKG-HA) (17). PfPKG-HA was immunoprecipitated from extracts of mature schizonts using anti-HA–conjugated magnetic beads and the eluates analyzed by quantitative tandem mass spectrometry (MS/MS). The most enriched protein coimmunoprecipitated with PfPKG-HA was identified with high confidence as Pf3D7_1231400, annotated as a putative amino acid transporter (www.plasmodb.org), but henceforth referred to as ICM1 (Fig. 1A and data S1). To corroborate these findings, we generated a second transgenic P. falciparum line expressing a green fluorescent protein (GFP)–tagged PKG (PKG-GFP) (fig. S1, A to E). Mass spectrometric analysis of pull-downs from extracts of these parasites using anti-GFP nanobodies similarly identified PfICM1 as a top hit (fig. S1F and data S1).
Fig. 1. PKG interacts with ICM1 in P. falciparum and P. berghei schizonts and in P. berghei gametocytes.

(A) Volcano plot depicting relative abundance of proteins identified by quantitative MS in pull-downs from P. falciparum PKG-HA compared to WT (control) parasites (n = 3). Significance (Student’s t test) is expressed as log10 of the P value (y axis). X axis, enrichment of interaction partners in the P. falciparum PKG-HA pull-down compared to controls. (B) Competitive chemoproteomic studies using Kinobeads on extracts of P. falciparum schizonts. (C) Mass spectrometric quantitation of proteins immunoprecipitated from extracts of P. berghei PKG-HA3 (n = 1) and ICM1-HA3 (n = 1) schizonts. ND, not detected (D) Mass spectrometric spectral count values for proteins copurifying with PbPKG-HA3 (n = 2) and PbICM1-HA3 (n = 2) gametocytes following IP and displayed in the first and second principal components compared with previously published IPs of CRK5, CDKrs, SOC2, CDPK4, SOC1, SOC3, and MCM5.
To independently validate the putative interaction between ICM1 and PKG, we took an orthogonal chemical proteomics approach. A potent PKG inhibitor (18) (ML10) was used on a Kinobead matrix (19) to identify the repertoire of P. falciparum proteins targeted by this compound. Proteomic profiling showed that ML10 prevented the binding of just four parasite proteins to kinase inhibitors; these were PfPKG, two calcium-dependent protein kinases (CDPK1 and CDPK4), and PfICM1. The selectivity of ML10 for Plasmodium PKG exploits the presence of a small threonine gatekeeper residue in the enzyme adenosine triphosphate binding pocket (18). As both CDPK1 and CDPK4 also have small serine gatekeeper residues (20), it is likely that ML10 binds these two kinases directly. However, identification of a nonkinase hit in the Kinobead experiments likely reflects either off-target effects of the compound or capture of kinase-associated proteins. These results, combined with the pull-down data, fully support a direct interaction between PKG and ICM1 (Fig. 1B and data S1).
To interrogate whether the interaction between PKG and ICM1 is conserved across Plasmodium species and developmental stages, we exploited a previously described transgenic Plasmodium berghei line expressing HA3-tagged PKG (PbPKG-HA3) (8) to immunoprecipitate PbPKG-HA3 from extracts of schizonts and gametocytes. Quantitative MS showed that in both stages, PbICM1 (PBANKA_1446100) was the most enriched protein coimmunoprecipitated with PbPKG-HA3 (Fig. 1, C and D, and data S1). To further validate these results in a reciprocal manner, we generated a PbICM1-HA3 line (fig. S1G). No signal was observed for PbICM1-HA3 in Western blot analysis of schizont or gametocyte extracts from this line, possibly due to low abundance of PbICM1. Consistent with this observation, no specific immunofluorescence signal could be observed in both schizonts and gametocytes. Nevertheless, immunoprecipitation (IP) of PbICM1-HA3 from extracts of both schizonts and gametocytes recovered multiple peptides, identified by MS to encompass up to 28% coverage of PbICM1. As expected, based on the PbPKG IPs, PbPKG was the most enriched protein coimmunoprecipitated with PbICM1-HA3 in both stages (Fig. 1, C and D, and data S1). Notably, no peptides from PbCDPK1 or PbCDPK4 were recovered from either immunoprecipitate, confirming that these kinases do not interact with PbPKG or PbICM1 as previously suggested (10).
To further characterize the specificity of PbPKG and PbICM1 co-IPs in gametocytes, we compared the relative abundance of proteins recovered in both immunoprecipitates with those of seven other proteins involved in gametogenesis (21). Nine-dimensional principal components analysis (PCA) of all detected proteins confirmed the clustering of PbPKG and PbICM1, consistent with the formation of a complex (Fig. 1D). Together, these results demonstrate a conserved interaction between ICM1 and PKG in at least two developmental stages of two different Plasmodium species.
Modeling of ICM1 shows resemblance to transporters and channels
ICM1 is a multipass membrane protein annotated as a putative amino acid transporter. A single ICM1 ortholog is present in all sequenced Plasmodium genomes, but no orthologs are evident in nonapicomplexan genomes. The N-terminal segment, which encompasses two conserved clusters of five and four transmembrane domains (TMDs), respectively, shows high levels of identity between the P. falciparum and P. berghei orthologs, and these nine TMDs are also conserved in Plasmodium yoelii, Plasmodium vivax, Plasmodium chabaudi, and Plasmodium knowlesi (data S2). This region is separated from a predicted C-terminal TMD (TMD10) by ~1100 amino acid residues that are less conserved between all species. Homology modeling using Phyre2 (22) suggests weak similarity of the ICM1 N terminus to cation transporters, amino acid transporters, or IP3 receptors (fig. S2A). To investigate this further, the Iterative Threading ASSEmbly Refinement (I-TASSER) approach was used to model the N-terminal 1500 residues of P. falciparum ICM1. Following structure assembly simulation, the Protein Data Bank (PDB) was screened for proteins with the closest structural similarity to the model. The top PDB hit was an IP3 receptor (rat cerebellum InsP3R1, PDB: 6MU1) with a template modelling score of 0.967, a root mean square deviation of 2.06 Å, and a coverage of 98.7%, although amino acid identity between the two proteins was very low (8.4%) (fig. S2B). However, our analyses did not identify any domains similar to other calcium channels such as IP3-binding domains or Ca2+ binding sites. Collectively, these findings suggest that ICM1 may represent a structurally divergent polytopic membrane protein with architectural similarities to mammalian channels, transporters, and IP3 receptors.
ICM1 is phosphorylated in a PKG-dependent manner
The physical interaction between PKG and ICM1 raised the possibility of PKG regulating the function of ICM1 through phosphorylation. Several global phosphoproteome studies in Plasmodium have identified multiple phosphorylation sites between TMD9 and TMD10 in both P. falciparum ICM1 (23–25) and PbICM1 (8, 26). In a previous study, pharmacological inhibition of PKG activity in mature schizonts did not detect any differential ICM1 phosphorylation under the tested conditions (25). However, a phosphoproteome analysis of P. berghei guanylyl cyclase beta (GCβ) mutant ookinetes, in which PKG activity is down-regulated (27), showed that PbICM1-S1288 was significantly less phosphorylated in this transgenic line (8). To shed more light on PKG-dependent phosphorylation of ICM1, we profiled the PKG-dependent phosphoproteomes in both gametocytes and schizonts. For the former, we examined phosphorylation events just 15 s after activation of gametocytes in the presence or absence of a selective PKG inhibitor (compound A) (10, 28), as PbPKG activity during gametogenesis is only required during the first 15 s following stimulation by xanthurenic acid (XA) (8, 10). Compound A was used at 1 μM, a concentration that did not impair exflagellation of a line expressing a resistant PKGT619Q-HA allele indicating the selectivity of this small molecule under these experimental conditions (10). In the case of P. falciparum schizonts, we generated a conditional knockout (cKO) of PKG to investigate the phosphoproteome changes in PKG-null mutants. Treatment of this line, termed pfpkg:cKO, with rapamycin (RAP) results in disruption of the pfpkg gene, expression of mCherry, and complete arrest of parasite replication due to the requirement of PKG for merozoite egress (fig. S3 and movie S1) (5).
Given the low abundance of PbICM1 in gametocytes, we used SDS–polyacrylamide gel electrophoresis (PAGE) fractionation to enrich for proteins of molecular mass between 180 and 220 kDa. Analysis of these proteins identified 957 phosphopeptides mapping onto 610 proteins (data S3). Upon pharmacological inhibition of PbPKG, 410 phosphopeptides were less phosphorylated, a large response amplitude that probably reflects the early requirement for PbPKG during gametogenesis (Fig. 2A). Conditional disruption of PfPKG had a similarly profound effect on the P. falciparum global schizont phosphoproteome, with more than 1000 phosphopeptides being significantly hypophosphorylated (Fig. 2B and data S4). Gene Ontology (GO) term enrichment analysis of hypophosphorylated proteins in P. berghei gametocytes suggested deregulation of both synthesis and mitosis cell cycle phases, which are initiated by calcium mobilization (Fig. 2C and data S3) (26, 29, 30). A similar analysis of the hypophosphorylated proteins in P. falciparum schizonts suggested potential defects in RNA processing and splicing (Fig. 2D and data S4).
Fig. 2. ICM1 is phosphorylated in a PKG-dependent manner.
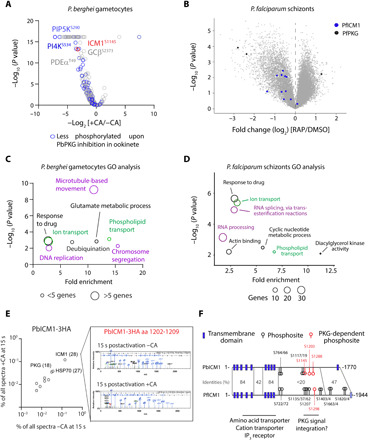
(A) Volcano plot showing the extent of differentially phosphorylated peptides in P. berghei gametocytes 15 s after XA stimulation in presence or absence of compound A (CA) (n = 3). (B) Volcano plots showing differentially phosphorylated peptides in DMSO- and RAP-treated pfpkg:cKO schizonts (n = 5). Blue dots correspond to identified PfICM1 phosphosites and black dots to PfPKG phosphosites. (C and D) GO term enrichment analysis for down-regulated phosphopeptides in 15-s activated gametocytes upon inhibition of PbPKG by compound A and of PfPKG-null schizonts. For the latter, a cutoff of Welch difference of <−1 and >1 with P <0.05 and a localization probability of >0.7 represent a significantly regulated peptide. Representative terms are shown (for a full list of GO term enrichment analysis in PfPKG-null schizonts, see data S4). (E) Relative abundance of proteins coprecipitated with PbICM1-HA3 in 15-s activated gametocytes in the presence or absence of compound A. Inset: Representative spectra indicating that phosphorylation of PbICM1-S1203 is only detected in absence of compound A. (F) Schematic of PbICM1 and PfICM1 indicating predicted TMDs and phosphosites detected in this and previous studies.
