

SUMMARY
Functional coupling between the amygdala and the dorsomedial prefrontal cortex (dmPFC) has been implicated in the generation of negative affective states; however, the mechanisms by which stress increases amygdala-dmPFC synaptic strength and generates anxiety-like behaviors are not well understood. Here we show that the mouse basolateral amygdala (BLA)-prelimbic prefrontal cortex (plPFC) circuit is engaged by stress and activation of this pathway in anxiogenic. Furthermore, we demonstrate that acute stress exposure leads to a lasting increase in synaptic strength within a reciprocal BLA-plPFC-BLA subcircuit. Importantly, we identify 2-arachidonoylglycerol (2-AG)-mediated endocannabinoid signaling as a key mechanism limiting glutamate release at BLA-plPFC synapses and the functional collapse of multimodal 2-AG signaling as a molecular mechanism leading to persistent circuit-specific synaptic strengthening and anxiety-like behaviors after stress exposure. These data suggest circuit-specific impairment in 2-AG signaling could facilitate functional coupling between the BLA and plPFC and the translation of environmental stress to affective pathology.
Keywords: 2-Arachidonoylglycerol, glutamate, prefrontal cortex, anxiety, cannabinoid, amygdala
eTOC blurb
Marcus et al. show that acute stress exposure induces a persistent collapse of multimodal endocannabinoid-mediated inhibition of a reciprocal BLA-plPFC-BLA glutamatergic subcircuit and that this collapse is causally related to the generation of stress-induced anxiety-like behaviors.
INTRODUCTION
Stress exposure is a major risk factor for the development and exacerbation of mental illnesses ranging from major depression to anxiety and substance use disorders (Arnsten, 2015; McEwen, 2012; Sharma et al., 2016). Furthermore, exposure to severe stress is required for the development of posttraumatic stress disorder (PTSD) (Fenster et al., 2018; Gillespie et al., 2009; Henigsberg et al., 2019; Kessler et al., 2005; Mark et al., 2018). In this context, understanding the molecular, cellular and circuit-level mechanisms by which stress exposure is translated into distinct pathological behavioral, emotional, and cognitive domains could have broad translational implications. Moreover, elucidating conserved molecular mechanisms linking stress to affective psychopathology could reveal novel therapeutic approaches to mitigate the adverse effects of stress on mental health. Although identification of a number of stress-regulatory neuromodulatory signaling systems has revealed potentially novel therapeutic targets for affective disorders, endogenous cannabinoid (eCB) signaling represents a leading drug-development candidate (McCormick et al. 2017; Hill et al., 2018; Hill and Patel, 2013; Lowe et al., 2018; Patel et al., 2017).
eCB signaling systems have been heavily implicated in stress-response physiology and pharmacological augmentation of eCB signaling has been suggested to represent a novel approach for the treatment of stress and trauma-related psychiatric disorders (Hill et al., 2018; Patel et al., 2017). At the synaptic level, 2-arachidonoylglycerol (2-AG)-mediated eCB signaling is a broadly expressed inhibitory retrograde signaling system. Specifically, 2-AG is canonically produced by postsynaptic neurons in an activity-dependent manner by diacylglycerol-lipase alpha (DAGLα) and activates presynaptic CB1 receptors to reduce neurotransmitter release probability (Kano et al., 2009; Stella et al., 1997; Wilson and Nicoll, 2001). 2-AG is degraded by monoacylglycerol lipase (MAGL) expressed within presynaptic terminals and glial cells (Dinh et al., 2004). Recent studies have implicated 2-AG signaling as a critical stress modulatory system and suggested 2-AG augmentation as a novel approach for the treatment of stress-related psychiatric disorders (Lisboa et al., 2017; Lutz et al., 2015). For example, 2-AG deficiency is associated with increased anxiety, impaired fear extinction, and increased susceptibility to stress-induced anxiety (Bluett et al., 2017; Cavener et al., 2018; Shonesy et al., 2014). Conversely, 2-AG augmentation promotes stress resilience and prevents stress-induced anxiety (Bedse et al., 2017; Bluett et al., 2017; Bosch-Bouju et al., 2016; McLaughlin et al., 2014; Patel et al., 2009; Qin et al., 2015; Sciolino et al., 2011; Sumislawski et al., 2011). Despite these data, the precise cellular and circuit-level mechanisms by which 2-AG interacts with environmental stress to affect emotional behavior are not well understood.
Studies over the past decade have elucidated distinct brain circuits connecting emotional and cognitive control centers in the modulation of stress responsivity, anxiety, and emotional regulation (Apps and Strata, 2015; McEwen et al., 2015; Tovote et al., 2015; Tye, 2018). For example, in humans, the dmPFC and amygdala show excitatory coupling during exposure to threatening stimuli, and enhanced activity in the amygdala-dmPFC circuit is correlated with subjective ratings of anxiety (Carlisi and Robinson, 2018; Cremers et al., 2010; Kim et al., 2011; Milad et al., 2009; Robinson et al., 2014). Consistent with these findings, activation of rodent basolateral amygdala (BLA) inputs to the prelimbic PFC (plPFC), a rodent homologue of the human dmPFC, generates anxiety-like behaviors and biases behavior toward fear responses in the face of uncertainty (Burgos-Robles et al., 2017; Felix-Ortiz et al., 2015; Senn et al., 2014). Recent studies have also demonstrated that BLA-plPFC glutamatergic synapses undergo presynaptic strengthening after stress exposure (Lowery-Gionta et al., 2018). Taken together, these data suggest enhanced amygdala-dmPFC (BLA-plPFC in rodents) coupling could represent a conserved circuit mechanism that translates stress exposure into anxiety-like emotional states. However, the molecular mechanisms subserving stress-induced strengthening of BLA-plPFC circuits and generation of anxiety-like behaviors after stress exposure are not known. Here we elucidate an eCB mechanism linking stress exposure to BLA-plPFC subcircuit-specific synaptic strengthening and persistent anxiety-like behavior.
RESULTS
The BLA-plPFC circuit is stress responsive and anxiogenic
In order to investigate the molecular mechanisms regulating BLA-plPFC connectivity and plasticity, we first verified that this circuit is engaged by exposure to stress (Figure 1a). Using in vivo fiber photometry, we observed that unpredictable foot-shock stress significantly increased presynaptic Ca2+ influx in BLA axon terminals innervating plPFC, time-locked to shock onset (Figure 1b,c). We next used in vivo single cell Ca2+ imaging to examine plPFC neuronal responses to foot-shock exposure. The bulk Ca2+ signal in the entire field of view was increased in response to shock exposure (Figure 1d,e), with subsequent single cell analysis revealing three distinct populations of neurons: stress excitatory (44.40%), stress inhibitory (38.43%) and stress non-responsive (17.16%) (Figure 1f,g). The peak excitatory response was greater in absolute magnitude than the peak inhibitory response (excitatory: |z| = 3.83, inhibitory: |z| = 2.62, p=0.0073 by two-tailed t test) and resultant average signal was excitatory (Z=1.09; Figure 1h), suggesting that stress-induced excitation of plPFC neurons predominates over inhibition. These data indicate that stress exposure engages the BLA-plPFC circuit and leads into enhanced activity of plPFC neurons.
Figure 1. Stress exposure activates an anxiogenic BLA-plPFC circuit.
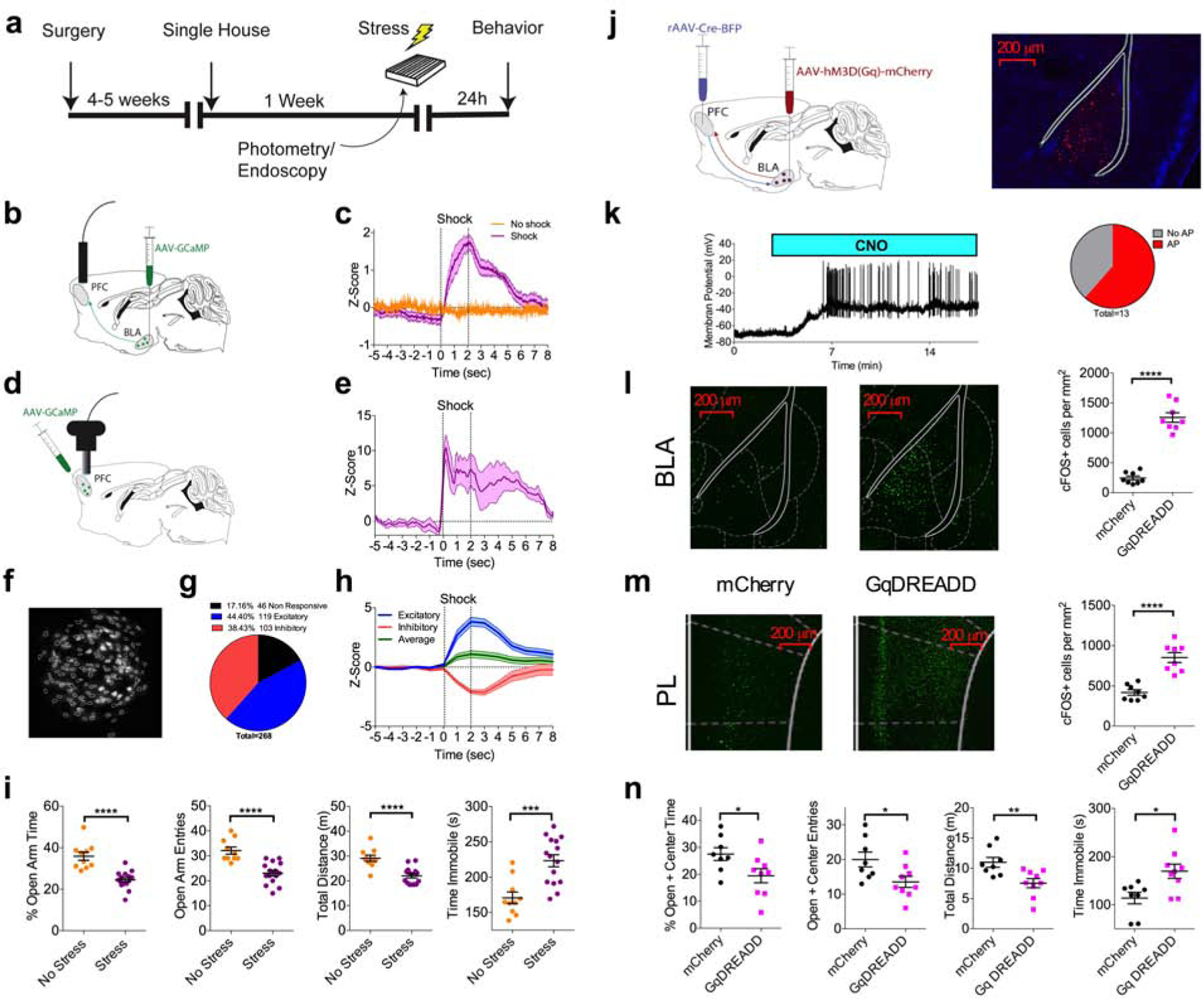
(a) Schematic and timeline for stress exposure and behavioral analysis.
(b) Schematic for in vivo fiber photometry recordings of BLA projections to the plPFC.
(c) Z-score of ∆F/F signal recorded from GCaMP6s expressing BLA terminals in the plPFC in response to a 2 second 0.5 mA foot shock. Traces represent mean of 4–5 mice, with each mouse trace derived from the average of 20 shock trials (Stress N=5, control N=4).
(d) Schematic for in vivo miniendoscopy-based Ca2+ imaging of plPFC neurons.
(e) Z-score of bulk ∆F/F recorded from total GCaMP7f signal in the field of view in response to a 2 second 0.5 mA foot shock. Trace represents mean of 4 mice, with each mouse trace derived from the average of 20 shock trials (N=4).
(f) Miniendoscopic maximal projection image of Ca2+ signal and extracted cell contours from representative mouse.
(g) Proportion of GCaMP7f expressing plPFC neurons displaying excitatory, inhibitory, or no responses to foot-shock (n=268 cells from N=4 mice).
(h) Z-score of excitatory (n=119), inhibitory (n=103), and averaged excitatory and inhibitory responses (n=222), across 1 second bins. Peak excitatory z-score=3.83, peak inhibitory z-score=−2.14, peak average=1.09.
(i) Effects of foot-shock stress on anxiety-like behavior in the elevated-zero maze (No Stress: N=10, Stress: N=15). % open-arm time (p<0.0001), open-arm entries (p<0.0001), total distance (p<0.0001), and immobility time (p=0.0003).
(j) Schematic of intersectional viral approach for hM3Dq (GqDREADD) expression in the BLA-plPFC circuit.
(k) Example trace and proportion of BLA neurons that displayed CNO-induced action potential firing (n=13, N=4).
(l) Representative images and quantification of cFOS expression in BLA neurons following in vivo administration of 10 mg/kg CNO (mCherry: N=8, GqDREADD: N=8). mCherry vs. GqDREADD p<0.0001.
(m) Representative images and quantification of cFOS expression in plPFC neurons following in vivo administration of 10 mg/kg CNO (mCherry: N=8, GqDREADD: N=8). mCherry vs. GqDREADD p<0.0001.
(n) Effects of Clozapine N-Oxide (5 mg/kg) on anxiety-like behavior in the elevated-plus maze (EPM)(GqDREADD: N=9, mCherry N=8). % open+center time (p=0.0378), open+center entries (p=0.0268), total distance (p=0.0067), and iimmobility time (p=0.0119).
