

Abstract
Cyclin-dependent kinases (CDKs) are the master regulators of the eukaryotic cell cycle. To become activated, CDKs require both regulatory phosphorylation and binding of a cognate cyclin subunit. We studied the activation process of the G1/S kinase Cdk2 in solution and developed a thermodynamic model that describes the allosteric coupling between regulatory phosphorylation, cyclin binding, and inhibitor binding. The results explain why monomeric Cdk2 lacks activity despite sampling an active-like state, reveal that regulatory phosphorylation enhances allosteric coupling with the cyclin subunit, and show that this coupling underlies differential recognition of Cdk2 and Cdk4 inhibitors. We identify an allosteric hub that has diverged between Cdk2 and Cdk4 and show that this hub controls the strength of allosteric coupling. The altered allosteric wiring of Cdk4 leads to compromised activity toward generic peptide substrates, and comparative specialization toward its primary substrate Retinoblastoma (RB).
Introduction
The cyclin-dependent kinases (CDKs) are the central regulators of the cell cycle in all eukaryotes. Cdk1 orchestrates progression through mitosis, Cdk2 controls the G1/S transition, and Cdk4 and Cdk6 trigger cell cycle entry in response to growth factor signaling1. The CDKs are inactive as monomeric kinases and require two separate steps for activation. In the canonical model developed for Cdk2, the kinase is first phosphorylated on the activation loop (A-loop) on a specific site (T160 in Cdk2) by a CDK-activating kinase (CAK) complex2 , and is subsequently activated by binding of a cognate cyclin subunit3. While CAK is active throughout the cell cycle, the expression of each cyclin subunit is restricted to a specific cell cycle stage, ensuring coordinated progression through the cell cycle.
Monomeric Cdk2 adopts an autoinhibited conformation in which the αC-helix swings out of the active site, breaking a catalytically-important salt bridge (“αC-out”), and the A-loop folds into the active site, blocking substrate binding (“Aloop-in”)4. Cyclin binding triggers rotation of the αC-helix into the active site (“αC-in”) and refolding of the A-loop into an extended conformation that permits substrate binding (“Aloop-out”)5. Cyclin binding also buries the phosphorylated T160 residue, blocking dephosphorylation and inactivation of the kinase6. This is thought to facilitate the abrupt switch-like activation of Cdk2 at the G1/S boundary.
The activation pathway of Cdk4/6 differs from that of the other cell cycle CDKs7. Expression of the cognate cyclin, cyclinD, is gradual rather than switch-like, and Cdk4:cyclinD complexes are continually phosphorylated and dephosphorylated on the A-loop. Reversible A-loop phosphorylation allows the activity of Cdk4:cyclinD complexes to be tuned by growth factors, which shift the balance towards phosphorylation by enhancing CAK activity. Reversible phosphorylation is possible because Cdk4, unlike other CDKs, does not switch to the active state when cyclinD binds8,9, leaving the activation loop accessible to phosphatases7. Thus, the degree to which cyclin binding is coupled to kinase activation has diverged between Cdk1/2 and Cdk4/6 in a manner that reflects a biological requirement for altered activation dynamics.
Here we study the dynamic activation process of Cdk2 in solution. We develop a quantitative model that describes the allosteric coupling between Cdk2 and the cyclin subunit, shows that this coupling is enhanced by phosphorylation on T160, and reveals that it is responsible for differential recognition of Cdk2 and Cdk4 inhibitors.
Results
Cyclin triggers a concerted structural change in Cdk2
We used double electron-electron resonance (DEER) spectroscopy to track conformational changes of Cdk2 in solution. One pair of spin labeling sites (A93C, R157C) was used to track the A-loop between the Aloop-in and the Aloop-out states (Fig. 1a, Supplementary Fig. 1). A second pair of labeling sites (A93C, S46C) was used to track the αC-helix between αC-out and αC-in states. Assignment of peaks in the spin-spin distance distributions10 to these structural states was achieved by reference to calculations performed on x-ray structures11 (see Methods and Supplementary Fig. 2).
Figure 1. Cyclin binding triggers a concerted conformational change in Cdk2.
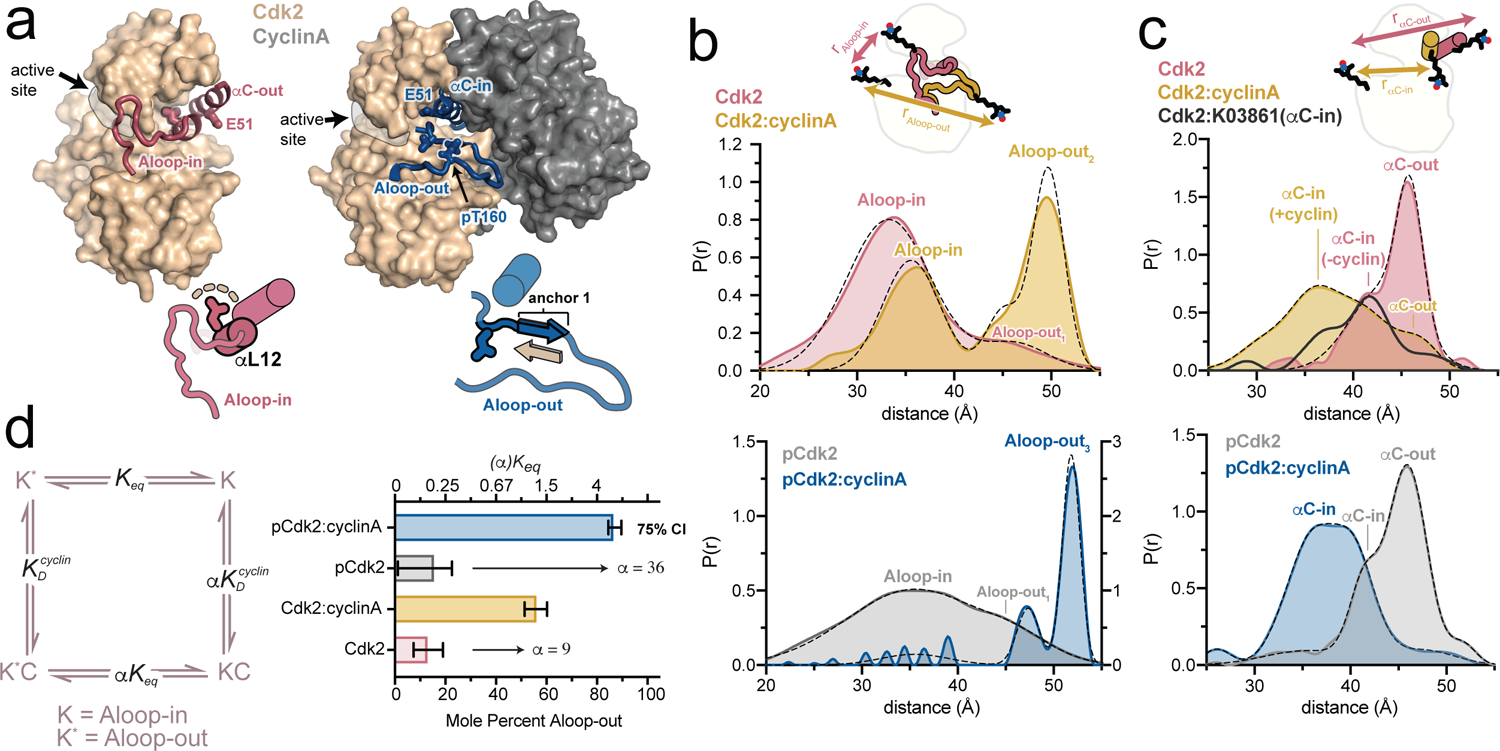
a) X-ray structures of Cdk2 in the Aloop-in/αC-out and Aloop-out/αC-in states (PDB IDs: 1HCK and 1JST), accompanied by schematics of the respective A-loop and αC-helix conformations. The segment of the A-loop highlighted in the schematics refolds from a helix (αL12) in the Aloop-in state to an anchoring β-strand in the Aloop-out state. b,c) DEER experiments tracking the A-loop (b) and αC-helix (c). Tikhonov-derived distance distributions are shown for monomeric Cdk2 and Cdk2:cyclinA dimer in the unphosphorylated state (top) and phosphorylated state (bottom). Dashed black lines represent Gaussian fits to the DEER data. Peak assignments are based on spin-spin distance calculations (see Supplementary Fig. 2 and Methods). The spin labeling schemes are represented schematically. Data for the αC-in inhibitor K03861 are shown in dark gray in c. Data for phosphorylated Cdk2 monomer are plotted on a different y-axis scale for clarity. d) Allosteric two-state model for cyclin binding to Cdk2, with K and K* representing the Aloop-in and Aloop-out states of the kinase, C representing the cyclin subunit, the Aloop-in/out equilibrium constant, and and representing microscopic equilibrium constants for cyclin binding to Aloop-out and Aloop-in states, respectively. The coupling parameter α describes the fold change in upon cyclin binding. The bar graph summarizes the values of and α derived from Gaussian fits of the DEER data. Error bars represent 75% confidence intervals, calculated from 50,000 simulations of Gaussian fits to the primary data.
In experiments tracking the A-loop, monomeric unphosphorylated Cdk2 (“Cdk2”) exhibited one predominant short-distance peak assigned to the Aloop-in state, and a minor longer-distance peak corresponding to an Aloop-out state (Fig. 1b). This demonstrates that monomeric Cdk2 samples the Aloop-out state to a substantial degree (13 ± 6%), as suggested by a recent molecular dynamics study12. Addition of cyclinA (“Cdk2:cyclinA”) resulted in the Aloop-out peak becoming dominant, but a subpopulation sampling the Aloop-in state was still present, indicating an incomplete shift to the Aloop-out state (Fig. 1b).
DEER data collected on T160-phosphorylated monomeric Cdk2 (“pCdk2”) were similar to the unphosphorylated case, although the Aloop-in peak was shifted and broadened (Fig. 1b), consistent with local disorder around the labeling site observed in the x-ray structure of phosphorylated Cdk213. However, addition of cyclinA to this sample (“pCdk2:cyclinA”) led to nearly homogeneous adoption of the Aloop-out state (Fig. 1b), indicating that phosphorylation enhances the cyclin-driven conformational shift.
