

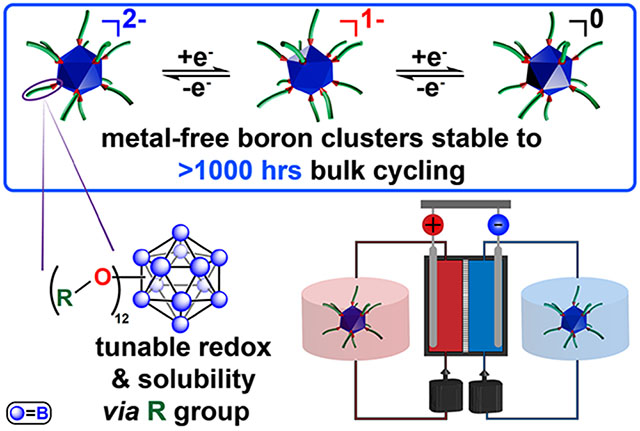
Abstract
We report a class of perfunctionalized dodecaborate clusters that exhibit high stability towards high concentration electrochemical cycling. These boron clusters afford several degrees of freedom in material design to tailor properties including solubility and redox potential. The exceptional stability of these clusters was demonstrated using a symmetric flow cell setup for electrochemical cycling between two oxidation states for 45 days, with post-run analysis showing negligible decomposition of the active species (<0.1%). To further probe the limits of this system, a prototype redox flow battery with two different cluster materials was used to determine mutual compatibility. This work effectively illustrates the potential of bespoke boron clusters as robust material platform for electrochemical energy conversion and storage.
Graphical Abstract
INTRODUCTION
The ability to reversibly shuttle electrons over a range of timescales is a ubiquitous feature found in both natural and synthetic molecules, and is essential for energy conversion and storage applications.1–10 Energy storage systems often utilize redox active metal cations as stable salts or coordination complexes, though inherent limitations hinder the degree to which solubility and redox potential of these species can be modified.11 Metal-free species can also undergo well-defined redox processes, though decomposition due to innate reactivity of radical intermediates formed during single electron shuttling is a common pitfall.12,13 To mitigate this undesired reactivity, molecular systems have been designed to delocalize radical density throughout neighboring bonds and/or to sterically protect the site of the unpaired electron (Figure 1a), which can lead to dramatic improvements in the cycle stability of these compounds.11,12,14
Figure 1.
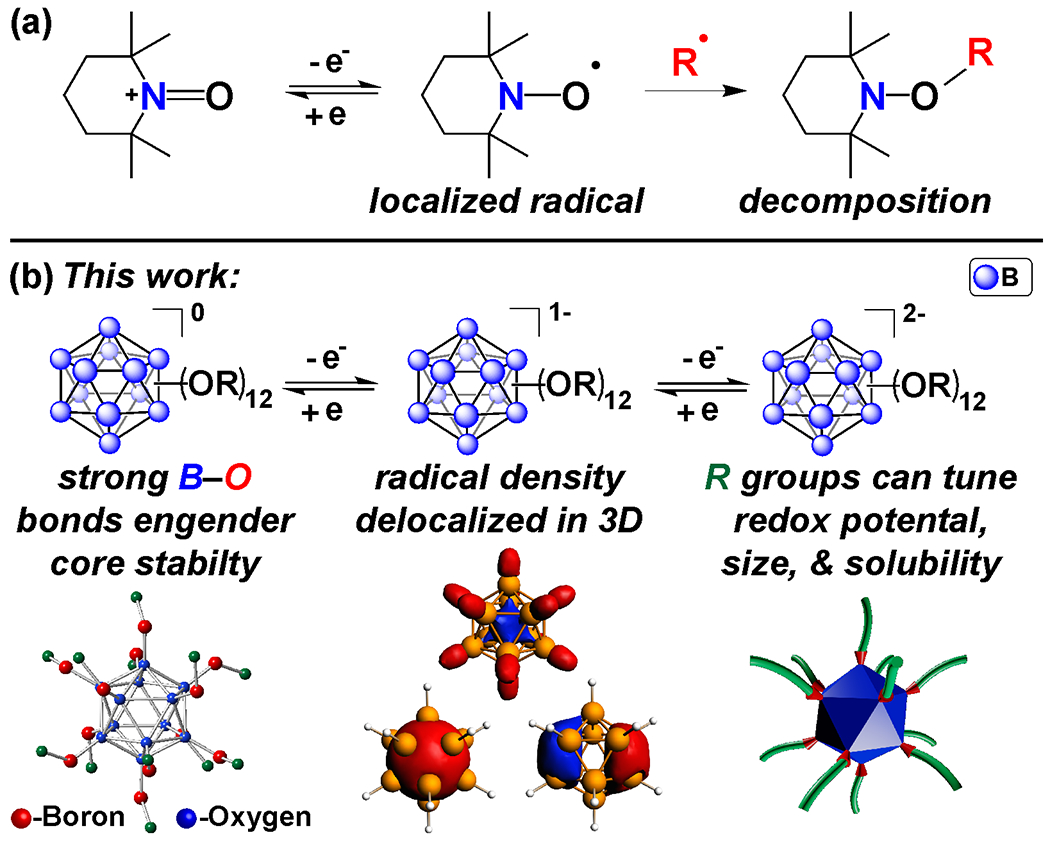
(a) Decomposition pathway of a prototypical redox-active organic radical (TEMPO) via a radical coupling. (b) Redox-active B12(OR)12 clusters do not undergo decomposition via radical coupling due to the 3D delocalization of the radical spin density.
