

Defective tRNA 2′-O-modification impairs translation efficiency at Phe codon in the brain and induces a neurological dysfunction.
Abstract
FtsJ RNA 2′-O-methyltransferase 1 (FTSJ1) gene has been implicated in X-linked intellectual disability (XLID), but the molecular pathogenesis is unknown. We show that Ftsj1 is responsible for 2′-O-methylation of 11 species of cytosolic transfer RNAs (tRNAs) at the anticodon region, and these modifications are abolished in Ftsj1 knockout (KO) mice and XLID patient–derived cells. Loss of 2′-O-methylation in Ftsj1 KO mouse selectively reduced the steady-state level of tRNAPhe in the brain, resulting in a slow decoding at Phe codons. Ribosome profiling showed that translation efficiency is significantly reduced in a subset of genes that need to be efficiently translated to support synaptic organization and functions. Ftsj1 KO mice display immature synaptic morphology and aberrant synaptic plasticity, which are associated with anxiety-like and memory deficits. The data illuminate a fundamental role of tRNA modification in the brain through regulation of translation efficiency and provide mechanistic insights into FTSJ1-related XLID.
INTRODUCTION
Transfer RNA (tRNA) is central to the decoding of the genetic information into polypeptide during translation. The nucleotides of tRNAs contain more than 100 species of modifications, which are posttranscriptionally introduced by specific enzymes (1, 2). tRNA modifications regulate structural integrity, stability, and codon-anticodon interaction and thus contribute to translation efficiency (TE) and fidelity. Accumulating evidence has shown that defective modifications can impair optimal protein synthesis in humans and cause a variety of diseases including cancer, type 2 diabetes, and mitochondrial and neurological diseases (3).
Intellectual disability (ID) is a neurological disorder that affects 2 to 3% of individuals worldwide (4). Clinical symptoms of ID include cognitive dysfunction, defined by an intelligence quotient of <70, and disability in adapting to social environments (5). The underlying cause of ID can be both genetically inherited and acquired (5). Defects in genes on the X chromosome are major contributors to ID (6), with X-linked ID (XLID) estimated to comprise 5 to 16% of all male patients with ID (6–8). To date, a total of 141 X-linked genes have been identified as causal genes for XLID (6). However, despite rapid progress in finding candidate genes, the pathogenic mechanisms underlying XLID have remained largely unknown.
The human FtsJ RNA 2′-O-methyltransferase 1 (FTSJ1) gene is localized at position Xp11.23 on the X chromosome, and pathogenic mutations of FTSJ1 have been associated with development of XLID in Belgian, Japanese, and Chinese families (8–12). Splice site mutations, nonsense mutations, and microdeletions have been identified in both coding and intron regions of FTSJ1, which results in production of abnormal FTSJ1 transcripts. Patients having a defective FTSJ1 gene manifest moderate to severe intellectual handicap characterized by delayed speech and disabilities in reading and extracting word meaning. Some patients display behavior problems such as aggressive outbursts, anxiety, and panic. Besides these intellectual and behavioral symptoms, these patients show no dysmorphism, indicating that patients having defective FTSJ1 gene develop nonsyndromic XLID.
Transfer RNA methyltransferase 7 (Trm7), the yeast homolog of Ftsj1, catalyzes 2′-O-methylation of cytidine at position 32 (C32) and guanosine at position 34 (G34) of tRNAPhe, C32 and C34 of tRNATrp, and uridine at position 34 (U34) of tRNALeu (13). At a cellular level, Trm7-deficient yeast are viable but show growth defects. Expression of human FTSJ1 in Trm7-deficient yeast restores 2′-O-methylation, leading to recovery of cell growth (14). Overexpression of tRNAPhe, but not other Trm7 substrate tRNAs, can rescue the growth defect of Trm7-deficient yeast cells (15). In human-derived cell lines, FTSJ1 appears to modify a broad number of tRNA species, and loss of FTSJ1 induces abnormal translation of a luciferase-based reporter (16, 17). Ftsj1-deficient animal models have been generated using Drosophila and mouse (18, 19). Drosophila with deficiency of Ftsj1 homologs shows defective 2′-O-methylation at tRNAPhe along with several other tRNAs and exhibits broad phenotypes including life-span reduction and dysfunction of small RNA pathways (18). Recently, a line of Ftsj1 mutant mice was generated using a gene-trapping method (19). Although the level of tRNA 2′-O-methylation in the Ftsj1 gene trapped mice has not been examined, the mutant male mice showed an abnormality in a place-learning paradigm (19). The mutant mice also showed a decrease in bone density and bodyweight (19). Despite these findings, two fundamental questions remain unsolved: (i) Why loss of FTSJ1 causes nonsyndromic ID despite the ubiquitous expression of FTSJ1, and (ii) how loss of functional Ftsj1 leads to the development of XLID.
