

Abstract
The preterm infant gut microbiota develops remarkably predictably1–7, with pioneer species colonizing after birth, followed by an ordered succession of microbes. The gut microbiota is vital to preterm infant health8,9 yet the forces underlying these predictable dynamics remain unknown. The environment, the host, and microbe-microbe interactions are all likely to shape microbiota dynamics, but in such a complex ecosystem identifying the specific role of any individual factor has remained a major challenge10–14. Here we use multi-kingdom absolute abundance quantitation, ecological modelling, and experimental validation to overcome this challenge. We quantify the absolute bacterial, fungal, and archaeal dynamics in a longitudinal cohort of 178 preterm infants. We uncover, with exquisite precision, microbial blooms and extinctions and reveal an inverse correlation between bacterial and fungal loads in the infant gut. We infer computationally and demonstrate experimentally in vitro and in vivo that predictable assembly dynamics may be driven by directed, context-dependent interactions between specific microbes. Mirroring the dynamics of macroscopic ecosystems15–17, a late-arriving member, Klebsiella, exploits the pioneer, Staphylococcus, to gain a foothold within the gut. Remarkably, we find that interactions between kingdoms can influence assembly, with a single fungal species, Candida albicans, inhibiting multiple dominant gut bacteria. Our work unveils the centrality of simple microbe-microbe interactions in shaping host-associated microbiota, critical for both our understanding of microbiota ecology and targeted microbiota interventions.
Humans are colonized by vast communities of microbes, particularly within the gastrointestinal tract, that play key roles in host health8,9. Infants are generally born uninhabited and their gut microbiota gradually assembles after birth1–7. Remarkably, this developmental process occurs in a predictable manner, with specific bacterial taxa establishing at distinct points in infant life18–22. The early life microbiota is critical to infant health, with microbiota composition linked to a range of diseases, morbidity and mortality, particularly within preterm infants1,23–27. Yet despite the importance of the infant microbiota, we do not understand what drives the patterned progressions of the infant gut community11–13. Gestational age, delivery mode, host epithelial and immune ontogeny, diet, antibiotics, and the interactions between individual microbes may each influence microbiota composition2,18–22,28–30. But with such complexity, the impact of any individual factor upon microbiota development has remained unclear. Indeed, disentangling how and why microbial communities change over time remains a major challenge both for human microbiota and for host-associated and environmental microbiomes more broadly.
Our ability to identify drivers of microbiota development has been hampered by the complexity of microbial ecosystems and also by fundamental limitations in how we quantify community composition10–13. First, while next-generation sequencing (NGS) has provided a comprehensive map of bacterial diversity within the human gut31,32, we still know little of the other microorganisms, such as fungi and archaea, that colonize the infant microbiota33–35, constraining our ability to identify inter-kingdom interactions driving ecosystem dynamics36. Second, NGS data typically chart only the relative abundances of taxa, providing the proportion of different microbes within a community, but not absolute amounts. If a species increases in relative abundance over time we cannot determine whether that species is blooming or others are dying out (Fig. 1a). The compositional nature of relative abundance data can thus mask community dynamics, undermining our ability to identify biotic and abiotic forces shaping microbiota change37–39. Here we used a scalable multi-kingdom quantitation method to map absolute microbiota dynamics in a longitudinal cohort of preterm infants. Combining ecological models, and in vitro and in vivo validation, we reveal that within and between-kingdom microbial interactions shape the predictability of early-life microbiome assembly.
Figure 1. Multiple Kingdom SpikeSeq (MK-SpikeSeq) enables robust quantitation of absolute abundances.
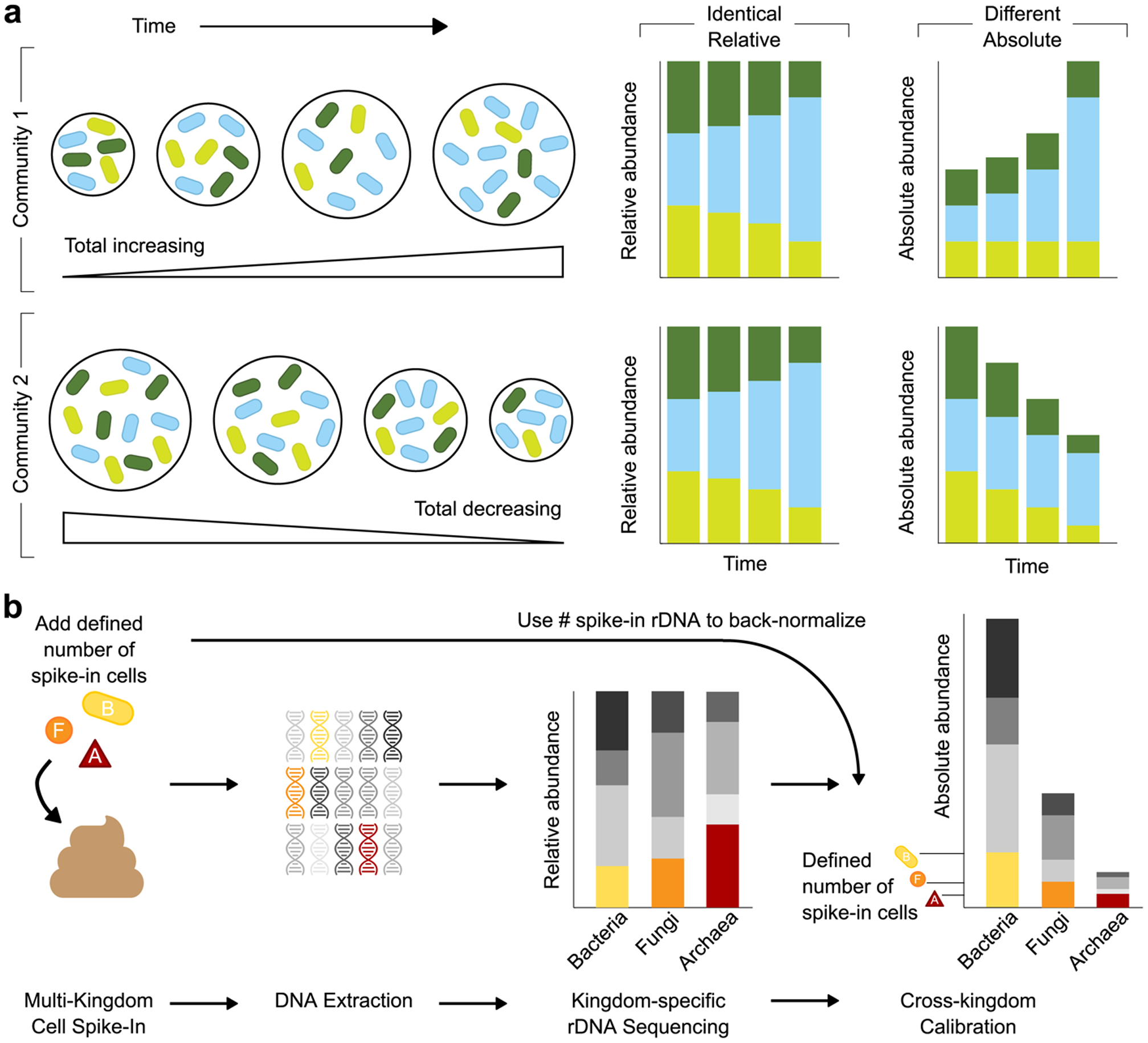
a, Schematic illustrating how relative abundance data can mask underlying community dynamics, rendering it challenging to distinguish different ecological scenarios. b, Overview of the MK-SpikeSeq pipeline. Prior to DNA extraction, defined amounts of each spike-in cell (bacteria (B), fungi (F) and archaea (A)) are added to each microbiome sample. Relative abundances of each microbial kingdom are then quantified using standard kingdom-specific rDNA amplicon sequencing. As the absolute abundances of each spike-in cell’s rDNA are known, these quantities can be used as back-normalization factors to calculate the absolute abundances of all other organisms present in each sample. The spike-in cells also serve as internal controls for the entire sample processing procedure, rendering the absolute quantification robust to factors such as sample-to-sample variability in DNA extraction efficiency.
NGS pipeline quantifies multi-kingdom abundances
To identify drivers of change within any microbial community one must quantify the absolute changes in community members over time. To achieve this, we developed a cell-based multiple kingdom spike-in method (MK-SpikeSeq) that quantifies the absolute abundances of bacteria, fungi and archaea simultaneously within any given microbiome (Fig. 1b, Supplemental Text). Specifically, we add to each sample defined numbers of exogenous microbial cells of each kingdom and perform kingdom-specific rDNA amplicon sequencing to obtain relative abundances in each kingdom. The spike-in cells serve as internal controls for sample processing and, as spike-in cell abundances are known, we can then back-normalize and calculate absolute abundances of all community members (Fig. 1b, Supplementary Fig. 1). As our primary objective was to study mammalian microbiota, our spike-in contained the bacterium Salinibacter ruber40, the fungus Trichoderma reesei and the archaeon Haloarcula hispanica, selected based on their absence or rarity in mammalian microbiomes (Supplemental Table 2). However, our approach can be adapted via spike-in choice to target any host-associated or environmental microbiome and can be combined with shotgun metagenomics to capture viruses and enable strain-level quantification. We validated MK-SpikeSeq’s ability to measure absolute abundances using a series of defined mock communities, then compared MK-SpikeSeq’s performance against existing approaches for absolute abundance quantification (total DNA, cytometry-based imaging, quantitative PCR and DNA-based spike-in) using a set of test samples (Supplemental Text, Extended Data Fig. 1–5). Together, these demonstrated that MK-SpikeSeq generates highly sensitive and robust absolute abundance measurements for individual taxa across multiple kingdoms, a key requisite for identifying drivers of microbiota dynamics.
Multi-kingdom dynamics during infant gut assembly
Having validated MK-SpikeSeq we built a high-resolution multi-kingdom picture of infant microbiota dynamics. Specifically, we assembled a prospective cohort of 178 preterm infants from a tertiary-care neonatal intensive care unit (NICU). The assembly of the preterm microbiota differs substantially from that of term infants. Most preterm infants are born via C-section and thus are seeded with skin and hospital-associated microbes, and devoid of key maternally derived bacteria7,21,29. The preterm microbiota also displays “delayed” maturity with prolonged membership of facultative anaerobic bacteria compared to that of the predominantly strict anaerobic community of term infants7,21,29. We focused on preterm infants due to their clinical relevance and because they are amenable to high-frequency longitudinal sampling with readily available clinical metadata. These features render the preterm gut an important and tractable system for establishing a proof-of-principle understanding of microbiota assembly. We sampled each infant within our cohort on approximately their first, 14th, 28th, and 42nd day of life, and for 13 infants we gathered nearly-daily stools for their first 6 weeks of life (940 samples in total). Together, this cohort enabled us to build a high-resolution picture of microbiota development within the preterm infant gut.
