

Abstract
The origins of low-temperature tissue storage research date back to the late 1800s. Over half a century later, osmotic stress was revealed to be a main contributor to cell death during cryopreservation. Consequently, the addition of cryoprotective agents (CPAs) such as dimethyl sulfoxide (DMSO), glycerol (GLY), ethylene glycol (EG), or propylene glycol (PG), although toxic to cells at high concentrations, was identified as a necessary step to protect against rampant cell death during cryopreservation. In addition to osmotic stress, cooling and thawing rates were also shown to have significant influence on cell survival during low temperature storage. In general, successful low-temperature cell preservation consists of the addition of a CPA (commonly 10% DMSO), alone or in combination with additional permeating or non-permeating agents, cooling rates of approximately 1ºC/min, and storage in either liquid or vapor phase nitrogen. In addition to general considerations, cell-specific recommendations for hepatocytes, pancreatic islets, sperm, oocytes, and stem cells should be observed to maximize yields. For example, rapid cooling is associated with better cryopreservation outcomes for oocytes, pancreatic islets, and embryonic stem cells while slow cooling is recommended for cryopreservation of hepatocytes, hematopoietic stem cells, and mesenchymal stem cells. Yields can be further maximized by implementing additional pre-cryo steps such as: pre-incubation with glucose and anti-oxidants, alginate encapsulation, and selecting cells within an optimal age range and functional ability. Finally, viability and functional assays are critical steps in determining the quality of the cells post-thaw and improving the efficiency of the current cryopreservation methods.
Keywords: cryopreservation, cryoprotectants, low temperature banking, freezing
Introduction
The origins of low-temperature tissue storage research date back to the late 1800s. Since then, numerous advancements related to cryopreservation have been made and protocol optimization continues to be an area of active research. Cryopreservation allows the banking of a large number of cells and tissues that can be utilized for scientific research and medical applications including hepatocyte and pancreatic islet transplantation, blood transfusion, bone marrow transplantation, artificial insemination, and in vitro fertilization. For these modes of treatment to be successful, however, large quantities of the desired cell type must be kept on standby and be available for use on-demand which is made possible through the process of cryopreservation. This review will discuss the history and basic physical principles of cryopreservation, including: permeating, and non-permeating cryoprotecting agents, vitrification mixtures, and cooling and thawing rates. In addition, unique aspects of hepatocytes, pancreatic islets, sperm, oocytes, and stem cells, as well as current methods in their preservation, and some novel agents and potential biomaterials that can be used to enhance cellular viability beyond current reports are discussed.
History of Cryopreservation
Following the discovery of the microscope, Spallanzani observed that sperm could maintain mobility even when exposed to cold temperature conditions in 17761. Research into the effects of cryopreservation on live tissue had its roots in late 1800s when scientists used this technology to preserve both spermatozoa and red blood cells (RBCs). During this time, research demonstrated weaknesses in the process which caused inconsistent results and frequent infertility caused by early embryonic death. A breakthrough occurred in the 1950s when James Lovelock discovered that the cryopreservation process caused osmotic stress in the cell by instantly freezing the liquid which directly contributed to the formation of ice crystals in RBCs2. In 1963, Mazur was able to characterize that process when they demonstrated that the rate of temperature change within a cell-containing medium controlled the movement of water across a cell membrane and thus the degree of intracellular freezing3. This together helped to improve the overall understanding of the mechanism associated with the cryoprotective process. During the 1980s, research surrounding the cryopreservation process revealed that the speed at which the freezing and thawing process occurred was the most important factor in determining the survivability of the cells4,5. It was demonstrated that small, slow increments in both the freezing and thawing processes prevented the rapid formation of ice crystals that increased membrane-bound solutes associated with early cell death6. Another initial advance in cryopreservation occurred in the late 1940s when researchers discovered that the use of glycerol as a medium increased the survivability of spermatozoa in subfreezing (−70ºC) temperatures7. Using glycerol as a medium effectively served to protect the cells from rapid formation of ice crystal during the preservation process. A commonly used cryoprotective agent currently employed is dimethyl sulfoxide (DMSO), which is added to cell media prior to the freezing process8,9. DMSO (10%) when added to the cell media, commonly at 2 M concentration, increases the porosity of the cellular membrane, which allows water to flow more freely through the membrane10,11. Additionally, like glycerol, DMSO is thought to help prevent the formation of water crystals by increasing intracellular solute concentration, thus aiding in the vitrification of water at low temperatures12.
Principles of Cryopreservation
In order to fully understand the role of cryoprotective agents (CPAs), we must first understand the effects of subzero temperatures on otherwise healthy tissue. Exposing cells to temperatures below 0ºC without the aid of cryoprotectants is typically lethal. Since water constitutes approximately 80% of tissue mass, the freezing of water, both intra and extracellularly, imposes the largest influence over harmful biochemical, and structural changes that are thought to result in unprotected freezing injury13. Two independent theories exist that attempt to explain the harmful effects of freezing on cells: (1) ice crystals mechanically disrupt cellular membranes thus making it impossible to obtain structurally-intact cells after thawing; and (2) deadly increases in solute concentration occur to the remaining liquid phase as ice crystals form intracellularly during cooling13. Whether the mechanical or osmotic effects of freezing dominate, the end result is the same; unprotected cooling and thawing of cells is a process incompatible with life. To mitigate these effects, two protective actions must be carried out: use of a cryoprotectant, and selection of an appropriate cooling and thawing rate.
Permeating Agents
A number of permeating agents (PAs) exist currently such as glycerol (the first agent discovered), dimethyl sulfoxide (DMSO), ethylene glycol (EG), and propanediol (propylene glycol). The ability of each of these compounds to protect a cell from mechanical and osmotic effects of freezing depends on several properties. Permeating agents must be highly water soluble at low temperatures, able to easily cross biological membranes, and ideally, be minimally toxic13.
The structures of four common permeating agents of the 100 that are known are represented in (Fig. 1). Their relatively small size (typically less than 100 daltons), and somewhat amphiphilic nature allows them to easily penetrate cell membranes where they can exert their effects14. The structures’ ability to hydrogen bond with water accounts for a large portion of their protective effects. Normally, as water freezes, the developing crystal structure excludes solutes as its lattice forms. Solutes are displaced to the diminishing liquid phase which effectively increases the solute concentration to lethal levels within the cell. Because permeating cryoprotectants interact strongly with water through hydrogen bonding, the freezing point of water is depressed, and less water molecules are available to interact with themselves to form critical nucleation sites required for crystal formation12. Formation of solid water with an irregular, amorphous structure is known as vitrification, and is achieved by utilizing a cryoprotectant accompanied by an appropriate cooling rate15. To minimize toxicity, vitrification mixtures are often added in a stepwise fashion at temperatures near 0ºC14. Thus, addition of permeating agents under these conditions allows successful storage of cells in a solid phase at the supercool temperatures required to halt biochemical processes without the formation of ice.
Figure 1.

Four common permeating cryoprotectants: glycerol (GLY), dimethyl sulfoxide (DMSO), ethylene glycol (EG), and propylene glycol (PG).
In addition to permitting vitrification, some PAs like DMSO are thought to increase cellular permeability by affecting membrane dynamics in a concentration dependent manner. At low concentrations (5%), evidence suggests DMSO decreases membrane thickness and, in turn, increases membrane permeability. At commonly used concentrations (10%), water pore formation in biological membranes is induced. Formation of pores can be advantageous as intracellular water can be more readily replaced by cryoprotectants that promote vitrification. At higher, toxic concentrations (40%) however, lipid bilayers begin to disintegrate16,17. It is evident then that selecting an appropriate cryoprotectant concentration is critical to the structural integrity, and therefore viability of cells after freezing.
