

Abstract
Introduction
Multiple myeloma (MM) is a plasma cell tumour with over 5800 new cases each year in the UK. The introduction of biological therapies has improved outcomes for the majority of patients with MM, but in approximately 20% of patients the tumour is characterised by genetic changes which confer a significantly poorer prognosis, generally termed high-risk (HR) MM. It is important to diagnose these genetic changes early and identify more effective first-line treatment options for these patients.
Methods and analysis
The Myeloma UK nine OPTIMUM trial (MUKnine) evaluates novel treatment strategies for patients with HRMM. Patients with suspected or newly diagnosed MM, fit for intensive therapy, are offered participation in a tumour genetic screening protocol (MUKnine a), with primary endpoint proportion of patients with molecular screening performed within 8 weeks. Patients identified as molecularly HR are invited into the phase II, single-arm, multicentre trial (MUKnine b) investigating an intensive treatment schedule comprising bortezomib, lenalidomide, daratumumab, low-dose cyclophosphamide and dexamethasone, with single high-dose melphalan and autologous stem cell transplantation (ASCT) followed by combination consolidation and maintenance therapy. MUKnine b primary endpoints are minimal residual disease (MRD) at day 100 post-ASCT and progression-free survival. Secondary endpoints include response, safety and quality of life. The trial uses a Bayesian decision rule to determine if this treatment strategy is sufficiently active for further study. Patients identified as not having HR disease receive standard treatment and are followed up in a cohort study. Exploratory studies include longitudinal whole-body diffusion-weighted MRI for imaging MRD testing.
Ethics and dissemination
Ethics approval London South East Research Ethics Committee (Ref: 17/LO/0022, 17/LO/0023). Results of studies will be submitted for publication in a peer-reviewed journal.
Trial registration number
ISRCTN16847817, May 2017; Pre-results.
Keywords: myeloma, clinical trials, statistics & research methods
Strengths and limitations of this study.
This is the first time in the UK that patients with newly diagnosed multiple myeloma may be entered into a clinical trial prospectively according to their genetic risk profile.
A flexible multiple outcome, multistage Bayesian design is used to enable early stopping for lack of efficacy.
No concurrent control arm is included due to the availability of near concurrent historical control data from the Myeloma XI trial.
Introduction
Multiple myeloma (MM) is a clonal disorder of plasma cells which accumulate in the bone marrow leading to cytopenias, bone resorption, renal impairment, infection and the production of a monoclonal protein.1 MM represents 1.5% of all malignant diseases, with an incidence of 9/100 000 per year accounting for around 5800 new cases each year in the UK (3000 deaths per year).2 Median age at diagnosis is 69 years but 37% of patients are diagnosed before the age of 65 (including 15%<55).3 Median overall survival (OS) of younger patients is approximately 10 years.4–11 However approximately 20% of patients have a significantly worse prognosis, with estimated survival of <3 years and are characterised as having high-risk (HR) disease.7 12 13 A number of genetic lesions and gene expression profiles (GEP) have been identified as associated with HR disease,7 and molecular risk models based on these markers can be used to predict HR disease in a clinical setting. Further research is ongoing to identify additional HR markers and to better understand the mechanisms driving this tumour biology.
Unfortunately, patients with HR disease have, in terms of absolute outcome, benefitted less from the introduction of novel therapies than standard risk (SR) patients.14–20 It is important to define the optimal way to treat this group of patients given the number of available novel agents with favourable toxicity profiles allowing the use of combination therapy, consolidation and maintenance therapy. Here, we describe the protocol for the MUKnine trial, a phase II study evaluating optimised combination of biological therapy in patients with newly diagnosed HRMM and plasma cell leukaemia (PCL), incorporating a screening and observational study for patients with SR disease. The trial has completed recruitment and is currently in follow-up.
Defining HR disease
In a recent meta-analysis of 1905 trial patients from the MRC Myeloma IX and NCRI Myeloma XI trials, recurrent chromosomal translocations t(4;14), t(14;16), t(14;20) and copy number aberrations (CNA) gain(1q) or del(17p) were independently associated with shorter progression-free survival (PFS) and OS. Presence of two or more such HR lesions, also termed double-hit,7 was associated with particularly adverse outcome and increased specificity of outcome prediction considering individual lesions in isolation. The cosegregation model is exclusively based on molecular features of the tumour cell and contrasts to risk predictors which require inclusion of clinical risk markers (renal function, age, performance status) or their proxies, such as the international staging system.12 For participants fit to receive intensive therapy, HR can thus be specifically defined by presence of two or more cytogenetically adverse lesions (t(4;14), t(14;16), t(14;20), del(1p32) gain(1q) or del(17p)).
The prognostic relevance of GEP risk signatures, in particular EMC-92, from which the SKY92 MMProfiler diagnostic assay was developed, has been demonstrated in the Myeloma IX trial dataset.21 A recent analysis including Myeloma IX and Myeloma XI trial patients demonstrated independent association of GEP SKY92 HR and genetic HR markers with adverse outcome in MM.11 13 21–24 Results suggest that both tests assay different clinically relevant qualities of HR biology. Combining GEP and double-hit genetic risk information identifies about 20%–30% of patients with markedly short PFS and OS.
The exact impact of single nucleotide variants on MM risk status is still under investigation. However, very recent evidence, published after design of MUKnine, seems to confirm that structural aberrations such as translocations and CNA are the dominant markers of HRMM, although detail on their assessment varies.25–27 The observation of poor prognosis associated with HR disease defined by such molecular criteria is consistent with clinical studies carried out by other trial groups.5–11 21 22 24 28 29 Clearly, a focused approach to improve the treatment and outcome of this poor performing subgroup of MM patients is essential.
Treatment
Recent data have demonstrated efficacy of the combination of multiple novel agents in HR disease.30 Until the molecular mechanisms contributing to HR biology can be directly targeted, combinations of multiple novel agents and ongoing therapy to induce and maintain remission are the most efficacious therapeutic principles.31
Maximising exposure to novel agents as an alternative to multidrug cytotoxic alkylating chemotherapy is hypothesised to benefit HR patients. Ongoing use of a combination of biological agents with favourable toxicity profiles can potentially minimise the chance of relapse due to sustained multiangled pressure on the MM repopulating cell pool.
Long-term exposure to thalidomide does not benefit HR patients.32 33 However, lenalidomide maintenance in newly diagnosed HR patients (t(4;14) or del 17p) does have a PFS and OS benefit.34 There is a substantial body of evidence suggesting that HR patients benefit from long-term exposure to proteasome inhibition such as bortezomib.35–39
The combination of bortezomib and lenalidomide as induction and consolidation therapy is safe and deliverable with a number of studies using this approach.40 Adding cyclophosphamide to this triplicate approach is safe, nevertheless the lenalidomide, cyclophosphamide, bortezomib and dexamethasone combination failed to show any additional benefit to lenalidomide, bortezomib and dexamethasone in the EVOLUTION study.41 However, this study evaluated all genetic risk groups and it is hypothesised that the addition of low-dose alkylating therapy may present an additional benefit in a HR population with highly proliferative subclones.