The previously identified cGMP-dependent phosphorylation of enzymes involved in the metabolism of inositol phospholipids in P. berghei ookinetes (8, 10) was confirmed here for both schizonts and gametocytes. We then focused our attention on ICM1 phosphorylation. Twelve PfICM1 phosphorylation sites were mapped in P. falciparum schizonts (Fig. 2B). Ten sites were hypophosphorylated in the absence of PfPKG, suggesting a general defect in phosphorylation of PfICM1 although only two sites (S1289 and S1875) exhibited a fold change of more than 1.8. In gametocytes, intensities of the eight nonphosphorylated peptides mapped onto PbICM1 were not affected by compound A, indicating that PbICM1 abundance is not dependent on PbPKG activity. We identified two PbICM1 phosphosites, of which S1145 was significantly down-regulated upon PbPKG inhibition. Phosphorylation of PbICM1-S1145 was previously shown to be up-regulated within seconds of gametocyte activation by XA (26) and so possibly represents an important event in PbICM1 regulation. We additionally interrogated the phosphorylation status of immunoprecipitated PbICM1-HA3 in gametocytes 15 s after activation, in the presence or absence of 1 μM compound A (Fig. 2E). Treatment with compound A had no effect on the interaction between PbPKG and PbICM1-HA3 or on the obtained coverage of PbICM1-HA3. However, S1203 was the only residue detected as phosphorylated following gametocyte activation by XA, and no phosphorylation of PbICM1-HA3 was detected upon inhibition of PbPKG. This suggests that ICM1-S1203 is also phosphorylated early during gametogenesis in a PKG-dependent manner.
Collectively, our results indicate that the phosphorylation of at least five ICM1 residues depends on PKG activation. Four of the detected PKG-dependent phosphorylation sites are clustered in a region encompassing residues Pf1162 (Pb1145) to Pf1357 (Pb1288), suggesting that this represents an important region for regulation of ICM1 function (Fig. 2F).
Stage-specific knockdown of PbICM1 reveals a crucial role in early calcium mobilization required to initiate gametogenesis
To study the function and essentiality of PbICM1 during gametogenesis, we first attempted to disrupt the pbicm1 gene. The gene was previously suggested to be resistant to disruption in a global gene KO study (31), although a transient nonclonal population of parasites lacking icm1 had been observed in the days following transfection in an independent study (32). We were here however unable to obtain a clonal icm1-KO population despite four independent attempts using highly efficient PlasmoGEM vectors (33, 34). We then tagged endogenous ICM1 with an auxin-inducible degron (AID) coupled to an HA epitope tag (fig. S4A) to allow degradation of the fusion protein in the presence of auxin in a strain expressing the Tir1 protein (35). Addition of auxin to PbICM1-AID/HA gametocytes did not lead to any defect in male gametogenesis. However, we could detect no Western blot signal for PbICM1-AID/HA; thus, we were unable to confirm successful degradation of the protein upon auxin treatment (fig. S4B). We thus opted for a stage-specific knockdown of PbICM1 by placing the endogenous pbicm1 gene under the control of the pbama1 promoter, which is active in schizonts but virtually silent in gametocytes (36). A Pama1ICM1 clone was readily obtained and reverse transcription quantitative polymerase chain reaction (PCR) analysis confirmed a two-fold reduction in pbicm1 expression in purified Pama1ICM1 gametocytes (Fig. 3A).
Fig. 3. Stage-specific knockdown of PbICM1 partially phenocopies PKG inhibition.
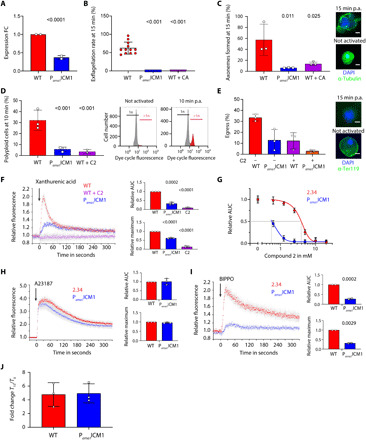
(A) Relative levels of pbicm1 mRNA in WT and Pama1ICM1gametocytes. FC, fold change. (B) Knockdown of PbICM1 impairs exflagellation. (C) Pama1ICM1 male gametocytes show a strong reduction in fully assembled axonemes. Insets: Representative images. Scale bars, 2 μm. (D) Proportion of male gametocytes undergoing DNA replication determined at 1 min postactivation (p.a.) and expressed as a percentage of polyploid (>1n) cells. Insets: Gating used for the analysis. (E) Egress from RBCs was quantified by flow cytometry based on the presence of the RBC membrane marker Ter-119 in gametocytes, 15 min p.a. Insets: IFA of WT and Pama1ICM1 gametocytes activated 15 min p.a. Scale bars, 2 μm. (F) Fluorescence response kinetics of gametocytes loaded with Fluo-4 AM upon stimulation with 100 μM XA. AUC, area under each curve. (G) Relative calcium response to 100 μM XA of WT and Pama1ICM1 gametocytes upon treatment with compound 2. (H) Fluorescence response of gametocytes loaded with Fluo-4 AM upon stimulation with A23187. (I) Fluorescence response of gametocytes loaded with Fluo-4 AM upon stimulation with BIPPO. (J) cGMP levels in WT and Pama1ICM1 gametocytes upon stimulation with BIPPO. Error bars, ±SD; numbers of independent replicates are indicated for each experiment; two-tailed unpaired t test.
To determine the requirement for PbICM1 during gametogenesis, we then compared the effect of pbicm1 down-regulation with PKG inhibition. Although Pama1ICM1 gametocytes were microscopically indistinguishable from their wild-type (WT) counterparts, they did not form active exflagellation centers upon treatment with XA, phenocopying PKG-specific inhibition by 1 μM compound A (Fig. 3B). A similar result was observed when pbicm1 was placed instead under the control of the clag9 promoter, which similarly down-regulates pbicm1 in gametocytes (fig. S4, C to E). In addition, PbICM1 down-regulation or PbPKG inhibition both led to significant decreases in axoneme formation, egress from the host RBC, and DNA replication, indicating an early requirement for PbICM1 in male gametogenesis (Fig. 3, C to E).
We previously showed that PKG is critical for mobilization of calcium from intracellular stores within seconds of gametocyte stimulation by XA (8). Consistent with a functional link between PbPKG and PbICM1, a strongly attenuated calcium mobilization was consistently observed in Pama1ICM1 and Pclag9ICM1 gametocytes (Fig. 3F and fig. S4F). The residual signal observed in Pama1ICM1 gametocytes could possibly be due to incomplete silencing of icm1 or due to PbICM1-independent pathways downstream of PbPKG also involved in calcium mobilization. The residual calcium mobilization observed in Pama1ICM1 gametocytes was 10 times more sensitive to PKG inhibition by compound 2 compared with WT parasites, further strengthening the evidence for a functional interplay between PbPKG and PbICM1 (Fig. 3G). Pama1ICM1 and Pclag9ICM1 gametocytes responded normally to the calcium ionophore A23187, indicating similar levels of mobilizable intracellular calcium in Pama1ICM1 and WT gametocytes (Fig. 3H and fig. S4G).
To define the functional requirement for PbICM1 in the first seconds of gametogenesis, we took advantage of a phosphodiesterase (PDE) inhibitor, BIPPO, which elevates levels of cGMP in Plasmodium by preventing its degradation and leading to PKG activation (37). As expected, elevation of cGMP by BIPPO led to calcium release in WT gametocytes. In stark contrast, the calcium response to PDE inhibition was strongly reduced in the Pama1ICM1 line (Fig. 3I and fig. S4H). However, chemical inhibition of PDEs and XA stimulation induced comparable cGMP levels in both WT and Pama1ICM1 gametocytes, indicating that PbICM1 is not required to control cGMP homeostasis upstream of PKG (Fig. 3J). Together, these results indicate that PbICM1 is required for PKG-dependent mobilization of intracellular calcium.
Loss of PfICM1 in asexual blood stages inhibits calcium mobilization leading to inefficient egress and invasion
To study the function of PfICM1 and its link to PKG function in P. falciparum asexual blood stages, we investigated the phenotypic consequences of PfICM1 disruption. Because pficm1 was predicted to be essential (38), we decided to disrupt the gene conditionally using the RAP-inducible DiCre system (39). Generation of the pficm1:cKO line was achieved in two steps using a marker-free Cas9-mediated strategy. In the first of these, we modified the 3′ end of the gene by fusing it to a C-terminal HA3 epitope tag followed by a loxN site, a T2A peptide and an out-of-frame GFP sequence (fig. S5, A and B). The pficm1:HA parasites displayed normal replication rates, indicating that the modification had no impact on parasite fitness (fig. S5C). In a second gene manipulation step, we replaced intron 1 of pficm1 in the pficm1:HA line with a 2loxPint element (5), creating line pficm1:cKO (fig. S5D). DiCre-mediated recombination between the two loxN sites (one within the 2loxPint and one downstream of the HA tag) was predicted to severely truncate the pficm1 gene while simultaneously positioning the downstream GFP in frame, enabling its expression as a reporter of successful excision (fig. S5, E and F). Immunofluorescence (IFA) analysis of dimethyl sulfoxide (DMSO)– and RAP-treated schizonts of line pficm1:cKO alongside a WT control did show evidence of protein regulation but the very low levels of protein expression prevented us from localizing the protein (fig. S5G).
Monitoring of DMSO- and RAP-treated pficm1:cKO parasites under standard static culture conditions showed complete arrest of growth in the RAP-treated cultures (Fig. 4A). RAP-treated parasites formed morphologically normal mature schizonts at the end of the erythrocytic cycle of treatment (cycle 0), indicating no effects on intracellular development. However, very few schizonts from the RAP-treated cultures underwent egress, resulting in a significant reduction in newly invaded rings (Fig. 4B). To seek further insights, we monitored the behavior of DMSO- and RAP-treated pficm1:cKO schizonts by time-lapse video microscopy. As shown in Fig 4C and movie S2, PfICM1-null schizonts exhibited an abnormal phenotype, with either complete loss of egress or rounding up with increased intracellular merozoite mobility (indicative of parasitophorous vacuole membrane rupture (40)) but no obvious rupture of the RBC membrane, with very few merozoites escaping the confines of the RBC. Quantification of the videos showed fewer egress events and a significant delay in time to egress for the PfICM1-null parasites (Fig. 4, D and E). No examples were observed of the explosive egress typical of WT parasites, although following prolonged incubation free PfICM1-null merozoites were visible (Fig. 4B and fig. S6A). To further characterize the phenotype, we compared egress of both DMSO- and RAP-treated pfpkg:cKO and pficm1:cKO schizonts by monitoring the appearance in culture supernatants of the parasitophorous vacuole protein Serine Repeat Antigen 5 (SERA5). No release of SERA5 occurred in cultures of PfPKG-null parasites, while in marked contrast processed SERA5 appeared in the medium of PfICM1-null parasites, albeit with a delay compared to DMSO-treated parasites (fig. S6, B and C). Because proteolytic processing of SERA5 requires discharge of the protease SUB1 from exonemes (41), this result suggests that protein discharge from these secretory organelles is not affected by loss of ICM1. No differences in cellular cGMP levels were detectable in DMSO- and RAP-treated pficm1:cKO parasites (Fig. 4F). Collectively, these results suggested a role for PfICM1 in egress downstream of PKG activation.
Fig. 4. PfICM1 is essential for parasite viability, microneme discharge, efficient egress, and invasion.
