

Abstract
Apoptosis is important for normal development and removal of damaged cells. Evasion of apoptosis by cancer cells is one of the key characteristics of many tumor types. Thus, discovering agents that promote apoptosis in tumor cells could have great therapeutic value. Marine natural products have demonstrated great potential as anticancer agents, and the proapoptotic activity of some of these products is emerging as a potentially useful property for cancer treatments. Using a tumor xenograft assay in rodents, we previously found that the marine alkaloid naamidine A is a potent antitumor agent. In this study, we further characterize the mechanism of action of naamidine A. In cultured tumor cells, we find that naamidine A induces cell death, which is accompanied with annexin V staining, disruption of the mitochondrial membrane potential, and cleavage and activation of caspases 3, 8, and 9, all of which are hallmarks of apoptosis. Furthermore, naamidine A-induced cell death is caspase dependent. We also find that under conditions where naamidine A inhibits tumor xenograft growth, it induces activation of caspase 3, suggesting that apoptosis is part of its antitumorigenic activity in vivo. Apoptosis is not dependent on extracellular signal-regulated kinase 1/2, previously characterized molecular targets of naamidine A, nor does it require functional p53. Our studies support the continued study of naamidine A and its target(s) for the potential development of better clinical treatments for cancer.
Keywords: antitumor agent, apoptosis, caspase, marine natural products, mitochondria
Introduction
Programmed cell death, termed apoptosis, is an important natural process used by the human body to remove unwanted cells. In the case of the immune system, these cells may react against self-antigens, whereas in other cases these cells may acquire harmful functions through the accumulation of genetic mutations. Deregulation of apoptosis contributes to many human diseases [1,2]. Of the six acquired capabilities of a malignant phenotype described by Hanahan and Weinberg, evasion of apoptosis is one of the critical hallmarks of most types of cancer [3,4]. Thus, discovering and developing therapeutics that either induce the proapoptotic or inhibit the antiapoptotic cellular machinery of cancer cells may have great therapeutic value [1,4].
Natural product scaffolds have an inherent ability to bind biological molecules that participate in a number of important processes, including apoptosis [5–7]. Currently, more than 78% of all chemotherapeutic agents used clinically are from natural sources or are derived from natural product scaffolds [8]. In this regard, marine natural products have shown great promise in the area of anticancer drug discovery, and currently there are 15 marine natural products in clinical trials in the United States [9–11]. An ongoing effort has been devoted to the preclinical development of the marine alkaloid naamidine A (NA) (Fig. 1a). NA was first isolated from the Red Sea sponge Leucetta chagosensis and reported in 1989 [12]. Approximately 10 years later, the Ireland laboratory isolated NA under bioassay-guided fractionation from the Fijian sponge Leucetta sp. Initial studies showed that NA was an inhibitor of epidermal growth factor (EGF)-mediated mitogenesis [13]. Furthermore, when tested in mice harboring human tumor xenografts, NA inhibited tumor growth by 87% at a maximum tolerated dose of 25 mg/kg [13]. Later studies suggested that the antitumor activity of NA might arise through its ability to promote G1 cell cycle arrest [14]. At a molecular level, NA promotes mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) activation, possibly through a direct mechanism [14]. As prolonged ERK activation is sometimes associated with cell cycle arrest [15,16], ERK might be a key mediator of NA-induced cell cycle arrest and the antitumor activity of NA in vivo.
Fig. 1.

Naamidine A (NA)-induced cell death in A431 cells. (a) The structure of naamidine A. (b) Fluorescence-activated cell sorter (FACS) cell cycle analysis quantifying the sub-G1 population of A431 cells treated with 1 and 30 μmol/l NA for 12 h. A431 cells treated with 30 μmol/l NA show a statistically significant 80% induction of a sub-G1 peak indicative of cell death. (c) A histogram depicting FACS cell cycle analysis of A431 cells after treatment with NA indicating the different phases of the cell cycle including the sub-G1 cell population. (d) FACS analysis quantifying annexin V-positive staining of A431 cells treated with 0.5, 1, 5, 15, and 30 μmol/l NA indicating apoptosis. The t-test statistical analysis was used to compare changes in percentage cell death (sub-G1) or percentage apoptosis (annexin V) between untreated control cells and cells treated with 5, 15, or 30 μmol/l NA for 12 h, which yielded a P value of 0.0001 (*).
This study has been devoted to further characterizing the mechanism by which NA exerts its antitumor activity. Here, we demonstrate that in addition to promoting cell cycle arrest, NA induces apoptosis in tumor cells grown in vitro. Apoptosis likely occurs through the disruption of the mitochondrial membrane potential (Δψm) and activation of the proapoptotic caspases 3, 8, and 9. Inhibitor studies indicate that MEK/ERK signaling is dispensable for this aspect of the antitumor activity of NA. Finally, we demonstrate that NA also induces caspase 3 cleavage in tumor cells growing in vivo as xenografts. Our results provide new insight into the antitumor activity of NA, and suggest the existence of an as yet undiscovered target(s) distinct from ERK that mediates the antitumor activity of NA in vivo.
Materials and methods
Antibodies, fluorescent dyes, and inhibitors
Primary antibodies to p21waf1/cip1, phospho-p42/44 (phospho-ERK1/2), p42/44 (total ERK1/2), cleaved poly-ADP ribose polymerase (PARP), cleaved caspases 3, 8, and 9, and the MEK inhibitor U0126 were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA). Blocking anti-Fas SM1/23 antibody was from Bender Medsystems (Burlingame, California, USA), and activating anti-Fas CH11 antibody was from Millipore (Billerica, Massachusetts, USA). Anti-tubulin antibody was from Sigma (St. Louis, Missouri, USA). Secondary anti-mouse and anti-rabbit horseradish peroxidase (HRP) antibodies were from Amersham (Buckinghamshire, UK) and secondary anti-rabbit conjugated to Alexa Fluor 594 (Molecular Probes/Invitrogen, Carlsbad, California, USA) was purchased from Invitrogen (Carlsbad, California, USA). Pan-caspase inhibitors z-VAD-FMK and z-DEVD-FMK were purchased from R&D Systems (Minneapolis, Minnesota, USA). Vybrant Apoptosis Assay Kit #2 and 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine (JC-1) were from Molecular Probes (Eugene, Oregon, USA), and propidium iodide was from Invitrogen. NA was isolated and purified from the Fijian sponge L. chagosensis as previously reported [13,14].
Tissue culture and cell treatments
A431, NR6, and HEK293 Epstein–Barr virus nuclear antigen 1 (EBNA) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS), and 1% penicillin, streptomycin, and glutamine and maintained in a 5% CO2 incubator at 37°C. Cells were plated at 1.5×106 cells per 60 mm dish in full media (10% FBS). The next day, cells were washed once with 1×phosphate-buffered saline (PBS) (pH = 7.4) and then starved for an additional 16–20 h in serum-free DMEM before treatment. All cell treatments with NA, U0126, UVB, z-VAD-FMK, z-DEVD-FMK, anti-Fas antibodies, and JC-1 were maintained in a 5% CO2 incubator at 37°C for the indicated times unless stated otherwise.
