

Abstract
Objective:
Statins lower cardiovascular event risk, yet they paradoxically increase coronary artery calcification, a marker consistently associated with increased cardiovascular risks. Since calcium deposits influence rupture risk due to stress from compliance mismatch at their surfaces, we hypothesized that statins may lower cardiovascular risk by altering the microarchitecture of calcium deposits. Thus, using mice with pre-existing vascular calcification, we tested whether pravastatin reduces the mineral surface area of calcium deposits.
Approach and Results:
Aged Apoe−/− mice were treated with pravastatin or vehicle for 20 weeks. Aortic calcification was assessed by in vivo 18F-NaF-μPET/μCT imaging at weeks 0, 10 and 20 and by histomorphometry at euthanasia. MicroCT analysis showed that, in both groups, the amount of vascular calcification increased significantly over the 20-week period, but pravastatin treatment did not augment over the controls. In contrast, the μPET analysis showed that, at week 10, the pravastatin group had less 18F uptake, suggesting reduced surface area of actively mineralizing deposits, but this decrease was not sustained at week 20. However, a significant difference in the mineral deposit size was found by histomorphometry. The pravastatin group had significantly more aortic microcalcium deposits (< 50 μm in diameter) than the controls. The pravastatin group also had more vascular cells positive for alkaline phosphatase activity than the controls. The amount of collagen and osteopontin, additional osteoblastic markers, were not significantly different between the two groups.
Conclusions:
These results suggest that pravastatin treatment alters the microarchitecture of aortic calcium deposits with potential effects on plaque stability.
Keywords: calcification, imaging, microarchitecture, aging
Subject terms: atherosclerosis, vascular disease, imaging
Graphical Abstract
INTRODUCTION
Cardiovascular calcification has been associated with increased morbidity and mortality.1 Particularly in the coronary arteries, calcification has been long established as a strong predictor of cardiovascular disease and poor prognosis2, 3 potentially due to plaque rupture.
Although 3-hydroxy-3-methylglutaryl (HMG) Co-A reductase inhibitors (statins), are associated with reduced cardiovascular risks,4–10 they are also associated with increased progression of coronary and aortic calcification,6, 8 which is associated with increased cardiovascular risk.11–13 In an effort to explain this paradoxical effect, some studies suggest that statin-induced progression of calcification may proceed in some as-of-yet unknown manner that increases the amount of calcification while reducing cardiovascular risk, such as by changing the size distribution or density of calcium deposits.5, 7, 14 It is conceivable that a change in microarchitecture may decrease rupture risk, such as reduced mineral surface area, which may occur by the coalescence of small deposits and/or decreased porosity. Surface area is expected to influence rupture because rupture stress is greatly magnified at the edges of calcium deposits facing solid stresses13 as a result of compliance mismatch.15 Thus, it is possible that coalesced calcium deposits with reduced surface area may pose less risk of debonding and subsequent plaque rupture. Since fluoride adsorbs to surfaces of actively mineralizing apatite mineral deposits with high affinity,16 18F-NaF micro-positron emission tomography (μPET) imaging may serve as a marker to quantify surface area of cardiovascular calcium deposits.
We previously found such a reduction in surface area of aortic calcium deposits in mice treated with parathyroid hormone (PTH)17 and those subjected to treadmill exercise.18 Given that previous clinical trials show increased levels of serum PTH with statin treatment,19, 20 we tested the effects of pravastatin on serum PTH levels in this report. We also assessed the progression and microarchitecture of vascular calcium deposits using a mouse model where pravastatin has previously been shown to increase plaque calcification.21
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals and treatments
Female apolipoprotein E-null (Apoe−/−) mice (C57BL/6J background, The Jackson Laboratory, Bar Harbor, ME) were fed a normal laboratory diet (Teklad 7013, Envigo, IN), and vascular calcification was allowed to develop as they aged. In order to have an adequate number of subjects and avoid confounding effects, female mice were chosen for this study, as they have been shown to have more vascular calcification than male mice.22 As shown in Figure 1, at 56 weeks of age, the mice were separated into two groups, a control group (n = 20) and a group treated with pravastatin (LKT Laboratories Inc., St. Paul, Minnesota) in the drinking water (dosage of 40 mg/kg of mouse body weight, n = 20) for 20 weeks (wk). One mouse assigned to the pravastatin group had to be euthanized due to ulcerative dermatitis. MicroPET/μCT (micro-computed tomography) imaging was performed on each mouse at 3 time points (wk 0, wk 10, and wk 20), and mice were euthanized 24 hours after the last scan to allow for the decay of 18F. Blood was collected immediately prior to euthanasia for serum PTH. Two additional mice from each group were euthanized due to ulcerative dermatitis during the 20-wk period. One additional mouse in the pravastatin group died after μPET/μCT imaging, prior to collection of the serum sample. The number of mice in each group is described in the figure legends. Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, Los Angeles.
Figure 1. Experimental design.
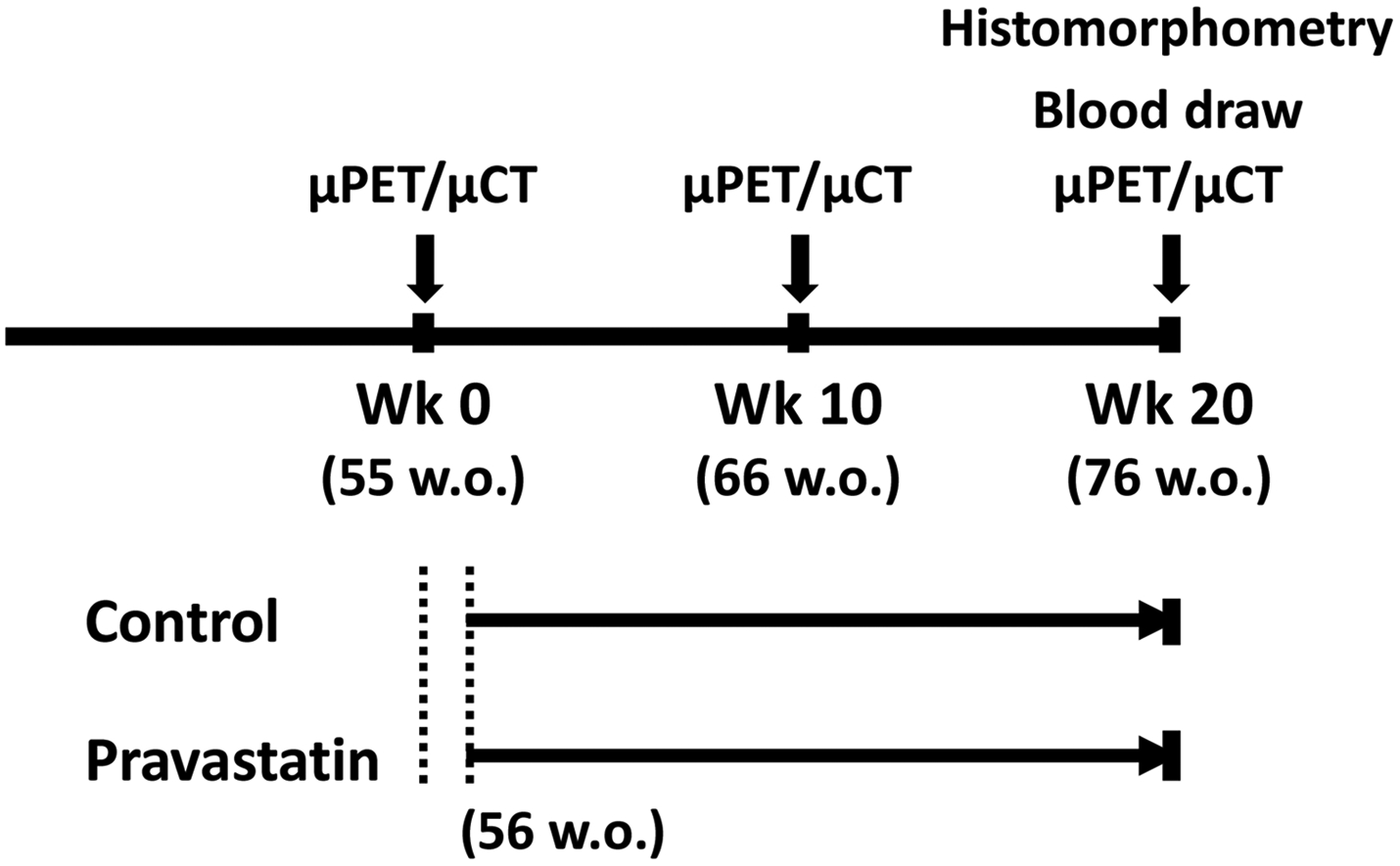
Schematic of the timing of interventions for control and pravastatin groups. Each mouse was imaged at weeks 0, 10 and 20. The treatment period started 1 week after the first μPET/μCT imaging at 56 weeks of age.