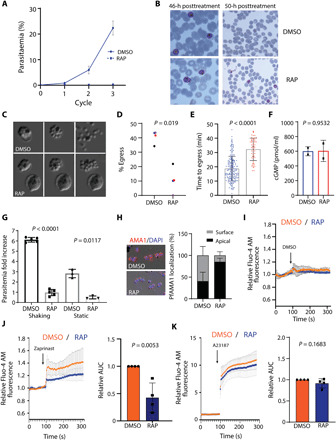
(A) Replication of DMSO- and RAP-treated pficm1:cKO parasites (n = 3). (B) Giemsa-stained blood films showing normal development of DMSO- and RAP-treated pficm1:cKO schizonts 46 hours after treatment. By 50 hours, most control parasites had successfully invaded while very few rings formed in RAP-treated cultures. (C) Stills from time-lapse differential inference contrast (DIC) video microscopy of DMSO/RAP-treated pficm1:cKO schizonts. (D) Egress quantification of DMSO- and RAP-treated pficm1:cKO schizonts. (n = 4). P value derived from paired t tests. (E) Quantification of time to egress for each schizont. P value, unpaired two-tailed t test (DMSO, 209 schizonts; RAP, 55 schizonts). Error bars, ±SD. (F) cGMP levels in DMSO- and RAP-treated pficm1:cKO schizonts (n = 2). P value, paired t test. (G) Invasion assays of DMSO- and RAP-treated pficm1:cKO schizonts. P values, paired t test. (H) Left: Representative IFA images of pficm1:cKO parasites showing translocated AMA1 in DMSO-treated or micronemal AMA1 in RAP-treated merozoites. Right: Quantification of AMA1 relocalization (n = 3). At least 150 schizonts were quantified in each. Values, means ± SD. (I to K) Relative Fluo-4 AM fluorescence response of DMSO- and RAP-treated pficm1:cKO schizonts after treatment with DMSO, zaprinast, or A21387 (n = 4). For the AUC, DMSO values were normalized to 1. Values, means ± SD. P values, two tailed t test.
Given the loss of long-term viability in static cultures of PfICM1-null parasites, we addressed whether the egress defect could be overcome by mechanical shear stress. As shown in Fig. 4G, even under shaking conditions, production of new rings was severely compromised in RAP-treated pficm1:cKO cultures. This prompted us to examine release of other secretory organelle proteins and in particular of AMA1, an essential invasion protein that translocates from micronemes to the surface of daughter merozoites at or just before the point of egress (42). As shown in Fig. 4H, comparative IFA revealed a severe defect in AMA1 discharge in the PfICM1-null schizonts. It was concluded that this likely explains the loss of invasion in the PfICM1-null parasites.
Release of micronemal contents has been linked to a calcium signaling pathway essential for egress and invasion, involving at least one calcium-dependent protein kinase, PfCDPK5 (43). To address the question of whether loss of ICM1 leads to a defect in calcium mobilization, we used the PDE inhibitor zaprinast to artificially activate PKG by prematurely increasing cGMP levels, forcing a PKG-dependent increase in cytosolic calcium levels (8). Addition of DMSO (vehicle control) to both WT and PfICM1-null parasites showed no increase in calcium levels (Fig. 4I). Calcium levels in PfICM1-null parasites exposed to zaprinast were significantly reduced compared to controls, similar to our findings with Pama1ICM1 gametocytes. In contrast, treatment with the calcium ionophore A23187 had similar effects in both DMSO- and RAP-treated pficm1:cKO schizonts, suggesting that intracellular calcium levels are unaffected by loss of ICM1 (Fig. 4, J and K). Our results indicate that PfICM1 is involved in downstream transduction of calcium signals essential for egress and invasion in asexual blood stages.
PfICM1 and PfPKG-dependent phosphoproteomes suggest functional overlap in schizonts
Given the physical interaction between ICM1 and PKG in P. falciparum schizonts and their roles in calcium mobilization, we further explored the functional link between the two proteins by determining the impact of ICM1 depletion on the global schizont phosphoproteome. We reasoned that if ICM1 acts downstream of PKG and if its function is regulated directly or indirectly by PKG-dependent phosphorylation, then an overlap would be expected between the two phosphoproteomes.
Using an approach similar to that previously used for PfPKG, we identified 106 phosphosites from 75 proteins that are significantly hypophosphorylated upon disruption of PfICM1 (Fig. 5A and data S4). Proteins involved in cyclic nucleotide-dependent signaling cascades were among this set, including adenylyl cyclase beta (ACβ), GCα, and CDPK1. Motif analysis of the hypophosphorylated phosphopeptides showed a strict preference for lysine in position −3, resembling a minimal cAMP-dependent protein kinase (PKA)/PKG recognition motif (KxxpS/pT) (Fig. 5B). A similar substrate preference was also described in a recent study on the impact of CDPK5 depletion on the schizont phosphoproteome (44). GO term enrichment analysis of this set of 75 proteins showed deregulation of cyclic nucleotide biosynthesis, ATPase activity and RNA splicing (GO:0000375), as also observed in the PfPKG phosphoproteome (Fig. 5C and data S4). Comparison of the two phosphoproteome sets identified 73 phosphosites in 67 proteins that are regulated by both PKG and ICM1 in schizonts (Fig. 5D and data S4). These include proteins involved in signaling as well as in egress and invasion, such as CDPK1, DGK1 (diacylglycerol kinase), ACβ, RhopH3 (high molecular weight rhoptry protein 3), and AMA1 (apical membrane antigen 1). Collectively, these data strongly support a functional link between the essential roles of PKG and ICM1.
Fig. 5. Impact of PfICM1 disruption on the schizont phosphoproteome.
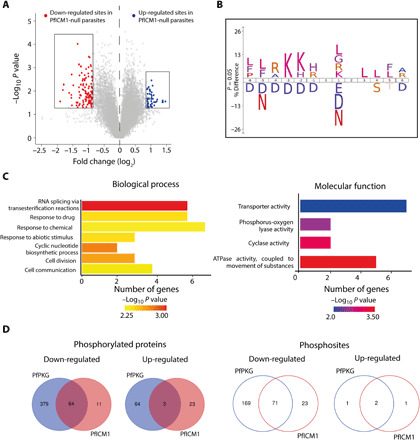
(A) Volcano plot showing distribution of phosphopeptide abundance in the presence or absence of PfICM1. Red circles correspond to significantly down-regulated peptides and blue circles to significantly up-regulated peptides. A cutoff of Welch difference of <−0.875 and >0.875 with P <0.05 and a localization probability of >0.7 represent significantly regulated peptides. (B) Motif analysis of the significantly down-regulated phosphosites in PfICM1-null parasites. All detected phosphopeptides were used as the reference dataset, with amino acids below the position line indicating residues unfavored for this position. Position 0 corresponds to the phosphorylated residue. (C) GO enrichment analysis of biological process and molecular function for proteins significantly hypophosphorylated in PfICM1-null parasites. Enriched terms with P < 0.01 are shown. (D) Venn diagram showing the number of phosphoproteins and the corresponding phosphopeptides significantly deregulated by PfPKG and PfICM1.
DISCUSSION
Many genetic and chemical genetic studies have shown a clear requirement for PKG in specific calcium signals or calcium-dependent pathways (7, 8, 10, 41, 43), but the molecular partners involved in calcium mobilization have remained elusive (3). Here, we have identified ICM1 as a tightly associated PKG partner protein that plays a key role in calcium signals in both schizonts and gametocytes. Our data suggest that ICM1 mainly acts downstream of PKG, because ICM1 does not seem to be required for calcium homeostasis during intraerythrocytic development but only for PKG-mediated calcium signals leading to egress and activation of gametogenesis. This suggests a temporally restricted role in calcium homeostasis across the Plasmodium life cycle. Future experiments will be needed to determine the impact of ICM1 ablation on CDPK activation in both asexual and sexual stages. Two putative orthologs of icm1 are predicted in Toxoplasma gondii, a related apicomplexan parasite, which also relies on PKG-dependent calcium signals upstream of CDPKs to control egress, motility, and invasion. Could ICM1 be the long-sought Plasmodium IP3 receptor or could it act as a calcium channel? Our modeling data suggest that ICM1 resembles a channel but the low amino acid sequence similarity indicates that if it is a calcium channel, then it is highly divergent from other well-studied calcium channels. ICM1 could possibly represent an evolutionary alternative pathway functionally replacing the IP3-R and the associated IRAG proteins that bridge PKG and calcium in human cells. Recombinant and/or heterologous expression and biophysical analysis will be needed to evaluate the function of ICM1 as a calcium channel and to investigate its regulation by IP3, other secondary messengers or even by PKG-dependent phosphorylation.
Interactions between kinases and other proteins are a common signaling mechanism, with the A-kinase anchoring proteins that bind to PKA or the mitogen-activated protein kinase scaffolding proteins being some of the best studied examples (45, 46). In contrast, little is known about proteins interacting with PKG in eukaryotes as few partners have been identified (13, 47–49). In Plasmodium, the only previous data available for a PKG-interacting protein came from an in vitro study using the kinase domain of PKG (50), while a putative interaction between PKG and ICM1 was detected in a very recent elegant study profiling a potent inhibitor of PKG. However, conditional knockdown of ICM1 in that work resulted only in a minor growth defect, perhaps due to the different conditional system used (51), the TetR-DOZI aptamer system, which may require more than one cycle to achieve efficient down-regulation (52). The phenotype observed in our study could be a consequence of the high efficiency of gene disruption obtained with the DiCre system, combined with the fact that gene disruption is at the genomic rather than the transcriptional level.
Our identification of the PKG-ICM1 interaction creates new and exciting questions regarding calcium homeostasis in apicomplexan parasites and further strengthens the notion that calcium regulation in this phylum could be regulated by a class of divergent channels.
MATERIALS AND METHODS
Ethics statement
All animal experiments were conducted with the authorization numbers GE/82/15 and GE/41/17, according to the guidelines and regulations issued by the Swiss Federal Veterinary Office.
Reagents
WR99210 was from Jacobus Pharmaceuticals (New Jersey, USA). RAP was from Sigma-Aldrich and used at 20 nM. The PKG inhibitors compound 2 (4-[7-[(dimethylamino)methyl]-2-(4-fluorphenyl)imidazol[1,2-α]pyridine-3-yl]pyrimidin-2-amine) and compound A (1,4-cyclohexanediamine, N-[3-[2-[(3-fluoro-2-pyridinyl)amino]-5-pyrimidinyl]imidazo[1,2-b]pyridazin-6-yl], trans-) were stored as a 10 mM stock in DMSO at −20°C, while working dilution was 1 μM. The calcium ionophore A23187 and zaprinast were both from Sigma-Aldrich and used at 10 and 100 μM, respectively.
P. falciparum culture, transfection, and synchronization
All P. falciparum lines were maintained at 37°C in human RBCs in RPMI 1640 containing AlbuMAX II (Thermo Fisher Scientific), and medium was supplemented with 2 mM l-glutamine. Parasite synchronization and transfections were performed as previously described (53). Clonal lines were obtained by serial limiting dilution in flat bottomed 96-well plates, and single plaques were selected and grown in the presence of 1 μM 5-fluorocytosine (provided as clinical grade Ancotyl) to select for Cas9 plasmid-free and marker-free parasites. For parasite genomic DNA (gDNA) extraction, the Qiagen DNeasy Blood and Tissue kit was used. Genotype analysis PCR was done using Phusion polymerase (New England Biolabs). For P. falciparum experiments, DiCre activity was induced by RAP treatment of early rings (2 to 3 hours after invasion) as previously described (54). Samples for excision PCRs were collected 24 hours after RAP treatment.