Non-permeating agents
The second category of cryoprotectant is non-permeating agents (NPAs). As the name suggests, they do not permeate intracellularly and therefore exert their protective influence outside of the cell. They are typically larger, and covalently linked as either polymers, dimers, or trimers. Some commonly-used agents in this class are: Polyethylene glycol (PEG), polyvinylpyrrolidone (PVP), raffinose, sucrose, and trehalose14,15,18. Non-permeating agents induce vitrification by the same mechanism as permeating agents but extracellularly and to a lesser extent.
Trehalose: unique structure and consequences
Unlike the non-permeating polymers, the disaccharide CPAs are naturally occurring. Trehalose, a less-well-known sugar, is produced by a wide variety of organisms including bacteria, fungi, yeast, insects, plants, and some invertebrates. Although it serves many functions, it has been demonstrated to act as a means to withstand freezing in these organisms19,20. The structure of trehalose gives it some useful properties. It is a glucose dimer linked via an α-1,1-glycosidic bond. Its acetal link prevents reduction of C-1 in each monomer which increases its stability under extreme temperatures and reduces its susceptibility to acid hydrolysis in low pH conditions. The structure of trehalose is shown in (Fig. 2).
Figure 2.
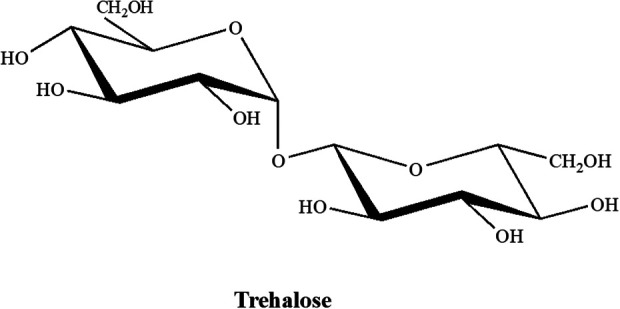
Naturally-occurring trehalose disaccharide. The unique α-1,1-glycosidic linkage prevents C-1 hydrolysis and increases stability under temperature extremes and low pH conditions.
Vitrification mixtures
Both permeating and non-permeating agents can prove toxic to cells at higher concentrations, although non-permeating agents in a lower degree. This undesirable feature of CPAs increases cell death and reduces viable yields. Given that donor tissue supply for most organ types represents the limiting factor for transplant procedures, CPA induced toxicity should be minimized as much as possible. As such, cells should be exposed to those agents for the shortest amount of time that still allows for consistent packaging. Since both PAs, and NPAs share the same vitrification mechanism, non-permeating agents can be added to solution to allow successful cryobanking but with lower concentrations of permeating agents. This has the benefit of a reduction in PA-induced toxicity and an increase in cellular viability and yields post-thaw. Another method that showed reduced CPA induced toxicity was demonstrated by Kojayan et al. in which multi-molar combinations of reduced concentrations of EG, and DMSO were used to cryopreserve both human and murine islet cells21. They proposed that the reduction in adverse effects was likely due to reduced exposure to each individual CPA even though overall osmolality of the different combinations was comparable to a standard 2 M DMSO-only solution21. Other studies have shown combinations of sucrose, propanediol, and DMSO to be effective at supporting post-warming survival of porcine blastocysts15. Although segregated use of EG and DMSO has been the paradigm in cryopreservation since the mid-1900s, more current research suggests combinations of these and other CPAs offer the highest cell viabilities post-cryopreservation.
Finally, to obtain consistency during cryopreservation, exposure time of cells to permeating and non-permeating agents during final packaging process have to be standardized. This is especially important when variable size of small (50 vials) or large (500 and more) lots of vials are being packaged. For instance, during hepatocyte packaging, if a 30-minute exposure time is required to make a 50-vial lot versus 2 hours to make a 500-vial lot, each lot will display variable cell viability and overall health. This then can impact metabolic qualities, thus further increasing variability already observed between hepatocyte donors.
Cooling and thawing rates
The second action that must be carried out during the cryopreservation process is the selection of cooling and thawing rates. Since the success of cryoprotection depends on the avoidance of intracellular ice formation, careful consideration must be paid to the movement of water across membranes during this process. Mazur demonstrated a dependence of survivability on the rate of cooling of various cells. Specifically, he provided evidence that the rate of cooling is proportional to the probability of intracellular ice formation, and therefore inversely proportional to survivability3. To decrease the probability of intracellular ice formation, water must leave the cell as its temperature is decreased. The driving force behind the outward movement of water is a pressure differential due to the supercooling of intracellular water. This water remains in liquid phase below 0ºC, a phenomenon enhanced by the freezing point depression induced by the presence of CPAs. Supercooled water in the cytoplasm has a relatively high vapor pressure compared to water in the external medium3. The resulting pressure difference would favor a net movement of water out of the cell, causing dehydration.
A pressure differential is necessary for a net movement of water to occur, but it is not the only factor involved. The amount of water remaining in the cell at the time of solidification is affected by the rate of water efflux during the cooling phase. Three variables that affect this rate are, the cell’s surface area to volume ratio (SA: V), the membrane’s permeability to water, and the rate of cooling3. The SA: V is a function of the inherent size of a cell and cannot be modified. It is important to acknowledge, however, because it has significant consequences; namely that dehydration of larger cells is inherently slow compared to the dehydration of smaller cells. In light of this, adjustments during cryopreservation should be considered based on cell size to allow a sufficient volume of water to exit before vitrification occurs. To make up for large cell size, for example in hepatocytes which have a diameter of 20-30 μm, the two remaining variables can be modified22. First, membrane permeability can be increased, as discussed earlier, by the addition of certain permeating cryoprotective agents like DMSO. Second, the rate of cooling can be slowed. The relationship between dehydration and cooling rate is less evident but critical, nonetheless. As temperature decreases, so too does intracellular kinetic energy. Rapid cooling rates do not allow sufficient time for water to leave before vitrification occurs, thus cellular water content remains high and crystallization is more likely to occur23. In order to ensure maximum survival, a cooling rate of approximately 1ºC/min is usually appropriate for most cells except exceptionally large ones3. This is typically obtained by using controlled rate freezers that modulate chamber temperatures via elaborate cooling programs to maintain steady 1oC/min drop of vial content5. After successful cryopreservation, it is expected that the cells will be stored and thawed for subsequent use. Compared to cooling, however, cell storage and thawing has been given less attention. Speculation as to why this is the case leads to several possible conclusions: theory surrounding storage conditions and thawing rates is already well-entrenched within the scientific community, and it is expected that primary cells are stored long-term at −140/−196°C. It is also important to mention that type of storage can impact cell health and recovery. For example, storage of primary hepatocytes in vapor phase LN2 tank at −140°C is preferred over standard liquid LN2 tanks at −196°C. It has been shown that vapor phase LN2 storage allows for far greater recovery of viable hepatocytes and maintains hepatic metabolic function24–29.
Thawing and warming of cells does not play as critical a role as cooling and therefore has not warranted the same level of investigation historically. Common theory holds that cells must be warmed rapidly to prevent recrystallization of ice30. The rationale for this idea is based on thermodynamic principles and is as follows: vitrified water exists in a higher-energy state compared to its crystallized form. It is only quasi-stable and therefore can rearrange itself into a more-stable, lower-energy crystal structure during thawing31. When measuring the effect of warming rate on viability, however, it is important to remember that two steps always precede thawing: addition of cryoprotectant(s), and subsequent cooling of cells. Both of these have a significant effect on viability and could confound warming survivability data. Nonetheless, current accepted protocols suggest cryovials be transported to the lab on vapor LN2 (not on ice or dry ice) and be warmed rapidly in a 37ºC water bath for 90-120 seconds to achieve maximum viability32–34. This translates to an approximate warming rate of 45–70ºC/min between −140ºC and 0ºC although warming rate is more or less rapid outside of this window due to its non-linear nature30. At least one study, however, provided evidence that warming cells anywhere between 2ºC/min to 100ºC/min had no effect on viability post-thaw35. If true, samples could be thawed at slower rates like those achieved with air thawing (6.2 ± 0.5ºC/ min)30. With this approach, viability of samples would be equal to those subjected to rapid-thaw water bath protocols. However, reducing water bath use would also have the added benefit of increasing sterility since they are one of the most common sources of contamination in the laboratory.