Here, we report comprehensive analyses of constitutive Ftsj1 knockout (KO) mice and a cell line derived from the patient with FTSJ1-related XLID, using biochemical, molecular, physiological, and behavioral approaches. We found that tRNAPhe, a Ftsj1 substrate tRNA, selectively decreased in the brain. Ftsj1 KO mice showed selective translational perturbation at Phe codons, leading to the abnormal translation of a subset of genes implicated in neuronal function. Furthermore, Ftsj1 KO mice showed morphological and electrophysiological deficits, which were associated with behavioral abnormalities that recapitulate symptoms of FTSJ1-related XLID.
RESULTS
Loss of tRNA 2′-O-methylation in Ftsj1 KO mouse and XLID patient–derived cells
We generated Ftsj1 KO mice (fig. S1A), which developed normally albeit with a slight but significant decrease in body weight compared to wild-type (WT) mice at 4 weeks of age (fig. S1B). According to the modification database (2), at least 11 species of tRNAs contain 2′-O-methylated nucleotides at position 32 and/or 34 in mammals. To investigate whether Ftsj1 is responsible for 2′-O-methylation of the 11 species of tRNAs, we isolated these tRNAs from the liver of 8-week-old WT and KO mice and examined by mass spectrometry. 2′-O-methylation at position 32 and/or 34 could not be detected for 11 species of tRNA in Ftsj1 KO mice including tRNAPhe, a known Ftsj1 substrate (Fig. 1, A to C, and fig. S1, C and D). tRNAPhe contains a hypermodified guanosine at position 37, the wybutosine (yW) modification in yeast and the hydroxywybutosine (OHyW) modification in human (2). G37 of tRNAPhe was modified to OHyW with 95% efficiency in WT mouse liver, but modification was reduced to 47.9% in KO mouse liver (Fig. 1, B and C). These results are consistent with previous studies showing that 2′-O-methylation at C32 and G34 is required for subsequent hypermodification of tRNAPhe at G37 in eukaryotic cells (14, 16, 17). In addition, we did not detect 2′-O-methylation at position 32 and/or 34 of all tRNALeu isoacceptors [tRNALeu(UAA), tRNALeu(CAA), tRNALeu(AAG), tRNALeu(UAG), and tRNALeu(CAG)], tRNATrp, tRNASec, tRNAVal(AAC), tRNAGln(CUG), and tRNAArg(ACG) in Ftsj1 KO mice (fig. S1C).
Fig. 1. Ftsj1/FTSJ1 is responsible for 2′-O-methylation of a subgroup of tRNAs at position 32 and/or 34 in mouse and human.
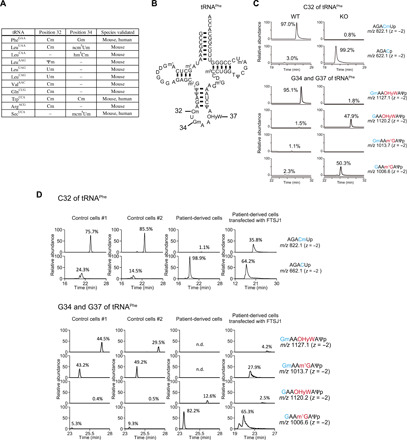
(A) List of Ftsj1 substrate tRNAs and modification sites in mouse and human. Gm, 2′-O-methylguanosine; Cm, 2′-O-methylcytidine; Um, 2′-O-methyluridine; Ψm, 2′-O-methylpseudouridine; ncm5Um, 5-carbamoylmethyl-2′-O-methyluridine; hm5Cm, 5-hydroxymethyl-2′-O-methylcytidine; mcm5Um, 5-methoxycarbonylmethyl-2′-O-methyluridine. (B) Secondary structure of mouse/human tRNAPhe. Numbers correspond to the positions of 2′-O-methylated cytidine and guanosine catalyzed by Ftsj1. (C) Representative mass chromatograms of tRNAPhe fragments (A29 ~ Ψ39) with and without Cm, Gm, and OHyW modification. Sequence of each tRNAPhe fragment is shown on the right side of chromatogram. Note that C32, G34, and G37 of tRNAPhe were fully modified to Cm, Gm, and OHyW in WT mouse, whereas Cm and Gm modification was not detected and OHyW showed a ~50% reduction in KO mice. m/z, mass/charge ratio. (D) Modifications of tRNAPhe at C32, G34, and G37 in XLID patient–derived lymphocytes and control lymphocytes from his parents. In the patient-derived lymphocytes, Cm and Gm modifications were absent, and OHyW modification reduced to 12.6% of levels in control cells. Transfection of human FTSJ1 cDNA in the patient-derived cells partially restored Cm, Gm, and OHyW modifications. n.d., not detected.