Consistent with previous studies18–23, we observed that preterm infant gut bacterial communities cluster primarily into four distinct community states, characterized by domination of one of four genera: Staphylococcus, Klebsiella, Escherichia or Enterococcus (Fig. 2a). In contrast to full-term infants, these microbiome clusters were independent of diet or delivery mode (Extended Data Fig. 6). Importantly, the bacterial community within our preterm cohort, as previously observed18–23, developed in a predictable and highly dynamic manner over time. Most infants were initially dominated by Staphylococcus, then transitioned to a state dominated by Klebsiella, Enterococcus or Escherichia as infants aged (Fig. 2a, b, Extended Data Fig. 6), with total bacterial load in the infant gut gradually increasing over time (Fig. 2f, g, Extended Data Fig. 8). Comparing the absolute and relative abundances of these dominant genera illustrated how compositional data can misattribute both how and when communities change. In several infants, relative abundances initially masked blooms in Klebsiella and Escherichia, and showed Staphylococcus and Enterococcus collapsing in the community when their abundances were instead comparatively stable (Fig. 2c, Supplementary Fig. 2–16). Such comparisons also indicated that, though the bacterial communities within our cohort were typically dominated by just one genus, often the other major genera remained stable at high levels within the preterm infant gut (Supplementary Fig. 2–16).
Figure 2. The preterm infant gut exhibits rich bacterial and fungal community dynamics.
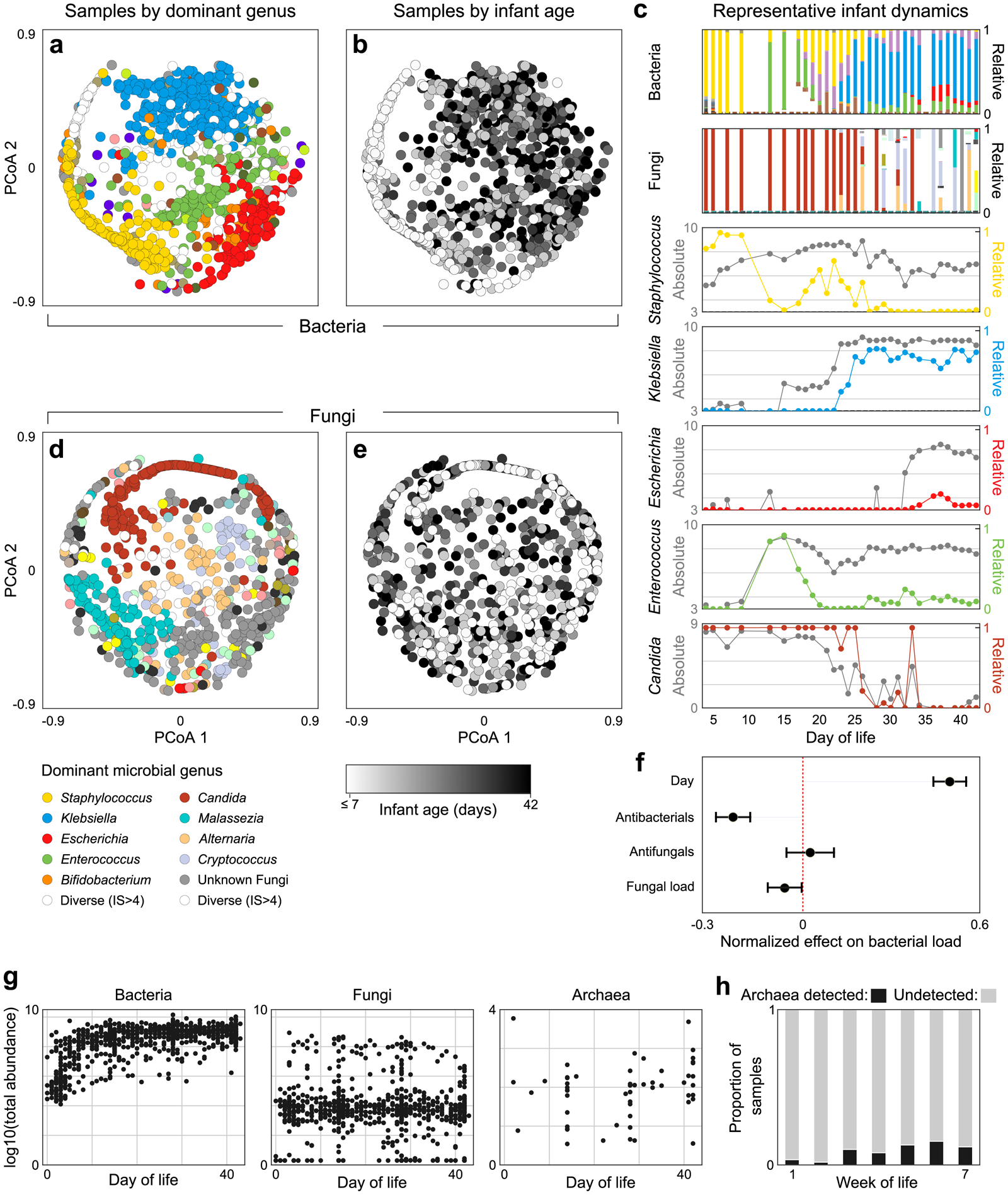
a, Principle Coordinate Analysis (PCoA) plot of Bray-Curtis dissimilarities between bacterial samples at the genus level. Each dot represents a sample, colored by the dominant genus present or white if diversity was high (Inverse Simpson index > 4). b, The same PCoA as panel a with samples instead colored by infant day of life, illustrating how bacterial community composition changes predictably over time. c, Microbiota dynamics of a single representative infant, highlighting the importance of gathering absolute abundances when studying microbiome ecology. Stacked bars represent total community composition (for full color schemes, see Extended Data Figs 3, 4). Line plots illustrate the relative (colored) and absolute (grey) abundances of individual genera. d-e, PCoA plots of fungal community composition colored by dominant genus (d) or infant age (e), indicating fungal community composition does not correlate with infant age. f, Effects of clinical and microbial factors on total bacterial load, quantified by a linear mixed effects model, suggesting a potential relationship between kingdoms within the preterm infant gut (centers and error bars indicate estimated fixed effects and 95% confidence intervals respectively). g, Total abundances of bacteria, fungi and archaea over time. h, Proportion of samples in which archaea could be detected during each week of life. For panels a/b/g left, number of samples n = 934, for d/e/g center n=772, for f n=770, for g right, h n = 596.
In contrast to the predictable dynamics of bacterial communities, we uncovered diverse but unpredictable fungal communities within the preterm infant gut. On average, fungal dynamics were noisier and exhibited less temporal structure than bacterial communities, with no clear correlation between fungal community composition or load and infant age (Fig. 2d, e, Extended Data Fig. 7). Notably, though rare in adults33, Cryptococcus was the dominant fungal genus in approximately 5% of samples; while despite being a common inhabitant of the adult gut, Saccharomyces species33 were detected in only 5 infants. As with bacterial communities, MK-SpikeSeq uncovered fungal blooms and collapses masked by relative abundances. For example, in several infants Candida stably maintained a high relative abundance, despite dropping multiple orders of magnitude in absolute load over time (Fig. 2c, Supplementary Fig. 4–18). However, though the fungal dynamics were themselves unpredictable, a linear mixed-effects model that accounted for infant age, anti-bacterials and anti-fungals, uncovered a weak negative correlation between bacterial and fungal loads (normalized effect size: −0.060, 95% Wald CI: [−0.119, −0.001], Fig. 2f, Extended Data Fig. 8). That is, when accounting for clinical covariates, samples with higher fungal loads tended to have lower bacterial loads. This inverse relationship led us to wonder whether cross-kingdom interactions might be influencing preterm microbiota dynamics.
Archaea were notably rare within our cohort, with most samples showing no archaeal signal. However, we detected a weak positive trend in both the frequency of archaeal detection (Chi Squared test for trend, p=0.002) and total archaeal load over time (Spearman’s R=0.13, p=0.002), with higher archaeal abundances generally detected in later weeks of life (Fig. 2g, h, Extended Data Fig. 8).
Ecological drivers of microbiota assembly
Having generated a high-resolution multi-kingdom map of infant microbiota assembly, we next sought to identify factors driving the predictable dynamics observed. To achieve this, we used Bayesian regularized regression to fit our longitudinal data to an extended generalized Lotka-Volterra (gLV) model, an approach only possible with absolute abundances. The gLV model assumes the growth rate of an individual taxon depends upon the taxon’s intrinsic growth rate and interaction with kin, the effect of clinically-administered antimicrobials, and interactions between the focal taxon and other community members (Fig. 3a, Supplemental Text)41–43. Specifically, the model allows microbes to interact in a number of different ways, from bidirectional competition (−/−) such as nutrient competition, to exploitation (+/−) wherein one microbe takes advantage of another, or not interact at all (0/0). The model also allows each clinically-administered antimicrobial agent to inhibit, promote or have no effect on each microbe. Together this yields a highly parameterized model of community dynamics, which we fit to our data using a conservative regularization framework. By doing so, we were able to identify those microbe-microbe or antibiotic-microbe interactions playing a strong, consistent role in shaping community dynamics – while avoiding overfitting and filtering weak interactions that do not influence community dynamics. This approach thus enabled us to disentangle the effects of different biotic and abiotic interactions on microbiota assembly, independently of missing community members (e.g. viruses), or underlying host variability.
Figure 3. Pairwise intra- and inter-kingdom interactions drive predictable patterns of infant microbiome assembly.
