

Abstract
Background:
Chronic fatigue syndrome (CFS) is an illness of unknown origin that may have familial risks. Low natural killer (NK) lymphocyte activity was proposed as a risk for familial CFS in 1998. Since then, there have been many studies of NK lymphocytes in CFS in general populations but few in familial CFS. Antibody-dependent cell-mediated cytotoxicity (ADCC) by NK lymphocytes helps control viral infections. ADCC is affected by variant CD16A receptors for antibody that are genetically encoded by FCGR3A.
Methods:
This report characterizes ADCC effector NK cell numbers, ADCC activities, and FCGR3A variants of five families each with 2–5 CFS patients, their family members without CFS and unrelated controls. The patients met the Fukuda diagnostic criteria. We determined: CD16Apositive blood NK cell counts; EC50s for NK cell recognition of antibody; ADCC lytic capacity; FCGR3A alleles encoding CD16A variants, ROC tests for biomarkers, and synergistic risks.
Results:
CFS patients and their family members had fewer CD16Apositive NK cells, required more antibody, and had ADCC that was lower than the unrelated controls. CFS family members were predominantly genetically CD16A F/F s for the variant with low affinity for antibodies. ROC tests indicated unsuitability of ADCC as a biomarker for CFS because of the low ADCC of family members without CFS. Familial synergistic risk vs. controls was evident for the combination of CD16Apositive NK cell counts with ADCC capacity.
Conclusions:
low ADCC may be a risk factor for familial CFS. Furthermore, characterization of familial CFS represents an opportunity to identify pathogenic mechanisms of CFS.
Keywords: Chronic Fatigue Syndrome, ADCC, antibody-dependent cell-mediated cytotoxicity, NK, CD16A, family studies
Graphical Abstract
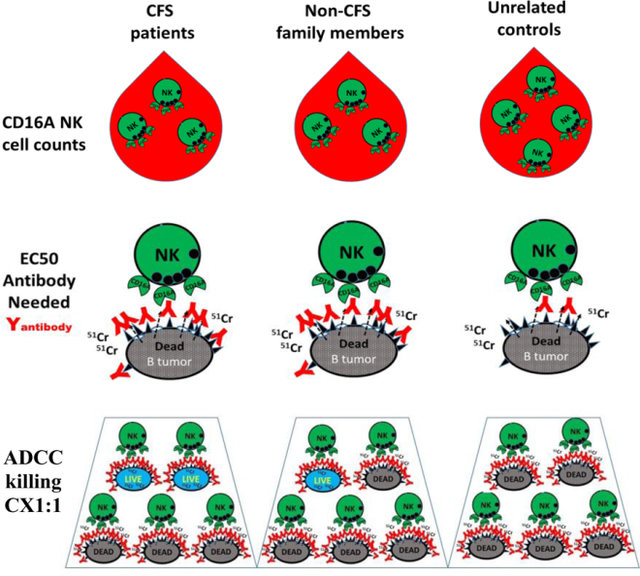
Introduction
Myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS) is disease of unknown etiology. Its nomenclature is still unresolved [1] despite concerted efforts [2]. ‘ME’ and ‘CFS’ are often used interchangeably; CFS will be used in this article. The disease is identified by debilitating chronic fatigue and diagnostic criteria, well-defined by the Centers for Disease Control USA in 1994 [3] and further delineated by the National Academy of Medicine USA in 2015 [1]. CFS affects 800,000 to 2.5 million adults in the USA and ~0.4% of the population worldwide [2]. Symptoms include severe fatigue for more than six months, long-lasting post-exertional malaise, un-refreshing sleep, ‘brain fog’ in the form of loss of memory and/or lessened ability to think, and chronic pain [3]. There are no known causes for most cases; however, CFS-like pathology can follow severe viral or bacterial infections [4]. CFS is receiving renewed attention as a distinct disease [5, 6] with high costs to society [7].
Subgroups of CFS patients [8, 9], have been proposed based on symptoms, candidate etiologies and potential disease-promoting mechanisms [10]. Familial CFS, defined by the occurrence of two or more CFS patients who are first-degree relatives within a family, represents a subgroup of CFS. Familial CFS was first reported in a 1998 study of natural killer (NK) lymphocytes of one family with 8 CFS patients [11]. A 2001 report found that 6/25 unrelated CFS patients (24%) had first-degree relatives with CFS [12]. Of these six patients, two had two other CFS-affected family members and four had one other CFS-affected family member (personal communication from author [12] Nor Zainal, Ph.D.) Both 1998 and 2001 reports used the Fukuda CDC 1994 diagnostic criteria for CFS. Two Studies of concordant twins add further evidence for familial CFS, reviewed [12]. The family described in 1998 with CFS patients with impaired NK activity also had non-CFS first degree relatives with low NK activity. NK activity has been reported to be low in many but not all studies of NK lymphocytes in non-familial CFS (reviewed, [13]). To our knowledge, the study of NK-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) reported here is the first study of immunity in familial CFS since 1998.
NK cells use both ‘natural cytotoxicity’ and ADCC to kill virally infected cells and tumor cells. ‘Natural cytotoxicity’ involves recognition of stress or other ligands present in ‘target’ cells and occurs in the absence of antibodies (reviewed, [14]). ADCC occurs only when specific antibodies are bound to infected cells or tumor cells. The NK cell receptor required for ADCC is CD16A which binds to antibodies and is present on most but not all NK cells. As a result of different recognition systems for natural cytotoxicity and ADCC, it is reasonable to postulate that ADCC activity could be altered without affecting natural cytotoxic activity. There are two common allelic variants of the gene FCGR3A that encode single amino acid differences in CD16A, with either phenylalanine (F) or valine (V) at AA158, that could affect ADCC. The V158 CD16A has twice the affinity for IgG antibody and higher cellular expression than the F158 CD16A [15, 16]. We postulated that low ADCC and the homozygous F/F form of CD16A could be familial CFS risk factors.
ADCC is an attractive consideration for CFS because NK cell-mediated ADCC helps control chronic herpes viral infections. Consideration of viral etiologies for CFS began in 1984 [17]. Viruses that have been proposed include the chronic herpes viruses Epstein Barr virus [18], human cytomegalovirus [19], herpes zoster [20], human herpes virus 6 (HHV6) [21], and a different DNA virus, parvovirus B19 [22]. The fatigue of CFS resembles the fatigue induced by gamma interferon during viral infections [23]. Elevated gamma interferon is detectable in the blood of patients with severe CFS [24]. In the face of rigorous research attempts and the high incidences of herpes viruses in the general population, proving that herpes viral infections are universally linked with CFS disease has been challenging and the issue remains unresolved [25].
The pilot study of ADCC in familial CFS reported here encompasses in vivo availability of NK cells that can mediate ADCC, ADCC functions assayed in vitro, and genetics of CD16A. We addressed: a) counts of NK cells with CD16A receptors; b) EC50s for the amount of antibody required for ADCC; c) NK cell ADCC capacity; and d) FCGR3A alleles. Two methods were developed: TruCount® bead enumeration of CD16A-positive(pos) NK cells and an assay to detect ADCC cytotoxic capacity regardless of FCGR3A genotypes [26]. The report focuses on five CFS families. The common genetic backgrounds within each family promote detection of specific alleles that might be preferentially inherited by CFS patients compared to their non-CFS siblings. Familial environments may direct environmentally stimulated subgroups of NK cells [27] and a background of similar NK development could favor detection of changes in ADCC specific to the CFS patients.
This study reports several novel observations. (1) CD16Apos NK cell counts of both CFS patients and their family members were lower than those of unrelated healthy controls. (2) There was lower ADCC for CFS patients compared to unrelated controls. (3) There was also lower ADCC of the family members without CFS compared to unrelated controls. (4) CFS family members (with or without the disease) were more likely to have a combination of low ADCC activity with low CD16A NK cell counts than the unrelated controls. (5) The CFS families were predominantly FCGR3A homozygous for CD16A F/F. Based on these observations, we suggest that low ADCC may be a risk factor for the familial form of CFS.
Methods
CFS patients, family members and unrelated healthy donors.
Five families were selected from many families afflicted with CFS. The patients were diagnosed at the Bateman Horne Center in Salt Lake City, UT, and met the Fukuda criteria [3] when first diagnosed. Selection was for families with several CFS patients and unaffected siblings of patients. Selection was also influenced by family members’ geographic availability to donate blood. The families had a total of 13 CFS patients with 2 to 5 CFS patients per family. Figure 1 illustrates the family pedigrees; participants in the study are indicated by their CD16A genotypes. Eleven CFS patients and 22 family members without CFS participated. Sixteen of the participating family members were first degree relatives of the patients and had 50% of all genes in common with a CFS patient in the family. Four non-CFS family members were the 2nd degree relatives and shared 25% genes with a CFS patient. The remaining two non-CFS family members were fathers of children included in the study and unrelated to the patients. Sixteen unrelated healthy control donors were matched by race, sex and age to the CFS patients. Healthy was defined as HIV-negative, without overt infections at the time of blood donation, and without CFS. All participants were Caucasian.