We also examined 2′-O-methylation of tRNAs in an immortal lymphocyte cell line established from a patient with XLID and control cell lines from their parents. This patient has a G-to-A substitution at the splice donor site in intron 8 (c.571+1G>A), which caused a retention of the entire intron 8 in FTSJ1 mRNA, leading to a premature termination codon at position 597 in exon 9 (fig. S2A). The c.571+1G>A mutation resulted in an ~80% reduction of FTSJ1 mRNA level in the patient-derived cell line compared to control cell lines (fig. S2B). Consequently, the 2′-O-methylation modifications at positions 32 and 34 of tRNAPhe were undetectable in the patient-derived cell line (Fig. 1D). Similar to Ftsj1 KO mice, loss of 2′-O-methylation led to a decrease in OHyW modification at G37 of tRNAPhe in the patient-derived cells (Fig. 1D). In addition to tRNAPhe, 2′-O-methylation at positions 32 and 34 of tRNATrp and position 34 of tRNASec was undetectable in the patient cells (fig. S2, C and D). Expression of human FTSJ1 complementary DNA (cDNA) in patient cells restored 2′-O-methylation at both positions 32 and 34 of tRNAPhe (Fig. 1D) and tRNATrp (fig. S2C). Upon FTSJ1 expression in patient-derived cells, OHyW modification of tRNAPhe increased slightly (Fig. 1D). The moderate effect of this rescue may be attributed to the poor transfection efficiency of lymphocytes.
We also performed an in vitro methylation assay using recombinant Flag-tagged FTSJ1 and Flag-tagged WD repeat domain 6 (WDR6) WD repeat domain 6 (WDR6), which is the cofactor that binds to FTSJ1 and is required for methylation of position 34 (fig. S3A) (16). The FTSJ1/WDR6 complex was incubated with hypomodified tRNATrp purified from patient cells, followed by mass spectrometry analysis. The enzyme complex was sufficient to catalyze 2′-O-methylation of human tRNATrp in vitro (fig. S3B).
Loss of Ftsj1 selectively reduces the steady-state level of tRNAPhe in the brain
We examined the transcriptional landscape of tRNAs purified from the forebrain of 8-week-old WT and Ftsj1 KO mice by next-generation sequencing [tRNA sequencing (tRNA-seq)] (Fig. 2A and fig. S4, A and B). Notably, among all tRNA species, tRNAPhe showed the largest reduction in Ftsj1 KO mouse brain (0.42 fold change relative to WT, P < 0.0001; also see the representative reads in fig. S4B), while other tRNA species only showed small differences between WT and KO mice regardless of the presence or absence of Ftsj1-mediated methylation (0.88 to 1.2 fold change; Fig. 2B). We also examined the profile of the ribosome-bound tRNA from WT and Ftsj1 KO brains (Fig. 2A) (20). Similar to the total tRNA fraction, ribosome-bound tRNAPhe also showed the largest reduction in the Ftsj1 KO brain when compared to WT brain (0.47 fold change relative to WT, P = 0.0019; Fig. 2C). Notably, in both WT and KO brains, the change in ribosomal tRNA abundance correlated with the change in total tRNA abundance (fig. S5A).
Fig. 2. Selective decrease in tRNAPhe in Ftsj1 KO mouse brain.
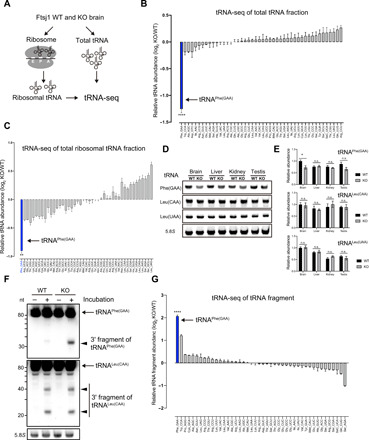
(A) Schematic illustration of tRNA-seq strategy for analysis of total tRNA and ribosome-bound tRNA isolated from WT and KO mouse brains at 8 weeks. (B) Relative abundance of individual tRNA in the KO mouse brain. tRNAPhe of the KO mouse brain showed the most pronounced decrease compared to that of WT mouse brain (n = 3 for each, ****P < 0.0001 by Student’s t test). (C) Relative abundance of individual ribosome-bound tRNA. Among all tRNAs in the KO mouse brain, tRNAPhe showed the largest reduction (n = 3 for each, **P = 0.0019 by Student’s t test). (D) Northern blotting of tRNAPhe, tRNALeu(CAA), and tRNALeu(UAA) from total RNA isolated from the brain, liver, kidney, and testis of 8-week-old WT and KO mice. 5.8S ribosomal RNA (rRNA) was used as loading control. (E) Quantitative analysis of tRNAPhe, tRNALeu(CAA), and tRNALeu(UAA) level in indicated tissues relative to the level in the WT brain. tRNAPhe was selectively and significantly decreased in the brain, but not in the liver, kidney, and testis (n = 4 for each, *P = 0.02 by Student’s t test). n.s., not significant. (F) Lysates of WT and KO mouse brains were briefly incubated on ice and subjected to RNA extraction and Northern blotting against tRNAPhe and tRNALeu(CAA). 5.8S rRNA was used as loading control. Note that a 3′ fragment of tRNAPhe was strongly detected in KO mouse brain lysate. (G) Relative abundance of tRNA fragments in WT and KO mouse brains were analyzed by tRNA-seq. Note that the tRNAPhe-derived fragment was the most abundant tRNA species that accumulated in the KO mouse brain when compared to WT (n = 3 for each, ****P < 0.0001 by Student’s t test). Error bars present SEM.