Daratumumab is a monoclonal antibody that targets the CD38 molecule and has multiple mechanisms of action against MM cells. It has demonstrated activity in MM as a single agent and in combinations with lenalidomide and dexamethasone where it enhances the potency of other drugs such as lenalidomide offering an interesting alternative to chemotherapy in MM.42 The addition of daratumumab to standard of care regimens improved outcome and combining with lenalidomide or bortezomib appears to improve the poor outcomes associated with HR disease.43 44
While tandem autologous stem cell transplantation (ASCT) may offer prolongation of response in comparison with single procedures, the comparative studies reported at time of design of MUKnine were undertaken in an era in which novel agents were not routinely incorporated in clinical practice.45 Recent exploratory analyses have suggested the potential advantage of tandem ASCT for patients with HR disease.46 Depth of response is associated with duration of response and therefore optimising the induction, consolidation and maintenance approach with a single ASCT is an alternative way to achieve minimal residual disease (MRD) negative disease state. Melphalan has been combined with bortezomib in phase II studies demonstrating safety and improvement in complete response (CR) rates compared with conventional high-dose melphalan conditioning.47 Although a recent report stated no PFS benefit of a Velcade-augmented ASCT in a randomised trial, results for an ultra-HR group such as double-hit MM are unknown.48 The highly proliferative behaviour of double-hit disease and GEP HR provides rationale for a bridging treatment for the 3 months recovery period post ASCT.
Rapid tumour evolution and associated early relapse are key characteristics of HRMM, even in patients who have achieved deep remission after ASCT.49 Maintaining multiagent treatment intensity around and long-term after ASCT to limit size of the clonal pool as well as molecular avenues for tumour escape seems currently one of the most promising treatment strategies for HRMM, with the aim of achieving sustained deep responses in at least some patients.50 Longitudinal MRD monitoring can predict remission status with higher sensitivity than standard biochemical/protein analyses and could be of use in identifying patients with HRMM benefitting most from treatment early. As bone marrow biopsy-based MRD assessment may be biased due to spatial disease heterogeneity, sensitive whole body imaging can be performed in parallel to capture residual disease in other bone marrow or soft tissue areas. Whole body diffusion weighted MRI is a particularly sensitive imaging modality for MM, and standardised image acquisition and interpretation guidelines make implementation in multicentre clinical trials feasible.51 52
In line with this, the MUKnine OPTIMUM trial has been designed to evaluate the following treatment regimen in patients with HRMM, the full schedule is given in table 1:
Table 1.
Treatment schedule
| Induction treatment | |||
| Regimen: CVRDd to maximum response (or a maximum of 6 cycles of bortezomib) | Cycle duration: 21 days | ||
| Drug | Dose | Route | Days |
| Cyclophosphamide | 500 mg | PO | 1 and 8 |
| Bortezomib | 1.3 mg/m2 | SC | 1, 4, 8, 11 |
| Lenalidomide | 25 mg | PO | 1–14 |
| Daratumumab | 16 mg/kg (actual body weight) | IV | 1, 8, 15† (cycles 1 and 2) |
| 1 only (cycle 3 onwards) | |||
| Dexamethasone* | 20–40 mg | PO/IV | 1, 4, 8, 11 |
|
Autologous stem cell transplant (ASCT)
Cyclophosphamide and GCSF mobilisation is recommended | |||
| Regimen: bortezomib HD-MEL+ASCT | |||
| Drug | Dose | Route | Days |
| Melphalan | 200 mg/m2 | IV | −1 |
| Bortezomib | 1.3 mg/m2 | SC | −1, (8–18 hours post Melphalan) |
| Autologous stem cell return | IV | 0 | |
| Bortezomib | 1.3 mg/m2 | SC | +5, +14, then weekly to consolidation 1 |
|
Consolidation treatment 1
To begin between 100 and 120 days post ASCT | |||
| Regimen: VRDd×6 cycles* | Cycle duration: 28 days | ||
| Drug | Dose | Route | Days |
| Bortezomib | 1.3 mg/m2 | SC | 1, 8,15, 22 |
| Lenalidomide | 25 mg | PO | 1–21 |
| Daratumumab | 16 mg/kg (actual body weight) | IV | 1 |
| Dexamethasone* | 20–40mg | PO/IV | 1, 8,15†, 22 |
| Consolidation treatment 2 | |||
| Regimen: VRD×12 cycles* | Cycle duration: 28 days | ||
| Drug | Dose | Route | Days |
| Bortezomib | 1.3 mg/m2 | SC | 1, 8,15 |
| Lenalidomide | 25 mg | PO | 1–21 |
| Daratumumab | 16 mg/kg (actual body weight) | IV | 1 |
| Maintenance treatment | |||
| Regimen: RD until progression | Cycle duration: 28 days | ||
| Drug | Dose | Route | Days |
| Lenalidomide | 10 mg | PO | 1–21 |
| Daratumumab | 16 mg/kg (actual body weight) | IV | 1 |
*On days where participants receive dexamethasone 40 mg at site (i.e. predaratumumab infusion), dexamethasone must not be self-administered at home too.
†On day 15, participants will receive premed as per SPC (eg, methylprednisolone).
CVRDd, cyclophosphamide, bortezomib (Velcade), lenalidomide (Revlimid), daratumumab (Darzalex), dexamethasone; GCSF, granulyte colony-stimulating factor; HD-MEL, high dose melphalan; IV, intravenous; RD, lenalidomide, daratumumab; SC, subcutaneous; SPC, summary of product characteristics; VRD, bortezomib, lenalidomide, daratumumab; VRDd, bortezomib, lenaliomide, daratumumab, dexamethasone.
-
CVRDd (induction)—cyclophosphamide, bortezomib (Velcade), lenalidomide (Revlimid), daratumumab (Darzalex), dexamethasone
Based on the EVOLUTION trial.41 Daratumumab doses are used in ongoing clinical trials.53
-
Melphalan—bortezomib ASCT
Melphalan 200 mg/m2 is standard practice in Europe for induction consolidation treatment.54 55 The addition of bortezomib in phase II studies demonstrated safety and improvement in CR rates compared with conventional high-dose melphalan conditioning.47 Velcade weekly monotherapy during the clinical recovery period from ASCT limits very early disease relapse in the HR population.
-
VRDd (consolidation 1)—bortezomib, lenaliomide, daratumumab, dexamethasone
Doses for VRd combination are based on IFM 2008-0140 and IFM 2009-02/DFCI. Daratumumab doses are used in current clinical trials.53
-
VRD (consolidation 2)—bortezomib, lenalidomide, daratumumab
The dose of VRD during consolidation 2 is used to minimise effects of long term corticosteroid use and risks of long-term neuropathy with weekly bortezomib with no break in treatment. Using existing daratumumab dosing schedules it is anticipated this will be a tolerable longer term combination.
-
RD (maintenance)—lenalidomide, daratumumab
The dose of lenalidomide is based on two pivotal studies34 56 and is the currrent dose used in the Myeloma XI trial.20 Daratumumab doses are used in current clinical trials.53
Current protocols
Current protocols: MUKnine a, v2.0, 25/07/2018. MUKnine b, v4.0, 14/05/2020.
Methods and analysis
Aims
To assess whether future trials in this setting are feasible and to determine risk status for participants with MM in order to deliver novel therapy to those deemed HR.
To determine whether it is possible to improve the outcome of HR patients by using multiple biological agents during induction, ASCT, consolidation and maintenance, and to provide evidence for the future evaluation of these high-cost interventions.
Primary objectives
Assess whether molecular risk-defining investigations can be turned around within 8 weeks.