Western blot analysis
Cells were cultured as described above. Starved cells were treated with 30 μmol/l NA in serum-free DMEM for 2–12 h. In experiments using the MEK inhibitor U0126, A431 cells were pretreated with 10 μmol/l U0126 for 2 h before NA treatment. When EGF was used as a positive control, cells were treated with either 20 pmol/l or 2 nmol/l concentrations as indicated. The medium was removed and cells were lysed with 1×lysis buffer containing 50 mmol/l Tris (pH = 8.0), 150 mmol/l NaCl, 1% Nonidet P-40, 20 mmol/l NaF, 2 mmol/l sodium orthovanadate, 20 mmol/l β-glycero-phosphate, 1 μg/ml antipain, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin A, and 20 μg/ml phenylmethyl-sulfonyl fluoride. Protein concentrations were measured by the bicinchoninic acid assay (Pierce, Rockford, Illinois, USA). Equal protein aliquots were subjected to SDS–polyacryl-amide gel electrophoresis. Subsequently, proteins were transferred to Immobilon-P membranes (Millipore) in transfer buffer (50 mmol/l Tris base, 40 mmol/l glycine, 0.04% sodium dodecyl sulfate, 10% methanol) using an Owl semidry transfer apparatus from Thermo Scientific (Rochester, New York, USA). The membranes were blocked in 5% bovine serum albumin-Tris-buffered saline Tween-20 (TBST) [50 mmol/l Tris–HCl, 150 mmol/l NaCl, 0.05% Tween-20 (pH = 8.0)] and probed with rabbit antibodies to cleaved PARP, cleaved caspase 3, 8, 9 or p44/42 (phospho-ERK1/2 and total ERK1/2) at 1 : 1000 or with a mouse anti-tubulin antibody at 1 : 10 000 for 1 h at room temperature (RT), then washed with TBST and probed with anti-rabbit-HRP-conjugated secondary antibodies at 1 : 6000 or anti-mouse-HRP-conjugated secondary antibodies at 1 : 20 000 for 1 h at RT. The blots were washed with TBST and developed using ECL Plus (Amersham).
Ultraviolet irradiation
A bank of four fan-cooled sun lamps (FS20T12-UVB; National Biological Corporation, Twinsburg, Ohio, USA) emitting 4 W/m2 and ultraviolet (UV) C filter were used for all experiments as described earlier [17]. Cells were exposed to either 50 mJ/cm2 (4 min) or 100 mJ/cm2 (8 min) before the indicated incubation times. All UV exposures were carried out in PBS (pH = 7.4) at RT.
G1 cell cycle arrest and cell death
A431 cells were cultured as described above. Cells were starved for a period of 16 h followed by treatment with 1 or 30 μmol/l concentrations of NA for 12 h. In experiments using the MEK inhibitor U0126, A431 cells were pretreated with 10 μmol/l U0126 for 2 h before NA treatment. Cell death and G1 arrest were assessed using propidium iodide staining and fluorescence-activated cell sorter (FACS) analysis as follows. Suspended cells were collected and adherent cells were trypsinized with 0.5 ml of 0.25% trypsin. Cells were collected by centrifugation (1000 rpm, 4°C, 5 min), washed twice with 2 ml of ice-cold 1 × PBS (pH = 7.4) and resuspended in 1 ml of ice-cold PBS and kept on ice. Cells were fixed by the drop-wise addition of 2 ml of cold methanol (− 20°C) with gentle vortexing. The fixed cells were then incubated at 4°C for 12 h, collected by centrifugation and suspended in 1 × PBS for 5 min to rehydrate the cells. Cells were collected and 1 ml of freshly prepared propidium iodide solution (50 μg/ml propidium iodide, 10 μg/ml RNase A, 0.1% Triton-X-100 and diluted with PBS) was added to each sample, resuspended, and incubated in the dark at 4°C for 30 min followed by FACS analysis. FACS analysis was performed with a FACScan analyzer using CellQuest software (BD Biosciences, Franklin Lakes, New Jersey, USA) for data acquisition. Data obtained for cell cycle analysis were then modeled using ModFit software (Verity Software House, Topsham, Maine, USA).
Evaluation of apoptosis by annexin V staining
A431 cells were serum starved for 16 h followed by treatment with 0.5, 1, 5, 15, or 30 μmol/l NA for 12 h. Cells in suspension were combined with adherent cells that had been trypsinized with 1 ml of 1× trypsin and pelleted by centrifugation (1500 rpm for 5 min at RT). Cells were washed once in cold PBS and stained for detection of apoptosis using the Vybrant Apoptosis Assay Kit #2. Briefly, cells were stained with 5 μl recombinant annexin V conjugated to Alexa Fluor 488 and 1 μl propidium iodide (100 μg/ml) in annexin-binding buffer (10 mmol/l HEPES, 140 mmol/l NaCl, 2.5 mmol/l CaCl2, pH 7.4) for 15 min followed by FACS analysis.
Evaluation of apoptosis independent of the Fas death receptor
Serum-starved A431 cells were pretreated with the blocking antibody anti-Fas SM1/23 (1 μg/ml) for 2 h at 37°C. Pretreated cells were then either treated with the activating antibody anti-Fas CH11 (1 μg/ml) or NA (15 μmol/l) and incubated for 72 h at 37°C. Cells in suspension were collected and the remaining adherent cells were trypsinized with 0.5 ml of 0.25% trypsin and combined with the suspended cells. Cells were pelleted by centrifugation (1000 rpm for 5 min at 4°C), fixed and stained with propidium iodide as described above, followed by FACS analysis.
Evaluation of mitochondrial membrane potential
JC-1 staining was used to assess the disruption of the Δψm in A431 cells after NA treatment. Serum-starved cells were treated with NA (15 μmol/l) or UVB (100 mJ/cm2) for 12 h. Cells in suspension were collected and the remaining adherent cells were trypsinized with 0.5 ml of 0.25% trypsin, combined with the suspended cells, followed by the addition of 1 ml DMEM containing 10% FBS to quench the trypsin. Cells were collected by centrifugation (1000 rpm for 5 min at RT), washed once with 2 ml of 1×PBS (pH = 7.4). Each sample was suspended in 2 ml of complete medium (10% FBS) at 37°C followed by the addition of 5 μl of JC-1 (2.5 μg/ml) under dim light. The samples were thoroughly mixed by gentle vortexing followed by incubation in the dark at 37°C for 10 min. Cells were collected by centrifugation, washed once with 2 ml of PBS (pH = 7.4) and resuspended in 500 μl of PBS (pH = 7.4) followed by immediate FACS analysis.
Evaluation of caspase-dependent apoptosis
A431 cells were serum starved for 16 h and then pretreated with 100 μmol/l of the pan-caspase inhibitor z-VAD-FMK for 1 h followed by treatment with 15 μmol/l NA for 12 h or with 1 μg/ml of anti-Fas CH11 for 24 h. Cells in suspension were collected and the remaining adherent cells were trypsinized with 0.5 ml of 1× trypsin and combined with the suspended cells. Cells were pelleted by centrifugation (1000 rpm for 5 min at 4°C), fixed and stained with propidium iodide as described above, followed by FACS analysis.
Evaluation of caspase 3-dependent apoptosis
A431 cells were serum starved for 16 h and then pretreated with 100 μmol/l of the caspase 3-specific inhibitor z-DEVD-FMK for 2 h followed by treatment with 5 μmol/l NA for 12 h. NA-induced apoptosis was evaluated by annexin V staining as described above.
A431 xenografts and immunofluorescence
A431 xenografts were prepared by subcutaneous injection of 5 × 105 cells, suspended in 100 μl of DMEM, into both the left and right flanks of female nude athymic mice nu/nu (n = 7). Tumors were grown to approximately 6×6 mm in diameter (measured using calipers) before treatment by intraperitoneal injection with dimethyl sulfoxide (DMSO) (n = 2) or NA (n = 5). NA was dissolved in DMSO and administered at a dose of 10 mg/kg, once daily five times per week for 2 weeks. Tumor volumes were measured daily using calipers. After 14 days of DMSO or NA treatments, tumors from both flanks were excised (DMSO, n = 3 and NA, n = 10) and fixed in 10% formalin for 12 h.