Serum PTH and lipid profile
Serum PTH levels were quantified by murine PTH (1–84) ELISA (Quidel, San Diego, CA), following the manufacturer’s instructions. Serum lipid levels were determined by the UCLA Atherosclerosis Research Unit Core Laboratory (Pointe Scientific Reagents; Fisher Scientific).
Serial in-vivo 18F-NaF μPET/μCT imaging
Aortic mineral content and surface area were quantified by in vivo 18F-NaF μPET/μCT imaging, as previously described,17 using a μPET scanner (Focus 200 μPET, Concorde, Microsystems, Knoxville, TN) and a high-resolution μCT scanner (CrumpCAT, UCLA). Each mouse was imaged at 3 time points: baseline (wk 0), mid-treatment (wk 10), and post-treatment (wk 20) at 55, 66, and 76 wks of age, respectively. AMIDE software was used to analyze both μPET and μCT images. A volumetric region of interest (ROI) of cardiovascular calcifications was drawn and isolated from the reminder of the body. Three-dimensional isocontour mapping was used to quantify ROI parameters. The minimum isocontour threshold for 18F-NaF was defined as 2.2% of injected dose per cubic centimeter (%ID/cc) and for μCT, it was defined as 250 Hounsfield units. To assess the total content of calcification, the volumetric Hounsfield unit was calculated as the product of mean density (Hounsfield units) and the volumetric size. One mouse in the control group was excluded from analysis due to technical difficulties with the 18F-NaF injections, and one mouse in the statin group was excluded from the μCT analysis due to undetectable calcification at wk 0 and wk 10.
Histomorphometric analyses
At euthanasia, hearts were harvested and embedded in optimal cutting temperature compound. Each aortic root was sectioned (10 μm thick) and stained for each analysis, as described below. For quantification, images of the sections were captured using the EXi Aqua camera under a 2X microscope magnification and processed on ImageJ software (NIH). By maximizing image contrast, areas of positive Alizarin red staining were isolated. The processed images were analyzed using customized MATLAB software to quantify the cross-sectional area and perimeter of deposits, as surrogate measures for the volume and surface area, respectively.
The calcium deposits were identified by Alizarin red staining. The microarchitecture of calcium deposits was quantified, as described above. For each mouse, 2 aortic root sections separated by ~ 300–500 μm were analyzed. According to previously defined criteria,23 calcium deposits were categorized as micro, intermediate, or large calcium deposits, according to diameter: ≤ 50 μm (corresponding cross-sectional area ≤ 2,000 μm2), 50 μm - 100 μm (cross-sectional area 2,000 μm2 – 7,850 μm2), and > 100 μm (cross-sectional area > 7,850 μm2), respectively.
Alkaline phosphatase activity was assessed by histochemical staining, using nitro blue tetrazolium/5-Bromo-4-chloro-3-indolyl phosphate solution (Fisher Scientific), collagen was assessed by Masson’s modified trichrome staining (International Medical Equip, CA), and osteopontin was assessed by immunohistochemistry using rabbit polyclonal anti-osteopontin antibody (Abcam). The positive staining was quantified as described above.
Statistical analysis
Statistical analysis was performed using Prism software (v.7). The data were first subjected to the D’Agostino-Pearson omnibus test for normality. A Student’s t-test (paired or unpaired, as appropriate) was performed for parametric (normally distributed) data, and the Mann-Whitney test was performed for non-parametric data. For comparison of more than two sets of data, the repeated-measures one-way ANOVA was performed for the parametric data and the Friedman test for the nonparametric data. Statistical significance was set at a p value ≤ 0.05. Values are expressed as mean ± SEM.
RESULTS
Effects of pravastatin on microarchitecture and total content of cardiovascular calcium deposits by in vivo imaging
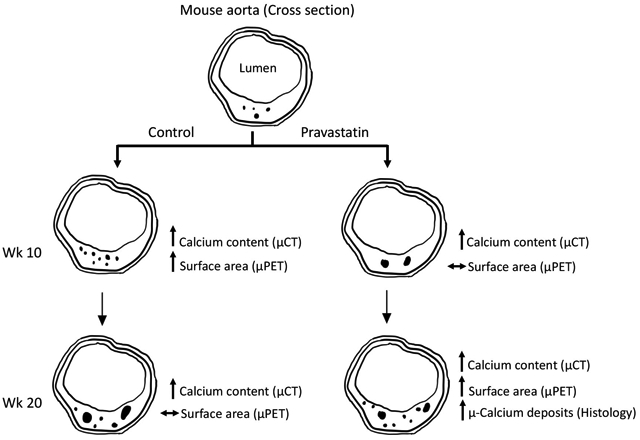
Effects of pravastatin on calcium deposits were tested in aged Apoe−/− mice, which spontaneously develop cardiovascular calcification on a normal laboratory diet with aging. Each mouse was imaged by μPET/μCT scanning at weeks 0, 10 and 20 (baseline pre-, mid- and post-treatment) to assess surface area and content of aortic calcium deposits, respectively (Fig. 2A). Analysis of serial μCT imaging revealed that aortic calcium content progressed significantly in both the control and pravastatin groups over the 20-wk period, but pravastatin treatment did not further augment aortic calcification over the controls at both time points (Fig. 2B). Analysis of serial μPET imaging revealed that 18F uptake in the control group increased significantly with aging from wk 0 to wk 10 (p = 0.03) but did not change from wk 10 to wk 20 (p = 0.77) (Fig. 2C). In contrast, 18F uptake in the pravastatin group did not change from wk 10 to wk 20 (p = 0.61) but increased significantly from wk 0 to wk 10 (p = 0.02) (Fig. 2C).