Figure 1. CFS family pedigrees.
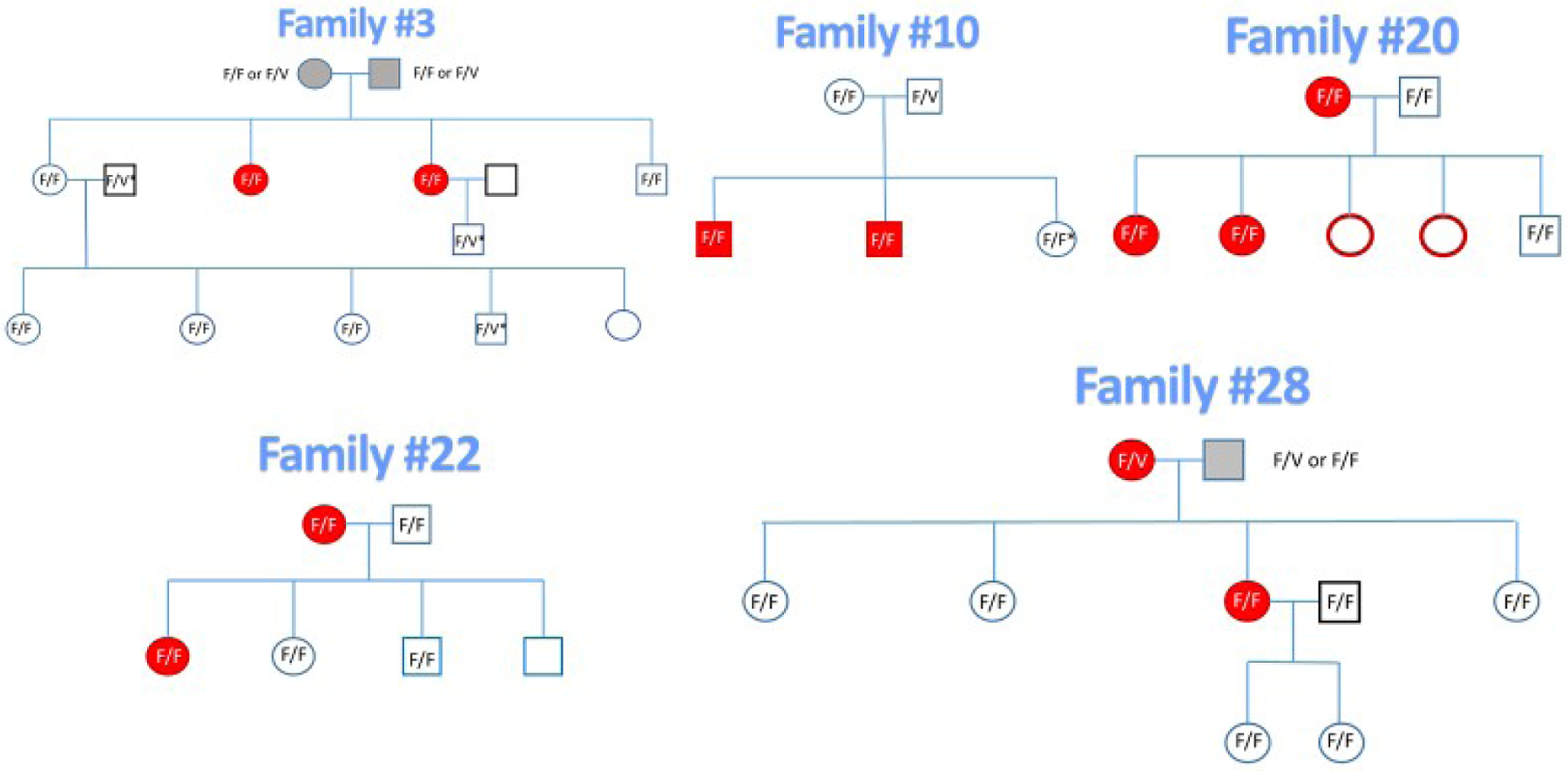
Red symbols indicate CFS patients and closed symbols indicate the 11 patients who participated. Study participants are indicated by their CD16A genotypes. The deceased parents are indicated with gray symbols. The 3 fathers of the 3rd generation were unrelated to the patients and are indicated with black bold squares. The genotypes marked F/V* were determined by flow cytometry and allelic inheritance.
Research with human subjects was approved by institutional review boards for the Bateman Horne Center and for the University of Nevada, Reno School of Medicine, IRB #2014B016. Written informed consent was obtained from the blood donors.
Questionnaires.
Patients and their family members without CFS answered the Rand-36 [28] (Rand Health Care, Santa Monica, CA) and Fibromyalgia Impact [29] (American Academy of Family Physicians, Leakwood, KS) questionnaires within weeks before blood donation. The timing of the questionnaires assured that symptoms reported were current with the evaluations of ADCC immunity.
Preparation of peripheral blood mononuclear cells (PBMCs).
Blood was drawn in Salt Lake City between 8–10 AM. Most overnight shipments to Reno, NV, contained blood from multiple CFS family members and two unrelated healthy controls. Blood samples were coded in Salt Lake City. The blood CD16A NK cell counts, EC50 assays, ADCC, and CD16A genotypes were run as coded samples and decoded after completion of the experiments.
For ADCC assays, PBMCs were isolated from 24 ml of blood by ficoll-hypaque density gradient centrifugation. The PBMCs were cultured overnight to ensure ADCC activity [30], at 1–2 x 106 cells/ml in complete assay media, 90% Dulbecco’ s media containing 4.5 g/L glucose and L-glutamine (Corning), 10% fetal calf serum (Atlanta Biologicals), 10 mM hepes (Sigma-Aldrich, St. Louis, MO), and 1% penicillin-streptomycin (Sigma-Aldrich). Culture and assay conditions were standardized using one lot of fetal calf serum and one lot of tissue culture flasks (Biolite, Thermo Scientific).
TruCounts® of CD16Apos NK cells
CD16Apos NK cells per µl blood
Fifty µl aliquots of blood were labeled on arrival with a panel of antibodies designed for no-wash analyses with TruCount® beads (Becton Dickenson no. 340334 [31]), The antibody panel contained FITC-anti-CD3e (clone UCHT1); PerCP-anti-CD16A (3G8); PacBlue anti-CD45 (clone HI30); and FITC-anti-CD91 (2MR-alpha), all purchased from BioLegend (San Diego, CA) except for anti-CD91 from Becton Dickenson. The flow cytometric gating is illustrated in supplement Figure S1. The ADCC effector cells were CD3negCD16posCD45posCD91neg.
CD16Apos NK cells per µl PBMCs
Fifty µl aliquots of PBMCs were labeled with a similar panel of antibodies in tubes with TruCount® beads, with the substitution of PE-Cy7 anti-CD33 (clone P67.6) for CD91 and inclusion of FITC-anti-CD7 (clone CD7–6B7) and APC-Cy7 anti-CD56 (clone HCD56). The NK ADCC effector cells were CD3negCD7posCD16posCD33negCD45posCD56variable. Anti-CD7 was critical to discriminate CD16AposCD7posCD56neg NK cells [32] from CD16AposCD56neg monocytes (that are all CD7neg and largely CD33pos) [33].
Cytometry for TruCounts®
Cells were analyzed with a BD Biosciences Special Order Research Product LSR II analytical flow cytometer. Analyses were with FlowJo software (FlowJo, LLC, Ashland, OR) to determine the CD16Apos NK cells and TruCount® beads in order to calculate the number of CD16Apos NK cells in the solutions [26].
ADCC assays
EC50 assay for antibody recognition by NK cells
The EC50s, effective concentrations of antibody needed for 50% of maximal ADCC [34], were determined with 4-fold dilutions of obinutuzumab that ranged from 0.04 to 625 ng/ml in the 51Cr assays described below. ADCC was determined at 4 hours, with duplicate or triplicate wells for each antibody concentration. The yields of PBMCs supported EC50 determinations for most, but not all, donors.
ADCC cell capacity and cooperativity
The ADCC assay has three critical features. 1) MHC class I-negative Daudi cells were used as ‘targets’ to avoid inter-donor variations caused by MHC I-dependent KIR inhibition The Daudi cells from the ATCC (Manassas, VA, catalog # CCL-213) were routinely tested and negative for mycoplasma. 2) A type 2 anti-CD20 monoclonal antibody obinutuzumab [35] was selected because it is poorly cleared from the membranes of B cells compared to type 1 anti-CD20 antibodies. Non-fucosylated obinutuzumab was obtained from the Roche Innovation Center, Zurich, Switzerland. 3) The persistence of the bound antibody on washed Daudi cells allowed use of unfractionated CD16A NK cells within PBMCs as effectors and avoided killing of normal B cells in PBMCs, thereby maximizing usage of the patients’ cells [26].