One such system employing stable charge carriers is the redox flow battery (RFB). RFBs are electrochemical energy storage technologies where solubilized charge storage materials are stored in external tanks and circulated through an electrochemical reactor where energy is extracted or stored. The majority of RFB literature, including their original development, exists within the aqueous domain, depending on metallic redox centers to store charge.15–17 More recently, RFB work has been extended into the nonaqueous domain, often with organic redox-active materials, primarily incentivized by the limited potential stability window of water and the cost of metal-based active species.18,19
Recent work has used process simulation software to demonstrate the low break-even price for the production of anthraquinone disulfonic acid, as considered to be a model organic material, largely due to the low-cost commercially-available feedstocks and limited number of process steps.20 Within this work, we demonstrate use of a new set of metal-free redox active materials for RFBs produced from commodity feedstocks with only 3 synthesis steps, implicitly suggesting the potential for low-cost production.21,22
Perfunctionalized dodecaborate clusters (B12(OR)12, R = H, alkyl, benzyl) are a promising new class of redox materials, which have demonstrated reversible redox activity when modified with different substituents (Figure 1b).23–25 Many of these clusters are extremely robust molecules, displaying thermal stability and resistance to harsh chemical environments.26 Several prior reports have developed metal coordination complexes or polyoxometalates, both of which have high levels of electron delocalization by design to promote stability.27–32 While superficially, the polyoxometalates appear to be structurally similar to the clusters presented here; however, the removal of metallic elements further can increase the stability such that these materials may be handled in the open air.29–30 Over a dozen B12(OR)12 clusters have been synthesized and electrochemically characterized by cyclic voltammetry (CV), exhibiting redox transitions that are reversibly accessible via sequential one-electron oxidation or reduction.23,24
In addition, the solubility and size of functionalized boron clusters can be modified to incorporate specific properties desirable for particular applications.24,33–35 Importantly, B12(OR)12 species can be isolated in a monoradical form (1−) and have shown good stability due to the complete delocalization of the radical spin density across the boron cluster cage (Figure 1b). Extending beyond voltammetric studies and ex-situ evaluation of stability, here we demonstrate the viability of these perfunctionalized boron clusters in complex electrolyte environments found in modern and emerging electrochemical systems. Specifically, we show several of these B12(OR)12 compounds can be electrochemically charged and discharged multiple times over 1000 hours without any apparent chemical degradation. Leveraging these attractive electrochemical properties, we have furthermore assembled a proof-of-concept redox flow battery, where performance of this metal-free cluster system was further demonstrated.
EXPERIMENTAL SECTION
Cyclic Voltammetry and Randles-Sevcik Analysis
All preparation and cyclic voltammetry (CV) measurements were performed in an argon-filled glovebox (MBraun Labmaster, O2 < 1 ppm, H2O < 5 ppm) at ca. 26 °C. The electrolyte used was 0.5 M TBAPF6 (Sigma-Aldrich, 98%) in MeCN (BASF, > 99.9%) (Figure S1). Solutions were prepared in a volumetric flask in order to account for the volume change from the high concentration of supporting salt (for full synthetic procedures and characterization, see SI). CV was performed in a 3-electrode cell with a 3 mm diameter glassy carbon disk working electrode (CH Instruments), a gold coil counter electrode (CH Instruments), and a fritted Ag/Ag+ reference electrode (fill solution: 0.05 M AgBF4 (Sigma-Aldrich, 98%), 0.5 M TEAPF6, propylene carbonate (BASF, >99.9%)). Before every CV, the working electrode was polished on a MicroCloth pad with an aqueous slurry of 0.05 μm alumina powder (Buehler Ltd.), rinsed with deionized water (Millipore), and dried with lens paper (VWR). In order to reference to ferrocene (Sigma-Aldrich, 98%), before every CV measurement, an additional CV was taken in the electrolyte with 0.001 M ferrocene as the active material. All CV data was collected on a VSP-300 potentiostat (Bio-Logic), and a 100% automated iR compensation was applied. The average resistance measured was about 20 Ω, leading to a total voltage compensation of about 0.3 mV for the highest currents. CV measurements were taken at scan rates of 10, 20, 30, 40, 50, 75, and 100 mV s−1 and were used to calculate the redox potential, peak separation, peak current ratio, and diffusion coefficient. The redox potential was calculated by taking the average of the potentials corresponding to the anodic and cathodic peak currents, while the peak separation was calculated by taking the difference between these two potentials. The peak current ratio was calculated by taking the ratio of the background corrected peak current for the anodic and cathodic peak.
In order to calculate the diffusion coefficient, the Randles-Sevcik relationship (1) was used for all of the scan rates:
| (1) |
where ip is the peak current (A), n is the number of electrons transferred (n = 1), F is Faraday’s constant (96485 C (mol e−)−1), A is the electrode area (0.0707 cm2), C is the bulk concentration (1 × 10−6 mol/cm3), R is the universal gas constant (8.314 J mol−1 K−1), T is the temperature (299.15 K), D is the diffusion coefficient (cm2 s−1) and ν is the scan rate (V s−1). Again the peak current was determined from the peak current from the voltammograms and subtracting an extrapolated background current.
Flow Cell Measurements.
Flow cell measurements employed a research-scale, 2.55 cm2 flow cell described in past work36,37. Flow fields were machined from impermeable graphite (Tokai G347B, MWI, Inc.) in-house and the engineering drawings are published in past work9. Sigracet® SGL 29AA carbon paper electrodes (190 ± 30 μm) were purchased from the Fuel Cell Store and used as received. Both the symmetric and full cells were tested with a flowrate of 10 mL min−1, a Daramic 175 porous separator, 2 layers of Gore® expanded polytetrafluoroethylene (ca. 250 μm thick) gaskets, and 2 layers of carbon paper per side. The Daramic separator was selected as it provides good wettability and consistent performance as a starting point for demonstration of these materials, and the design of a high-performance RFB membrane is beyond the scope of this work. A Cole-Parmer Masterflex® pump drive with an Easy-Load® II pump head was used to control the flowrate. Perfluoroalkoxy reservoirs (10 mL) were purchased from Savillex, and the volumetric capacities presented within this work only accounted for the total volume of electrolyte within the system (20 mL). For the symmetric flow cell, each 10 mL reservoir contained 0.05 M 11− / 0.05 M 12− / 0.5 M TBAPF6 in MeCN initially. For the full cell, each 10 mL reservoir contained of 0.05 M 20 / 0.05 M 12− / 0.5 M TBAPF6 in MeCN. Premixing was employed to mitigate confounding effects of rapid crossover due to the use of Daramic 175, a nonselective porous separator.
All flow cell experiments were controlled with a Bio-Logic VMP3 potentiostat. After setting up the initial cell and turning the pump on, the cell was allowed to sit for 30 min prior to electrochemical measurements. We then cell was then subjected to electrochemical impedance spectroscopy (EIS), followed by galvanostatic cycling with potential limitations (GCPL). EIS measurements were taken with frequencies ranging from 200 kHz to 10 mHz with 6 points per decade, 5 measures per frequency, and an absolute (sine) amplitude of 10 mV. The EIS is shown in Figures S3 and S4 for the symmetric and full cell, respectively. GCPL was performed at 5 mA cm−2, without any constant potential holds. Voltage limitations were set as −0.3 V – 0.3 V and 0.10 V – 0.58 V for the symmetric and full cells respectively. After the symmetric flow cell cycling of [1]2− & [1]1−, the solution containing the two materials and the electrolyte (TBAPF6) was recollected and the solvent was removed via rotary evaporation. The mixture was analyzed by 11B{1H}, 19F, and 1H NMR spectroscopy for evidence of any chemical degradation (easily identified by the presence of any borate decomposition products with characteristic resonances around δ 20.0 or 0.0 in the 11B{1H} NMR).