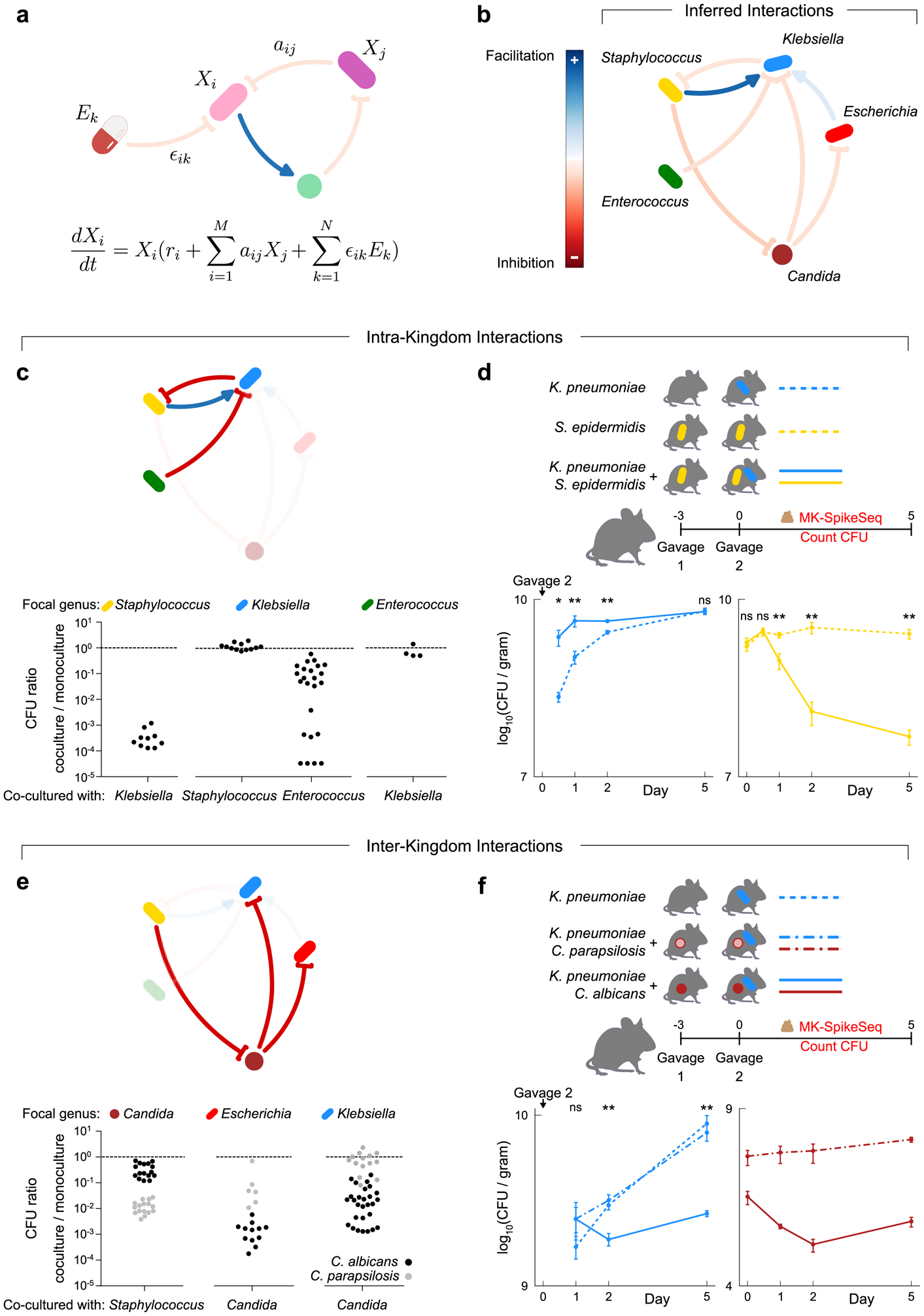
a, Schematic illustrating the generalized Lotka-Volterra (gLV) model used to identify causative drivers of bacterial and fungal dynamics within the infant gut. This model assumes the growth rate of each taxon, is determined by its own intrinsic growth rate ri, its interaction with other community members aijXj, and any environmental perturbation ϵikEk. b, The gLV model identified a network of microbe-microbe interactions occurring between dominant members of the preterm gut that are predicted to affect microbiota dynamics. c, in vitro growth effects of infant isolates upon one another using monoculture and pairwise co-culture, testing the predicted within-kingdom interactions. d, CFU counts quantifying microbial fitness in vivo in a specific pathogen free (SPF) mouse model, reproducing the predicted exploitation of Staphylococcus by Klebsiella. Gavage 2 indicates day of inoculation with K. pneumoniae. e, in vitro growth of infant isolates when growing alone and in co-culture, testing the predicted cross-kingdom interactions. Black and grey dots indicate co-cultures with C. albicans and C. parapsilosis respectively. f, CFU counts quantifying microbial fitness in vivo in a SPF mouse model, reproducing the species-specific differences in the cross-kingdom inhibition observed in vitro. Gavage 2 indicates day of inoculation with K. pneumoniae. For panels c/e: each dot denotes one unique pair of strains of the indicated genera, with each pair replicated at least once (Supplemental Table 14a). For panels d/f: Klebsiella was undetected at time 0 upon gavage; n=5 per group, error bars denote the SEM; * p<0.05, ** p<0.01, ns not significant, by two-tailed student’s t test; see Supplemental Table 15 for exact p-values; see repeat in vivo experiments in Extended Data Fig 10c–d.
Our ecological inference predicted that strong intra- and inter-kingdom interactions between specific microbial genera play a pivotal role in shaping infant gut assembly (Fig. 3b, Extended Data Fig. 9, Supplemental Text). Remarkably, we inferred that the early colonizing Staphylococcus enhanced growth of Klebsiella within the infant gut but was itself inhibited by Klebsiella. Thus our model suggests the characteristic transition from Staphylococcus to Klebsiella domination observed in preterm infants (Fig. 2a, b) is shaped, in part, by Klebsiella exploiting the early colonizer. We also inferred that Klebsiella itself was inhibited by another dominant genus Enterococcus, suggesting the distinct domination states of these two genera may be partly driven by one excluding the other. Most strikingly, consistent with the inverse correlation between bacterial and fungal loads, our analyses suggested that between-kingdom interactions play a key role in community dynamics. Specifically, we inferred that the fungal genus Candida inhibited both Klebsiella and Escherichia, but was itself inhibited by Staphylococcus. These results suggested that not only do preterm infants harbor diverse fungal communities, but that members of these communities play a role in influencing overall community dynamics.
Notably, we discovered that a substantial proportion of the interactions shaping preterm infant assembly are exploitative (+/−), with these asymmetric interactions comprising over 20% of inferred microbe-microbe interactions (Extended Data Fig. 9c). The importance of these directed, asymmetric interactions in shaping microbiota assembly underlines the power of our absolute abundance-based inference. Without absolute abundances, ecological inferences are limited to correlational analyses. These analyses identify positive or negative correlations between taxa, but cannot determine directionally who is interacting with whom, nor identify asymmetric interactions37,44–46. As a consequence, these cannot identify exploitative, commensal, or amensal interactions. Indeed, when applied to our dataset, correlational relative abundance analyses47 erroneously inferred that Staphylococcus inhibited Klebsiella and promoted Candida (Extended Data Fig. 9d). In other words, relative abundances and correlation analysis not only misrepresented the dynamics of infant microbiome assembly (Fig. 2), but also misclassified the ecological processes underlying these dynamics.
Validation of interactions shaping assembly
Our ecological inference indicated that microbe-microbe interactions are central to predictable infant microbiome assembly. However, though we employed a conservative regularization framework to ensure robustness to spurious correlations, our predictions may still be vulnerable to unobserved confounding factors not incorporated in the model, such as diet, viruses, or the host. Indeed, though a number of studies have used similar modelling approaches to infer interactions, few predictions have been experimentally validated43,48,49. We therefore sought to determine whether we could reproduce our inferred interactions in a reductionist experimental system. Focusing first on our predicted within-kingdom interactions, we isolated Staphylococcus, Klebsiella, Escherichia and Enterococcus strains from five infants in our cohort, capturing several distinct species of each genus (Supplemental Table 13). We then performed monoculture and pairwise co-culture of these strains and used colony forming units (CFU) to determine the pairwise fitness effects of strains upon one another (Supplemental Table 14). Remarkably, we were able to reproduce in vitro all of the inhibitory interactions predicted by our model, with growth effects largely conserved within genera (Fig. 3c). Klebsiella strongly inhibited Staphylococcus, reducing Staphylococcus yields by over 1000-fold, while Enterococcus variably but consistently inhibited Klebsiella (Fig. 3c). Importantly, as predicted, Klebsiella showed no effect on Enterococcus, consistent with this interaction being amensalism rather than bi-directional competition, validating the directionality of our inference.
In contrast to our predictions that Staphylococcus benefitted Klebsiella during microbiome assembly, we did not observe a positive effect of Staphylococcus on Klebsiella in vitro (Fig. 3c). Given the strength of the predicted Klebsiella-Staphylococcus exploitation, we hypothesized that this interaction may be context dependent. That is, we hypothesized that, due to differing environments in vitro versus in vivo, Klebsiella might only benefit from Staphylococcus within the gut. To investigate this, we used two co-resident NICU isolates, Klebsiella pneumoniae and Staphylococcus epidermidis, to test if Klebsiella benefited from Staphylococcus in vivo in the mammalian gut, using a mouse model of intestinal colonization (Fig. 3d). Specifically, using CFU counts and MK-SpikeSeq we measured the fitness of K. pneumoniae in mice pre-colonized with or without S. epidermidis (Fig. 3d, Extended Data Fig. 10a, c). Strikingly, as predicted, S. epidermidis significantly enhanced the ability of K. pneumoniae to colonize the mouse gut, with K. pneumoniae exhibiting faster colonization if the gut was pre-colonized with S. epidermidis (Fig. 3d). Moreover, mice colonized with K. pneumoniae had significantly reduced levels of S. epidermidis than those without K. pneumoniae, with S. epidermidis declining alongside the rise in K. pneumoniae (Fig. 3d). These in vivo data recapitulated the dynamics observed in infant assembly (Fig. 2c) and suggested that the predictable patterns in infant microbiome assembly may indeed be due to exploitation of an early pioneer by a late colonizer. These data also underlined the importance of context when studying microbiota interactions; illustrating how taxa may interact differently in vitro versus within a host.
Having validated our inferred within-kingdom interactions, we sought to validate the between-kingdom interactions predicted to influence preterm gut assembly (Fig. 3e). Again using infant isolates, each of our predicted between-kingdom interactions could also be reproduced within our in vitro system (Fig. 3e). As predicted, Candida members caused a ~100–1000-fold inhibition of each Enterobacteriaceae and experienced a ~10–100-fold reduction in growth when co-cultured with Staphylococcus, consistent with previous observations50. Notably though, we observed a bimodal distribution in the strength of inhibition of different Candida isolates by Staphylococcus (Fig. 3e), with the two modes corresponding to two Candida species, C. albicans and C. parapsilosis. Moreover, these two species also exerted differing inhibitory effects on Enterobacteriaceae; overall C. albicans both resisted Staphylococcus and inhibited Enterobacteriaceae more than C. parapsilosis (Fig. 3e). To examine if this species-specific inhibition of Enterobacteriaceae by Candida occurred in vivo, we pre-colonized mice with either C. albicans, C. parapsilosis or vehicle control, then introduced K. pneumoniae and measured microbial colonization dynamics. We observed a significantly reduced colonization of K. pneumoniae in the presence of C. albicans, compared to vehicle-control or C. parapsilosis pre-colonization, validating both the species-specificity in the fungi-bacteria interaction and its occurrence within the mammalian gut (Fig. 3f, Extended Data Fig. 10b, d). Together, our data demonstrated a novel species-specific cross-kingdom interaction that appears to shape the preterm infant microbiota.
Discussion
The de novo assembly of the infant gut microbiome is remarkably ordered, with pioneer microbes colonizing first, followed by predictable waves of other microbes. To date, the forces driving these predictable transitions have remained elusive. Priority effects, diet and antibiotics, and the developing immune system are all thought to impact microbiota dynamics. However, with multiple interacting factors at play, disentangling the role of any individual process has remained a formidable task. Here we demonstrate the power of combining multi-kingdom absolute abundance quantitation, ecological modelling, and experimental validation to overcome this challenge. We have demonstrated that the predictable patterns of preterm infant gut assembly can be driven by direct, context-dependent interactions between microbes. Our findings suggest a common mechanism of assembly between the infant microbiota and macroscopic ecological succession. Just as in macroscopic ecosystems15–17, microbes may exploit one another to establish within the infant gut, and direct interactions between kingdoms appear to play a central role in community dynamics. The reducibility of gut microbiota assembly to simple, pair-wise interactions has profound implications for understanding and ultimately manipulating microbial ecosystems in health and disease.
Extended Data
Extended Data Figure 1. MK-SpikeSeq reliably measures absolute abundances across kingdoms.