ADCC was measured by 51Cr-release [26]. Daudi cells were labeled with Na51CrO4 (Perkin Elmer, Waltham, MA), pretreated with 1 µg/ml obinutuzumab for 0.5 hour at room temperature and washed 5 times to remove unbound antibody and unincorporated chromium. The PBMC solutions containing the CD16A NK cells were diluted 2-fold serially in V-bottom plates (Costar 3894, 96 well) in 0.1 ml to create six CD16Apos NK:Daudi target cell (effector:target, E:T) ratios, 4 wells per ratio. Daudi cells, with or without mAb, 1 x 104 per well in 0.1 ml, were added, for ADCC or NK background activities, respectively. Plates were centrifuged for 3 minutes at 1000 rpm and incubated for 4 hours at 5% CO2 and 37oC. After incubation, plates were centrifuged for 10 minutes at 1200 rpm and 0.1 ml of cell-free supernatants were removed and counted in a Packard Cobre II gamma counter. The percent specific release (SR) was calculated using the formula
% SR = [(Experimental counts - Spontaneous Release)/(Max - Spontaneous Release)] x 100.
Spontaneous release was the leak rate of targets without PBMCs and the Max was the radioactivity released by targets lysed with 1% SDS. There was negligible ‘natural cytotoxicity’ in the absence of antibody for all donors.
We report separately the lytic slopes (CX-slope) and ADCC capacities used to compare patients with the two other groups of donors. This distinction was necessary because there were intra-donor differences in lytic slopes. These differences prevented application of lytic units to compare ADCC activities. CX1:1 [26] measurement of ADCC capacity is the % of the targets killed at a CD16Apos NK cells to Daudi ratio of 1:1. It was calculated as follows. Percentages of ADCC from the assay were plotted as linear cytotoxicity with y = % specific 51Cr release vs. x = the log10 of the 6 CD16Apos NK cells (in the PBMCs) to Daudi ratios. The linear cytotoxicity was used to determine y = mx + b, with the CX-lytic slope = m, x = log10 of the E:T and b = the y intercept. The P values for linear fit were <0.05, with R2s >0.8. To determine CX1:1, y was calculated with x =1 and log10(1) = 0.
FCGR3A Genotyping
Genotyping by DNA sequences.
The FCGR3A genotypes encoding CD16A at AA158 were determined by PCR and DNA sequence analysis at the Frederick National Laboratory for Cancer Research, Frederick, MD, by Stephen K. Anderson, Ph.D. Amplicons specific for the FCGR3A gene and that exclude the FCGR3B gene (which encodes neutrophil CD16B) were generated with forward and reverse PCR primers, (5’ to 3’) for FCGR3A (TCCTACTTCTGCAGGGGGCTTGT) and (CCAACTCAACTTCCCAGTGTGATTG), respectively. The amplicons were directly sequenced using Sanger methodology.
Genotyping by flow cytometry.
This genotyping was done for all donors and was the sole method used for a few of the control donors. The homozygous F/F genotype was distinguished from F/V & V/V genotypes using the MEM-154 clone of anti-CD16 mAb (PE-labeled, Pierce Chemical Co, Rockford, IL). MEM154 reacts with the CD16A 158 V but not the 158 F [36). PBMCs were labeled with: FITC-anti-CD3e (cloneOKT3); PE-anti-CD16A (3G8) or PE anti-CD16A 158V selective-(MEM154); BV605-anti-CD19 (HIB19.11); PacBlue anti-CD45 (HI30); FITC-anti-CD91 (2MR-alpha) and APC-Cy7-anti-CD56 (HCD56), purchased from BioLegend (San Diego, CA) with the exception of MEM154. CD16A F/F cells were negative with clone MEM154 and positive with clone 3G8.
Health and Safety.
BSL level 2 laboratory safety procedures were maintained throughout the experiments.
Statistical analyses
Cytotoxic activities were determined with the Excel Analysis Tool Pack, using best fit for linear regressions. Student’s t-tests were used to compare the different groups of subjects. Excel and GraphPad Prism 7 (San Diego, CA) were used for illustrations. The UCSF website http://www.sample-size.net/sample-size-proportions/ was used to determine the 95% confidence limits for Table 4 (calculation: ‘CI for Proportion’). The quadrants for the analyses of synergy were divided by the median values of the variables CX1:1 or CX-slope vs. CD16Apos NK cell counts. Logistic regression using interaction terms was applied for comparisons of distributions in quadrants. Results were expressed as the odds of being in the low-low quadrant for CFS patients & family members vs. unrelated healthy controls, determined with SAS software (version 9.2 Institute Inc., Cary, NC, USA). Biomarkers were evaluated using receiver operating characteristics (ROC) analyses [37] with SAS version 9.4. P values were determined by maximum likelihood tests for difference of AUCs from 0.5. For Table 4, P values for the frequencies of CD16A F/F parents were determined using the Appendix to Chapter 5: Exact Binomial Probability Calculator available at http://vassarstats.net/textbook/ch5apx.html. Unavailability of the flow cytometer impacted data collection for a few donors. As a result, there are more measurements of CX-slopes available for statistical comparison than CX1:1s, as the CX1:1s are dependent on TruCount®s.
Table 4.
Frequencies of CD16A F/F homozygosity
| Populations | Number donors | % F/F | 95% confidence limits+ | ||
|---|---|---|---|---|---|
| F/F | F/V or V/V | All | |||
| CFS patients | 10 | 1 | 11 | 90.9 | 58.7–99.8% |
| Non-CFS family members | 18 | 4 | 22 | 81.8 | 59.7 –94.8% |
| Unrelated healthy donors | 3 | 13 | 16 | 18.8 | 4–45.6% |
| CFS family parents, highest possible %F/F# | 8 | 2 | 10 | 80@ | 44.4–97.5% |
| CFS family parents, lowest possible %F/F# | 5 | 5 | 10 | 50.0 | 18.7–81.3% |
| Reference Group: Northern European origin^ | 385 | 534 | 919 | 41.9 | 38.7–45.2% |
Binomial “exact” calculations of the confidence intervals for proportions
Ten CFS family parents: living 5 F/F, 2 F/V, and 0 V/V; 3 deceased.
P <0.05, for 8 or more F/F, calculated with 0.419 as the probability that a parent will be F/F.
From reference (40); the FCGR3A allele encoding V158 CD16A had a frequency of 0.363.
Results
Study participants
The study participants were divided into 3 groups: CFS patients; their family members without CFS; and unrelated healthy controls. The pedigrees of the 5 families are illustrated in Figure 1. Each family had 2 to 5 CFS patients. Eleven of the 13 patients (85%) participated. Six of the 13 patients (46%) were second-generation CFS and four participated, so 36% of the CFS patients in the study were children of CFS patients. The participating CFS patients were 82% female, with a mean age of 23.4 years at first diagnosis and mean duration of illness of 22 years. Twenty-two of 27 (81%) of the eligible family members without CFS participated. The participants are indicated by the FCGR3 genotypes in Figure 1. The controls matched the CFS patients by gender and age. Table 1 indicates the age and gender distributions of the three groups. The patients and controls were gender-matched and predominantly female as is the disease CFS but there were only slightly more women than men among the non-CFS family members. Overall, the three groups share dominant gender, average age, and Caucasian race.
Table 1.
Characteristics of the Three Study Populations
| Characteristic | CFS Cases | Family members w/o CFS | Unrelated healthy controls | |||
|---|---|---|---|---|---|---|
| Number of participants | 11 | 22 | 16 | |||
| Number of families | 5 | 5 | 16 | |||
| Age in years, mean +/− SD | 45.5 +/− 21.3 | 46.2 +/− 14.7 | 42.8 +/− 18.6 | |||
| Sex | Number | % | Number | % | Number | % |
| Female | 9 | 81.8 | 12 | 54.5 | 12 | 75.0 |
| Male | 2 | 18.2 | 10 | 45.5 | 4 | 25.0 |
The CFS patients and their non-CFS family members differed in the symptoms used to diagnose CFS (Table 2). Information excerpted from Rand36 Questionnaires, Fibromyalgia Impact Questionnaires, and Bateman Horne Center medical histories indicates that the patients were ‘moderately’ affected. They were able to walk and lift groceries. All experienced post-exertional malaise. The differences between the patients and their non-CFS family members were significant (P>0.05) for all the CFS diagnostic symptoms.
Table 2A.