All error bars represent ± SEM. “n” represents number of neurons, “N” represents number of mice. P values reported from two-tailed unpaired t-test (h,k,l,m)
Stress exposure is a ubiquitous risk factor for the development of anxiety disorders and stress exposure in rodents can model many psychopathological domains relevant to affective disorders. Indeed, 24 hours following foot-shock stress exposure, we observed an increase in anxiety-like behavior in the elevated-zero maze (EZM) (Figure 1i). To test whether a ‘stress-like’ state could be recapitulated through direct activation of the BLA-plPFC circuit we used an intersectional chemogenetic approach to specifically enhance the excitability of BLA neurons that project to the plPFC (Figure 1j). We used whole-cell current clamp electrophysiology to show that plPFC-projecting hM3D (GqDREADD)-expressing BLA pyramidal neurons fire action potentials following CNO application (Figure 1k). Administration of CNO induced robust cFOS expression in both the BLA (Figure 1l) and the mPFC (Figure 1m). Furthermore, although stress led to neuronal activation in the plPFC (Figure 1d,e), stress did not lead to a further increase in cFOS expression in the BLA (N=6, p=0.6418) or plPFC (N=6, p=0.0855) after GqDREADD activation of BLA-plPFC neurons, suggesting that stress and GqDREADD activation recruit overlapping BLA-plPFC neural circuits (data not shown). In the elevated-plus maze (EPM), chemogenetic activation of this circuit significantly enhanced anxiety-like behavior (Figure 1n). These data indicate the BLA-plPFC circuit is stress responsive and its activation is anxiogenic in mice, suggesting that stress-induced anxiety like behavior could be mediated via long-term enhancement of BLA-plPFC circuit function.
Stress exposure potentiates excitatory signaling in a BLA-plPFC reciprocal circuit
Our data thus far suggest enhanced BLA-plPFC circuit activity may be a relevant substrate for the translation of environmental stress into anxiety-like behaviors. To examine stress-induced synaptic adaptations within the BLA-plPFC circuit, we used a combination of anterograde ChR2-assisted projection-targeting and retrograde tracing approaches, followed by ex-vivo electrophysiology (Figure 2a–b). Four to six weeks after co-injection of AAV5-CaMKII-ChR2-eYFP (ChR2) and retrograde rAAV2-CAG-td-tomato (rAAV) into the BLA, mice were sacrificed for ex vivo electrophysiological studies. Monosynaptic BLA-originating, optically-evoked excitatory post-synaptic currents (oEPSCs) were recorded from rAAV-positive (rAAV+) and rAAV-negative (rAAV-) pyramidal neurons in both L2/3 and L5 in the plPFC (Figure 2 and S1a–c). Consistent with previous studies, we found BLA inputs made stronger synaptic connections onto reciprocally projecting rAAV+ plPFC neurons than non-reciprocally projecting rAAV-neurons (Figure S1d–i) (Figure S1d–i) (Little and Carter, 2013).
Figure 2: Stress exposure enhances glutamatergic signaling in the BLA-L2/3 plPFC reciprocal circuit.
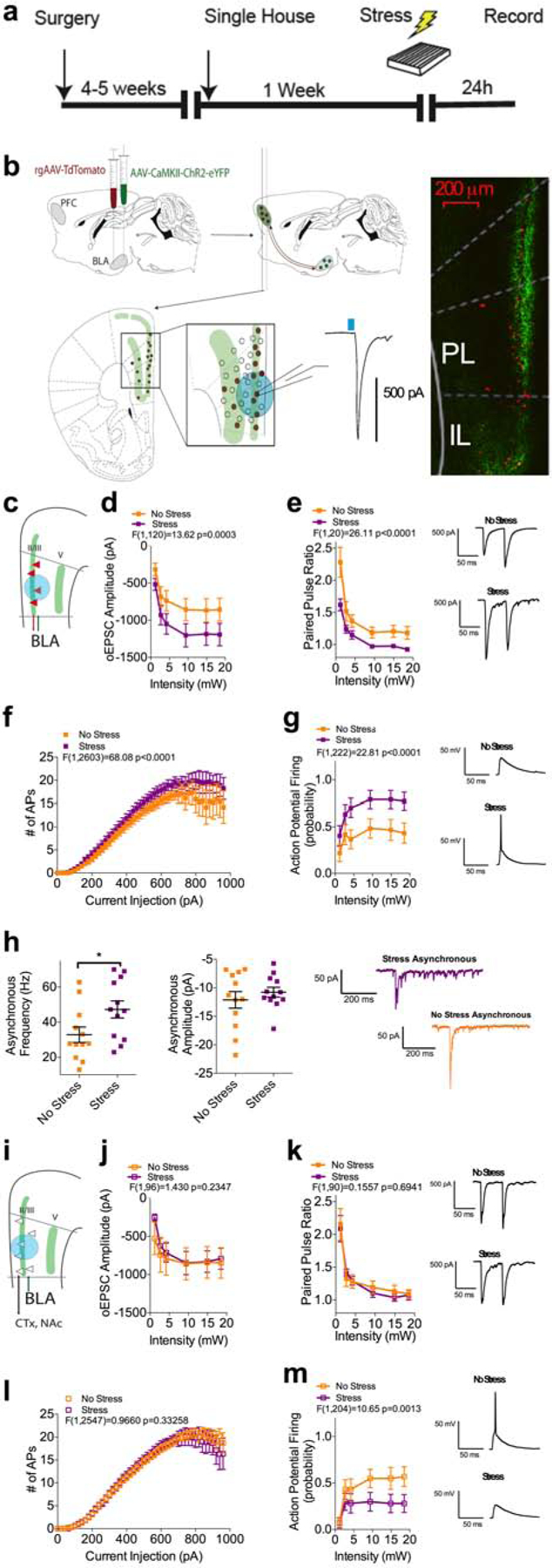
(a) Experimental timeline.
(b) Schematic for stereotaxic delivery of AAV5-CaMKII-ChR2(H147R)-eYFP and rAAV2-CAG-tdTomato into the BLA and recordings of oEPSCs from retrogradely labeled plPFC neurons.
(c) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV-positive (rAAV+) neurons.
(d) Optically-evoked input/output curve from L2/3 rAAV+ neurons from non-stressed (n=11; N=4) and stressed (n=11; N=4 mice) mice.
(e) Effects of stress on Paired Pulse Ratio (PPR) at BLA-L2/3 rAAV+ synapses (No Stress: n=11, N=4; Stress: n=10, N=4).
(f) Intrinsic excitability of L2/3 rAAV+ neurons following stress exposure (No Stress: n=37, N=12; Stress: n=33, N=14).
(g) Optically-evoked spiking in L2/3 rAAV+ neurons in non-stressed (n=20, N=7) and stressed mice (n=19, N=7).
(h) Effects of stress on asynchronous EPSC frequency (No Stress: n=12, N=4; Stress: n=12, N=5; p=0.0394) and amplitude (NS, p=0.4311) at BLA-L2/3 rAAV+ synapses.
(i) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV-negative (rAAV-) neurons.
(j) Optically-evoked input/output curve from L2/3 rAAV- neurons from non-stressed (n=9, N=4) and stressed (n=9, N=4) mice.
(k) Effects of stress on PPR at BLA-L2/3 rAAV- synapses (No Stress: n=8, N=4; Stress: n=9, N=4).
(l) Intrinsic excitability of L2/3 rAAV- neurons following stress exposure (No Stress n=35, N=11; Stress n=28, N=10).
(m) Optically-evoked spiking in L2/3 rAAV- neurons in non-stressed (n=17, N=7) and stressed mice (n=19, N=7).
All error bars represent ± SEM, “n” represents number of neurons, “N” represents number of mice. P values reported from two-tailed unpaired t-test (h). F and P values for two-way ANOVA shown in relevant panels.
24 hours after foot-shock exposure, we observed an increase in BLA-originating oEPSC amplitude and a decrease in the paired-pulse ratio (PPR) specifically in rAAV+ L2/3 plPFC neurons (Figure 2c–e), suggesting increased glutamate release probability within a reciprocal BLA-plPFC-BLA subcircuit after stress. This enhanced excitatory drive was accompanied by an increase in the probability of synaptically-driven action potential firing in rAAV+ L2/3 neurons, and a small but significant increase in the intrinsic excitability of these neurons (Figure 2f–g). We also observed an increase in the frequency, but not amplitude, of optogenetically-elicited asynchronous EPSCs (o-aEPSCs) onto L2/3 rAAV+ neurons, confirming an increase in presynaptic release probability at BLA-plPFC synapses 24 hours after stress exposure (Figure 2h). In contrast, there was no significant change in excitatory input (Figure 2j), PPR (Figure 2k), or somatically driven AP firing (Figure 2l) in L2/3 rAAV- neurons after stress. The only change observed in L2/3 rAAV- neurons was a significant decrease in optogenetically-elicited action potential firing (Figure 2m).
No stress-induced changes were found in L5 rAAV+ or rAAV- neurons (Figure S2a–h), suggesting both subcircuit- (BLA-plPFC reciprocal vs. non-reciprocal circuits) and laminar-specific synaptic strengthening occurs after acute stress exposure. Additionally, no stress-induced changes in either resting membrane potential or input resistance were found in any population of neurons (Figure S2i–t). Lastly, to determine the specificity of stress-induced synaptic strengthening in the plPFC, we examined whether projections from the mediodorsal thalamus (MDT) were modulated by stress exposure (Matyas et al., 2014; Vertes, 2004). We observed that stress did not cause persistent alterations at MDT to L2/3 or to L6 synapses, suggesting that there is selective enhancement of excitatory signaling in the BLA-L2/3 plPFC-BLA reciprocal circuit (Figure S2u–dd).
Similar effects were observed in male mice exposed to the predator odor 2-methyl-thiazoline (2MT) and in foot-shock exposed female mice, suggesting that stress induced enhancement of presynaptic glutamate release at BLA-plPFC-BLA synapses is not modality- or sex-specific (Figure S3a–c,e,f). We next determined whether stress induced presynaptic strengthening at BLA-L2/3 plPFC-BLA synapses was dependent on stressor intensity. Fiber photometry approaches revealed an increase in BLA-plPFC terminal activity at 0.25 but not 0.1 mA shock intensity (Figure S3h–j). Similarly, 24 hours later, we observed a trend towards increased oEPSC amplitude and significantly reduced PPR in the 0.25 mA, but not 0.1mA shock group, suggesting stress induced BLA-plPFC terminal activation and persistent strengthening both scale dynamically with the intensity of the stressor (Figure S3k,l). Lastly, to determine the duration of these synaptic modifications, we also performed electrophysiological recordings at 3 and 10 days following exposure to 20 0.5 mA foot-shocks. At 3 days post-stress, oEPSC amplitudes remained higher and PPR remained reduced, while these values were normalized 10 days post-stress, indicating that these stress-induced synaptic alterations are relatively persistent but not permanent (Figure S3n,o).
Endocannabinoid signaling broadly inhibits glutamatergic input from the BLA to the plPFC
Given that the observed stress-induced increase in BLA-plPFC glutamatergic transmission is mediated via enhanced presynaptic release, we next sought to determine the mechanism driving this effect. The retrograde acting eCBs, namely anandamide (AEA) and 2-AG, are known to be regulated by stress and regulate synaptic transmission in the PFC, raising the possibility that functional impairment in this neuromodulatory system could contribute to increased presynaptic drive at BLA-plPFC synapses after stress exposure (Katona and Freund, 2012; Lafourcade et al., 2007; Manduca et al., 2017; McLaughlin et al., 2014). To test this hypothesis, we first demonstrated that the cannabinoid receptor agonist CP55,940 robustly depressed BLA-evoked oEPSC amplitude in rAAV+ and rAAV- L2/3 plPFC neurons, indicating that BLA projections to the plPFC are broadly regulated by cannabinoid receptors (Figure 3a–b,e–f). To determine whether eCBs regulate BLA-plPFC glutamatergic transmission, we analyzed depolarization-induced suppression of excitation (DSE), a well characterized form of 2-AG mediated short-term synaptic depression, in which brief post-synaptic depolarization leads to 2-AG production and inhibition of glutamate release through binding to presynaptic CB1 receptors (Kano et al., 2009; Wilson and Nicoll, 2001). We found that DSE, induced by 10 second post-synaptic depolarization to +30mV, was expressed at both L2/3 rAAV+ and rAAV-neurons, and could be blocked by rimonabant, a CB1 inverse agonist, or DO34, an inhibitor of the rate-limiting enzyme in 2-AG biosynthesis, diacylglycerol lipase (DAGL) (Figure 3c,g). Interestingly, these two compounds alone induced a dramatic reduction in the PPR, suggesting tonic inhibition of glutamate release at BLA-plPFC synapses by 2-AG-CB1 signaling (Figure 3d,h). Similar results were found in L5 rAAV+ and rAAV- neurons, although DSE could not be elicited efficiently in L5 rAAV- neurons (Figure S4a–h). To ensure that the effect of rimonabant on PPR was due to blockade of tonic eCB signaling and not its inverse agonist properties, we also repeated this experiment with the neutral CB1 antagonist NESS0327 (NESS). Both NESS and rimonabant blocked DSE, reduced the PPR, and led to greater oEPSC amplitude, further supporting tonic eCB control over BLA-plPFC glutamatergic synapses (Figure S5j–k,l,m,p,q). Given that tonic eCB signaling has generally been ascribed to AEA, rather than 2-AG (Kim and Alger, 2010), we augmented our PPR experiments by demonstrating that both rimonabant and DO34 increased the frequency, but not amplitude, of asynchronous oEPSCs, confirming tonic 2-AG-CB1 signaling at these synapses (Figure 3i–k). These data provide the first evidence of broadly expressed multimodal tonic and phasic 2-AG-mediated regulation of presynaptic neurotransmitter release at BLA-plPFC synapses. To examine the input-specificity of eCB signaling in the plPFC, we performed the same key experiments while examining MDT projections to the plPFC. The MDT is known to have low CB1 expression (Herkenham et al., 1990), and as such, we observed little CP55,940-induced depression of oEPSC amplitude and no DSE, suggesting there is circuit-level specificity to eCB regulation of excitatory transmission in the plPFC (Figure S4i–k).
Figure 3: Phasic and tonic 2-AG signaling broadly regulate BLA-L2/3 plPFC glutamatergic synapses.