Figure 2. Effects of pravastatin on calcium deposits by μCT/μPET imaging.
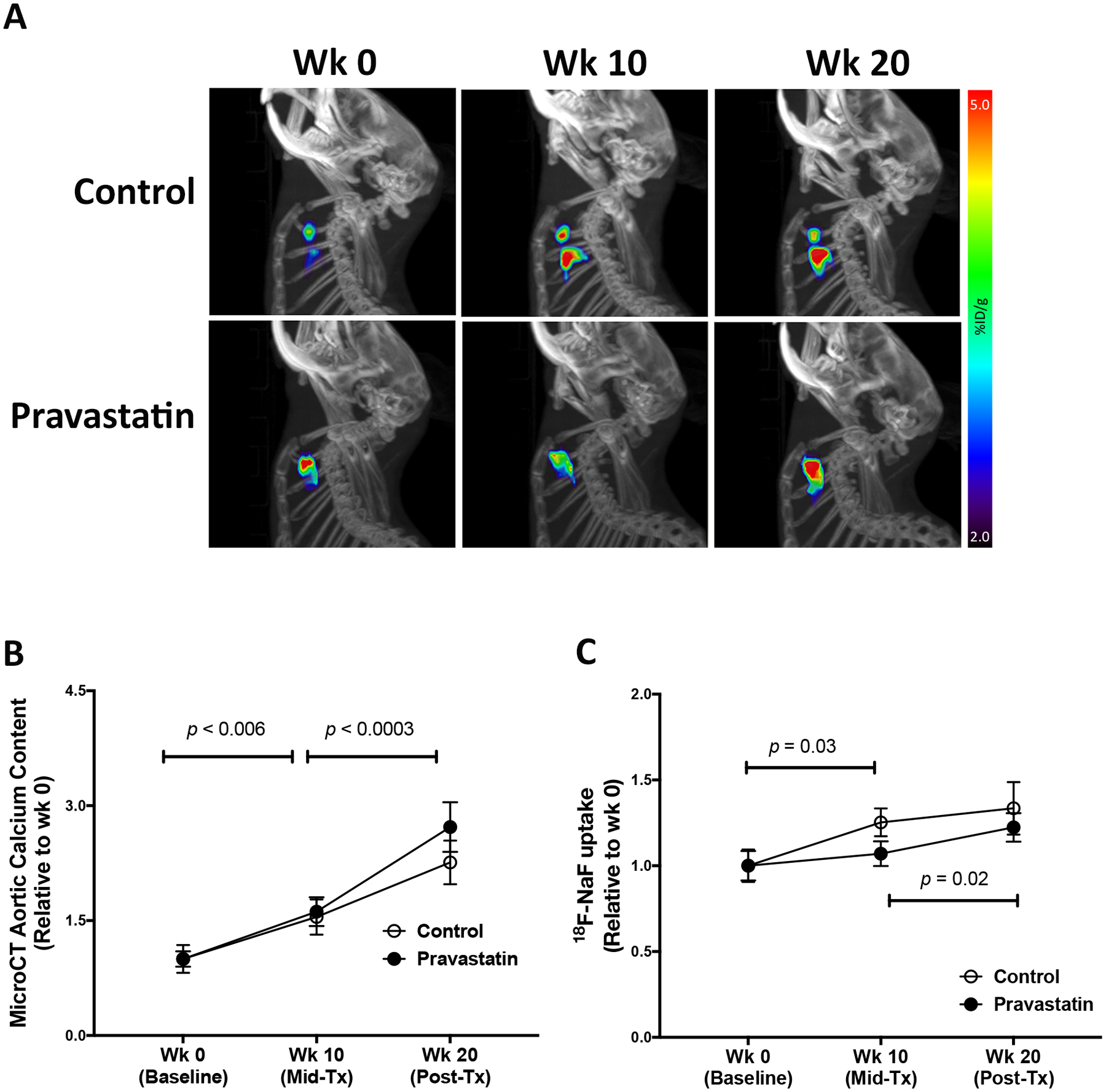
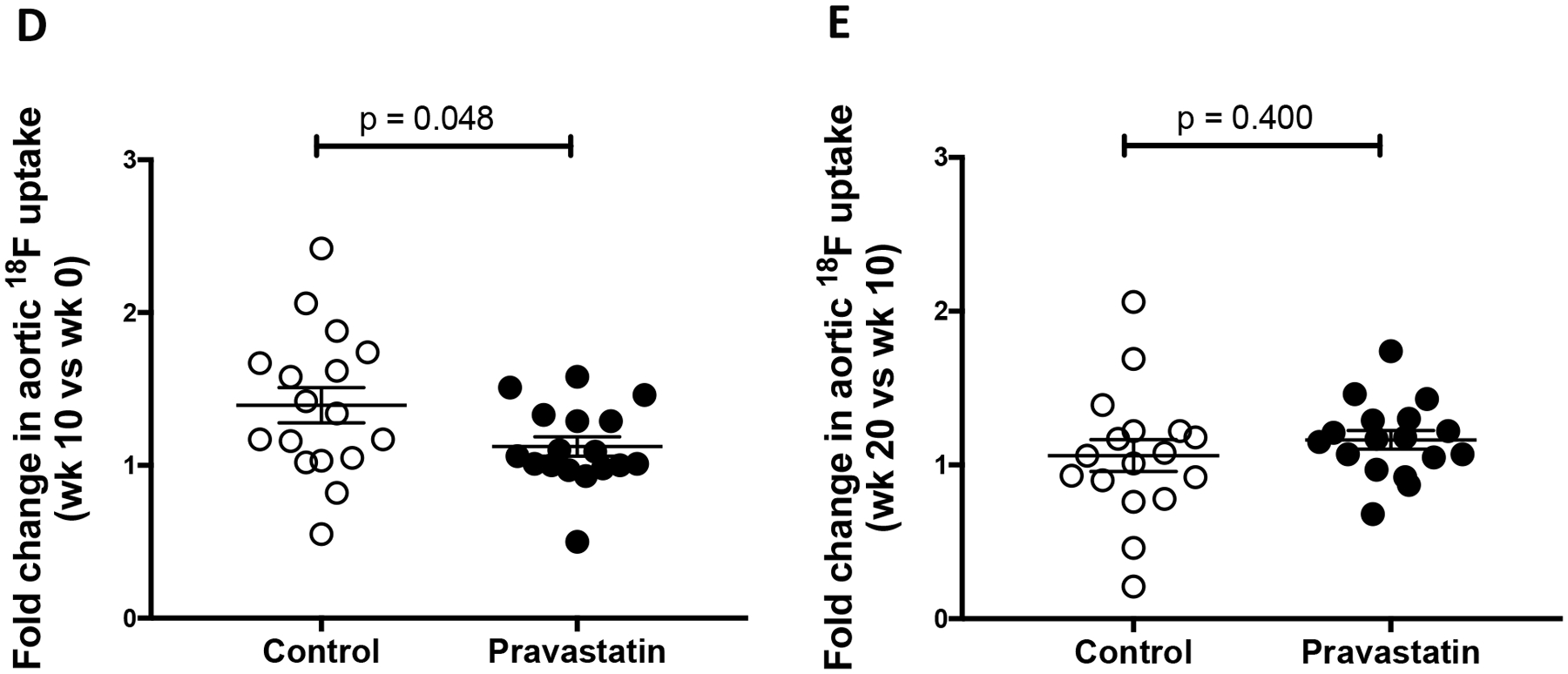
(A) A representative lateral view of the μPET maximum-intensity projection (pseudo-color) superimposed on the μCT image of the skeleton in the control and pravastatin-treated mice at baseline (wk 0), mid-treatment (wk 10), and post-treatment (wk 20). (B) MicroCT analysis of aortic calcium content in the control (n = 18) and pravastatin-treated (n = 16) mice. (C) MicroPET analysis of aortic 18F-NaF uptake in the control (n = 17) and pravastatin-treated (n = 17) mice. *p = 0.03 (Control wk 10 vs. wk 0), **p = 0.02 (Pravastatin wk 20 vs. wk 10). (D) Fold-change in 18F-NaF uptake over the first 10-wk period (wk 10 vs. wk 0) in the control (n = 17) and pravastatin-treated (n = 17) mice. (E) Fold change in 18F-NaF uptake over the second 10-wk period (wk 20 vs. wk 10) in the control (n = 17) and pravastatin-treated (n = 17) mice.
Since baseline calcification differed among the mice, we calculated fold-changes between the two 10-wk periods for each mouse. The μCT analysis showed that the fold changes between the control and pravastatin groups were not significantly different between the first 10-wk period (control, 1.72 ± 0.15 vs. pravastatin-treated, 1.68 ± 0.12; p = 0.95) or the second 10-wek period (control, 1.53 ± 0.11 vs. pravastatin-treated, 