Characteristics of the CFS Patients
| Donor numbere | Bateman Horne Center (BHC) Patient Historya | Rand36b Qs re Fatigue; 1 worse >3 | Rand36b (5 worse>1) | Postexertional Malaise (PEM)c | BHC Historya | Fibromyalgia Impact Questionnaired | DNA Sequence | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family # | Gender | Agef | Age of first diagnosis | Duration of CFS, yrs | Q5g Lifting groceries | Q7h Climbing stairs | Q11i Difficulty, walk 1 block | General health Q36j | pem_onsetk : 1 minutes; 2 hours; 3 one day after | pem_durationl: 1 = 1 day; 2=several days; 3 = at least a week | Fibro-myalgia | Head aches | Unrefresh ed sleepm (100% worst) | Memoryn (100% worst) | Muscle paino (100% worst) | Balancep (100% worst) | CD16A Genotype | |
| 3 | 10 | M | 23 | 15 | 8 | 2 | 2 | 3 | 4 | 3 | 3 | Yes | No | 55 | 19 | 51.0 | 14 | F/F |
| 4 | 10 | M | 26 | 6 | 20 | 3 | 3 | 3 | 2 | 1 | 2 | No | No | 14 | 7 | 14.0 | 5 | F/F |
| 13 | 28 | F | 61 | 31 | 30 | 2 | 3 | 3 | 5 | 2 | 2 | Yes | No | 71 | 16 | 37.0 | 21 | F/F |
| 14 | 28 | F | 86 | 53 | 33 | 3 | 3 | 3 | 4 | 2 | 1 | No | Yes | 0 | 56 | 0.0 | 64 | V/F |
| 20 | 20 | F | 30 | 11 | 19 | 2 | 2 | 3 | 5 | 3 | 2 | No | Yes | 82 | 85 | 47.0 | 65 | F/F |
| 21 | 20 | F | 60 | 33 | 27 | 2 | 3 | 3 | 4 | 2 | 1 | No | Yes | 46 | 21 | 64.0 | 62 | F/F |
| 23 | 20 | F | 27 | 6 | 21 | 2 | 2 | 3 | 5 | 2 | 2 | Yes | Yes | 88 | 100 | 54.0 | 62 | F/F |
| 25 | 22 | F | 50 | 18 | 32 | 1 | 1 | 1 | 5 | 1 | 2 | Yes | Yes | 100 | 98 | 77.0 | 94 | F/F |
| 27 | 22 | F | 21 | 11 | 10 | 2 | 2 | 2 | 4 | 1 | 2 | Yes | Yes | 30 | 20 | 70.0 | 0 | F/F |
| 36 | 3 | F | 57 | 24 | 33 | 2 | 2 | 3 | 5 | 2 | 2 | Yes | Yes | 100 | 82 | 58.0 | 70 | F/F |
| 37 | 3 | F | 60 | 49 | 11 | 2 | 2 | 2 | 5 | 2 | 1 | Yes | Yes | 91 | 94 | 74.0 | 73 | F/F |
| Average | 46 | 23.4 | 22.2 | 2.1** | 2.3** | 2.6* | 4.4** | 1.9 | 1.8 | 64%α | 73%β | 61.5* | 54.4* | 49.6** | 48.2** | 91% F/F | ||
| std deviation | 21 | 16.4 | 9.5 | 0.5 | 0.6 | 0.7 | 0.9 | 0.7 | 0.6 | 35.2 | 38.1 | 24.4 | 31.9 | |||||
| median | 50 | 18 | 21 | 2 | 2 | 3 | 5 | 2 | 2 | 71 | 56 | 54 | 62 | |||||
| minimum | 21 | 6 | 8 | 1 | 1 | 1 | 2 | 1 | 1 | 0 | 7 | 0 | 0 | |||||
| maximum | 86 | 53 | 33 | 3 | 3 | 3 | 5 | 3 | 3 | 100 | 100 | 77 | 94 | |||||
| Table 2B. Characteristics of the Family Members without CFS | ||||||||||||||||||
| Number of donors | Family # | % F | Agef | NA | NA | Rand36b Qs re Fatigue; 1 worse >3 | Rand36b (5 worse>1) | NA | NA | BHC Historya | Fibromyalgia Impact Questionnaired | DNA Sequence | ||||||
| 22 | 3, 10, 20, 22, 28 | 55 | Q5g Lifting groceries | Q7h Climbing stairs | Q11i Difficulty, walk 1 block | General health Q36j (5 worse>1) | Fibro-myalgia | Head-aches | Unrefresh ed sleepm (100% worst) | Memoryn (100% worst) | Muscle paino (100% worst) | Balancep (100% worst) | CD16A Genotype | |||||
| Average | 46 | 2.9 | 2.9 | 3.0 | 2.3 | 0% | 23% | 35.5 | 25.3 | 10.0 | 1.9 | 82% F/F | ||||||
| std deviation | 15 | 0.4 | 0.3 | 0.2 | 1.3 | 31.4 | 25.8 | 11.5 | 3.4 | |||||||||
| median | 47 | 3.0 | 3.0 | 3.0 | 2.0 | 22.5 | 17.0 | 7.0 | 0.0 | |||||||||
| minimum | 21 | 2.0 | 2.0 | 2.0 | 1.0 | 0.0 | 0.0 | 0.0 | 0 | |||||||||
| maximum | 69 | 3.0 | 3.0 | 3.0 | 5.0 | 100.0 | 92.0 | 50.0 | 9 | |||||||||
P<0.05 &
P<0.01 by T-test
P<0.05 by Fisher exact test
P<0.05 by Chi square.
BHC data from patient records
Rand36 questionnaire
The Canadian Consensus Criteria defines Post-Exertional Malaise (PEM) as an inappropriate loss of physical and mental stamina, rapid muscular and cognitive fatigability.
Fibromyalgia Impact Questionnaire
Numbers assigned at site of ADCC assays.
Age at time of blood donation
Difficulty lifting or carrying groceries. 1, Yes, limited a lot; 2, Yes, limited a little; 3, No, not limited at all
Climbing one flight of stairs. 1, Yes, limited a lot ; 2, Yes, limited a little; 3, No, not limited at all
Walking one block. 1, Yes, limited a lot ; 2, Yes, limited a little; 3, No, not limited at all
My health is excellent. 1, Definitely true; 2, Mostly true; 3, Don’t know; 4, Mostly false; 5, Definitely false
Does PEM happen …1, minutes after exertion; 2, hours after exertion; 3, a day or more after exertion; 4, not at all
Duration How long does it take you to recover from PEM? 1, minutes after exertion; 2, hours after exertion; 3, a day or more after exertion; 4, not at all
Please rate the quality of your sleep. Slider scale, 0–100: Awoke well rested vs. Awoke very tired
Please rate your level of memory problems. Slider scale, 0 –100: Good memory vs. Very poor memory
Please rate your level of pain. Slider scale, 0 –100: No pain vs. Unbearable pain
Please rate your level of balance problems. Slider scale, 0 –100: No imbalance vs. Severe imbalance
Comparisons of CD16Apos NK blood cell counts
An individual’s in vivo ADCC will be affected by the number of available CD16Apos NK cells and by the activity of the individual’s killer cells. Only the CD16Apos set of NK cells mediates ADCC and most of these CD16Apos NK cells are located in blood. Tissue NK cells lack CD16A (38). In this section we compare CD16Apos NK blood cell counts and in the following sections we compare the ADCC functions of these cells.
Counts of CD16Apos NK cells per ul of blood for the 3 groups of donors are illustrated in Figure 2A. The counts were lower (74%) for CFS patients vs. unrelated healthy controls. However, the counts were indistinguishable between CFS patients and their family members without CFS, 74% and 78% of controls, respectively. These results are suggestive of a family trait rather than a feature specific for CFS patients. The lower counts of each of the two family groups were statistically insignificant compared to the controls but, when the family members were combined, the CD16Apos NK cells were 77% of controls and the family-wide difference was statistically significant P <0.05 by one-tailed analysis. Within each family, the CD16Apos NK cell counts of CFS patients and non-CFS family members were evenly distributed (high and low) (supplement Figure S2A), also consistent with a trait that affected all family members rather than CFS patients preferentially. Three of four families evaluated had low CD16Apos NK counts. One family (#3 which included four 2nd degree relatives) was similar to the unrelated healthy controls. Thus, there is a difference in CD16Apos NK ADCC cell counts between several CFS families and healthy controls. The difference is of unknown cause though it is compatible with genetic inheritance.
Figure 2. ADCC by CFS patients, family members without CFS, and unrelated healthy controls.