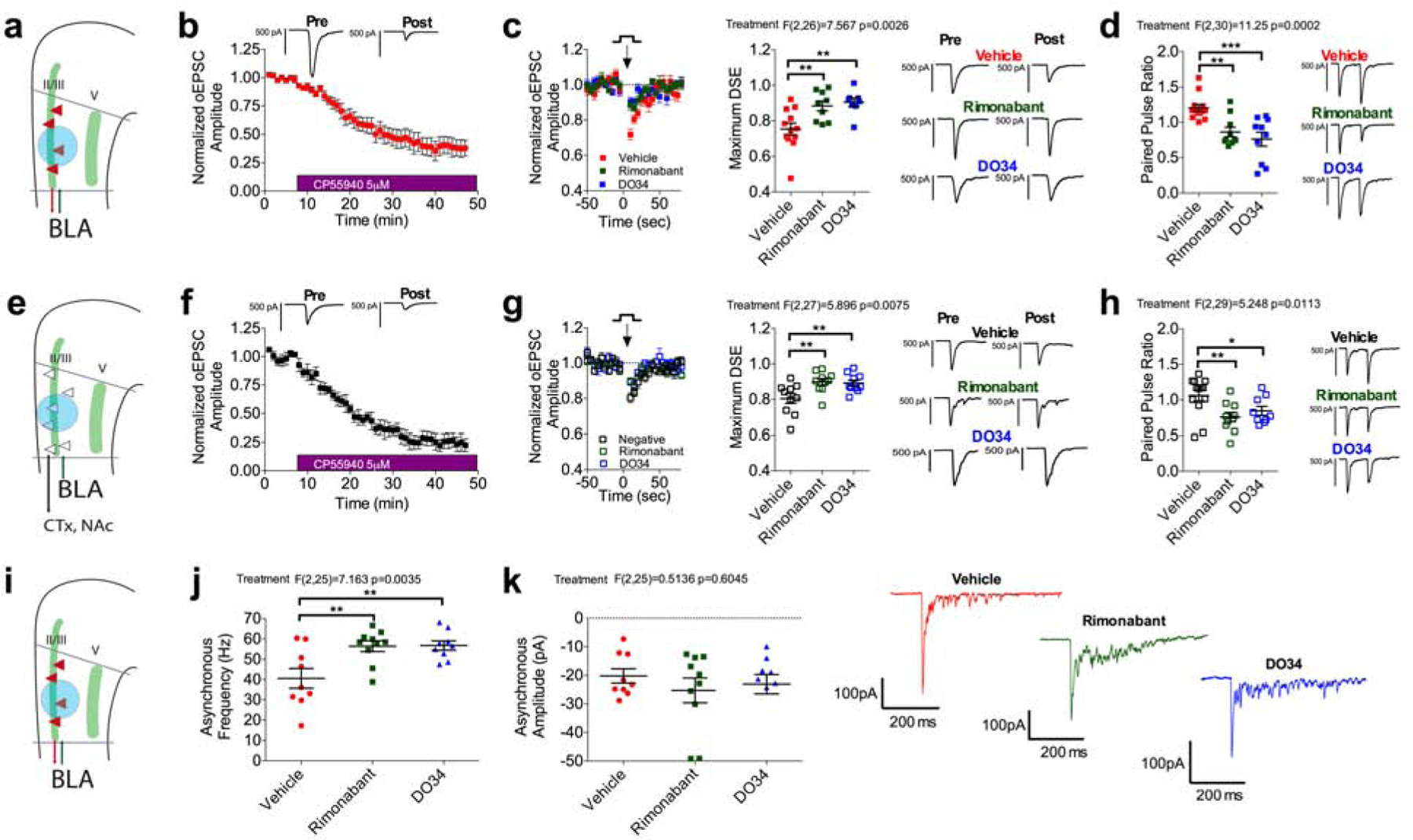
(a) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV+ neurons.
(b) Effect of 5 μM CP55,940 on oEPSC amplitude at BLA-L2/3 rAAV+ synapses (n=6, N=3).
(c) Depolarization-induced suppression of excitation (DSE) at BLA-L2/3 rAAV+ synapses (n=12, N=6) is blocked by 10 μM rimonabant (n=9, N=5; p=0.0049) and 2.5 μM DO34 (n=8, N=4; p=0.0039).
(d) PPR at BLA-L2/3 rAAV+ synapses (n=13, N=6) is reduced by 10 μM rimonabant (n=10, N=6; p=0.0034) and 2.5 μM DO34 (n=10, N=4; p=0.0002).
(e) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV- neurons.
(f) Effect of CP55,940 on oEPSC amplitude at BLA-L2/3 rAAV- synapses (n=5, N=4).
(g) DSE at BLA-L2/3 rAAV- synapses (n=10, N=5) is blocked by rimonabant (n=10, N=4; p=0.0095) and DO34 (n=10, N=4; p=0.0095).
(h) PPR at BLA-L2/3 negative synapses (n=13, N=5) is reduced by rimonabant (n=10, N=5; p=0.0084) and DO34 (n=9, N=4; p=0.0409).
(i) Schematic for voltage-clamp recordings of asynchronous EPSCs from L2/3 rAAV+ neurons.
(j) Asynchronous EPSC frequency at BLA-L2/3 rAAV+ synapses (n=9, N=2) is increased by rimonabant (n=10, N=2; p=0.0058) and DO34 (n=9, N=2; p=0.0058).
(k) Asynchronous EPSC amplitude at BLA-L2/3 rAAV+ synapses (n=9, N=2) is not altered by rimonabant (n=10, N=2; p=0.5392) or DO34 (n=9, N=2; p=0.5785).
All error bars represent ± SEM. “n” represents number of neurons, “N” represents number of mice. All post-hoc p values derived from one-way ANOVA with Holm-Sidak multiple comparisons (c,d,g,h,j,k). F and P values for ANOVA shown in relevant panels.
Stress impairs 2-AG inhibition of the BLA-plPFC reciprocal glutamatergic circuits
Following the observation that stress increased presynaptic release probability within the BLA-plPFC reciprocal circuit and that BLA inputs to the plPFC are highly regulated by 2-AG signaling, we next sought to determine whether stress-induced synaptic strengthening within this circuit was mediated via dynamic remodeling of BLA-plPFC 2-AG signaling. Using mass spectrometry, we observed that stress exposure decreased 2-AG levels in the mPFC 24 hours later, consistent with previous studies in other brain regions (Qin et al., 2015) (Figure 4a). Furthermore, bulk levels of arachidonic acid (AA), a primary degradative product of 2-AG hydrolysis, were also significantly reduced in the mPFC of stressed mice (Figure 4b). These data suggest that stress exposure could downregulate 2-AG synthesis in the mPFC, as levels of both 2-AG and AA are similarly reduced following stress exposure; in support of this, inhibition of 2-AG hydrolysis with the monoacylglycerol lipase (MAGL) inhibitor JZL184 increased 2-AG and reduced AA (Figure 4a–b). Importantly, elevating 2-AG levels with JZL184 reversed, while depleting 2-AG levels with DO34 exacerbated, stress-induced increases in anxiety-like behavior in the EZM, supporting the notion that 2-AG signaling can bidirectionally modulate stress-induced anxiety (Figure 4c). Lastly, we found the anxiolytic effects of JZL184 required CB1 receptor availability (Figure S5c).
Figure 4: Stress impairs 2-AG signaling within the BLA-L2/3 plPFC reciprocal circuit.
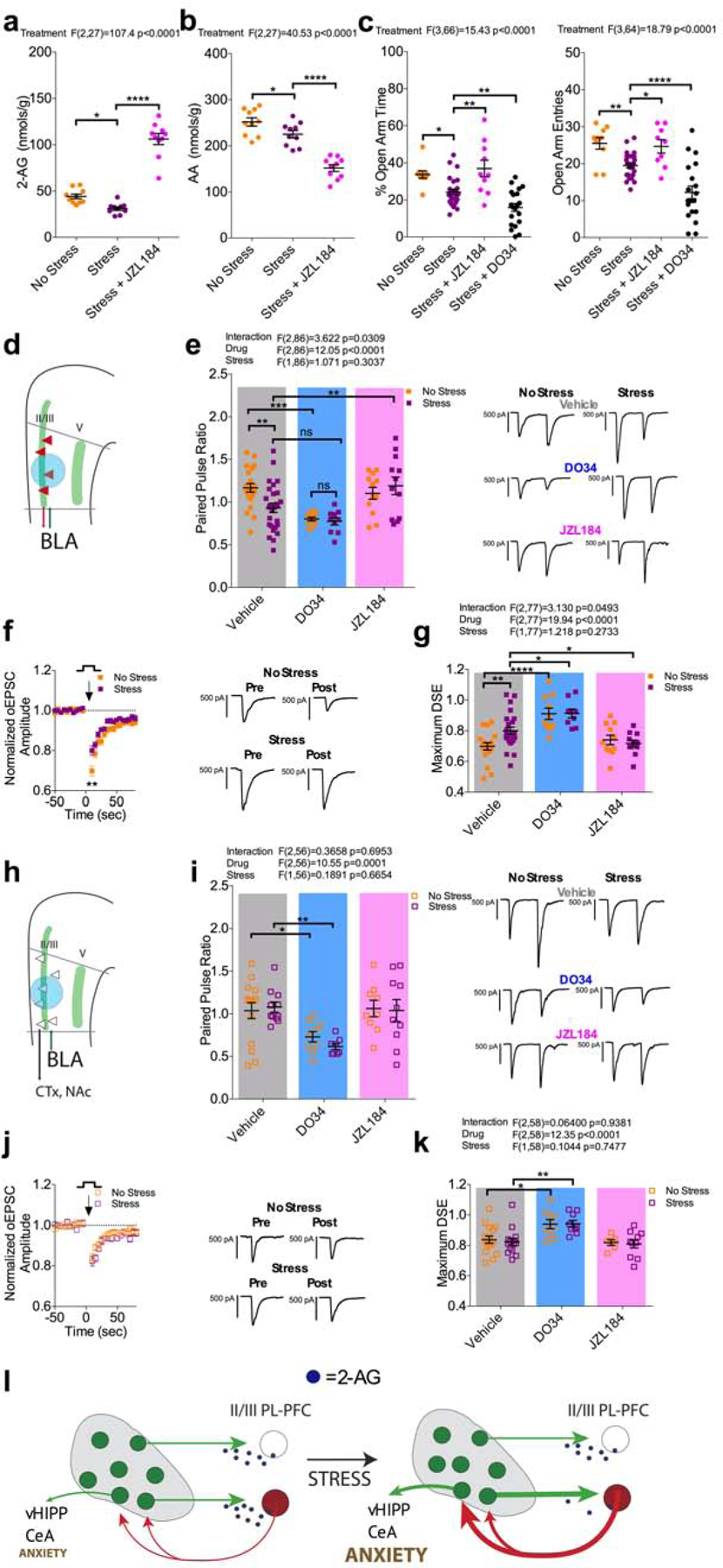
(a) Effect of stress and JZL184 on mPFC 2-AG levels. Stress exposure decreased mPFC 2-AG levels (No Stress: N=10, Stress: N=10; p=0.0233). Treatment with 15 mg/kg JZL184 (N=10) increased mPFC 2-AG levels in stressed mice (p<0.0001).
(b) Effect of stress and JZL184 on mPFC AA levels. Stress exposure decreased mPFC levels of AA (No Stress: N=10, Stress: N=10, p=0.0290). Treatment with 15 mg/kg JZL-184 (N=10) reduced mPFC AA levels in stressed mice (p<0.0001).
(c) Effect of stress on anxiety like behavior in the EZM. Stress exposure decreased % time spent in the open-arms (p=0.0110) and open-arm entries (p=0.0099; No Stress: N=10, Stress: N=10). 15 mg/kg JZL184 (N=10) reversed the stress induced decrease in % open-arm time (p=0.0012) and open-arm entries (p=0.0278). 50 mg/kg DO34 exacerbated the stress induced decrease in % open-arm time (p=0.0077) and open-arm entries (p<0.0001).
(d) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV+ neurons.
(e) Effect of stress, 2.5 μM DO34, and 1 μM JZL on PPR at L2/3 rAAV+ synapses. Stress exposure decreased PPR (No Stress: n=21 N=8, Stress: n=27, N=11; p=0.0053). DO34 decreased PPR in non-stressed mice (n=10, N=4; p=0.0008) but not in stressed mice (n=10, N=4; p=0.0904). JZL184 reversed the stress induced decrease PPR (n=13, N=4; p=0.0075) but did not affect PPR in non-stressed mice (n=12, N=4; p=0.4778).
(f) Effect of stress on DSE at BLA-L2/3 rAAV+ synapses.
(g) Effect of stress, DO34, and JZL184 on maximum DSE at BLA-L2/3 rAAV+ synapses. Stress exposure impaired DSE (No Stress: n=18, N=7, Stress: n=24, N=11; p=0.0072). DO34 blocked DSE in both non-stressed (n=10 N=4; p<0.0001) and stressed (n=9 N=4; p=0.0129) mice. JZL184 selectively reversed the stress-induced impairment of DSE (n=11 N=4; p=0.0312).
(h) Schematic for voltage-clamp recordings of oEPSCs from L2/3 rAAV- neurons.
(i) Effect of stress, DO34, and JZL184 on PPR at BLA-L2/3-rAAV- synapses. Stress exposure did not alter PPR (No Stress: n=15 N=8, Stress: n=11 N=7; p=0.9249). DO34 significantly decreased PPR in both non-stressed (n=9 N=4; p=0.0451) and stressed (n=8, N=4; p=0.0035) mice.
(j) Effect of stress on DSE at BLA-L2/3 rAAV- synapses.