RESULTS AND DISCUSSION
Voltammetry.
While voltammetric analysis of molecular compounds can be useful for initial analysis of redox processes, during these tests the materials are only charged for short durations, with only a small fraction of the active material charged at any given time. In order to better evaluate and understand the stability of the B12(OR)12 system to electrochemical cycling associated with bulk charge storage, we decided to test these molecules further under controlled conditions that more closely mimic energy storage applications. We started by identifying two model B12(OR)12 cluster systems with high (1) and low (2) redox potentials relative to each other (table 1). The high-potential cluster 1 features 12 benzyl substituents with perfluorinated aryl groups, which, due to their inherent inductive electron-withdrawing nature, increase the redox potential for the 2−/1− redox couple.24 For a cluster system exhibiting redox properties at lower potential, we designed 2, which contains 12 inductively electron-donating alkyl groups with terminating OMe moieties that improve solubility in polar solvents. Both species have a solubility ≥0.5 M in MeCN in all relevant states-of-charge. First, CV on a glassy carbon electrode was conducted to provide in-depth information about the electrochemical and transport metrics for the two cluster systems (Figure 2). The first oxidation of 1 and the first reduction of 2 occur at 0.074 V vs. Fc/Fc+ and −0.256 V vs. Fc/Fc+, respectively. Both materials show excellent chemical reversibility on the CV timescale (~seconds-minutes) as evidenced by a peak current ratio close to one across all scan rates (1: 0.97 ± 0.01, 2: 1.00 ± 0.02). Additionally, both redox couples show electrochemical reversibility with a peak separation of 61 ± 1 mV (Nernstian is 60 mV at ambient glovebox temperature), which is invariant of scan rate (Figure S5).38
| Cluster | Potential (V vs Fc/Fc+) | Peak Separation (mV) | Peak Current Ratio | Diffusion Coefficient (× 10−6cm2s−1) |
|---|---|---|---|---|
| 1−2 | 0.074 ± 0.001 | 61 ± 1 | 0.97 ± 0.01 | 3.3 ± 0.1 |
| 20 | − 0.256 ± 0.001 | 61 ± 1 | 1.00 ± 0.02 | 3.3 ± 0.1 |
Figure 2.
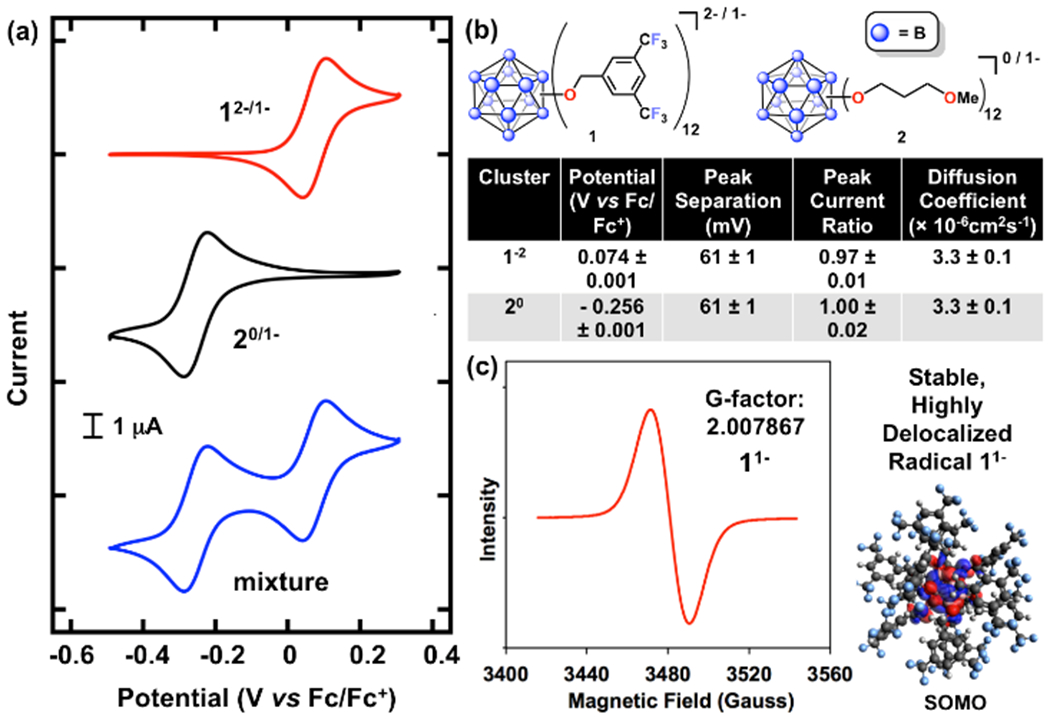
(a) Cyclic voltammograms of 12−/1− (top, red), 20/1− (middle, black), and a mixture of the two clusters (bottom, blue) at 1 mM in 0.5 M TBAPF6 in MeCN at a scan rate of 10 mV s−1, referenced to ferrocene/ferrocenium at 0 V. (b) Measured electrochemical parameters for 12−/1− and 20/1− calculated at 10 mV s−1. (c) EPR spectrum and calculated SOMO energy state representation of 1−1.22
Lastly, the diffusion coefficients of the starting materials were calculated using Randles-Sevcik analysis (Figure S2) with scan rates of 10 to 100 mV s−1, and were determined to be 3.3 × 10−6 cm2 s−1. As expected based on the borate cluster sizes and the Stokes-Einstein relation (diffusivity is inversely proportional to molecular radius), these diffusion coefficients are about an order of magnitude lower than those of small organic molecules in similar electrolyte solutions, but are still sufficiently large to support a range of electrochemical applications.39,40 Importantly, clusters 1 and 2 appear to be non-interacting on the CV timescale as the voltammogram with both materials present represents a superposition of the two independent scans with no changes in original peak shape, height, or position, and no new peaks appearing as a function of cycling.
Flow Cell Results.
We then assembled a symmetric flow cell with 1 (Figure 3). The symmetric flow cell allows for cycling of both the reduced and oxidized species in the cluster redox pairs to determine the active species stability without the convoluting effects of side products generated at the counter electrode or the mass transport enforced concentration limitations, which are both associated with conventional bulk electrolysis apparatus.41–43 Figure 3a shows the initial active species distribution within the reservoirs and redox reactions occurring during the charging step. During the discharge step, these reactions occur in reverse. Although it is generally desirable to test at higher active species concentrations (>0.1 M), to both reduce mass transport resistance and explore more practical electrolyte formulation, we sought to first demonstrate behavior at lower concentrations. Consequently, we have used a low current density of 5 mA cm−2, to mitigate the effect of mass transfer limitations on the accessed capacity. Figure 3b shows capacity and colombic efficiency as a function of cycle number and illustrates a low fade rate due to high material stability.