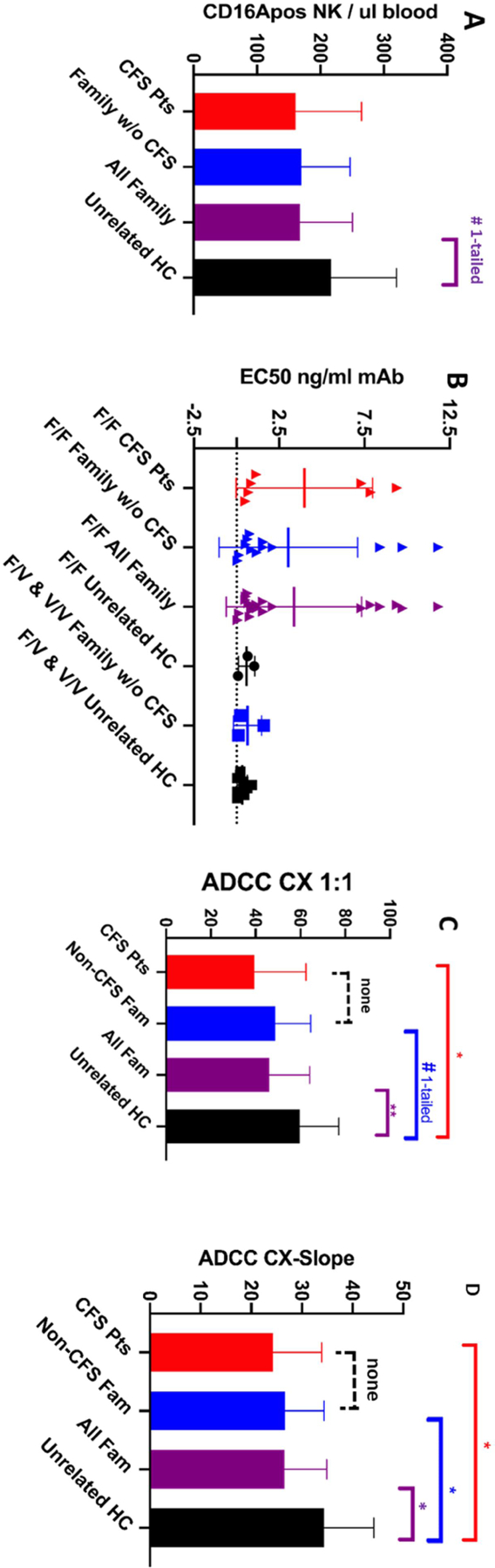
Mean values with standard deviations are indicated. Single asterisks above the brackets indicate Ps<0.05, two asterisks Ps <0.01, for two-tailed T-tests. A hashtag indicates P<0.05 for one-tailed T tests. Unbracketed mean values indicate the absence of statistical significance compared with other means within the data set. A. CD16Apos NK cells per ul blood. B. EC50s (effective concentrations of antibody to support 50% of maximal lysis). CD16A F/F donors are represented separately from the F/V and V/V donors in order to compare genotypes with and without high affinity CD16A V receptors. The statistically significant differences (P<0.05) between CFS patients or CFS family members and controls, are not illustrated because of biases in comparisons of predominantly F/F groups with a predominantly F/V control group. C. ADCC CX1:1s (Cells killed by ADCC at a CD16Apos NK to target cell ratio of 1:1). The CX1:1 data are from families #3, 10 & 28. D. ADCC CX-slopes. CX-slopes are the % increase in dead cells per 10-fold increase in CD16A NK cells. The data are from families #3, 10, 20, 22 & 28.
EC50 NK cell recognition of antibodies
ADCC function is affected by an initial step of NK cell recognition of antibodies and by a subsequent step, the extent of killing that occurs after recognition. EC50s measure the first step. EC50s are the concentrations of antibody needed to support 50% of the maximal ADCC that the lymphocytes can effect. The lower the EC50, the better the NK cells recognize antibody bound to target cells.
High EC50s were required by the CFS patients and by their family members (Figure 2B). Average EC50s were 4.0 +/− 4.0 and 2.6 +/− 3.8 ng/ml obinutuzumab, respectively. The average unrelated control EC50 was 0.39 +/− 0.35 ng/ml (P<0.05 for differences from the CFS groups). The EC50s of the CFS family members were bimodal, with a lower mode greater than the unimodal unrelated controls.
Interpretation of the high EC50s is complicated by differences in FCGR3A genotypes between the families and the controls. To assess if high EC50s are CFS-associated, it is appropriate to compare EC50s of donors with the same FCGR3A genotypes, since the FCGR3A V allele encodes high affinity CD16A which is associated with low EC50s. The CFS family members were 82% CD16A F/F while 80.2% of the unrelated controls had a V allele, data presented later in the results. Only three unrelated controls were F/F, with average EC50s of 0.6 ng/ml, but there were too few unrelated donors to support inter-group statistical comparisons of only F/F genotypes. The high EC50s were common to both CFS patients and non-CFS family members and, as a consequence, disqualify EC50s as a CFS-specific biomarker.
ADCC capacities
ADCC capacity was evaluated using CX1:1and CX-slopes that are illustrated in supplement Figure S3. In contrast to the EC50s, these assessments of ADCC capacity were made with excess concentrations of antibody (625 ng/ml), a concentration that saturated both low and high affinity CD16A receptors. Both CX1:1 and CX-slope are unaffected by CD16A genotypes under these experimental conditions [26]. CX1:1 measures the percentage of ‘target’ cells that are killed at a 1:1 ratio of CD16Apos NK cells to target cells. It measures net capacity: not every cell with CD16A receptors will kill while a few cells are capable of killing more than one target. CX-slope indicates cellular cooperativity when several lymphocytes attack a single target cell.
CX1:1 and CX-slopes were significantly lower for the CFS patients compared to controls (Figure 2C&D). These measurements were lower for both the patients and their non-CFS family members compared to controls: 66% & 82% of controls for CX1:1 and 71% & 78% for CX-slopes, respectively. When all the family members were combined into one group, there was statistically significant lower ADCC compared to the unrelated healthy controls (P<0.01). When each family was considered separately, the lower ADCC occurred in all five families (supplement Figure S2B & C). The CFS patients ran the gamut from high to low ADCC activities within each family. In aggregate, the data are consistent with low ADCC as a potential risk factor for CFS families.
ROC assessment of biomarker potential
We used receiver operating characteristic (ROC) plots to test the validity of low ADCC as a biomarker for CFS. The CFS patients were the true positives. The true negatives were either: non-CFS family members; unrelated controls; or these two groups combined to represent all donors without CFS. Figure 3 illustrates the ROCs. Table 3 indicates the areas under the curves (AUCs). For both CX1:1 and CX-slope tests, AUCs were large and test validity was good for unrelated healthy controls (Ps = 0.06 & 0.02, respectively). However, the AUCs were also greater than 0.5 for the family members without CFS. These AUCs indicate that ADCC is unsuitable as a CFS biomarker when applied to family members without CFS: some of these family members would test false positive for CFS. Ps for test validity for this group of true negatives were insignificant, 0.29 & 0.92 for CX1:1 and CX-slope, respectively). When all donors without CFS were combined into a true negative group with a larger sample number, the ROC test indicated non-validity. Overall, the ROC tests indicate that low ADCC may be a risk factor but is unsuitable as a diagnostic biomarker for CFS, particularly for close relatives of CFS patients.
Figure 3. ROC tests of ADCC to diagnose CFS.