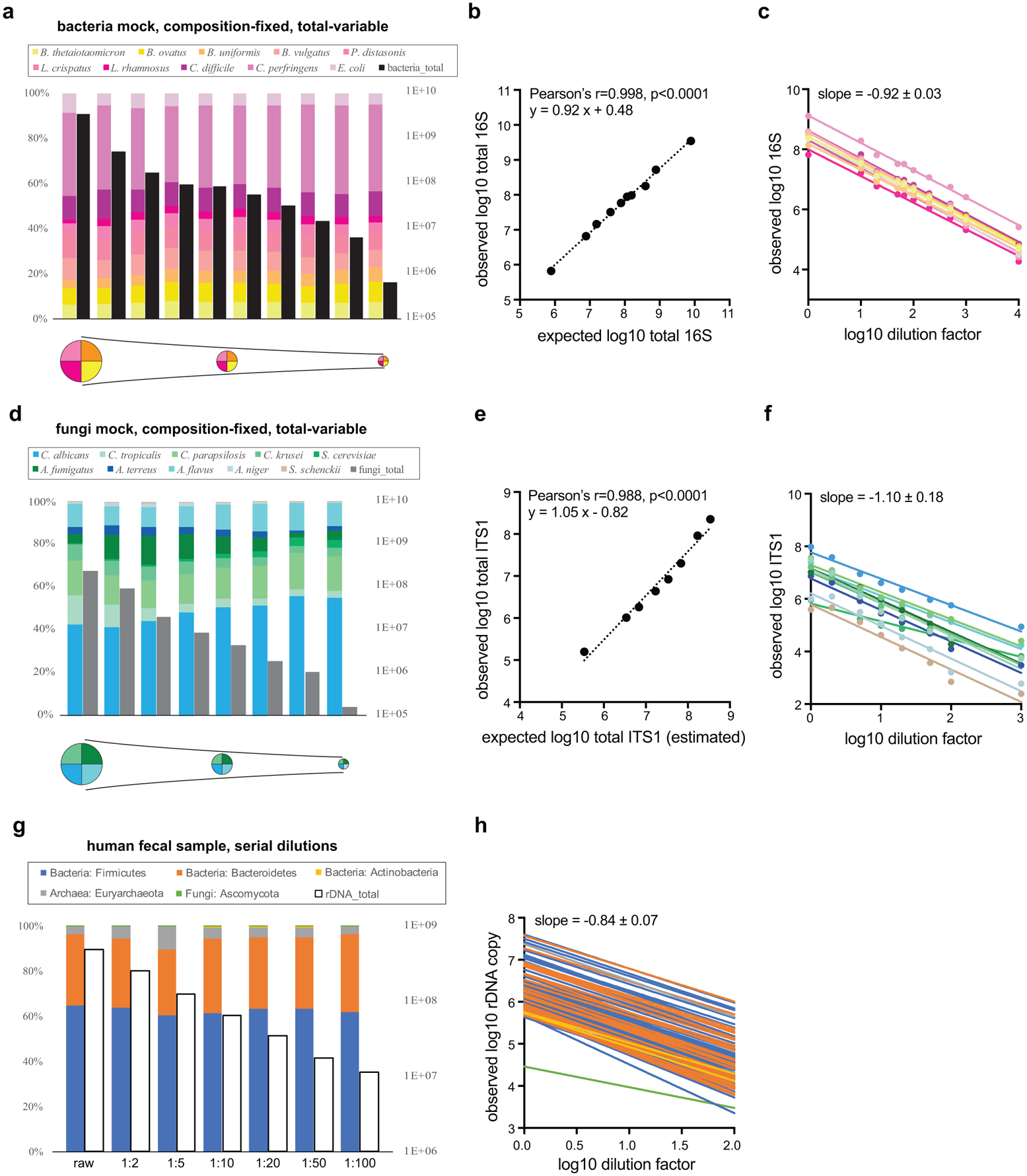
A set of single-kingdom mock communities with a fixed composition of 10 bacterial (a) or 10 fungal (d) species and variable total microbial loads (indicated by the pie chart schematics underneath), were quantified using MK-SpikeSeq for relative composition (colored bars) and absolute abundance (black/grey bars). b, e, Correlations between expected (based on initial microbial densities and known dilution factors) and MK-SpikeSeq-measured total absolute abundances show that MK-SpikeSeq reliably detects absolute abundances of bacteria and fungi. Note that for e, as exact rDNA copy numbers per fungal cell are undefined, the expected total ITS1 abundances are only estimates (here using 200 rDNA copies per fungal cell). c, f, Absolute abundance changes for individual members (color coded same as a, d) in the bacterial and fungal mock communities are largely consistent with known dilution factors. g, A set of serial dilutions of a human fecal sample was quantified using MK-SpikeSeq for relative composition (colored bars, shown are the phylum level taxa) and absolute abundance (empty bars). h, Absolute abundance changes for individual OTUs (color coded in phyla same as g) across kingdoms are largely consistent with known dilution factors.
Extended Data Figure 2. MK-SpikeSeq reliably captures key ecological dynamics in multi-kingdom mock communities.
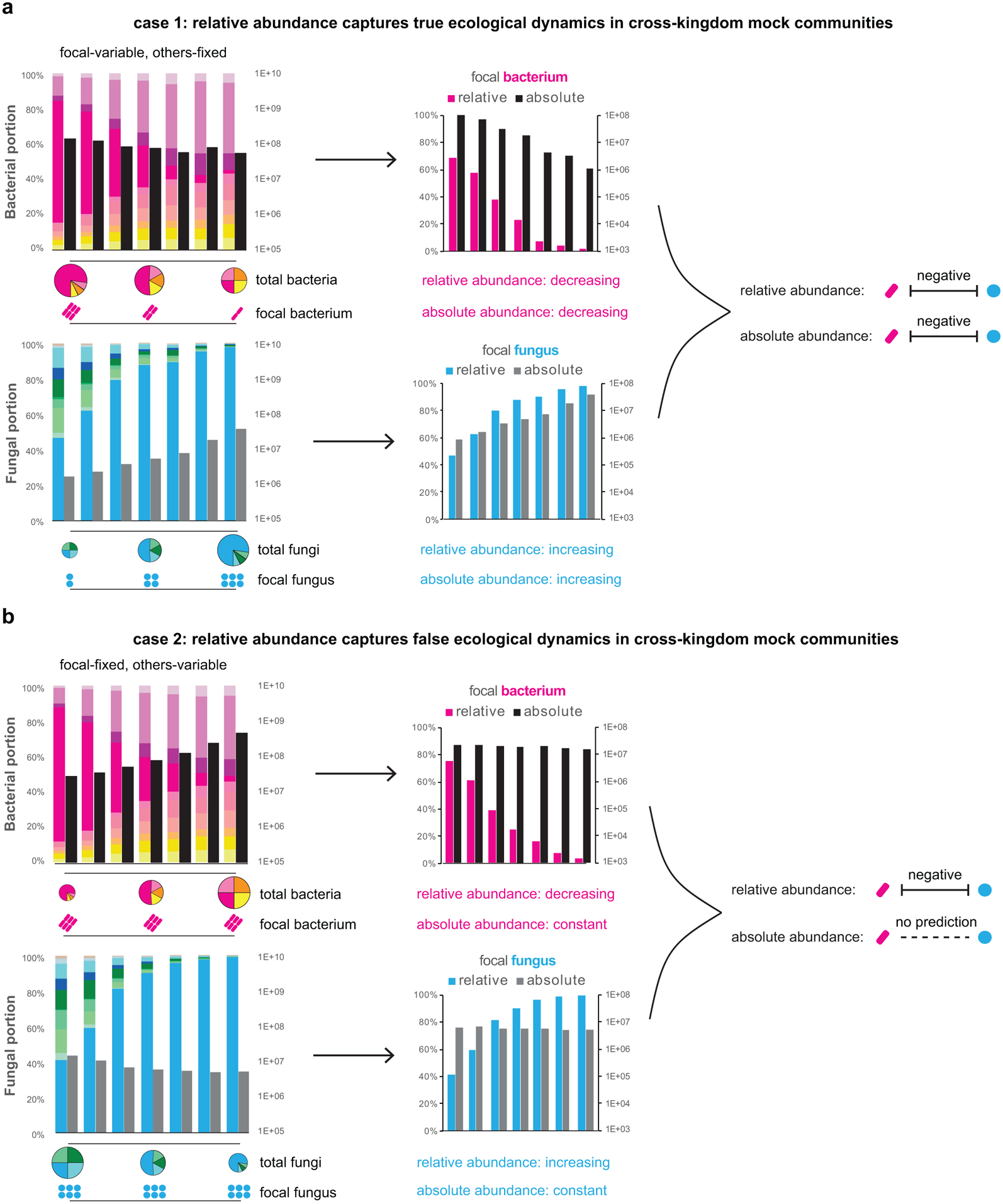
Two sets of defined multi-kingdom consortia, including ten bacteria and ten fungi (left panels, color coded same as Extended Data Fig. 2a/d), were assembled to model a “true” (a) and a “false” (b) negative interaction between one focal bacterium and one focal fungus, by varying the abundances of either these focal species or other background members. The MK-SpikeSeq quantitations of focal species highlight either consistent (a) or distinct (b) patterns between relative abundance and absolute abundance (middle panels). Relative abundance data may lead to a false prediction of cross-kingdom interaction between the focal species, while absolute abundance data measured by MK-SpikeSeq could disentangle these distinct mock ecological dynamics (right panels).
Extended Data Figure 3. MK-SpikeSeq outperforms other quantitation methods in cross-kingdom specificity.