Fixed tissues were embedded in paraffin as described earlier [18] followed by immunofluorescence analysis for cleaved caspase 3. Samples were deparaffinized in xylene and rehydrated in a 30–100% ethanol ddH2O. Antigen retrieval was performed by boiling the samples in 10 mmol/l citrate buffer, pH 6.0, in a microwave oven. The slides were then washed with PBS for 5 min at RT. Sections were blocked in 5% BSA in PBS for 30 min at RT in a humidity chamber. Sections were incubated with a primary antibody for cleaved caspase 3 in blocking buffer overnight in a humidity chamber at 4°C. Slides were washed with PBS and a secondary antibody conjugated to Alexa Fluor 594 (diluted in blocking buffer) was added to the samples for 30 min at RT. Slides were washed in PBS and then mounted with Prolong-Gold and cover slips. All slides were processed simultaneously, and all of the photomicrographs were performed with identical camera settings and exposure times to ensure uniformity. All images were obtained at RT with a fluorescence microscope (AX70; Olympus, Tokyo, Japan) that was equipped with the following objectives: ×4 (0.16 NA), ×20 (0.7 NA), ×40 (0.85 NA), ×60 oil (1.4 NA), and ×100 oil (0.5–1.35 NA). The images were digitally recorded by an AxioCam (Carl Zeiss MicroImaging Inc., Oberkuchen, Germany) and saved using the AxioVision program (Carl Zeiss MicroImaging Inc.) and imported into Illustrator (Adobe, San Jose, California, USA). Overlays of the immunofluorescence images were performed in Photoshop (Adobe). To quantify the percentage of cells positive for cleaved caspase 3, three fields from each tumor tissue section were randomly selected. Cells displaying red fluorescence were considered positive for cleaved caspase 3 and apoptosis.
Statistics
Data were subjected to unpaired one-tailed t-test statistical analysis using Prism (Graphpad software, San Diego, California, USA) and P values ≤ 0.05 (*) were considered statistically significant.
Results
Naamidine A induces apoptosis in tumor cells
During the course of our studies with NA-treated A431 epidermoid carcinoma cells, we noticed significant changes in cell morphology. These morphological changes, most notably cell rounding and detachment, are consistent with cells undergoing cell death or apoptosis [19]. To investigate the possibility of induction of apoptosis by NA, A431 cells were treated with 1 and 30 μmol/l concentrations of NA for 12 h. Cells were then fixed, incubated with the DNA stain propidium iodide, and subjected to FACS cell cycle analysis [20]. The percentage of potential apoptotic cells was determined by measuring the fraction of nuclei that contained a sub-diploid (sub-G1) population. Sub-G1 populations occurred after treatment with either 1 or 30 μmol/l NA (Fig. 1b and c). Quantitation of the sub-G1 populations indicated that when A431 cells were treated with 1 μmol/l NA, there was a slight induction of cell death (from 5 to 20%), and that when A431 cells were treated with 30 μmol/l NA, 80% of cells died (Fig. 1b). Propidium iodide staining, indicative of cell destruction, is a late stage marker for cell death, but is not a specific marker for programmed cell death/apoptosis. To assess whether death induced by NA was a result of programmed cell death, we performed annexin V staining, which detects exposed phosphatidyl serine, an early event in apoptosis [21]. A431 cells were treated with NA concentrations ranging from 0.5 to 30 μmol/l for 12 h, followed by staining with annexin V coupled to the fluorescent dye Alexa Fluor 488 (Fig. 1d). FACS analysis showed that A431 cells treated with 5, 15, and 30 μmol/l NA were positive for annexin V staining with 80% induction of apoptosis. The magnitude of this result correlates with the observed 80% cell death using propidium iodide staining, and indicates that NA induces cell death by apoptosis.
Naamidine A can induce G1 arrest, as well as apoptosis, in tumor cells
Our earlier data demonstrated that NA could induce G1 arrest in A431 cells [14]. We speculated that NA has the ability to induce both G1 arrest and apoptosis and that in our earlier experiments, the effect of NA on apoptosis was not observed because the experimental conditions were not optimized to measure apoptosis. To verify that under our new conditions NA still affected cell cycle progression, we measured the cell cycle profile in the cells that remained viable after 12 h of NA treatment. After 12 h of treatment with 30 μmol/l NA, FACS analysis showed a significant increase in the G1 phase cell population with a complete withdrawal of cells from the S phase (Fig. 2a). To further verify whether cells were arresting in the G1 phase of the cell cycle, we examined the expression of the G1 phase CDK inhibitor, p21. As a positive control, we treated A431 cells with a high dose of EGF (2 nmol/l) that has been previously reported to inhibit mitogenesis and induce p21 expression in A431 cells [22]. We found that EGF induced p21 expression, which was maintained out till our last time point of 6 h (Fig. 2b). In agreement with our cell cycle data, NA also induced p21 expression, which peaked at 2 h, and declined thereafter (Fig. 2b). Thus, under conditions where NA induces death in the majority of the cells, most of the living cells are no longer cycling, and have arrested in the G1 phase.
Fig. 2.

Naamidine A (NA)-induced G1 cell cycle arrest. (a) Fluorescence-activated cell sorter (FACS) cell cycle analysis after NA treatment of A431 cells for 12 h. When treated with 30 μmol/l NA, an increase in percentage of cells in the G1 phase of the cell cycle occurs with a concomitant withdrawal of cells in the S phase. The t-test statistical analysis was used to compare the changes in percentage G1 and percentage S phase populations to untreated control cells, which yielded a P ≤ 0.005 (*). (b) Western blot analysis of cell cycle inhibitor p21waf1/cip1 protein expression levels after NA (30 μmol/l) or epidermal growth factor (EGF) (2 nmol/l) treatment. Induction of p21 protein levels occurs within 1 h of NA treatment with maximum induction after 2 h. Full-length western blots are presented in Supplemental Figure S1.
Naamidine A promotes apoptosis through a MEK-ERK-independent mechanism
The molecular mechanism(s) by which NA exerts its antitumor activity(s) in vivo is unknown. Our prior work suggested that NA might cooperate with epidermal growth factor receptor (EGFR)-mediated signaling to promote ERK activation either directly or indirectly [14]. To investigate the possible involvement of ERK activity in the proapoptotic function of NA, we first sought to investigate the mechanism of NA-induced ERK activation in A431 cells. A431 cells were serum starved and then stimulated with either EGF or NA. EGF and NA each induced robust activation of ERK1/2 (Fig. 3a). Preincubation of cells with U0126, an inhibitor of MEK, the major activator of ERK kinase, completely blocked activation of ERK1/2 by either EGF or NA (Fig. 3b). This result indicates that MEK activity is absolutely required for NA to promote ERK activation in A431 cells, and suggests that NA is not a potent direct activator of ERK in intact cells.
Fig. 3.