Figure 3.
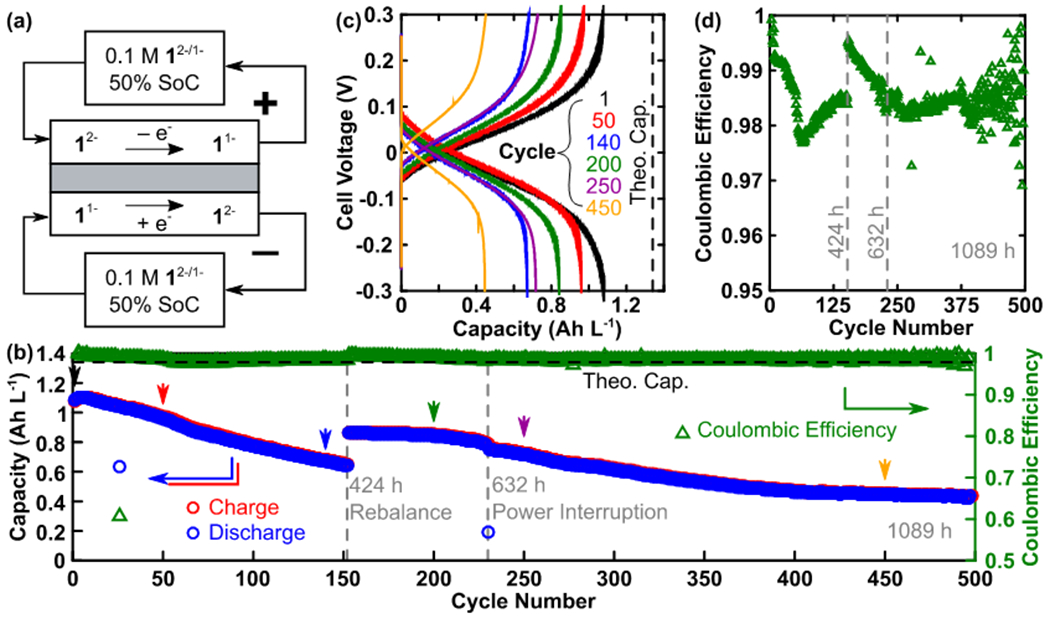
Symmetric flow cell containing cluster 1. (a) Cell setup. (b) Charge and discharge capacity plotted (left) as a function of cycle number in along with coulombic efficiency (right). Arrowheads (colors match c) indicate cycles for which the voltage profile is shown in (c). (c) Charge and discharge voltage profiles are shown for select cycles. (d) Detailed view of coulombic efficiency as a function of cycle number. Gray dashed lines in (b) and (d) indicate interruptions of the experiment due to intentional electrolyte rebalancing (424 h) and a building power outage (632 h).
The cell retained high coulombic efficiency (> 96%) and 40% of the initial capacity after 1089 h (45 days) of cycling, which exceeds the duration of many published nonaqueous RFB stability studies.44–47 We suspect that a portion of the cell fade at the later stages of this experiment, specifically the decrease in accessed capacity and the increase coulombic efficiency instability, are due to a combination of imperfect sealing and pressure driven crossover. Although compression fittings and expanded PTFE gaskets are used in the cell construction, we inevitably lose some redox active electrolyte via evaporative leaking. Loss of volatile components, specifically the solvent, gradually increases the solution viscosity and promotes further leaks (observed) via an increased pressure within the cell. Dashed gray vertical lines indicate experimental interruptions. The cycling was stopped briefly to rebalance the electrolyte reservoirs at 424 h as the reservoir levels were visibly mismatched (ca. 5 mL vs. 15 mL), which is partially enabled by our use of a non-selective separator (Daramic 175). A second (unintentional) interruption occurred at 632 h when the building lost power. Voltage profiles from select cycles, as indicated by arrowheads in Figure 3b, are shown in Figure 3c, illustrating the appreciable fraction of the theoretical capacity that is repeatedly accessible for select cycles over all uninterrupted segments of the experiment. Note that at least one cycle is shown from each uninterrupted segment of the cycling protocol. The third cycle shown, cycle 200, takes place after the electrolyte rebalancing at 424 h, and the increase in capacity (30%) from cycle 140 is indicative of a crossover imbalance within our experimental setup. We attribute the limited approach to theoretical capacity (80% for cycle 1) to the mass-transfer limitations at extreme states of charge, which are exacerbated by the relatively low total concentration (0.1 M). For the entire experiment the coulombic efficiency remained high (> 96%), indicating high material stability (Figure 3d).
Importantly, post-mortem B 1s XPS analysis of [1] after cycling (see Figure 3) revealed that there were no decomposition products (borates) (Figure 4).
Figure 4.
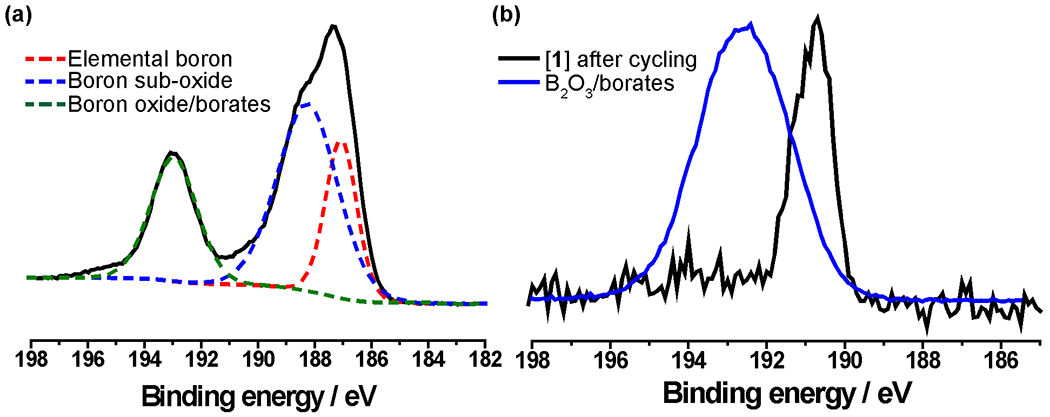
(a) B 1s XPS region for elemental boron (control). (b) Post-mortem B 1s XPS analysis of [1] after 45 days of cycling indication no presence of borates. Additional controls can be found in the SI (Figure S7).