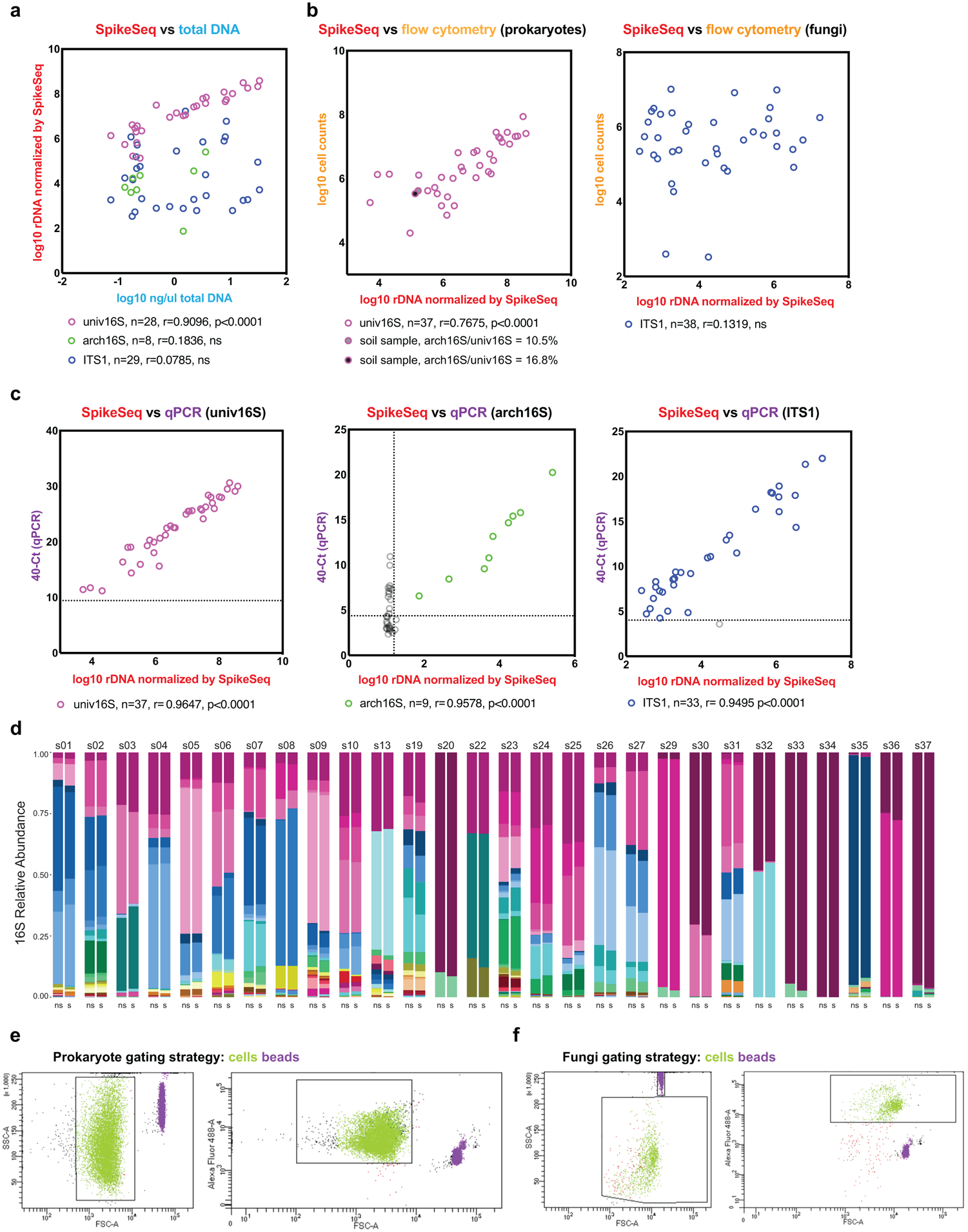
A set of 40 test samples including human stools and soil samples were used to compare kingdom-specific absolute abundance quantifications. a, MK-SpikeSeq compared with total DNA yields. Pearson correlation tests show that total DNA yields mostly only reflect bacterial community abundances. b, MK-SpikeSeq compared with flow cytometry cell enumerations using gating strategies targeted for either prokaryotes or fungi. For prokaryotic enumerations, two soil samples are highlighted due to their high archaeal loads that cannot be distinguished from bacterial counts by flow cytometry. For fungi enumerations, shown are results using one gating strategy; attempts using two additional gating strategies show similar over-estimation of fungal counts (Supplemental Table 4). c, MK-SpikeSeq compared with kingdom-specific qPCR. Horizontal dashed lines show the limit of detection using qPCR, based on the negative control (DNA extraction of water); vertical dashed line shows the limit of detection using MK-SpikeSeq, based on minimal one non-spike-in arch16S read normalized against the average arch16S sequencing depth. Samples below limit of detection are excluded from correlational tests. Note that some samples with arch16S below MK-SpikeSeq limit of detection showed arch16S qPCR signals higher than the negative control, likely due to bacterial signals bleeding into archaea-specific qPCR. For a-c: Pearson correlation r and two-sided p values were shown (no adjustment for multiple comparisons); ns, not significant. d, Comparison of 16S genus-level profiles sequenced with (s) or without (ns) spike-in shows largely unaltered community compositions having exogenous spike-in. e, f, Flow cytometry gating strategies used in prokaryote and fungi cell counting (b), with green showing bacterial and fungal cells and purple showing microsphere particles provided in the bacteria counting kit. Note that higher voltage settings were used in flow cytometry for prokaryote cell counting than for fungi cell counting.
Extended Data Figure 4. MK-SpikeSeq outperforms qPCR in the sensitivity of detection and robustness to sample background.
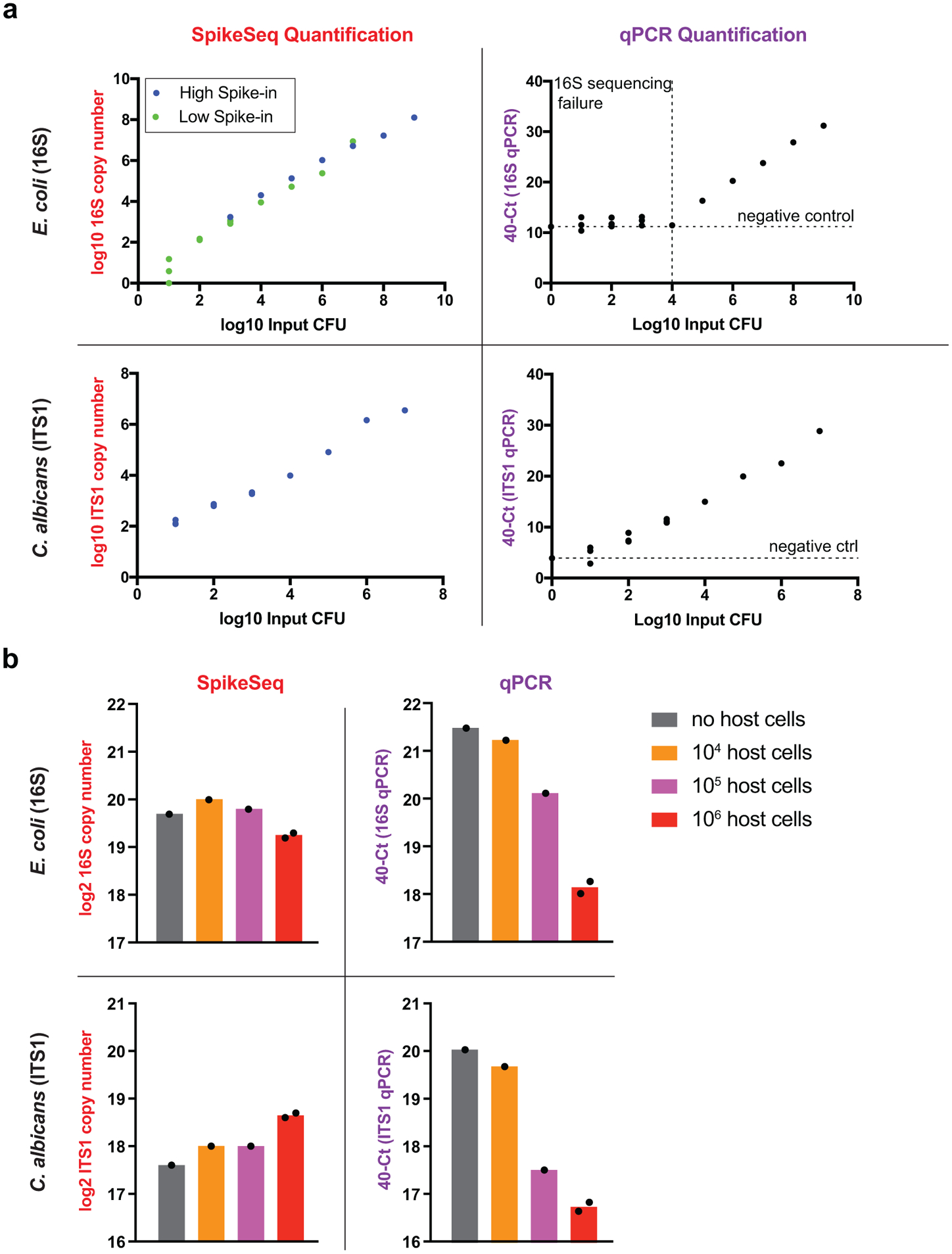
a, Comparison of sensitivity between MK-SpikeSeq and qPCR using 10-fold serial dilutions of E. coli and C. albicans. MK-SpikeSeq showed 100~1000-fold increased sensitivity over qPCR in low bacterial abundance samples (detecting as few as 10 bacterial cells). For MK-SpikeSeq of E. coli samples, two levels of spike-in were used to cover the whole range of detection under the sequencing depth of 10~100k reads per sample. For qPCR, horizontal dashed lines indicate the negative control (DNA extraction of water) and vertical dashed line shows the threshold below which pooled 16S sequencing yielded <100 reads (sequencing failed likely due to too low signal). b, Comparison of robustness to host cell background between MK-SpikeSeq and qPCR using test samples with fixed amount of E. coli and C. albicans and variable number of Caco-2 colonic cells. MK-SpikeSeq detected consistent (< 2-fold variations) microbial abundances in samples with high host-cell background whereas qPCR under-measured microbial abundances by 10-fold (deltaCt > 3.3). n=2 for the 106 host cells group, n=1 for the other groups.
Extended Data Figure 5. MK-SpikeSeq identifies errors in sample processing of fungal communities.