We also performed Northern blotting to investigate the expression level of tRNAPhe in brain and peripheral tissues, including the liver, kidney, and testis. Similar to tRNA-seq, the steady-state level of tRNAPhe was significantly decreased in the brains of Ftsj1 KO mice (70% of WT brain). The level of tRNAPhe in the liver, kidney, and testis did not show a significant change between WT and KO mice (Fig. 2, D and E). In contrast, the steady-state level of tRNALeu(CAA) and tRNALeu(UAA) did not differ between WT and KO mice in any of these tissues (Fig. 2, D and E). We also examined the level of tRNAPhe and tRNALeu(CAA) in the brain and liver from newborn (P0) mice. The level of tRNAPhe, but not tRNALeu(CAA), was decreased in the brain of P0 KO mice (fig. S5B). Notably, the extent of decrease in tRNAPhe in the Ftsj1 KO brain was moderate (80% of WT brain) at P0 when compared to 8 weeks (fig. S5C). These results demonstrate that loss of Ftsj1 caused a selective decrease in the abundance of brain tRNAPhe, with the degree of reduction progressing with age.
The selective decrease in tRNAPhe in the Ftsj1 KO brain suggested that hypomodified tRNAPhe is susceptible to fragmentation. Brief incubation of brain lysate without ribonuclease (RNase) inhibitor quickly yielded a short fragment [~35 nucleotides (nt)] of tRNAPhe, which was abundantly accumulated in the Ftsj1 KO mice when compared to WT mice (Fig. 2F). In contrast, the degradation pattern of tRNALeu(CAA) did not differ between WT and KO brain lysates (Fig. 2F). Next, we extracted small RNA (30 to 40 nt) from WT and Ftsj1 KO brains and performed tRNA-seq. The unbiased analysis showed that both WT and Ftsj1 KO mouse brains contain some extent of tRNA fragments regardless of tRNA species (fig. S5, D and E) (for full dataset, see GSE153758). Notably, the most abundant tRNA fragment accumulated in the KO brain were derived from the 3′ half of tRNAPhe (4.3 fold change relative to WT, P < 0.0001; Fig. 2G and fig. S5D). Furthermore, tRNA-seq of tRNAPhe in the KO mouse brain revealed that a large portion of the 3′ half tRNAPhe fragment was cleaved at position G37, with the rest of the fragment being cleaved at positions 34 to 36 (fig. S5D).
We also examined whether loss of Ftsj1-mediated 2′-O-methylation affected aminoacylation of tRNAPhe. The level of aminoacylated tRNAPhe in the Ftsj1 KO brain exhibited a marked decrease (fig. S5F). However, the relative aminoacylation level of tRNAPhe in the brain, liver, and kidney, which is calculated by normalizing aminoacylated tRNAPhe to nonaminoacylated tRNAPhe in each tissue, did not differ between WT and KO mice (fig. S5G). Neither the steady-state level of aminoacylated tRNALeu(UAA) nor the relative aminoacylation level of tRNALeu(UAA) differed between WT and KO mice (fig. S5, H and I). These results suggest that the decrease in aminoacylated tRNAPhe in the Ftsj1 KO brain can be attributed to the decrease in the steady-state level of tRNAPhe, but not to aminoacylation efficiency.
Translational perturbation at the Phe codon in Ftsj1 KO brain
We next investigated whether decoding was impaired at Phe codons by ribosome profiling analysis of ribosome-protected mRNA footprints from WT and KO mouse brains at 8 weeks of age (Fig. 3A) (21). The relative frequency of each codon (herein referred to as ribosome occupancy) of Phe UUC and UUU codons at the ribosomal A site exhibited the highest up-regulation among all codons in the Ftsj1 KO brain (UUC, 1.36-fold, P = 0.0003; UUU, 1.31-fold compared to WT, P = 0.0024) (Fig. 3B). When the relative change of ribosome occupancy was plotted against the relative abundance of ribosome-bound tRNA, ribosome occupancy at any given codon, except Phe codons, was positively correlated with the abundance of ribosome bound tRNA (P = 0.0062, Pearson r = 0.3583) (Fig. 3C).
Fig. 3. Impaired translation at Phe codons in Ftsj1 KO brain.