(k) Effect of stress, DO34, and JZL184 on maximum DSE at BLA-L2/3 rAAV- synapses. Stress exposure did not alter DSE (No Stress: n=16 N=8, Stress: n=14 N=7; p=0.9476). DO34 blocked DSE in both non-stressed (n=9 N=4; p=0.0155) and stressed (n=9 N=4; p=0.0035) mice.
(l) Stress impairs 2-AG regulation of the BLA-L2/3 plPFC-BLA synapses, leading to strengthening of BLA-L2/3 reciprocal circuits involved in generating anxiety-like responses via activation of BLA output neurons to the ventral hippocampus for example.
All error bars represent ± SEM. “n” represents number of neurons, “N” represents number of mice. All post-hoc p values derived from one-way ANOVA (a,b,c) or two-way ANOVA (e,g,i,k) with Holm-Sidak multiple comparisons. F and P values for ANOVA shown in relevant panels.
We next tested the hypothesis that stress-induced strengthening of the BLA-plPFC reciprocal circuit was mediated by collapse of 2-AG-mediated inhibition of BLA-plPFC glutamatergic transmission. We found once again that stress increased presynaptic release probability selectively onto L2/3 rAAV+ neurons, as indicated by a decrease in the PPR 24 hours after stress exposure (Figure 4d–e). Interestingly, while bath application of DO34 reduced PPR in non-stressed mice, this effect was occluded in stressed animals. Furthermore, DO34 application occluded further stress-induced reductions in PPR. Conversely, pharmacological augmentation of 2-AG signaling via bath application of JZL184 was able to selectively rescue this stress-induced decrease in PPR at BLA-L2/3 rAAV+ synapses in a CB1-dependent manner (Figure 4e) and (Figure S5a). Lastly, a similar pattern was observed with regard to phasic 2-AG signaling, as stress reduced DSE magnitude in L2/3 rAAV+ neurons, an effect rescued by incubation with JZL184 (Figure 4f–g), and which was dependent upon stressor intensity and recovered by three days after stress exposure (Figure S3m,p). As observed with the PPR, the effect of JZL184 on DSE could be blocked by rimonabant pre-application (Figure S5b). No stress-induced changes were observed in L2/3 rAAV- neurons (Figure 4h–k) or L5 neurons (data not shown). These data suggest stress-induced collapse of phasic and tonic 2-AG signaling at BLA-plPFC synapses could contribute to stress-induced BLA-plPFC circuit strengthening and subsequent expression of anxiety-like behaviors (Figure 4l). Interestingly, female mice did not show a stress-induced reduction in DSE magnitude, indicating that stress effects on tonic vs. phasic 2-AG signaling could differ between sexes (Figure S3d). However, DSE was impaired by predator odor exposure, indicating that 2-AG signaling collapse is not stressor modality-specific (Figure S3g). To further support stress-induced functional collapse of 2-AG-CB1 regulation of excitatory input from the BLA, we demonstrated that stress occludes the ability of rimonabant and NESS to increase oEPSC amplitude (Figure S5n,r), and fully occludes the ability of NESS to decrease the PPR while partially occluding the ability of rimonabant to decrease the PPR at BLA-L2/3 plPFC rAAV+ synapses (Figure S5o,s).
Lastly, our mass spectrometry data indicated that AEA levels were also decreased in the mPFC following stress exposure (Figure S5d). This hints at the possibility that stress could also impair AEA regulation of the BLA-plPFC circuit. However, incubation with an inhibitor of AEA degradation, PF3845, affected neither basal PPR nor rescued the stress induced decrease in PPR or DSE magnitude, suggesting that AEA does not strongly regulate BLA-plPFC glutamatergic transmission (Figure S5e–g). Furthermore, stress exposure did not affect cannabinoid agonist-induced synaptic depression at BLA-L2/3 plPFC synapses, suggesting stress impairs 2-AG levels rather than affecting CB1 receptor sensitivity (Figure S5h,i).
Prelimbic DAGLα deletion increases BLA-plPFC synaptic strength and anxiety-like behavior
Our data thus far indicate that 2-AG plays a crucial role in limiting excitatory input from the BLA to the plPFC and that stress exposure compromises the efficacy of this signaling in a circuit-specific manner, contributing to synaptic strengthening after stress. These data suggest 2-AG signaling deficiency within the plPFC could contribute to the generation of anxiety-like behaviors via enabling enhanced BLA-plPFC glutamatergic coupling after stress. To address this hypothesis experimentally, we took advantage of conditional DAGLα knock-out mice (Bluett et al., 2017). We first demonstrated that stereotaxic injection of AAV-Cre into the plPFC of DAGLαf/f mice resulted in selective reduction in DAGLα protein in the plPFC but not the adjacent infralimbic PFC (ilPFC) (Figure 5a). Using plPFC-specific DAGLα knockout combined with the aforementioned injections of ChR2 and rAAV2-tdTomato into the BLA, we found significantly impaired DSE and a robust decrease in the PPR at BLA-plPFC glutamatergic synapses, recapitulating the synaptic phenotype observed after stress exposure (Figure 5b–d). Consistent with this stress-like synaptic phenotype, plPFC-specific DAGLα deletion elicited an anxiety-like behavioral phenotype that was similar to the anxiogenic effects observed after stress exposure (Figure 5e). This behavioral profile persisted following exposure to stress, as examined in a separate cohort of mice, suggesting a crucial role for plPFC 2-AG signaling in regulating anxiety-like behavior (Figure 5f). These data suggest that impaired plPFC 2-AG signaling results in a stress-like synaptic phenotype at BLA-plPFC glutamatergic synapses and is sufficient to induce anxiety-like behavior, recapitulating the effects of stress exposure (Figure 5g). Taken together our data support the hypothesis that stress-induced impairment in 2-AG signaling at BLA-plPFC synapses could represent an important mechanism translating the effects of stress into anxiety-like behavior.
Figure 5: plPFC-specific DAGLα deletion phenocopies synaptic and behavioral effects of stress.
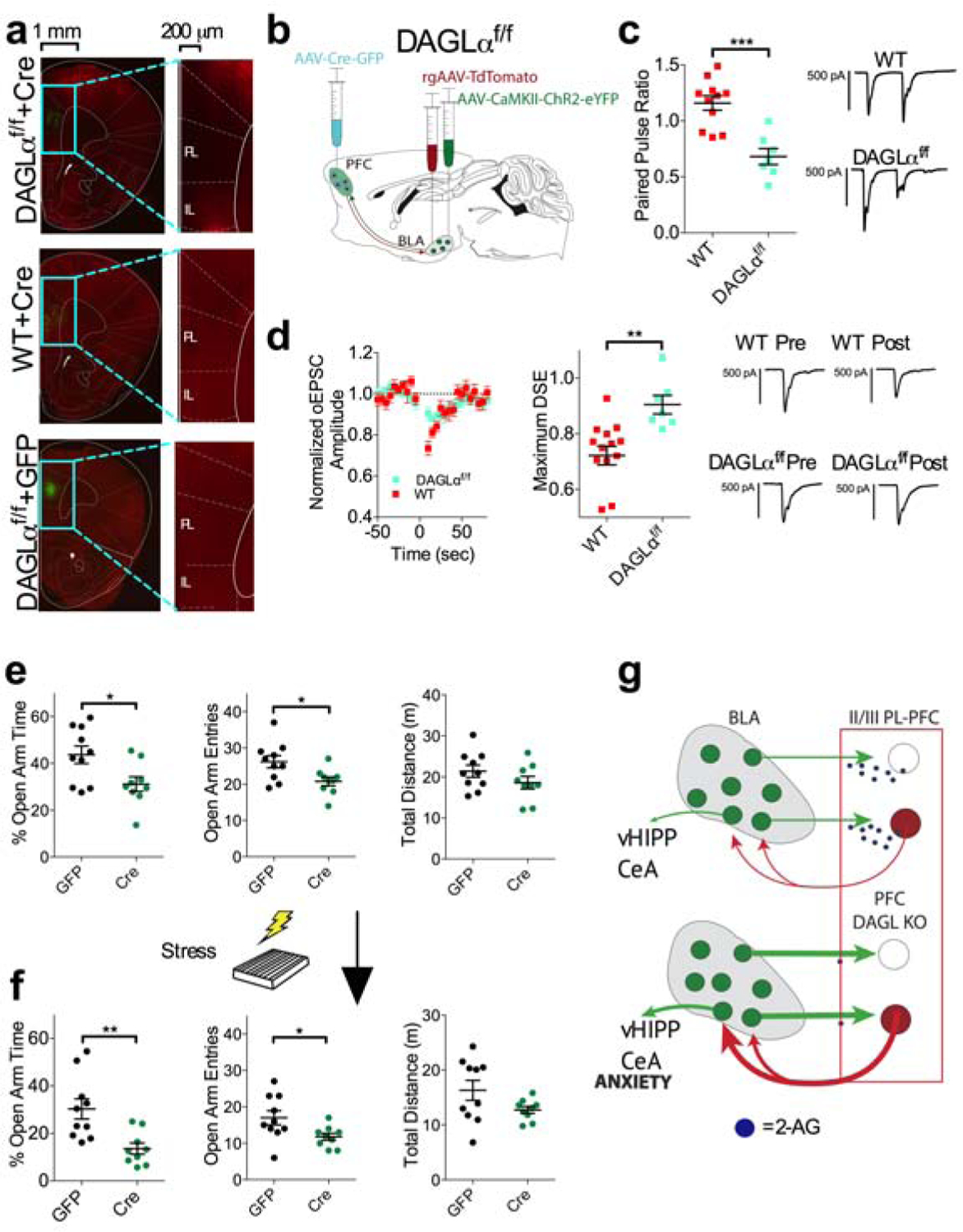
(a) Immunohistochemical validation of plPFC conditional knockout of DAGLα; AAV-Cre injection (green) and DAGLα expression (red).
(b) Schematic approach for electrophysiological examination of BLA-plPFC circuit after plPFC-specific DAGLα deletion.
(c) Effect of plPFC DAGLα deletion on PPR at BLA-L2/3 plPFC rAAV+ synapses (WT: n=11 N=5, DAGLαf/f: n=7 N=3; p=0.0002).
(d) Effect of plPFC DAGLα deletion on DSE at BLA-L2/3 plPFC rAAV+ synapses (WT: n=14 N=6, DAGLαf/f: n=7, N=3; p=0.0026).
(e) Effect of plPFC DAGLα deletion on anxiety-like behavior in the EZM. Mice with plPFC DAGLα deletion (n=9) show decreased % open-arm time (p=0.0213), decreased open-arm entries (p=0.0197), and similar distance traveled (p=0.2006) compared to GFP injected controls (N=10).
(f) Effect of plPFC DAGLα deletion on anxiety-like behavior in the EZM 24 hours following stress exposure. Mice with plPFC DAGLα deletion (N=9) show decreased % open-arm time (p=0.0039), decreased open-arm entries (p=0.0322), and similar distance traveled (p=0.0941) compared to GFP injected control (N=10) following stress exposure.
(g) Deletion of DAGLα from plPFC neurons induces a stress-like synaptic phenotype and results in increased anxiety-like behavior.
All error bars represent ± SEM. “n” represents number of neurons, “N” represents number of mice. P values reported from two-tailed unpaired t-test (c,d,e,f).
Circuit-specific CB1 deletion increases synaptic strength and stress-induced anxiety-like behavior
To further solidify the role of 2-AG-CB1 signaling specifically at BLA-plPFC synapses in the regulation of stress-induced anxiety, we utilized an INTRSECT approach to selectively delete the CB1 receptor from BLA neurons projecting to the plPFC (Fenno et al., 2017). Using a mouse in which exon 2 of Cnr1 is flanked by loxP sites (CB1f/f; Figure S6a–e), we injected a retrograde virus that drives the expression of Flp recombinase and td-Tomato into the plPFC and a virus that drives expression of Cre recombinase in a Flp-dependent manner into the BLA. Injection of both of these viruses into CB1f/f mice would be predicted to delete the CB1 receptor specifically from plPFC-projecting BLA neurons (Figure 6a,e). Using electrophysiological approaches, we first showed that this approach leads to almost complete functional removal of CB1 from BLA neurons projecting to the plPFC, as oEPSCs are completely insensitive to the cannabinoid agonist CP55,940 compared to wild-type mice injected with the same viral combinations (Figure 6b). The circuit-selectivity of this manipulation was verified by demonstrating intact cannabinoid receptor function at BLA-nucleus accumbens synapses after the BLA-plPFC-specific CB1 deletion (Figure S6f–h).
Figure 6: BLA-plPFC-specific CB1 deletion phenocopies stress-induced synaptic strengthening and exacerbates stress-induced anxiety.