1.67 ± 0.10; p = 0.12). Interestingly, 18F-NaF uptake was significantly lower in the pravastatin group than the control group over the first (0–10-wk) period (Fig. 2D) but not over the second (10–20-wk) period (Fig. 2E).
Effects of pravastatin on cardiovascular calcium deposits by histomorphometric analysis
We further analyzed the calcium deposits in the aortic roots via histomorphometry. Results showed that consistent with μCT analysis, the cross-sectional area of calcium deposits was similar between the two groups (data not shown). Interestingly, the pravastatin group had a greater total number of calcium deposits, identified by Alizarin red positivity (Fig. 3A). Further analysis of the calcium deposits, categorized into micro (<50 μm in diameter), intermediate (50–100 μm in diameter), or large deposits (>100 μm in diameter),23 revealed that the greater number of deposits in the pravastatin-treated mice, compared with controls, were in the size range of microdeposits (Fig. 3B).
Figure 3. Effects of pravastatin on sizes of calcium deposits by histomorphometric analysis.
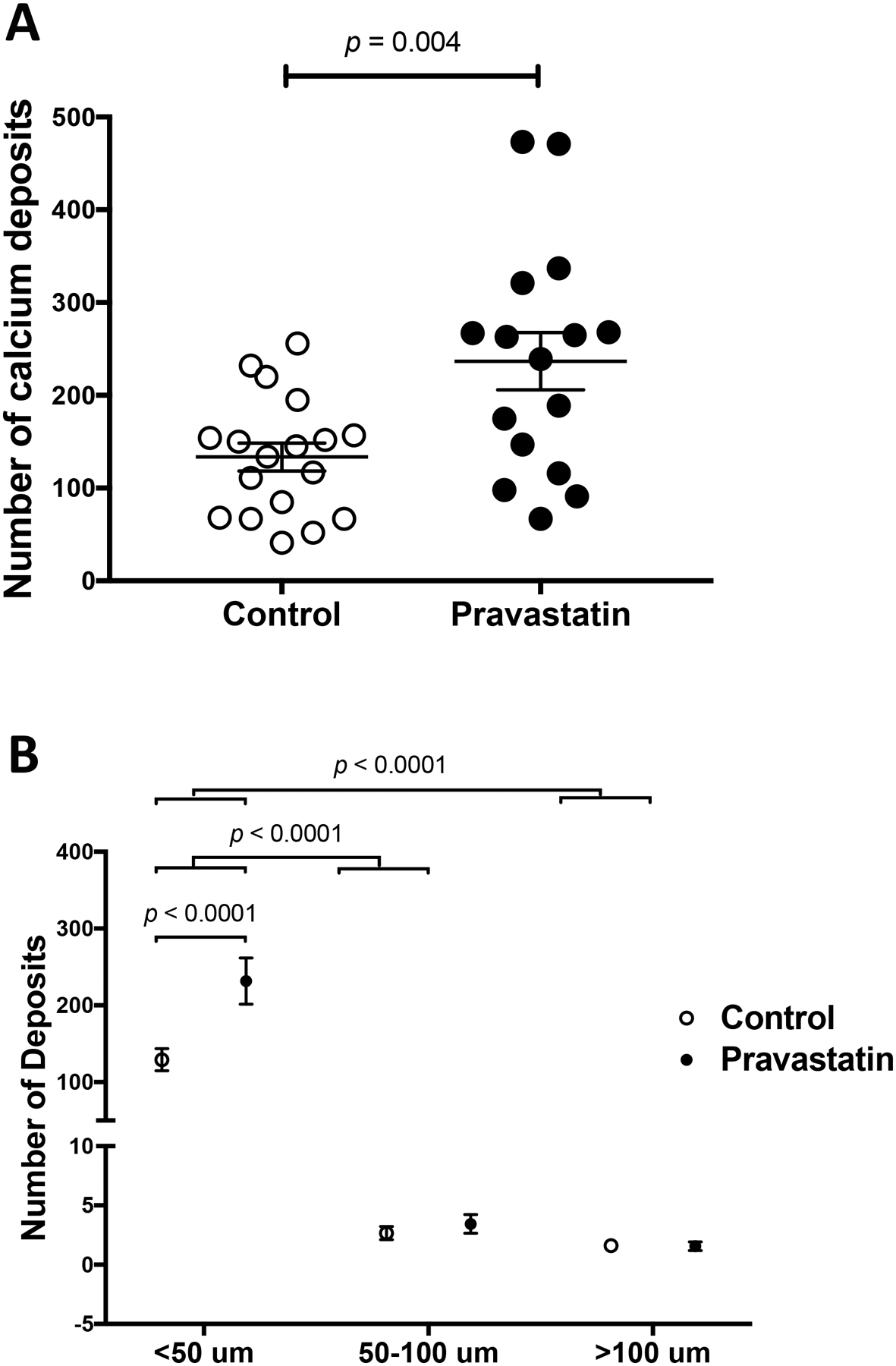
(A) The total number of calcium deposits in the control (n = 18) and pravastatin-treated (n = 16) mice. (B) The number of deposits categorized by size: microcalcium (< 50 μm in diameter), intermediate (50 – 100 μm in diameter), and macrocalcium (> 100 μm in diameter) deposits.