Finally, it is important to note that no matter how well the cells are stored or how they are thawed, a decrease in post-thaw viability is inevitable. As such it is important to clean recovered cells using increased density media (Percoll®, Ficoll®, or similar) to remove dead cells and maximize viability of cells prior to their use. Standardization of this approach will increase quality of recovered cells and reduce variability when they are used for metabolic studies or for clinical treatment.
Cryopreservation of Hepatocytes, Pancreatic Islets, Gametes, and Stem Cells
Cryobiological responses can vary greatly depending on cell type due to variations in cell and tissue composition. Therefore, cell-specific biophysiological and biological characteristics should be considered during cryopreservation to maximize post-cryo viability for each cell type36. The following sections discuss cell-specific cryopreservation considerations, including optimal cooling and thawing rates, and selection of appropriate CPAs for hepatocytes, pancreatic islets, sperm, oocytes, and stem cells.
Hepatocytes
Hepatocyte transplantation, which involves the infusion of mature adult hepatocytes in the portal system of a recipient, is a novel therapeutic approach that can be used to treat various types of liver disease. Hepatocyte cryopreservation is a critical step in this treatment method since there is often a necessary delay between the initial cell isolation and the patient’s transplantation procedure. In addition, most patients require multiple cell transplantations over an extended period making optimization of cryopreservation techniques all the more important to help maintain cell functionality37,38.
Several factors influence cryopreservation outcomes including: the quality of the tissue that hepatocytes are derived from, the isolation procedure itself, preservation medium, and freeze/thaw rates. Hepatocyte quality is adversely affected by the presence of steatotic or otherwise diseased tissue as well as prolonged warm ischemia times, both of which could compromise the quality of the isolated hepatocytes25. Next, the cell isolation procedure itself can induce oxidative stress which, in turn, adversely affects cryopreservation outcomes. It has been suggested that pre-incubation under non-attached culture conditions in media supplemented with 2 mM N-acetyl-cystein (an antioxidant) and 15 mM glucose could provide antioxidant protection after isolation and further optimize post-cryopreservation outcomes including cell viability, attaching capacity, and functionality, particularly GSH, glycogen levels, and drug-metabolizing cytochrome P450 enzymes39. Cryopreservation medium can also influence storage outcomes, and the University of Wisconsin (UW) is currently considered the standard medium for hepatocyte preservation. As an alternative to UW solution, HypoThermosol (HTS) freezing solution supplemented with 10% DMSO at 4ºC has been shown to produce high cellular viability, long-term hepatocyte function, and good-quality response to cytokine challenge post-thawing24,40,41. Furthermore, the combination of an NPA such as trehalose and a CPA like DMSO has been shown to significantly increase both human and rat hepatocyte viability post-thaw42. Thawing and freezing rates also play important roles in determining cryopreservation outcome. The slow cooling rate is the best technique for cryopreservation of mature isolated hepatocytes. Different studies that utilized DMSO as their CPA have proposed various rates of cooling ranging from −1 to −5ºC/min up to −40ºC or −80ºC before storing at −196ºC in liquid nitrogen43. Rapid thawing at 37ºC is recommended to avoid intracellular ice formation, minimize cellular damage, and enhance post-thaw viability.
After thawing, it is important to evaluate the viability and functionality of cryopreserved hepatocytes. Attachment of hepatocytes post-thaw to collagen-coated plates provides a quick, qualitative measure of cell viability, and assays such as trypan blue exclusion, lactate dehydrogenase release, mitochondrial function, and measurement of necrotic or apoptotic markers allow for quantitative assessments of viability and functionality. Assays that assess post-thaw CYP450 enzyme functionality, like the 7-ethoxyresorufin-O-deethylase and testosterone hydroxylation assays, as well as phase II metabolism through resorufin conjugation can be utilized as well. These assays can be used to establish whether or not post-thaw hepatocyte drug metabolism capabilities remain intact. To further enhance yield and viability, hepatocytes can also be encapsulated to guard against mechanical stressors during cryopreservation24. One study reported no significant difference between pre- and post-cryopreservation viability when human hepatocytes were encapsulated with alginate/poly-L-lysine microcapsules44. Encapsulation of hepatocytes also offers protection from host immune defenses, an attractive feature for cells destined for transplantation after cryopreservation.
Pancreatic islets
Islet transplantation is an effective therapy for the treatment of patients with complicated type I diabetes mellitus. Advances in clinical islet isolation and transplantation have been made since the development of the Edmonton protocol in 200045. It is reported that clinical islet transplantation can establish insulin independence for up to 5 years post-transplant with minimal complications46. However, limited availability of donor pancreas and suitable islets for transplantation has remained a challenge45. Cryogenic banking of islet cells from multiple donors allows for long-term storage of islets and addresses the issue of donor availability for islet transplantation47. Islets can be cryopreserved at −80 to −196ºC with DMSO48,49. Freezing and thawing rates can affect islet morphology and function. An optimal freezing rate should provide high yield and viability while keeping the immunostimulatory molecules low47. Foreman et al. reported rapid freezing with the rate of 20 ºC/min and 70ºC/min when islets were cultured in 1 M DMSO for 30 minutes at room temperature, followed by 2 M DSMO exposure for 10 minutes at 0 ºC, improved in vitro insulin secretory ability of the islets cells after thawing48. Current standard islet thawing protocols involve rapid thawing at 150-200ºC/min in a 37 ºC water bath50. Evidence suggests that exposure to 50% oxygen during the thawing process could reduce the injury of cryopreserved human islets51. Glycerol or DMSO can be used as CPAs for long-term storage of islet cells at subzero temperatures (typically less than −100°C)52. Chandravanshi et al. have suggested supplementation with docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), and metformin could provide a higher islet recovery from −196°C storage and allow for proper islet banking53. In addition to the selection of CPAs and optimization of freezing and thawing rate, other methods have been developed to improve islet cryopreservation outcomes. Alginate encapsulation of islets before cryopreservation has shown to be a promising approach for future transplants. A study demonstrated that, compared to non-encapsulated frozen islets, islets encapsulated in 1.75% alginate had higher a survival and function post-thawing as well as an improved graft response54.
Post-thaw islet survival can be assessed by multiple methods including fluorescein diacetate (FDA), calcein AM, or SYTO Green stain in conjunction with a propidium iodide (PI), or ethidium bromide (EtBr) counter stain to measure viability55–57. Additionally, post-thaw damage to general baseline islet health can be monitored through PCR-based quantitation of mRNA transcripts including: HIF1A (activated in hypoxic conditions), SLC2A1 (GLUT1 transporter), VEGFA (vascularization), and ACTB (β actin/ cytoskeleton)51. Impairment of metabolic ability and islet function can be assessed through careful comparison of post-thaw oxygen consumption rate (OCR), and insulin production (GSIR) to pre-cryo baseline levels51. Ultimately, optimizing cryopreservation parameters could reduce the gap between transplantation from the donor to the recipient and further advance the clinical transplantation of islet cells for the treatment of type I diabetes47.