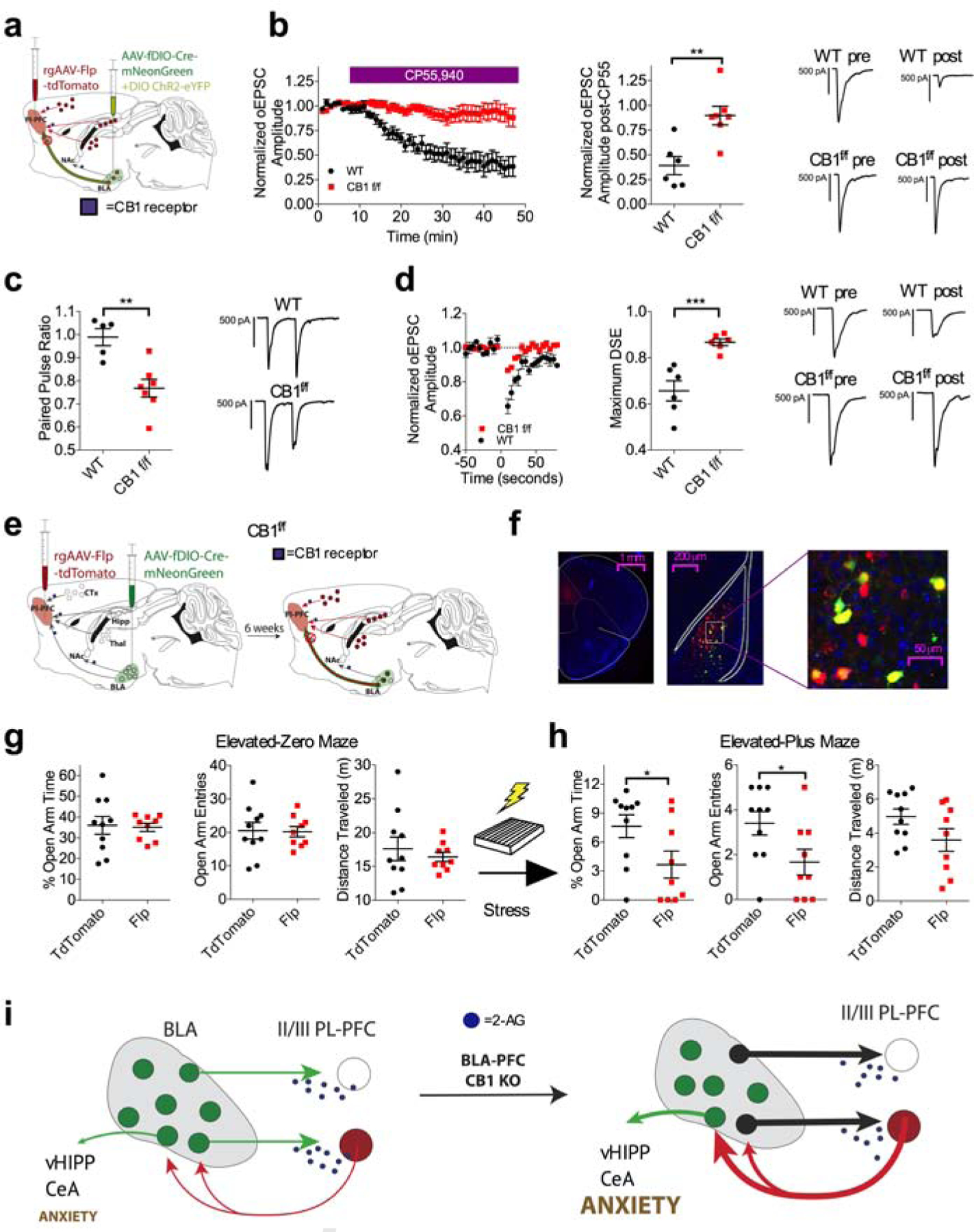
(a) Schematic for physiological validation of the INTRSECT approach for deletion of the CB1 receptor from plPFC projecting BLA neurons.
(b) Effect of 5 μM CP55,940 on oEPSC amplitude at BLA-L2/3 plPFC synapses. CP55,940-induced depression of oEPSC amplitude is dramatically attenuated in CB1f/f INTRSECT mice (n=7, N=3) compared to WT control INRSECT mice (n=6, N=2; p=0.0028).
(c) Effect of BLA-plPFC CB1 deletion on PPR at BLA-L2/3 plPFC synapses (WT: n=5, N=2; CB1f/f: n=7, N=3; p=0.0026).
(d) Effect of BLA-plPFC CB1 deletion on DSE at BLA-L2/3 plPFC synapses (WT: n=6, N=2; CB1f/f: n=6, N=3; p=0.0010).
(e) Schematic of INTRSECT approach for deletion of the CB1 receptor from plPFC projecting BLA neurons for behavioral characterization.
(f) Representative images showing injection site of rAAV2-EF1-Flp-tdTomato in the plPFC and AAV5-fDIO-Cre-mNeonGreen in the BLA, showing co-expression of rAAV+ neurons (red) and Cre+ neurons (green) in the BLA.
(g) Effect of BLA-plPFC CB1 deletion on anxiety-like behavior in the EZM in stress-naive mice. There were no significant differences in anxiety-like behavior in Flp injected CB1f/f mice (N=9) compared to Td-tomato injected CB1f/f mice (N=10).
(h) Effect of BLA-plPFC CB1 deletion on anxiety-like behavior in the EPM following stress exposure. Flp injected CB1f/f mice (N=9) show significantly decreased % open-arm time (p=0.0421) and decreased open-arm entries (p=0.0391), without changes in total distance travelled (p=0.1020) compared to control virus-injected littermates (N=10).
(i) Deletion of CB1 from BLA projections to the plPFC induces a stress-like synaptic phenotype and exacerbates the anxiogenic effects of stress exposure.
All error bars represent ± SEM. “n” represents number of neurons, “N” represents number of mice. P values reported from two-tailed unpaired t-test (b,c,d,g,h).
Similar to plPFC DAGLα deletion, we found that BLA-plPFC-specific CB1 deletion resulted in a synaptic phenotype comparable to that observed after stress exposure, evidenced by a lower PPR and reduced DSE magnitude (Figure 6c–d). We next determined the behavioral consequences of BLA-plPFC-specific CB1 deletion at baseline and after stress exposure (Figure 6e–f). Prior to stress exposure, BLA-plPFC-specific CB1 deletion did not affect anxiety-like behavior in the EZM compared to control virus (plPFC rAAV-td-Tomato combined with fDIO-Cre-mNeonGreen in the BLA) injected CB1f/f littermates (Figure 6g). However, deletion of CB1 from the BLA-plPFC pathway enhanced anxiety-like behavior 24 hours after foot-shock stress exposure in the EPM in the same cohort of mice, suggesting pivotal role for this CB1 population in regulating stress-induced anxiety like behavior (Figure 6h). Together, these data support our global hypothesis that stress-induced strengthening of BLA-plPFC circuits is mediated by circuit-specific impairment in 2-AG-CB1 signaling (Figure 6i).
DISCUSSION
The amygdala and dmPFC are crucial components of the negative valence system in humans and their functional connectivity is integral to threat reactivity (Carlisi and Robinson, 2018; Kalisch and Gerlicher, 2014; Robinson et al., 2014). Here we demonstrate circuit-specific 2-AG signaling collapse at BLA-plPFC synapses links environmental stress exposure to anxiety-like behavior in mice. Specifically, we found that stress caused a selective strengthening of glutamatergic transmission within a reciprocal BLA-plPFC-BLA subcircuit, which was driven by collapse of multimodal retrograde 2-AG-mediated eCB signaling. Importantly, experimentally-induced circuit-specific impairment in BLA-plPFC 2-AG-CB1 signaling phenocopied both stress-induced circuit strengthening and persistent anxiety-like behavior. These studies support the critical role of the BLA-plPFC circuit in the generation of anxiety-like states after stress and reveal an eCB-mediated mechanism subserving stress-induced BLA-plPFC circuit strengthening and behavioral adaptation. Our data are consistent with the notion that the amygdala and dmPFC comprise an “aversive amplification circuit” associated with elevated threat processing during stress (Robinson 2014) and the anxiogenic effects of BLA-plPFC circuit activation in mice (Felix-Ortiz et al., 2015; Lowery-Gionta et al., 2018). Taken together, these data suggest that aberrant activity and/or plasticity in this circuit could contribute to the pathogenesis of stress-related and affective disorders and that increased amygdala-dmPFC connectivity/signaling represents a conserved translationally relevant mechanism linking environmental stress to its behavioral, emotional, and cognitive consequences.
It is important to note that the BLA-plPFC circuit is not a closed loop. Indeed, both the BLA and plPFC send excitatory projections to brain regions that are directly involved with invigorating behavioral and motor responses. For example, BLA-projecting plPFC neurons, in addition to synapsing onto reciprocally projecting BLA-plPFC neurons, also strongly synapse onto open-loop BLA projection neurons innervating the ventral hippocampus and central amygdala (McGarry and Carter, 2017), providing a potential mechanism where by the BLA-plPFC-BLA reciprocal circuit gains access to limbic output structures to generate behavioral responses to stress. Supporting this hypothesis, optogenetic activation of BLA-ventral hippocampal projections increases anxiety and induces social avoidance in mice (Felix-Ortiz et al., 2013).
Although there is a prominent role of prefrontocortical eCB signaling in regulating presynaptic glutamate release and anxiety-like behaviors (Lafourcade et al., 2007; Lisboa et al., 2014; Lutz et al., 2015; Manduca et al., 2017; Puente et al., 2011; Rubino et al., 2008), whether this modulation is ubiquitous or circuit-specific is not known. Our data indicate that CB1 receptors and retrograde 2-AG signaling are present at BLA-plPFC but not MDT-plPFC synapses, suggesting that 2-AG regulates plPFC glutamatergic transmission in a circuit-specific manner. Interestingly, our data indicate that 2-AG modulates BLA-plPFC glutamatergic transmission in both phasic and tonic modes. Specifically, although prototypical phasic 2-AG signaling in the form of DSE is present at BLA-plPFC synapses and blocked by inhibitors of CB1 receptors and DAGL, both inhibitors also decrease the PPR and increase the frequency of BLA-elicited asynchronous EPSCs, revealing tonic inhibition of BLA-plPFC glutamatergic transmission by 2-AG-CB1 signaling. Take together with our data that BLA-plPFC-specific CB1 deletion and a conditional plPFC DAGLα deletion also decrease PPR, these data strongly indicate that 2-AG exerts both tonic and phasic presynaptic inhibition of BLA-plPFC glutamatergic transmission.
Given that that BLA projections to the plPFC are multimodally inhibited by 2-AG signaling and that some forms of stress impair eCB signaling (Bluett et al., 2017; Hill et al., 2009; McLaughlin et al., 2012; Patel et al., 2004; Rademacher et al., 2008; Wamsteeker et al., 2010), we next determined whether 2-AG signaling collapse could drive the stress-induced strengthening of BLA-plPFC glutamatergic synapses. In support of this hypothesis, we found impairment in both tonic and phasic 2-AG modulation of BLA to L2/3 plPFC reciprocally-projecting neuron synapses. This impairment was occluded by DAGL and CB1 inhibition and could be reversed by pharmacological inhibition of MAGL, which increases 2-AG levels, but not by inhibition of FAAH, which selectively increases AEA levels. Paralleling these data, systemic MAGL inhibition reduced, while DAGL inhibition increased, stress-induced anxiety-like behaviors. Given these findings, in tandem with our data demonstrating that activation of the BLA-plPFC circuit is sufficient to induce anxiety-like behavior, we suggest 2-AG-CB1 signaling collapse within the BLA-plPFC-BLA reciprocal circuit could represent a mechanistic link between stress exposure, BLA-plPFC synaptic strengthening, and stress-induced anxiety-like behaviors.
Previous studies have shown that prolonged stress exposure can compromise eCB signaling via reductions in CB1 receptor signaling and/or expression (Hill et al., 2005; Hillard, 2014; Patel et al., 2009; Wamsteeker et al., 2010). However, our acute stress manipulations did not affect CB1 receptor function at BLA-plPFC synapses. Therefore, the most likely mechanism subserving 2-AG signaling collapse after acute stress is a reduction in 2-AG signaling availability, which could occur via reduced 2-AG synthesis or enhanced degradation. Our data support an impairment in 2-AG synthesis over changes in degradation, as both 2-AG and free AA levels were reduced and only the maximal magnitude (but not recovery) of DSE was reduced in the PFC 24 hours after stress exposure. If increased 2-AG degradation were the primary driver of reduced 2-AG signaling, one would expect elevated levels of AA and a faster DSE decay (Pan et al., 2009; Zhong et al., 2014), neither of which were observed. However, additional mechanisms such as impaired 2-AG transport, or combinatorial effects of multiple mechanisms, cannot currently be excluded. Similarly, it is unclear from the present results what the initiating factors for 2-AG signaling collapse are. Although we hypothesize that the increase in excitatory input from the BLA, and potentially other brain regions, during the stressor serves as the trigger that leads to a persistent reduction in 2-AG signaling capacity in reciprocally projecting L2/3 plPFC neurons, this remains to be tested experimentally and the subsequent molecular cascades resulting in impaired 2-AG signaling remain to be established. In addition, our single-cell calcium imaging and cFOS data suggest broad plPFC neuronal activation after stress exposure, suggesting mechanisms must exist to guide the postsynaptic specificity of stress-induced 2-AG signaling impairment to BLA-plPFC-BLA reciprocal circuit, which likewise remain to be elucidated.
To investigate whether BLA-plPFC 2-AG signaling collapse is causally related to circuit strengthening and behavioral adaptations to stress, we used two different genetic strategies to impair BLA-plPFC 2-AG-CB1 signaling. We hypothesized that, because stress exposure reduces mPFC 2-AG levels and leads to anxiety-like behavior, then direct depletion of plPFC 2-AG, via plPFC-specific DAGLα deletion, would recapitulate stress-induced BLA-plPFC circuit strengthening and increase anxiety-like behavior. Consistent with this hypothesis, we found that plPFC DAGLα deletion increased release probability at BLA-plPFC synapses, impaired DSE, and elicited a corresponding increase in anxiety-like behavior, mimicking the effects of stress at the synaptic and behavioral level. Interestingly, a different behavioral pattern was observed following deletion of the CB1 receptor selectively from BLA neurons projecting to the plPFC. Although this circuit-specific manipulation again phenocopied stress-induced synaptic strengthening, the deletion had a minimal effect on basal anxiety, but increased anxiety-like behavior following stress exposure. A possible explanation for lack of robust behavioral effects of BLA-plPFC CB1 deletion under basal conditions is that 2-AG-CB1 signaling plays a minimal role in regulating the activity of the quiescent circuit. However, following stress-induced activation of the BLA-plPFC circuit, attenuation of 2-AG-CB1 negative feedback signaling impairs the ability of L2/3 plPFC neurons to reduce excitatory input from the BLA, leading to an exacerbation of stress-induced anxiety-like behaviors. The differential effects of plPFC DAGLα deletion and BLA-plPFC-specific CB1 deletion in terms of effect on basal anxiety could also be explained by the fact that plPFC DAGLα deletion impairs 2-AG signaling at all synapses (including other limbic inputs not examined here) resulting in a more robust behavioral phenotype. Future studies should be aimed at elucidating the roles of additional afferents and local eCB-sensitive circuits in the regulation of stress adaptation and anxiety-like behavior.