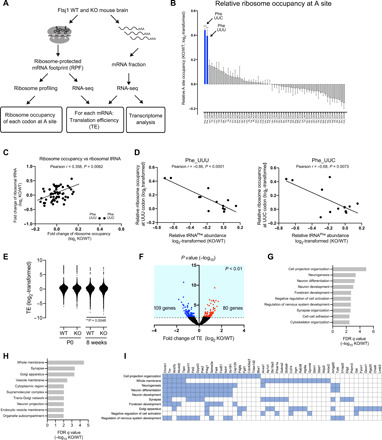
(A) Strategy for ribosomal profiling and RNA-seq. (B) Ribosomal profiling of 8-week-old WT and KO mouse brains. Relative ribosome occupancy of each codon at the A site is shown. Phe UUC and UUU codons showed the highest occupancy in KO mouse brain (UUC, 1.36-fold change versus WT, ***P = 0.0003; UUU, 1.32-fold change versus WT, **P = 0.002 by Student’s t test; n = 3 for each). Error bars present SEM. (C) Correlation between relative ribosome occupancy at the A site and relative tRNA abundance in 8-week-old WT and KO mouse brains. Pearson r = 0.358, P = 0.0062. (D) Correlation between tRNAPhe abundance and relative ribosome occupancy at UUU (left) and UUC (right) codons (UUU, Pearson r = −0.86, ****P < 0.0001; UUC, Pearson r = −0.68, P = 0.0073). Data are from the brain of P0 (n = 2 for each genotype) and 8-week-old mice (n = 3 for each genotype) and the liver of P0 mice (n = 2 for each genotype). (E) Scatter plot of TE for each mRNA in P0 and 8-week-old WT and KO mouse brains (WT and KO genes at P0, 4474 genes; WT and KO genes at 8 weeks, 5577 genes; **P = 0.0048 by Wilcoxon rank test). Each data point represents the average TE of three mice for each mRNA. (F) Volcano plot of TE in 8-week-old KO and WT mouse brains. Red dots represent significantly up-regulated (P < 0.01) genes, and blue dots represent significantly (P < 0.01) down-regulated genes in KO brain. (G and H) Gene ontology analysis [biological process (G) and cellular component (H)] of genes down-regulated TE in the KO mouse brain. (I) Gene/gene set overlap matrix based on gene ontology analysis (G and H). Blue boxes represent genes enriched in each pathway.
We also performed ribosome profiling using P0 mouse brain and liver (fig. S6, A and B). In the Ftsj1 KO mouse brain, the Phe UUU codon showed the highest up-regulation of ribosome occupancy among all codons (1.20-fold compared to WT, P = 0.02), whereas the Phe UUC codon only showed 1.05-fold up-regulation (P = 0.6) (fig. S6A). In contrast, ribosome occupancy at Phe UUU and UUC codons only showed slight or no change, respectively, in the Ftsj1 KO liver when compared to WT (UUU, 1.07-fold, P = 0.009; UUC, 1.01-fold, P = 0.87) (fig. S6B). When ribosome occupancy at Phe codons in all genotypes and tissues (brain and liver) were plotted to the corresponding tRNAPhe levels, ribosome occupancy at the UUU codon was significantly and negatively correlated with abundance of tRNAPhe (Spearman r = −0.66, P = 0.0121; Pearson r = −0.86, P < 0.0001), whereas the correlation between ribosome occupancy at the UUC codon and tRNAPhe abundance was ambiguous (Spearman r = −0.31, P = 0.29; Pearson r = −0.68, P = 0.0073) (Fig. 3D). Together, these results suggest that loss of Ftjs1 selectively perturbed decoding at Phe codons and that the degree of perturbation may be influenced by the abundance of tRNAPhe and the type of codon.
Defective translation in Ftsj1 KO brain and its link to brain dysfunction
Dysregulation of decoding can affect gene expression at both transcriptional and translational levels. We performed RNA-seq using WT and Ftsj1 KO mouse brains at P0 and 8 weeks of age and compared the transcriptome (Fig. 3A). At both developmental stages, only a small subset of genes showed change at the mRNA level in Ftsj1 KO mouse brain, and the degree of change was small compared to WT (fig. S6, C to F). Among genes that showed significant changes (P < 0.01) in the Ftsj1 KO brain at P0 stage, 27 genes showed a 0.72- to 0.88-fold decrease, and seven genes showed a 1.11- to 1.25-fold increase compared to the WT brain at P0 (fig. S6C). Gene ontology analysis of differentially regulated genes revealed that these genes are involved in metabolic processes not specifically limited to neuronal function (fig. S6D). Likewise, among genes that showed significant changes (P < 0.01) in 8-week-old KO mouse brain, 55 genes showed 0.7- to 0.9-fold decrease, and 19 genes showed 1.09- to 1.40-fold increase compared to the WT brain (fig. S6E). The down-regulated genes were enriched in pathways that affect neuronal development and structure, while the up-regulated genes had only limited enrichment in pathways related to neuron functions (fig. S6F).