Given the high sensitivity of XPS, the lack of any observable borates after cycling clearly demonstrates the stability of these clusters, further confirmed by NMR spectroscopy and mass spectrometry (see SI). In the NMR spectra, only resonances matching both the 2− and 1− oxidation states of [1] along with the presence of TBA-PF6 (Figure S6) were visible. No borates were observed in the 11B NMR spectrum (Figure S6), further corroborating that no chemical decomposition of the boron cage framework has occurred. Lastly, electrospray mass spectrometry on the post-mortem electrolyte confirmed the presence of intact [1] in multiple oxidation states further highlighting the robustness of this system (Figure S7).
We then sought to evaluate the use of 1 and 2 in a full redox flow battery (RFB).17 A flow cell containing 12−/1− (high potential) and 20/1− (low potential) was cycled at 5 mA cm−2 (Figure 5 and SI). Both reservoirs initially contained 10 mL of 0.05 M 20 / 0.05 M 12− / 0.5 M TBAPF6 in MeCN. Utilizing the two materials together leads to a cell voltage of 0.33 V. Indeed, this is lower than desirable for a RFB, but full cell cycling can highlight the stability of the highly tunable borate clusters.24
Figure 5.
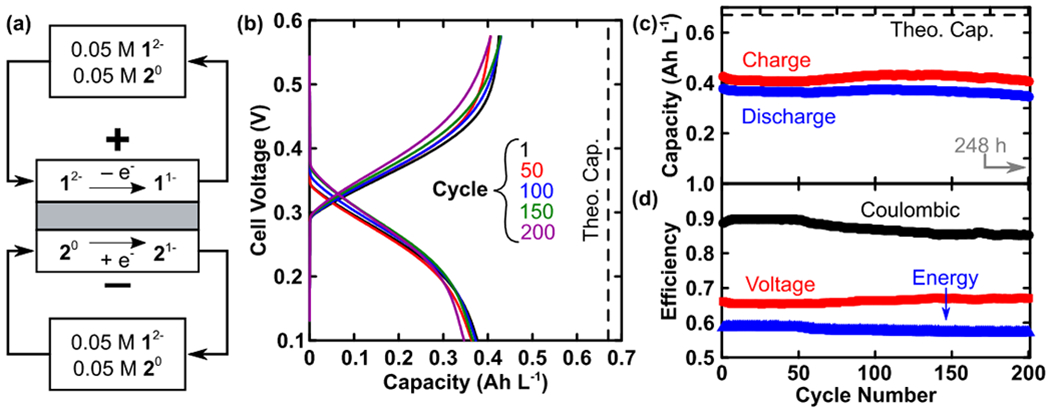
Full RFB cell cycling results. (a) Flow cell setup for cycling at 5 mA cm−2 showing premixed redox-active electrolytes. (b) Voltage profiles for selected cycles. (c) Capacity and (d) efficiency as a function of cycle number.
Specifically, this borate cluster pairing was selected based on preliminary assessments of stability, solubility, and simplicity of synthesis in spite of the low average discharge voltage, 0.258 V. The cell configuration, which used pre-mixed electrolytes, similar to an Fe-Cr RFB, to mitigate crossover-driven capacity fade, is shown in Figure 4a17,48. Detailed experimental conditions and electrochemical impedance spectra are included in the SI (Figure S4). Figure 4b shows the charge and discharge profiles for cycles 1, 50, 100, 150, and 200, spanning 248 h of experiment time. Figure 4c shows the capacity as a function of cycle number, which remains relatively stable and has a mean value of 0.366 Ah L−1 (55% of theoretical) based on discharge. Energy, coulombic, and voltage efficiencies are shown in Figure 4d. The low average voltage efficiency, 66%, can be primarily attributed to the low cell voltage, as well as significant contributions from ohmic and mass-transfer overpotentials due to low active species concentration. Specifically, the voltage losses within the cell are not atypically large as compared to prior non-aqueous flow battery literature46,49,50; however, they constitute a larger fraction of the total cell voltage, thus significantly impacting the voltaic efficiency. The ohmic resistance, as determined through impedance analysis, was 3.6 Ω cm2, and contributes 23% of the voltage losses (i.e., 8% of the average charging voltage). The combination of stable capacity along with moderate average coulombic efficiency (87%) suggests that the cell experiences significant yet non-destructive parasitic charge-transfer pathways, presumably crossover coupled to self-discharge. These losses may be greatly diminished through ongoing membrane research for RFBs40,51,52. In particular, these clusters appear sufficiently large53 (ca. 2-3 nm) as compared to smaller redox-active organic molecules (ca. 0.6 nm), to enable the use of nanoporous size-selective separators for crossover mitigation. This, in turn, would obviate the need for functionalized membranes, reducing both component cost and research time52,54–56.
CONCLUSION
In summary, we have demonstrated the design versatility and bulk electrochemical stability and cyclability of several perfunctionalized dodecaborate clusters, B12(OR)12. This work expands upon the repertoire of cluster-based materials amenable to electrochemical cycling,28,29,32,57,58 with flow cell cycling validating the exceptional stability of this material class as it is cycled between 2 oxidation states for 45 days showing no apparent chemical degradation. Given the favorable and tunable properties of perfunctionalized boron clusters21, we envision further optimization of this system to enable more practical energy storage uses.