The experiments tracking the αC-helix revealed conformational shifts that were correlated to those of the A-loop, with a subpopulation of monomeric Cdk2 sampling the αC-in state, and a larger cyclin-driven shift to the αC-in state when the kinase is phosphorylated (Fig. 1c, Supplementary Fig. 2). The inhibitor K03861, which has an αC-in binding mode14, was used to confirm the assignment of the αC-in peak in monomeric Cdk2 (Fig. 1c).
These results are consistent with an allosteric conformational shift model15 (Fig. 1d), in which the kinase transitions in a concerted manner between Aloop-in/αC-out and Aloop-out/αC-in states. The cyclin-driven population shift is described by the allosteric coupling parameter α. The value of α can be estimated from the DEER data to be 9 for unphosphorylated Cdk2, and 36 for phosphorylated Cdk2, indicating that phosphorylation on T160 increases the cyclin-driven conformational shift by ~4-fold.
Cdk2 samples catalytically inactive Aloop-out states
We used paramagnetic relaxation enhancement (PRE) NMR experiments16 to further probe the A-loop movements. The A-loop of Cdk2 is invisible in NMR spectra due to exchange broadening. Nonetheless, by incorporating a spin label on the same A-loop site used for DEER, the position of the A-loop can be inferred through the effects of the paramagnetic label on the spin relaxation of nearby elements of the kinase. In monomeric Cdk2 the observed PRE effects were localized to the αG helix, the β3-αC loop, the CMGC insert region and the αEF and αF helices, and were not affected by phosphorylation (Fig. 2a, Supplementary Fig. 3 and Supplementary Fig. 4). PRE calculations (see Methods) revealed that the effects on the αG helix and part of the β3-αC loop were consistent with the Aloop-in state, but the other effects were not. Calculations performed on three Aloop-out x-ray structures of Cdk2, differing in the position of the labeled segment of the A-loop, indicated that these latter PRE effects were most consistent with the conformation observed in the structure of Cdk2 bound to KAP (kinase associated phosphatase)17. This structure is a rare example of a CDK adopting an Aloop-out state in the absence of a cyclin subunit. Rather than being induced by KAP binding, it appears that Cdk2 dynamically samples this Aloop-out state in which the T160 site is presented for efficient dephosphorylation.
Figure 2. Monomeric Cdk2 dynamically samples a catalytically inactive Aloop-out state.
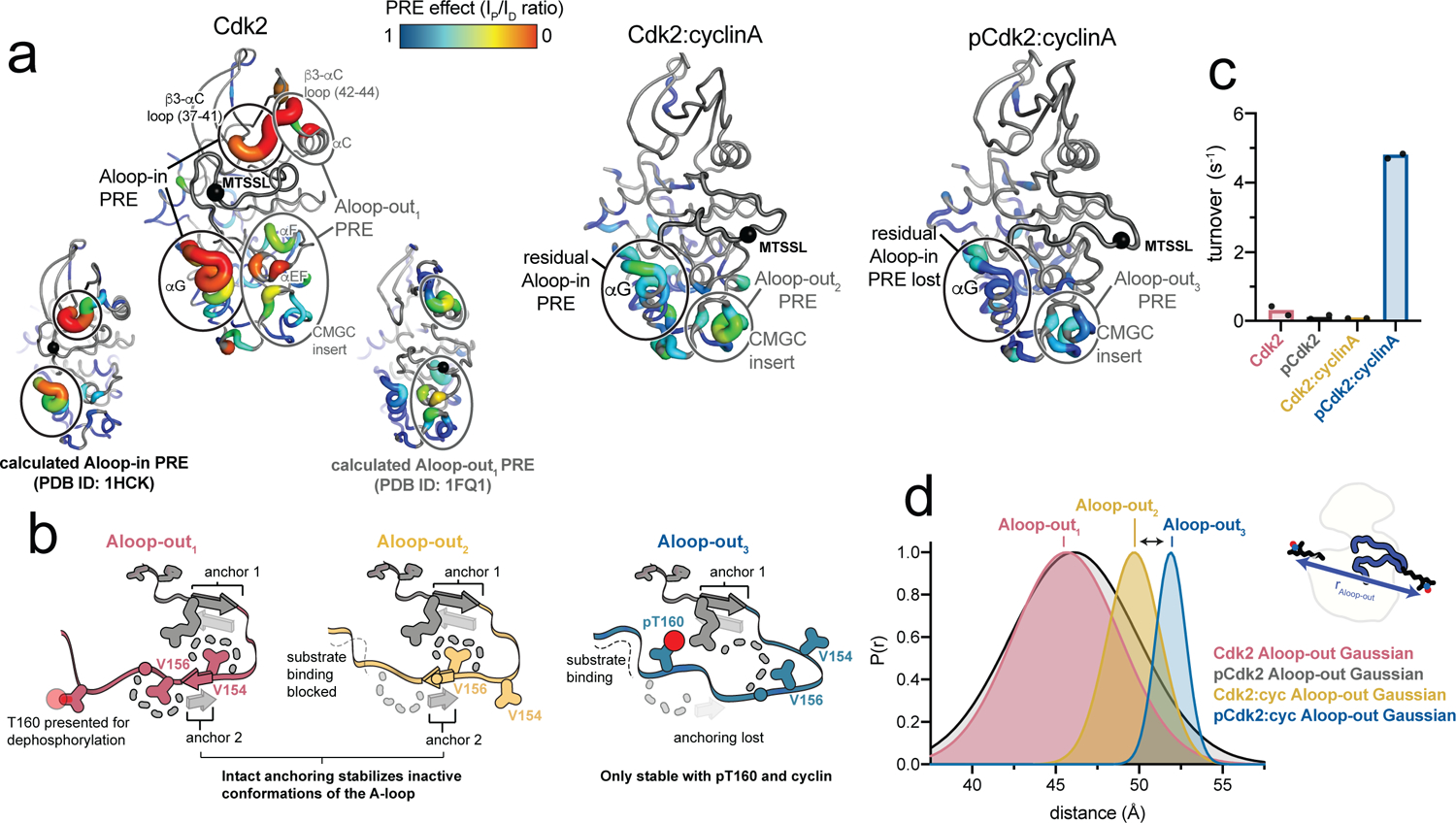
a) NMR PRE data for monomeric unphosphorylated Cdk2 (left), the unphosphorylated Cdk2:cyclinA dimer (middle) and the phosphorylated Cdk2:cyclinA dimer (right) are mapped onto the respective x-ray structures. Data for phosphorylated monomeric Cdk2 are shown in Supplementary Fig. 4. Ovals represent assignments of PRE effects to specific structural states based on PRE calculations (see Supplementary Fig. 4 and Methods). Calculated PRE effects are shown for monomeric Cdk2 as insets on the left. Black spheres represent the R157C spin labeling site. PRE Ip/Id values are shown in Supplementary Fig. 4. b) Schematics representing the three Aloop-out states detected in the PRE experiments. The spin labeling site on the A-loop is shown as a circle. c) Kinase assays showing catalytic activity for four biochemical forms of Cdk2 (with and without phosphorylation and cyclinA). d) The Aloop-out Gaussian fits to the DEER experiments shown in Fig. 1, highlighting the detection of the three different Aloop-out states.
For the Cdk2:cyclinA dimer, the PRE effects were more muted (Fig. 2a). In the unphosphorylated dimer, they were localized predominantly to the CMGC insert region, as predicted by PRE calculations, although there was a remnant of the αG helix PRE effect seen in monomeric Cdk2 (Supplementary Fig. 4), consistent with the dimer sampling the Aloop-in state as indicated by the DEER experiments. In the phosphorylated Cdk2:cyclinA dimer, both the residual αG PRE effect and the CMGC insert PRE effect largely disappeared, and calculations confirmed this to be consistent with the homogeneous adoption of the Aloop-out conformation seen in the x-ray structure of the phosphorylated dimer6.
Together with the DEER results, the PRE experiments help clarify that Cdk2 can sample multiple Aloop-out states. These Aloop-out states all share the characteristic β-sheet formed in the N-terminal segment of the A-loop (anchor 1), but they differ in the positioning of the C-terminal segment of the A-loop (Fig. 2b). In the Aloop-out state adopted by monomeric Cdk2 (termed “Aloop-out1”), this segment of the A-loop is anchored onto the C-terminal lobe of the kinase through a second β-sheet interaction (anchor 2). This feature is common in activated kinases18, but in Cdk2 the β-sheet is out of register, and instead stabilizes an inactive conformation in which the T160 residue is presented for dephosphorylation. In the unphosphorylated Cdk2:cyclinA dimer, anchor 2 is also formed, but its register is shifted by two amino acids, allowing more extensive interactions with the cyclin subunit, while simultaneously blocking the substrate binding site (“Aloop-out2”). Finally, in the phosphorylated Cdk2:cyclinA dimer, anchor 2 is broken, and the A-loop is further shifted toward the cyclin subunit and locked in place by ionic interactions of the pT160 residue with three arginine residues (“Aloop-out3”). Only in this Aloop-out3 state is the peptide substrate binding site formed. Kinase activity assays confirmed that this is the only state that demonstrates appreciable kinase activity (Fig. 2c). Comparison of the long-distance subpopulations measured by DEER revealed that these experiments also distinguish between the three Aloop-out states (Fig. 2d). This provides a simple explanation for why monomeric Cdk2 remains catalytically inactive when phosphorylated on T160, as the Aloop-out1 conformation cannot support substrate binding.
Cdk2 inhibitors drive conformational shifts upon binding
We used DEER to probe the conformational effects of four ATP-competitive Cdk2 inhibitors: flavopiridol, roscovitine, AZD5438, and dinaciclib19–21. All of the inhibitors increased the Aloop-out1 subpopulation when bound to monomeric Cdk2, and also promoted the Aloop-out2 subpopulation when bound to the Cdk2:cyclinA dimer (Fig. 3a, Supplementary Fig. 5). Corresponding population shifts toward the αC-in state were also observed (Fig. 3b, Supplementary Fig. 6), demonstrating that the Cdk2 inhibitors promote concerted conformational shifts from the Aloop-in/αC-out state to the Aloop-out/αC-in state. These shifts can be described by an allosteric coupling parameter β, analogous to the parameter α for cyclin (Fig 3c).