Determine whether the combination of three novel agents bortezomib, lenalidomide and daratumumab in combination with low-dose cyclophosphamide and dexamethasone is sufficiently active in terms of PFS in a HR population to take forward to a phase III trial.
Secondary objectives
Secondary objectives include evaluating safety and toxicity profiles of trial treatment, evaluating additional measures of treatment activity and assessing quality of life. In patients not identified as having HR disease, secondary objectives are to summarise treatment pathways and clinical outcomes in this setting.
Exploratory objectives
To explore novel molecular biomarkers associated with treatment activity, and evaluate germline variability/mutations, genomic instability and clonal evolution.
An exploratory imaging substudy is included to explore the association of imaging MRD status with clinical outcomes and to assess patterns of disease distribution by whole body diffusion-weighted (DW)-MRI.
Trial design
The MUKnine OPTIMUM trial is comprised of two components, MUKnine a and MUKnine b, as outlined in figure 1. MUKnine a is a genetic screening component, where patients with suspected symptomatic MM will be screened to determine their risk status. Patients identified as not having HR disease will receive treatment as standard of care and will have data collected on their treatment and survival. Patients who are identified as having HR disease or PCL are invited to take part in the second component, MUKnine b, a single arm phase II, multicentre trial. MUKnine b incorporates interim assessments for futility using a Bayesian strategy for monitoring multiple outcomes proposed by Thall et al 57 58 and extended by Thall and Sung.59 The trial is single arm to ensure a feasible sample size given the availability of molecularly matched individual participant data from currently running trials (Myeloma XI/XI+). This provides a body of almost concurrent control data available for the purpose of exploratory statistical comparison.
Figure 1.
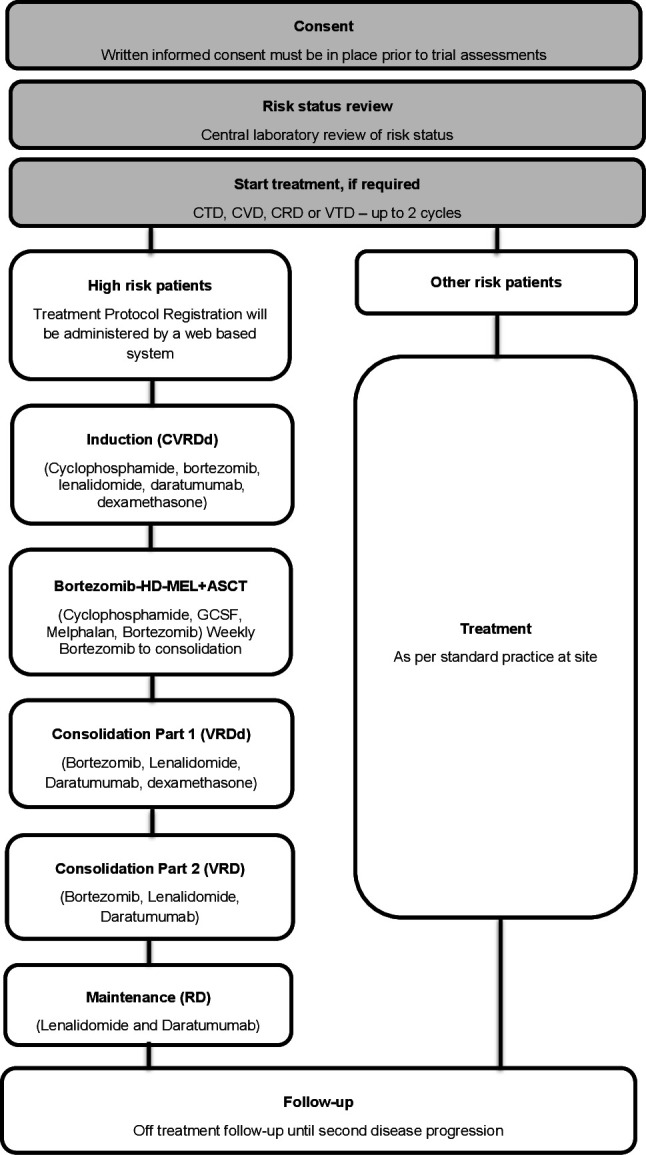
MUKnine OPTIMUM trial design. ASCT, autologous stem cell transplantation; CRD, cyclophosphamide, lenalidomide, dexamethasone; CTD, cyclophosphamide, thalidomide, dexamethasone; CVD, cyclophosphamide, bortezomib, dexamethasone; GCSF, granulyte colony-stimulating factor; HD-MEL, high dose melphalan; VTD, bortezomib, thalidomide, dexamethasone.
Whole-body DW-MRI is a functional method capable of detecting small-volume disease activity in MM,60 61 being used in standard practice at several academic UK hospitals already, demonstrating excellent performance in guiding therapy on a day-to-day basis. An exploratory substudy is incorporated in MUKnine using DW-MRI for disease distribution assessment and imaging MRD in combination with cellular (bone marrow) MRD.
Sample size
Recent data from the Myeloma XI trial demonstrate a median PFS for patients with HR disease in the intensive pathway of 19.7 months (598 patients12). With a median PFS of 19–20 months in the control arm, we require 92–94 patients to observe a 25% difference in median PFS (corresponding to a difference of 4.8–5.0 months) in the 85% credible interval. Allowing for slight changes in the actual count data, we require 95 HR patients to be registered.
A sample size re-estimation using individual patient data from Myeloma XI/XI+, when available, allows the number of HR patients required to detect a 25% difference in median PFS to be increased to 105. In order to include 105 HR patients, approximately 620 patients with MM would need to be registered at diagnosis, assuming approximately 10%–15% failed diagnostic tests, and approximately 20% patients identified as HR.
The trial design includes interim analyses after every cohort of 10 MUKnine b participants have been followed-up to 120 days post ASCT. Until recruitment is complete, the trial could be terminated early for futility on the basis of MRD status and PFS at 100 days post ASCT.
Consent, eligibility, screening and registration
Participants are recruited from UK National Health Service (NHS) hospitals. Hospital sites delivering the HR treatment are approved sites within the Myeloma UK Early Phase Clinical Trials Network62 and patients recruited from sites outside of the network sites are referred to receive treatment, to ensure sufficient patient reach to achieve target sample size. The imaging substudy is undertaken at select sites with appropriate radiology capacity. Assenting patients will provide written informed consent and be registered.
Patients presenting who are likely to have symptomatic MM (identified by pretests performed as standard) are approached prior to having a bone marrow biopsy for diagnosis or confirmation of MM. A full list of inclusion and exclusion criteria is in table 2. No age cut-off is incorporated for transplant eligibility, as per Myeloma XI/XI+ and standard practice.
Table 2.