Supplementary Material
ACKNOWLEDGMENTS
J. L. B., J. A. K., and F. R. B. gratefully acknowledge support from the Joint Center for Energy Storage Research, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. J. L. B. acknowledges additional funding from the ExxonMobil-MIT Energy Fellowship (2017-2018). A. I. W., E. A. Q., and D. J. acknowledge the University of California, Los Angeles (UCLA) Graduate Division for the dissertation year fellowship. A. M. S. acknowledges the University of California, Los Angeles (UCLA) Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, the Alfred P. Sloan Foundation for a research fellowship in chemistry, Research Corporation for Science Advancement for a Cottrell Scholar Award and the NIGMS for the Maximizing Investigators Research Award (MIRA, R35GM124746). The authors thank the MRI program of the National Science Foundation (NSF grant no. 1532232 and 1625776). We thank UCLA Molecular Instrumentation Center for mass spectrometry and NMR spectroscopy (NIH grant 1S10OD016387-01).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Synthesis details, NMR spectra, elemental analysis, and mass spectra of the dodecaborate clusters along with additional voltammetry and flow cell data is provided. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL, “Nature of biological electron transfer”. Nature 1992, 355, 796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- (2).Gust D, Moore TA, Moore AL, “Molecular mimicry of photosynthetic energy and electron transfer”. Acc. Chem. Res. 1993, 26, 198–205. doi: 10.1021/ar00028a010. [DOI] [Google Scholar]
- (3).Kalyanasundaram K, Grätzel M, “Applications of functionalized transition metal complexes in photonic and optoelectronic devices”. Coord. Chem. Rev. 1998, 177, 347–414. doi: 10.1016/S0010-8545(98)00189-1. [DOI] [Google Scholar]
- (4).Ji X, Zhang B, Tritt TM, Kolis JW, Kumbhar A, “Solution-chemical syntheses of nano-structured Bi2Te3 and PbTe thermoelectric materials”. J. Electron. Mater. 2007, 36, 721–726. doi: 10.1007/s11664-007-0156-y. [DOI] [Google Scholar]
- (5).Fukuzumi S, “Bioinspired energy conversion systems for hydrogen production and storage”. Eur. J. Inorg. Chem. 2008, 2008, 1351–1362. doi: 10.1002/ejic.200701369. [DOI] [Google Scholar]
- (6).Medlin DL, Snyder GJ, “Interfaces in bulk thermoelectric materials: a review for current opinion in colloid and interface science”. Curr. Opin. Colloid Interface Sci. 2009, 14, 226–235. doi: 10.1016/j.cocis.2009.05.001. [DOI] [Google Scholar]
- (7).Kalyanasundaram K, Graetzel M, “Artificial photosynthesis: biomimetic approaches to solar energy conversion and storage”. Curr. Opin. Biotechnol. 2010, 21, 298–310. doi: 10.1016/j.copbio.2010.03.021. [DOI] [PubMed] [Google Scholar]
- (8).Milshtein JD, Kaur AP, Casselman MD, Kowalski JA, Modekrutti S, Zhang PL, Harsha Attanayake N, Elliott CF, Parkin SR, Risko C, Brushett FR, Odom SA, “High current density, long duration cycling of soluble organic active species for non-aqueous redox flow batteries”. Energy Environ. Sci. 2016, 9, 3531–3543. doi: 10.1039/C6EE02027E. [DOI] [Google Scholar]
- (9).Milshtein JD, Tenny KM, Barton JL, Drake J, Darling RM, Brushett FR, “Quantifying mass transfer rates in redox flow batteries”. J. Electrochem. Soc. 2017, 164, E3265–E3275. doi: 10.1149/2.0201711jes. [DOI] [Google Scholar]
- (10).Gür TM, “Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage”. Energy Environ. Sci. 2018, 11, 2696–2767. doi: 10.1039/C8EE01419A. [DOI] [Google Scholar]
- (11).Winsberg J, Hagemann T, Janoschka T, Hager MD, Schubert US, “Redox-flow batteries: from metals to organic redox-active materials”. Angew. Chem. - Int. Ed. 2017, 56, 686–711. doi: 10.1002/anie.201604925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kowalski JA, Su L, Milshtein JD, Brushett FR, “Recent advances in molecular engineering of redox active organic molecules for nonaqueous flow batteries”. Curr. Opin. Chem. Eng. 2016, 13, 45–52. doi: 10.1016/j.coche.2016.08.002. [DOI] [Google Scholar]
- (13).Darling RM, Gallagher KG, Kowalski JA, Ha S, Brushett FR, “Pathways to low-cost electrochemical energy storage: a comparison of aqueous and nonaqueous flow batteries”. Energy Environ. Sci. 2014, 7, 3459–3477. doi: 10.1039/C4EE02158D. [DOI] [Google Scholar]
- (14).Chen H, Cong G, Lu Y-C, “Recent progress in organic redox flow batteries: active materials, electrolytes and membranes”. J. Energy Chem. 2018, 27, 1304–1325. doi: 10.1016/j.jechem.2018.02.009. [DOI] [Google Scholar]
- (15).Thaller LH, Electrically rechargeable redox flow cells, NASA, Cleveland, Ohio, United States, 1974. https://ntrs.nasa.gov/search.jsp?R=19740013575. [Google Scholar]
- (16).Skyllas-Kazacos M, Cao L, Kazacos M, Kausar N, Mousa A, “Vanadium electrolyte studies for the vanadium redox battery—a review”. ChemSusChem 2016, 9, 1521–1543. doi: 10.1002/cssc.201600102. [DOI] [PubMed] [Google Scholar]
- (17).Weber AZ, Mench MM, Meyers JP, Ross PN, Gostick JT, Liu Q, “Redox flow batteries : a review”. J. Appl. Electrochem. 2011, 41, 1137–1164. doi: 10.1007/s10800-011-0348-2. [DOI] [Google Scholar]
- (18).Brushett FR, Vaughey JT, Jansen AN, “An all-organic non-aqueous lithium-ion redox flow battery”. Adv. Energy Mater. 2012, 2, 1390–1396. doi: 10.1002/aenm.201200322. [DOI] [Google Scholar]
- (19).Sevov CS; Brooner REM; Chenard E; Assary RS; Moore JS; Rodriguez-López Sanford, M. S. “Evolutionary design of low molecular weight organic anolyte materials for appications in nonaqueous redox flow batteries” J. Am. Chem. Soc. 2015, 137, 14465–14472. doi: 10.1021/jacs.5b09572. [DOI] [PubMed] [Google Scholar]