We next estimated TE based on RNA-seq and ribosomal profiling data, a measure that is generally in proportion with protein translation rate (Fig. 3A). TE was determined by dividing the normalized read number of ribosome-bound mRNA footprints to normalized read number of the corresponding mRNA for each gene. We selected 4474 genes from datasets from P0 brain and 5577 genes from datasets from 8-week-old brain based on the following criteria: (i) Genes are protein-encoding genes, and (ii) genes are consistently detected across WT and KO brain samples (three biological replicates each) at each developmental stage. The mean TE of gene products from the KO brain was significantly lower than that of the WT brain at 8 weeks (WT, 1.452; KO, 1.428; P = 0.0048), while the mean TE did not differ between WT and KO brains at P0 (WT, 2.077; KO, 2.070, P = 0.63) (Fig. 3E). Deficiency of Trm7 in yeast induced a strong general amino acid control (GAAC) pathway and is involved in the growth defect (22). However, genes related to the GAAC pathway in mammals did not show a significant increase in the Ftsj1 KO mouse brain at both 8 weeks of age and P0 stage (fig. S6G), as well as mutant lymphocyte cells derived from the patient (fig. S6H).
We performed gene ontology analysis on genes that showed significant (P < 0.01) changes in TE at both P0 and 8 weeks of age. In P0 KO mouse brain, 74 genes showed a 0.38- to 0.84-fold decrease in TE, and 70 genes showed a 1.20- to 4.09-fold increase in TE compared to the WT brain (fig. S6I). These differentially changed genes in P0 KO brain were not specifically related to neuronal functions (fig. S6J). On the other hand, in 8-weeks-old KO mouse brain, 109 genes showed a 0.28- to 0.75-fold decrease in TE, and 80 genes showed a 1.34- to 3.26-fold increase in TE (Fig. 3F). The translationally down-regulated genes are actively involved the brain functions including cell projection, neuronal development, and synapse organization and correspond to membrane and synaptic proteins (Fig. 3, G and H). In contrast, the translationally up-regulated genes are not enriched in any particular biological processes (fig. S6K).
A close inspection of the translationally down-regulated genes revealed that some are involved in Wnt signaling [e.g., Ctnnb1 (β-catenin 1), Wnt7b, and Pak2] (Fig. 3I) and are important for brain development and maturation (23–25). Notably, deficiency of Ctnnb1 has been implicated in neurological disorders including ID and autism in humans (26, 27). Furthermore, genes related to neuron adhesion or synaptic spine formation, such as tenascin R (Tnr) and discs large MAGUK scaffold protein 2 (Dlg2), also exhibited a decrease in TE in 8-week-old KO mouse brain. Deficiencies of these genes have been implicated in cognitive deficits in animal models (28, 29). The 109 genes with significantly down-regulated TE in the KO mouse brain belong to a group of genes that are efficiently translated in the WT mouse brain (mean TE of the 109 genes in WT: 2.326 versus mean background TE of WT: 1.442, P < 0.0001; fig. S6L).
Altered neuron ultrastructure in the hippocampus and cortex of Ftsj1 KO brain
We next examined the morphology of cortex and hippocampus at histological, cellular, and synaptic levels using 8-week-old mouse brain. Nissl staining revealed that the macroscopic morphology of cortex and hippocampus was comparable between WT and KO mice (fig. S7, A and B). There was no obvious difference in the population of hippocampal and cortical neurons and glial cells between WT and KO mice (fig. S7, C to E). We also performed histological examination of the liver, kidney, heart, and testis by hematoxylin and eosin (H&E) staining; however, there were no morphological changes in these tissues in KO mice (fig. S7F).
We also investigated the morphology of dendritic spines in cortical and hippocampal neurons by Golgi staining. Dendritic spine ultrastructure and density have been implicated in many aspects of brain functions including learning and memory (30). Dendritic spines can be categorized into distinct types depending on their shape, which has been correlated with neuronal function. In 8-week-old Ftsj1 KO brain, both hippocampal and cortical neurons exhibited a significant increase in thin dendritic spines and a concomitant decrease in mushroom type spines when compared to the WT brain, an indication of immature spine formation (Fig. 4, A and B). In addition, we also examined striatal neurons that are also implicated learning and memory (31, 32). Ftsj1 KO mice exhibited an increase in the immature filopodia-like spines and a concomitant decrease in mushroom-type spines when compared to the WT brain (fig. S7, G and H). Moreover, the density of dendritic spines was significantly decreased in Ftsj1 KO hippocampal neurons compared to WT (Fig. 4C). We also analyzed the morphology of the postsynaptic density (PSD) using electron microscopy (Fig. 4D). In both hippocampal and cortical neurons, there was a significant decrease in the length and width of the PSD (Fig. 4, E and F).
Fig. 4. Impaired synaptic morphology in cortex and hippocampus of Ftsj1 KO mouse brain.