Figure 3. Cdk2 inhibitors drive conformational shifts upon binding.
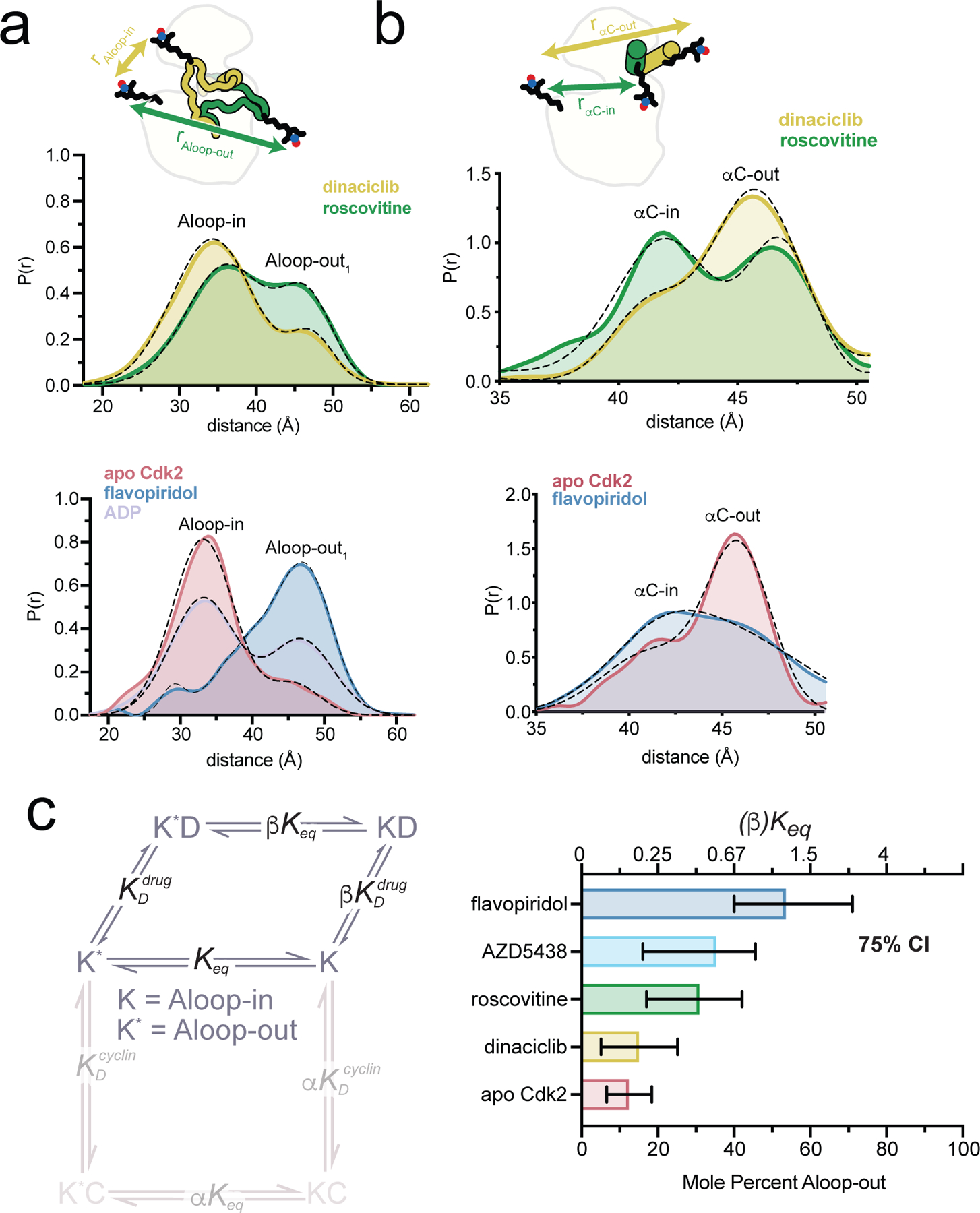
a, b) DEER experiments tracking the A-loop (a) and αC-helix (b). Tikhonov-derived distance distributions are shown for monomeric Cdk2 bound to either dinaciclib or roscovitine (top) and to flavopiridol (bottom). For the A-loop experiments data are also shown for Cdk2 bound to ADP (bottom). Dashed black lines represent the Gaussian fits to the DEER data. Peak assignments are based on spin-spin distance calculations. The spin labeling schemes are represented schematically at the top. Data for AZD5438 are shown in Supplementary Fig. 5 and 6. c) Allosteric two-state model for inhibitor binding to Cdk2, with K and K* representing the Aloop-in and Aloop-out states of the kinase, D representing the drug/inhibitor, Keq the Aloop-in/out equilibrium constant, and and the microscopic equilibrium constants for inhibitor binding to Aloop-out and Aloop-in states, respectively. The coupling parameter β describes the fold change in Keq upon inhibitor binding. The bar graph summarizes the values of Keq and β derived from Gaussian fits of the A-loop DEER data. Error bars represent 75% confidence intervals, calculated from 50,000 simulations of Gaussian fits to the primary data.
Although dinaciclib and roscovitine have similar chemical structures, roscovitine triggers a larger conformational shift. The x-ray structures of Cdk2 bound to dinaciclib and roscovitine are nearly identical, with the notable exception of a 3.5 angstrom shift of the ε-amino group of the catalytic lysine residue K33 (Extended Data Fig. 1, top)22,23. In the dinaciclib structure, K33 forms a salt bridge with D145 of the catalytic DFG motif, but the larger 9-isopropyl substituent of roscovitine compared to the 3-ethyl group of dinaciclib causes K33 to shift away from D145, breaking the salt bridge. Breakage of this salt bridge has been linked to conformational transitions away from the CDK/Src-like inactive state24, and may explain the larger shift triggered by roscovitine. A similar mechanism may also explain the shift triggered by AZD5438, the 2-methylimidazole group of which, like the 9-isopropyl group of roscovitine, would clash with the position of the K33 lysine residue seen in the dinaciclib complex (Supplementary Fig. 7).
Flavopiridol promotes the Aloop-out/αC-in state by a different mechanism. In the crystal structure of flavopiridol bound to the Cdk2:cyclinA dimer25, the inhibitor forms hydrogen bonds with the K33 residue, the catalytic glutamate E51 and a water molecule (Extended Data Fig. 1, bottom). Water-mediated hydrogen bond networks at this site are important for catalytic function and inhibitor recognition26,27, and structures of Cdk2:cyclinA28 bound to nucleotides show a strikingly similar hydrogen bonding geometry (Extended Data Fig. 1, bottom). In DEER experiments, ADP binding also promoted the Aloop-out state (Fig 3a), suggesting that flavopiridol and nucleotide trigger conformational shifts by stabilizing the active state.
Inhibitor and cyclin binding are allosterically coupled
Several Cdk2 inhibitors bind more tightly in the presence of the cyclin subunit25. To simultaneously track inhibitor binding and conformational changes, we labeled Cdk2 with donor and acceptor dyes on the same sites used for conjugating spin labels for the A-loop DEER experiments, yielding high FRET for the Aloop-in state and low FRET for the Aloop-out state (Fig. 4a). Titrations of cyclinA and Cdk2 inhibitors both resulted in decreased FRET, consistent with shifts to the Aloop-out state (Fig. 4b and Supplementary Fig. 8), and the decrease triggered by cyclinA was larger for phosphorylated than unphosphorylated Cdk2, in agreement with the larger shift observed by DEER. The FRET data were analyzed by global fitting to an extended allosteric two-state model15,29 (Fig. 4a) that describes the binding of cyclin and inhibitor and the effects thereof on the equilibrium between Aloop-in and Aloop-out states (see Methods). To map the FRET signal onto the conformational equilibrium, the model was parameterized using the Aloop-in/out equilibrium constants measured by DEER for apo and cyclin-bound Cdk2, thereby constraining the allosteric coupling parameter α. The resulting fits allowed the remaining thermodynamic parameters of the model to be defined, including the microscopic equilibrium constants for cyclin and inhibitor binding to Aloop-in and Aloop-out states, and the allosteric coupling parameters β,γ and δ. The values of β obtained from the model are similar to those measured by DEER and confirm that the Cdk2 inhibitors promote the Aloop-out state (Supplementary Fig. 9).
Figure 4. Tight binding of Cdk2 inhibitors arises from allosteric coupling with the cyclin subunit.