To assess the mechanism of pravastatin effects on microcalcium deposits, we tested whether pravastatin induced calcification through osteogenic differentiation. Based on histochemical staining, the pravastatin group had greater numbers of alkaline phosphatase positive cells (Fig. 4A–B). We also examined the extracellular matrix, a key determinant of biomineralization. Our histochemical staining showed no significant difference between the two groups with respect to the amount of collagen present in these mice (Fig. 4A; control, 0.44 ± 0.04 vs. pravastatin-treated, 0.50 ± 0.05 mm2; p = 0.33), which is in agreement with another study in mice,24 although statin therapy has been shown to promote collagen degradation in humans.25 An additional osteoblastic differentiation marker, osteopontin, also showed similar expression between the two groups (Fig. 4A; control, 0.23 ± 0.45 vs. pravastatin-treated, 0.41 ± 0.88 mm2; p = 0.13).
Figure 4. Effects of pravastatin on osteoblastic markers by histomorphometric analysis.
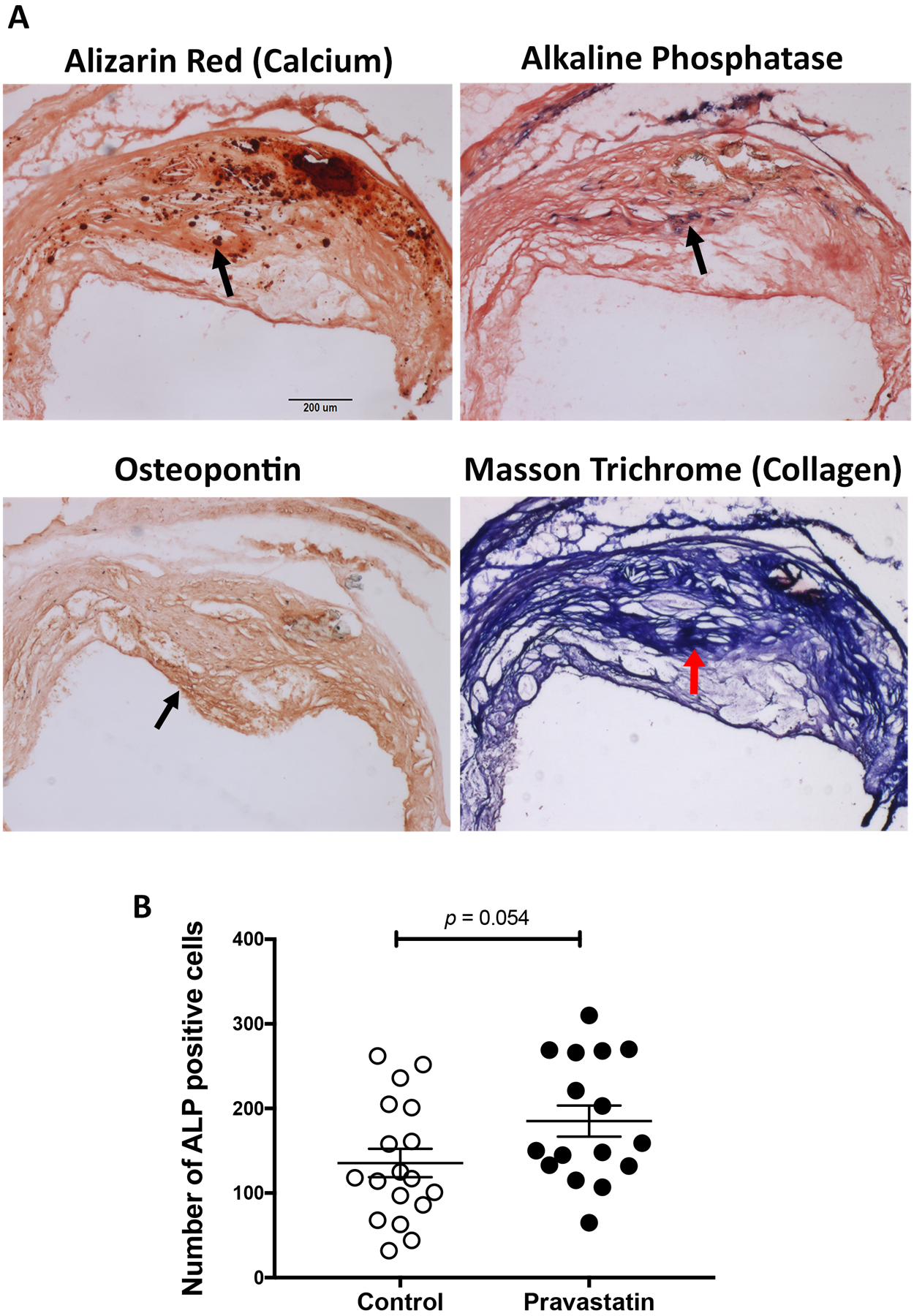
(A) Representative images of calcium deposits in a control mouse by alizarin red histochemistry, alkaline phosphatase (purple) histochemistry, osteopontin (brown) immunohistochemistry and collagen (blue) histochemistry. An arrow indicates the location of each marker. Scale bar, 200 μm. (B) Quantitation of the number of alkaline phosphatase (ALP) positive cells in the control (n = 18) and pravastatin-treated (n = 16) mice.
Consistent with a prior study,21 our results also showed an increase in total cholesterol level (control, 146 ± 8 vs. pravastatin, 202 ± 12 mg/dL; p < 0.01) with pravastatin treatment for 4.5 months. Consistent with the increase in cholesterol levels, the lesion area, stained by oil red O, in the aortic root was also greater in the pravastatin group (control, 0.29 ± 0.04 vs. pravastatin-treated, 0.40 ± 0.04 mm2, p = 0.04). Serum levels of PTH, however, did not significantly differ between the two groups (control, 187 ± 32 vs. pravastatin, 211 ± 39 pg/mL; p = 0.54).
DISCUSSION
Since statin therapy, which is widely accepted as having positive cardiovascular outcomes, paradoxically increases vascular calcification,4–10 we tested whether the cardiovascular benefits of statins are in part explained by an alteration in the morphological features of calcium deposits, specifically a reduction of surface area, which may reduce the potential sites of rupture. Several of the many pleiotropic mechanisms of statins may have a role in such a phenomenon, including those acting via HMG-CoA reductase inhibition to improve dyslipidemia, HMG-CoA reductase inhibition that is independent of the effects on circulating lipids, and/or off-target effects that are independent of HMG-CoA reductase inhibition.