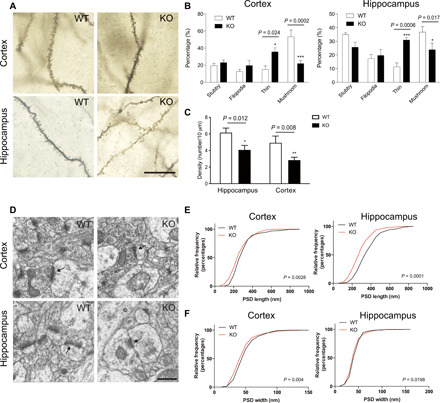
(A) Representative Golgi staining of cortical and hippocampal neurons from 8-week-old Ftsj1 WT and KO mice. Scale bar, 20 μm. (B) Quantitative analysis of dendritic spine morphology. Data were from 128 WT cortical spines, 169 WT hippocampal spines, 169 KO cortical spines, and 118 KO hippocampal spines of three mice for each genotype. Cortical spines: *P = 0.023 and ***P = 0.0002 by two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. Hippocampal spines: *P = 0.017 and ***P = 0.0006 by two-way ANOVA followed by Bonferroni post hoc test. Error bars present SEM. (C) Density of cortical and hippocampal spines in WT and KO mouse brains. Data were from 235 spines and 366-μm dendrite in WT hippocampal neurons, 159 spines and 388-μm dendrite in KO hippocampal neurons, 163 spines and 456-μm dendrite in WT cortical neurons, and 190 spines and 703-μm dendrite in KO cortical neurons. Three mice were used for each genotype. *P = 0.012 and **P = 0.008 by two-way ANOVA followed by Bonferroni post hoc test. Error bars present SEM. (D) Representative electron microscopic images of cortical and hippocampal PSD (arrows). Scale bar, 500 nm. (E and F) Cumulative frequency distribution of length (E) and width (F) of PSDs from cortex and hippocampus of WT and KO mouse brains. Data were from 240 WT cortical PSD, 251 KO cortical PSD, 253 WT hippocampal PSD, and 270 KO hippocampal PSD of three mice for each genotype. The statistical difference between WT and KO shown in the graphs was analyzed by Mann-Whitney test.
Impaired long-term potentiation and long-term depression in the hippocampus of Ftsj1 KO mice
We performed electrophysiological analysis using hippocampal slices prepared from WT and Ftsj1 KO brains from mice at 8 weeks of age. A paired-pulse facilitation protocol was applied to the hippocampal slices to examine the presynaptic release of neurotransmitter by measuring the relative increase in population excitatory postsynaptic potential (pEPSP) (fig. S7I). The amplitude of facilitation did not differ between WT and Ftsj1 KO mice (fig. S7J). We also investigated the relationship between the intensity of presynaptic stimulus input with the pEPSP output (I/O curve) using the same hippocampal slices (fig. S7K). There was no significant difference of I/O curve between WT and KO mice (fig. S7L). These results suggest that neurotransmitter release and its corresponding postsynaptic response were well preserved in the Ftsj1 KO brain at the basal level.
Repeated electrical stimuli can produce a persistent increase or decrease in the efficiency of synaptic transmission known as long-term potentiation (LTP) or long-term depression (LTD), respectively. LTP and LTD are associated with learning and memory and are mediated by complex molecular signaling including transcriptional, posttranscriptional, translational, and posttranslational mechanisms. We found that theta burst stimulation (TBS) induced a robust LTP in WT mouse hippocampal slices (Fig. 5, A and B). In hippocampal slices from KO mice, the normalized pEPSP slope of TBS-evoked LTP showed a 50% reduction compared to WT (WT, 1.66 ± 0.05; KO, 1.33 ± 0.06; ***P < 0.0001, WT versus KO; Fig. 5C). In LTD experiments, the low-frequency stimulation (LFS) effectively suppressed the pEPSP slope in hippocampal slices of the WT mouse brain (0.75 ± 0.02 related to baseline; Fig. 5, D to F). The LFS induced only a slight decrease in the pEPSP slope in hippocampal slices of the KO mouse brain (0.90 ± 0.02 related to baseline; WT versus KO, ***P < 0.001; Fig. 5, D to F), suggesting that LTD formation was impaired in the hippocampus of the KO mouse. Notably, both LTP and LTD were effectively blocked by application of D-2-amino-5-phosphonopentanoate (AP5), an inhibitor of the N-methyl-d-aspartate (NMDA) receptor, in WT and KO hippocampal slices (Fig. 5, C and F).
Fig. 5. Impaired synaptic plasticity in Ftsj1 KO brain.