Sperm
Sperm cryopreservation is an effective method of fertility preservation in humans, and is particularly useful to patients prior to cancer treatment and in other mammals including endangered species58–60. Sperm are so amenable to this method of storage that one study reported a batch of cells kept for 21 years was used to successfully fertilize an oocyte ultimately resulting in a live birth61. Long-term cold storage of these cells usually involves glycerol and/or egg yolk as CPAs, and an increase in the concentration of glycerol at a constant cooling rate has been shown to increase sperm survival rates62. For cryopreservation of human sperm cells, an initial cooling rate of 0.5–1 ºC/min is recommended when freezing the cells from room temperature to 5 ºC. Afterwards, an increase in the freezing rate to 10 ºC/min when cooling from 5 to −80°C, is recommended to maximize sperm cryosurvival. When thawing samples, rates of 43.5ºC/min in 20ºC air or 55.2ºC/min in 35ºC air on a dry surface are suggested to result in optimal sperm viability and motility post-thaw63.
In general, post-thaw damage common to sperm include reduced acrosome integrity, motility, fertilization capability, and an overall decrease in viability regardless of the species of origin.58 More specifically, chromatin disruption through protamine translocations, DNA fragmentation, and lesions to genes involved in fertilization capability and embryonic development (ADD1, ARNT, PEG1/MEST, SNORD116/PWSAS) are known consequences of the cryopreservation process64. Although the exact mechanism is unknown, oxidative stress common during the cryo process is thought to underlie increased DNA fragmentation in human sperm thereby resulting in reduced final yield and viability65. Yeste et al. has suggested supplementing freezing media with vitamin E, hypotaurine, or other natural antioxidants to help mitigate oxidative effects58. While cryo-induced genetic alterations in sperm have been documented, data surrounding potential epigenetic changes imparted by cryopreservation are limited and warrant further study58.
In addition, pre-cryo sperm motility and viability can help predict post-thaw outcomes and cryosurvival. Degl’Innocenti et al. assessed these characteristics in samples from patients with oligospermia, cancer, and other pathologies. They reported the lowest recovery rates occurred when pre-cryo basal numbers, motility, and viability fell below the World Health Organization (WHO) fifth percentile reference values (39 × 106 sperm per ejaculate, total motility: 40%, progressive motility: 32%, and viability: 58%)66,67. In addition to the intrinsic characteristic and quality of the semen sample, the holding period of the semen before freezing is critical. It is recommended that the sample should be processed within an hour post-delivery for optimal post-thaw motility. Other factors that should be considered include initial sperm motility of at least 55% at one hour post-delivery with most cells in forward motility, less than 15% decay in sperm motility, and no significant decrease in forward motility 4 hours after delivery68.
Oocyte
Oocyte cryopreservation is a reliable method for the maintenance of fertility in women undergoing treatment for medical conditions such as cancer or autoimmune diseases. Treatment of these diseases could lead to ovarian insufficiency, which indicates the importance of fertility preservation. Ethical and legal concerns regarding embryo cryopreservation have led to research advancements in oocyte cryopreservation during the past few years as an alternative approach. Freezing eggs provides autonomy for women since it allows for elective fertility preservation compared to embryo preservation. Moreover, oocyte cryopreservtation provides women who have concerns about age-related reproductive decline with the option to delay pregnancy69,70.
Cryostorage of human oocytes is almost exclusively performed at the Metaphase II stage when both nuclear and cytoplasmic maturation has completed. The large size, high water content, and unique intracellular composition of oocytes makes cryopreservation challenging, however. Damage to the meiotic spindles, actin filaments, and DNA in addition to chromosome dispersion, microtubule depolymerization, and increased polyspermy rate are among specific challenges that that arise with oocyte preservation71. Like sperm, oocytes also appear to be susceptible to epigenetic changes as a result of low-temperature storage, although research in this area is ongoing70.
Currently, two approaches to oocyte cryopreservation dominate. They include slow cooling (equilibrium freezing protocols) and vitrification (ultra-rapid cooling or non-equilibrium protocols). Briefly, slow cooling involves gradual cell dehydration by adding low concentrations of a permeating agent such as DMSO (typically ≤ 1.5 M) and a non-permeating agent (commonly sucrose or trehalose, ≤ 0.3 M) with controlled slow cooling rates. After cooling to −150 oC, the cells are stored in liquid nitrogen at −196oC until they are needed for use. To thaw the cells, solutions with decreasing concentrations of NPAs are used to obtain gradual rehydration70. Cryosurvival of oocytes has increased with the improvement in slow cooling protocols, especially with the introduction of sucrose concentrations of greater than 0.1 M during pre-freeze dehydration. However, evidence from widespread practice indicates survival rates plateau around 70%–80% with this method72.
Vitrification protocols, however, are currently considered the best method for cryopreservation of oocytes. The most common method of vitrification involves the stepwise addition of CPAs in cryomedia. In the first equilibrium phase, oocytes are added to a solution containing 7.5% v/v EG and 7.5% v/v DMSO for 5 to 15 minutes. The cells are then exposed to a vitrification solution with 15% v/v EG and 15% v/v DMSO, plus 0.5 M sucrose. After a brief incubation (≤ 1 minute), the cells are t stored in liquid nitrogen at −196oC. Warming should be performed rapidly to avoid ice crystal formation, and after gradual removal of the CPA, the cells should be incubated in culture medium until use70,73,74. A systematic review reported that the rates of oocyte cryosurvival and fertilization were higher in vitrified oocytes than slow-cooled oocytes. Vitrification also resulted in a higher top-rate quality embryo (22.4% vs 8.0%) and a higher cleavage rate (day 2: 64.6% vs 47.7%) when compared to slow rate cooling. Moreover, the rates of ongoing pregnancy, top-quality embryo, embryo cleavage, and fertilization were not significantly different between vitrified and fresh oocytes. These findings suggest vitrification is the better procedure for the cryopreservation of oocytes75.
Stem cells
Stem cells’ ability to differentiate into various cell phenotypes provides the foundation for regenerative cell therapies including treatment of degenerative diseases and traumatic injuries. Stem cell-based therapies allow the restoration of tissue structure and functional recovery. Moreover, the biomolecules synthesized by stem cells can aid in tissue repair76. Hematopoietic stem cells (HSCs) can be used for the treatment of hematological as well as non-hematological diseases77. The immunomodulatory and immunosuppressive properties of mesenchymal stem cells (MSCs) make this cell type ideal for allogenic transplantation.78,79 In addition to their clinical applications in tissue regeneration and transplantation, human embryonic stem cells (ESCs) are used in research to study basic developmental processes and cell signaling pathways80. In order to further extend the clinical applications of stem cells, long-term cryogenic banking protocols should be followed that are suitable for each stem cell type. Cryopreservation protocols for HSCs and MSCs follow traditional slow cooling methods, whereas ESC protocols follow a rapid cooling/ vitrification approach. The standard approach for cryopreservation of HSCs includes the use of DMSO as a CPA, controlled rate of freezing at 1 to 2 ºC/min, and rapid thawing77,81. MSCs can be cryopreserved similarly using slow freezing protocols and DMSO as a CPA82.
Conventional methods (DMSO and slow cooling rate), however, have shown low efficiency for cryopreservation of ESCs. As a result, vitrification protocols have been developed for cryopreservation of ESCs. A higher recovery rate has been reported for vitrified ESCs compared to the cells cryopreserved by slow cooling protocols (75% compared to about 5%)83–85. A detailed protocol for the vitrification of human ESCs has been described. Briefly, human ESCs are exposed to the stepwise addition of two vitrification solutions of increasing concentration of CPA. The common components of both solutions include DMSO and EG. The composition of the vehicle solution can vary with differences in the concentration of sucrose and the presence or absence of serum and the buffer used. Human ESC colonies are exposed to the two vitrification solutions sequentially for 60 and 25 seconds respectively at room temperature or 37 ºC. In addition, it has been suggested that the higher cooling rate in non-equilibrium vitrification processes could allow a lower total concentration of CPA to be used, and a combination of CPAs could help to reduce their toxic effects. Reubinoff et al. suggested a mixture of 20% DMSO, 20% EG, and 0.5 M sucrose with rapid cooling rates72,83,86.