Eligibility criteria for trial entry and continuing treatment through each stage
| Inclusion criteria | Exclusion criteria |
Screening
|
Only those taking part in the imaging sub study have these exclusions:
|
Treatment
|
Only those taking part in the imaging sub study have these exclusions:
|
Autologous stem cell transplant
|
|
Consolidation part 1
|
|
Consolidation part 2
|
|
Maintenance
|
|
ALT, alanine transaminase; AST, aspartate transaminase; CR, complete response; CRD, cyclophosphamide, lenalidomide, dexamethasone; CTCAE, common terminology criteria for adverse events; CTD, cyclophosphamide, thalidomide, dexamethasone; CVD, cyclophosphamide, bortezomib, dexamethasone; CVRDd, Cyclophosphamide, bortezomib (Velcade), lenalidomide (Revlimid), daratumumab (Darzalex), dexamethasone; DW, diffusion-weighted; HDM-V, high dose mephalan with Velcade; HMDS, Haematological Malignancy Diagnostic Service; ICR, Institute of Cancer Research, London; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes; VRD, bortezomib, lenalidomide, daratumumab; VRDd, bortezomib, lenaliomide, daratumumab, dexamethasone; VTD, bortezomib, thalidomide, dexamethasone.
Patients are provided with information about the trial and if agreeable are consented for the bone marrow biopsy to allow samples to be sent to central laboratories and for screening. This consent allows follow-up data to be collected under the MUKnine a protocol if the patient is found not to have HR disease. Patients are registered to the trial via a web-based system (provided by University of Leeds) prior to any trial-specific assessments being conducted. Participants can also optionally consent to the imaging substudy. Participants retain the right to withdraw at any time without giving reasons and without their further treatment being prejudiced.
Bone marrow and blood samples are taken as per standard care and sent to the Institute of Cancer Research, London (ICR) by next day postal delivery for genetic molecular risk profiling.
HR status is determined by the presence of one or more of the following, based on the International Myeloma Working Group guidelines,63 the Myeloma IX trial and the EMC92 GEP model3 5 10 21 64:
Two or more adverse lesions (t(4;14), t(14;16), t(14;20), gain(1q), del(17p), del(1p)).
GEP—HR score as per EMC92/SKY92 GEP model.
PCL, defined as the presence of more than 2×109/L peripheral blood plasma cells or a plasmacytosis accounting for >20% of the differential white cell count.
Patients identified as having HR disease are provided with a patient information sheet detailing the HR treatment schedule in MUKnine b and consented if willing to participate. A further registration documents all patients going on to HR treatment. If the patient does not wish to receive HR treatment they continue with standard treatment and data collected through the MUKnine a protocol.
For all patients at screening, bone marrow samples are sent to Haematological Malignancy Diagnostic Service, Leeds, for MRD monitoring. Blood and urine samples are sent to Clinical Immunology Service, University of Birmingham for disease response assessments. A cell-free DNA peripheral blood sample is sent to the ICR.
Interventions
On first consent, treatment with standard local treatment may commence for up to 2 cycles (up to 8 weeks) while central molecular risk profiling is performed. Treatment may be with cyclophosphamide, thalidomide, dexamethasone, cyclophosphamide, lenalidomide, dexamethasone, bortezomib, thalidomide, dexamethasone or cyclophosphamide, bortezomib, dexamethasone to further take part in the MUKnine trial. This allows participants to start treatment for MM while awaiting results from risk-defining genetic investigations.
MUKnine a: participants not identified as having HR disease continue to receive standard treatment or treatment as directed by their clinician and are followed up regularly, with information on their treatment pathway and outcomes collected.
MUKnine b: participants identified as having HR disease and who consent to take part in the HR treatment schedule receive treatment as in table 1. Eligibility criteria to continue treatment through each stage of ASCT, consolidation part 1 and 2, and maintenance, are detailed in table 2.
Each individual drug in the schedule may be dose reduced if toxicity is experienced, as deemed necessary by the treating physician and in line with standard reductions used for these treatments (table 3). Dose reductions can be made for grade 1 toxicity (eg, neuropathy) to maximise long-term tolerability and treatment effect in this patient group. Dose reductions from pretrial treatment may be continued at induction treatment. The majority of treatment is delivered in hospital; therefore, adherence is as per protocol. Patients are reminded of treatment scheduling for oral medication at each cycle prescription.
Table 3.
Dose modifications
| Cyclophosphamide Modifications are at the discretion of the investigator Renal impairment—a dose reduction of 50% for creatinine clearance of 10 mL/min is recommended Hepatic impairment—a dose reduction to 350 mg is recommended with a serum bilirubin of >2.5 times the upper limit of normal (ULN) | ||||||
| Bortezomib | ||||||
| Induction dose reductions | ||||||
| Regimen: first dose reduction CVRDd | Cycle duration: 21 days | |||||
| Drug | Dose | Route | Days | |||
| Cyclophosphamide | 500 mg | PO | 1 and 8 | |||
| Bortezomib | 1.3 mg/m2 | SC | 1, 8, 15 | |||
| Lenalidomide | 25 mg | PO | 1–14 | |||
| Daratumumab | 16 mg/kg (actual body weight) | IV | 1, 8, 15 (cycles 1 and 2) | |||
| 1 only (cycle 3 onwards) | ||||||
| Dexamethasone | 20–40 mg | PO/IV | 1, 8, 15 | |||
| Postinduction dose reductions | ||||||
| Bortezomib schedule | Dose levels | |||||
| 0 | −1 | −2 | −3 | −4 | ||
| Twice weekly schedules | 1.3 mg/m2 d 1, 4, 8, 11 | 1.3 mg/m2 d 1, 8, 15 | 1.0 mg/m2 d 1, 8, 15 | 1.3 mg/m2 d 1, 15 | Stop | |
| Once weekly schedules | 1.3 mg/m2 d 1, 8, 15, (22) | 1.0 mg/m2 d 1, 8, 15 (22) | 1.0 mg/m2 d 1, 15 | 0.7 mg/m2 d 1, 15, | Stop | |
| Consolidation 1 | 1.3 mg/m2 d 1, 8, 15, 22 | 1.0 mg/m2 d 1, 8, 15, 22 | 1.0 mg/m2 d 1, 15 | 0.7 mg/m2 d 1, 15, | Stop | |
| Consolidation 2 | 1.3 mg/m2 d 1, 8, 15 | 1.0 mg/m2 d 1, 8, 15 | 1.0 mg/m2 d 1, 15 | 0.7 mg/m2 d 1, 15, | Stop | |
| Neuropathy- CTCAE Grade 1 with pain or grade 2—withhold bortezomib until returns to baseline. Dose reduce 1 level; CTCAE Grade 2 with pain or grade 3—withhold bortezomib until returns to baseline. Dose reduce 2 levels; CTCAE Grade 4—discontinue treatment Renal impairment—dose reduce at the discretion of the clinician Hepatic impairment—moderate or severe impairment (>1.5–3×ULN) should start on a reduced dose of 0.7 mg/m2 during the first cycle of treatment and dose escalate to 1.0 mg/m2 or dose reduce to 0.5 mg/m2 may be considered Grade 3 Non-haematological toxicity—withhold until symptoms of toxicity resolve and reduce one dose. Grade 4 haematological toxicity—withhold until symptoms of toxicity resolve and reduce one dose. Support may be given. | ||||||
| Lenalidomide schedule | Dose levels | |||||
| 0 | −1 | −2 | −3 | −4 | −5 | |
| 25 mg | 20 mg | 15 mg | 10 mg | 5 mg | 2.5 mg | |
| Thrombocytopenia—<25×109/L stop lenalidomide for the remainder of the cycle. Return to ≥50 × 109/L decrease by 1 dose level to resume the next cycle. Neutropenia—first fall to <0.5×109/L omit lenalidomide until a return to ≥0.5 × 109/L when neutropenia is the only toxicity. Resume lenalidomide at one dose lower. For each subsequent drop to ≥0.5 × 109/L omit lenalidomide, resume lenalidomide decreased by 1 dose level at the next cycle. Renal impairment—30–50 mL/min 10 mg daily;<30 mL/min, not requiring dialysis 7.5 mg daily or 15 mg every other day; <30 mL/min, requiring dialysis 5 mg daily administered following dialysis Other non-haematological toxicities: CTCAE grade 3 and 4 related to lenalidomide should be stopped and started 1 dose lower when toxicity has resolved to grade 2 at clinicians discretion. Rash—interrupt or discontinue for grade 2 or 3. Grade 4 rash discontinue including angioedema, exfoliative or bullous rash or Steven Johnson syndrome or toxic epidermal necrosis. | ||||||
| Daratumumab schedule | Frequency | Dose held | Dosing restart | |||
| Induction cycles 1 and 2 | Weekly | >3 days | Next planned weekly dose | |||
| Induction cycles 3–6 | Monthly | >1 week | Next planned weekly dose | |||
| Consolidation 1, Consolidation 2, Maintenance | Monthly | >2 weeks | Next planned weekly dose | |||
Follow the daratumumab SPC. The daratumumab infusion must be withheld to allow for recovery from toxicity ONLY where any of the following criteria are met and the event cannot be ascribed to lenalidomide or cyclophosphamide.