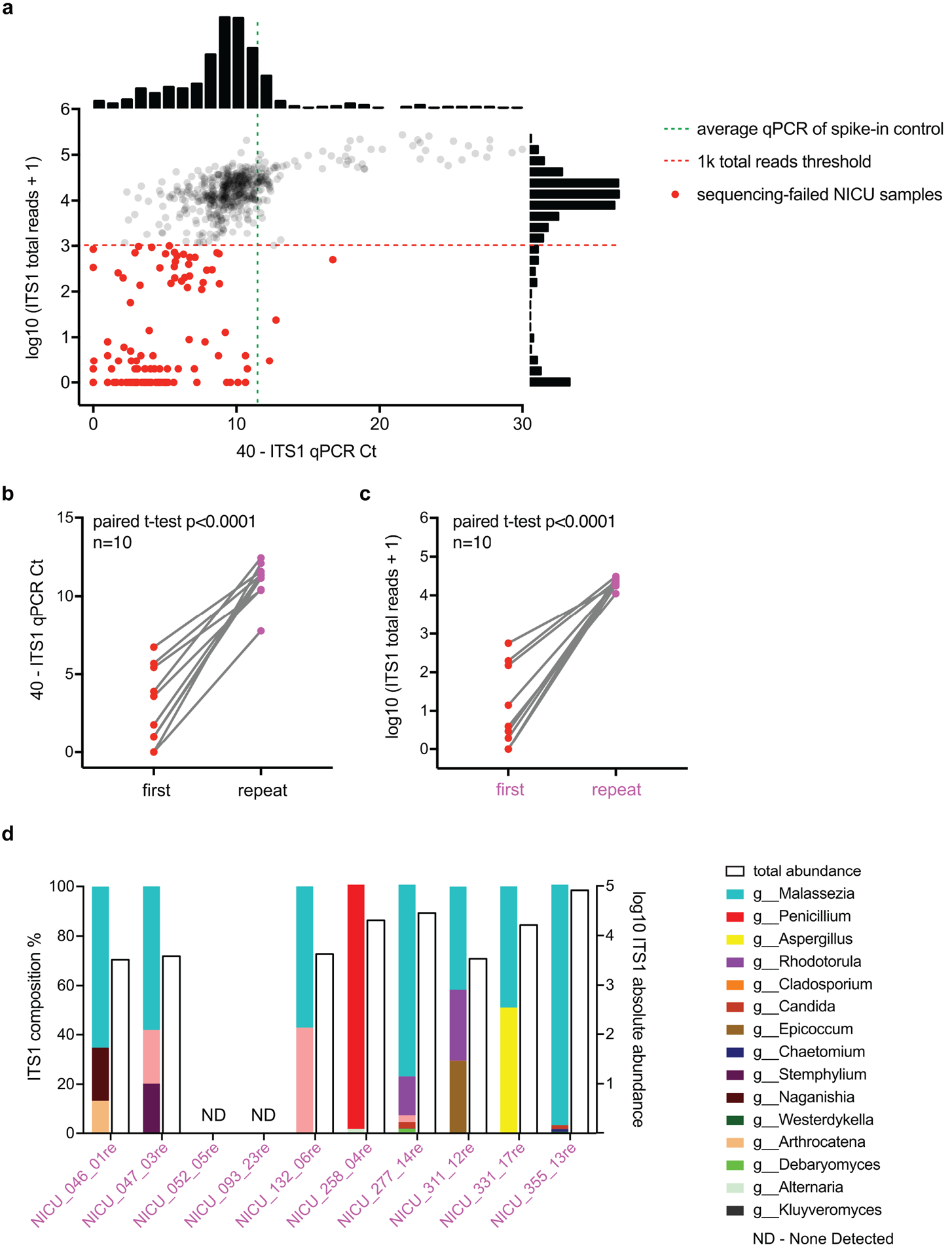
a, In our first phase of NICU sequencing (see Supplemental Text), we identified a number of samples, highlighted in red dots, that failed to yield >1k ITS1 reads per sample post quality filtering (red dashed line). Many of these sequencing-failed samples showed much lower (>5 deltaCt) ITS1 qPCR signals than the spike-in control (green dashed line), indicating poor DNA extractions of fungi in these samples. Shown next to the axes are frequency histograms of measurements. b, Reprocessing of 10 of these sequencing-failed samples led to increased ITS1 qPCR signals, indicating improved DNA extractions. c, These reprocessed samples also yielded desired >10k ITS1 reads, passing our rDNA sequencing criteria. For b/c, two-tailed paired student’s t test. d, Eight of the reprocessed samples showed non-zero fungal communities, and only two had no detectable fungal signal. Shown are the composition (colored bars) and total abundance (empty bars) of fungal communities in these reprocessed samples.
Extended Data Figure 6. Bacterial samples cluster based on composition and infant age, but not diet, delivery mode, or gender.
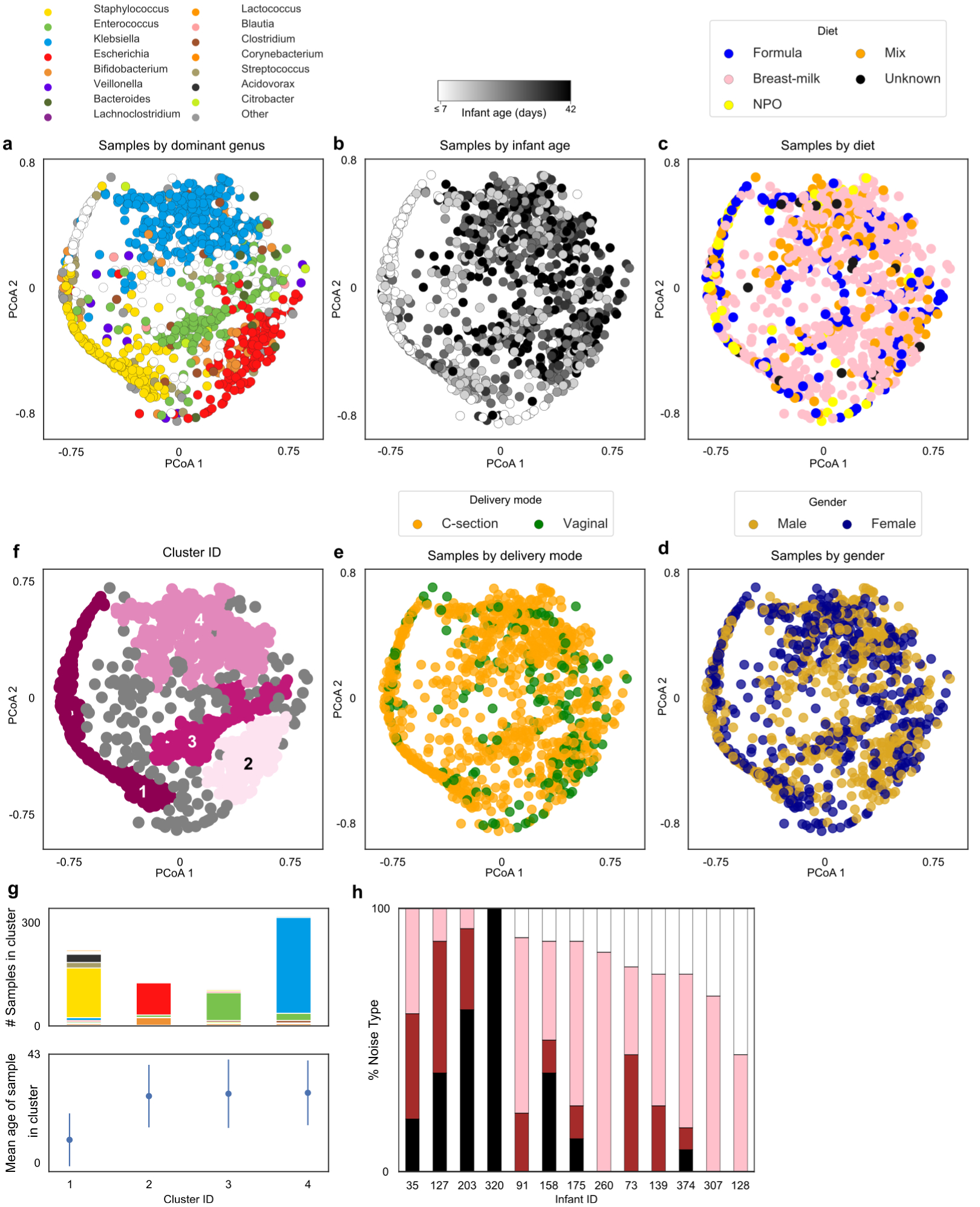
a, Principle Coordinate Analysis (PCoA) based on Bray-Curtis dissimilarities of bacterial community composition between samples (genus level). Samples colored by dominant taxa or white when diversity is high (IS>4). b, PCoA colored by infant age. c, PCoA colored by infant diet close. d, PCoA with samples colored by infant gender. e, PCoA with samples colored by delivery mode. f, PCoA with samples colored by cluster membership, calculated using DBSCAN algorithm. g, Stacked bars represent distribution of dominant genus within each cluster and dot plots illustrate average day of life of samples within each cluster. Kruskal-Wallis test with Bonferroni correction showed statistically significant differences in day of life of samples between clusters (Chi square = 254, p-value << 0.0001, df = 3), error bars show mean +/− standard deviation. h, Stacked bars indicate the proportion of genera exhibiting each noise type per infant. Dark noise indicates increasing temporal dependence, with white noise suggesting temporal dynamics are entirely random.
Extended Data Figure 7. Fungal community composition does not map to infant age, diet, gender or delivery mode.
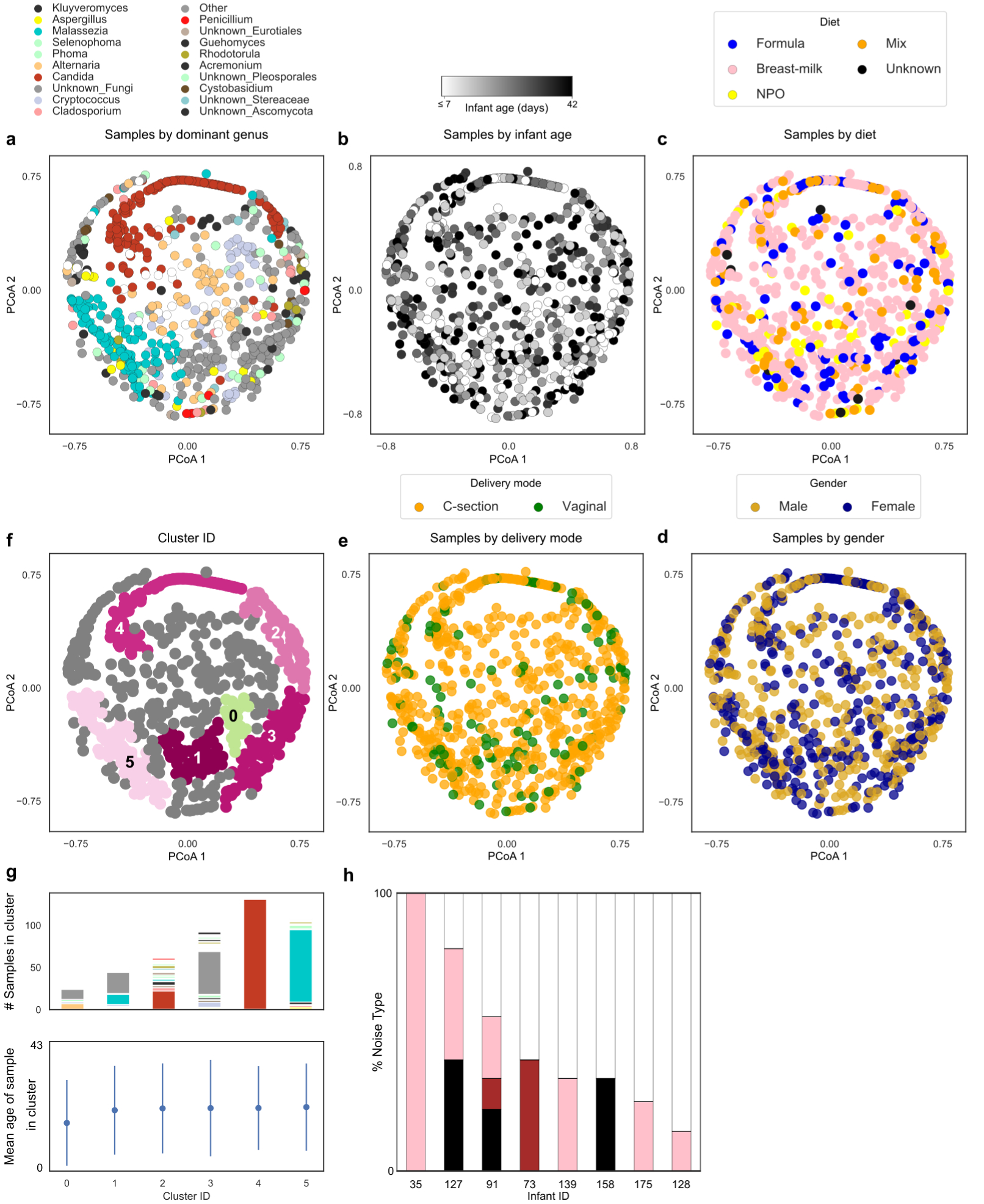
a, Principle Coordinate Analysis (PCoA) based on Bray-Curtis dissimilarities of fungal community composition between samples (genus level). Samples colored by dominant taxa or white when diversity is high (IS>4). b, PCoA colored by infant age. c, PCoA colored by infant diet close. d, PCoA with samples colored by infant gender. e, PCoA with samples colored by delivery mode. f, PCoA with samples colored by cluster membership, calculated using DBSCAN algorithm. g, Stacked bars represent distribution of dominant genus within each cluster and dot plots illustrate average day of life of samples within each cluster. Kruskal-Wallis test with Bonferroni correction showed no statistically significant differences in day of life of samples between clusters (Chi square = 3.06, p-value = 0.69, df = 5), error bars show mean +/− standard deviation. h, Stacked bars indicate the proportion of genera exhibiting each noise type per infant. Dark noise indicates increasing temporal dependence, with white noise suggesting temporal dynamics are entirely random. Notably, 5 infants’ mycobiomes could not be classified.