Studies show that, in humans, increased coronary calcification has been observed starting after about 6 years of statin treatment.6, 8 In our study, the aortic calcium mineral content assessed by μCT imaging showed a significant progression of calcification with aging in both the control and pravastatin-treated groups. Interestingly, in the pravastatin group, the progression appears to be greater at week 20 than week 10. Histological analysis showed that the number of microcalcifications was greater in statin-treated mice at the 20-week timepoint (equivalent to about 15.5 years in humans, based on lifespan26). Since microcalcifications are associated with progression of calcium content,27 one may predict that, if the treatment were to continue longer, the calcium content may exceed that of controls. A 35-wk treatment with pravastatin in mice has been shown to increase the percentage of plaques having calcification.21
Given that mineral surface area is expected to influence biomechanical vulnerability, we used serial in vivo μPET imaging to assess changes in surface area of aortic calcium deposits. In the control group, the 18F-NaF uptake increased significantly with aging during the first 10-wk period then plateaued during the second 10-wk period. This latter finding of increased calcification by μCT without a corresponding increase in surface area by μPET suggests that calcium deposits may coalesce spontaneously with aging (in the control group). Interestingly, the pravastatin group showed the opposite results with μPET imaging. The 18F-NaF uptake failed to increase during the first 10-wk period, becoming significantly less than in controls, suggesting they had significantly less increase in surface area despite the significant increase in the calcium mineral content by μCT. Interestingly, this difference of PET tracer uptake between the two groups was not sustained during the second 10-wk period since the pravastatin group showed a trend toward a greater increase, which may be, in part, attributable to the greater number of microdeposits since they have greater surface area.
Hydrophilic statins (such as pravastatin and rosuvastatin) have been shown to penetrate and modulate the biology of the vasculature both at the level of vascular cells and at the level of intact vessel physiology.28–30 Pravastatin also inhibits HMG-CoA reductase in myocytes, resulting in cholesterol depletion.31, 32 Two potential mechanisms for pravastatin effects on calcification include effects on vitamin K and/or lipid levels (Fig. 5). Statin inhibition of vitamin K availability33, 34 may prevent activation (by gamma-carboxylation) of the calcification delimiters, matrix-Gla protein and osteocalcin,35, 36 allowing juxtaposed deposits to coalesce, reducing the surface area, and thus 18F-NaF uptake at week 10. Such coalescence is supported by the finding of mismatch between μCT and μPET at week 10. The increase in cholesterol levels in the statin-treated mice may contribute to oxidized lipid accumulations, causing increased macrophage production of procalcific cytokines24 that we previously showed to induce alkaline phosphatase and promote calcification.37–39 The observed increase in the number of alkaline phosphatase-positive cells may increase the number of niduses from which microcalcium deposits develop, thus increasing 18F-NaF uptake at week 20. If the two mechanisms are staggered, they may account for the observed biphasic effects.
Figure 5. Schematic of the pravastatin effects on calcification.
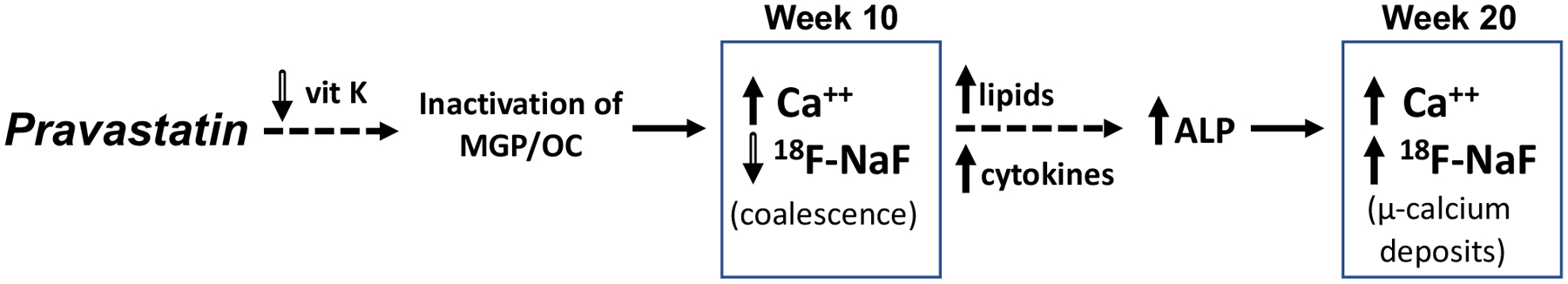
Potential mechanisms are depicted. 1) At week 10, statin inhibition of vitamin K availability33, 34 may prevent activation (by gamma-carboxylation) of the calcification delimiters, matrix-Gla protein (MGP) and osteocalcin (OC),35, 36 allowing juxtaposed deposits to coalesce, reducing the surface area, and thus 18F-NaF uptake compared with controls. Such coalescence is supported by the finding of mismatch between μCT and μPET at week 10. 2) The increase in cholesterol levels in the statin-treated mice may contribute to oxidized lipid accumulations, causing increased macrophage production of procalcific cytokines24 that we previously showed to induce alkaline phosphatase and promote calcification.37–39 The observed increase in the number of ALP-positive cells may increase the number of microcalcium deposits, thus increasing 18F-NaF uptake at week 20 compared with week 10. The staggered mechanisms may account for the observed biphasic effects.
Our previous experiments with intermittent PTH injections17 and treadmill exercise,18 which also elevated serum PTH levels, showed a reduction in mineral surface area and increased size of vascular calcium deposits by 18F-NaF μPET/μCT imaging and histological analysis. However, in this study, we did not observe increased serum PTH levels with pravastatin treatment. Given some evidence that statins increase serum PTH levels,19, 20 one possibility is that an initial transient increase in PTH blunted the expansion of vascular mineral surface area with aging, but once PTH levels normalized, the effect on surface area returned to control levels. Further studies are required to test whether statins affect serum PTH levels at various time points.
Although statins lower lipids and reduce vascular calcification when mice are on a high-fat diet,40–42 we applied a regimen, which used a normal laboratory diet, previously shown to increase vascular calcification in order to test our hypothesis. This regimen happens to also increase or did not lower the cholesterol levels in other studies,21, 42–44 which is one limitation of this study.
To our knowledge, there are no studies showing a window of time when humans undergoing statin treatment have increased susceptibility to cardiovascular events. However, our findings suggest that pravastatin treatment in mice has a biphasic effect on the microarchitecture of the calcium deposits in a time dependent manner – reducing surface area initially and recovering to near control levels later, possibly from de novo formation of smaller deposits. Thus, the ability of statins to confer clinical benefit while increasing coronary calcification may relate to complex time-dependent changes in microarchitecture of the calcium deposits.