- (20).Dieterich V, Milshtein JD, Barton JL, Carney TJ, Darling RM, Brushett FR, “Estimating the cost of organic battery active materials : a case study on anthraquinone disulfonic acid”. Trans. Mater. Res. 2018, 5, 034001. [Google Scholar]
- (21).Bondarev O, Khan AA, Tu X, Sevryugina YV, Jalisatgi SS, Hawthorne MF, “Synthesis of [closo-B12(OH)11NH3]−: a new heterobifunctional dodecaborane scaffold for drug delivery applications”. J. Am. Chem. Soc. 2013, 135, 13204–13211. doi: 10.1021/ja4069613. [DOI] [PubMed] [Google Scholar]
- (22).Wixtrom AI, Shao Y, Jung D, Machan CW, Kevork SN, Qian EA, Axtell JC, Khan SI, Kubiak CP, Spokoyny AM, “Rapid synthesis of redox-active dodecaborane B12(OR)12 clusters under ambient conditions”. Inorg. Chem. Front. 2016, 3, 711–717. doi: 10.1039/C5QI00263J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee MW, Farha OK, Hawthorne MF, Hansch CH, “Alkoxy derivatives of dodecaborate: discrete nanomolecular ions with tunable pseudometallic properties”. Angew. Chem. - Int. Ed. 2007, 46, 3018–3022. doi: 10.1002/anie.200605126. [DOI] [PubMed] [Google Scholar]
- (24).Wixtrom AI, Parvez Z; Savage MA; Qian EA; Jung D, Khan SI; Rheingold AL; Spokoyny AM, “Tuning the electropotential of perfunctionalized dodecaborate clusters through vertex differentiation”. Chem. Commun. 2018, 54, 5867–5870. doi: 10.1039/C8CC03477J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jung D, Saleh LMA, Berkson ZJ, El-Kady MF, Hwang JY, Mohamed N, Wixtrom AI, Titarenko E, Shao Y, McCarthy K, Guo J, Martini IB, Kraemer S, Wegener EC, Saint-Cricq P, Ruehle B, Langeslay RR, Delferro M, Brosmer JL, Hendon CH, Gallagher-Jones M, Rodriguez J, Chapman KW, Miller JT, Duan X, Kaner RB, Zink JI, Chmelka BF, Spokoyny AM, “A molecular cross-linking approach for hybrid metal oxides”. Nat. Mater. 2018, 17, 341–348. doi: 10.1038/s41563-018-0021-9. [DOI] [PubMed] [Google Scholar]
- (26).Muetterties EL, Balthis JH, Chia YT, Knoth WH, Miller HC, “Chemistry of boranes. VIII. Salts and acids of B10 H10 −2 and B12H12−2”. Inorg. Chem. 1964, 3, 444–451. doi: 10.1021/ic50013a030. [DOI] [Google Scholar]
- (27).Andersson M, Persson JL, Rosén A, “Reactivity of Fen, Con , and Cun clusters with O2 and D2 studied at single-collision conditions”. J. Phys. Chem. 1996, 100, 12222–12234. doi: 10.1021/jp960889z. [DOI] [Google Scholar]
- (28).Sevov CS, Fisher SL, Thompson LT, Sanford MS, “Mechanism-based development of a low-potential, soluble, and cyclable multielectron anolyte for nonaqueous redox flow batteries”. J. Am. Chem. Soc. 2016, 138, 15378–15384. doi: 10.1021/jacs.6b07638. [DOI] [PubMed] [Google Scholar]
- (29).VanGelder LE, Kosswattaarachchi AM, Forrestel PL, Cook TR, Matson EM, “Polyoxovanadate-alkoxide clusters as multi-electron charge carriers for symmetric non-aqueous redox flow batteries”. Chem. Sci. 2018, 9, 1692–1699. doi: 10.1039/C7SC05295B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).VanGelder LE, Matson EM, “Heterometal functionalization yields improved energy density for charge carriers in nonaqueous redox flow batteries”. J. Mater. Chem. A. 2018, 6, 13874–13882. doi: 10.1039/c8ta03312a. [DOI] [Google Scholar]
- (31).Stauber JM, Zhang S, Gvozdik N, Jiang Y, Avena L, Stevenson KJ, Cummins CC, “Cobalt and Vanadium Trimetaphosphate Polyanions: Synthesis, Characterization and Electrochemical Evaluation for Non-Aqueous Redox-Flow Battery Applications”. J. Am. Chem. Soc. 2018, 140, 538–541. doi: 10.1021/jacs.7b08751. [DOI] [PubMed] [Google Scholar]
- (32).Chen J-J, Symes MD, Cronin L, “Highly reduced and protonated aqueous solutions of [P2W18O62]6− for on-demand hydrogen generation and energy storage”. Nat. Chem. 2018, 10, 1042–1047. doi: 10.1038/s41557-018-0109-5. [DOI] [PubMed] [Google Scholar]
- (33).Aubry TJ, Axtell JC, Basile VM, Winchell KJ, Lindemuth JR, Porter TM, Liu J-Y, Alexandrova AN, Kubiak CP, Tolbert SH, Spokoyny AM, Schwartz BJ “Dodecaborane-based dopants designed to shield anion electrostatics lead to increased carrier mobility in a doped conjugated polymer”, Adv. Mater. 2019, online. doi: 10.1002/adma201805647. [DOI] [PubMed] [Google Scholar]
- (34).Messina MS, Axtell JC, Wang Y, Chong P, Wixtrom AI, Kirlikovali KO, Upton BM, Hunter BM, Shafaat OS, Khan SI, Winkler JR, Gray HB, Alexandrova AN, Maynard HD, Spokoyny AM, “Visible-light-induced olefin activation using 3D aromatic boron-rich cluster photooxidants”. J. Am. Chem. Soc. 2016, 138, 6952–6955. doi: 10.1021/jacs.6b03568. [DOI] [PubMed] [Google Scholar]
- (35).Axtell JC, Saleh LMA, Qian EA, Wixtrom AI, Spokoyny AM, “Synthesis and applications of perfunctionalized boron clusters”. Inorg. Chem. 2018, 57, 2333–2350. doi: 10.1021/acs.inorgchem.7b02912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Milshtein JD, Barton JL, Carney TJ, Kowalski JA, Darling RM, Brushett FR, “Towards low resistance nonaqueous redox flow batteries”. J. Electrochem. Soc. 2017, 164, A2487–A2499. doi: 10.1149/2.0741712jes. [DOI] [Google Scholar]
- (37).Huang J; Pan B; Duan W; Wei X; Assary RS; Su L; Brushett FR; Cheng L; Liao C; Ferrandon MS; Wang W; Zhang Z; Burrell AK; Curtiss LA; Shkrob IA; Moore JS; Zhang L, “The lightest organic radical cation for charge storage in redox flow batteries”. Sci. Rep. 2016, 6, 32102. doi: 10.1038/srep32102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bard AJ, Faulkner LR, Electrochemical methods fundamentals and applications, 2nd ed., John Wiley & Sons, Inc., New York, 2001. [Google Scholar]