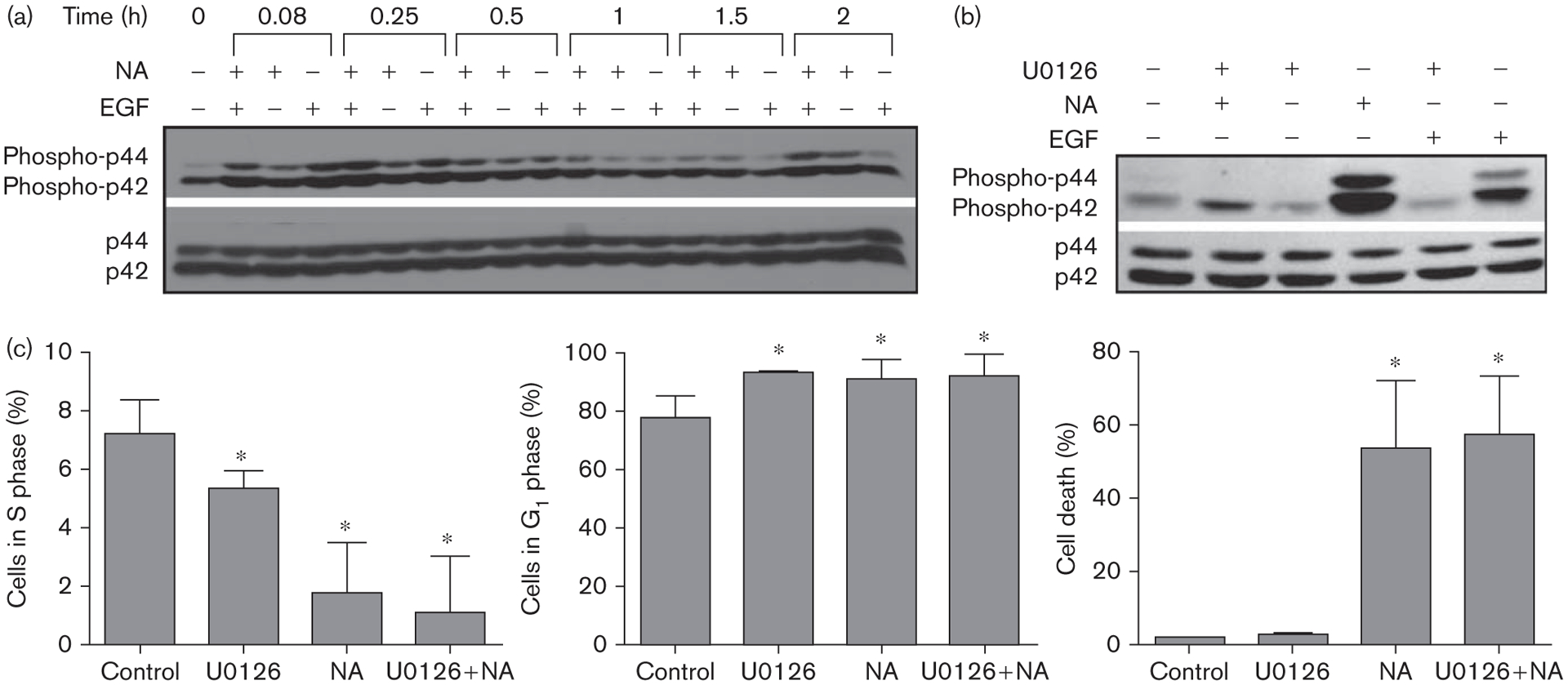
Naamidine A (NA) induces cell death independent of the MEK/extracellular signal-regulated kinase (ERK) pathway. (a) Western blot analysis of ERK1/2 activation in A431 cells treated with either 3 pmol/l epidermal growth factor (EGF) and or 30 μmol/l NA over a 2-h time course. NA induces ERK1/2 activation within 5 min that steadily increases over a 2 h period. (b) Western blot analysis of ERK1/2 activation in A431 cells pretreated with the MEK inhibitor U0126 (10 μmol/l) for 2 h before NA (30 μmol/l) or EGF (3 pmol/l) treatment. U0126 completely blocked ERK1/2 activation by both EGF (control) and NA. (c) A431 cells were pretreated with the MEK inhibitor U0126 (10 μmol/l) before NA (30 μmol/l) treatment followed by FACS cell cycle analysis. Assessment of the sub-G1 (cell death) phase of the cell cycle showed that a statistically significant 50% increase in cell death occurred in cells treated with NA (*P ≤ 0.005; right bar graph). Of the remaining viable cells, a statistically significant decrease in the S phase population occurred [*P ≤ 0.05; left bar graph; U0126 treatment (25%), NA treatment (71%), U0126 + NA treatment (71%)], as well as a statistically significant 16% increase in the G1 phase population (*P ≤ 0.05; middle bar graph). Full-length western blots are presented in Supplemental Figure S2.
To determine whether the MEK–ERK module is necessary for NA to promote apoptosis, we preincubated A431 cells with the same dose of MEK inhibitor (U0126) that completely blocks NA-induced ERK activation (Fig. 3b) before NA treatment, and examined both the cell cycle and apoptosis using FACS analysis (Fig. 3c). Despite its ability to block ERK activation, the MEK inhibitor did not impair the ability of NA to induce apoptosis. In fact, the MEK inhibitor alone caused A431 cells to undergo G1 arrest, indicating the MEK–ERK module is necessary for cell cycle progression of A431 cells, and that the MEK inhibitor is biologically and biochemically active during the 12-h period of the cell death studies. Thus, NA most likely acts through targets distinct from the MEK–ERK module to promote apoptosis.
Naamidine A induces apoptosis independent of the Fas death receptor
Apoptosis can be triggered through extracellular signal-mediated activation of an extrinsic signaling pathway, or direct intracellular activation of an intrinsic mitochondrial-signaling pathway. CD95, also called Fas, is a member of the tumor necrosis factor super family of death receptors, which drive extracellular signal-mediated apoptosis [23]. The binding of Fas ligand to Fas results in the formation of a complex between the Fas-associated death domain protein and pro-caspase 8. This interaction leads to the activation of caspase 8 and other caspases, and the subsequent induction of apoptosis [2]. To investigate the potential role of Fas in NA-induced cell death, A431 cells were pretreated with the anti-Fas-blocking antibody SM1/23 [24], before stimulation with NA. Preincubation of A431 cells with the anti-Fas-blocking antibody did not inhibit the ability of NA to induce cell death (Fig. 4). In contrast, when cell death was triggered with the anti-Fas-activating antibody CH11 [24], most of the cells died, but this cell death was completely suppressed by preincubation with the anti-Fas-blocking antibody (Fig. 4). Thus, A431 cells express Fas and all of the components necessary for Fas-dependent cell death, but NA induces cell death independently of this pathway.
Fig. 4.
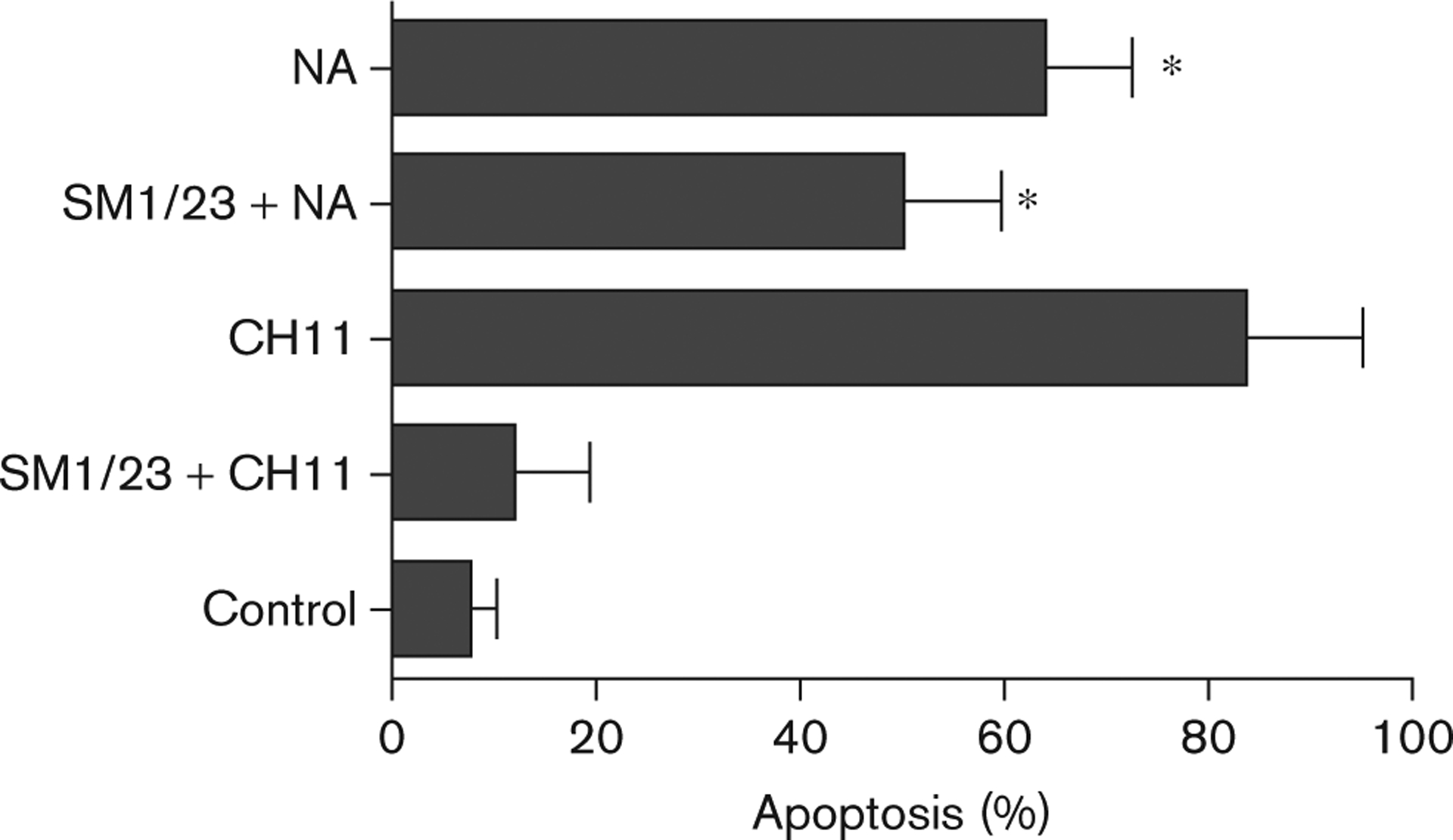
Naamidine A (NA) induces apoptosis independent of the Fas death receptor. A431 cells were pretreated with 1 μg/ml SM1/23 (blocking antibody) for 2 h before 1 μg/ml CH11 (activating antibody) or 15 μmol/l NA treatment for 72 h, followed by fluorescence-activated cell sorter analysis to assess the induction of apoptosis. SM1/23 does not block the induction of apoptosis induced by NA. The t-test analysis was used to compare changes in the percentage apoptosis among untreated cells, cells treated with SM1/23 + NA, and cells treated with NA alone, *P ≤ 0.002.