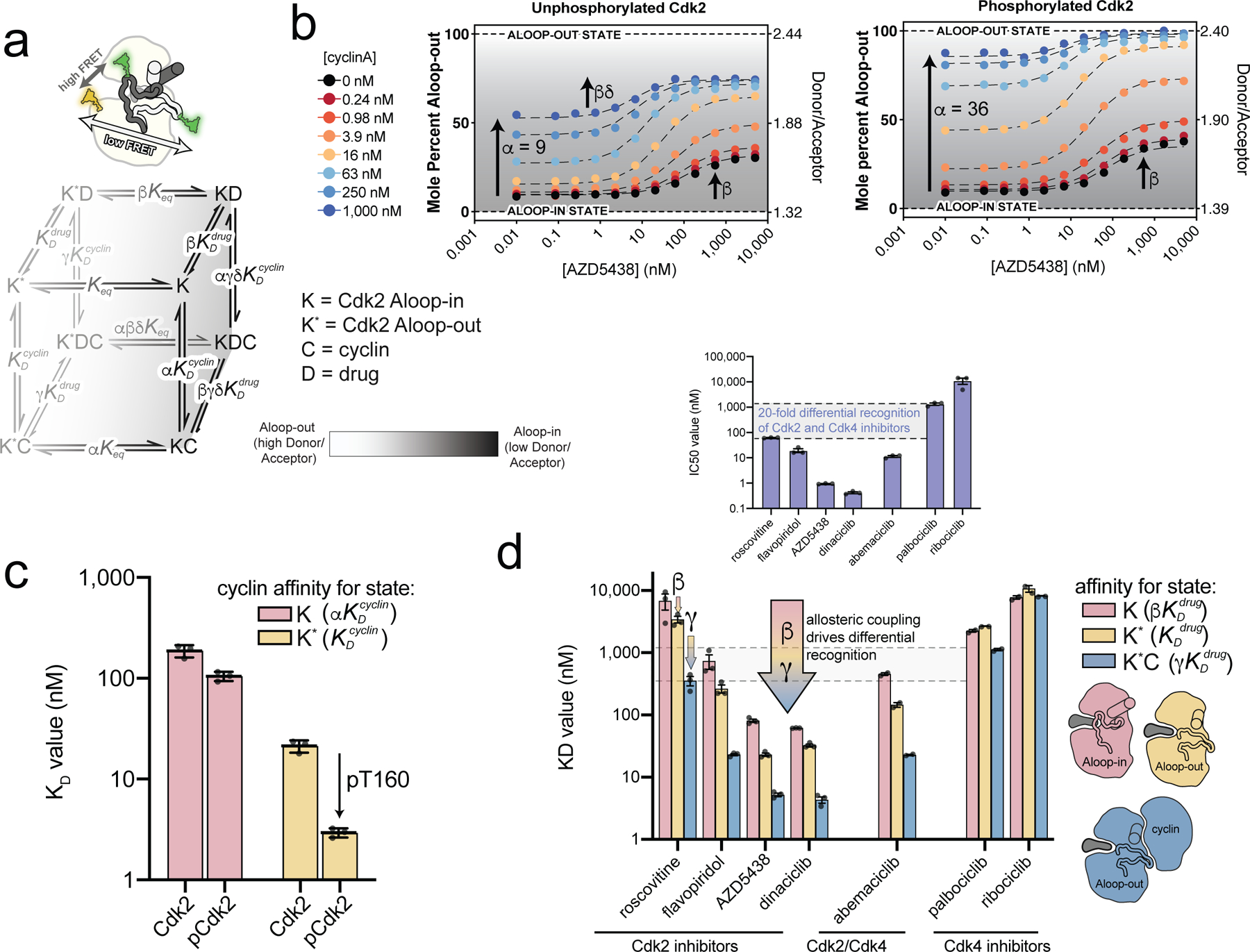
a) A schematic of the FRET sensor is shown above the allosteric model used to fit the FRET data. The model describes the binding of cyclin and inhibitor to the Aloop-in and Aloop-out states, with representing the Aloop-in/out equilibrium, and representing microscopic equilibrium constants for cyclin and inhibitor binding to the Aloop-out state, and the allosteric coupling described with parameters α, β, γ, δ. The Aloop-in/out equilibrium is represented as a gray scale. b) FRET measurements for AZD5438 binding to Cdk2 (left) and phosphorylated pCdk2 (right) at different cyclinA concentrations. The black dashed lines are the global fit to the allosteric model shown in a. The FRET values are shown on the right ordinate and the corresponding values of the conformational equilibrium on the left ordinate. Data for all inhibitors are shown in Supplementary Fig. 8. c) Microscopic equilibrium constants for cyclin binding to Aloop-in ( and Aloop-out states of unphosphorylated and phosphorylated Cdk2. Values are mean ± S.E.M; n = 3 independent experiments. d) Microscopic equilibrium constants for inhibitor binding to three different structural states of phosphorylated Cdk2. The arrow represents the allosteric coupling effects that drive cooperative binding of inhibitor and cyclin. Values are mean ± S.E.M; n = 3 independent experiments. Corresponding data for the unphosphorylated kinase are shown in Supplementary Fig. 9. The inset shows IC50 values determined for the same set of inhibitors using phosphorylated pCdk2:cyclinA dimer.
Several insights emerge from this analysis. First, cyclin affinity for the Aloop-out state is enhanced ~8-fold by phosphorylation (Fig. 4c). In contrast, phosphorylation has a modest effect on the affinity for cyclin binding to the Aloop-in state. This explains why phosphorylation enhances the allosteric shift triggered by cyclin binding. Second, the model recapitulates the positive binding cooperativity between cyclin and the Cdk2 inhibitors25 and reveals that it can be partitioned into two allosteric effects, quantified by the allosteric coupling parameters β and γ (Fig. 4d). These can be understood as follows: the conformational shifts triggered by the inhibitors (β) arise from higher binding affinity for the Aloop-out state; since this state predominates when cyclin is bound, the inhibitor affinity is also enhanced by cyclin binding. However, even within the Aloop-out state there is allosteric cooperativity between inhibitor and cyclin (γ). This may arise from overlapping effects of the two ligands on the kinase conformational entropy, consistent with previous ITC experiments that showed a smaller entropic penalty for AZD5438 binding to the Cdk2:cyclin dimer than monomeric Cdk225. Together, the two allosteric effects give rise to strong binding cooperativity: >10-fold in all four cases, and 50-fold for flavopiridol binding unphosphorylated Cdk2. Without this cooperativity, these molecules would lack sufficient potency to effectively inhibit Cdk2 in cells, where they must compete with high concentrations of ATP.
We also tested the Cdk4 inhibitors abemaciclib, palbociclib and ribociclib in our FRET assay (Fig. 4d). Palbociclib and ribociclib are selective for Cdk4, while abemaciclib inhibits both Cdk4 and Cdk230,31. Abemaciclib behaved like the Cdk2 inhibitors, promoting the Aloop-out state and exhibiting binding cooperativity with cyclin that was similarly partitioned between β and γ effects, and without which the drug would likely lack efficacy against Cdk2. In striking contrast, palbociclib and ribociclib did not promote the Aloop-out state and lacked substantial binding cooperativity with cyclin.
While the Cdk4 inhibitors palbociclib and ribociclib are readily distinguished from the Cdk2 inhibitors by their weak affinities for the Cdk2:cyclinA dimer, the distinction between the two classes of inhibitor becomes blurred when considering their binding affinities for the different conformational states of Cdk2. For example, the Cdk2 inhibitor roscovitine and the Cdk4 inhibitor ribociclib are predicted to bind with similar affinity to the Aloop-in state of monomeric Cdk2, and the same holds for flavopiridol and palbociclib. Thus, the clear separation between the Cdk4 and Cdk2 inhibitor classes apparent in IC50 inhibition values (Fig. 4d inset) is attributable to the strong allosteric coupling of the Cdk2 inhibitors.
A divergent hub controls allosteric coupling in Cdk2
The weak allosteric coupling of the Cdk4 inhibitors might reflect the unusual allosteric wiring of Cdk4, in which cyclinD binding is uncoupled from the Aloop-in/out structural change8,9. To investigate the origins of the distinct allosteric wiring of Cdk2 and Cdk4, we performed a phylogenetic analysis of Cdk1, Cdk2 and the Cdk4/6 family across metazoan lineages (Supplementary Fig. 10). Mapping the metazoan sequence conservation within the respective CDK families onto the structures of Cdk2 and Cdk4 in the Aloop-in state revealed that the entire kinase domain interior was conserved in the Cdk1 and Cdk2 families, but that there was a hotspot of conservative amino acid substitutions in Cdk4/6 clustered in and around the αL12 helical turn of the A-loop (Fig. 5a,b). The variable residues are hydrophobic and interact with one another across the interface between the αL12 helix and the N-terminal lobe. These interactions are broken when the αL12 helix unfolds to form anchor 1 in the Aloop-out state. We refer to these residues as the hub. The residue at the center of the hub is on the αL12 helix and is strictly conserved as an alanine in Cdk1 and Cdk2 (A151 in human Cdk2). In vertebrate Cdk4/6, this residue is instead conserved as an isoleucine (I164 in human Cdk4) and engages in more extensive hydrophobic interactions with the rest of the hub than the corresponding alanine residue of Cdk2 (Fig. 5b). The isoleucine in vertebrate Cdk4/6 is substituted in more basal clades with either valine or threonine (Fig. 5c). An enlarged β-branched residue at this position is thus a characteristic feature of Cdk4/6 kinases conserved across most metazoan life.
Figure 5. A divergent hub controlling allosteric coupling in Cdk2.