P. berghei maintenance, purification, and transfection
P. berghei maintenance, purification, and transfection were performed as previously described (21). P. berghei ANKA strain (36) derived clones 2.34 (27) and 615 (35), together with derived transgenic lines, were grown and maintained in CD1 outbred mice. Six- to 10-week-old mice were obtained from Charles River Laboratories, and females were used for all experiments. Mice were specific pathogen free (including Mycoplasma pulmonis) and subjected to regular pathogen monitoring by sentinel screening. They were housed in individually ventilated cages furnished with a cardboard mouse house and Nestlet, maintained at 21° ± 2°C under a 12-hour light/dark cycle, and given commercially prepared autoclaved dry rodent diet and water ad libitum. The parasitemia of infected animals was determined by microscopy of methanol-fixed Giemsa-stained thin blood smears.
For gametocyte production, parasites were grown in mice that had been phenyl hydrazine-treated 3 days before infection. One day after infection, sulfadiazine (20 mg/liter) was added in the drinking water to eliminate asexually replicating parasites. Microgametocyte exflagellation was measured 3 or 4 days after infection by adding 4 μl of blood from a superficial tail vein to 70 μl of exflagellation medium [RPMI 1640 containing 25 mM Hepes, 4 mM sodium bicarbonate, 5% fetal calf serum (FCS), and 100 μM XA (pH 7.4)]. To calculate the number of exflagellation centers per 100 microgametocytes, the percentage of RBCs infected with microgametocytes was assessed on Giemsa-stained smears. For gametocyte purification, parasites were harvested in suspended animation (SA) medium [RPMI 1640 containing 25 mM Hepes, 5% FCS, and 4 mM sodium bicarbonate (pH 7.20)] and separated from uninfected RBCs on a Histodenz (Sigma-Aldrich) cushion made from 48% of a Histodenz stock [27.6% (w/v) Histodenz in 5.0 mM tris-HCl, 3.0 mM KCl, and 0.3 mM EDTA (pH 7.20)] and 52% SA (final pH 7.2). Gametocytes were harvested from the interface. To induce ICM1-AID/HA degradation, 1 mM auxin dissolved in ethanol was added to purified gametocytes for 1 hour.
Schizonts for transfection were purified from overnight in vitro culture on a Histodenz cushion made from 55% of the Histodenz stock and 45% phosphate-buffered saline (PBS). Parasites were harvested from the interface and collected by centrifugation at 500g for 3 min, resuspended in 25 μl of Amaxa Basic Parasite Nucleofector solution (Lonza), and added to 10 to 20 μg of DNA dissolved in 10 μl of H2O. Cells were electroporated using the FI-115 program of the Amaxa Nucleofector 4D. Transfected parasites were resuspended in 200 μl of fresh RBCs and injected intraperitoneally into mice. Parasite selection with pyrimethamine (0.07 mg/ml; Sigma-Aldrich) in the drinking water (pH ~4.5) was initiated 1 day after infection.
Plasmid construction and genotyping of P. falciparum transgenic lines
Oligonucleotides used in this study are shown in table S1. To create line PKG-GFP, the triple HA tag from plasmid pHH1-PfPKG-HA (17) was replaced by enhanced GFP (eGFP). Briefly, eGFP was PCR-amplified from plasmid pBAT-SIL6 (55) using primers GFP_For and GFP_Rev. The PCR product was digested with Avr II and Xho I and ligated to vector pHH1-PfPKG-HA previously digested with the same enzymes. Plasmid was transfected into 3D7 P. falciparum, and growth medium was replaced 24 hours after transfection with fresh medium containing 2.5 nM WR99210. Parasites were subjected to culture in the absence of WR99210 (3 cycles) followed by culturing with drug to select for transgenic parasites. Clonal lines were confirmed by PCR using primers wtpkg_For and 3utr_Rev and maintained in the presence of WR99210.
The DiCre-expressing B11 line (39) was used to generate line icm:cKO in two steps. The first step aimed at modifying the 3′ end of the gene. To achieve that, a commercially obtained construct (GeneArt, Thermo Fisher Scientific) contained in tandem: (i) a 5′ homology arm of 456–base pair (bp) endogenous and 255 bp of synthetic sequence comprising the 3′ end of exon 2 and the whole of exon 3; (ii) a fragment comprising a triple HA tag, loxN, and the 3′ 46 bp of the sera2 intron; and lastly, (iii) the first 408 bp of pficm1 3′ untranslated region (3′UTR) as a 3′ homology arm. This plasmid (pMX_icm1-int) was digested with Sal I/Sac I and a T2A peptide followed by a GFP gene (fragment obtained from plasmid pT2A_D1_cKO, a gift from E. Deu) was cloned in after the 3′ end of the sera2 intron, resulting in plasmid pMX_icm1_2. A single guide RNA (sgRNA) targeting sequence TTGATTAAATAAATATATAA was inserted into a previously described pDC2 plasmid expressing Cas9, resulting in plasmid pDC2-icm1_2g. The repair plasmid (pMX_icm1_2) was linearized overnight with Bgl II and transfected in B11 parasites together with plasmid pDC2_icm1_2g. Integration was confirmed by PCR, using primer pairs L2_For/L2_Rev and L2_For/L2_HA. This clonal line (icm1:HA) was obtained via limiting dilution and was used for the second modification step, which involved replacing the endogenous intron 1 with the 2loxPint module (5). A commercially obtained construct (GeneArt, Thermo Fisher Scientific) was used (pMX_icm1_2loxPint), containing the 2loxPint module flanked by approximately 400 bp of endogenous sequence as homology arms. This vector was linearized with Bgl II and cotransfected with a pDC2-based Cas9 plasmid (56), harboring the sgRNA targeting sequence ATTTCAAAGGACTTACTTTA, in line icm1_HA. Clonal lines were obtained as described above, and integration was confirmed using primer pairs L1_For/L1_Rev.
To create a conditional KO of PfPKG, vector pDC_loxnPKG:lox22mCherry was digested with Xba I and Nhe I. The latter restriction site was blunted and the vector was religated resulting in vector pDC_lox22mCherry. This plasmid was linearized overnight with Sca I and transfected together with the pDC2-pkg Cas9 plasmid into P. falciparum line pfpkg_2lox (5). Correct 5′ and 3′ integration were verified by PCR using primers wtpkg_For/5int_Rev. The absence of the endogenous locus was confirmed using primers exon1_For/PKGutr_Rev.
Plasmid construction and genotyping of P. berghei transgenic lines
The oligonucleotides used to generate and genotype the mutant parasite lines are in table S1.
Restriction/ligation cloning
To generate the Pama1ICM1 line, the plasmid pOB116-ama1 was used (36). The first 1000 bp from PbICM1 gene was amplified from gDNA using the primers 453 and 458. The PCR product was digested with Xho I and Not I and ligated into the pOB116-ama1 plasmid previously digested with the same enzymes, resulting in the plasmid pAB025. The last 750 bp of PbICM1 5′UTR were amplified using primers 459 and 464, and the resulting fragment was digested with Hind III and Pst I and ligated into pAB026 previously digested with the same enzymes. The same strategy was used to generate the line Pclag9ICM1, with the plasmid pOB116-clag9 resulting in plasmid pAB027. Both plasmids were digested with the enzymes Eco RV and Hind III to be transfected in P. berghei.
Recombineering
ICM1-HA3 and ICM1-AID/HA tagging constructs were generated using phage recombineering in Escherichia coli TSA strain with PlasmoGEM vectors (http://plasmogem.sanger.ac.uk/) as previously described (33, 34). The modified library inserts were then released from the plasmid backbone using Not I. The ICM1-AID/HA (PbGEM-645751) targeting vector was transfected into the 615 line (35). The ICM1-HA3 (PbGEM-121338) and ICM1-KO (PbGEM-265644) vectors were transfected into the 2.34 line.
Each mutant parasite was genotyped by PCR using three combinations of primers, specific for either the WT or the modified locus on both sides of the targeted region (experimental designs are shown in figs. S1 and S4). Controls using WT DNA were included in each genotyping experiment; parasite lines were cloned when indicated.
P. falciparum growth assays
Growth rates in P. falciparum mutant lines were performed as follows: synchronous ring-stage parasites at 0.1% parasitemia and 2% hematocrit were dispensed in triplicate into 12-well plates. Samples (50 μl) from each well were collected at 0, 2, 4, and 6 days, stained with SYBR green, and analyzed by flow cytometry on a BD FACSVerse using BD FACSuite software. Data acquired were analyzed using FlowJo software.
P. falciparum egress and invasion assays
Mature schizonts were isolated by Percoll centrifugation and incubated for a further 3 hours in complete medium containing the reversible PKG inhibitor compound 2 (1 μM). After removal of the inhibitor, schizonts were immediately resuspended in fresh serum-free RPMI 1640 at 37°C to allow egress. Schizont pellets and culture supernatants at t = 0 were collected as a control sample, while culture supernatants were collected by centrifugation at the relevant time points.
For invasion assays, mature schizonts were incubated at 2 to 4% parasitemia in 2% hematocrit under static or shaking conditions for 4 hours. Unruptured schizonts were removed by Percoll gradient, parasitemia was measured again, and the fold increase was calculated.
Calcium measurements
P. falciparum schizonts
Changes in calcium levels were measured using Fluo-4 acetoxymethyl ester (AM)–loaded (Thermo Fisher Scientific) parasites, and differences in fluorescence were measured over time on a BD FACSVerse flow cytometer using BD FACSuite software. Briefly, schizonts were washed twice in phenol red– and serum-free RPMI medium (11835030, Thermo Fisher Scientific). Fluo-4 AM (5 μM) was added to 1 ml of parasite preparation. Cells were incubated with Fluo-4 AM at 37°C for 45 min and washed twice in phenol red– and serum-free RPMI as above. Parasites were incubated for 30 min for deesterification followed by a further two washes. Schizonts were resuspended in 1 ml of warm phenol red– and serum-free RPMI. For Fluo-4 AM detection a 488-nm Blue Laser was used with a 530/30 filter. Fluorescence levels were measured for a period of 90 s to obtain a baseline read. Compounds were then added onto the cells, and reads were recorded for a further 3 min. Data analysis was done with FlowJo software.
P. berghei gametocytes
To measure changes in the calcium level, P. berghei gametocytes were harvested in coelenterazine loading buffer (29) [CLB; PBS containing 20 mM Hepes, 20 mM glucose, 4 mM NaHCO3, 1 mM EDTA, and 0.1% bovine serum albumin (BSA) (pH 7.2)]. Gametocytes were purified as described above and resuspended in 1 ml of CLB and loaded with 5 μM of Fluo-4 AM for an hour. The parasites were then washed twice with CLB and resuspended in 500 μl, and 20 μl was added per well in a 96-well plate. Compound dispense and simultaneous fluorescence reading were performed by a FDSS/μCELL (Hamamatsu Photonics) for a period of 5 min. The fluorescence value of each time point was first normalized to the initial fluorescence value at 0 s (Ft = n/Ft = 0) and then by the vehicle control (Ft = n/Ft = 0)/(Fvehicle t = n/Fvehicle t = 0).