- (39).Orella MJ; Román-Leshkov Y; Brushett FR “Emerging opportunities for the electrochemical processing to enable sustainable chemical manufacturing.” Curr. Op. Chem. Eng. 2018, 20, 159–167. doi: 10.1016/j.coche.2018.05.002. [DOI] [Google Scholar]
- (40).Hendriks KH, Robinson SG, Braten MN, Sevov CS, Helms BA, Sigman MS, Minteer SD, Sanford MS, “High-performance oligomeric catholytes for effective macromolecular separation in nonaqueous redox flow batteries”. ACS Cent. Sci. 2018, 4, 189–196. doi: 10.1021/acscentsci.7b00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kowalski JA, Casselman MD, Kaur AP, Milshtein JD, Elliott CF, Modekrutti S, Attanayake NH, Zhang N, Parkin SR, Risko C, Brushett FR, Odom SA, “A stable two-electron-donating phenothiazine for application in nonaqueous redox flow batteries”. J. Mater. Chem. A. 2017, 5, 24371–24379. doi: 10.1039/C7TA05883G. [DOI] [Google Scholar]
- (42).Milshtein JD, Barton JL, Darling RM, Brushett FR, “4-Acetamido-2,2,6,6-tetramethylpiperidine-1-oxyl as a model organic redox active compound for nonaqueous flow batteries”. J. Power Sources. 2016, 327, 151–159. doi: 10.1016/j.jpowsour.2016.06.125. [DOI] [Google Scholar]
- (43).Goulet M, Aziz MJ, “Flow battery molecular reactant stability determined by symmetric cell cycling methods”. J. Electrochem. Soc. 2018, 165, A1466–A1477. doi: 10.1149/2.0891807jes. [DOI] [Google Scholar]
- (44).Huang H, Howland R, Agar E, Nourani M, Golen JA, Cappillino PJ, “Bioinspired, high-stability, nonaqueous redox flow battery electrolytes”. J. Mater. Chem. A. 2017, 5, 11586–11591. doi: 10.1039/C7TA00365J. [DOI] [Google Scholar]
- (45).Wei X, Xu W, Huang J, Zhang L, Walter E, Lawrence C, Vijayakumar M, Henderson WA, Liu T, Cosimbescu L, Li B, Sprenkle V, Wang W, “Radical compatibility with nonaqueous electrolytes and its impact on an all-organic redox flow battery”. Angew. Chem. Int. Ed. 2015, 54, 8684–8687. doi: 10.1002/anie.201501443. [DOI] [PubMed] [Google Scholar]
- (46).Wei X, Duan W, Huang J, Zhang L, Li B, Reed D, Xu W, Sprenkle V, Wang W, “A high-current, stable nonaqueous organic redox flow battery”. ACS Energy Lett. 2016, 1, 705–711. doi: 10.1021/acsenergylett.6b00255. [DOI] [Google Scholar]
- (47).Sevov CS, Hickey DP, Cook ME, Robinson SG, Barnett S, Minteer SD, Sigman MS, Sanford MS, “Physical organic approach to persistent, cyclable, low-potential electrolytes for flow battery applications”. J. Am. Chem. Soc. 2017, 139, 2924–2927. doi: 10.1021/jacs.7b00147. [DOI] [PubMed] [Google Scholar]
- (48).Zeng YK, Zhou XL, An L, Wei L, Zhao TS, “A high-performance flow-field structured iron-chromium redox flow battery”. J. Power Sources. 2016, 324, 738–744. doi: 10.1016/j.jpowsour.2016.05.138. [DOI] [Google Scholar]
- (49).Duan W, Huang J, Kowalski JA, Shkrob IA, Vijayakumar M, Walter E, Pan B, Yang Z, Milshtein JD, Li B, Liao C, Zhang Z, Wang W, Liu J, Moore JS, Brushett FR, Zhang L, Wei X, ““Wine-Dark Sea” in an organic flow battery: storing negative charge in 2,1,3-benzothiadiazole radicals leads to improved cyclability”. ACS Energy Lett. 2017, 2, 1156–1161. doi: 10.1021/acsenergylett.7b00261. [DOI] [Google Scholar]
- (50).Gong K, Fang Q, Gu S, Li SFY, Yan Y, “Nonaqueous redox-flow batteries: organic solvents, supporting electrolytes, and redox pairs”. Energy Environ. Sci. 2015, 8, 3515–3530. doi: 10.1039/C5EE02341F. [DOI] [Google Scholar]
- (51).Su L, Darling RM, Gallagher KG, Xie W, Thelen JL, Badel AF, Barton JL, Cheng KJ, Balsara NP, Moore JS, Brushett FR, “An investigation of the ionic conductivity and species crossover of lithiated nafion 117 in nonaqueous electrolytes”. J. Electrochem. Soc. 2016, 163, A5253–A5262. doi: 10.1149/2.03211601jes. [DOI] [Google Scholar]
- (52).Doris SE, Ward AL, Baskin A, Frischmann PD, Gavvalapalli N, Chénard E, Sevov CS, Prendergast D, Moore JS, Helms BA, “macromolecular design strategies for preventing active-material crossover in non-aqueous all-organic redox-flow batteries”. Angew. Chem. Int. Ed. 2017, 56, 1595–1599. doi: 10.1002/anie.201610582. [DOI] [PubMed] [Google Scholar]
- (53).Majewski MB, Howarth AJ, Farha OK, “Organomimetic clusters: precision in 3D”. Nat. Chem. 2017, 9, 299–301. doi: 10.1038/nchem.2756. [DOI] [PubMed] [Google Scholar]
- (54).Montoto EC, Nagarjuna G, Hui J, Burgess M, Sekerak NM, Hernández-Burgos K, Wei T-S, Kneer M, Grolman J, Cheng KJ, Lewis JA, Moore JS, Rodríguez-López J, “Redox active colloids as discrete energy storage carriers”. J. Am. Chem. Soc. 2016, 138, 13230–13237. doi: 10.1021/jacs.6b06365. [DOI] [PubMed] [Google Scholar]
- (55).Montoto EC, Nagarjuna G, Moore JS, Rodríguez-López J, “Redox active polymers for non-aqueous redox flow batteries: validation of the size-exclusion approach”. J. Electrochem. Soc. 2017, 164, A1688–A1694. doi: 10.1149/2.1511707jes. [DOI] [Google Scholar]
- (56).Milshtein JD, Darling RM, Drake J, Perry ML, Brushett FR, “The critical role of supporting electrolyte selection on flow battery cost”. J. Electrochem. Soc. 2017, 164, A3883–A3895. doi: 10.1149/2.1031714jes. [DOI] [Google Scholar]
- (57).Hu B, DeBruler C, Rhodes Z, Liu TL, “Long-cycling aqueous organic redox flow battery (AORFB) toward sustainable and safe energy storage”. J. Am. Chem. Soc. 2017, 139, 1207–1214. doi: 10.1021/jacs.6b10984. [DOI] [PubMed] [Google Scholar]
- (58).Friedl J, Lebedeva MA, Porfyrakis K, Stimming U, Chamberlain TW, “All-fullerene-based cells for nonaqueous redox flow batteries”. J. Am. Chem. Soc. 2018, 140, 401–405. doi: 10.1021/jacs.7b11041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.