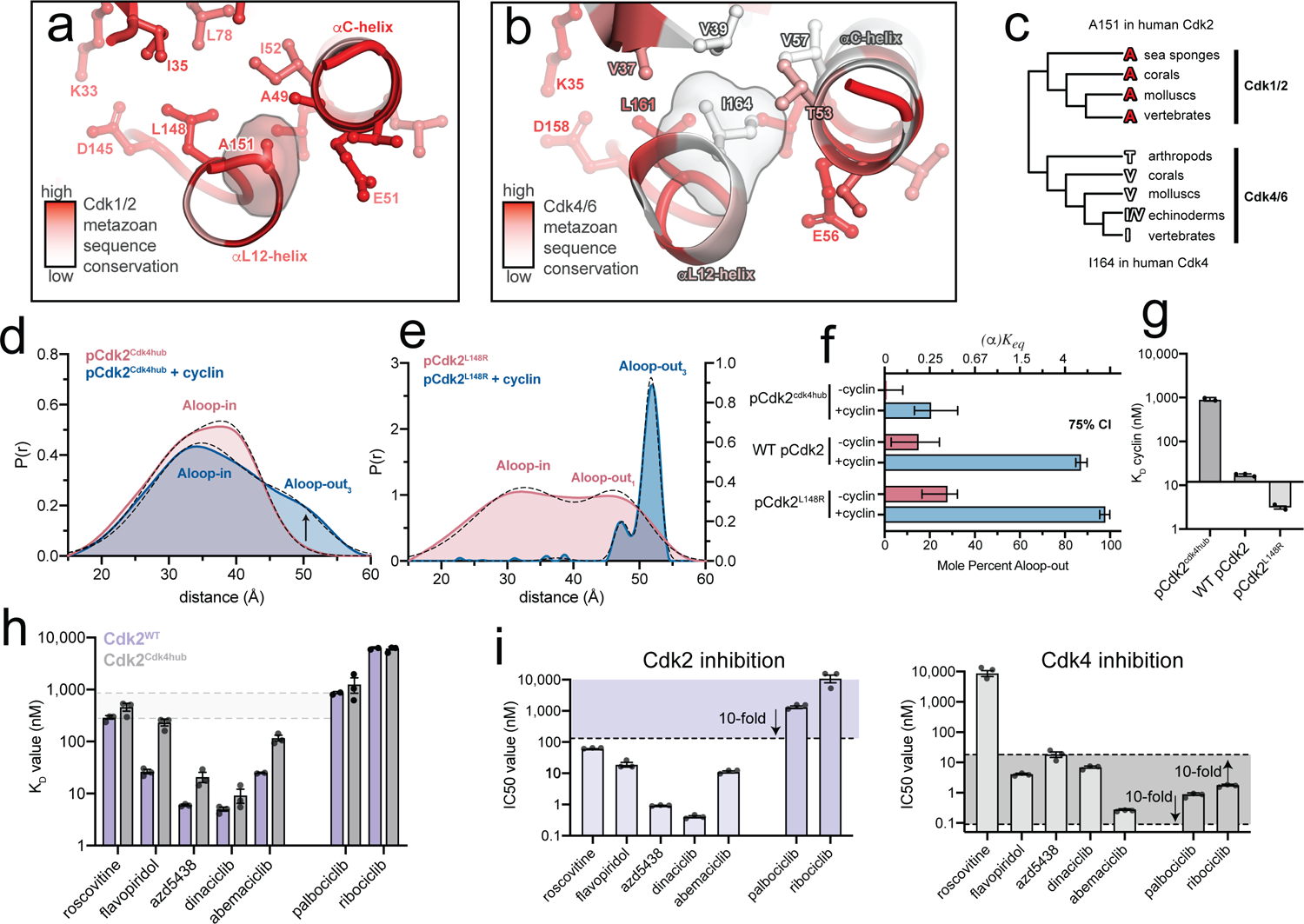
a,b) Metazoan sequence conservation in Cdk1/2 and Cdk4/6, determined from a set of 20 sequences (Supplementary Fig. 10), is mapped onto the structures of Cdk2 (left) and Cdk4 (right) in the Aloop-in conformation. Red represents strict conservation and white represents lower conservation. c) Divergence of the central hub residue (A151 in Cdk2, I164 in Cdk4) between Cdk1/2 and Cdk4/6 lineages. d) DEER data for the phosphorylated pCdk2cdk4hub mutant. Data for the unphosphorylated mutant are shown in Supplementary Fig. 12. e) DEER data for the phosphorylated pCdk2L148R mutant. The monomer data are plotted on a different y-axis scale for clarity. Data for the unphosphorylated mutant are shown in Supplementary Fig. 12. f) Conformational equilibrium values for WT Cdk2, pCdk2L148R and pCdk2cdk4hub. Error bars represent 75% confidence intervals, calculated from 50,000 simulations of Gaussian fits to the primary data. g) Cyclin binding affinities measured by FRET for WT Cdk2, Cdk2L148R and Cdk2cdk4hub mutants. Values are mean ± S.E.M; n = 3 independent experiments. h) Inhibitor KD values measured by FRET for WT pCdk2:cyclinA and pCdk2cdk4hub:cyclinA. Values are mean ± S.E.M; n = 3 independent experiments. Data for the monomeric kinases are shown in Supplementary Fig. 13. i) IC50 values measured in kinase assays for the 7 inhibitors for pCdk2:cyclinA (left) and pCdk4:cyclinD (right). The shaded areas represent the range of IC50 values within 10-fold of the IC50 values for palbociclib and ribociclib. Values are mean ± S.E.M; n = 3 independent experiments.
We prepared a mutant of Cdk2 in which five hub residues were replaced with those of human Cdk4 (Cdk2cdk4hub). We also prepared a second mutant in which the L148 residue was replaced with arginine to destabilize the hub (Cdk2L148R), inspired by a similar oncogenic mutation in EGFR32. DEER experiments revealed opposite effects of these mutations on the kinase conformation (Fig. 5d,e,f). In the monomeric form, the pCdk2cdk4hub mutant was fully in the Aloop-in state, with no evidence of the Aloop-out subpopulation observed in WT Cdk2 (Fig. 5d). Addition of saturating cyclinA resulted in the appearance of an Aloop-out subpopulation, but it remained the minor population (19 ± 9 %). Results with the phosphorylated and unphosphorylated Cdk2cdk4hub mutant were similar (Supplementary Fig. 11 and Supplementary Fig. 12), indicating that phosphorylation was no longer coupled to the structural change. Thus, the Cdk2cdk4hub mutant remains predominantly in the Aloop-in state even when bound to cyclin and phosphorylated on the A-loop, as has been reported for Cdk48,9. In contrast, monomeric pCdk2L148R displayed an increased Aloop-out subpopulation compared to WT Cdk2, and addition of cyclinA resulted in homogeneous adoption of the Aloop-out state with no evidence of a residual Aloop-in subpopulation, unlike WT Cdk2 (Fig. 5e, Supplementary Fig. 12). The two sets of mutations also had opposite effects on the affinity of cyclin binding measured by FRET (Fig. 5g). These results demonstrate that the residues of the hub play an important role in controlling the Aloop-in/out equilibrium and the degree to which cyclin binding is coupled to this equilibrium. The hub residues found in Cdk4/6 appear to weaken this allosteric coupling.
For the Cdk2cdk4hub mutant we also probed the effects of the hub mutations on inhibitor affinities. The affinities of the Cdk2 inhibitors and abemaciclib for the cyclin-bound mutant were markedly decreased compared to WT Cdk2, whereas the affinities of the Cdk4 inhibitors were unaffected (Fig. 5h). Consequently, the discrimination between the Cdk2 and Cdk4 inhibitor classes, so apparent with WT Cdk2, was diminished. These changes in affinity arise specifically from loss of allosteric coupling with the cyclin subunit, as they were not observed with the monomeric Cdk2cdk4hub mutant (Supplementary Fig. 13).
We measured IC50 values for all 7 inhibitors against both pCdk2:cyclinA and pCdk4:cyclinD. The IC50 values obtained for Cdk2 show the expected >20-fold discrimination between the Cdk2 and Cdk4 inhibitors (Fig. 5i), but the values measured for Cdk4 show a strikingly different pattern. As expected, palbociclib, ribociclib and abemaciclib inhibit Cdk4 potently, but so do the majority of the Cdk2 inhibitors (Fig. 5i). With the exception of roscovitine, the Cdk2 inhibitors inhibit Cdk4 with IC50 values within 10-fold of palbociclib and ribociclib. Unlike Cdk2, Cdk4 does not differentiate clearly between Cdk2 and Cdk4 inhibitors. Thus, by uncoupling cyclin binding from the structural change, the altered hub residues of Cdk4 have also removed a major mechanism for distinguishing the two inhibitor classes.
In kinase assays, WT Cdk2, the Cdk2cdk4hub mutant, and Cdk4 itself showed similar KM values toward the physiological substrate RB (Extended Data Fig. 2a). However, the KM value of Cdk4 for a short peptide substrate was 8-fold higher than that of Cdk2, and the KM value of the Cdk2cdk4hub mutant was in between that of WT Cdk2 and Cdk4. This is likely attributable to Cdk4 and the Cdk2cdk4hub mutant favoring the Aloop-in state, in which the peptide binding site is disassembled. In DEER experiments with the Cdk2cdk4hub mutant, the addition of the short peptide substrate increased the population of the Aloop-out3 state (Extended Data Fig. 2b). This indicates that when binding to the Cdk2cdk4hub mutant, and presumably to Cdk4 itself, substrates pay an energetic penalty to induce the Aloop-out3 state, leading to compromised substrate recognition. RB possesses a secondary docking site specific for Cdk4 and essential for phosphorylation by Cdk4, but not Cdk233. A central function of this site may be to compensate for the intrinsically poor substrate binding of Cdk4, driving the comparative specialization of Cdk4 toward RB relative to Cdk234.
Discussion
In cycling cells, monomeric Cdk2 is subject to a continuous cycle of phosphorylation and dephosphorylation on T16035. Our results show that regardless of its phosphorylation state, monomeric Cdk2 samples the Aloop-out1 conformation that is optimized for dephosphorylation rather than for substrate binding, thereby avoiding any residual kinase activity arising from the Aloop-out subpopulation. This permits a small energy barrier for the global structural change, promoting facile regulation while preventing kinase activity from being uncoupled from the expression of cyclins A and E in G1 and S phase. The effects of T160 phosphorylation are only unmasked in the presence of the cyclin subunit, where it enhances the cyclin-driven conformational shift and favors the Aloop-out3 state, trapping the kinase in the catalytically active form. This explains why cyclin binding renders Cdk2 resistant to dephosphorylation in cells, a property that is thought to be important for switch-like activation of the kinase at the G1/S boundary7. In contrast, the graded activation of Cdk4 by growth factor signaling has been linked to the A-loop phosphorylation site remaining accessible to both CAK and phosphatases in Cdk4:cyclinD complexes7. Our results with the Cdk2cdk4hub mutant suggest that this is due to the altered allosteric wiring of Cdk4, in which the cyclin-driven conformational shift is reduced and the enhancement of this shift by A-loop phosphorylation, observed in Cdk2, is lost. The resulting disassembly of the peptide binding site in Cdk4:cyclinD complexes also leads to weak activity toward generic substrates and dependence on the secondary docking site of RB. We have traced the contrasting allosteric properties of Cdk2 and Cdk4 to evolutionary divergence within the allosteric hub, and shown that these differences have important implications for inhibitor recognition.