Extended Data Figure 8. Trends in total microbial loads for all three kingdoms.
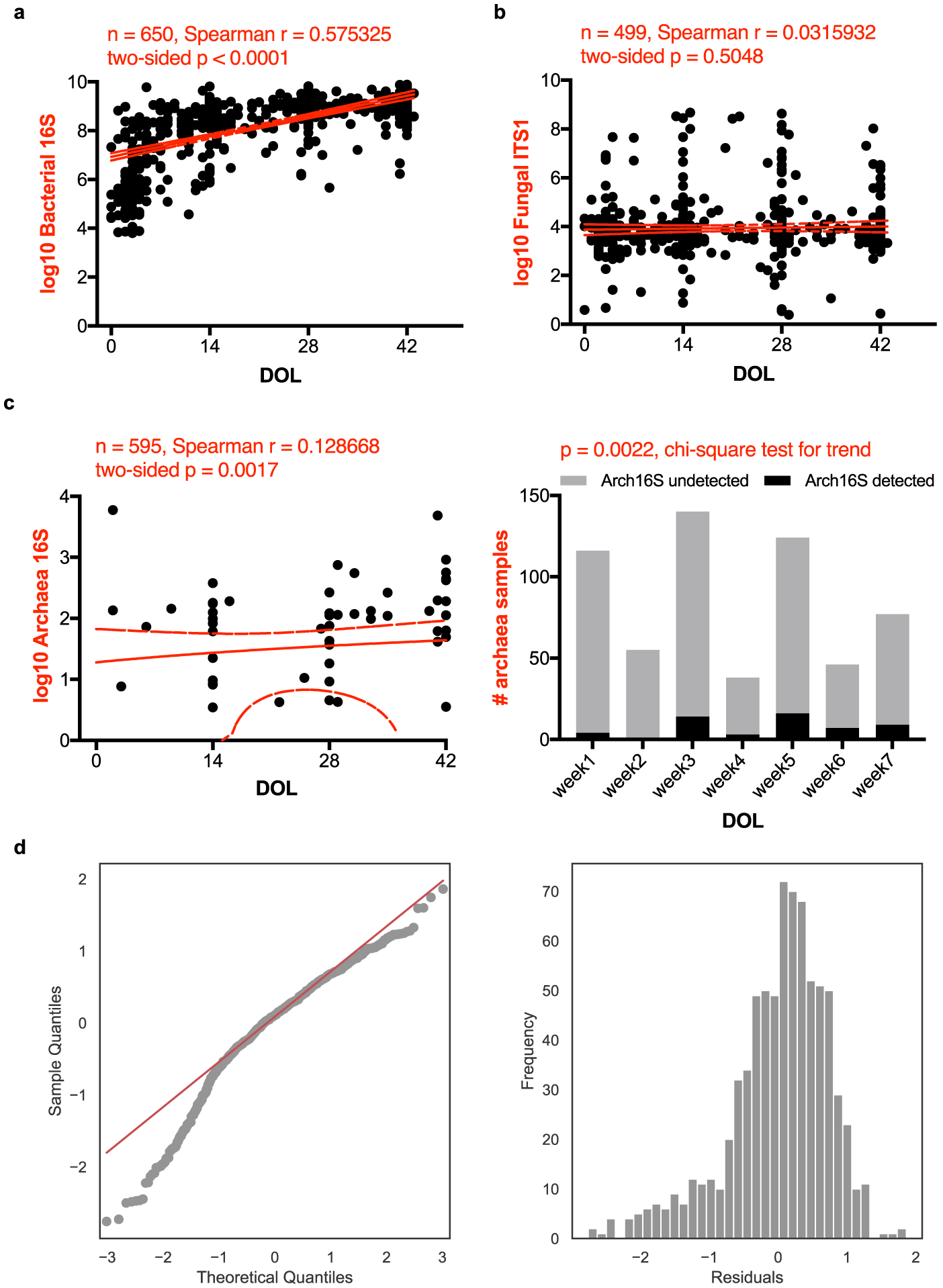
Scatter plots of rDNA-based total abundances of bacteria (a), fungi (b) and archaea (c) against infant day of life (DOL), measured by MK-SpikeSeq in the first phase Nextseq sequencing. The red lines denote the linear regression fit and the 90% confidence bands of the best-fit line of absolute abundances in log scale. Spearman correlations show that bacterial and archaeal, but not fungal, loads are positively associated with infant age. Samples with undetectable kingdom-specific rDNA signal are not plotted. For archaea that show scarce signal in the cohort (c, left), a separate presence/absence plot and chi-square test of binned samples (c, right) also show a positive correlation between archaeal loads and infant age. d, Diagnostics for linear mixed effects model.
Extended Data Figure 9. Microbe-microbe interactions are predominantly asymmetric, while inferring interactions from relative abundance data generates misleading results.
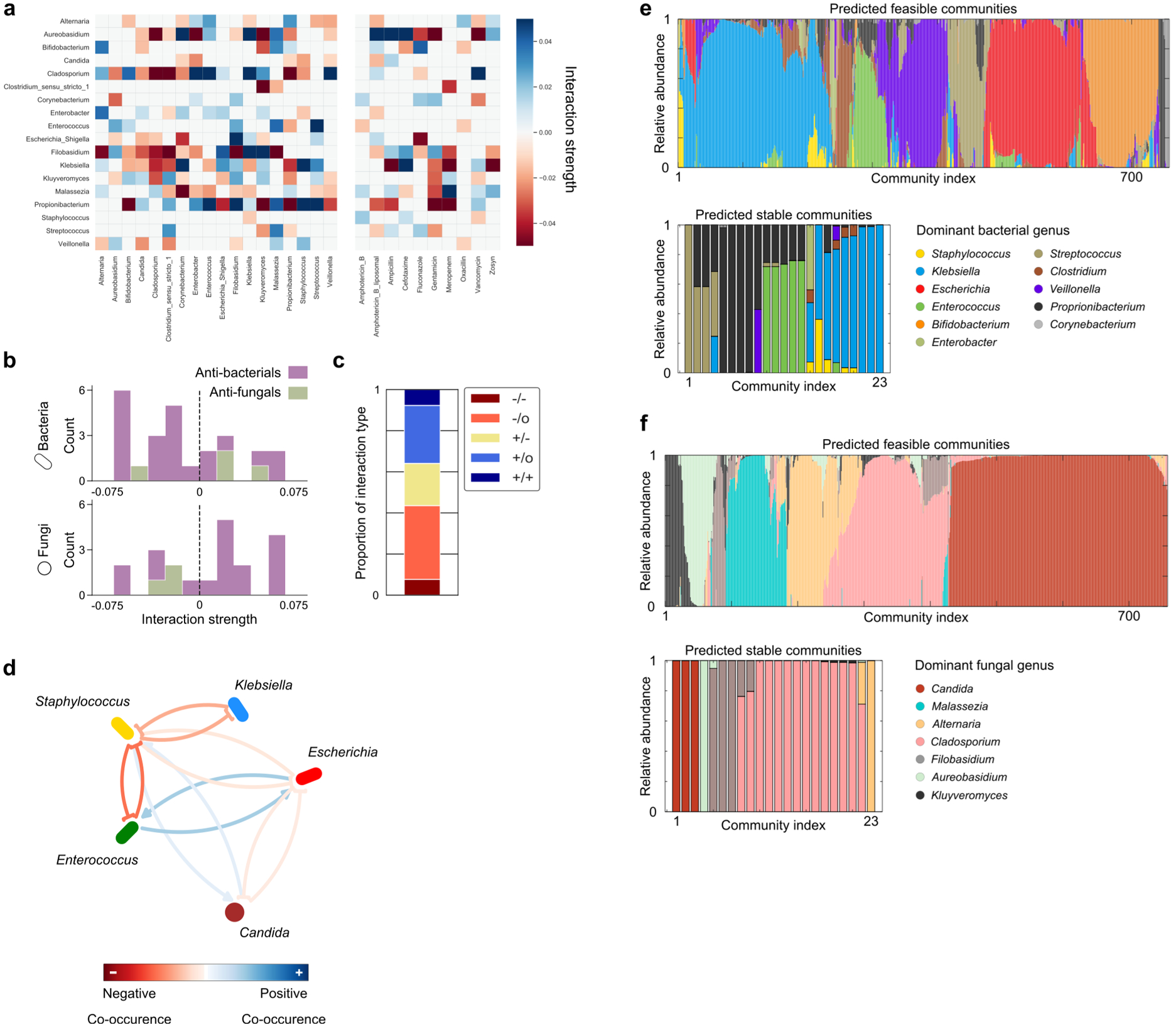
a, Heatmap plotting interactions inferred by the gLV model. Each row of the heatmap illustrates the effect upon the target genera by other members of the gut community (left columns) or documented usage of antimicrobials according the clinical metadata (right columns). b, Histogram of individual antibacterial (purple) or antifungal (green) interaction strengths, split by kingdom. Antibacterials primarily inhibit bacteria, and antifungals primarily inhibit fungi, however there is not a significant bias in the likelihood of either antimicrobial inhibiting their target kingdom (exact binomial tests, H0: P(Inhibition) = 0.5, p>0.05). c, Stacked bar illustrates the proportion of different interaction types occurring between genera. Over 80% of interactions are asymmetric, being either exploitative (+/−), commensal (+/0), or amensal (-/0). d, To confirm the value of our absolute abundance methods, we inferred inter-genus interactions from relative abundance data alone using the FastSpar47 algorithm. This approach robustly identifies co-occurrence relationships between different microbial taxa in a manner that accounts for the compositional nature of relative abundance data. Notably, correlation networks cannot infer asymmetric interactions thus this approach cannot detect the exploitation of Staphylococcus by Klebsiella. It also erroneously infers that Staphylococcus increases the growth of Candida, and cannot detect the inhibition of Klebsiella by Candida or Enterococcus. e, Steady-state bacterial relative abundances of those subcommunities predicted to be feasible and/or linearly asymptotically stable. f, Steady-state fungal relative abundances of those subcommunities predicted to be feasible and/or linearly asymptotically stable.
Extended Data Figure 10. MK-SpikeSeq measurement and repeat experiments of in vivo colonization.