Here we explored the neurobiological substrate by which stress exposure is translated into anxiety-like behavior and identified collapse of 2-AG-CB1 signaling within a reciprocally connected BLA-plPFC-BLA circuit as a molecular mechanism subserving stress-induced circuit strengthening and anxiety-like behavior. These data suggest that the enhancing 2-AG-CB1 signaling, via MAGL inhibition for example, could represent an attractive therapeutic approach for the treatment of stress-induced psychiatric disorders (Chanda et al., 2019; Lisboa et al., 2017; Patel et al., 2017). Furthermore, our data suggest that analysis of functional connectivity in the amygdala-dmPFC circuit could represent a useful and conserved circuit-based biomarker bridging preclinical studies to MAGL inhibitor efficacy trials and facilitate optimal patient selection for future clinical studies.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sachin Patel (Sachin.patel@vanderbilt.edu).
All unique/stable reagents generated in this study are available to academic researchers from the Lead Contact, but may require completion of a Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committees and were conducted in accordance with the National Institute of Health guidelines for the Care and Use of Laboratory Animals. 8–14 week-old male and female C57BL/6J mice obtained from Jackson Labs were used for electrophysiological and behavioral experiments. 8–14 week-old male DAGLαf/f and WT controls were used for electrophysiological and behavioral experiments in figure 5. 10–18-week old male CB1f/f and WT controls were used for electrophysiological and behavioral experiments in figure 6. Mice were housed in a temperature and humidity-controlled housing facility under a 12h light/dark cycle with ad libitum access to food. All behavioral and physiological experiments were run on littermate-matched stressed or non-stressed mice that had been singly housed ≥1 week. DAGLαf/f and CB1f/f mice bred in house on a homozygous x homozygous breeding scheme, and littermate-matched controls were used for all behavioral experiments.
Littermate-matched mice were randomly assigned to treatment (e.g. stress vs. no stress or virus vs. control virus) for all behavioral and electrophysiological experiments performed, excluding the physiological validation for the DAGLαf/f and CB1f/f knockouts. For behavioral cohorts, we used a homozygous/homozygous-breeding scheme, as preliminary reports from our lab and others that suggest that the CB1f/f mice have a basal phenotype in various behavioral assays that differentiates them from their wild-type littermates. Therefore, we used a homozygous/homozygous breeding scheme to generate only CB1f/f homozygotes, so that both the experimental and control groups had the same genetic background. However, this breeding scheme precluded our ability to perform physiological validation on littermate-matched controls, due to the fact that Cre expression would always lead to CB1 deletion in addition to allowing for expression of DIO ChR2. Therefore, we used non-littermate matched WT controls for physiological validation to determine whether the CB1 receptor or DAGLα was functionally deleted in the experimental group.
Generation of CB1f/f mice.
To produce a conditional knockout mouse of the cannabinoid receptor 1 (Cnr1 gene), we created a targeting construct centered around exon 2, the single coding exon of the gene (Figure S7). A first loxP site was inserted 141 bp upstream of the exon, whereas a DNA fragment containing a neomycin-resistance gene cassette flanked by frt and loxP sites was inserted 622 bp downstream of exon 2. Relatively large arms of recombination: 6.5 kb (5’ end) and 3.1 kb (3’ end) were then added to the construct (Figure S7). The cnr1 construct was electroporated into 129/SvEvTac embryonic stem cells and 540 neomycin resistant clones were picked and analyzed by Southern blot analysis. Genomic DNA was digested with MfeI, run on agarose gel, and transferred to nitrocellulose membranes to identify a 16 kb control fragment (wild-type allele) and a 7 kb mutant fragment (targeted allele) by Southern blot analysis. Out of the 540 clones, three clones were identified containing the mutant allele (Figure S7). Presence of the three loxP sites in ES cell clone 3H3 was verified by PCR and sequencing. The ES cell clone was injected into C57BL6 blastocysts which were implanted into pseudo-pregnant females to produce cnr1 chimeric mice. Three chimeras were produced from the 3H3 clone and one of these chimeras went germline and produced 9 pups, five of which carried the 3 loxP allele. After 2 backcrossing into C57BL6/J mice, the neomycin-resistance gene cassette was successfully removed by breeding a 3 loxP male mouse with two females carrying a FlpE allele (Figure S7). Functionality of the remaining 2 loxP sites was demonstrated by crossing a male carrying an E2a-CRE allele with two females carrying a cnr1 2 loxP allele. Seven out of 16 pups carried the E2a-CRE allele and out of them, four pups demonstrated successful elimination of exon 2 (Figure S7). The Cnr1 line was then backcrossed to C57BL/6J mice for an additional 5–6 generations prior to breeding to homozygosity.
DAGLf/f mice.
See (Bluett et al., 2017).
METHOD DETAILS
Surgeries.
Mice were initially anesthetized with 5% isoflurane and then transferred to the stereotax (Kopf Instruments, Tujunga, CA) and kept under 2% isoflurane anesthesia. The hair over the incision cite was trimmed and the skin was prepped with alcohol and iodine scrub. The skull was exposed via a midline sagittal incision and treated with the local anesthetic, benzocaine (Medline Industries, Brentwood, TN). For all surgeries, we used a motorized digital software (NeuroStar; Stoelting Co., Wood Dale, IL) to guide a 10μL microinjection syringe (Hamilton Co., Reno, NV) driven by a Micropump Controller (World Precision Instruments, Sarasota, FL). Virus was delivered bilaterally into the plPFC (AP:+2.42, ML:±0.35, DV: 2.09), BLA (AP: −1.25, ML: ±3.30, DV: 5.10), or MDT: (AP:−1.1, ML.: ±0.59, DV: 3.4). Following completion of each surgery, 10mg/kg ketoprofen (AlliVet, St. Hialeah, FL) was administered as an analgesic, and post-operative treatment with ketoprofen was administered 24 and 48 hours after the surgery.
Ex Vivo Electrophysiology.
Coronal brain slices were prepared at 250μM on a vibrating Leica VT1000S microtome using standard procedures. Mice were anesthetized using isoflurane, and transcardially perfused with ice-cold and oxygenated cutting solution consisting of (in mM): 93 N-Methyl-D-glucamine (NMDG), 2.5 KCL, 20 HEPES, 10 MgSO4•7H20, 1.2 NaH2PO4, 0.5 CaCl2•2H20, 25 glucose, 3 Na+-pyruvate, 5 Na+-ascorbate, and 5 N-acetylcysteine. Following collection of coronal sections, the brain slices were transferred to a 34°C chamber containing oxygenated cutting solution for a 10-minute recovery period. Slices were then transferred to a holding chamber consisting of (in mM) 92 NaCl, 2.5 KCl, 20 HEPES, 2 MgSO4•7H20, 1.2 NaH2PO4, 30NaHCO3, 2 CaCl2•2H20, 25 glucose, 3 Na-pyruvate, 5 Na-ascorbate, 5 N-acetylcysteine and were allowed to recover for ≥30 min. For recording, slices were placed in a perfusion chamber (Warner Instruments RC-27L) and perfused with oxygenated artificial cerebrospinal fluid (ACSF; 31–33°C) consisting of (in mM): 113 NaCl, 2.5 KCl, 1.2 MgSO4•7H20, 2.5 CaCl2•6H20, 1 NaH2PO4, 26 NaHCO3, 20 glucose, 3 Na+-pyruvate, 1 Na+-ascorbate, at a flow rate of 2–3ml/min. All drugs were stored in DMSO stocks and then included in ACSF containing 1:2000 (w/v) Bovine Serum Albumin (Fisher Scientific) and ≤1:2000 (v/v) DMSO.
Fluorescently labeled neurons in the mPFC were identified using a series 120Q X-cite lamp at 40X magnification with an immersion objective with differential interference contrast microscopy (DIC). The plPFC was visually distinguished from the ilPFC by packing density of L2/3 neurons and termination of BLA projections to L2/3. L2/3 plPFC neurons were differentiated from L5 neurons by packing density and a change in the laminar distribution of BLA projections, where a prominent gap in BLA projections is observed between L2/3 and deep layer 5. For investigation of thalamo-cortical circuitry (Figures S4 and S6), L6 was identified by expression of strong reciprocal thalamo-cortical projections (i.e. eYFP expressing axon terminals from the MDT and rAAV positive reciprocally projecting plPFC neurons). plPFC neurons were voltage clamped in whole-cell configuration using borosilicate glass pipettes (2–4 MΩ) filled with internal solution containing (in mM): 125 K+-gluconate, 4 NaCl, 10 HEPES, 4 MgATP, 0.3 Na-GTP, and 10 Na-phosphocreatine (pH 7.30–7.35). For all experiments other than those shown in Figure S2, neurons were clamped at −70mV and 50μM picrotoxin (Cayman Chemical, Ann Arbor, Michigan) was included in the patch pipette. Following break-in to the cell, we waited ≥3 minutes to allow for exchange of internal solution and stabilization of membrane properties. Neurons with an access resistance of >20MΩ or that exhibited greater than a 20% change in access resistance during the recording were not included in our data sets.
Ex vivo optogenetics.
For electrophysiological interrogation of the BLA-plPFC circuit, mice were bilaterally injected with 250 μL of AAV5-CaMKIIα-ChR2(H134R)-eYFP and rAAV2-CAG-tdTomato in a 2:1 ratio into the BLA or MDT. 3–5 weeks of viral expression was allowed prior to sacrificing the mice. ≥1 week prior to electrophysiological analysis, littermate-matched mice were singly housed. 24 hours before electrophysiological analysis, littermate-matched mice were randomly assigned to receive either 20 randomly interspersed 0.5 mA shocks over a 10-minute period or no shocks.
For optogenetic recordings of input/output curves, we used a Thorlabs LEDD1B T-Cube driver and obtained separate recordings of 470nm wavelength oEPSCs at 200, 400, 600, 800, 1000, and 1200 mA of LED intensity. The same stimulation paradigm was used for current clamp input/output curves. Current clamp recordings of somatic current injection induced AP firing were obtained by initially injecting enough current to hold the neuron at −70mV and then applying sequential depolarizing steps that increase by 20pA. PPR recordings of oEPSCs were obtained in voltage-clamp with an inter-stimulus interval of 50ms. PPR is reported as a ratio between the amplitude of the second oEPSC divided by the first. In Figure 2, PPR values are shown across the entire range of stimulus intensities. PPR effects are not dependent on the stimulus intensity; therefore, in subsequent figures, PPR values are shown at the maximum stimulus intensity. For optogenetically elicited AP firing, three 2ms light stimulations were given at each intensity, and the probability was calculated as the % of neurons firing APs (e.g. spiking on 2 of 3 stimulations =0.67). Recordings of DSE were obtained following at 10 second voltage step to +30mV. A baseline of 10 oEPSCs were taken prior to the depolarizing step, and all data is plotted as an oEPSC amplitude normalized to the baseline period. A light exposure time of 2ms was used for all optogenetic experiments. For recordings of asynchronous neurotransmitter release, the CaCl2•6H20 in the ACSF was replaced with SrCl2•6H20. Asynchronous release events were analyzed in a 500ms window following optogenetic stimulation. A Clampfit template was made by selecting individual asynchronous release events and averaging them. The template was then used to analyze asynchronous events.
Fiber Photometry.
Mice were placed on a stereotaxic frame and unilateral holes drilled over PL (AP = +2.0 mm, ML = ± 0.3 mm) and BLA (AP = −1.75 mm, ML = ±3.3 mm). A 10 μL Nanofil syringe (World Precision Instruments) fitted with a 33-gauge beveled needle and connected to an infusion pump was used to microinject 200nl of AAV1.Syn.GCaMP6s.WPRE.SV40 (Penn Vector Core) into the BLA (DV = 4.6 mm) at a rate of 50 nl/min. A 400 μm diameter optical fiber (Doric) was implanted into the PL (DV = −1.6 mm) and secured to the skull with Metabond (Parkell). Two to four weeks after surgery, mice underwent the acute shock exposure protocol with concurrent recording of GCaMP6s signal. The fiber photometry rig was based on a previously described design (Cui et al., 2013; Gunaydin et al., 2014); briefly, to induce GCaMP6s fluorescence, 470 nm wavelength light emitted from an LED (Thorlabs) was passed first through a filter (Semrock, FF02–472/30) and then connected to the fiber implant with a 0.48 NA fiber optic patch cord (Doric). Activity-dependent GCaMP6s fluorescent signal was then transmitted through the fiber optic patch cord and separated from the excitation light with a dichroic (Semrock, FF495-Di03) and then passed through a single band filter (Semrock, FF01–535/50) and focused on a photodetector (Newport, Model 2151) until finally being recorded by a real-time processor (Tucker Davis Technologies). A pulse from the behavioral set-up (Video Freeze) was used to time lock fluorescent signal recordings with shock presentations. To confirm fiber placement and GCaMP6s expression, mice were perfused, and brains were sectioned for histological verification of injection sites and fiber optic placement using Stereo Investigator software (MBF Bioscience) with a fluorescent microscope (Nikon Eclipse 80i).
Miniendoscopy:
For in vivo single cell Ca2+ imaging, mice were injected with AAV expressing GCaMP7f into the plPFC at 2 different levels (1.6 and 1.9 DV, 300 nL per injection). Following or prior to injection, a 0.5 mm diameter tract was created over PL with a blunt needle connected to a vacuum line stereotaxically driven to DV 1.55. Thereafter, a 0.5 mm diameter GRIN lens (Inscopix, Palo Alto, CA) was introduced into the tract and slowly lowered into position (1.8 DV). Animals were allowed to recover for at least 2 weeks, after which a baseplate was installed over the lens in order to dock the miniaturized microscope at an empirically optimized working distance. Mice were habituated to the microscope for at least 2 days. On test day, mice were exposed to 20 shocks as described below (Foot-shock stress) and the recording system (nVista, Inscopix, Palo Alto, CA) was synchronized to the fear conditioning software (FreezeFrame, actimetrics) via a TTL pulse. Data was acquired at a frame rate of 10 Hz. Laser power, gain, and lens focus were empirically adjusted to maximize the quality of the recordings. Data was acquired continuously throughout the duration of the session.