Flow cytometry analysis of gametocytes
To analyze their DNA content, gametocytes were harvested in SA and purified as described above. Gametocytes were resuspended in 100 μl and activated with 100 μl of modified exflagellation medium [RPMI 1640 containing 25 mM Hepes, 4 mM sodium bicarbonate, 5% FCS, and 200 μM XA (pH 8.0)]. Gametogenesis was stopped by ice-cold PBS containing Vybrant dye cycle violet (Life Technologies), and cells were stained for 30 min at 4°C. DNA content was determined using a Beckman Coulter Gallios 4 and analyzed with the Kaluza software. Per sample, >10,000 cells were analyzed.
Immunoblotting and immunofluorescence assays
P. falciparum
Synchronized P. falciparum schizonts were isolated by Percoll gradient centrifugation and washed in RPMI 1640 without Albumax. Parasites were extracted into a Triton X-100 buffer [20 mM tris-HCl (pH 7.4), 150 mM NaCl, 0.5 mM EDTA, and 1% Triton X-100, supplemented with 1× protease inhibitors, Roche]. Extracts were incubated on ice for 30 min then clarified by centrifugation at 12,000g for 15 min at 4°C. Supernatants were mixed with SDS sample buffer containing dithiothreitol (DTT) and incubated for 5 min at 95°C before fractionation by SDS-PAGE analysis on 4 to 15% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad). Transfer to nitrocellulose membranes and probing for Western blot analysis were as described previously. A rabbit polyclonal human-PKG antibody (Enzo Life Sciences) was used for PKG detection at 1:1000. A GFP-specific monoclonal antibody 11814460001 (Roche) was used at 1:1000, as was a polyclonal rabbit anti-mCherry (ab167453; Abcam). A polyclonal rabbit anti-SERA5 antibody was used at 1:2000, and an anti-HSP70 antibody (a gift from E. Knuepfer, Francis Crick Institute) was used at 1:2000.
For IFA analysis, thin blood smears of wt, pficm1:cKO DMSO-, and RAP-treated Percoll-enriched schizonts were fixed in 4% (w/v) paraformaldehyde and permeabilized in 0.1% (v/v) Triton X-100. After blocking with 3% BSA, smears were probed with a rat anti-HA (3F10; 1:100) and a rabbit anti-SERA5 (1:500). For AMA1 relocalization, vehicle (DMSO) or RAP-treated Percoll-enriched pficm1:cKO schizonts were probed with a rabbit anti-AMA1 (1:200). Subsequently, slides were probed with the required Alexa Fluor 594–conjugated secondary antibody diluted (1:1000). Images were collected using a Nikon Eclipse Ni-E wide-field microscope with a Hamamatsu C11440 digital camera and 100×/1.45 numerical aperture (NA) oil immersion objective. Identical exposure conditions were used at each wavelength for each pair of mock- and RAP-treated samples under comparison. Images were processed using Fiji software (57).
P. berghei
Gametocyte immunofluorescence assays were performed as previously described (58). For HA and α-tubulin staining, purified cells were fixed with 4% paraformaldehyde and 0.05% glutaraldehyde in PBS for 1 hour, permeabilized with 0.1% Triton X-100/PBS for 10 min, and blocked with 2% BSA/PBS for 2 hours. Primary antibodies were diluted in blocking solution (rat anti-HA clone, 3F10, 1:1000; mouse anti–α-tubulin clone, DM1A, 1:1000; both from Sigma-Aldrich). Anti-rat Alexa 594, anti-mouse Alexa 488, anti-rabbit Alexa 488, and anti-rabbit Alexa 594 were used as secondary antibodies together with DAPI (4′,6-diamidino-2-phenylindole) (all from Life Technologies), all diluted at 1:1000 in blocking solution. Confocal images were acquired with a LSM700 or a LSM800 scanning confocal microscope (Zeiss).
Time-lapse and live fluorescence microscopy
Viewing chambers were constructed as previously described (41). Images were recorded on a Nikon Eclipse Ni light microscope fitted with a Hamamatsu C11440 digital camera and Nikon N Plan Apo λ 63×/1.45 NA oil immersion objective. For time-lapse video microscopy, vehicle (DMSO) or RAP-treated schizonts were synchronized by incubation with the reversible PKG inhibitor compound 2 and then washed, mixed in equal proportions, and monitored for egress. Differential inference contrast (DIC) images were taken at 10-s intervals over 45 min, while fluorescence (GFP or mCherry) images were taken every 5 to 10 min to prevent bleaching. Time-lapse videos were analyzed and annotated using Fiji (57).
Measurement of intracellular cyclic nucleotide levels
Relative intracellular cGMP levels in mature schizonts were measured using the Direct cGMP ELISA kit (Enzo). RAP- or DMSO-treated icm1:cKO schizonts were Percoll-purified and lysed in the presence of 0.1 ml of 0.1 M HCl to harvest cGMP. Samples were incubated at room temperature for 5 min with intermittent vortexing. Standards were fitted to a sigmoidal curve and used to determine cyclic nucleotide concentrations in parasite samples.
ICM1 protein structure prediction
The homology models of PfICM1 were constructed initially using the Phyre2 Server in intensive mode (22). The I-TASSER meta-threading procedure (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was run to generate a molecular model of PfICM1, using the first 1500 amino acids of the protein (I-TASSER maximum allowed residues) and covering domains of the protein predicted to be highly helical (59–61). The local Meta-Threader Server identified PDB 6MU1, 6JXC, 6RLB, and 6C08 as the top four threading templates for protein structure prediction, three of them being transporter proteins.
IP and MS analysis in P. falciparum
For pull-down analysis, frozen schizont preparations from lines PfPKG-HA, PKG-GFP, and 3D7 were resuspended in 100 μl of lysis buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100, complete protease inhibitors and PhosSTOP] and extracted at 4°C for 30 min with intermittent vortexing. Extracts were clarified by centrifugation at 16,000g for 20 min at 4°C, filtered through 0.22 Corning Costar Spin-X centrifuge tube filters, and then incubated with 25 μl of anti-HA magnetic beads (Pierce) or with 25 μl of GFP-Trap Magnetic Agarose (ChromoTek) overnight at 4°C. Sample processing followed the manufacturer’s protocol. Protein complexes were eluted from beads with the addition of 2× volumes of reduced 2× SDS sample buffer. Samples were incubated at 95°C for 5 min and run on 12% SDS-PAGE gels. Band slices covering the whole separation profile were excised and washed. Reduced and alkylated proteins were in-gel digested with 100 ng of trypsin (modified sequencing grade, Promega) overnight at 37°C. Supernatants were dried in a vacuum centrifuge and resuspended in 0.1% trifluoroacetic acid (TFA). Digested protein (1 to 10 μl) acidified to a final concentration of 0.1% TFA was loaded on an UltiMate 3000 nanoRSLC HPLC (Thermo Fisher Scientific) at 15 μl/min of 0.1% TFA onto a 2 mm by 0.3 mm Acclaim PepMap C18 trap column (Thermo Fisher Scientific) before the trap being switched to elute at 0.25 μl/min through a 50 cm by 75 μm EASY-Spray C18 column. A 90′ gradient of 9 to 25% B over 37′ and then 25 to 40% B over 18′ was used followed by a short gradient to 100% B and back down to 9% B and a 20′ equilibration in 9% B [A = 2% acetonitrile (ACN), 0.1% formic acid (FA); B = 80% ACN, 0.1% FA].
The Orbitrap was operated in “Data Dependent Acquisition” mode with a survey scan at a resolution of 120,000 from mass/charge ratio (m/z) of 300 to 1500, followed by MS/MS in “TopS” mode. Dynamic exclusion was used with a time window of 20 s. The Orbitrap charge capacity was set to a maximum of 1 × 106 ions in 10 ms, while the LTQ was set to 1 × 104 ions in 100 ms. Raw files were processed using MaxQuant 1.3.0.5 and Perseus 1.4.0.11 against a recent version of PlasmoDB (www.plasmodb.org). A decoy database of reversed sequences was used to filter false positives, at a peptide false detection rate of 1%.
IP and MS analysis in P. berghei
Sample preparation
Co-IPs of proteins were performed with purified gametocytes. The following IPs were performed: ICM1-HA3 in schizonts or gametocytes (15 s post activation +/− compound A) and PKG-HA3 in schizonts or gametocytes (15 s post activation +/− compound A). IPs from WT nonactivated gametocytes lacking an epitope tag were used as controls.
Samples were fixed for 10 min with 1% formaldehyde. Parasites were lysed in RIPA buffer [50 mM tris-HCl (pH 8), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS], and the supernatant was subjected to affinity purification with anti-HA antibody (Sigma-Aldrich) conjugated to magnetics beads. Beads were resuspended in 100 μl of 6 M urea in 50 mM ammonium bicarbonate (AB). Two microliters of 50 mM dithioerythritol (DTE) were added, and the reduction was carried out at 37°C for 1 hour. Alkylation was performed by adding 2 μl of 400 mM iodoacetamide for 1 hour at room temperature in the dark. Urea was reduced to 1 M by addition of 500 μl of AB, and overnight digestion was performed at 37°C with 5 μl of freshly prepared trypsin (0.2 μg/μl; Promega) in AB. Supernatants were collected and completely dried under speed vacuum. Samples were then desalted with a C18 microspin column (Harvard Apparatus) according to the manufacturer’s instructions, completely dried under speed vacuum, and stored at −20°C.
Liquid chromatography electrospray ionization tandem mass spectrometry
Samples were diluted in 20 μl of loading buffer [5% acetonitrile (AcN) (CH3CN) and 0.1% FA], and 2 μl was injected onto the column. Liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) was performed either on a Q-Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC 1000 liquid chromatography system (Thermo Fisher Scientific) or an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC 1200 liquid chromatography system (Thermo Fisher Scientific). Peptides were trapped on an Acclaim PepMap 100, 3-μm C18, 75 μm by 20 mm nano-trap column (Thermo Fisher Scientific) and separated on a 75 μm by 250 mm (Q-Exactive) or 500 mm (Orbitrap Fusion Lumos), 2-μm C18, 100 Å EASY-Spray column (Thermo Fisher Scientific). The analytical separation used a gradient of H2O/0.1% FA (solvent A) and CH3CN/0.1% FA (solvent B). The gradient was run as follows: 95% A and 5% B for 0 to 5 min, then to 65% A and 35% B for 60 min, then to 10% A and 90% B for 10 min, and lastly to 10% A and 90% B for 15 min. Flow rate was 250 nl/min for a total run time of 90 min.
Data-dependent analysis (DDA) was performed on the Q-Exactive Plus with MS1 full scan at a resolution of 70,000 full width at half maximum (FWHM) followed by MS2 scans on up to 15 selected precursors. MS1 was performed with an automatic gain control (AGC) target of 3 × 106, a maximum injection time of 100 ms, and a scan range from 400 to 2000 m/z. MS2 was performed at a resolution of 17,500 FWHM with an AGC target at 1 × 105 and a maximum injection time of 50 ms. Isolation window was set at 1.6 m/z, and 27% normalized collision energy was used for higher-energy collisional dissociation (HCD). DDA was performed on the Orbitrap Fusion Lumos with MS1 full scan at a resolution of 120,000 FWHM followed by as many subsequent MS2 scans on selected precursors as possible within a 3-s maximum cycle time. MS1 was performed in the Orbitrap with an AGC target of 4 × 105, a maximum injection time of 50 ms, and a scan range from 400 to 2000 m/z. MS2 was performed in the ion trap with a rapid scan rate, an AGC target of 1 × 104, and a maximum injection time of 35 ms. Isolation window was set at 1.2 m/z, and 30% normalized collision energy was used for HCD.