| ||||||
| Dexamethasone Occasionally patients will not be able to tolerate because of corticosteroid effects. Dose reductions from 40 to 20 mg daily. Further dose reductions to 10 mg daily is acceptable followed by the omission of dexamethasone If the bortezomib schedule changes, dexamethasone should change in line with it. | ||||||
| Melphalan Dose may be adjusted based on performance status and clinical judgement in discussion with the Chief Investigator GFR measured by Cockcroft & Gault formula or EDTA—>50 mL/min 200 mg/m2; 30–50 mL/min 140 mg/m2;<30 mL/min 100 mg/m2 | ||||||
CTCAE, common terminology criteria for adverse events; CVRDd, Cyclophosphamide, bortezomib (Velcade), lenalidomide (Revlimid), daratumumab (Darzalex), dexamethasone; GFR, glomerular filtration rate; IV, intravenous; SC, subcutaneous; SPC, summary of product characteristics.
Trial assessments
During treatment
MUKnine a: for non-HR participants, a summary of treatment received in each phase of treatment is collected. Central samples are collected at the end of any line of standard treatment for response assessment. For patients participating in the imaging study a DW-MRI scan is performed at 100–120 days and 21 months post ASCT, along with bone marrow, peripheral blood and urine samples for disease assessment.
MUKnine b: for HR participants, trial assessments are performed in line with the schedule of assessments in table 4. Data are collected at each cycle of treatment and at the end of each phase of treatment, thus limiting loss to follow-up. All adverse events will be collected for all participants from the first investigational medicinal product (IMP) dose until 90 days after the date of the last dose of study drugs.
Table 4.
Trial assessments
| Investigations | All Patients | Non-HR patients | HR patients | |||||||||
| Screening—all participants | Prior to any new line of treatment | Post any line of treatment | First and second disease progression | Before starting MUKnine treatment* | Prior to each cycle of induction treatment CVRDd† | End of induction treatment | Autologous stem cell transplant¶ | 100–120 days post transplant | Prior to each cycle of consolidation part 1 (VRDd), consolidation part 2 (VRD) and maintenance treatment (RD) | End of consolidation part 1 (VRDd), consolidation part 2 (VRD) and maintenance treatment (RD) | First and second disease progression | |
| Consent | X | X | ||||||||||
| Medical history | X | X | ||||||||||
| Symptom-directed physical exam (including weight, ECOG) | X | X | X | X | X | X | X | X | X | X | ||
| Haematology and biochemistry test | X | X | X | X | X | X | X | X | X | X | ||
| Disease assessment‡ | X | X | X | X | X | X | X | X | X | X | X | X |
| DW-MRI Imaging†† | X | X | X (Part 2 only) | |||||||||
| ECG | X | |||||||||||
| Pregnancy testing as required | X | X | X | X | X | X | ||||||
| Participant questionnaires | X | X | X | X** | X | |||||||
| Details of treatment | X | X | X | X | X | X | X | |||||
| Clinical assessment of treatment benefit | X | X | X | |||||||||
| Central laboratory samples | ||||||||||||
| Bone marrow aspirate | X | X | X | X | X | X | ||||||
| Peripheral blood | X | X | X | X § | X | X | X§, ** | X | X | |||
| Urine sample | X | X | X | X § | X | X | X§, ** | X | X | |||
*Treatment must start within 14 days of registration to MUKnine treatment.
†All assessments must be performed within 72 hours prior to day 1 of each cycle of treatment.
‡Response assessments must be made in line with the International Myeloma Working Group criteria.
§Cycle 1 day 1 only.
¶ Autologous stem cell transplant will be performed as per local practice with local monitoring of adverse events and haematology tests. Participants will be given weekly bortezomib until 100–120 days post transplant, the assessments will be performed monthly during this time for the trial.
**3 monthly during treatment.
††If site and participant taking part in the imaging substudy.
CVRDd, Cyclophosphamide, bortezomib (Velcade), lenalidomide (Revlimid), daratumumab (Darzalex); DW, diffusion-weighted; ECOG, Eastern Cooperative Oncology Group; HR, high risk; RD, lenalidomide, daratumumab; VRD, bortezomib, lenalidomide, daratumumab; VRDd, bortezomib, lenaliomide, daratumumab, dexamethasone.
Central laboratory investigations include:
-
Bone marrow aspirate and peripheral blood for molecular profiling:
Multiplex ligation-dependent probe amplification (MLPA) or equivalent platform for CNA (del(17p), gain(1q), del(1p)).28
Real-Time Quantitative Polymerase Chain Reaction (RQ-PCR) translocation assay or equivalent tool for prediction of HR translocations (t(4;14), t(14;16) and t(14;20)).65
Gene expression profiling based on Affymetrix HG-U133 Plus 2.0 or equivalent platform with risk profile determined as per EMC92 model23
-
Exploratory molecular analyses to identify potentially targetable mutations
Whole exome or whole genome next-generation sequencing.
Gene expression profiling (GEP).
Epigenetic analyses.
Germline variant analysis.
Bone marrow aspirate for MRD analyses.
-
Peripheral blood for disease assessment
Disease parameters, for example, paraprotein, for serum response assessments.
Beta-2-microglobulin.
Albumin.
Quality of life questionnaires, EQ-5D, QLQ-C30 and QLQ-MY20, are collected from all participants at baseline, and for participants who go on to HR treatment these are completed at:
End of induction treatment.
100 days post ASCT then 3-monthly thereafter until disease progression.
Follow-up
On completion of treatment, patients are followed-up at 3 months, and then 6 monthly during standard of care visits, until second disease progression, death or withdrawal. Assessment via standard of care visits promotes participant retention and complete follow-up.
Imaging assessments
All patients participating in the DW-MRI substudy have whole body DW-MRI scan performed at baseline, 100–120 days post ASCT and at end of consolidation part 2.
Outcomes
Primary endpoint
MUKnine a:
The proportion of patients with molecular risk-defining investigations performed within 8 weeks.