Lipid analysis.
Mice underwent cervical dislocation immediately followed by decapitation. The brain was quickly removed and placed in a brain matrix. 2 mm thick coronal sections containing the target brain regions were frozen on a metal block in dry ice. Dissections were performed on the frozen tissue using a 2 mm diameter metal micropunch. Samples were stored at −80 °C until extraction. Lipids were extracted from brain tissue by sonication in 1 ml of acetonitrile (ACN). The samples were sonicated at 60% power for 1 min while incubated in an ice bath to prevent sample heating (the sonicator was a Hielscher UP100H ultrasonic device – 100W, 30kHz). The ACN contained the following internal standards: 2-AG-d5 (1 nmol), AEA-d4 (2 pmol), AA-d8 (2 nmol), OG-d5 (0.25 nmol) and OEA-d4 (25 pmol) was included for selected samples. The sonicated homogenate was stored at −20°C overnight and then centrifuged at 4°C for 5 min at 3,000 rcf. The supernatant was dried under nitrogen. Samples were re-suspended in 200 μl of methanol:water (50:50), followed by brief centrifugation to pellet any solid material. The cleared samples were transferred to autosampler vials and analyzed via LC-MS/MS as described in Bedse et 2017 (Biol Psychiatry. 2017 Oct 1;82(7):488–499).
Foot-shock stress.
Foot-shock stress occurred 24 hours before behavioral testing and consisted of twenty unpredictable 0.1, 0.25, or 0.5 mA foot-shocks within 10 min using a MED Associates fear-conditioning chamber (St. Albans, VT, USA). 24 hours after foot-shock stress mice were tested in elevated zero maze or elevated plus maze test.
Elevated-zero maze.
The elevated-zero maze (EZM, San Diego Instruments, San Diego, California, USA) is an annular white platform and divided four equal quadrants. It consisted of two open arms and two closed arms. The outer and inner diameters of the EZM were 60.9 cm and 50.8 cm, respectively. The apparatus was elevated 60.9 cm from the floor. Light levels in the open arms were approximately 200 lux, while the closed arms were <100 lux. Mice were placed in the closed arm of the maze and allowed to explore for 5 min. ANY-maze (Stoelting, Wood Dale, Illinois, USA) video-tracking software was used to monitor and analyze behaviors during the test.
Elevated-plus maze.
The elevated plus maze was custom built (Vanderbilt Machine Shop, Nashville, TN) and consists of two pairs of open and closed arms which intersect in an open center platform. The total length of each set of arms is 27”, and the closed arms are bordered with 6” walls. The maze platform is elevated to 15.5”. Mice were placed in the maze center, facing the entrance to the closed arm away from the experimenter. Light was set to a lux value of 150–200 for the open arms. Mice were allowed to roam for five minutes, and movement was measured with ANY-maze software (Stoelting Co., Wood Dale, IL).
INTRSECT behavior.
Littermate-matched homozygous CB1f/f mice were bilaterally injected with 500μL AAV5-CMV-fDIO-Cre-mNeonGreen into the BLA and 200μL rAAV2-EF1α-mCherry-IRES-Flp or mCherry control virus (rAAV2-EF1α-mCherry) into the plPFC. 6 weeks after stereotaxic surgery, mice were singly housed and allowed to acclimate for > 1 week. Prior to testing, mice were transported from a housing room to a nearby experimental room. We then allowed at least 10 minutes for mice to habituate to the experimental room. Mice were then run through EZM and Light/Dark box test (data not shown) on non-sequential days. One day following the last behavioral test, mice were exposed to the aforementioned 20 foot-shock stress protocol, and 24 hours later, run through the EPM test. For both the EZM and EPM, open arm light levels were set to 150 lux, and anxiety-like behaviors were analyzed by examining open arm entries and % open arm time using ANY-Maze software (Stoelting Co., Wood Dale, IL). The experimenter was blinded to the experimental group of the mice.
Chemogenetic behavior.
Littermate-matched WT C57 mice were bilaterally injected with rAAV2-pmSyn1-EBFP-Cre in the plPFC, and AAV5-hSyn-DIO-hM3D(Gq)-mCherry or AAV5-hSyn-DIO-mCherry control virus in the BLA. A minimum of six weeks was allowed for virus expression. Thirty minutes prior to testing, mice were given I.P. administration of Clozapine N-Oxide (CNO) (MilliporeSigma, St. Louis, MO) at 5mg/kg. Mice were transported from a housing room to a nearby experimental room. We then allowed at least 20 minutes for mice to habituate to the experimental room. The EPM assay was run as described above.
Immunohistochemistry and Imaging.
Mice were anesthetized using isoflurane and transcardially perfused with ice-cold phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA) solution in PBS. Brains were dissected and stored overnight in 4% PFA and transferred to a 30% sucrose solution for four days. 40μm brain sections were taken using a Leica CM3050 S cryostat (Leica Microsystem, Weitzlar, Germany). Brain sections were then washed in Tris-Buffered Saline (TBS) 3X for 10 minutes. Slices were then directly mounted on glass slides and VectaShield H-1200 DAPI mounting medium (Vector Laboratories, Burlingame, CA) was applied before coverslipping. Images were taken using an Axio Imager M2 epifluorescent microscope. Whole slice images were acquired using a 5x objective while zoomed in images of the BLA or plPFC were acquired using a 10 or 20x objective. Brightness and contrast were adjusted using Adobe CS4 software for presentation in figures.
cFOS staining.
Four to six weeks after intracranial viral injection surgery, both GqDREADD and mCherry mice were given an IP injection of 10mg/kg CNO-HCl dissolved in saline. Two hours after IP injection, mice were perfused as previously described. 40 μm sections were cut, and immediately washed in TBS 3X for 10 minutes. Slices were then washed in a blocking buffer 1x TBS containing 4% horse serum and 0.2% Triton X-100 (Fisher Scientific)(TBS+). Slices were then placed into 1.5 mL Eppendorf tubes containing the blocking buffer and 1:500 Rabbit-α-cFOS antibody (abcam) and covered and put on a nutator overnight. The following day, slices were washed 3X in TBS+, and then put into Eppendorf tubes containing TBS+ and 1:1000 Alexa Fluor 488 Donkey-α-Rabbit and allowed to incubate for two hours. Slices were subsequently washed and mounted for image analysis.
Viruses.
For chemogenetic manipulation of the BLA-plPFC circuit, we used rAAV2-Syn1-eBFP-Cre (200μL) in the plPFC and AA5-hSyn-DIO-hM3d(Gq)-mCherry (500μL)(Addgene, Cambridge MA) in the BLA. For electrophysiological interrogation of the BLA-plPFC circuit, we used AAV5-CaMKII-ChR2(H134R)-eYFP (UPenn Vector Core, Philadelphia, PA) and rAAV2-CAG-tdTomato (Addgene). We combined these viruses in a 2:1 ratio and injected a total volume of 250μL into the BLA. For plPFC deletion of DAGLα, we used AAV5-CMV-Cre-eGFP (200μL)(Addgene). For deletion of the CB1 receptor from BLA cells projecting to the plPFC, we designed a custom Flp recombinase dependent Cre virus, AAV5-CMV-fDIO-Cre-P2A-mNeonGreen(500μL into bilateral BLA)(Catalogue# VB180530–1030aad, VectorBuilder, Shenandoah, TX) and used a commercially available retrograde Flp virus, rAAV2-EF1α-mCherry-IRES-Flp (200μL into bilateral plPFC). For electrophysiological validation of this approach, we injected a 600μL combination of custom fDIO-Cre virus with AAV5-EF1α-DIO-ChR2(H134R)-eYFP in a 3:1 ratio. Finally, for in vivo fiber photometry experiment, we used AAV1.Syn.GCaMP6s.WPRE.SV40 (Penn Vector Core) which was bilaterally injected into the BLA at a volume of 200μL. Also see Key Resources table.
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit-α-cFOS | Abcam | 190289 RRID: AB_443538 |
| Alexafluor 488 Donkey-α-Rabbit | Invitrogen | A21206 RRID: AB_221544 |
| Rabbit-α-DAGLα | Gift from Ken Mackie | N/A |
| Bacterial and Virus Strains | ||
| AAV5-CMV-fDIO-Cre-mNeonGreen-wPRE | This paper | VB180530-1030aad |
| rAAV2-CAG-tdTomato | (Chan et al., 2017) | RRID: Addgene_59462 |
| AAV5-CaMKII-ChR2(H134R)-eYFP-wPRE | (Lee et al., 2010) | RRID: Addgene_26969 |
| rAAV2-EF1a-mCherry-IRES-Flpo | (Fenno et al., 2014) | RRID: Addgene_55634 |
| AAV5-CMV-Cre-eGFP-SV40 | Gift from James M. Wilson | RRID: Addgene_105545 |
| rAAV2-pmSyn1-EBFP-Cre | (Madisen et al., 2015) | RRID: Addgene_51507 |
| AAV5-EF1a-DIO-ChR2(H134R)-eYFP-wPRE | Gift from Karl Deisseroth | RRID: Addgene_20298 |
| AAV5-hSyn-DIO-hM3D(Gq)-mCherry | (Krashes et al., 2011) | RRID: Addgene_44361 |
| rAAV2-hSyn-GCaMP7f-wPRE | (Dana et al., 2016) | RRID: Addgene_104488 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rimonabant | Cayman Chemical | 9000484 |
| DO34 | Glixx Laboratories | GLXC-09757 |
| JZL184 | Cayman Chemical | 13158 |
| CP55,940 | Cayman Chemical | 13608 |
| PF3845 | Cayman Chemical | 13279 |
| NESS0327 | Cayman Chemical | 10004184 |
| CNO-HCl | Cayman Chemical | 25780 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| CB1f/f mice | Dr. Eric Delpire | N/A |
| DAGLf/f mice | Dr. Sachin Patel | N/A |
| C57 WT mice | Jackson Laboratory | IMSR_JAX000664 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Prism 6 | Graphpad | www.graphpad.com |
| pClamp10.5 | Molecular Devices | www.moleculardevices.com |
| MATLAB | Mathwords | www.mathworks.com |
| ANY-maze | Stoelting Co | www.Stoelting.com |
| Video Freeze | Med associates | www.med-associates.com |
| Other | ||
| Fear Conditioning Chamber | Med associates | MED-VFC-SCT-M |
| Elevated Plus Maze | San Diego Instruments | |
| Elevated Zero Maze | San Diego Instruments | |
QUANTIFICATION AND STATISTICAL ANALYSIS
Electrophysiology
Electrophysiological data was initially analyzed using ClampFit 10.5 software (Molecular Devices, San Jose, California). Data sets were organized in Microsoft Excel and then transferred to GraphPad Prism 6.0 for generation of graphs and statistical analyses. All statistical tests are reported in the figure legends. For analysis of two groups, an unpaired Student’s t-test was used, with error bars indicating the mean ± SEM. For analysis of three more groups, a one-way ANOVA with Holm-Sidak post-hoc correction was used, with error bars indicating the mean ± SEM. For analysis of two or more groups across two or more treatments or time points, a two-way ANOVA with Holm-Sidak post-hoc correction was used. For all data sets, significance was defined by a p value of <0.05. The Grubbs outlier test was run on each data set individually and outliers were excluded from our data. Mice were excluded from physiological experiments if there was improper targeting of the BLA or plPFC or if there was no viral expression in plPFC terminals. Neurons were excluded from physiological experiments for four reasons. 1: if the holding current dropped below −200 pA at any time during the recording. 2: if the access resistance was > 20 MΩ. 3: if the access resistance fluctuated by more than 20% throughout the recording. 4: There was no optogenetically-evoked response. Paired-pulse ratios were only taken from neurons in which both the first and the second oEPSC had an amplitude ≥ 50pA.
Behavior
All behavior was analyzed via ANY-maze (Stoelting, Wood Dale, Illinois, USA) software. All statistical tests are reported in the figure legends. For analysis of two groups, an unpaired Student’s t-test was used, with error bars indicating the mean ± SEM. For analysis of three more groups, a one-way ANOVA with Holm-Sidak post-hoc correction was used. For analysis of two or more groups across two or more treatments or time points, a two-way ANOVA with Holm-Sidak post-hoc correction was used. For all data sets, significance was defined by a p value of <0.05. The Grubbs outlier test was run on each data set individually and outliers were excluded from our data. Mice were excluded from behavioral experiments if any of the viral injections were misplaced or if there was no viral expression. Furthermore, mice were excluded if there was a technical issue during the behavioral experiment (e.g. mouse fell out the EZM/EPM or there was a malfunction on the behavioral hardware or software).
cFOS analysis
For cFOS imaging, a set exposure time of 400 ms for the GFP channel was used to ensure that there was no bias in the image acquisition process. Scale bars were included on each image to allow for subsequent processing. Raw 10x images were then opened with ImageJ, and the scale was set using the aforementioned scale bar. Images were then thresholded, and the region of interest was drawn using the Image J software with the Allen Brain Atlas being used to ensure the correct region was selected. Image J particle counting software was then used to analyze the number of cFOS cells in the region of interest. This number was then divided by the area of the region of interest to get the number of cFOS cells per square mm. Data were analyzed via an unpaired Student’s t-test.