Like fully-differentiated cells, non-or partially committed stem cells are subject to adverse alterations in structure and function after cryopreservation. A recent systematic review compared post-thaw assessments of bone marrow-derived MSCs across ten species from 41 separate studies87. The authors reported an overall consensus that morphology, immunophenotype, differentiation and proliferation potential were largely unaffected by cryopreservation, but that two thirds of experiments reported decreased metabolic activity post-cryopreservation. In addition, reported changes in viability varied with approximately 43% of studies reporting no change and 27% reporting significantly decreased viability87. Post-cryo decreases in viability and metabolic activity are often the result of physical and molecular cell damage. Physical injuries include intracellular ice formation, solution effects, cryo solution toxicity, and molecular damage that manifests as alterations in gene expression, stress response induction, and epigenetic alterations88,89,90. The molecular mechanisms behind genetic responses to cryopreservation in stem cells, much like gametic responses, are not completely understood and are still an area of active research87.
Finally, several assays exist to assess cell viability after cryopreservation which includes membrane integrity test, metabolic and mechanical activity assays, and mitotic activity assays such as “plating” tests. To determine the in vivo functionality of the cryopreserved cells, fertilization and development assays as well as transplantation assays can be performed91.
Novel agents, and biomaterials to improve viability
More recent approaches to cell cryopreservation demonstrate increased post-thaw viability and yield when cells are encapsulated prior to freezing. One example used alginate encapsulation of hepatocytes to increase cryopreservation success. In combination with University of Wisconsin (UW) solution + 10% DMSO + 5% glucose, encapsulated cell microbeads showed better viability post-thawing than non-encapsulated cells, and better than the standard cryopreservation solution (Bambanker®). The same approach could be tested on other cells types to improve cryopreservation yields. This effect is further improved when using pan-caspase inhibitor (benzyloxycarbonyl-Val-Ala-dl-Asp-fluoromethylketone [ZVAD])92. A more recent study bypasses the need for alginate, instead, freezing the hepatocytes as droplets in the CPA mixture (UW supplemented with 2 mg/mL BSA, 32.5% v/v DMSO, 32.5% v/v EG, and 800 mM sucrose). The droplets were frozen rapidly in liquid nitrogen, unusual in comparison to the slow freezing normally used for cryopreservation. Bulk-droplet-vitrified hepatocytes had significantly higher viability, better morphology, and higher metabolic activity than non-droplet frozen hepatocytes93. Aside from changing the cryopreservation techniques, addition of cell-survival signal, such as myricetin (inhibitor of mitogen-activated protein kinase kinase 4 (MKK4) in culture after thawing cryopreserved hepatocytes aids both the cell survival in vitro and after transplantation in immunodeficient mice94.
Novel agents to improve post-cryo cell recovery
Great effort is made to preserve cell functionality and viability throughout the cryopreservation process since each step, primary cell isolation, initial purification, culture, CPA selection, and freeze-thaw rates carries with it the potential to damage cells and decrease overall cell function. In spite of these efforts, cryopreserved cells invariably experience a decrease in viability post-thaw. Current strategies for identifying cells that remain viable after preservation utilize organic fluorophores, and dyes95. And, as discussed previously, density gradients can be utilized to increase viable cell density although this method often involves exposing cells to additional, potentially-harmful centrifugation. More involved methods to separate viable and non-viable cells include magnetic affinity cell separation (MACS), and fluorescence-activated cell sorting (FACS) although the cost and practicality sometimes limit their utility95,96. The emerging field of nanoscience could provide the next step forward in cryobiology by offering alternative non-destructive cell sorting and live cell imaging methods95. One recent study utilized annexin V-conjugated magnetic nanoparticles to enrich viable sperm content in fresh boar semen via selective binding to the exposed phosphatidylserine present in apoptotic cells97,98. This sorting scheme could be utilized as a simple high throughput alternative to enrich post-thaw cell viability in an array of cell types.
Conclusion
Significant improvements in our understanding of the cryopreservation principles and techniques for long-term storage and cryobanking of the cells have been made since the late 1800s when the research on the effect of cryopreservation on live tissue first began. Long-term storage of the cells at −196°C halts cellular metabolism which results in inevitable alternations in lipids and proteins that could impair cell function and structure. Ideally, a higher concentration of CPAs could allow the cells to be preserved perfectly. However, increasing the concentrations of CPAs could also be damaging to the cells. DMSO has remained the gold standard CPA for many different cell types. New techniques such as a combination of CPAs or the use of new CPAs has been investigated to address the toxicity effect of some known agents99–101. In addition, alginate encapsulation of cells prior to freezing has been shown to improve preservation yields through non-chemical means. Regardless of cell type, the success of any cryopreservation protocol is dictated by careful selection of a few common variables: cryoprotecting agent type including permeating and non-permeating agents or a combination of both, as well as appropriate cooling and thawing rates. More recent approaches such as alginate encapsulation and nanotechnology-based cell sorting may offer enhanced post-cryopreservation viability and could eventually become a part of standard cryopreservation milieu. Understanding the principles behind the chemistry and biology of freezing and thawing processes could allow the development of more efficient procedures for cryopreservation of cells and further expand their clinical applications.
Acknowledgments
The authors would like to acknowledge the support of the University of California, Irvine Department of Surgery, and Ambys Medicines, for providing their expertise and support throughout the writing of this manuscript.
Statement of Human and Animal Rights: This article does not contain any studies with human or animal subjects.