MUKnine b:
The primary endpoints to determine whether to terminate the trial early for futility are
MRD at 100 days post-ASCT
Progression-free survival at 100 days post-ASCT
The primary endpoint to assess efficacy of HR treatment if the trial is not stopped early for futility is PFS at 18 months post registration to screening.
Secondary endpoints
MUKnine a: recruitment rates; PFS; OS; second PFS (PFS2); treatment received; overall response;
MUKnine b:
Safety and toxicity (adverse reactions (ARs), serious adverse events, serious ARs and suspected unexpected serious ARs graded by common terminology criteria for adverse events v5.0).
MRD at the end of induction therapy, and postconsolidation part 2.
Overall survival
Maximum and overall response at the end of induction therapy, 100 days post-ASCT and postconsolidation part 2.
Time to progression and time to maximum response.
PFS2
Overall treatment benefit and clinician assessment of treatment benefit at the end of induction therapy and 100 days post ASCT.
Quality of life as assessed by the EQ-5D, EORTC QLQ-C30 and EORTC QLQ-MY20.
Treatment compliance.
Exploratory endpoints
Genomic instability, mutation rates and clonal evolution.
Imaging substudy
PFS; OS; response; patterns of disease distribution and discreet ‘3D phenotypes’.
Statistical analysis
The MUKnine b trial is designed using a Bayesian approach to enable assessment of multiple outcomes and incorporating multiple interim analyses.
The experimental treatment will be evaluated on an ongoing basis based on assessment of MRD status and PFS. Interim assessments are made after cohorts of 10 participants have been followed up to 100–120 days post ASCT, and data reviewed by an independent Data Monitoring and Ethics Committee (DMEC). The trial may be terminated early for futility on the basis of MRD status and PFS at 100–120 days post ASCT, using initial predefined stopping boundaries based on Myeloma IX data. Following updated prior information becoming available from Myeloma XI/XI+, these stopping boundaries were recalculated to provide updated decision criteria.
If the trial is not terminated early, up to 105 newly diagnosed patients with molecular HR disease will be registered to treatment. With the availability of molecularly matched individual participant data from currently running trials (Myeloma XI/XI+) a body of almost concurrent control data is available to use for the purpose of exploratory statistical comparison.
The experimental treatment arm will be compared with control in terms of PFS at 18 months post registration to screening, expressed as a binary outcome, within the Bayesian framework. Further analyses of PFS at 18 months will be performed outside of the Bayesian framework using Kaplan-Meier estimation.
MUKnine a endpoints, and secondary and exploratory endpoints will be analysed using summary statistics alongside confidence intervals where appropriate. All analyses are fully detailed in a statistical analysis plan prior to being undertaken. Full statistical analysis for MUKnine is provided in online supplemental file 1, and discussed in the MUKnine statistical methods paper (in preparation).
bmjopen-2020-046225supp001.pdf (72KB, pdf)
Trial conduct
Data are collected via electronic case report forms. Site monitoring of source data is performed by University of Leeds Clinical Trials Research Unit (CTRU) following the trial monitoring plan. The trial is conducted in accordance with the principles of Good Clinical Practice and in line with the relevant Research Governance Framework within the UK through adherence with CTRU standard operating procedures. All information collected during the course of the trial will be kept strictly confidential. Information will be held securely on paper and electronically at the CTRU. An independent DMEC reviews safety data on a regular basis to identify any safety concerns or trends. An independent Trial Steering Committee periodically reviews safety data and discusses recommendations made by the DMEC.
Statement of indemnity
This trial is sponsored by The University of Leeds and the University of Leeds will be liable for negligent harm caused by the design of the trial. The NHS has a duty of care to participants treated, whether or not the participant is taking part in a clinical trial, and the NHS remains liable for clinical negligence and other negligent harm to participants under this duty of care.
As this is a clinician-led trial, there are no arrangements for no-fault compensation. As this is a clinician-led trial, there are no arrangements for no-fault compensation; however, usual product liability will be covered by the manufacturer under the Consumer Protection Act 1987.
Patient and public involvement
Patients were involved in review and development of trial design, protocol and patient information sheet (model consent form provided in online supplemental file 2).
bmjopen-2020-046225supp002.pdf (1.4MB, pdf)
Ethics and dissemination
The trial has national research ethics approval from the NHS National Research Ethics Service, London South East Research Ethics Committee (Ref: 17/LO/0022, 17/LO/0023). All patients provide written informed consent prior to take part in the trial at the hospital site where they are recruited. Any required protocol amendments will be submitted to ethics and the Medicines and Healthcare products Regulatory Agency (MHRA) (as appropriate), and will be implemented at the relevant sites once approved. Information on amendments will be reported to the DMEC and Trial Steering Committee (TSC).
A manuscript with results of the MUKnine b study will be published in a peer-reviewed journal. Separate manuscripts will be written for results of MUKnine a and each of the exploratory objectives; these will also be submitted for publication in peer-reviewed journals. Credit for the main results will be given to all those who have collaborated in the trial, through authorship and contributorship. Uniform requirements for authorship for manuscripts submitted to medical journals will guide authorship decisions. Professional writers are not intended to be used. On publication of the final long-term results of the study, requests for use of data may be made to the CTRU and will be reviewed by the Trial Management Group.
Discussion
This is the first time in the UK genetic risk has been used prospectively in MM to identify participants to be treated in an academically-led clinical trial and select treatment based solely on this. It is hoped this trial will bring improved survival and longer term disease control for patients with HRMM in the future by providing an intensive treatment regimen specifically targeted at this difficult to treat disease subgroup. In addition, the trial will provide important evidence regarding feasibility of multicentre molecular-risk stratified trials in MM at the point of diagnosis, using central molecular tumour investigations.
Intensive treatment in HR patients has been used outside the UK with some promising results but access to drugs in the UK has been challenging. This trial is designed to work within the UK NHS system and provide the best treatment for HR patients. The availability of novel targeted molecular therapies helps in treating the highly heterogeneous disease of MM. Ultimately data generated through this trial aim to support the case for access to combination therapies of expensive agents to patient subgroups with a high unmet need such as HR disease.
Supplementary Material
Acknowledgments
Janssen and Celgene provided funding to Myeloma UK and University of Leeds who in turn fund the MUKnine trial. Janssen and Celgene approved the design of the study but have no input in the collection, analysis or interpretation of data as this is a fully academically sponsored trial. Contact details of the sponsor, the University of Leeds, are accessible via the trial registration. Through the Myeloma UK Clinical Trials Network, Myeloma UK were involved in the study design and are actively involved in the collection and interpretation of data, as well as in the review of manuscripts arising from the study publishing trial outcomes.
Footnotes
Twitter: @DrSRBrown
SB and DS contributed equally.
MJ and MK contributed equally.
Contributors: SB and DS contributed equally as joint first authors. MJ and MK contributed equally as joint last authors. MJ, MK, GP, SB, LF, SH and DS designed the trial. SH, AH, KW and SB developed the statistical analysis plan and are responsible for the ongoing statistical monitoring, analysis and interpretation of data. DS, SH, MK and MJ wrote the manuscript. MJ, MK, CM and GP perform the research and collect data. DS, LF and SR perform trial and data management. All authors reviewed and approved the final manuscript.
Funding: This trial is funded by Janssen, Celgene and Myeloma UK. Award/Grant number is not applicable.