Naamidine A disrupts the mitochondrial membrane potential in tumor cells
As our data suggested that the activation of an extrinsic apoptotic pathway is not a key event triggered by NA, we speculated that NA-induced apoptosis might result from direct action on the mitochondria. Mitochondria play a pivotal role in regulating apoptosis by harboring proapoptotic factors such as cytochrome C [2,25]. The disruption of the Δψm leads to the release of these proapoptotic factors and is a key event in the induction of intrinsic apoptosis [26]. However, a decrease in Δψm can also be associated with necrosis [27]. To determine whether NA disrupts Δψm, we used staining with the mitochondrial-specific cationic dye JC-1 [28,29]. In healthy cells, JC-1 enters the mitochondrial matrix, aggregates, and emits strong red fluorescence. However, in apoptotic cells, the disruption of Δψm prevents JC-1 from entering and aggregating in the mitochondrial matrix. Instead, JC-1 accumulates in the cytosol as monomers and fluoresces green. Exposure of A431 cells to UVB light, a known inducer of intrinsic apoptosis in these cells, followed by JC-1 staining [30], caused a large increase in the percentage of green-fluorescing cells (Fig. 5d), thus validating the JC-1 assay for studying cell death in A431 cells. The treatment of A431 cells with 15 μmol/l NA caused a time-dependent shift from red fluorescence to green fluorescence (Fig. 5a–c). Cells treated with NA for 5 h showed an increase in green fluorescence from 5 to 20%, whereas at 12 h, 80% of cells were green-shifted. These results demonstrate that NA treatment leads to the disruption of Δψm, and that this disruption correlates with the induction of apoptosis by NA in A431 cells.
Fig. 5.

Naamidine A (NA) induced disruption of the mitochondrial membrane potential. (a) JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine) analysis showing the formation of J-aggregates (red fluorescence) in A431 untreated control cells as assessed by fluorescence-activated cell sorter analysis. (b) A431 cells treated with 15 μmol/l NA for 5 h resulting in a 25% increase in green fluorescence (JC-1 monomer). (c) A431 cells treated with 15 μmol/l NA for 12 h resulting in a 97% increase in green fluorescence (JC-1 monomer). There is a clear shift in fluorescence from red (J-aggregates) to green (JC-1 monomers) after NA treatment for 12 h, which is indicative of the disruption of the mitochondrial membrane potential. (d) A431 cells treated with NA show an increase in cell populations displaying green fluorescence as compared with untreated control cells. NA induces a 20% green shift within 5 h of treatment. However, within 12 h of NA treatment, there is an 80% green shift. The t-test statistical analysis was used to compare differences in the percentage green fluorescence between untreated cells and cells treated with NA for 12 h, which yielded a P ≤ 0.002 (*). UVB, ultra violet B.
Naamidine A induces caspase-dependent apoptosis in tumor and immortalized cells
Cleavage and activation of caspases are considered to be specific hallmarks of apoptosis [31]. During the induction of apoptosis, disruption of the Δψm and the subsequent release of cytochrome C is typically accompanied by cleavage and activation of caspases 3 and 9 [27]. To determine whether NA induces activation of these proapoptotic caspases, we used western blotting to monitor the appearance of the cleavage products associated with their activation. A431 cells were treated with 30 μmol/l NA over a 12-h time course. Cleavage of caspases 3, 8, and 9 occurred as early as 6 h and increased thereafter during the 12-h time course (Fig. 6a). To further verify that caspase-mediated apoptosis was occurring, we examined NA-treated A431 cells for the appearance of cleaved PARP, a well-known substrate of activated proapoptotic caspases [32]. Consistent with the NA-induced appearance of cleaved caspases truly reflecting caspase activation, PARP cleavage was also induced by NA treatment (Fig. 6a).
Fig. 6.

Caspase activation by naamidine A (NA)-induced apoptosis. (a) Western blot analysis of A431 cells treated with 30 μmol/l NA showing cleavage products of poly-ADP ribose polymerase (PARP) and caspases 3, 8, and 9. (b and c) Western blot analysis of HEK293 Epstein–Barr virus nuclear antigen-1 (EBNA) and NR6 cells showing cleaved caspase 3 and PARP. Full-length western blots are presented in Supplemental Figure S3. UVB, ultra violet B.
We earlier found that NA was antimitogenic in other cell lines, including EGFR-expressing NIH3T3 cells [13]. To determine whether the proapoptotic activity of NA also extends beyond A431 epidermoid carcinoma cells, we examined the ability of NA to induce caspase 3 activation in other cell lines. We found that NA can induce caspase 3 activation, as well as PARP cleavage, in HEK293 EBNA tumor cells, which carry transforming DNA from adenovirus type 5 and also express EBNA-1 (Fig. 6b). We also found that NA could induce activation of caspase 3 in the immortalized EGFR-negative variant of murine Swiss 3T3 cells, also called NR6 (Fig. 6c). These results suggest that the mechanism through which NA induces apoptosis is conserved across multiple cell types, and is not unique to A431 cells.
To determine whether activated caspases are the major drivers of apoptosis in cells exposed to NA, we performed FACS cell cycle analysis after pretreating A431 cells with an irreversible pan-caspase inhibitor. Pretreatment of A431 cells with the cell-permeable pan-caspase inhibitor z-VAD-FMK [27] completely blocked the control anti-Fas-activating antibody (CH11) and NA induction of apoptosis (Fig. 7a). To determine the role of caspase 3, a specific effector of the extrinsic and intrinsic apoptosis pathways, we pretreated A431 cells with the caspase 3-specific inhibitor z-DEVD-FMK [33] before NA treatment. Twelve hours after NA treatment, apoptosis was measured by annexin V staining (Fig. 7b). z-DEVD inhibited NA-induced apoptosis by almost 50%, suggesting that caspase 3 specifically plays a significant role in the induction of apoptosis by NA.
Fig. 7.