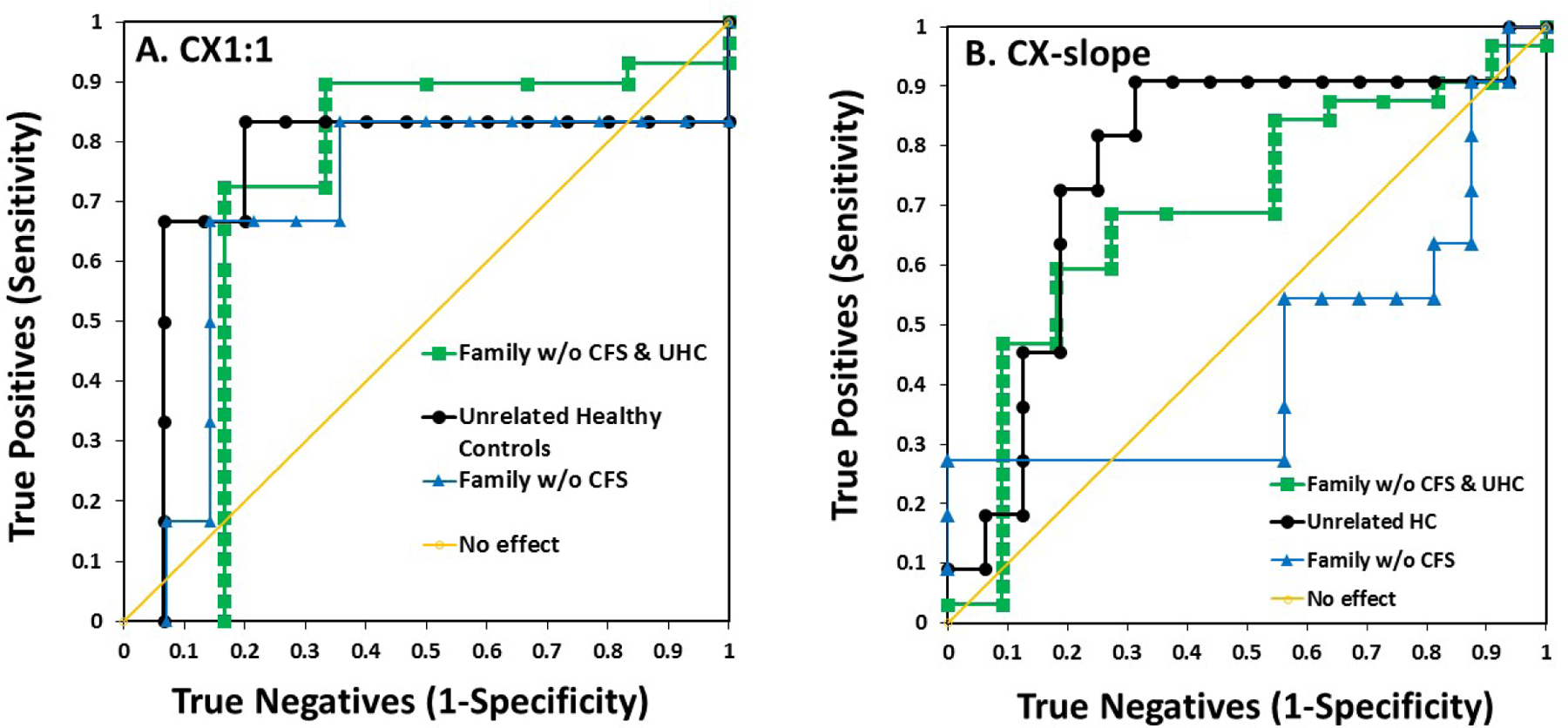
The areas under the curve (AUC) indicate suitability of a test. For the ROC analyses, the true positives were defined as the CFS patients. The true negatives were defined as either: 1) family members without CFS; 2) the unrelated healthy controls; or 3) all donors without CFS, the unrelated donors and the family members without CFS combined. The diagonal line marks an AUC of 0.5 that indicates a test without the ability to distinguish true positives from true negatives.
Table 3.
ROC Assessments of Low ADCC as a Biomarker for CFS
| True vs. False Test Groups | ADCC | Area Under Curve | P |
|---|---|---|---|
| CFS patients vs. Family w/o CFS | CX1:1 | 0.69 | 0.29 |
| CFS patients vs. All Donors w/o CFS | CX1:1 | 0.72 | 0.09 |
| CFS patients vs. Unrelated Healthy Donors | CX1:1 | 0.76 | 0.06 |
| CFS patients vs. Family w/o CFS | CX-slope | 0.45 | 0.92 |
| CFS patients vs. All Donors w/o CFS | CX-slope | 0.69 | 0.08 |
| CFS patients vs. Unrelated Healthy Donors | CX-slope | 0.77 | 0.02 |
Synergistic risk of low ADCC plus low CD16Apos NK cell numbers
The combination of low CD16Apos NK blood cell counts plus low ADCC capacity could represent familial synergistic risk for CFS. Quadrant analyses offer a simple means to determine if a group of subjects is over-represented within the quadrant with combined low activities for two variables compared to the other 3 quadrants. First, we assessed if ADCC and the cell counts varied independently, which would affect synergy. Even though there were positive correlations of the CX1:1s and the CX-slopes with the cell counts (not illustrated), the R2s for linearity were low, consistent with independence. Figure 4A indicates that CFS patients and family members were over-represented in the low-low quadrant for low CX1:1 combined with low CD16Apos NK blood cell counts. These group of all family members together was 24-fold more likely than unrelated healthy donors to be in this low-low quadrant (P = 0.02). All family members together were also 12.5-fold more likely than controls to be in the low-low quadrant for CX-slope vs. cell counts (Figure 4B, P<0.05). Overall, the quadrant analyses indicate that synergistic risks may exist for all members of a family that includes multiple CFS patients.
Figure 4. Assessment of synergistic risks for CFS.
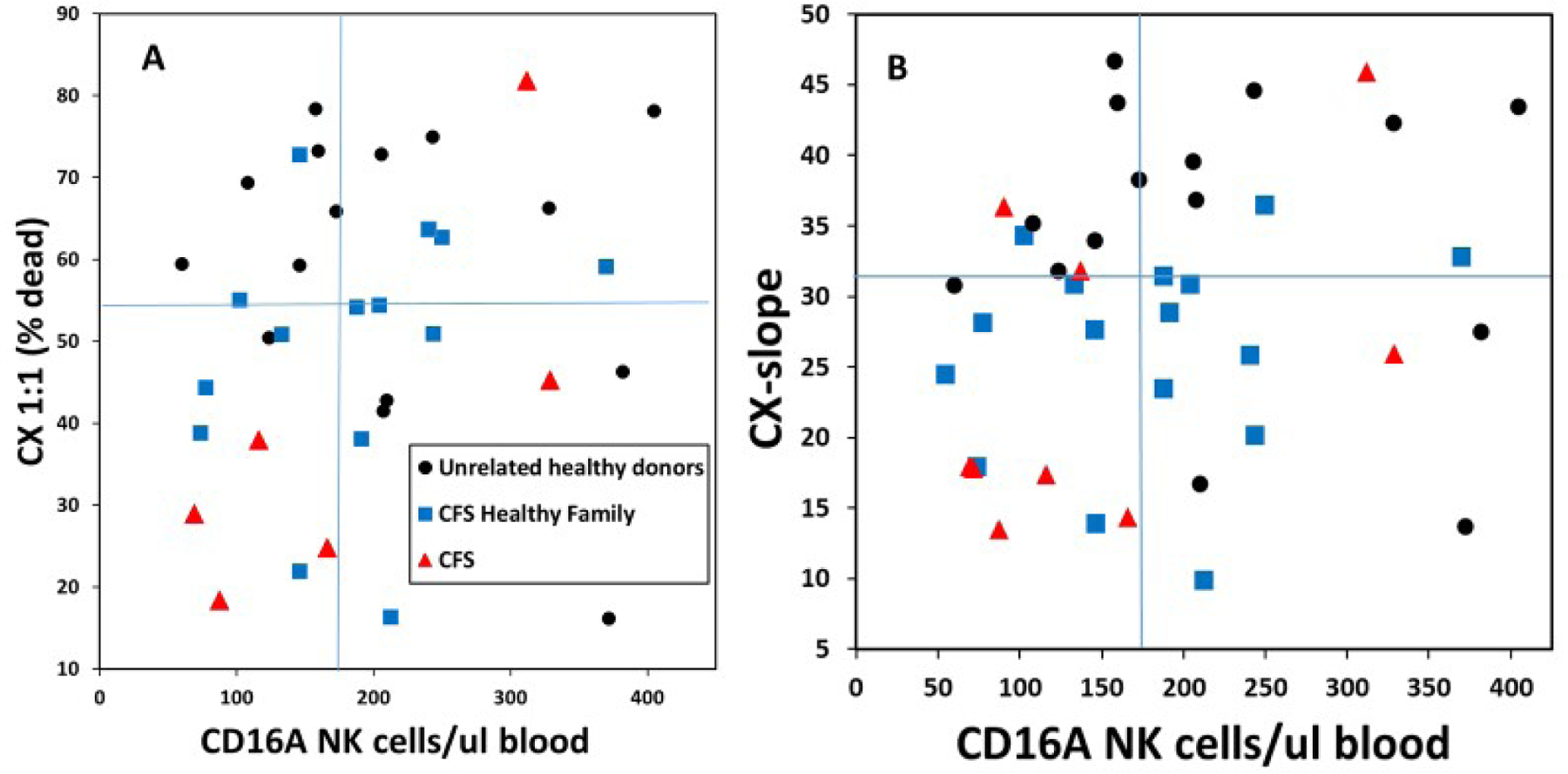
Synergy was evaluated for low CD16A NK effector cell counts combined with low ADCC capacities. CFS family members, with CFS and without CFS, were compared to unrelated controls. Division of the quadrants was based on the median values of each variable. A. CX1:1. Data are from families #3, 10 & 28. B. CX-slopes. Data are from families #3, 10, 22 & 28. Ps <0.05 for family members in the low-low quadrants.