Competing interests: SH, DS, LF, SR, AH, KW and SB declare they have no competing interests. MK has served as a consultant and received honoraria from Celgene, Takeda, Amgen, Chugai, BMS, AbbVie and Janssen, and has received research funding from Celgene. GP served as a consultant and received honoraria from Janssen Oncology, Celgene, Amgen, Takeda, Gilead Sciences and Binding Site. MJ has served as a consultant and received honoraria from Celgene, Janssen, Takeda, Amgen, AbbVie and Novartis.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
References
- 1. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009;23:3–9. 10.1038/leu.2008.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cancer Research UK . Myeloma statistics cancer research UK, 2015. Available: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/myeloma#heading-Two
- 3. Ries LAG YJ, Keel GE, Eisner MP. Seer survival monograph: cancer survival among adults: U S. SEER program, 1988-2001, patient and tumor characteristics. National Cancer Institute, SEER Program 2007;NIH Pub. No. 07-6215. [Google Scholar]
- 4. Avet-Loiseau H. Ultra high-risk myeloma. Hematology 2010;2010:489–93. 10.1182/asheducation-2010.1.489 [DOI] [PubMed] [Google Scholar]
- 5. Avet-Loiseau H, Attal M, Campion L, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol 2012;30:1949–52. 10.1200/JCO.2011.36.5726 [DOI] [PubMed] [Google Scholar]
- 6. Avet-Loiseau H, Durie BGM, Cavo M, et al. Combining fluorescent in situ hybridization data with ISS staging improves risk assessment in myeloma: an international myeloma Working group collaborative project. Leukemia 2013;27:711–7. 10.1038/leu.2012.282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boyd KD, Ross FM, Chiecchio L, et al. A novel prognostic model in myeloma based on co-segregating adverse fish lesions and the ISS: analysis of patients treated in the MRC myeloma IX trial. Leukemia 2012;26:349–55. 10.1038/leu.2011.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chng WJ, Kuehl WM, Bergsagel PL, et al. Translocation t(4;14) retains prognostic significance even in the setting of high-risk molecular signature. Leukemia 2008;22:459–61. 10.1038/sj.leu.2404934 [DOI] [PubMed] [Google Scholar]
- 9. Morgan GJ, Davies FE, Gregory WM, et al. First-Line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC myeloma IX): a randomised controlled trial. The Lancet 2010;376:1989–99. 10.1016/S0140-6736(10)62051-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morgan GJ, Davies FE, Gregory WM, et al. Cyclophosphamide, thalidomide, and dexamethasone (CTD) as initial therapy for patients with multiple myeloma unsuitable for autologous transplantation. Blood 2011;118:1231–8. 10.1182/blood-2011-02-338665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shaughnessy JD, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007;109:2276–84. 10.1182/blood-2006-07-038430 [DOI] [PubMed] [Google Scholar]
- 12. Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2018;32:102–10. 10.1038/leu.2017.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shah V, Sherborne AL, Johnson DC, et al. Predicting ultrahigh risk multiple myeloma by molecular profiling: an analysis of newly diagnosed transplant eligible myeloma XI trial patients. Leukemia 2020;34:3091–6. 10.1038/s41375-020-0750-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med Overseas Ed 2016;375:1319–31. 10.1056/NEJMoa1607751 [DOI] [PubMed] [Google Scholar]
- 15. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med Overseas Ed 2016;375:754–66. 10.1056/NEJMoa1606038 [DOI] [PubMed] [Google Scholar]
- 16. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (endeavor): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016;17:27–38. 10.1016/S1470-2045(15)00464-7 [DOI] [PubMed] [Google Scholar]
- 17. Stewart AK. Carfilzomib for the treatment of patients with relapsed and/or refractory multiple myeloma. Future Oncology 2015;11:2121–36. 10.2217/fon.15.123 [DOI] [PubMed] [Google Scholar]
- 18. Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2018;32:102–10. 10.1038/leu.2017.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chng WJ, Dispenzieri A, Chim C-S, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014;28:269–77. 10.1038/leu.2013.247 [DOI] [PubMed] [Google Scholar]
- 20. Jackson GH, Davies FE, Pawlyn C, et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2019;20:57–73. 10.1016/S1470-2045(18)30687-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuiper R, Broyl A, de Knegt Y, et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012;26:2406–13. 10.1038/leu.2012.127 [DOI] [PubMed] [Google Scholar]
- 22. Decaux O, Lodé L, Magrangeas F, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone Du Myélome. J Clin Oncol 2008;26:4798–805. 10.1200/JCO.2007.13.8545 [DOI] [PubMed] [Google Scholar]
- 23. Dickens NJ, Walker BA, Leone PE, et al. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome. Clin Cancer Res 2010;16:1856–64. 10.1158/1078-0432.CCR-09-2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mulligan G, Mitsiades C, Bryant B, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007;109:3177–88. 10.1182/blood-2006-09-044974 [DOI] [PubMed] [Google Scholar]
- 25. Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol 2015;33:3911–20. 10.1200/JCO.2014.59.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Samur MK, Aktas Samur A, Fulciniti M, et al. Genome-Wide somatic alterations in multiple myeloma reveal a superior outcome group. JCO 2020;38:3107–18. 10.1200/JCO.20.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perrot A, Lauwers-Cances V, Tournay E, et al. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J Clin Oncol 2019;37:1657–65. 10.1200/JCO.18.00776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar SK, Uno H, Jacobus SJ, et al. Impact of gene expression profiling-based risk stratification in patients with myeloma receiving initial therapy with lenalidomide and dexamethasone. Blood 2011;118:4359–62. 10.1182/blood-2011-03-342089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meissner T, Seckinger A, Rème T, et al. Gene expression profiling in multiple myeloma--reporting of entities, risk, and targets in clinical routine. Clin Cancer Res 2011;17:7240–7. 10.1158/1078-0432.CCR-11-1628 [DOI] [PubMed] [Google Scholar]
- 30. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015;372:142–52. 10.1056/NEJMoa1411321 [DOI] [PubMed] [Google Scholar]
- 31. Palumbo A, Gay F, Cavallo F, et al. Continuous therapy versus fixed duration of therapy in patients with newly diagnosed multiple myeloma. Journal of Clinical Oncology 2015;33:3459–66. 10.1200/JCO.2014.60.2466 [DOI] [PubMed] [Google Scholar]
- 32. Brioli A, Kaiser MF, Pawlyn C, et al. Biologically defined risk groups can be used to define the impact of thalidomide maintenance therapy in newly diagnosed multiple myeloma. Leuk Lymphoma 2013;54:1975–81. 10.3109/10428194.2012.760736 [DOI] [PubMed] [Google Scholar]
- 33. Morgan GJ, Gregory WM, Davies FE, et al. The role of maintenance thalidomide therapy in multiple myeloma: MRC myeloma IX results and meta-analysis. Blood 2012;119:7–15. 10.1182/blood-2011-06-357038 [DOI] [PubMed] [Google Scholar]