Database searches
Peak lists (MGF file format) were generated from raw data using the MS Convert conversion tool from ProteoWizard. The peak list files were searched against the PlasmoDB_P.berghei ANKA database (PlasmoDB.org, release 38, 5076 entries) combined with an in-house database of common contaminants using Mascot (Matrix Science, London, UK; version 2.5.1). Trypsin was selected as the enzyme, with one potential missed cleavage. Precursor ion tolerance was set to 10 parts per million (ppm) and fragment ion tolerance to 0.02 Da for Q-Exactive Plus data and to 0.6 for Lumos data. Variable amino acid modifications were oxidized methionine and deamination (asparagine and glutamine) as well as phosphorylated serine, threonine, and tyrosine. Fixed amino acid modification was carbamidomethyl cysteine. The Mascot search was validated using Scaffold 4.8.4 (Proteome Software) with 1% of protein false discovery rate (FDR) and at least two unique peptides per protein with a 0.1% peptide FDR.
PCA analysis
Enrichment and PCA were performed in the statistical programming package “R” (www.r-project.org). Quantitative values were analyzed as log-transformed spectral count values and displayed in principal components with greatest degrees of variance.
Chemoproteomics
Kinobeads were prepared as described (62, 63). The chemoproteomic affinity capturing experiments were performed as previously described (62, 64). Briefly, Kinobeads were incubated with P. falciparum extract (65), which was preincubated with compound or DMSO (vehicle control). The experimental setup was such that 10 samples are measured in parallel [TMT 10-plex (66)] to generate values for the affinity of the beads to the bound proteins (“depletion” values, four samples) and to generate median inhibitory concentration values based on dose-dependent competition (six samples, 20 μM, 1:3 dilutions) in a single experiment. Beads were transferred to Filter plates (Durapore polyvinylidene difluoride membrane, Merck Millipore), washed extensively, and eluted with SDS sample buffer (67).
Proteins were digested according to a modified single pot solid-phase sample preparation (SP3) protocol (68, 69). Proteins were digested by resuspending in 0.1 mM Hepes (pH 8.5) containing tris(2-carboxyethyl)phosphine (TCEP), chloracetamide, trypsin, and LysC following overnight incubation.
Peptides were labeled with isobaric mass tags (TMT10, Thermo Fisher Scientific, Waltham, MA) using the 10-plex TMT reagents, enabling relative quantification of 10 conditions in a single experiment (66, 70). Labeled peptide extracts were combined to a single sample per experiment, lyophilized, and subjected to LC-MS analysis.
Samples were injected into an UltiMate 3000 nanoRSLC (Dionex) coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific). Peptides were separated on reversed-phase columns (Reprosil) at 55°C by gradient elution (3.5 to 29% AcN in aqueous 0.1% FA, 3.5% DMSO) in 120 min. The instruments were operated with Tune 2.4 and Xcalibur 3.0 build 63.
Mascot 2.4 (Matrix Science, Boston, MA) was used for protein identification by using a 10 parts per million mass tolerance for peptide precursors and 20 mD (HCD) mass tolerance for fragment ions. To create the FASTA file for mascot searching, all proteins corresponding to the taxonomy “Plasmodium falciparum (isolate 3D7)” were downloaded from UniProt (release 20170621) and supplemented with common contaminant protein sequences of BSA, porcine trypsin, and mouse, rat, sheep, and dog keratins. To assess the FDR, “decoy” proteins (reverse of all protein sequences) were created and added to the database, resulting in a database containing a total of 14,266 protein sequences, 50% forward and 50% reverse.
Unless stated otherwise, we accepted protein identifications as follows: (i) For single-spectrum to sequence assignments, we required this assignment to be the best match and a minimum Mascot score of 31 and a 10× difference of this assignment over the next best assignment. On the basis of these criteria, the decoy search results indicated <1% FDR. (ii) For multiple spectrum to sequence assignments and using the same parameters, the decoy search results indicated <0.1% FDR. Quantified proteins were required to contain at least two unique peptide matches. FDR for quantified proteins was <0.1%.
P. falciparum phosphoproteomics
Parasite pellets were treated as previously described (71). Briefly, schizont pellets were resuspended in 1 ml of 8 M urea in 50 mM Hepes (pH 8.5), containing protease and phosphatase inhibitors and benzonase (100 U/ml; Sigma-Aldrich). Proteins were extracted from the pellets by sonication. Samples were incubated on ice for 10 min and centrifuged for 30 min at 14,000 rpm at 4°C. Protein content was estimated by a BCA protein assay, and 200 μg of each sample was reduced with 10 mM DTT for 25 min at 56°C and then alkylated with 20 mM iodoacetamide for 30 min at room temperature. Samples were digested initially with LysC (Wako) at 37°C for 2.5 hours and subsequently with 10 μg of trypsin (modified sequencing grade, Promega) overnight. After acidification, C18 MacroSpin columns (Nest Group) were used to clean up the digested peptide solutions and the eluted peptides dried by vacuum centrifugation. Samples were resuspended in 50 mM Hepes and labeled using the 0.8 mg Tandem Mass Tag 10plex isobaric reagent kit (Thermo Fisher Scientific) following the manufacturer’s instructions. The eluted TMT-labeled peptides were dried by vacuum centrifugation, and phosphopeptide enrichment was subsequently carried out using the sequential metal oxide affinity chromatography strategy with High Select TiO2 and Fe-NTA enrichment kits (Thermo Fisher Scientific). Eluates were combined before fractionation with the Pierce High pH Reversed-Phase Peptide Fractionation kit (Thermo Fisher Scientific). The dried TMT-labeled phosphopeptide fractions generated were resuspended in 0.1% TFA for LC-MS/MS analysis using a UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) interfaced with an Orbitrap Fusion Lumos (Thermo Fisher Scientific). Each peptide fraction was preconcentrated on an Acclaim PepMap 100 trapping column before separation on a 50-cm, 75-μm I.D. EASY-Spray PepMap column over a 3-hour gradient run at 40°C, eluted directly into the mass spectrometer. The instrument was run in data-dependent acquisition mode with the most abundant peptides selected for MS/MS fragmentation. Two replicate injections were made for each fraction with different fragmentation methods based on the MS2 HCD and MSA SPS MS3 strategies described (72). The acquired raw mass spectrometric data were processed in MaxQuant (73) (v1.6.2.10) and searched against UniProt Homo sapiens complete proteome canonical sequences (September 2019) and PlasmoDB. Fixed modifications were set as Carbamidomethyl (C) and variable modifications set as Oxidation (M) and Phospho (STY). The estimated FDR was set to 1% at the peptide, protein, and site levels. A maximum of two missed cleavages were allowed. Reporter ion MS2 or Reporter ion MS3 was appropriately selected for each raw file. Other parameters were used as preset in the software. The MaxQuant output file PhosphoSTY Sites.txt, an FDR-controlled site-based table compiled by MaxQuant from the relevant information about the identified peptides, was imported into Perseus (v1.4.0.2) for data evaluation. Sequence logos were generated by IceLogo (https://iomics.ugent.be/icelogoserver/). Paired t tests were performed using R (R Core Team, 2017; www.R-project.org/). Significantly down-regulated genes from PfPKG and PfICM1 phosphoproteomes were used for enrichment of GO terms in PlasmoDB using a cutoff P value of 0.05. Results were visualized using REVIGO (74) with the P. falciparum database and similarity: 0.7, semantic similarity measure: SimRel. Plots were made using R (R Core Team, 2017; www.R-project.org/) package ggplot2 (75).
P. berghei phosphoproteomics
Sample preparation
Samples (about 100 μl of packed cell volume) were diluted with 25 μl of Laemmli 5× buffer and sonicated 6× 30 s at 70% amplitude and 0.5 pulse (30 s on ice between each cycle). Samples were heated 5 min at 65°C and loaded onto a Novex Wedgewell 12% tris-glycine gel (Invitrogen). Each sample was loaded on two different wells, and protein’s migration was then performed according to the manufacturer’s instructions. Proteins were then stained with Coomassie blue. For each lane, a band between 180 and 220 kDa was cut and destained by incubation in 100 μl of 30% AcN in 50 mM AB for 15 min at room temperature. Proteins were reduced by incubation of gel pieces for 35 min at 56°C in 100 μl of 10 mM DTE in 50 mM AB. DTE solution was then replaced by 100 μl of 55 mM iodoacetamide in 50 mM AB, and proteins were alkylated by incubation of the gel pieces for 30 min at room temperature in the dark. Gel pieces were then washed for 10 min with 100 μl of 50 mM AB and dehydrated for 10 min with 100 μl of 30% AcN. Gel pieces were then dried for 30 min in a vacuum centrifuge. Dried pieces of gel were rehydrated for 45 min at 4°C with 70 μl of trypsin (6.25 ng/μl in AB 50 mM) and then incubated overnight at 37°C. The supernatant was transferred to a new polypropylene tube, and two additional peptide extraction were performed with 70 μl of 1% TFA and 100 μl of 50% ACN, respectively; 0.1% TFA. Extractions were pooled and dried under speed vacuum.
Titanium dioxide enrichment
Phosphopeptide enrichment was performed as previously described (76). Briefly, stage tips were prepared using Glass Microfiber Filters (Whatman) and 5-μm TiO2 beads (GL Sciences). Beads were suspended with 0.1% TFA in 80% AcN and inserted onto a gel-loading tip to create a 5-mm-long column. Peptides samples were dissolved in 1 M glycolic acid 2% TFA in 80% AcN and loaded onto dedicated TiO2 columns by centrifugation 1 min at 6000 rpm. Columns were then washed four times with 1 M glycolic acid 2% TFA in 80% AcN and four times with 0.1% TFA in 80% AcN. Bounded peptides were eluted first with 3% NH4OH and then with 0.1% TFA in 80% AcN. Eluates were then acidified with 5% TFA and dried under speed vacuum.
Fe-NTA enrichment
Flow through from TiO2 beads were enriched using homemade stage tips as described before but containing beads from the High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Fisher Scientific). Enrichment was performed according to the manufacturer’s instructions.
Liquid chromatography electrospray ionization tandem mass spectrometry
Samples were diluted in 10 μl of loading buffer (5% CH3CN and 0.1% FA), and 6 μl was injected on column. LC-ESI-MS/MS was performed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) equipped with an Easy nLC1200 liquid chromatography system (Thermo Fisher Scientific). Peptides were trapped on an Acclaim PepMap 100, 3-μm C18, 75 μm by 20 mm nano-trap column (Thermo Fisher Scientific) and separated on a 75 μm by 500 mm, C18 ReproSil-Pur from Dr. Maisch GmBH, 1.9 μm and 100 Å, homemade column. The analytical separation was run for 90 min using a gradient of H2O/FA 99.9%/0.1% (solvent A) and CH3CN/FA 80%/0.1% (solvent B). The gradient was run as follows: 95% A and 5% B for 0 to 5 min, then to 65% A and 35% B for 60 min, and lastly to 5% A and 95% B for 10 min with a stay for 15 min at this composition. Flow rate was of 250 nl/min. DDA was performed with MS1 full scan at a resolution of 120,000 FWHM followed by as many subsequent MS2 scans on selected precursors as possible within 3-s maximum cycle time. MS1 was performed in the Orbitrap with an AGC target of 4 × 105, a maximum injection time of 50 ms, and a scan range from 375 to 1500 m/z. MS2 was performed in the Orbitrap at a resolution of 30,000 FWHM with an AGC target at 5 × 104 and a maximum injection time of 54 ms. Isolation windows was set at 1.6 m/z, and 30% normalized collision energy was used for HCD.