Supplementary Material
HIGHLIGHTS.
Aortic calcification progresses in control and pravastatin-treated mice to a similar degree over a 20-week period.
Pravastatin treatment was associated with significantly less progression in apparent mineral surface area at early time points.
Progression of aortic calcification without corresponding progression in surface area suggests the coalescence of calcium deposits.
The number of microcalcium deposits was greater in pravastatin-treated mice than in controls.
Surface area of aortic calcium deposits changed in a biphasic manner in pravastatin-treated mice.
Pravastatin may alter progression and microarchitectural features of vascular calcium deposits.
Acknowledgements:
The authors thank the UCLA Atherosclerosis Research Unit for lipid analysis.
Sources of Funding: This work was supported, in part, by funding from the National Institute of Aging (NIA) and National Heart, Lung, and Blood Institute (NHLBI) (AG061586 and HL137647) of the National Institutes of Health.
ABBREVIATIONS
- ALP
Alkaline phosphatase
- HMG Co-A
3-hydroxy-3-methylglutaryl coenzyme A
- μCT
Micro-computed tomography
- μPET
Micro-positron emission tomography
- 18F-NaF
Sodium fluoride labeled with fluoride 18 isotope
- PTH
Parathyroid hormone
Footnotes
Disclosures: None
REFERENCES
- 1.Gepner AD, Young R, Delaney JA, Tattersall MC, Blaha MJ, Post WS, Gottesman RF, Kronmal R, Budoff MJ, Burke GL, Folsom AR, Liu K, Kaufman J, Stein JH. Comparison of coronary artery calcium presence, carotid plaque presence, and carotid intima-media thickness for cardiovascular disease prediction in the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging. 2015;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw LJ, Giambrone AE, Blaha MJ, Knapper JT, Berman DS, Bellam N, Quyyumi A, Budoff MJ, Callister TQ, Min JK. Long-term prognosis after coronary artery calcification testing in asymptomatic patients: A cohort study. Ann Intern Med. 2015;163:14–21 [DOI] [PubMed] [Google Scholar]
- 3.Polonsky TS, McClelland RL, Jorgensen NW, Bild DE, Burke GL, Guerci AD, Greenland P. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010;303:1610–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knopp RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341:498–511 [DOI] [PubMed] [Google Scholar]
- 5.Mujaj B, Bos D, Selwaness M, Leening MJG, Kavousi M, Wentzel JJ, van der Lugt A, Hofman A, Stricker BH, Vernooij MW, Franco OH. Statin use is associated with carotid plaque composition: The rotterdam study. Int J Cardiol. 2018;260:213–218 [DOI] [PubMed] [Google Scholar]
- 6.Henein M, Granasen G, Wiklund U, Schmermund A, Guerci A, Erbel R, Raggi P. High dose and long-term statin therapy accelerate coronary artery calcification. Int J Cardiol. 2015;184:581–586 [DOI] [PubMed] [Google Scholar]
- 7.Puri R, Nicholls SJ, Shao M, Kataoka Y, Uno K, Kapadia SR, Tuzcu EM, Nissen SE. Impact of statins on serial coronary calcification during atheroma progression and regression. J Am Coll Cardiol. 2015;65:1273–1282 [DOI] [PubMed] [Google Scholar]
- 8.Dykun I, Lehmann N, Kalsch H, Mohlenkamp S, Moebus S, Budde T, Seibel R, Gronemeyer D, Jockel KH, Erbel R, Mahabadi AA. Statin medication enhances progression of coronary artery calcification: The heinz nixdorf recall study. J Am Coll Cardiol. 2016;68:2123–2125 [DOI] [PubMed] [Google Scholar]
- 9.Saremi A, Bahn G, Reaven PD. Progression of vascular calcification is increased with statin use in the veterans affairs diabetes trial (vadt). Diabetes Care. 2012;35:2390–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banach M, Serban C, Sahebkar A, Mikhailidis DP, Ursoniu S, Ray KK, Rysz J, Toth PP, Muntner P, Mosteoru S, Garcia-Garcia HM, Hovingh GK, Kastelein JJ, Serruys PW, Lipid, Blood Pressure Meta-analysis Collaboration G. Impact of statin therapy on coronary plaque composition: A systematic review and meta-analysis of virtual histology intravascular ultrasound studies. BMC Med. 2015;13:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abedin M, Tintut Y, Demer LL. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–1170 [DOI] [PubMed] [Google Scholar]
- 12.Criqui MH, Denenberg JO, Ix JH, McClelland RL, Wassel CL, Rifkin DE, Carr JJ, Budoff MJ, Allison MA. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA. 2014;311:271–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshino T, Chow LA, Hsu JJ, Perlowski AA, Abedin M, Tobis J, Tintut Y, Mal AK, Klug WS, Demer LL. Mechanical stress analysis of a rigid inclusion in distensible material: A model of atherosclerotic calcification and plaque vulnerability. Am J Physiol Heart Circ Physiol. 2009;297:H802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houslay ES, Cowell SJ, Prescott RJ, Reid J, Burton J, Northridge DB, Boon NA, Newby DE, Scottish Aortic S, Lipid Lowering Therapy IoRtI. Progressive coronary calcification despite intensive lipid-lowering treatment: A randomised controlled trial. Heart. 2006;92:1207–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett HE, Van der Heiden K, Farrell E, Gijsen FJH, Akyildiz AC. Calcifications in atherosclerotic plaques and impact on plaque biomechanics. J Biomech. 2019;87:1–12 [DOI] [PubMed] [Google Scholar]
- 16.Czernin J, Satyamurthy N, Schiepers C. Molecular mechanisms of bone 18f-naf deposition. J Nucl Med. 2010;51:1826–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu JJ, Lu J, Umar S, Lee JT, Kulkarni RP, Ding Y, Chang CC, Hsiai TK, Hokugo A, Gkouveris I, Tetradis S, Nishimura I, Demer LL, Tintut Y. Effects of teriparatide on morphology of aortic calcification in aged hyperlipidemic mice. Am J Physiol Heart Circ Physiol. 2018;314:H1203–H1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu JJ, Fong F, Patel R, Qiao R, Lo K, Soundia A, Chang CC, Le V, Tseng CH, Demer LL, Tintut Y. Changes in microarchitecture of atherosclerotic calcification assessed by (18)f-naf pet and ct after a progressive exercise regimen in hyperlipidemic mice. J Nucl Cardiol. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rejnmark L, Buus NH, Vestergaard P, Andreasen F, Larsen ML, Mosekilde L. Statins decrease bone turnover in postmenopausal women: A cross-sectional study. Eur J Clin Invest. 2002;32:581–589 [DOI] [PubMed] [Google Scholar]