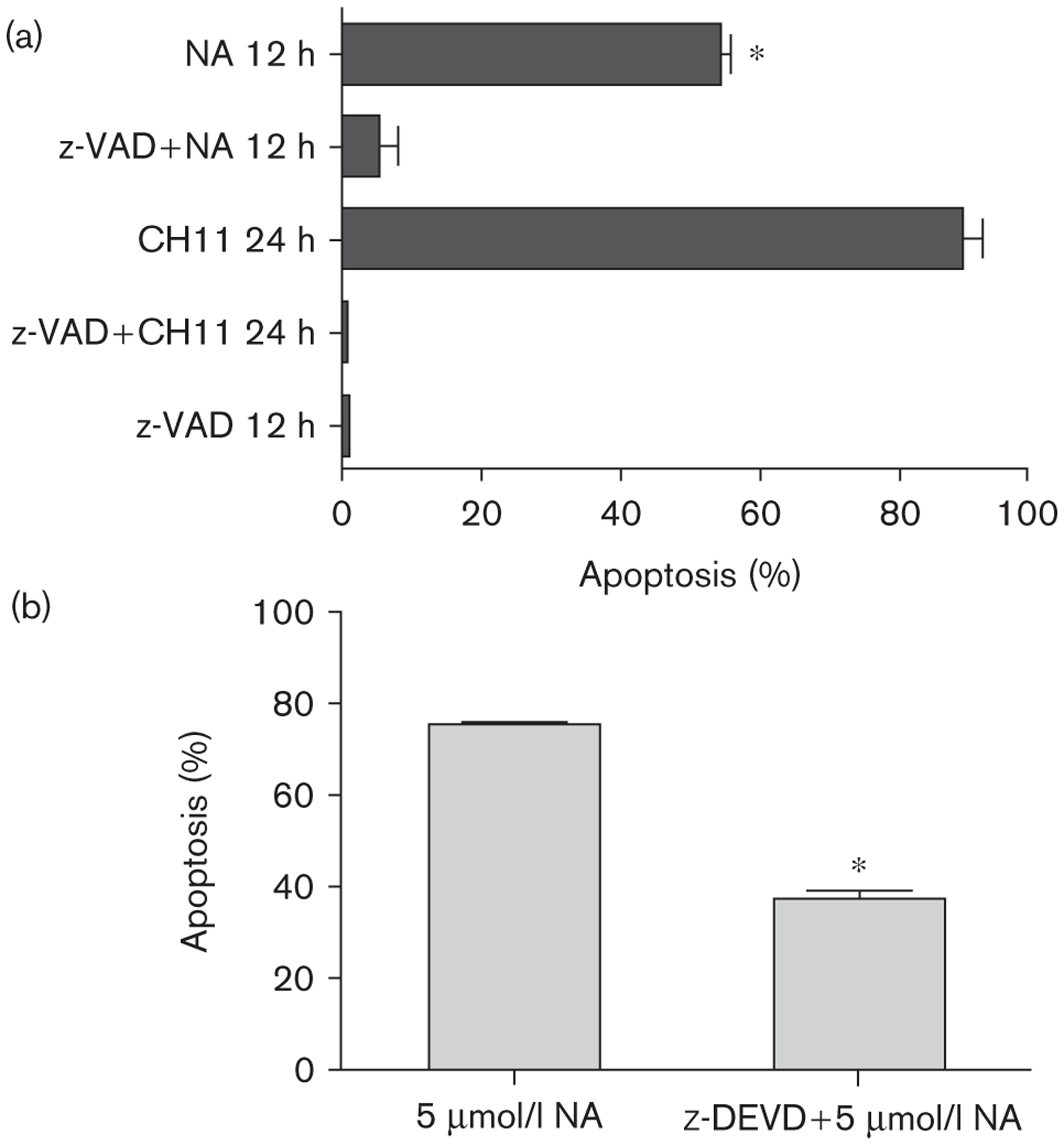
Caspase-dependent naamidine A (NA) induced apoptosis. (a) A431 cells were pretreated with 100 μmol/l z-VAD-FMK (pan-caspase inhibitor) for 1 h before treatment with 1 μg/ml CH11 (Fas-activating antibody) or 15 μmol/l NA. The activity of z-VAD-FMK completely blocked NA-induced apoptosis (measured by the presence of a sub-G1 population by fluorescence-activated cell sorter analysis) as compared with cells treated with NA alone (t-test; *P ≤ 0.002). (b) A431 cells were pretreated with 100 μmol/l z-DEVD-FMK (caspase 3 inhibitor) for 2 h before treatment with 5 μmol/l NA. The activity of z-DEVD-FMK blocked 40% of NA induced apoptosis (measured by annexin V staining) as compared with cells treated with NA alone (t-test; *P ≤ 0.0001).
Naamidine A induces caspase 3 cleavage in vivo
We have previously found that when NA was coadministered with injected A431 tumor cells in mice, NA could inhibit tumor xenograft growth by 87% at its maximum-tolerated dose [13]. To assess whether apoptosis could be part of the mechanism by which NA exerts its antitumor activity, we modified our previous xenograft study to maximize our ability to detect molecular evidence of apoptosis and to more accurately mimic a clinical situation. We first injected A431 tumor cells subcutaneously into the flanks of athymic nude mice. After tumors grew to approximately 6×6 mm, NA was administered intraperitoneally, once daily, five times per week for 2 weeks, at a dose of 10 mg/kg, which is lower than the maximal tolerated dose [13]. Under these conditions, despite the antitumor activity of NA, we expected to have sufficient tumor mass in the NA-treated samples for the examination of apoptotic markers. Tumor volume measurements were taken regularly. After 2 weeks of NA treatment, tumors were excised for immunofluorescence analysis. Tumors from NA-treated animals demonstrated significant staining for the activated cleaved form of caspase 3 (Fig. 8a), a necessary effector of NA-mediated apoptosis in vitro (Fig. 7b). Quantitation of the percentage of cleaved caspase 3-positive cells indicated that approximately 50% of tumor cells in NA-treated animals were apoptotic, a 10-fold increase compared with vehicle-treated control animals (Fig. 8b). Although this particular experiment was not optimized for a therapeutic assessment of NA, even under these conditions, NA-treated animals tended to have reduced tumor growth, especially between 8 and 12 days of treatment (Fig. 8c). These data are consistent with the in-vitro data and suggest that apoptosis is part of the antitumor activity of NA in vivo.
Fig. 8.
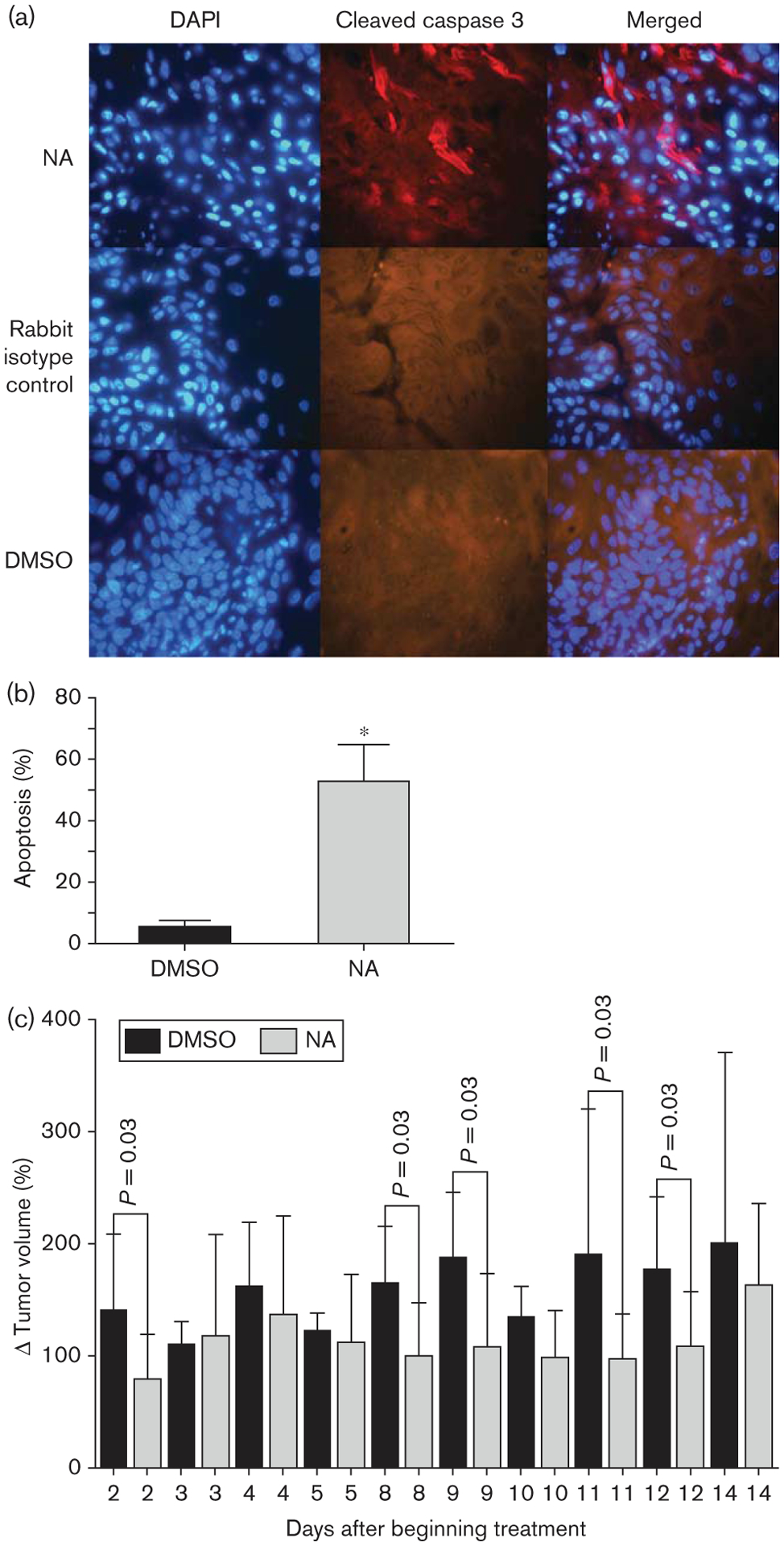
Naamidine A (NA) induced apoptosis in vivo. (a) A431 xenograft tumors were excised from athymic nude mice that had been treated with either 10 mg/kg NA (n = 10) for 2 weeks or with the vehicle dimethyl sulfoxide (DMSO) (n = 3), and were subjected to immunofluorescent staining for the proapoptotic marker cleaved caspase 3 (red). DAPI (4′,6-diamidino-2-phenylindole) staining was used to detect nuclei (blue), the middle panel represents tissues treated with a nonspecific rabbit isotype antibody as a control for the rabbit polyclonal anti-cleaved caspase 3 antibody. Representative photographs are shown. (b) Three fields were chosen randomly (100–200 cells total per field determined by counting nuclei stained with DAPI) from each tumor section and cells displaying red fluorescence were counted as positive for cleaved caspase 3 and apoptosis. The t-test statistical analysis was used to compare differences in the percentage of apoptotic cells between xenograft tumor tissues from mice treated with either NA or DMSO, which yielded a P value of 0.0005 (*). (c) Graphical representation of the percent change in tumor volume over time. These data indicate a trend where NA-treated mice tended to have less tumor growth as compared with DMSO-treated mice.