Database search
Raw data were processed using Proteome Discoverer 2.3 software (Thermo Fisher Scientific). Briefly, spectra were extracted and searched against the P. berghei ANKA database (PlasmoDB.org, release 38, 5076 entries) combined with an in-house database of common contaminants using Mascot (Matrix Science, London, UK; version 2.5.1). Trypsin was selected as the enzyme, with one potential missed cleavage. Precursor ion tolerance was set to 10 ppm and fragment ion tolerance to 0.02 Da. Carbamidomethylation of cysteine was specified as fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine, as well as phosphorylation of serine, threonine, and tyrosine were specified as variable modifications. Peptide-spectrum matches were validated using the target decoy PSM validator node with a target FDR of 0.01 and a Delta Cn of 0.5. For label-free quantification, features and chromatographic peaks were detected using the “Minora Feature Detector” node with the default parameters. PSM and peptides were filtered with a FDR of 1% and then grouped to proteins with again an FDR of 1% and using only peptides with a high confidence level. Both unique and razor peptides were used for quantitation, and protein abundances are calculated as the average of the three most abundant distinct peptide groups. The abundances were normalized on the “total peptide amount,” then “protein abundance–based” option was selected for protein ratio calculation, and associated P values were calculated with an analysis of variance (ANOVA) test (individual proteins).
Acknowledgments
We thank the excellent service at the bioimaging and flow cytometry core facilities at the Faculty of Medicine of the University of Geneva. M.B. is grateful to O. Billker for support during the initial steps of this project. We would also like to acknowledge E. Fernández-Álvaro at GSK for support in the initial stages of the project and S. Osborne at LifeArc who was the lead chemist on the project that generated ML10. Funding: This work was supported by the Swiss National Science Foundation (starting grant BSSGI0_155852), by the Novartis Foundation (19C189), and by the Fondation privée des Hôpitaux Universitaires de Genève (CONFIRM grant RC05-10) to M.B. M.B. is an INSERM and EMBO young investigator. This work was also supported by Wellcome Trust grant 106239/Z/14/A (to K.K., A.J.P., and M.J.B.), Wellcome Trust grant 106240/Z/14/Z (to D.A.B.), MRC Developmental pathway funding scheme grant (ref G10000779) (to D.A.B.), and Wellcome ISSF2 funding to the London School of Hygiene & Tropical Medicine. The work was also supported by funding to M.J.B. from the Francis Crick Institute (www.crick.ac.uk/), which receives its core funding from Cancer Research UK (FC001043; www.cancerresearchuk.org), the UK Medical Research Council (FC001043; www.mrc.ac.uk/), and the Wellcome Trust (FC001043; https://wellcome.ac.uk/). This research was funded, in part, by the Wellcome Trust. For the purpose of Open Access, the authors have applied a CC BY public copyright license to any author accepted manuscript version arising from this submission. Author contributions: Conceptualization: K.K., D.A.B., M.J.B., and M.B. Methodology, formal analysis, and investigation: A.C.B., K.K., N.K., S.A.H., H.R.F., M.B., C.P., A.J.P., L.B., P.A., O.S., L.P.-B.C., C.W.-M., and A.H. Writing—original draft: A.C.B., K.K., D.A.B., M.J.B., and M.B. Writing—review and editing: A.C.B., K.K., S.A.H., H.R.F., A.J.P., C.W.-M., S.G.-D., D.A.B., M.J.B., and M.B. Funding acquisition: D.A.B., M.J.B., and M.B. Resources: S.G.-D., A.P.S., D.A.B., M.J.B., and M.B. Supervision; S.G.-D., A.P.S, D.A.B., M.J.B., and M.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions vin the paper are present in the paper and/or the Supplementary Materials. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (77) partner repository (http://proteomecentral.proteomexchange.org) with the dataset identifiers PXD020210 (P. berghei IP), PXD020189 (P. berghei proteomics), PXD020447 (P. falciparum IP), and PXD020381 (P. falciparum proteomics). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabe5396/DC1
REFERENCES AND NOTES
- 1.WHO, World malaria report (2019).
- 2.Baker D. A., Drought L. G., Flueck C., Nofal S. D., Patel A., Penzo M., Walker E. M., Cyclic nucleotide signalling in malaria parasites. Open Biol. 7, 170213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brochet M., Billker O., Calcium signalling in malaria parasites. Mol. Microbiol. 100, 397–408 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Taylor H. M., McRobert L., Grainger M., Sicard A., Dluzewski A. R., Hopp C. S., Holder A. A., Baker D. A., The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot. Cell 9, 37–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koussis K., Withers-Martinez C., Baker D. A., Blackman M. J., Simultaneous multiple allelic replacement in the malaria parasite enables dissection of PKG function. Life Sci. Alliance 3, e201900626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falae A., Combe A., Amaladoss A., Carvalho T., Menard R., Bhanot P., Role of Plasmodium berghei cGMP-dependent protein kinase in late liver stage development. J. Biol. Chem. 285, 3282–3288 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McRobert L., Taylor C. J., Deng W., Fivelman Q. L., Cummings R. M., Polley S. D., Billker O., Baker D. A., Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 6, e139 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brochet M., Collins M. O., Smith T. K., Thompson E., Sebastian S., Volkmann K., Schwach F., Chappell L., Gomes A. R., Berriman M., Rayner J. C., Baker D. A., Choudhary J., Billker O., Phosphoinositide metabolism links cGMP-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol. 12, e1001806 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govindasamy K., Jebiwott S., Jaijyan D. K., Davidow A., Ojo K. K., van Voorhis W. C., Brochet M., Billker O., Bhanot P., Invasion of hepatocytes by Plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 102, 349–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang H., Gomes A. R., Klages N., Pino P., Maco B., Walker E. M., Zenonos Z. A., Angrisano F., Baum J., Doerig C., Baker D. A., Billker O., Brochet M., Epistasis studies reveal redundancy among calcium-dependent protein kinases in motility and invasion of malaria parasites. Nat. Commun. 9, 4248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francis S. H., Busch J. L., Corbin J. D., Sibley D., cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L., Liu G., Zakharov S. I., Bellinger A. M., Mongillo M., Marx S. O., Protein kinase G phosphorylates Cav1.2 alpha1c and beta2 subunits. Circ. Res. 101, 465–474 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Schlossmann J., Ammendola A., Ashman K., Zong X., Huber A., Neubauer G., Wang G. X., Allescher H. D., Korth M., Wilm M., Hofmann F., Ruth P., Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 404, 197–201 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Xia C., Bao Z., Yue C., Sanborn B. M., Liu M., Phosphorylation and regulation of G-protein-activated phospholipase C-β3 by cGMP-dependent protein kinases. J. Biol. Chem. 276, 19770–19777 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Koller A., Schlossmann J., Ashman K., Uttenweiler-Joseph S., Ruth P., Hofmann F., Association of phospholamban with a cGMP kinase signaling complex. Biochem. Biophys. Res. Commun. 300, 155–160 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Baldauf S. L., The deep roots of eukaryotes. Science 300, 1703–1706 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Hopp C. S., Flueck C., Solyakov L., Tobin A., Baker D. A., Spatiotemporal and functional characterisation of the Plasmodium falciparum cGMP–dependent protein kinase. PLOS ONE 7, e48206 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker D. A., Stewart L. B., Large J. M., Bowyer P. W., Ansell K. H., Jiménez-Díaz M. B., el Bakkouri M., Birchall K., Dechering K. J., Bouloc N. S., Coombs P. J., Whalley D., Harding D. J., Smiljanic-Hurley E., Wheldon M. C., Walker E. M., Dessens J. T., Lafuente M. J., Sanz L. M., Gamo F. J., Ferrer S. B., Hui R., Bousema T., Angulo-Barturén I., Merritt A. T., Croft S. L., Gutteridge W. E., Kettleborough C. A., Osborne S. A., A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat. Commun. 8, 430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., Reader V., Sweetman G., Bauer A., Bouwmeester T., Hopf C., Kruse U., Neubauer G., Ramsden N., Rick J., Kuster B., Drewes G., Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Tewari R., Straschil U., Bateman A., Böhme U., Cherevach I., Gong P., Pain A., Billker O., The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 8, 377–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balestra A. C., Zeeshan M., Rea E., Pasquarello C., Brusini L., Mourier T., Subudhi A. K., Klages N., Arboit P., Pandey R., Brady D., Vaughan S., Holder A. A., Pain A., Ferguson D. J. P., Hainard A., Tewari R., Brochet M., A divergent cyclin/cyclin-dependent kinase complex controls the atypical replication of a malaria parasite during gametogony and transmission. eLife 9, e56474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solyakov L., Halbert J., Alam M. M., Semblat J. P., Dorin-Semblat D., Reininger L., Bottrill A. R., Mistry S., Abdi A., Fennell C., Holland Z., Demarta C., Bouza Y., Sicard A., Nivez M. P., Eschenlauer S., Lama T., Thomas D. C., Sharma P., Agarwal S., Kern S., Pradel G., Graciotti M., Tobin A. B., Doerig C., Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2, 565 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Treeck M., Sanders J. L., Elias J. E., Boothroyd J. C., The Phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii Reveal Unusual Adaptations Within and Beyond the Parasites’ Boundaries. Cell Host Microbe 10, 410–419 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alam M. M., Solyakov L., Bottrill A. R., Flueck C., Siddiqui F. A., Singh S., Mistry S., Viskaduraki M., Lee K., Hopp C. S., Chitnis C. E., Doerig C., Moon R. W., Green J. L., Holder A. A., Baker D. A., Tobin A. B., Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion. Nat. Commun. 6, 7285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Invergo B. M., Brochet M., Yu L., Choudhary J., Beltrao P., Billker O., Sub-minute phosphoregulation of cell cycle systems during Plasmodium gamete formation. Cell Rep. 21, 2017–2029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moon R. W., Taylor C. J., Bex C., Schepers R., Goulding D., Janse C. J., Waters A. P., Baker D. A., Billker O., A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLOS Pathog. 5, e1000599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green J. L., Moon R. W., Whalley D., Bowyer P. W., Wallace C., Rochani A., Nageshan R. K., Howell S. A., Grainger M., Jones H. M., Ansell K. H., Chapman T. M., Taylor D. L., Osborne S. A., Baker D. A., Tatu U., Holder A. A., Imidazopyridazine inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 also target cyclic GMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob. Agents Chemother. 60, 1464–1475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Billker O., Dechamps S., Tewari R., Wenig G., Franke-Fayard B., Brinkmann V., Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 117, 503–514 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Fang H., Klages N., Baechler B., Hillner E., Yu L., Pardo M., Choudhary J., Brochet M., Multiple short windows of calcium-dependent protein kinase 4 activity coordinate distinct cell cycle events during Plasmodium gametogenesis. eLife 6, e26524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bushell E., Gomes A. R., Sanderson T., Anar B., Girling G., Herd C., Metcalf T., Modrzynska K., Schwach F., Martin R. E., Mather M. W., McFadden G. I., Parts L., Rutledge G. G., Vaidya A. B., Wengelnik K., Rayner J. C., Billker O., Functional profiling of a Plasmodium genome reveals an abundance of essential genes. Cell 170, 260–272.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayers C. P., Mollard V., Buchanan H. D., McFadden G. I., Goodman C. D., A genetic screen in rodent malaria parasites identifies five new apicoplast putative membrane transporters, one of which is essential in human malaria parasites. Cell. Microbiol. 20, (2018). [DOI] [PubMed] [Google Scholar]