Miniendoscopy
Recordings were spatially downsampled by a factor of 2, bandpass filtered, and motion corrected using Inscopix Data Processing software V1.2. Individual Ca2+ traces were extracted using Constrained Nonnegative Matrix Factorization for miniendoscopic data (CNMFE)(Zhou et al., 2018). For CNMFE, we used the following parameters: min corr= 0.9, min pnr= 20, gSiz= 20, gSig= 10. For our analysis we used the raw extracted values rather than the denoised values. Individual extracted traces and corresponding identified neurons were visually confirmed and traces that corresponded to artifacts rather than neurons were excluded. For bulk fluorescence analysis, the total fluorescence for the full field of view was extracted from recordings prior to band pass filtering. The baseline and stimulus response periods were defined as the 10 seconds preceding shock onset and the 10 seconds following tone onset. For bulk fluorescence signal, the total fluorescence was averaged across the 20 stimulus response periods. Similarly, for single cell analysis, data from each individual cell was averaged across the 20 stimulus trials and then binned into 1 second intervals. Both total fluorescence signals and individual traces were quantified using custom code in MATLAB. To normalize the data, z-scores were calculated using the 10 second pre-shock period as the baseline. Traces exceeding a z-score value of 1.645 or −1.645 (p<0.05, two-tails) for any of the following 3 1-second bins following shock onset were considered excitatory shock-responsive or inhibitory shock-responsive, respectively.
Fiber Photometry
Fiber photometry data was analyzed using custom code in MATLAB, where ∆F/F was calculated to normalize fluorescent signal data from each mouse and z-scored to account for between-subject variability in signal magnitude. To control for photobleaching, median fluorescence during a rolling window of 80 seconds (40 seconds before and 40 seconds after every given data point) was calculated and subtracted from each data point across the recording session.
DATA AND CODE AVAILABILITY
The codes generated during this study are available at https://github.com/dmarcus22/NEURON-D-19-00796R2 or by request from lead contact (sachin.patel@vanderbilt.edu).
Supplementary Material
Highlights.
The BLA-plPFC circuit is engaged by stress exposure and its activation is anxiogenic
Stress enhances glutamate release in a reciprocal BLA-plPFC-BLA subcircuit
BLA-plPFC glutamatergic drive is constrained by multimodal 2-AG signaling
2-AG signaling collapse mediates stress-induced circuit strengthening and anxiety
ACKNOWLEDGEMENTS
These studies were supported by NIH grants MH107435 (SP), MH114363 (DM), DA043982 (KM), NS052819 (FSL) and a NARSAD Young Investigator Award (GB). The CB1 floxed mouse generation was supported by the Integrative Neuroscience Initiative on Alcoholism (INIA stress) grant AA9013514 (ED). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLERATION OF INTERESTS
SP has received research support from H. Lundbeck A/S in the past 3 years. S.P. is a consultant for Psy therapeutics.
REFERENCES
- Apps R, and Strata P (2015). Neuronal circuits for fear and anxiety - the missing link. Nat Rev Neurosci 16, 642. [DOI] [PubMed] [Google Scholar]
- Arnsten AF (2015). Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci 18, 1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedse G, Hartley ND, Neale E, Gaulden AD, Patrick TA, Kingsley PJ, Uddin MJ, Plath N, Marnett LJ, and Patel S (2017). Functional Redundancy Between Canonical Endocannabinoid Signaling Systems in the Modulation of Anxiety. Biol Psychiatry 82, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluett RJ, Baldi R, Haymer A, Gaulden AD, Hartley ND, Parrish WP, Baechle J, Marcus DJ, Mardam-Bey R, Shonesy BC, et al. (2017). Endocannabinoid signalling modulates susceptibility to traumatic stress exposure. Nat Commun 8, 14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch-Bouju C, Larrieu T, Linders L, Manzoni OJ, and Laye S (2016). Endocannabinoid-Mediated Plasticity in Nucleus Accumbens Controls Vulnerability to Anxiety after Social Defeat Stress. Cell Rep 16, 1237–1242. [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Kimchi EY, Izadmehr EM, Porzenheim MJ, Ramos-Guasp WA, Nieh EH, Felix-Ortiz AC, Namburi P, Leppla CA, Presbrey KN, et al. (2017). Amygdala inputs to prefrontal cortex guide behavior amid conflicting cues of reward and punishment. Nat Neurosci 20, 824–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisi CO, and Robinson OJ (2018). The role of prefrontal-subcortical circuitry in negative bias in anxiety: Translational, developmental and treatment perspectives. Brain Neurosci Adv 2, 2398212818774223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavener VS, Gaulden A, Pennipede D, Jagasia P, Uddin J, Marnett LJ, and Patel S (2018). Inhibition of Diacylglycerol Lipase Impairs Fear Extinction in Mice. Front Neurosci 12, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sanchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, et al. (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda D, Neumann D, and Glatz JFC (2019). The endocannabinoid system: Overview of an emerging multi-faceted therapeutic target. Prostaglandins Leukot Essent Fatty Acids 140, 51–56. [DOI] [PubMed] [Google Scholar]
- Cremers HR, Demenescu LR, Aleman A, Renken R, van Tol MJ, van der Wee NJ, Veltman DJ, and Roelofs K (2010). Neuroticism modulates amygdala-prefrontal connectivity in response to negative emotional facial expressions. Neuroimage 49, 963–970. [DOI] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, and Costa RM (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. (2016). Sensitive red protein calcium indicators for imaging neural activity. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Kathuria S, and Piomelli D (2004). RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol 66, 1260–1264. [DOI] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, and Tye KM (2013). BLA to vHPC inputs modulate anxiety-related behaviors. Neuron 79, 658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, and Tye KM (2015). Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience 321 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno LE, Mattis J, Ramakrishnan C, and Deisseroth K (2017). A Guide to Creating and Testing New INTRSECT Constructs. Curr Protoc Neurosci 80, 4 39 31–34 39 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster RJ, Lebois LAM, Ressler KJ, and Suh J (2018). Brain circuit dysfunction in post-traumatic stress disorder: from mouse to man. Nat Rev Neurosci 19, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CF, Phifer J, Bradley B, and Ressler KJ (2009). Risk and resilience: genetic and environmental influences on development of the stress response. Depress Anxiety 26, 984–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, et al. (2014). Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henigsberg N, Kalember P, Petrovic ZK, and Secic A (2019). Neuroimaging research in posttraumatic stress disorder - Focus on amygdala, hippocampus and prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry 90, 37–42. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, and Rice KC (1990). Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87, 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Campolongo P, Yehuda R, and Patel S (2018). Integrating Endocannabinoid Signaling and Cannabinoids into the Biology and Treatment of Posttraumatic Stress Disorder. Neuropsychopharmacology 43, 80–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, and Gorzalka BB (2009). Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 34, 2733–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, and Patel S (2013). Translational evidence for the involvement of the endocannabinoid system in stress-related psychiatric illnesses. Biol Mood Anxiety Disord 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, and Gorzalka BB (2005). Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 30, 508–515. [DOI] [PubMed] [Google Scholar]
- Hillard CJ (2014). Stress regulates endocannabinoid-CB1 receptor signaling. Semin Immunol 26, 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch R, and Gerlicher AM (2014). Making a mountain out of a molehill: on the role of the rostral dorsal anterior cingulate and dorsomedial prefrontal cortex in conscious threat appraisal, catastrophizing, and worrying. Neurosci Biobehav Rev 42, 1–8. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, and Watanabe M (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89, 309–380. [DOI] [PubMed] [Google Scholar]
- Katona I, and Freund TF (2012). Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35, 529–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, and Walters EE (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 62, 593–602. [DOI] [PubMed] [Google Scholar]
- Kim J, and Alger BE (2010). Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci 13, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Gee DG, Loucks RA, Davis FC, and Whalen PJ (2011). Anxiety dissociates dorsal and ventral medial prefrontal cortex functional connectivity with the amygdala at rest. Cereb Cortex 21, 1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, and Manzoni OJ (2007). Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One 2, e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, Fenno LE, Ramakrishnan C, and Deisseroth K (2010). Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature 465, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisboa SF, Camargo LH, Magesto AC, Resstel LB, and Guimaraes FS (2014). Cannabinoid modulation of predator fear: involvement of the dorsolateral periaqueductal gray. Int J Neuropsychopharmacol 17, 1193–1206. [DOI] [PubMed] [Google Scholar]
- Lisboa SF, Gomes FV, Terzian AL, Aguiar DC, Moreira FA, Resstel LB, and Guimaraes FS (2017). The Endocannabinoid System and Anxiety. Vitam Horm 103, 193–279. [DOI] [PubMed] [Google Scholar]
- Little JP, and Carter AG (2013). Synaptic mechanisms underlying strong reciprocal connectivity between the medial prefrontal cortex and basolateral amygdala. J Neurosci 33, 15333–15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DJE, Sasiadek JD, Coles AS, and George TP (2018). Cannabis and mental illness: a review. Eur Arch Psychiatry Clin Neurosci [DOI] [PMC free article] [PubMed]
- Lowery-Gionta EG, Crowley NA, Bukalo O, Silverstein S, Holmes A, and Kash TL (2018). Chronic stress dysregulates amygdalar output to the prefrontal cortex. Neuropharmacology 139, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B, Marsicano G, Maldonado R, and Hillard CJ (2015). The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci 16, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manduca A, Bara A, Larrieu T, Lassalle O, Joffre C, Laye S, and Manzoni OJ (2017). Amplification of mGlu5-Endocannabinoid Signaling Rescues Behavioral and Synaptic Deficits in a Mouse Model of Adolescent and Adult Dietary Polyunsaturated Fatty Acid Imbalance. J Neurosci 37, 6851–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KM, Stevelink SAM, Choi J, and Fear NT (2018). Post-traumatic growth in the military: a systematic review. Occup Environ Med 75, 904–915. [DOI] [PubMed] [Google Scholar]
- Matyas F, Lee J, Shin HS, and Acsady L (2014). The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci 39, 1810–1823. [DOI] [PubMed] [Google Scholar]
- McCormick MC, Abrams DI, Alegria M, Checkley W, Collins RL, Cooper ZD, du Plessis AJ, Ewing SF, Hennessy S, Hutchison K, et al. (2017). The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research (Washington (DC)).
- McEwen BS (2012). Brain on stress: how the social environment gets under the skin. Proc Natl Acad Sci U S A 109 Suppl 2, 17180–17185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, and Nasca C (2015). Mechanisms of stress in the brain. Nat Neurosci 18, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry LM, and Carter AG (2017). Prefrontal Cortex Drives Distinct Projection Neurons in the Basolateral Amygdala. Cell Rep 21, 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, and Gorzalka BB (2012). Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol 22, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RJ, Hill MN, and Gorzalka BB (2014). A critical role for prefrontocortical endocannabinoid signaling in the regulation of stress and emotional behavior. Neurosci Biobehav Rev 42, 116–131. [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, and Rauch SL (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry 66, 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, and Liu QS (2009). Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther 331, 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Hill MN, Cheer JF, Wotjak CT, and Holmes A (2017). The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci Biobehav Rev 76, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, and Winder DG (2009). Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology 34, 2699–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, and Hillard CJ (2004). Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 145, 5431–5438. [DOI] [PubMed] [Google Scholar]
- Puente N, Cui Y, Lassalle O, Lafourcade M, Georges F, Venance L, Grandes P, and Manzoni OJ (2011). Polymodal activation of the endocannabinoid system in the extended amygdala. Nat Neurosci 14, 1542–1547. [DOI] [PubMed] [Google Scholar]
- Qin Z, Zhou X, Pandey NR, Vecchiarelli HA, Stewart CA, Zhang X, Lagace DC, Brunel JM, Beique JC, Stewart AF, et al. (2015). Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron 85, 1319–1331. [DOI] [PubMed] [Google Scholar]
- Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, and Hillard CJ (2008). Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology 54, 108–116. [DOI] [PubMed] [Google Scholar]
- Robinson OJ, Krimsky M, Lieberman L, Allen P, Vytal K, and Grillon C (2014). Towards a mechanistic understanding of pathological anxiety: the dorsal medial prefrontal-amygdala ‘aversive amplification’ circuit in unmedicated generalized and social anxiety disorders. Lancet Psychiatry 1, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino T, Realini N, Castiglioni C, Guidali C, Vigano D, Marras E, Petrosino S, Perletti G, Maccarrone M, Di Marzo V, et al. (2008). Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex 18, 1292–1301. [DOI] [PubMed] [Google Scholar]
- Sciolino NR, Zhou W, and Hohmann AG (2011). Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res 64, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn V, Wolff SB, Herry C, Grenier F, Ehrlich I, Grundemann J, Fadok JP, Muller C, Letzkus JJ, and Luthi A (2014). Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81, 428–437. [DOI] [PubMed] [Google Scholar]
- Sharma S, Powers A, Bradley B, and Ressler KJ (2016). Gene x Environment Determinants of Stress- and Anxiety-Related Disorders. Annu Rev Psychol 67, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonesy BC, Bluett RJ, Ramikie TS, Baldi R, Hermanson DJ, Kingsley PJ, Marnett LJ, Winder DG, Colbran RJ, and Patel S (2014). Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep 9, 1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, and Piomelli D (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature 388, 773–778. [DOI] [PubMed] [Google Scholar]
- Sumislawski JJ, Ramikie TS, and Patel S (2011). Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology 36, 2750–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, and Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16, 317–331. [DOI] [PubMed] [Google Scholar]
- Tye KM (2018). Neural Circuit Motifs in Valence Processing. Neuron 100, 436–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP (2004). Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51, 32–58. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, and Bains JS (2010). Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci 30, 11188–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, and Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410, 588–592. [DOI] [PubMed] [Google Scholar]
- Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, Zhang HT, Cravatt BF, and Liu QS (2014). Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 39, 1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Resendez SL, Rodriguez-Romaguera J, Jimenez JC, Neufeld SQ, Giovannucci A, Friedrich J, Pnevmatikakis EA, Stuber GD, Hen R, et al. (2018). Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes generated during this study are available at https://github.com/dmarcus22/NEURON-D-19-00796R2 or by request from lead contact (sachin.patel@vanderbilt.edu).