Discussion
Although the isolation of NA was first reported in 1989 [12], its antitumor activity was only reported in 1998 [13]. Furthermore, despite some exciting results in our initial preclinical study [13], such as preferential inhibition of EGF-dependent mitogenesis over insulin-dependent mitogenesis and an almost 90% inhibition of EGFR-expressing A431 tumor xenografts, few studies have been conducted to characterize its mechanism of action and potential clinical benefit. In this study, we confirm that one potential mechanism for the antitumor activity of NA likely stems from its ability to promote G1 arrest, as determined by FACS cell cycle analysis and by the induction of the p21waf1/cip1 CDK inhibitor (Fig. 2). We now find that NA is also a potent inducer of cell death in transformed and immortalized cells. The induction of cell death was established by the appearance of a sub-G1 population during FACS analysis, as well as a decrease in the Δψm (Figs 1 and 5). Several experimental results support the model that cell death likely occurs through an apoptotic mechanism, rather than necrosis. First, similar percentages of cells score positive for cell death by propidium iodide staining and annexin V staining (Fig. 1b and d). Second, NA induces the appearance of cleaved forms of the proapoptotic caspases 3, 8, and 9, as well as the caspase substrate PARP-1, all of which are associated with apoptotic caspase activation (Fig. 6). Finally, treatment of cells with either the pan-caspase inhibitor z-VAD-FMK or the caspase 3-specific inhibitor z-DEV D-FMK significantly blocks NA-induced apoptosis (Fig. 7). NA-induced apoptosis is likely caused by both intrinsic and extrinsic signaling pathways. NA activates both caspase 8 and caspase 9, which are the initiating caspases for the intrinsic and extrinsic pathways, respectively (Fig. 6) [34]. Although NA activates caspase 8, and the Fas-dependent apoptotic pathway is intact in A431 cells (Figs 4 and 6), NA-mediated apoptosis does not require Fas activation (Fig. 4). The SM1/23 anti-Fas-blocking antibody completely blocks anti-Fas CH11 antibody-stimulated apoptosis in A431 cells, but does not block NA-induced apoptosis. Thus, it is possible that NA treatment stimulates an alternative death receptor-mediated pathway, such as the tumor necrosis receptor pathway, which is known to be cytotoxic to A431 cells.
Evasion of apoptosis is a major clinical hurdle for effective cancer treatments. The most common loss of proapoptotic regulation is through the mutation or inactivation of p53 [4], which is estimated to be nonfunctional in 50% of all cancer types [35]. Significantly, our studies were conducted with the A431 cell line, which expresses a dominant-negative p53 mutant (R273H), which is one of the most frequently observed p53 mutants in human cancer [36,37]. Thus, NA-induced G1 cell cycle arrest and apoptosis are independent of p53 function, and could be beneficial for patients with inactive p53. Importantly, we find that NA also induces the appearance of the cleaved activated form of caspase 3 in tumor cells growing in vivo (Fig. 8a and b). Caspase 3 is the major effector caspase downstream of both intrinsic and extrinsic apoptotic pathways [34], and is necessary for almost half of the proapoptotic activity of NA in tumor cells grown in vitro (Fig. 7b). When mice harboring A431 tumor xenografts are treated with even a submaximal tolerated dose of NA, there is a 10-fold increase in the number of tumor cells expressing activated caspase 3 (Fig. 8b). Although our studies were optimized to detect potential apoptotic cells in tumors in vivo and not to assess the therapeutic potential of NA, NA treatment at this dose did cause a statistically significant reduction in tumor volume compared with vehicle alone at multiple time points (Fig. 8c).
The molecular details of how NA promotes G1 arrest and apoptosis are currently unknown. In A431 cells, NA is a potent activator of ERK1/2 (Fig. 3a) [14]. We also previously showed that NA could directly stimulate ERK activity in vitro [14]. However, through the use of the MEK inhibitor U0126 in intact A431 cells, we find that NA-mediated activation of ERK1/2 requires MEK activity. Thus, in vivo, the target(s) of NA is likely to be upstream of ERK. Although ERK1/2 can be activated by NA, the ERKs are not likely to be mediators of the growth arresting or apoptotic activity of NA. Under conditions where U0126 fully inhibits ERK activity, NA-treated cells still undergo growth arrest and apoptosis (Fig. 3b and c). Thus, it is possible that the NA target is in the EGFR pathway and that under some conditions, the interaction with NA can simultaneously promote both ERK activation and growth arrest/apoptosis. This model is consistent with our previous results showing cooperative effects of low doses of NA and EGF on ERK activation and inhibition of DNA synthesis [14]. The notion that the NA target can be regulated by EGF is also consistent with the observation that high doses of EGF, by itself, can promote growth arrest of A431 cells (Fig. 2) [22].
Finally, our in-vitro studies indicate that the proapoptotic activity of NA can be extended to other cell types including HEK293 human tumor cells and immortalized NR6 mouse fibroblasts (Fig. 6b and c). We do not believe that NA is generally cytotoxic, as our previous data indicate that in an in-vivo setting, at the maximum tolerated dose, there is still an almost 90% inhibition of tumor growth [13]. Thus, it is possible that immortalized and transformed cells might be more sensitive to the effects of NA than normal cells. Certain oncogenic or immortalizing mutations might cooperate with NA to enhance the growth arrest or apoptotic response. As our initial data indicated that in a mitogenesis assay, NA was a more potent inhibitor of EGF activity than insulin activity [13], and given the cooperativity between NA and EGF in regulating ERK activation and growth arrest, one of the sensitizers to NA could be the EGFR. However, the EGFR is unlikely to be the only major sensitizer, as NA induces caspase 3 cleavage in the EGFR-negative NR6 cells (Fig. 6c). To further explore this issue, it would be interesting to test whether lung cancer cells, which harbor activating mutations in the EGFR [38,39], display enhanced sensitivity to NA either alone or in combination with other drugs. For example, patients harboring such activating mutations initially respond to soluble EGFR kinase domain inhibitors, such as erlotinib and gefitinib [40], but overall survival is not dramatically increased [41], and patients tend to acquire resistance to the inhibitor when administered as mono-therapy [42]. Further studies will be aimed at characterizing the determinants of NA sensitivity and to explore potential therapeutic applications.