Statement of Informed Consent: There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Jonathan RT Lakey  https://orcid.org/0000-0001-8553-4287
https://orcid.org/0000-0001-8553-4287
References
- 1. Walters EM, Benson JD, Woods EJ, Crister JK. The history of sperm cryopreservation. In: Pacey AA, Tomlinson MJ, eds. Sperm Banking Theory and Practice. London (UK: ): Cambridge University Press; 2009. p. 1–17. [Google Scholar]
- 2. Lovelock JE. The haemolysis of human red blood-cells by freezing and thawing. Biochim Biophys Acta. 1953;10(3):414–426. [DOI] [PubMed] [Google Scholar]
- 3. Mazur P. Kinetics of water loss from cells at subzero temperatures and the likelihood of intracellular freezing. J Gen Physiol. 1963;47(2):347–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chesné C, Guillouzo A. Cryopreservation of isolated rat hepatocytes: a Critical evaluation of freezing and thawing conditions. Cryobiology. 1988;25(4):323–330. [DOI] [PubMed] [Google Scholar]
- 5. Al-Hasani S, Diedrich K, Wan der Ven H, Reinecke A, Hartje M, Krebs D. Cryopreservation of human oocytes. Hum Reprod. 1987;2(8):695–700. [DOI] [PubMed] [Google Scholar]
- 6. Mazur P, Leibo SP, Chu EHY. A two-factor hypothesis of freezing injury: evidence from chinese hamster tissue-culture cells. Exp Cell Res. 1972;71(2):345–355. [DOI] [PubMed] [Google Scholar]
- 7. Polge C, Smith AU, Parkes AS. Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature. 1949;164(4172):666. [DOI] [PubMed] [Google Scholar]
- 8. Siebzehnruebl E, Todorow S, Van Uem J, Koch R, Wildt L, Lang N. Cryopreservation of human and rabbit oocytes and one-cell embryos: a Comparison of DMSO and propanediol. Hum Reprod. 1989;4(3):312–317. [DOI] [PubMed] [Google Scholar]
- 9. Friedler S, Giudice LC, Lamb EJ. Cryopreservation of embryos and ova. Fertil Steril. 1988;49(5):743–764. [DOI] [PubMed] [Google Scholar]
- 10. Ock SA, Rho GJ. Effect of dimethyl sulfoxide (DMSO) on cryopreservation of porcine mesenchymal stem cells (pMSCs). Cell Transplant. 2011;20(8):1231–1239. [DOI] [PubMed] [Google Scholar]
- 11. Boquet M, Selva J, Auroux M. Effects of cooling and equilibration in DMSO, and cryopreservation of mouse oocytes on the rates of in vitro fertilization, development, and chromosomal abnormalities. Mol Reprod Dev. 1995;40(1):110–115. [DOI] [PubMed] [Google Scholar]
- 12. Mandumpal JB, Kreck CA, Mancera RL. A molecular mechanism of solvent cryoprotection in aqueous DMSO solutions. Phys Chem Chem Phys. 2010;13(9):3839–3842. [DOI] [PubMed] [Google Scholar]
- 13. Pegg DE. Principles of cryopreservation. In: Day JG, Stacey GN, eds. Cryopreservation and Freeze-Drying Protocols. Methods in Molecular Biology. Totowa (NJ: ): Humana Press; 2007. p. 39–75. [Google Scholar]
- 14. Wowk B. How cryoprotectants work. cryonics. scottsdale (AZ): Alcor life extension foundation;2007. Cited. 2019;27. [Google Scholar]
- 15. Bartolac LK, Lowe JL, Koustas G, Grupen CG, Sjöblom C. Effect of different penetrating and non-penetrating cryoprotectants and media temperature on the cryosurvival of vitrified in vitro produced porcine blastocysts. Anim Sci J. 2018;89(9):1230–1239. [DOI] [PubMed] [Google Scholar]
- 16. Gurtovenko AA, Anwar J. Modulating the structure and properties of cell membranes: The molecular mechanism of dimethyl sulfoxide. J Phy Chem B. 2007;111(35):10453–10460. [DOI] [PubMed] [Google Scholar]
- 17. He F, Liu W, Zheng S, Zhou L, Ye B, Qi Z. Ion transport through dimethyl sulfoxide (DMSO) induced transient water pores in cell membranes. Mol Membr Biol. 2012;29(3–4):107–113. [DOI] [PubMed] [Google Scholar]
- 18. Eroglu A. Cryopreservation of mammalian oocytes by using sugars: intra- and extracellular raffinose with small amounts of dimethylsulfoxide yields high cryosurvival, fertilization, and development rates. Cryobiology. 2010;60(3 suppl):S54–S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13(4):17R–27R. [DOI] [PubMed] [Google Scholar]
- 20. Reis FS, Barros L, Martins A, Ferreira IC. Chemical composition and nutritional value of the most widely appreciated cultivated mushrooms: an inter-species comparative study. Food Chem Toxicol. 2012;50(2):191–197. [DOI] [PubMed] [Google Scholar]
- 21. Kojayan GG, Whaley D, Alexander M, Rodriguez S, Lee S, Lakey JR. Improved cryopreservation yield of pancreatic islets using combination of lower dose permeable cryoprotective agents. Cryobiology. 2019;88:23–28. [DOI] [PubMed] [Google Scholar]
- 22. Standring S., Gray’s Anatomy: The anatomical basis of clinical practice, 41st ed. Philadelphia (PA: ): Elsevier Limited; 2016. [Google Scholar]
- 23. Sieme H, Oldenhof H, Wolkers WF. Mode of action of cryoprotectants for sperm preservation. Anim Reprod Sci. 2016;169:2–5. [DOI] [PubMed] [Google Scholar]
- 24. Stéphenne X, Najimi M, Sokal EM. Hepatocyte cryopreservation: Is it time to change the strategy? World J Gastroenterol. 2010;16(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li AP, Gorycki PD, Hengstler JG, Kedderis GL, Koebe HG, Rahmani R, de Sousas G, Silva JM, Skett P. Present status of the application of cryopreserved hepatocytes in the evaluation of xenobiotics: Consensus of an international expert panel. Chem Biol Interact. 1999;121(1):117–123. [DOI] [PubMed] [Google Scholar]
- 26. Rijntjes PJ, Moshage HJ, Van Gemert PJ, De Waal R, Yap SH. Cryopreservation of adult human hepatocytes. The influence of deep freezing storage on the viability, cell seeding, survival, fine structures and albumin synthesis in primary cultures. J Hepatol. 1986;3(1):7–18. [DOI] [PubMed] [Google Scholar]
- 27. Dou M, de Sousa G, Lacarelle B, Placidi M, Lechene de la Porte P, Domingo M, Lafont H, Rahmani R. Thawed human hepatocytes in primary culture. Cryobiology. 1992;29:454–469. [DOI] [PubMed] [Google Scholar]
- 28. De Sousa G, Langouët S, Nicolas F, Lorenzon G, Placidi M, Rahmani R, Guillouzo A. Increase of cytochrome P-450 1A and glutathione transferase transcripts in cultured hepatocytes from dogs, monkeys, and humans after cryopreservation. Cell Biol Toxicol. 1996;12(4–6):351–358. [DOI] [PubMed] [Google Scholar]
- 29. Ostrowska A, Bode DC, Pruss J, Bilir B, Smith GD, Zeisloft S. Investigation of functional and morphological integrity of freshly isolated and cryopreserved human hepatocytes. Cell Tissue Bank. 2000;1(1):55–68. [DOI] [PubMed] [Google Scholar]
- 30. Baboo J, Kilbride P, Delahaye M, Milne S, Fonseca F, Blanco M, Meneghel J, Nancekievill A, Gaddum N, Morris GJ. The impact of varying cooling and thawing rates on the quality of cryopreserved human peripheral blood T cells. Sci Rep. 2019;9(1):3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benson JD, Woods EJ, Walters EM, Critser JK. The cryobiology of spermatozoa. Theriogenology. 2012;78(8):1682–1699. [DOI] [PubMed] [Google Scholar]
- 32. Thompson M, Nemits M, Ehrhardt R. Rate-controlled cryopreservation and thawing of mammalian cells [method article]. https://protocolexchange.researchsquare.com/article/nprot-2085/v1 (accessed 28 January 2021). [Google Scholar]
- 33. Yokoyama WM, Thompson ML. Ehrhardt RO. Cryopreservation and thawing of cells. Curr Protoc Immunol. 2012;99:A.3G.1–A.3G.5. [DOI] [PubMed] [Google Scholar]
- 34. Terry C, Dhawan A, Mitry RR, Lehec SC, Hughes RD. Optimization of the cryopreservation and thawing protocol for human hepatocytes for use in cell transplantation. Liver Transpl. 2010;16(2):229–237. [DOI] [PubMed] [Google Scholar]