- 33.Pfander C., Anar B., Brochet M., Rayner J. C., Billker O., Recombination-mediated genetic engineering of Plasmodium berghei DNA. Methods Mol. Biol. 923, 127–138 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Pfander C., Anar B., Schwach F., Otto T. D., Brochet M., Volkmann K., Quail M. A., Pain A., Rosen B., Skarnes W., Rayner J. C., Billker O., A scalable pipeline for highly effective genetic modification of a malaria parasite. Nat. Methods 8, 1078–1082 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philip N., Waters A. P., Conditional degradation of Plasmodium calcineurin reveals functions in parasite colonization of both host and vector. Cell Host Microbe 18, 122–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sebastian S., Brochet M., Collins M. O., Schwach F., Jones M. L., Goulding D., Rayner J. C., Choudhary J. S., Billker O., A Plasmodium calcium-dependent protein kinase controls zygote development and transmission by translationally activating repressed mRNAs. Cell Host Microbe 12, 9–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howard B. L., Harvey K. L., Stewart R. J., Azevedo M. F., Crabb B. S., Jennings I. G., Sanders P. R., Manallack D. T., Thompson P. E., Tonkin C. J., Gilson P. R., Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem. Biol. 10, 1145–1154 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Zhang M., Wang C., Otto T. D., Oberstaller J., Liao X., Adapa S. R., Udenze K., Bronner I. F., Casandra D., Mayho M., Brown J., Li S., Swanson J., Rayner J. C., Jiang R. H. Y., Adams J. H., Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, eaap7847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrin A. J., Collins C. R., Russell M. R. G., Collinson L. M., Baker D. A., Blackman M. J., The actinomyosin motor drives malaria parasite red blood cell invasion but not egress. MBio 9, e00905-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas J. A., Tan M. S. Y., Bisson C., Borg A., Umrekar T. R., Hackett F., Hale V. L., Vizcay-Barrena G., Fleck R. A., Snijders A. P., Saibil H. R., Blackman M. J., A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat. Microbiol. 3, 447–455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins C. R., Hackett F., Strath M., Penzo M., Withers-Martinez C., Baker D. A., Blackman M. J., Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLOS Pathog. 9, e1003344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Healer J., Crawford S., Ralph S., McFadden G., Cowman A. F., Independent translocation of two micronemal proteins in developing Plasmodium falciparum merozoites. Infect. Immun. 70, 5751–5758 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Absalon S., Blomqvist K., Rudlaff R. M., De Lano T. J., Pollastri M. P., Dvorin J. D., Calcium-dependent protein kinase 5 is required for release of egress-specific organelles in Plasmodium falciparum. MBio 9, e00130-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blomqvist K., Helmel M., Wang C., Absalon S., Labunska T., Rudlaff R. M., Adapa S., Jiang R., Steen H., Dvorin J. D., Influence of Plasmodium falciparum calcium-dependent protein kinase 5 (PfCDPK5) on the late schizont stage phosphoproteome. mSphere 5, e00921-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlisle Michel J. J., Dodge K. L., Wong W., Mayer N. C., Langeberg L. K., Scott J. D., PKA-phosphorylation of PDE4D3 facilitates recruitment of the mAKAP signalling complex. Biochem. J. 381, 587–592 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meister M., Tomasovic A., Banning A., Tikkanen R., Mitogen-activated protein (MAP) kinase scaffolding proteins: A recount. Int. J. Mol. Sci. 14, 4854–4884 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuasa K., Omori K., Yanaka N., Binding and phosphorylation of a novel male germ cell-specific cGMP-dependent protein kinase-anchoring protein by cGMP-dependent protein kinase Iα. J. Biol. Chem. 275, 4897–4905 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Casteel D. E., Zhuang S., Gudi T., Tang J., Vuica M., Desiderio S., Pilz R. B., cGMP-dependent protein kinase Iβ physically and functionally interacts with the transcriptional regulator TFII-I. J. Biol. Chem. 277, 32003–32014 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., Mendelsohn M. E., Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286, 1583–1587 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Govindasamy K., Khan R., Snyder M., Lou H. J., Du P., Kudyba H. M., Muralidharan V., Turk B. E., Bhanot P., Plasmodium falciparum cyclic GMP-dependent protein kinase interacts with a subunit of the parasite proteasome. Infect. Immun. 87, e00523-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanaerschot M., Murithi J. M., Pasaje C. F. A., Ghidelli-Disse S., Dwomoh L., Bird M., Spottiswoode N., Mittal N., Arendse L. B., Owen E. S., Wicht K. J., Siciliano G., Bösche M., Yeo T., Kumar T. R. S., Mok S., Carpenter E. F., Giddins M. J., Sanz O., Ottilie S., Alano P., Chibale K., Llinás M., Uhlemann A.-C., Delves M., Tobin A. B., Doerig C., Winzeler E. A., Lee M. C. S., Niles J. C., Fidock D. A., Inhibition of resistance-refractory P. falciparum kinase PKG delivers prophylactic, blood stage, and transmission-blocking antiplasmodial activity. Cell Chem. Biol. 27, 806–816.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganesan S. M., Falla A., Goldfless S. J., Nasamu A. S., Niles J. C., Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat. Commun. 7, 10727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das S., Hertrich N., Perrin A. J., Withers-Martinez C., Collins C. R., Jones M. L., Watermeyer J. M., Fobes E. T., Martin S. R., Saibil H. R., Wright G. J., Treeck M., Epp C., Blackman M. J., Processing of Plasmodium falciparum merozoite surface protein MSP1 activates a spectrin-binding function enabling parasite egress from RBCs. Cell Host Microbe 18, 433–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collins C. R., Das S., Wong E. H., Andenmatten N., Stallmach R., Hackett F., Herman J. P., Müller S., Meissner M., Blackman M. J., Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 88, 687–701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kooij T. W., Rauch M. M., Matuschewski K., Expansion of experimental genetics approaches for Plasmodium berghei with versatile transfection vectors. Mol. Biochem. Parasitol. 185, 19–26 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Knuepfer E., Napiorkowska M., van Ooij C., Holder A. A., Generating conditional gene knockouts in Plasmodium – a toolkit to produce stable DiCre recombinase-expressing parasite lines using CRISPR/Cas9. Sci. Rep. 7, 3881–3881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Volkmann K., Pfander C., Burstroem C., Ahras M., Goulding D., Rayner J. C., Frischknecht F., Billker O., Brochet M., The alveolin IMC1h is required for normal ookinete and sporozoite motility behaviour and host colonisation in Plasmodium berghei. PLOS ONE 7, e41409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y., The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roy A., Kucukural A., Zhang Y., I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y., I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A. M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Dümpelfeld B., Eberhard D., Huthmacher C., Mathieson T., Poeckel D., Reader V., Strunk K., Sweetman G., Kruse U., Neubauer G., Ramsden N. G., Drewes G., Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Bergamini G., Bell K., Shimamura S., Werner T., Cansfield A., Müller K., Perrin J., Rau C., Ellard K., Hopf C., Doce C., Leggate D., Mangano R., Mathieson T., O’Mahony A., Plavec I., Rharbaoui F., Reinhard F., Savitski M. M., Ramsden N., Hirsch E., Drewes G., Rausch O., Bantscheff M., Neubauer G., A selective inhibitor reveals PI3Kγ dependence of TH17 cell differentiation. Nat. Chem. Biol. 8, 576–582 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Matralis A. N., Malik A., Penzo M., Moreno I., Almela M. J., Camino I., Crespo B., Saadeddin A., Ghidelli-Disse S., Rueda L., Calderon F., Osborne S. A., Drewes G., Böesche M., Fernández-Álvaro E., Martin Hernando J. I., Baker D. A., Development of chemical entities endowed with potent fast-killing properties against Plasmodium falciparum malaria parasites. J. Med. Chem. 62, 9217–9235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paquet T., le Manach C., Cabrera D. G., Younis Y., Henrich P. P., Abraham T. S., Lee M. C. S., Basak R., Ghidelli-Disse S., Lafuente-Monasterio M. J., Bantscheff M., Ruecker A., Blagborough A. M., Zakutansky S. E., Zeeman A. M., White K. L., Shackleford D. M., Mannila J., Morizzi J., Scheurer C., Angulo-Barturen I., Martínez M. S., Ferrer S., Sanz L. M., Gamo F. J., Reader J., Botha M., Dechering K. J., Sauerwein R. W., Tungtaeng A., Vanachayangkul P., Lim C. S., Burrows J., Witty M. J., Marsh K. C., Bodenreider C., Rochford R., Solapure S. M., Jiménez-Díaz M. B., Wittlin S., Charman S. A., Donini C., Campo B., Birkholtz L. M., Hanson K. K., Drewes G., Kocken C. H. M., Delves M. J., Leroy D., Fidock D. A., Waterson D., Street L. J., Chibale K., Antimalarial efficacy of MMV390048, an inhibitor of Plasmodiumphosphatidylinositol 4-kinase. Sci. Transl. Med. 9, eaad9735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Werner T., Sweetman G., Savitski M. F., Mathieson T., Bantscheff M., Savitski M. M., Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 86, 3594–3601 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Eberl H. C., Werner T., Reinhard F. B., Lehmann S., Thomson D., Chen P., Zhang C., Rau C., Muelbaier M., Drewes G., Drewry D., Bantscheff M., Chemical proteomics reveals target selectivity of clinical Jak inhibitors in human primary cells. Sci. Rep. 9, 14159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hughes C. S., Foehr S., Garfield D. A., Furlong E. E., Steinmetz L. M., Krijgsveld J., Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moggridge S., Sorensen P. H., Morin G. B., Hughes C. S., Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J. Proteome Res. 17, 1730–1740 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Werner T., Becher I., Sweetman G., Doce C., Savitski M. M., Bantscheff M., High-resolution enabled TMT 8-plexing. Anal. Chem. 84, 7188–7194 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Patel A., Perrin A. J., Flynn H. R., Bisson C., Withers-Martinez C., Treeck M., Flueck C., Nicastro G., Martin S. R., Ramos A., Gilberger T. W., Snijders A. P., Blackman M. J., Baker D. A., Cyclic AMP signalling controls key components of malaria parasite host cell invasion machinery. PLoS Biol. 17, e3000264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang X., Bomgarden R., Brown J., Drew D. L., Robitaille A. M., Viner R., Huhmer A. R., Sensitive and accurate quantitation of phosphopeptides using TMT isobaric labeling technique. J. Proteome Res. 16, 4244–4252 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 74.Supek F., Bošnjak M., Škunca N., Šmuc T., REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.H. Wickham, in ggplot2: Elegant Graphics for Data Analysis (Springer International Publishing, 2016), pp. 11–31. [Google Scholar]
- 76.Jensen S. S., Larsen M. R., Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun. Mass Spectrom. 21, 3635–3645 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yılmaz Ş., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., Vizcaíno J. A., The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabe5396/DC1