Acknowledgements
The authors thank Dr Wayne F. Green and H. Christopher Leukel, University of Utah flow cytometry core facility, for their help in collecting and interpreting flow cytometric data. This study was supported by NIH grants CA36622 (C.M.I.), AR050102 (D.G.), T32 Training Grant CA093247 (D.V.L. and K.M.), core facilities supported by P30CA042014, and Veterans Administration merit grant (S.K.K.). N.M. is the recipient of an investigator award from the Ontario Institute for Cancer Research.
Footnotes
Supplementary data
Supplementary data are available at The Anti-Cancer Drugs Journal online (www.anti-cancerdrugs.com).
References
- 1.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 2005; 5:876–885. [DOI] [PubMed] [Google Scholar]
- 2.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov 2002; 1:111–121. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008; 13:472–482. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57–70. [DOI] [PubMed] [Google Scholar]
- 5.Catassi A, Cesario A, Arzani D, Menichini P, Alama A, Bruzzo C, et al. Characterization of apoptosis induced by marine natural products in non small cell lung cancer A549 cells. Cell Mol Life Sci 2006; 63:2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagle DG, Zhou YD, Mora FD, Mohammed KA, Kim YP. Mechanism targeted discovery of antitumor marine natural products. Curr Med Chem 2004; 11:1725–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod 2003; 66:1022–1037. [DOI] [PubMed] [Google Scholar]
- 8.Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem 2008; 51:2589–2599. [DOI] [PubMed] [Google Scholar]
- 9.Butler MS. Natural products to drugs: natural product-derived compounds in clinical trials. Nat Prod Rep 2008; 25:475–516. [DOI] [PubMed] [Google Scholar]
- 10.Newman DJ, Cragg GM. Natural products from marine invertebrates and microbes as modulators of antitumor targets. Curr Drug Targets 2006; 7:279–304. [DOI] [PubMed] [Google Scholar]
- 11.Simmons TL, Andrianasolo E, McPhail K, Flatt P, Gerwick WH. Marine natural products as anticancer drugs. Mol Cancer Ther 2005; 4:333–342. [PubMed] [Google Scholar]
- 12.Carmely S, Ilan M, Kashman Y. 2-amino imidazole alkaloids from the marine sponge Leucetta chagosensis. Tetrahedron 1989; 45:2193–2200. [Google Scholar]
- 13.Copp BR, Fairchild CR, Cornell L, Casazza AM, Robinson S, Ireland CM. Naamidine A is an antagonist of the epidermal growth factor receptor and an in vivo active antitumor agent. J Med Chem 1998; 41:3909–3911. [DOI] [PubMed] [Google Scholar]
- 14.James RD, Jones DA, Aalbersberg W, Ireland CM. Naamidine A intensifies the phosphotransferase activity of extracellular signal-regulated kinases causing A-431 cells to arrest in G1. Mol Cancer Ther 2003; 2:747–751. [PubMed] [Google Scholar]
- 15.Pumiglia KM, Decker SJ. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A 1997; 94:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 1997; 17:5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cotter MA, Thomas J, Cassidy P, Robinette K, Jenkins N, Florell SR, et al. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin Cancer Res 2007; 13:5952–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones RG, Li X, Gray PD, Kuang J, Clayton F, Samowitz WS, et al. Conditional deletion of beta1 integrins in the intestinal epithelium causes a loss of Hedgehog expression, intestinal hyperplasia, and early postnatal lethality. J Cell Biol 2006; 175:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hacker G. The morphology of apoptosis. Cell Tissue Res 2000; 301:5–17. [DOI] [PubMed] [Google Scholar]
- 20.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, et al. Features of apoptotic cells measured by flow cytometry. Cytometry 1992; 13:795–808. [DOI] [PubMed] [Google Scholar]
- 21.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184:39–51. [DOI] [PubMed] [Google Scholar]
- 22.Johannessen LE, Knardal SL, Madshus IH. Epidermal growth factor increases the level of the cyclin-dependent kinase (CDK) inhibitor p21/CIP1 (CDK-interacting protein 1) in A431 cells by increasing the half-lives of the p21/CIP1 transcript and the p21/CIP1 protein. Biochem J 1999; 337 (Pt 3):599–606. [PMC free article] [PubMed] [Google Scholar]
- 23.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281:1305–1308. [DOI] [PubMed] [Google Scholar]
- 24.Dong C, Li Q, Lyu SC, Krensky AM, Clayberger C. A novel apoptosis pathway activated by the carboxyl terminus of p21. Blood 2005; 105:1187–1194. [DOI] [PubMed] [Google Scholar]
- 25.Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 26.Belizario JE, Alves J, Occhiucci JM, Garay-Malpartida M, Sesso A. A mechanistic view of mitochondrial death decision pores. Braz J Med Biol Res 2007; 40:1011–1024. [DOI] [PubMed] [Google Scholar]
- 27.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998; 60:619–642. [DOI] [PubMed] [Google Scholar]
- 28.Chaoui D, Faussat AM, Majdak P, Tang R, Perrot JY, Pasco S, et al. JC-1, a sensitive probe for a simultaneous detection of P-glycoprotein activity and apoptosis in leukemic cells. Cytometry B Clin Cytom 2006; 70:189–196. [DOI] [PubMed] [Google Scholar]
- 29.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A 1991; 88:3671–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohan S, Dhanalakshmi S, Mallikarjuna GU, Singh RP, Agarwal R. Silibinin modulates UVB-induced apoptosis via mitochondrial proteins, caspases activation, and mitogen-activated protein kinase signaling in human epidermoid carcinoma A431 cells. Biochem Biophys Res Commun 2004; 320:183–189. [DOI] [PubMed] [Google Scholar]
- 31.Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 2002; 9:459–470. [DOI] [PubMed] [Google Scholar]
- 32.McGinnis KM, Gnegy ME, Park YH, Mukerjee N, Wang KK. Procaspase-3 and poly(ADP)ribose polymerase (PARP) are calpain substrates. Biochem Biophys Res Commun 1999; 263:94–99. [DOI] [PubMed] [Google Scholar]
- 33.Schon M, Bong AB, Drewniok C, Herz J, Geilen CC, Reifenberger J, et al. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J Natl Cancer Inst 2003; 95:1138–1149. [DOI] [PubMed] [Google Scholar]
- 34.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol 2003; 15:725–731. [DOI] [PubMed] [Google Scholar]
- 35.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307–310. [DOI] [PubMed] [Google Scholar]
- 36.Walker DR, Bond JP, Tarone RE, Harris CC, Makalowski W, Boguski MS, et al. Evolutionary conservation and somatic mutation hotspot maps of p53: correlation with p53 protein structural and functional features. Oncogene 1999; 18:211–218. [DOI] [PubMed] [Google Scholar]
- 37.Kwok TT, Mok CH, Menton-Brennan L. Up-regulation of a mutant form of p53 by doxorubicin in human squamous carcinoma cells. Cancer Res 1994; 54:2834–2836. [PubMed] [Google Scholar]
- 38.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 39.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497–1500. [DOI] [PubMed] [Google Scholar]
- 40.Inoue A, Suzuki T, Fukuhara T, Maemondo M, Kimura Y, Morikawa N, et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006; 24:3340–3346. [DOI] [PubMed] [Google Scholar]
- 41.Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer-molecular and clinical predictors of outcome. N Engl J Med 2005; 353:133–144. [DOI] [PubMed] [Google Scholar]
- 42.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008; 14:2895–2899. [DOI] [PubMed] [Google Scholar]