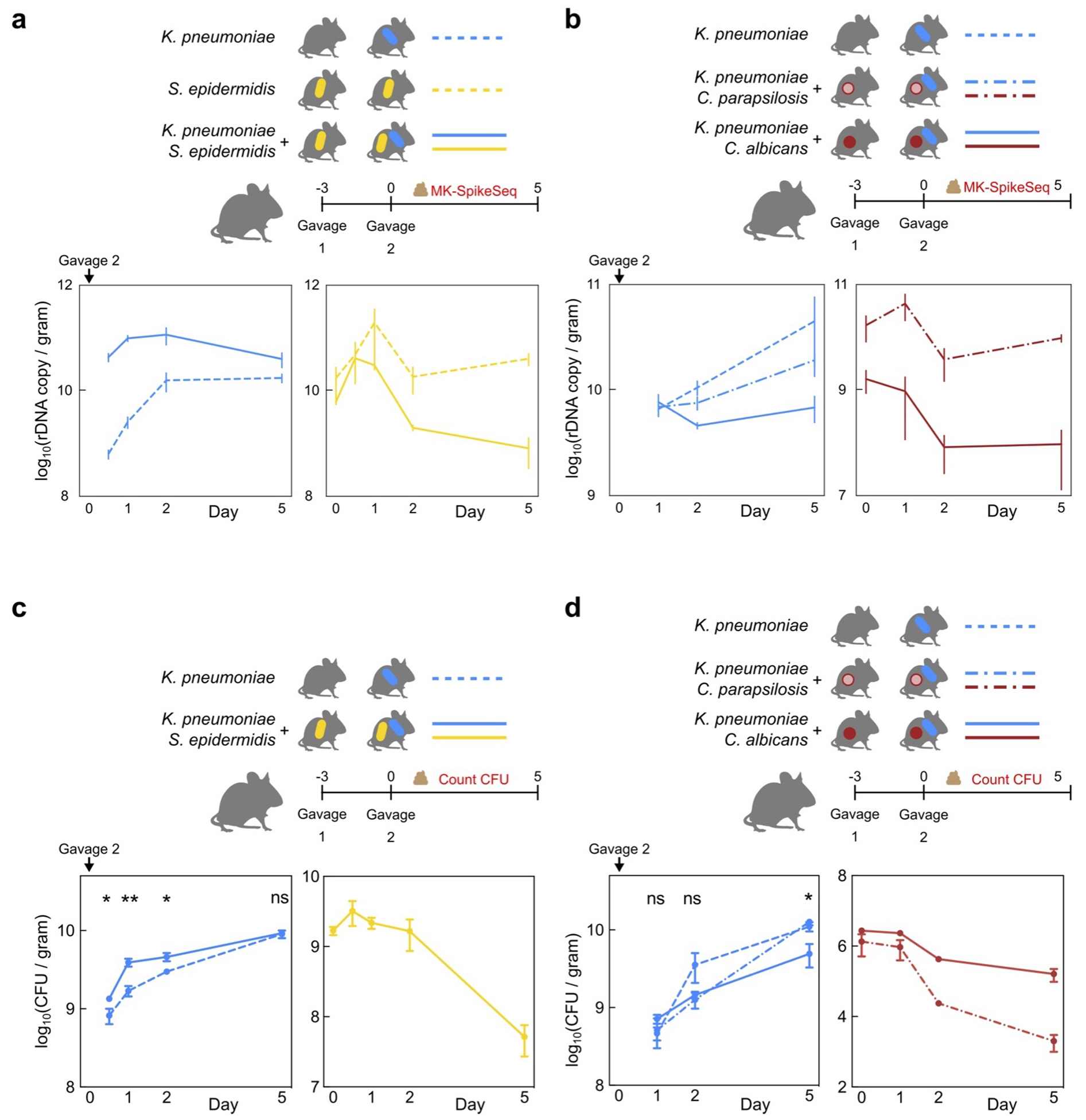
a/b, Biological replicate samples of mouse stools characterized by CFU counting of strains of interest in Fig. 3d/f were subjected to MK-SpikeSeq to determine rDNA-based absolute abundances of the specified strains. c/d, Repeat in vivo colonization experiments. Error bars denote the standard error of the mean (SEM); * p<0.05, ** p<0.01, ns not significant, by two-tailed student’s t test. For panel c: n=5 per group. For panel d: n=4 for C. albicans/K. pneumoniae group, n=3 for the other two groups; t test of K. pneumoniae CFU between C. albicans and C. parapsilosis groups. See Supplemental Table 15 for exact p-values.
Supplementary Material
Acknowledgements
We thank all of the infants and their families who participated in the study. We also thank Joao Xavier, Jose Ordovas-Montanes, Olivier Cunrath, and members of the Rakoff-Nahoum Lab for helpful discussions, and Linnea Martin for assistance with sample collection. We thank the reviewers for their helpful comments and suggestions. K.Z.C. is funded by a Sir Henry Wellcome Postdoctoral Research Fellowship (grant 201341/A/16/Z) and a University of Manchester Presidential Fellowship. RSG is supported by grants 1R01AI153257-01 and 5R01AI139633-03. S.R-N. is supported by a Career Award for Medical Scientists from the Burroughs Wellcome Fund, a Pew Biomedical Scholarship, a Basil O’Connor Starter Scholar Award from the March of Dimes, P30DK040561, K08AI130392-01, and a NIH Director’s New Innovator Award DP2GM136652.
Footnotes
Data and Code availability
All Illumina sequencing raw read, including cohort samples and validation samples, have been deposited at the European Nucleotide Archive (ENA) under study accession no. PRJEB36435. Custom scripts for microbiome analyses and interaction inference are available at https://github.com/katcoyte/MK-SpikeSeq. Source data for all figures are included in Supplemental Tables. Public rDNA databases SILVA (119 release, www.arb-silva.de/documentation/release-119/) and UNITE (2017-12-01 release, unite.ut.ee/repository.php) were used to annotate OTU tables.
Competing interests
CRM receives grant funding from Mead Johnson Nutrition. CRM also provides consulting services for Mead Johnson Nutrition, Alcresta and Fresenius Kabi, and sits on the Scientific Advisory Boards of Plakous Therapeutics and LUCA Biologics. All other authors declare no competing interests.
References
- 1.Charbonneau MR et al. Human developmental biology viewed from a microbial perspective. Nature 535, 48–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäckhed F et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Stewart CJ et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derrien M, Alvarez AS & de Vos WM The Gut Microbiota in the First Decade of Life. Trends in Microbiology 27, 997–1010 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Yatsunenko T et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim ES et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med 21, 1228–1234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer C, Bik EM, DiGiulio DB, Relman DA & Brown PO Development of the human infant intestinal microbiota. PLoS Biol. 5, 1556–1573 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch SV & Pedersen O The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med 375, 2369–2379 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Honda K & Littman DR The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Fischbach MA Microbiome: Focus on Causation and Mechanism. Cell 174, 785–790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widder S et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME Journal 10, 2557–2568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vrancken G, Gregory AC, Huys GRB, Faust K & Raes J Synthetic ecology of the human gut microbiota. Nature Reviews Microbiology 17, 754–763 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Walter J, Armet AM, Finlay BB & Shanahan F Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 180, 221–232 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Wolfe BE, Button JE, Santarelli M & Dutton RJ Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 158, 422–433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connell JH & Slatyer RO Mechanisms of Succession in Natural Communities and Their Role in Community Stability and Organization. The American Naturalist 111, 1119–1144 [Google Scholar]
- 16.Bertness MD & Callaway R Positive interactions in communities. Trends in Ecology and Evolution 9, 191–193 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Shade A et al. Macroecology to Unite All Life, Large and Small. Trends in Ecology and Evolution 33, 731–744 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Gregory KE et al. Influence of maternal breast milk ingestion on acquisition of the intestinal microbiome in preterm infants. Microbiome 4, 68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson MK et al. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol 1, 16024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dibartolomeo ME & Claud EC The Developing Microbiome of the Preterm Infant. Clinical Therapeutics 38, 733–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.La Rosa PS et al. Patterned progression of bacterial populations in the premature infant gut. Proc. Natl. Acad. Sci 111, 12522–12527 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costello EK, Carlisle EM, Bik EM, Morowitz MJ & Relman DA Microbiome assembly across multiple body sites in low-birthweight infants. MBio 4, e00782–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart CJ et al. Temporal bacterial and metabolic development of the preterm gut reveals specific signatures in health and disease. Microbiome 4, 67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pammi M et al. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: A systematic review and meta-analysis. Microbiome 5, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasparrini AJ et al. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol 4, 2285–2297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynolds LA & Finlay BB Early life factors that affect allergy development. Nature Reviews Immunology 17, 518–528 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Gensollen T, Iyer SS, Kasper DL & Blumberg RS How colonization by microbiota in early life shapes the immune system. Science 352, 539–544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bokulich NA et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shao Y et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer C, Bik EM, DiGiulio DB, Relman DA & Brown PO Development of the human infant intestinal microbiota. PLoS Biol. 5, 1556–1573 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasolli E et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Proctor LM et al. The Integrative Human Microbiome Project. Nature 569, 641–648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nash AK et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5, 153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Limon JJ, Skalski JH & Underhill DM Commensal Fungi in Health and Disease. Cell Host and Microbe 22, 156–165 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koskinen K et al. First insights into the diverse human archaeome: Specific detection of Archaea in the gastrointestinal tract, lung, and nose and on skin. MBio 8, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durán P et al. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 175, 973–983.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carr A, Diener C, Baliga NS & Gibbons SM Use and abuse of correlation analyses in microbial ecology. ISME J. 1 (2019). doi: 10.1038/s41396-019-0459-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Contijoch EJ et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vandeputte D et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Stämmler F et al. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4, 28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishwaran H & Rao JS SPIKE AND SLAB VARIABLE SELECTION: FREQUENTIST AND BAYESIAN STRATEGIES. Ann. Stat 33, 730–773 (2005). [Google Scholar]
- 42.Gonze D, Coyte KZ, Lahti L & Faust K Microbial communities as dynamical systems. Curr. Opin. Microbiol 44, (2018). [DOI] [PubMed] [Google Scholar]
- 43.Bucci V et al. MDSINE: Microbial Dynamical Systems INference Engine for microbiome time-series analyses. Genome Biol. 17, 121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freilich MA, Wieters E, Broitman BR, Marquet PA & Navarrete SA Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology 99, 690–699 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Friedman J & Alm EJ Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol 8, e1002687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faust K et al. Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Comput. Biol 8, e1002606 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watts SC, Ritchie SC, Inouye M & Holt KE FastSpar: rapid and scalable correlation estimation for compositional data. Bioinformatics 35, 1064–1066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stein RR et al. Ecological Modeling from Time-Series Inference: Insight into Dynamics and Stability of Intestinal Microbiota. PLoS Comput. Biol 9, e1003388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisher CK & Mehta P Identifying Keystone Species in the Human Gut Microbiome from Metagenomic Timeseries Using Sparse Linear Regression. PLoS One 9, e102451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pammi M, Liang R, Hicks J, Mistretta TA & Versalovic J Biofilm extracellular DNA enhances mixed species biofilms of Staphylococcus epidermidis and Candida albicans. BMC Microbiol. 13, 257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.