FCGR3A genetics
To qualify the FCGR3A F/F genotype as a risk factor for CFS, this genotype would have to be significantly greater for patients or families than what would occur randomly. Qualification is problematic because the F allele has high frequency. The Utah population of this family study is of non-Finnish northern European descent [39] with an expected 41.9% F/F frequency [40]. The frequency of F/F was 91% for the CFS patients and 82% for the family members without CFS, while the unrelated healthy controls were 18.8% F/F (Table 4). Interpretation of these data is impacted by the fact that F/F by F/F parents will produce only F/F offspring. This scenario of homozygous F/F parents applied to the families of this study (Figure 1). To make matters more challenging to interpret, 3 parents were deceased. The genotypes of the 7 living parents were 5 F/F and 2 F/V. The deceased parents were either F/F or F/V, and not V/V as determined from the genes inherited by their progeny. Two estimates (for the highest and lowest possible F/F frequencies) were used to assess the significance of the parental F/F homozygosity families, see Table 4. The highest estimate of 8 (80%) F/F parents was statistically significantly different from northern European controls (P = 0.02). The lowest estimate of 5 (50%) F/F parents was insignificant (P = 0.42). These results indicate that F/F homozygosity could affect CFS families but may not. The results are simply inconclusive.
Discussion
This pilot report provides five findings that indicate that low ADCC may be of importance in familial CFS. (1) The CD16Apos NK cell blood counts were lower for all family members vs. unrelated healthy controls. Family members, both patients and non-CFS, had ~75% of cytotoxic cells compared to the unrelated healthy controls. (2) Patients and family members required high amounts of antibody (EC50s) for NK cell recognition of cells bound with antibodies. This finding suggests that family members might need to produce more antibodies than controls in order to control infections. (3) ADCC capacity of CFS patients and their family members without CFS was lower than that of unrelated controls. The modest reduction, to ~75% of unrelated control ADCC, may be of unexpected importance because NK cells lose their CD16A receptors during killing. After only a few rounds of killing [41), the NK cells’ ability to mediate ADCC is lost. Re-synthesis of receptors is insufficient to rapidly replenish a depleted supply of CD16Apos NK cells. (4) Synergistic combinations of low CD16A NK cell numbers and low ADCC occurred in the CFS families. Simplistically, a multiplier combination of two reductions, ~0.75 times ~0.75, could potentially decrease the ADCC component of immunity for some individuals to ~0.56 of controls. (5) Parents in the CFS-affected families had a high incidence of homozygous low affinity CD16A F/F antibody receptors. The low sample number contributed to a lack of statistical significance. However, if familial CFS were to have multiple genetic risks, CD16A F/F homozygosity would be a candidate for future investigation.
The findings above resemble and contrast with previous studies. The similar low ADCC of the patients and their unaffected family members resembles observations reported by Dr. Paul Levine et al. [11]. Their study monitored antibody-independent natural killing towards K562 tumor cells. They reported a lack of difference in NK activities (P=0.38) for 8 family members with CFS compared to 12 unaffected family members. The rank order of NK activity was lowest for CFS patients, intermediate for non-CFS family members and greatest for controls. We observed the same rankings for ADCC. For perspective, the low ADCC may apply only to familial CFS. An early report found low ADCC in unrelated CFS patients [42]. However, the ADCC of unrelated CFS patients in Stockholm was lower than healthy controls [43] but statistically insignificant. The investigators of the Scandinavian study used CD107A expression by effector NK lymphocytes rather than cytotoxicity to monitor ADCC, which is a less sensitive way to monitor ADCC and may have affected detection of low ADCC. The issue of low ADCC in unrelated CFS patients (and their family members) warrants further attention. Thus, the present report considerably extends research concerning the role of ADCC in CFS, including consideration of FCGR3A genetics for the first time, and raises new questions.
The synergistic ADCC risks applied to all the CFS family members. Individuals with combined risks could have prolonged clearance times for viral infections while cytotoxic T cells may exert secondary control of viral infections. As observed with type 1A autoimmune diabetes [44], multiple immunological risk factors can predispose individuals to a disease without causing the disease.
This report has several limitations: numbers of families and donors, inclusion of only familial CFS (with the exclusion of families with only one case of CFS), only a single blood sample per donor; one race; one geographic location; and CFS patients with only moderate rather than severe disease. The effects of low ADCC in CFS for other locations, races and in extreme CFS disease are un-addressed. The continuation of research presents opportunities to address these limitations and to benefit the patients by guiding therapies. Identification of CFS patients with low NK and/or low ADCC may indicate those patients most likely to respond to immunomodulatory therapies such as poly(I:C) [45] that promotes NK cell cytotoxic functions. In summary, this report supports a role for low ADCC as a risk factor for familial CFS and suggests aspects of immunology that may also apply to other subgroups of CFS patients.
Supplementary Material
Acknowledgements
We are deeply grateful to the patients and families who made this study possible. Suzanne D. Vernon, Ph.D., at the Bateman Horne Center contributed valuable guidance. Nor Zainal, Ph.D., examined her records to obtain specifics from her study of familial CFS [12]. We thank Roche Pharmaceutical Research & Early Development for obinutuzumab. DNA was isolated by Ms. Laura Meadows and FCGR3A alleles sequenced at the NIH by Stephen K. Anderson, Ph.D. We thank Lynn B. Jorde, Ph.D., University of Utah School of Medicine, for help with the genetic data and Parker Hoshizaki, Terry Woodin, Ph.D., and Stephen K. Anderson, Ph.D. for manuscript editing.
Funding
The research was supported in part by NIH R21 AI117491 awarded to Dr. D Hudig as a co-investigator, by NIH P30 GM GM110767 (Cytometry Center), and by an anonymous generous private donor to the Bateman Horne Center who helped pay for the collection and shipment of blood samples. We would also thank the Nevada INBRE (NIH GM103440) for undergraduate research scholarships (to AP Sung and J-J Tang). The American Association of Immunologists awarded a predoctoral fellowship to AP Sung.
Abbreviations:
- AA
amino acid
- ADCC
antibody-dependent cell-mediated cytotoxicity
- AUC
area under the curve
- CD
cluster of differentiation
- CD16A
IgG Fc-receptor of NK cells
- CFS
chronic fatigue syndrome
- CX1:1
the % cells killed at a 1:1 ratio of CD16Apositive NK cells to Daudi ‘target’ cells
- CX-slope
linear slope of cytotoxicity with increased killer cells
- E:T
effector to target cell ratio
- EC50
the effective concentration of antibody required for 50% of maximal ADCC
- FcR
cellular receptor for the Fc region of immunoglobulin (antibody)
- FCGR3A
the gene encoding CD16A
- KIR
killer cell immunoglobulin-like receptor
- ME
myalgic encephalomyelitis
- NK
natural killer lymphocyte
- PBMC
peripheral blood mononuclear cells
- ROC
receiver-operating characteristic plot
- UHC
unrelated healthy control subject
Biographies
Author Biographies
Alexander P. Sung, M.S., is an MD-PhD student class of 2022 at the University of Nevada, Reno (UNR), School of Medicine (SoM) with Dr. Hudig as his thesis advisor.
Jennifer J-J Tang, M.S., is currently a student at the William S. Boyd School of Law, University of Nevada, Las Vegas. She performed research as an M.S. Biotechnology student at UNR.
Michael J. Guglielmo is a research associate II in the Hudig lab and an M.S. student in Biotechnology at UNR.
Julie Smith-Gagen, Ph.D., is an associate professor of Community Health Sciences. She is a member of the American College of Epidemiology. Her current research focuses on diagnostics and treatments for patients with cancers of unknown primary origin.
Lucinda Bateman, M.D., is a general internist who has specialized in the diagnosis and management of ME/CFS for 20 years. She has participated in many ME/CFS clinical research projects throughout this period.
Lydia Navarrete-Galvan, B.S., is a 2019 UNR graduate with majors in Microbiology and Nutrition. She is currently a research assistant in the Hudig lab and plans to become an MD.
Doug D. Redelman, Ph.D., deceased. He was a research professor in the Department of Physiology, UNR SoM, a lifelong member of the Am. Assn. of Immunologists, and head of the UNR Cytometry Center.
Dorothy Hudig, Ph.D., is a professor in the Department of Microbiology and Immunology, UNR SoM and a lifelong member of the Am. Assn. of Immunologists. Her research interests are granzyme serine proteases and cytotoxic T & NK cell-mediated killing. Dr. Hudig is currently working on aspects of serial NK lymphocyte-mediated killing.
Footnotes
Geolocation (Reno, NV and Salt Lake City, UT, USA)
Competing interests
The authors lack financial and non-financial competing interests in this study.
Data availability
Additional clinical information may be obtained upon request.