- 20.Zarei B, Mousavi M, Mehdizadeh S, Mehrad-Majd H, Zarif M, Erfanian Z, Moradi A. Early effects of atorvastatin on vitamin d and parathyroid hormone serum levels following acute myocardial infarction. J Res Pharm Pract. 2019;8:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Xiao S, Li Q. Pravastatin polarizes the phenotype of macrophages toward m2 and elevates serum cholesterol levels in apolipoprotein e knockout mice. J Int Med Res. 2018;46:3365–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marek I, Canu M, Cordasic N, Rauh M, Volkert G, Fahlbusch FB, Rascher W, Hilgers KF, Hartner A, Menendez-Castro C. Sex differences in the development of vascular and renal lesions in mice with a simultaneous deficiency of apoe and the integrin chain itga8. Biol Sex Differ. 2017;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly-Arnold A, Maldonado N, Laudier D, Aikawa E, Cardoso L, Weinbaum S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc Natl Acad Sci U S A. 2013;110:10741–10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Healy A, Berus JM, Christensen JL, Lee C, Mantsounga C, Dong W, Watts JP Jr., Assali M, Ceneri N, Nilson R, Neverson J, Wu WC, Choudhary G, Morrison AR. Statins disrupt macrophage rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscler Thromb Vasc Biol. 2020;40:714–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tziakas DN, Chalikias GK, Stakos DA, Papanas N, Chatzikyriakou SV, Mitrousi K, Maltezos E, Boudoulas H. Effect of statins on collagen type i degradation in patients with coronary artery disease and atrial fibrillation. Am J Cardiol. 2008;101:199–202 [DOI] [PubMed] [Google Scholar]
- 26.Dutta S, Sengupta P. Men and mice: Relating their ages. Life Sci. 2016;152:244–248 [DOI] [PubMed] [Google Scholar]
- 27.Doris MK, Meah MN, Moss AJ, Andrews JPM, Bing R, Gillen R, Weir N, Syed M, Daghem M, Shah A, Williams MC, van Beek EJR, Forsyth L, Dey D, Slomka PJ, Dweck MR, Newby DE, Adamson PD. Coronary (18)f-fluoride uptake and progression of coronary artery calcification. Circ Cardiovasc Imaging. 2020;13:e011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irwin JC, Fenning AS, Vella RK. Statins with different lipophilic indices exert distinct effects on skeletal, cardiac and vascular smooth muscle. Life Sci. 2020;242:117225. [DOI] [PubMed] [Google Scholar]
- 29.Rawlings R, Nohria A, Liu PY, Donnelly J, Creager MA, Ganz P, Selwyn A, Liao JK. Comparison of effects of rosuvastatin (10 mg) versus atorvastatin (40 mg) on rho kinase activity in caucasian men with a previous atherosclerotic event. Am J Cardiol. 2009;103:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamanouchi D, Banno H, Nakayama M, Sugimoto M, Fujita H, Kobayashi M, Kuwano H, Komori K. Hydrophilic statin suppresses vein graft intimal hyperplasia via endothelial cell-tropic rho-kinase inhibition. J Vasc Surg. 2005;42:757–764 [DOI] [PubMed] [Google Scholar]
- 31.Osaki Y, Nakagawa Y, Miyahara S, Iwasaki H, Ishii A, Matsuzaka T, Kobayashi K, Yatoh S, Takahashi A, Yahagi N, Suzuki H, Sone H, Ohashi K, Ishibashi S, Yamada N, Shimano H. Skeletal muscle-specific hmg-coa reductase knockout mice exhibit rhabdomyolysis: A model for statin-induced myopathy. Biochem Biophys Res Commun. 2015;466:536–540 [DOI] [PubMed] [Google Scholar]
- 32.Sanyour HJ, Li N, Rickel AP, Torres HM, Anderson RH, Miles MR, Childs JD, Francis KR, Tao J, Hong Z. Statin-mediated cholesterol depletion exerts coordinated effects on the alterations in rat vascular smooth muscle cell biomechanics and migration. J Physiol. 2020;598:1505–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Z, Qureshi AR, Parini P, Hurt-Camejo E, Ripsweden J, Brismar TB, Barany P, Jaminon AM, Schurgers LJ, Heimburger O, Lindholm B, Stenvinkel P. Does statins promote vascular calcification in chronic kidney disease? Eur J Clin Invest. 2017;47:137–148 [DOI] [PubMed] [Google Scholar]
- 34.Harshman SG, Shea MK, Fu X, Grusak MA, Smith D, Lamon-Fava S, Kuliopulos A, Greenberg A, Booth SL. Atorvastatin decreases renal menaquinone-4 formation in c57bl/6 male mice. J Nutr. 2019;149:416–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cranenburg EC, Brandenburg VM, Vermeer C, Stenger M, Muhlenbruch G, Mahnken AH, Gladziwa U, Ketteler M, Schurgers LJ. Uncarboxylated matrix gla protein (ucmgp) is associated with coronary artery calcification in haemodialysis patients. Thromb Haemost. 2009;101:359–366 [PubMed] [Google Scholar]
- 36.Cranenburg EC, Vermeer C, Koos R, Boumans ML, Hackeng TM, Bouwman FG, Kwaijtaal M, Brandenburg VM, Ketteler M, Schurgers LJ. The circulating inactive form of matrix gla protein (ucmgp) as a biomarker for cardiovascular calcification. J Vasc Res. 2008;45:427–436 [DOI] [PubMed] [Google Scholar]
- 37.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the camp pathway. Circulation. 2000;102:2636–2642 [DOI] [PubMed] [Google Scholar]
- 38.Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL. High-density lipoprotein regulates calcification of vascular cells. Circ Res. 2002;91:570–576 [DOI] [PubMed] [Google Scholar]
- 39.Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655 [DOI] [PubMed] [Google Scholar]
- 40.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850 [DOI] [PubMed] [Google Scholar]
- 41.Gronros J, Wikstrom J, Brandt-Eliasson U, Forsberg GB, Behrendt M, Hansson GI, Gan LM. Effects of rosuvastatin on cardiovascular morphology and function in an apoe-knockout mouse model of atherosclerosis. Am J Physiol Heart Circ Physiol. 2008;295:H2046–2053 [DOI] [PubMed] [Google Scholar]
- 42.Wang YX, Martin-McNulty B, Huw LY, da Cunha V, Post J, Hinchman J, Vergona R, Sullivan ME, Dole W, Kauser K. Anti-atherosclerotic effect of simvastatin depends on the presence of apolipoprotein e. Atherosclerosis. 2002;162:23–31 [DOI] [PubMed] [Google Scholar]
- 43.Yin M, Liu Q, Yu L, Yang Y, Lu M, Wang H, Luo D, Rong X, Tang F, Guo J. Downregulations of cd36 and calpain-1, inflammation, and atherosclerosis by simvastatin in apolipoprotein e knockout mice. J Vasc Res. 2017;54:123–130 [DOI] [PubMed] [Google Scholar]
- 44.Navab M, Anantharamaiah GM, Hama S, Hough G, Reddy ST, Frank JS, Garber DW, Handattu S, Fogelman AM. D-4f and statins synergize to render hdl antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein e-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–1432 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.