- 35. Thorpe P, Knight SC, Farrant J. Optimal conditions for the preservation of mouse lymph node cells in liquid nitrogen using cooling rate techniques. Cryobiology. 1976;13(2):126–133. [DOI] [PubMed] [Google Scholar]
- 36. Gao D, Critser JK. Mechanisms of cryoinjury in living cells. ILAR J. 2000;41(4):187–196. [DOI] [PubMed] [Google Scholar]
- 37. Najimi M, Sokal E. Liver cell transplantation. Minerva Pediatr. 2005;57(5):243–257. [PubMed] [Google Scholar]
- 38. Gramignoli R, Dorko K, Tahan V, Skvorak KJ, Ellis E, Jorns C, Ericzon BG, Fox IJ, Strom SC. Hypothermic storage of human hepatocytes for transplantation. Cell Transplant. 2014;23(9):1143–1151. [DOI] [PubMed] [Google Scholar]
- 39. Gómez-Lechón MJ, Lahoz A, Jiménez N, Bonora A, Castell JV, Donato MT. Evaluation of drug-metabolizing and functional competence of human hepatocytes incubated under hypothermia in different media for clinical infusion. Cell Transplant. 2008;17(8):887–897. [DOI] [PubMed] [Google Scholar]
- 40. Sosef MN, Baust JM, Sugimachi K, Fowler A, Tompkins RG, Toner M. Cryopreservation of isolated primary rat hepatocytes: enhanced survival and long-term hepatospecific function. Ann Surg. 2005;241(1):125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hengstler JG, Utesch D, Steinberg P, Platt KL, Diener B, Ringel M, Swales N, Fischer T, Biefang K, Gerl M, Böttger T, et al. Cryopreserved primary hepatocytes as a constantly available in vitro model for the evaluation of human and animal drug metabolism and enzyme induction. Drug Met Rev. 2000;32(1):81–118. [DOI] [PubMed] [Google Scholar]
- 42. Miyamoto Y, Suzuki S, Nomura K, Enosawa S. Improvement of hepatocyte viability after cryopreservation by supplementation of long-chain oligosaccharide in the freezing medium in rats and humans. Cell Transplant. 2006;15(10):911–919. [DOI] [PubMed] [Google Scholar]
- 43. Lloyd TD, Orr S, Skett P, Berry DP, Dennison AR. Cryopreservation of hepatocytes: a review of current methods for banking. Cell Tissue Bank. 2003;4(1):3–15. [DOI] [PubMed] [Google Scholar]
- 44. Aoki T, Koizumi T, Kobayashi Y, Yasuda D, Izumida Y, Jin Z, Nishino N, Shimizu Y, Kato H, Murai N, Niiya T, et al. A novel method of cryopreservation of rat and human hepatocytes by using encapsulation technique and possible use for cell transplantation. Cell Transplant. 2005;14(9):609–620. [DOI] [PubMed] [Google Scholar]
- 45. Bruni A, Gala-Lopez B, Pepper AR, Abualhassan NS, Shapiro AJ. Islet cell transplantation for the treatment of type 1 diabetes: recent advances and future challenges. Diabetes Met Syndr Obes. 2014;7:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54(7):2060–2069. [DOI] [PubMed] [Google Scholar]
- 47. Kojayan GG, Alexander M, Imagawa DK, Lakey JR. Systematic review of islet cryopreservation. Islets. 2018;10(1):40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Foreman J, Moriya H, Taylor MJ. Effect of cooling rate and its interaction with pre-freeze and post-thaw tissue culture on the in vitro and in vivo function of cryopreserved pancreatic islets. Transpl Int. 1993;6(4):191–200. [DOI] [PubMed] [Google Scholar]
- 49. Liu F, Tian W, Yang Y, Zhang Q, Zhu M, Yang L, Yang L, Li J, Liu J, Wu P, Yang K, et al. Optimal method for short-term or long-term islet preservation: Comparison of islet culture, cold preservation and cryopreservation. J Artif Organs. 2014;17(4):337–343. [DOI] [PubMed] [Google Scholar]
- 50. Rajotte RV, Warnock GL, Kneteman NM, Erickson C, Ellis DK. Optimizing cryopreservation of isolated islets. Transplant Proc. 1989;21(1 Pt 3):2638–2640. [PubMed] [Google Scholar]
- 51. Komatsu H, Barriga A, Medrano L, Omori K, Kandeel F, Mullen Y. Oxygenated thawing and rewarming alleviate rewarming injury of cryopreserved pancreatic islets. Biochem Biophys Res Commun. 2017;486(3):817–823. [DOI] [PubMed] [Google Scholar]
- 52. Taylor MJ, Baicu S. Review of vitreous islet cryopreservation: some practical issues and their resolution. Organogenesis. 2009;5(3):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chandravanshi B, Dhanushkodi A, Bhonde R. High recovery of functional islets stored at low and ultralow temperatures. Rev Diabet Stud. 2014;11(3–4):267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kojayan GG, Flores A, Li S, Alexander M, Lakey JR. Cryopreserved alginate-encapsulated islets can restore euglycemia in a diabetic animal model better than cryopreserved non-encapsulated islets. Cell Med. 2019;11:2155179019876641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boyd V, Cholewa OM, Papas KK. Limitation in the use of fluorescein diacetate/propidium iodide (FDA/PI) and cell permeable nucleic acid stains for viability measurements of isolated islets of langerhans. Curr Trends Biotechnol Pharm. 2008;2(2):66–84. [PMC free article] [PubMed] [Google Scholar]
- 56. Lau H, Corrales N, Alexander M, Mohammadi MR, Li S, Smink AM, de Vos Paul, Lakey JRT. Necrostatin-1 supplementation enhances young porcine islet maturation and in vitro function. Xenotransplantation. 2020;27(1);e12555. [DOI] [PubMed] [Google Scholar]
- 57. Barnett MJ, McGhee-Wilson D, Shapiro AM, Lakey JR. Variation in human islet viability based on different membrane integrity stains. Cell Transplant. 2004;13(5):481–488. [DOI] [PubMed] [Google Scholar]
- 58. Yeste M. Sperm cryopreservation update: Cryodamage, markers, and factors affecting the sperm freezability in pigs. Theriogenology. 2016;85(1):47–64. [DOI] [PubMed] [Google Scholar]
- 59. Loren AW, Mangu PB, Beck LN, Brennan L, Magdalinski AJ, Partridge AH, Quinn G, Wallace WH, Oktay K, American Society of Clinical Oncology. Fertility preservation for patients with cancer: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2013;31(19):2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Comizzoli P, Wildt DE. Mammalian fertility preservation through cryobiology: Value of classical comparative studies and the need for new preservation options. Reprod Fertil Dev. 2013;26:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Horne G, Atkinson AD, Pease EHE, Logue JP, Brison DR, Lieberman BA. Live birth with sperm cryopreserved for 21 years prior to cancer treatment: Case report, Hum Reprod. 2004;19(6):1448–1449. [DOI] [PubMed] [Google Scholar]
- 62. Anger JT, Gilbert BR, Goldstein M. Cryopreservation of sperm: indications, methods and results. J Urol. 2003;170(4 Pt 1):1079–1084. [DOI] [PubMed] [Google Scholar]
- 63. Mahadevan M, Trounson AO. Effect of cooling, freezing and thawing rates and storage conditions on preservation of human spermatozoa. Andrologia. 1984;16(1):52–60. [DOI] [PubMed] [Google Scholar]
- 64. Valcarce DG, Cartón-García F, Riesco MF, Herráez MP, Robles V. Analysis of DNA damage after human sperm cryopreservation in genes crucial for fertilization and early embryo development. Andrology. 2013;1(5):723–730. [DOI] [PubMed] [Google Scholar]
- 65. Thomson LK, Fleming SD, Aitken RJ, De Iuliis GN, Zieschang JA, Clark AM. Cryopreservation-induced human sperm DNA damage is predominantly mediated by oxidative stress rather than apoptosis. Hum Reprod. 2009;24(9):2061–2070. [DOI] [PubMed] [Google Scholar]