References
- 1.Jason LA, Johnson M. Solving the ME/CFS criteria and name conundrum: the aftermath of IOM. Fatigue: Biomedicine,Health & Behavior 2020;8:97–107. [Google Scholar]
- 2.Institute, Medicine of. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness: National Academies Press, Washington, DC; 2015. [PubMed] [Google Scholar]
- 3.Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med 1994;121(12):953–9. [DOI] [PubMed] [Google Scholar]
- 4.Hickie I, Davenport T, Wakefield D, Vollmer-Conna U, Cameron B, Vernon SD, et al. Post-infective and chronic fatigue syndromes precipitated by viral and non-viral pathogens: prospective cohort study. BMJ 2006;333(7568):575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komaroff AL. Advances in Understanding the Pathophysiology of Chronic Fatigue Syndrome. Jama 2019. [DOI] [PubMed]
- 6.Friedberg F Legitimizing myalgic encephalomyelitis/chronic fatigue syndrome: indications of change over a decade. Fatigue: Biomedicine, Health & Behavior 2019;8:24–31. [Google Scholar]
- 7.Reynolds KJ, Vernon SD, Bouchery E, Reeves WC. The economic impact of chronic fatigue syndrome. Cost Eff Resour Alloc 2004;2(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimosako N, Kerr JR. Use of single-nucleotide polymorphisms (SNPs) to distinguish gene expression subtypes of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J Clin Pathol 2014;67(12):1078–83. [DOI] [PubMed] [Google Scholar]
- 9.Janal MN, Ciccone DS, Natelson BH. Sub-typing CFS patients on the basis of ‘minor’ symptoms. Biol Psychol 2006;73(2):124–31. [DOI] [PubMed] [Google Scholar]
- 10.Nagy-Szakal D, Barupal DK, Lee B, Che X, Williams BL, Kahn EJR, et al. Insights into myalgic encephalomyelitis/chronic fatigue syndrome phenotypes through comprehensive metabolomics. Sci Rep 2018;8(1):10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine PH, Whiteside TL, Friberg D, Bryant J, Colclough G, Herberman RB. Dysfunction of natural killer activity in a family with chronic fatigue syndrome. Clin Immunol Immunopathol 1998;88(1):96–104. [DOI] [PubMed] [Google Scholar]
- 12.Walsh CM, Zainal NZ, Middleton SJ, Paykel ES. A family history study of chronic fatigue syndrome. Psychiatr Genet 2001;11(3):123–8. [DOI] [PubMed] [Google Scholar]
- 13.Eaton-Fitch N, du Preez S, Cabanas H, Staines D, Marshall-Gradisnik S. A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst Rev 2019;8(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–25. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest 1997;100(5):1059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daugherty SA, Henry BE, Peterson DL, Swarts RL, Bastien S, Thomas RS. Chronic fatigue syndrome in northern Nevada. Rev Infect Dis 1991;13 Suppl 1:S39–44. [DOI] [PubMed] [Google Scholar]
- 18.Loebel M, Strohschein K, Giannini C, Koelsch U, Bauer S, Doebis C, et al. Deficient EBV-specific B- and T-cell response in patients with chronic fatigue syndrome. PLoS ONE. 2014;9(1):e85387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beqaj SH, Lerner AM, Fitzgerald JT. Immunoassay with cytomegalovirus early antigens from gene products p52 and CM2 (UL44 and UL57) detects active infection in patients with chronic fatigue syndrome. J Clin Pathol 2008;61(5):623–6. [DOI] [PubMed] [Google Scholar]
- 20.Shapiro JS. Does varicella-zoster virus infection of the peripheral ganglia cause Chronic Fatigue Syndrome? Med Hypotheses 2009;73(5):728–34. [DOI] [PubMed] [Google Scholar]
- 21.Komaroff AL. Is human herpesvirus-6 a trigger for chronic fatigue syndrome? J Clin Virol 2006;37 Suppl 1:S39–S46. [DOI] [PubMed] [Google Scholar]
- 22.Chapenko S, Krumina A, Logina I, Rasa S, Chistjakovs M, Sultanova A, et al. Association of active human herpesvirus-6, −7 and parvovirus b19 infection with clinical outcomes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Adv Virol 2012;2012:205085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerr JR, Tyrrell DA. Cytokines in parvovirus B19 infection as an aid to understanding chronic fatigue syndrome. Curr Pain Headache Rep 2003;7(5):333–41. [DOI] [PubMed] [Google Scholar]
- 24.Montoya JG, Holmes TH, Anderson JN, Maecker HT, Rosenberg-Hasson Y, Valencia IJ, et al. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc Natl Acad Sci U S A 2017;114(34):E7150–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soto NE, Straus SE. Chronic Fatigue Syndrome and Herpesviruses: the Fading Evidence. Herpes 2000;7(2):46–50. [PubMed] [Google Scholar]
- 26.Sung AP, Tang JJ, Guglielmo MJ, Redelman D, Smith-Gagen J, Bateman L, et al. An improved method to quantify human NK cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) per IgG FcR-positive NK cell without purification of NK cells. J Immunol Methods. 2018;452:63–72. [DOI] [PubMed] [Google Scholar]
- 27.Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med 2015;7(297):297ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hays RD, Sherbourne CD, Mazel RM. The RAND 36-Item Health Survey 1.0. Health Econ 1993;2(3):217–27. [DOI] [PubMed] [Google Scholar]
- 29.Burckhardt CS, Clark SR, Bennett RM. The fibromyalgia impact questionnaire: development and validation. J Rheumatol 1991;18(5):728–33. [PubMed] [Google Scholar]
- 30.Mata MM, Mahmood F, Sowell RT, Baum LL. Effects of cryopreservation on effector cells for antibody dependent cell-mediated cytotoxicity (ADCC) and natural killer (NK) cell activity in (51)Cr-release and CD107a assays. J Immunol Methods. 2014;406:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langenskiold C, Mellgren K, Abrahamsson J, Bemark M. Determination of blood cell subtype concentrations from frozen whole blood samples using TruCount beads. Cytometry B Clin Cytom 2016. [DOI] [PubMed]
- 32.Milush JM, Lopez-Verges S, York VA, Deeks SG, Martin JN, Hecht FM, et al. CD56negCD16(+) NK cells are activated mature NK cells with impaired effector function during HIV-1 infection. Retrovirology. 2013;10:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudig D, Hunter KW, Diamond WJ, Redelman D. Properties of human blood monocytes. I. CD91 expression and log orthogonal light scatter provide a robust method to identify monocytes that is more accurate than CD14 expression. Cytometry B Clin Cytom 2014;86(2):111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung S, Lin YL, Reed C, Ng C, Cheng ZJ, Malavasi F, et al. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies - impact of effector cells. J Immunol Methods. 2014;407:63–75. [DOI] [PubMed] [Google Scholar]
- 35.Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottcher S, Ritgen M, Bruggemann M, Raff T, Luschen S, Humpe A, et al. Flow cytometric assay for determination of FcgammaRIIIA-158 V/F polymorphism. J Immunol Methods. 2005;306(1–2):128–36. [DOI] [PubMed] [Google Scholar]
- 37.Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem 1993;39(4):561–77. [PubMed] [Google Scholar]
- 38.Moller MJ, Kammerer R, von Kleist S. A distinct distribution of natural killer cell subgroups in human tissues and blood. Int J Cancer. 1998;78(5):533–8. [DOI] [PubMed] [Google Scholar]
- 39.McLellan T, Jorde LB, Skolnick MH. Genetic distances between the Utah Mormons and related populations. Am J Hum Genet 1984;36(4):836–57. [PMC free article] [PubMed] [Google Scholar]
- 40.Tsang A-SMW, Nagelkerke SQ, Bultink IE, Geissler J, Tanck MW, Tacke CE, et al. Fc-gamma receptor polymorphisms differentially influence susceptibility to systemic lupus erythematosus and lupus nephritis. Rheumatology (Oxford). 2016;55(5):939–48. [DOI] [PubMed] [Google Scholar]
- 41.Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. 2014;124(22):3241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.See DM, Broumand N, Sahl L, Tilles JG. In vitro effects of echinacea and ginseng on natural killer and antibody-dependent cell cytotoxicity in healthy subjects and chronic fatigue syndrome or acquired immunodeficiency syndrome patients. Immunopharmacology. 1997;35(3):229–35. [DOI] [PubMed] [Google Scholar]
- 43.Theorell J, Bileviciute-Ljungar I, Tesi B, Schlums H, Johnsgaard MS, Asadi-Azarbaijani B, et al. Unperturbed Cytotoxic Lymphocyte Phenotype and Function in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients. Front Immunol 2017;8:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 2011;91(1):79–118. [DOI] [PubMed] [Google Scholar]
- 45.Strayer DR, Carter WA, Brodsky I, Cheney P, Peterson D, Salvato P, et al. A controlled clinical trial with a specifically configured RNA drug, poly(I).poly(C12U), in chronic fatigue syndrome. Clin Infect Dis 1994;18 Suppl 1:S88–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional clinical information may be obtained upon request.