The advent of Cdk4 inhibitors transformed the treatment of hormone-responsive breast cancer36,37, but the clinical experience with Cdk2 inhibitors has been less favorable. This has been attributed in part to the greater selectivity and therapeutic window of Cdk4 inhibitors. Our results show that the active sites of Cdk2 and Cdk4 do not inherently differentiate between Cdk2 and Cdk4 inhibitors. Instead, the distinction between these two drug classes arises from their differential recognition of particular conformational states. The allosteric properties of the Cdk2 inhibitors reflect the fact that cyclin binding is tightly coupled to the Aloop-out/αC-in state in Cdk2. By similarly coupling to this conformational state, Cdk2 inhibitors are able to target the catalytically active Cdk2:cyclin complexes with high affinity despite binding monomeric Cdk2 relatively weakly. The Cdk4 inhibitors palbociclib and ribociclib are not allosterically coupled in this way because cyclin binding is not tightly coupled to the structural change in Cdk4. This lack of allosteric coupling ensures that palbociclib and ribociclib bind both monomeric Cdk2 and Cdk2:cyclin complexes weakly, and is likely the result of the development process of these drugs in which selectivity for Cdk4 over Cdk2 was prioritized38,39. The importance of allostery is underscored by the contrasting example of abemaciclib, which has greater potency for Cdk4 than either palbociclib or ribociclib, but possesses the allosteric properties of a Cdk2 inhibitor and consequently targets Cdk2 as well. This ability of abemaciclib to target CDKs other than Cdk4/6 has been associated with a superior cytotoxicity profile and the ability to override clinical resistance to palbociclib arising from overexpression of cyclinE31.
The allosteric effects of the Cdk2 inhibitors and abemaciclib have implications for CDK scaffolding interactions, analogous to those reported for palbociclib40. The cognate pairing of CDK and cyclin subunits is relatively labile in cells. For example, enhancing or blocking A-loop phosphorylation of Cdk1 shunts cyclinA onto Cdk1 or cyclinB onto Cdk2, respectively35. The enhancement of cyclinA affinity by Cdk2 inhibitors is likely to apply to other cyclin subunits, which could become sequestered in non-cognate Cdk2:cyclin complexes. Sequestration of cyclinB in such a manner may explain why abemaciclib blocks Cdk1 substrate phosphorylation and triggers a G2 arrest in cells31 despite lacking potent activity against Cdk1 in vitro30. Thus, the contrasting allosteric properties of Cdk4 and Cdk2 inhibitors reported here may play a central role in the therapeutic efficacy of these drugs.
Online Methods
Expression and purification of Cdk2, cyclinA, and RB
Human Cdk2 (residues 1–298 with a TEV-cleavable N-terminal hexahistidine tag in pCDFduet) and mutants were expressed in BL21(DE3) E.coli overnight at 18 ºC. The Cdk2cdk4hub mutant contained the I35V, I52V, L76V, A151I and F152Y mutations derived from the sequence of human Cdk4. Coexpression with yeast CAK was used to prepare T160-phosphorylated Cdk2, and mass spectrometry confirmed homogeneous single phosphorylation in these samples (see Supplementary Fig. 1 and 11). Harvested cell pellets were lysed using an Emulsiflex C3 homogenizer (Avestin). Lysates were clarified by centrifugation and loaded onto HisTrap HP IMAC columns (GE), washed with lysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 10% glycerol) and eluted with elution buffer (1x PBS, 500 mM imidazole, 10% glycerol). Imidazole was immediately removed by desalting, and the hexahistidine tag was cleaved overnight with TEV protease. Protein was further purified using a Superdex 75 10/300 GL size exclusion column (GE).
Bovine CyclinA3 (residues 171–432 with a non-cleavable C-terminal hexahistidine tag) was expressed overnight in BL21(DE3)pLysS E.coli. Cell pellets were lysed as above, lysates clarified, loaded onto HisTrap HP columns, and eluted using a 0–500 mM linear imidazole gradient (50 mM Tris pH 8.25, 300 mM NaCl, 10% glycerol). CyclinA was stabilized with the immediate addition of 100 mM MgCl2 and further purified using a HiLoad 16/600 Superdex 200 size exclusion column (50 mM Tris pH 8.25, 100 mM MgCl2, 5 mM 2-mercaptoethanol).
Human RB (residues 771–928 in a PGEX-3T vector) was expressed overnight in BL21 (DE3) E.coli. Harvested cells were resuspended in 1XPBS, 10% glycerol, 1mM DTT, pH 7.4, lysed with a homogenizer, and lysates clarified by centrifugation at 20,000 rpm. Clarified lysate was loaded onto 2 5mL GSTrap columns (GE), washed with 1XPBS, 10% glycerol, 1mM DTT, pH 7.4, and eluted with 50mM Tris, 10% glycerol, 20mM reduced glutathione, 5mM DTT, pH 8. Eluted protein was desalted into 20mM sodium phosphate pH 7.4, loaded onto 2 5mL SP columns and eluted with 20mM sodium phosphate, 1M NaCl, pH 7.4.
DEER spectroscopy
DEER samples were prepared by labeling “Cys-lite” constructs of Cdk2 (C118A, C177S, A93C, and either R157C or S46C for A-loop and αC-helix experiments, respectively) with a 2.25-fold excess of MTSSL spin label, at a protein concentration of 50–100 µM. Labeled kinase was purified by size exclusion chromatography and concentrated to 60–80 µM, buffered in 1x PBS pH 7.4, 10 mM MgCl2 and 10% d8-glycerol in D2O and rapidly frozen in 1.1 mm ID/1.6 mm OD glass capillary tubes. For samples containing inhibitors, deuterated DMSO was used and inhibitor concentrations were 250 µM. DEER spectra were acquired at 65 K using the standard deadtime-free four-pulse sequence (π/2 = 16 ns and π = 32 ns)41 on an Elexsys E580 spectrometer (Bruker) equipped with an EN5107 resonator operating at Q-band frequencies (~34 GHz). The pump frequency was set to the maximum of a two-pulse echo detected field sweep and the observe frequency was set to 24 G up-field. DEER waveforms were analyzed using Venison (github.com/thompsar/Venison), a custom program written in Python based on DeerAnalysis 2017. Briefly, distance distributions were fit to the background-corrected waveforms using unconstrained Tikhonov regularization, with smoothing parameter λ chosen using the L-curve and leave-one-out cross validation (LOOCV). Features of the spectrum that were tightly coupled to the background model and contributed to unstable populations that were both distinct from the primary distance populations and beyond the limit of sensitivity of the 6 µs evolution time (~58 Å) were suppressed by incorporating them into the background model. Tikhonov regularization was repeated on the corrected waveform. The Tikhonov distribution was used to initialize fitting of the waveforms to a sum of Gaussians model in order to determine the population centers of the spin-spin distances, as well as the widths and mole fractions. The appropriate number of Gaussian subpopulations was determined by selecting the fewest number of Gaussian centers that met the RMSD minimization threshold calculated by the Bayesian information criterion42. Confidence intervals for Gaussian fits were calculated with Monte Carlo simulations of Gaussian fits to the background-corrected waveforms. All uncertainties quoted in main text and figures represent 75% confidence intervals.
Predicted spin-spin distance distributions were calculated from x-ray structures using the PyMOL plugin mtsslWizard11 with the thorough conformational search setting for generating MTSSL spin probe ensembles. Calculated distance distributions agreed well with experiment allowing unambiguous assignment of peaks in the experimental distance distributions to individual structural states (Supplementary Fig. 2). In particular, these calculations confirmed that the Aloop-out1, Aloop-out2, and Aloop-out3 states should give rise to increasingly longer spin-spin distances in A-loop DEER experiments, and that the 41 Å peak observed in the αC-helix DEER experiments with monomeric Cdk2 corresponds to the αC-in state.
NMR sample preparation and spectroscopy
Cdk2 constructs were expressed in M9 minimal media with 15N- ammonium chloride to yield 15N-labeled protein, and additionally with 2D-glucose, and 100% 2D2O to yield 2D-15N-labeled protein. T160-phosphorylated Cdk2 was prepared by coexpression with yeast CAK in E. coli grown in M9 minimal media with 15N-ammonium chloride, 2D-glucose, and 50% 2D2O to yield 2D-15N-labeled phosphorylated protein with ~53% 2D incorporation. Isotope labeled proteins were purified as described above. Constructs were labeled on R157C with MTSSL (paramagnetic) or dMTSSL (diamagnetic) as described above (see Supplementary Figure 1). 1H-15N TROSY-HSQC spectra were collected on 850 and 900 MHz Bruker Avance III spectrometers equipped with triple resonance cryoprobes in the Minnesota NMR Center. Monomeric Cdk2 NMR experiments were collected in 10 mM NaPO4 pH 7.4, 140 mM NaCl, 1 mM roscovitine, 10 mM MgCl2, and 10% D2O. Dimeric Cdk2:CyclinA NMR experiments were collected in 10 mM NaPO4 pH 7.4, 300 mM NaCl, 1 mM roscovitine, 10 mM MgCl2, 500 μM p27 derived peptide, and 10% D2O. NMR spectra were processed using NMRPipe43 and analyzed using Sparky44.
For PRE measurements, 1H-15N TROSY-HSQC spectra were collected for each sample with an MTSSL probe for paramagnetic spectra and dMTSSL probe for diamagnetic spectra (see Supplementary Fig. 3). Because the spectra lacked sufficient signal-to-noise for accurate determination of Γ2 values, peak intensities were extracted from each spectrum using Sparky, and the ratio of paramagnetic intensities (IP) to diamagnetic intensities (ID) was mapped onto the relevant x-ray structures using a b-factor replacement script for PyMOL. Peak intensities were normalized within each spectrum to residues distal to the probe site (residues 282–289). Standard errors in the IP/ID values were calculated from the signal-to-noise ratio of each spectrum45. Peaks with errors > 0.2 were excluded from the analysis.