- 66. Degl’Innocenti S, Filimberti E, Magini A, Krausz C, Lombardi G, Fino MG, Rastrelli G, Maggi M, Baldi E. Semen cryopreservation for men banking for oligospermia, cancers, and other pathologies: Prediction of post-thaw outcome using basal semen quality. Fertil Steril. 2013;100(6):1555–1563. [DOI] [PubMed] [Google Scholar]
- 67. World Health Organization. WHO laboratory manual for the examination and processing of human semen. 5th ed. Geneva (Switzerland; ): Department of Reproductive Health and Research; 2010. [Google Scholar]
- 68. Haim Y, Yogev L, Homonnai Z, Paz G. Prerequisites for successful human sperm cryobanking: Sperm quality and prefreezing holding time. Fertil Steril. 1991;55(4):812–816. [DOI] [PubMed] [Google Scholar]
- 69. Rienzi L, Ubaldi FM. Oocyte versus embryo cryopreservation for fertility preservation in cancer patients: guaranteeing a women’s autonomy. J Assist Reprod Genet. 2015;32(8):1195–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Prentice JR, Anzar M. Cryopreservation on mammalian oocyte for conservation of animal genetics. Vet Med Int. 2011;2011:146405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Iussig B, Maggiulli R, Fabozzi G, Bertelle S, Vaiarelli A, Cimadomo D, Ubaldi FM, Rienzi L. A brief history of oocyte cryopreservation: Arguments and facts. Acta Obstet Gynecol Scand. 2019;98(5):550–558. [DOI] [PubMed] [Google Scholar]
- 72. Edgar DH, Gook DA. A critical appraisal of cryopreservation (slow cooling versus vitrification) of human oocytes and embryos. Hum Reprod Update. 2012;18(5):536–554. [DOI] [PubMed] [Google Scholar]
- 73. Kuwayama M, Vajta G, Kato O, Leibo SP. Highly efficient vitrification method for cryopreservation of human oocytes. Reprod biomed Online. 2005;11(3):300–308. [DOI] [PubMed] [Google Scholar]
- 74. Kuwayama M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: The Cryotop method. Theriogenology. 2007;67(1):73–80. [DOI] [PubMed] [Google Scholar]
- 75. Cobo A, Diaz C. Clinical application of oocyte vitrification: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2011;96(2):277–285. [DOI] [PubMed] [Google Scholar]
- 76. Baraniak PR, McDevitt TC. Stem cell paracrine actions and tissue regeneration. Regen Med. 2010;5(1):121–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hunt CJ. Cryopreservation of human stem cells for clinical application: a review. Transfus Med Hemother. 2011;38(2):107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110(10):3499–3506. [DOI] [PubMed] [Google Scholar]
- 79. De Miguel MP, Fuentes-Julián S, Blázquez-Martínez A, Pascual CY, Aller MA, Arias J, Arnalich-Montiel F. Immunosuppressive properties of mesenchymal stem cells: advances and applications. Curr Mol Med. 2012;12(5):574–591. [DOI] [PubMed] [Google Scholar]
- 80. Ha SY, Jee BC, Suh CS, Kim HS, Oh SK, Kim SH, Moon SY. Cryopreservation of human embryonic stem cells without the use of a programmable freezer. Hum Reprod. 2005;20(7):1779–1785. [DOI] [PubMed] [Google Scholar]
- 81. Berz D, McCormack EM, Winer ES, Colvin GA, Quesenberry PJ. Cryopreservation of hematopoietic stem cells. Am J Hematol. 2007;82(6):463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Thirumala S, Goebel WS, Woods EJ. Clinical grade adult stem cell banking. Organogenesis. 2009;5(3):143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Reubinoff BE, Pera MF, Vajta G, Trounson AO. Effective cryopreservation of human embryonic stem cells by the open pulled straw vitrification method. Hum Reprod. 2001;16(10):2187–2194. [DOI] [PubMed] [Google Scholar]
- 84. Zhou CQ, Mai QY, Li T, Zhuang GL. Cryopreservation of human embryonic stem cells by vitrification. Chin Med J. 2004;117(7):1050–1055. [PubMed] [Google Scholar]
- 85. Richards M, Fong CY, Tan S, Chan WK, Bongso A. An efficient and safe xeno-free cryopreservation method for the storage of human embryonic stem cells. Stem Cells. 2004;22(5):779–789. [DOI] [PubMed] [Google Scholar]
- 86. Hunt CJ, Timmons PM. Cryopreservation of human embryonic stem cell lines. In: Day JG, Stacey G, editors. Cryopreservation and Freeze-Drying Protocols. Methods in Molecular Biology. Totowa (NJ: ): Humana Press; 2007. p. 261–270. [DOI] [PubMed] [Google Scholar]
- 87. Bahsoun S, Coopman K Akam EC. The impact of cryopreservation on bone marrow-derived mesenchymal stem cells: a systematic review. J Transl Med. 2019;17(1):397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Woods EJ, Thirumala S, Badhe-Buchanan SS, Clarke D, Mathew AJ. Off the shelf cellular therapeutics: factors to consider during cryo-preservation and storage of human cells for clinical use. Cytotherapy. 2016;18(6):697–711. [DOI] [PubMed] [Google Scholar]
- 89. Baust JM, Corwin W, Snyder KK, Van Buskirk R, Baust JG. Cryopreservation: evolution of molecular based strategies. Biobanking and cryopreservation of stem cells. Cham: Springer; 2016. p. 13–29. [DOI] [PubMed] [Google Scholar]
- 90. Chatterjee A, Saha D, Niemann H, Gryshkov O, Glasmacher B, Hofmann N. Effects of cryopreservation on the epigenetic profile of cells. Cryobiology. 2017;74:1–7. [DOI] [PubMed] [Google Scholar]
- 91. Pegg DE. Viability assays for preserved cells, tissues, andorgans. Cryobiology. 1989;26(3):212–231. [DOI] [PubMed] [Google Scholar]
- 92. Jitraruch S, Dhawan A, Hughes RD, Filippi C, Lehec SC, Glover Leanne, Mitry RR. Cryopreservation of hepatocyte microbeads for clinical transplantation. Cell Transplant. 2017;26(8):1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. De Vries RJ, Banik PD, Nagpal S, Weng L, Ozer S, Van Guilk TM, Toner M, Tessier SN, Uygun K. Bulk droplet vitrification: an approach to improve large-scale hepatocyte cryopreservation outcome. Langmuir. 2019;35(23):7354–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cui C, Enosawa S, Matsunari H, Nagashima H, Umezawa A. Natural flavinol, myricetin, enhances the function and survival of cryopreserved hepatocytes in vitro and in vivo. Int J Mol Sci. 2019;20(24):6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Feugang JM. Novel agents for sperm purification, sorting, and imaging. Mol Reprod Dev. 2017;84(9):832–841. [DOI] [PubMed] [Google Scholar]
- 96. Okumura N, Kusakabe A, Hirano H, Inoue R, Okazaki Y, Nakano S, Kinoshita S, Koizumi N. Density-gradient centrifugation enables the purification of cultured corneal endothelial cells for cell therapy by eliminating senescent cells. Sci Rep. 2015;5:15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Durfey CL, Swistek SE, Liao SF, Crenshaw MA, Clemente HJ, Thirumalai RVKG, Steadman CS, Ryan PL, Willard ST, Feugang JM. Nanotechnology-based approach for safer enrichment of semen with best spermatozoa. J Anim Sci Biotechnol. 2019;10:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Logue S, Elgendy M, Martin S. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc. 2009;4(9):1383–1395. [DOI] [PubMed] [Google Scholar]
- 99. Jang TH, Park SC, Yang JH, Kim JY, Seok JH, Park US, Choi CW, Lee SR, Han J. Cryopreservation and its clinical applications. Integr Med Res. 2017;6(1):12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Karlsson JO, Toner M. Long-term storage of tissues by cryopreservation: Critical issues. Biomaterials. 1996;17(3):243–256. [DOI] [PubMed] [Google Scholar]
- 101. Best BP. Cryoprotectant toxicity: facts, issues, and questions. Rejuvenation Res. 2015;18(5):422–436. [DOI] [PMC free article] [PubMed] [Google Scholar]