- 34. Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med Overseas Ed 2012;366:1782–91. 10.1056/NEJMoa1114138 [DOI] [PubMed] [Google Scholar]
- 35. Barlogie B, Anaissie E, van Rhee F, et al. Incorporating bortezomib into upfront treatment for multiple myeloma: early results of total therapy 3. Br J Haematol 2007;138:176–85. 10.1111/j.1365-2141.2007.06639.x [DOI] [PubMed] [Google Scholar]
- 36. Bergsagel PL, Mateos M-V, Gutierrez NC, et al. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013;121:884–92. 10.1182/blood-2012-05-432203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harousseau J-L, Attal M, Avet-Loiseau H, et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. J Clin Oncol 2010;28:4621–9. 10.1200/JCO.2009.27.9158 [DOI] [PubMed] [Google Scholar]
- 38. Neben K, Lokhorst HM, Jauch A, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood 2012;119:940–8. 10.1182/blood-2011-09-379164 [DOI] [PubMed] [Google Scholar]
- 39. Shaughnessy JD, Zhou Y, Haessler J, et al. TP53 deletion is not an adverse feature in multiple myeloma treated with total therapy 3. Br J Haematol 2009;147:347–51. 10.1111/j.1365-2141.2009.07864.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roussel M, Lauwers-Cances V, Robillard N, et al. Front-Line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone Du Myélome. J Clin Oncol 2014;32:2712–7. 10.1200/JCO.2013.54.8164 [DOI] [PubMed] [Google Scholar]
- 41. Kumar S, Flinn I, Richardson PG, et al. Randomized, multicenter, phase 2 study (evolution) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood 2012;119:4375–82. 10.1182/blood-2011-11-395749 [DOI] [PubMed] [Google Scholar]
- 42. Plesner T, Arkenau H-T, Gimsing P, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood 2016;128:1821–8. 10.1182/blood-2016-07-726729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katja Weisel C C, Cook G, Leiba M. Efficacy of daratumumab in combination with lenalidomide plus dexamethasone (DRd) or bortezomib plus dexamethasone (DVd) in relapsed or refractory multiple myeloma (RRMM) based on cytogenetic risk status. 2017 ASCO Annual Meeting 2017. J Clin Oncol 2017;35, no. 15_suppl:8006. [Google Scholar]
- 44. Farooqui AA, Tariq M, Nabeel S, et al. Daratumumab-based three drug regimens for high-risk multiple myeloma: a meta-analysis. JCO 2020;38:e20549–e49. 10.1200/JCO.2020.38.15_suppl.e20549 [DOI] [Google Scholar]
- 45. Cavo M, Tosi P, Zamagni E, et al. Prospective, randomized study of single compared with double autologous stem-cell transplantation for multiple myeloma: bologna 96 clinical study. J Clin Oncol 2007;25:2434–41. 10.1200/JCO.2006.10.2509 [DOI] [PubMed] [Google Scholar]
- 46. Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol 2020;7:e456–68. 10.1016/S2352-3026(20)30099-5 [DOI] [PubMed] [Google Scholar]
- 47. Barlogie B, Tricot GJ, van Rhee F, et al. Long-Term outcome results of the first tandem autotransplant trial for multiple myeloma. Br J Haematol 2006;135:158–64. 10.1111/j.1365-2141.2006.06271.x [DOI] [PubMed] [Google Scholar]
- 48. Roussel M, Moreau P, Huynh A, et al. Bortezomib and high-dose melphalan as conditioning regimen before autologous stem cell transplantation in patients with de novo multiple myeloma: a phase 2 study of the Intergroupe Francophone Du Myélome (IFM). Blood 2010;115:32–7. 10.1182/blood-2009-06-229658 [DOI] [PubMed] [Google Scholar]
- 49. Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the medical Research Council myeloma IX study. J Clin Oncol 2013;31:2540–7. 10.1200/JCO.2012.46.2119 [DOI] [PubMed] [Google Scholar]
- 50. Goicoechea I, Puig N, Cedena M-T, et al. Deep MRD profiling defines outcome and unveils different modes of treatment resistance in standard- and high-risk myeloma. Blood 2021;137:49–60. 10.1182/blood.2020006731 [DOI] [PubMed] [Google Scholar]
- 51. Croft J, Riddell A, Koh D-M, et al. Inter-Observer agreement of baseline whole body MRI in multiple myeloma. Cancer Imaging 2020;20:48. 10.1186/s40644-020-00328-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Messiou C, Hillengass J, Delorme S, et al. Guidelines for acquisition, interpretation, and reporting of whole-body MRI in myeloma: myeloma response assessment and diagnosis system (MY-RADS). Radiology 2019;291:5–13. 10.1148/radiol.2019181949 [DOI] [PubMed] [Google Scholar]
- 53. Clinical Trials.gov website, 2021. Available: https://clinicaltrials.gov/ct2/results?recrs=&cond=&term=daratumumab&cntry=&state=&city=&dist=
- 54. Harousseau J-L, Avet-Loiseau H, Attal M, et al. Achievement of at least very good partial response is a simple and robust prognostic factor in patients with multiple myeloma treated with high-dose therapy: long-term analysis of the IFM 99-02 and 99-04 trials. J Clin Oncol 2009;27:5720–6. 10.1200/JCO.2008.21.1060 [DOI] [PubMed] [Google Scholar]
- 55. Moreau P, Hullin C, Garban F, et al. Tandem autologous stem cell transplantation in high-risk de novo multiple myeloma: final results of the prospective and randomized IFM 99-04 protocol. Blood 2006;107:397–403. 10.1182/blood-2005-06-2573 [DOI] [PubMed] [Google Scholar]
- 56. McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med Overseas Ed 2012;366:1770–81. 10.1056/NEJMoa1114083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thall PF, Simon RM, Estey EH. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Stat Med 1995;14:357–79. 10.1002/sim.4780140404 [DOI] [PubMed] [Google Scholar]
- 58. Thall PF, Simon RM, Estey EH. New statistical strategy for monitoring safety and efficacy in single-arm clinical trials. J Clin Oncol 1996;14:296–303. 10.1200/JCO.1996.14.1.296 [DOI] [PubMed] [Google Scholar]
- 59. Thall PF, Sung H-G. Some extensions and applications of a Bayesian strategy for monitoring multiple outcomes in clinical trials. Stat Med 1998;17:1563–80. [DOI] [PubMed] [Google Scholar]
- 60. Wale A, Pawlyn C, Kaiser M, et al. Frequency, distribution and clinical management of incidental findings and extramedullary plasmacytomas in whole body diffusion weighted magnetic resonance imaging in patients with multiple myeloma. Haematologica 2016;101:e142–4. 10.3324/haematol.2015.139816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Messiou C, Kaiser M. Whole body diffusion weighted MRI – a new view of myeloma. Br J Haematol 2015;171:29–37. 10.1111/bjh.13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brown SR, Sherratt D, Booth G. Experiences of establishing an academic early phase clinical trials unit. Clinical trials 2017;1740774517710250. [DOI] [PubMed] [Google Scholar]
- 63. Rajkumar SV, Harousseau J-L, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International myeloma workshop consensus panel 1. Blood 2011;117:4691–5. 10.1182/blood-2010-10-299487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Office for National Statistics . Office for national statistics: cancer statistics registrations, England (series MB1), no. 42, 2011 2013.
- 65. Kaiser MF, Walker BA, Hockley SL, et al. A Tc classification-based predictor for multiple myeloma using multiplexed real-time quantitative PCR. Leukemia 2013;27:1754–7. 10.1038/leu.2013.12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2020-046225supp001.pdf (72KB, pdf)
bmjopen-2020-046225supp002.pdf (1.4MB, pdf)