Calculation of PRE effects from x-ray structures
The PyMOL plugin mtsslWizard11 was used to model an ensemble of MTSSL spin probe conformations, and predicted distance distributions were generated by calculating the distances between the spin probe ensemble and the backbone nitrogen atoms of each amino acid residue. The weighted mean distance was then used to estimate the paramagnetic relaxation enhancement (Γ2) for each amide nitrogen using the following equation46:
where is the distance between the spin probe and the nitrogen atom; μ0 is the permeability of vacuum; γI is the nuclear gyromagnetic ratio; g is the electron g-factor; μB is the Bohr magneton; S is the electron spin quantum number; τc is the PRE correlation time; and ωH/2π is the nuclear Larmor frequency. Computational IP/ID ratios were calculated according to Iwahara et al.46 :
where T is the delay time in the experiment. For these calculations T was set to 15 ms and IP(0)/ID(0) was set to 1.
FRET experiments tracking the A-loop
FRET samples were prepared by labeling “Cys-lite” constructs of Cdk2 (C118A, C177S, A93C, R157C) with a 0.7:1 ratio of donor (AF488, Fluoroprobes) followed by a threefold excess of acceptor (AF568; Fluoroprobes) to ensure complete labeling. Labeled samples were purified by size exclusion chromatography into 1X PBS pH 7.4, 10 mM MgCl2, 5 mM DTT and supplemented with 0.5 mg/ml bovine gamma globulins (Sigma) and 0.02% Tween-20. Inhibitor dose response titrations were prepared in 384-well plates using a mosquito liquid handling robot (ttp Labtech). Cdk2 FRET sensor (24 μL) was added to each well containing 1 μL of inhibitor in DMSO and incubated for 30 minutes prior to data collection. Steady-state fluorescence emission spectra were recorded with a fluorescence plate reader (Fluorescence Innovations), and the contributions of the donor and acceptor emission peaks to the measured emission spectra were unmixed by fitting each spectrum to basis functions for AF488, AF568 and the water Raman band47. For each inhibitor, binding experiments were performed using eight concentrations of cyclinA and twelve concentrations of inhibitor.
Global fit analysis of FRET data
FRET data were fit globally, using the numerical simulation package Kintek Explorer, to the extended allosteric two state model15,29 shown in Figure 4a. In this model, and represent the microscopic equilibrium constants for binding to the Aloop-out state of cyclinA and inhibitor, respectively, and the respective equilibrium constants for binding to the Aloop-in state, represents the conformational equilibrium constant for monomeric Cdk2, and α, β, γ, and δ represent the allosteric coupling factors that describe the coupling between cyclin binding and the conformational equilibrium (α), drug binding and the conformational equilibrium (β), the binding cooperativity between cyclin and drug in the Aloop-out state (γ) and the difference in binding cooperativity between the Aloop-in and Aloop-out states (δ).
The measured ratiometric FRET signal was mapped onto the conformational equilibrium using one fluorescence coefficient, , to represent the Aloop-in state, and one coefficient, , for the Aloop-out state,
where and represent monomeric Cdk2 in the Aloop-in and Aloop-out states, respectively, and Cdk2:cyclinA in the Aloop-in and Aloop-out states, and Cdk2:drug in the Aloop-in and Aloop-out states, and and Cdk2 bound to both cyclinA and inhibitor in the Aloop-in and Aloop-out states. The values of and were constrained to their values measured in the DEER experiments ( for Cdk2, for pCdk2, for Cdk2:cyclinA and for pCdk2:cyclinA, see Figure 1d). For unphosphorylated Cdk2, fitting yielded well-constrained values for all remaining parameters of the model (β, γ, δ, , and ) as judged by one- and two-dimensional error surface analysis (see Supplementary Fig. 14). For phosphorylated Cdk2, the value of the allosteric coupling parameter δ, which determines the cyclin and inhibitor affinities for the inactive Aloop-in state of the pCdk2:cyclin dimer, was not well constrained in some datasets. We therefore chose not to interpret the value of δ. The values of , and shown in Figure 4c,d represent the averages from three independent experiments.
Kinase activity assays
Kinase activities of WT and mutant recombinant pCdk2:cyclinA and commercial pCdk4:cyclinD (Thermo Fisher) were determined using the ADP Quest accumulation assay (Eurofins). Reactions were performed with 5 nM kinase and 200 μM ATP in 384-well plates using a fluorescence plate reader and the Magellan Standard software (Tecan) at 20 ºC. Rates were determined from time-dependent fluorescence traces by linear regression using Prism 8 (Graphpad). Reactions were performed in the presence and absence of substrate, and rates were corrected for nonspecific ATP turnover by subtracting the rates measured in the absence of substrate. Substrates used include RB residues 771–928, which includes the C-terminal Cdk4 docking site, and the histone H1-derived short peptide substrate PKTPKKAKKL48,49. IC50 values for the 7 inhibitors were measured using a γ−33P radioactivity assay with RB as a substrate and 10 μM ATP (Reaction Biology).
Extended Data
Extended Data Fig. 1. Cdk2 inhibitors drive conformational shifts upon binding.
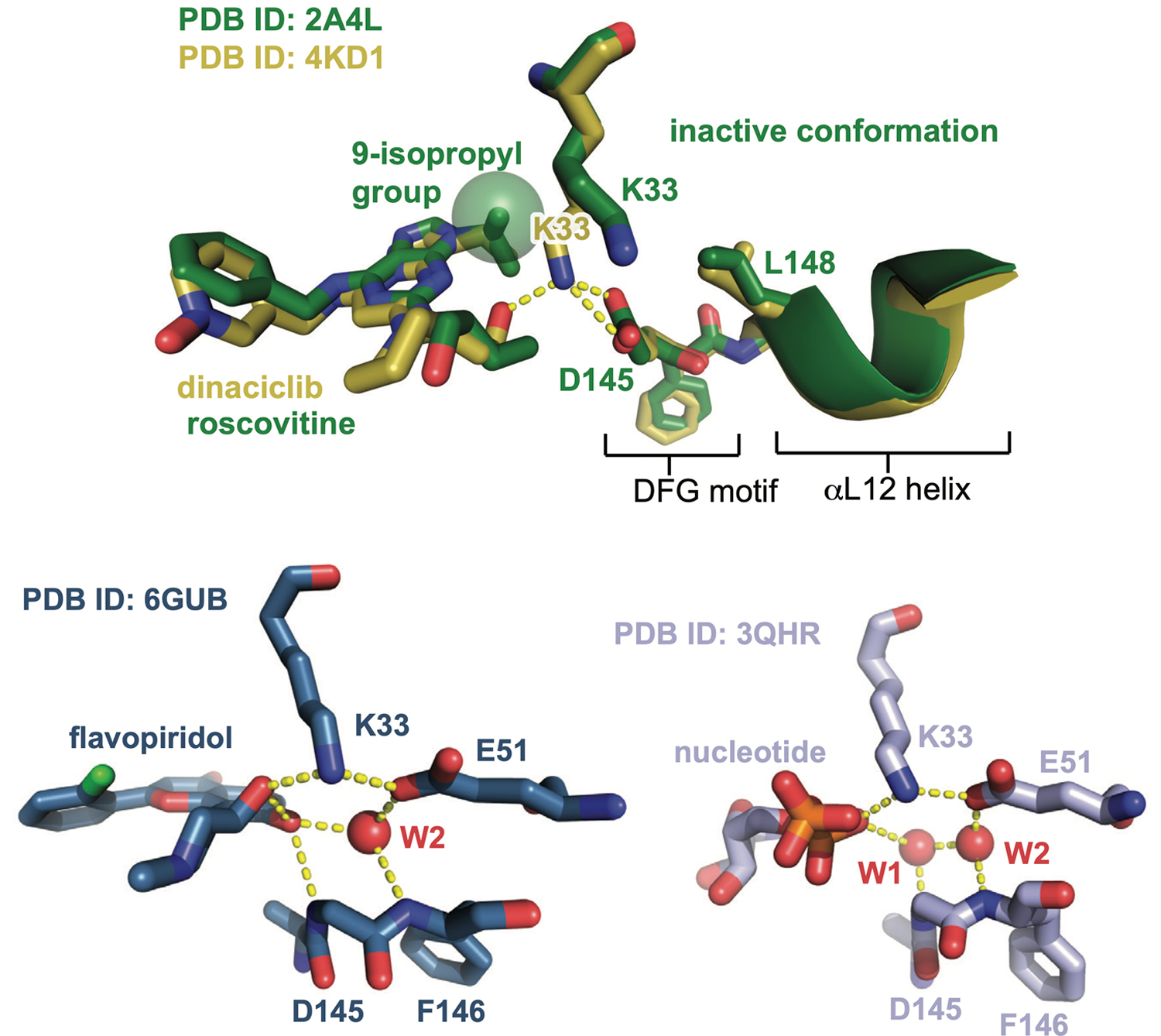
Aligned crystal structures of Cdk2 bound to dinaciclib and roscovitine (top), and structures of Cdk2:cyclinA bound to flavopiridol and ADP shown side by side (bottom). Hydrogen bonds are shown as yellow dashed lines, and structured water molecules as red spheres. The structure of Cdk2 bound to AZD5438 is shown in Supplementary Fig. 7.
Extended Data Fig. 2. A divergent hub controlling allosteric coupling in Cdk2.
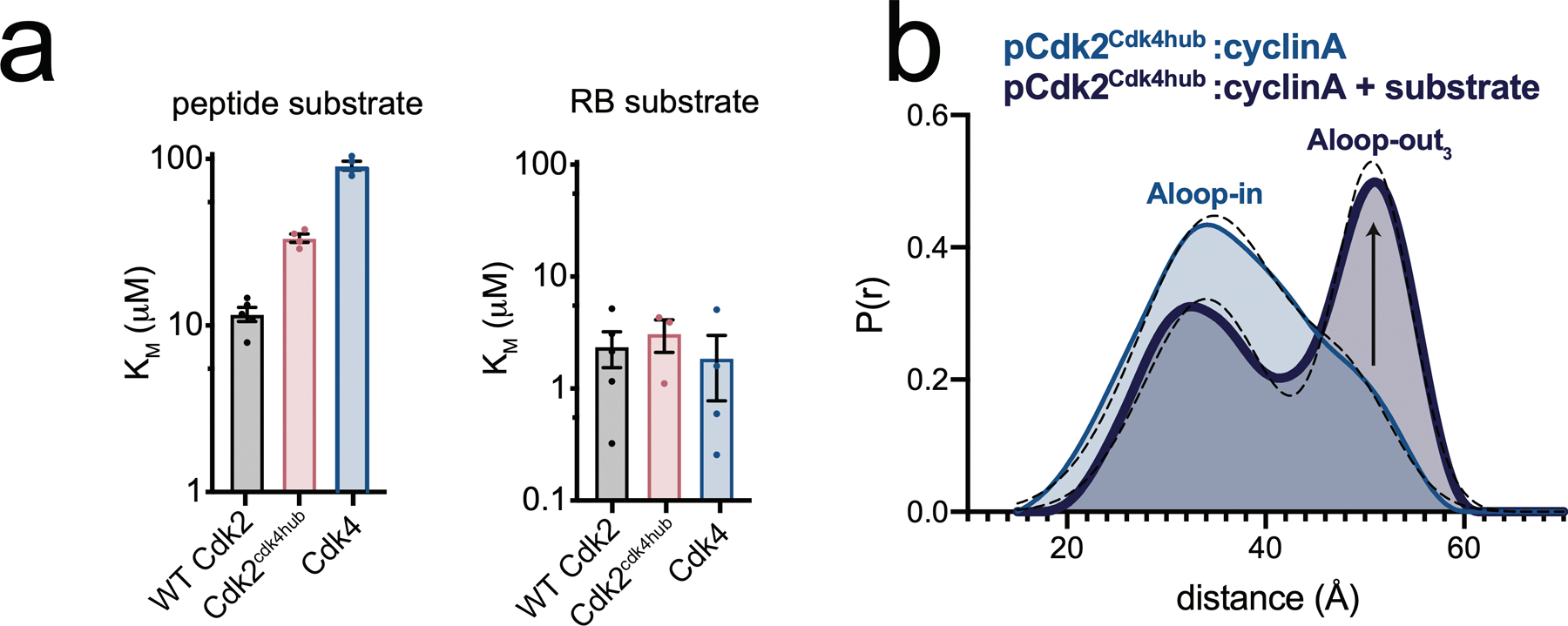
a) KM values for phosphorylation of a short peptide substrate (left) and RB (right), measured for WT Cdk2, Cdk2cdk4hub and Cdk4. Values are mean ± S.E.M; n = 4 independent experiments. b) DEER data for the Cdk2cdk4hub mutant bound to cyclinA with and without addition of saturating peptide substrate and AMPPNP.
Supplementary Material
Acknowledgements
We thank Matt Young for the Cdk2 and yeast CAK constructs, Jane Endicott for the bovine cyclinA construct, and Seth Rubin for the RB771−928 construct. We thank Joseph Dalluge for assistance with mass spectrometry. This work was supported in part by grants from the National Institutes of Health (R01 GM121515, N.M.L.) and the National Institutes of Health Cancer Biology Training Grant (T32 CA009138, A.M.) and Chemistry-Biology Interface Training Grant (T32 GM132029, D.M.R.).
Footnotes
Competing Financial Interests
The authors report no competing financial interests
Code Availability
The program used to analyze DEER data for this study, Venison, is available for download from https://github.com/thompsar/Vension.
Data Availability
The data that support the findings in this study are available within the paper (main and supplementary sections).
References
- 1.Morgan DO Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13, 261–291, doi: 10.1146/annurev.cellbio.13.1.261 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Fisher RP & Morgan DO A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78, 713–724, doi: 10.1016/0092-8674(94)90535-5 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Desai D, Wessling HC, Fisher RP & Morgan DO Effects of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol Cell Biol 15, 345–350 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Bondt HL et al. Crystal structure of cyclin-dependent kinase 2. Nature 363, 595–602, doi: 10.1038/363595a0 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Jeffrey PD et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376, 313–320, doi: 10.1038/376313a0 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Russo AA, Jeffrey PD & Pavletich NP Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol 3, 696–700 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Schachter MM et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell 50, 250–260, doi: 10.1016/j.molcel.2013.04.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Day PJ et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci U S A 106, 4166–4170, doi:0809645106 [pii] 10.1073/pnas.0809645106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takaki T et al. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A 106, 4171–4176, doi:0809674106 [pii] 10.1073/pnas.0809674106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiang YW, Borbat PP & Freed JH The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J Magn Reson 172, 279–295, doi: 10.1016/j.jmr.2004.10.012 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Hagelueken G, Ward R, Naismith JH & Schiemann O MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl Magn Reson 42, 377–391, doi: 10.1007/s00723-012-0314-0 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisani P, Caporuscio F, Carlino L & Rastelli G Molecular Dynamics Simulations and Classical Multidimensional Scaling Unveil New Metastable States in the Conformational Landscape of CDK2. PLoS One 11, e0154066, doi: 10.1371/journal.pone.0154066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown NR et al. Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J Biol Chem 274, 8746–8756 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Alexander LT et al. Type II Inhibitors Targeting CDK2. ACS Chem Biol 10, 2116–2125, doi: 10.1021/acschembio.5b00398 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Tsai CJ & Nussinov R A unified view of “how allostery works”. PLoS Comput Biol 10, e1003394, doi: 10.1371/journal.pcbi.1003394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battiste JL & Wagner G Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 39, 5355–5365, doi: 10.1021/bi000060h (2000). [DOI] [PubMed] [Google Scholar]
- 17.Song H et al. Phosphoprotein-protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol Cell 7, 615–626, doi: 10.1016/s1097-2765(01)00208-8 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Levinson NM The multifaceted allosteric regulation of Aurora kinase A. Biochem J 475, 2025–2042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Tourneau C et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur J Cancer 46, 3243–3250, doi: 10.1016/j.ejca.2010.08.001 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Ghia P et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 129, 1876–1878, doi: 10.1182/blood-2016-10-748210 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Shapiro GI Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res 10, 4270s–4275s, doi: 10.1158/1078-0432.CCR-040020 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Martin MP, Olesen SH, Georg GI & Schonbrunn E Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem Biol 8, 2360–2365, doi: 10.1021/cb4003283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Azevedo WF et al. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem 243, 518–526, doi: 10.1111/j.1432-1033.1997.0518a.x (1997). [DOI] [PubMed] [Google Scholar]
- 24.Levinson NM et al. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol 4, e144, doi:05-PLBI-RA-1226R3 [pii] 10.1371/journal.pbio.004014 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood DJ et al. Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem Biol 26, 121–130 e125, doi: 10.1016/j.chembiol.2018.10.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levinson NM & Boxer SG A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat Chem Biol 10, 127–132, doi: 10.1038/nchembio.1404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cyphers S, Ruff EF, Behr JM, Chodera JD & Levinson NM A water-mediated allosteric network governs activation of Aurora kinase A. Nat Chem Biol, doi: 10.1038/nchembio.2296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao ZQ, Jacobsen DM & Young MA Briefly bound to activate: transient binding of a second catalytic magnesium activates the structure and dynamics of CDK2 kinase for catalysis. Structure 19, 675–690, doi: 10.1016/j.str.2011.02.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall DA Modeling the functional effects of allosteric modulators at pharmacological receptors: an extension of the two-state model of receptor activation. Mol Pharmacol 58, 1412–1423, doi: 10.1124/mol.58.6.1412 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Chen P et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol Cancer Ther 15, 2273–2281, doi: 10.1158/1535-7163.MCT-16-0300 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Hafner M et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol 26, 1067–1080 e1068, doi: 10.1016/j.chembiol.2019.05.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paez JG et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500, doi: 10.1126/science.1099314 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Topacio BR et al. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol Cell 74, 758–770 e754, doi: 10.1016/j.molcel.2019.03.020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chi Y et al. Identification of CDK2 substrates in human cell lysates. Genome Biol 9, R149, doi: 10.1186/gb-2008-9-10-r149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merrick KA et al. Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol Cell 32, 662–672, doi: 10.1016/j.molcel.2008.10.022 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finn RS et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16, 25–35, doi:S1470–2045(14)71159–3 [pii] 10.1016/S1470-2045(14)71159-3 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Hortobagyi GN Ribociclib for HR-Positive, Advanced Breast Cancer. N Engl J Med 376, 289, doi: 10.1056/NEJMc1615255 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Toogood PL et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem 48, 2388–2406, doi: 10.1021/jm049354h (2005). [DOI] [PubMed] [Google Scholar]
- 39.VanderWel SN et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem 48, 2371–2387, doi: 10.1021/jm049355+ (2005). [DOI] [PubMed] [Google Scholar]
- 40.Guiley KZ et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 41.Pannier M, Veit S, Godt A, Jeschke G & Spiess HW Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson 142, 331–340, doi: 10.1006/jmre.1999.1944 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Priestley MB Spectral Analysis and Time Series (Academic Press, 1981). [Google Scholar]
- 43.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293, doi: 10.1007/BF00197809 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Lee W, Tonelli M & Markley JL NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327, doi: 10.1093/bioinformatics/btu830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warren C et al. Dynamic intramolecular regulation of the histone chaperone nucleoplasmin controls histone binding and release. Nat Commun 8, 2215, doi: 10.1038/s41467-017-02308-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwahara J, Tang C & Marius Clore G Practical aspects of (1)H transverse paramagnetic relaxation enhancement measurements on macromolecules. J Magn Reson 184, 185–195, doi: 10.1016/j.jmr.2006.10.003 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaaf TM, Peterson KC, Grant BD, Thomas DD & Gillispie GD Spectral Unmixing Plate Reader: High-Throughput, High-Precision FRET Assays in Living Cells. SLAS Discov 22, 250–261, doi: 10.1177/1087057116679637 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hagopian JC et al. Kinetic basis for activation of CDK2/cyclin A by phosphorylation. J Biol Chem 276, 275–280, doi: 10.1074/jbc.M007337200 M007337200 [pii] (2001). [DOI] [PubMed] [Google Scholar]
- 49.Stevenson-Lindert LM, Fowler P & Lew J Substrate specificity of CDK2-cyclin A. What is optimal? J Biol Chem 278, 50956–50960, doi: 10.1074/jbc.M306546200 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this study are available within the paper (main and supplementary sections).