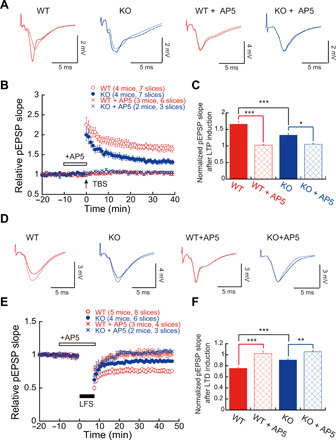
(A) Representative traces of pEPSPs recorded in CA1 region with and without N-methyl-d-aspartate (NMDA) receptor antagonist AP5 (50 μM). Traces with dashed lines represent basal pEPSPs, and traces with solid lines represent pEPSPs after TBS. (B) TBS-evoked LTP in hippocampal slices from WT and KO mice with and without 50 μM AP5. Seven slices from four WT mice and seven slices from four KO mice were used for LTP experiment. Six slices from three WT mice and three slices from two KO mice were used for LTP experiments in the presence of 50 μM AP5. (C) Impairment of NMDA receptor–dependent LTP in the KO mouse brain. *P < 0.05 and ***P < 0.001 by two-way ANOVA followed by Bonferroni post hoc test. Error bars present SEM. (D) Representative traces of pEPSPs recorded in CA1 region using LTD protocol. Traces with dashed lines represent basal pEPSPs, and traces with solid lines represent pEPSPs after LFS. (E) LFS-evoked LTD in hippocampal slices from WT and KO mice with and without 50 μM AP5. Eight slices from five WT mice and six slices from four KO mice were used for LTD experiment. Four slices from three WT mice and three slices from two KO mice were used for LTD experiments in the presence of 50 μM AP5. (F) NMDA receptor–dependent LTD was impaired in the KO mouse brain. **P < 0.01 and ***P < 0.001 by two-way ANOVA followed by Bonferroni post hoc test. Error bars present SEM.
Ftsj1 KO mice show behavioral abnormality
We performed a battery of behavioral tests in 8-week-old WT and KO mice. First, mice were subjected to open-field and elevated plus maze tests to examine locomotor activity and anxiety-like behavior in a novel environment (Fig. 6A) because anxiety is a symptom observed in some patients with FTSJ1-related XLID (9). Ftsj1 KO mice spent less time and traveled less distance in the center of the open-field chamber than WT mice, while the total travel distance in the chamber did not differ between WT and Ftsj1 KO mice (Fig. 6A). In the elevated plus maze, Ftsj1 KO mice spent less time and traveled less distance in the open arm than WT mice, while the total travel distance in both open arm and closed arm was longer in KO mice than in WT (Fig. 6B). The results from the open-field and elevated plus maze tests suggest that loss of Ftsj1 induced anxiety-like behavior in mouse.
Fig. 6. Ftsj1 KO mouse display behavioral abnormalities.
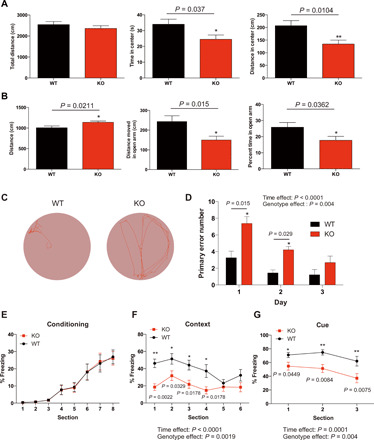
(A) WT and KO mice were subjected to open-field test. Total distance traveled (left), time spent in the center (middle), and distance traveled in the center (right) are shown. Statistical significance was analyzed by Mann-Whitney test. n = 10 for WT and n = 11 for KO. (B) WT and KO mice were subjected to an elevated plus maze test. Total distance traveled (left), distance traveled in open arm (middle), and time spent in the open arm (right) are shown. n = 20 for WT and n = 22 for KO. Statistical significance was analyzed by Mann-Whitney test. (C) Representative movement during goal seeking behavior of WT and KO mice in a Barnes maze task. The tracking data show that a KO mouse could not efficiently find the goal compared to a WT mouse. (D) Ftsj1 KO mice made significantly more errors in finding the goal than WT mice. n = 18 for WT and n = 16 for KO. Statistical significance was analyzed by two-way ANOVA followed by Holm-Sidak post hoc test. (E to G) WT and KO mice were subject to a fear conditioning paradigm. Percentage of freezing behavior during conditioning period did not differ between WT and KO (E). Ftsj1 KO mice showed significantly less contextual (F) and cued (G) fear behavior than WT. n = 18 for WT and n = 20 for KO. Statistical significance was analyzed by repeated-measures, two-way ANOVA followed by Holm-Sidak post hoc test. Error bars represent SEM.
Next, we investigated whether loss of Ftsj1 would affect spatial learning using the Barnes maze test, in which mice were trained to find a hidden chamber using spatial cues as guidance (Fig. 6C). The number of mistakes until the mouse found the goal chamber was counted as a primary error number to evaluate the ability of spatial leaning. Compared to WT mice, Ftsj1 KO mice showed slower spatial learning, with the primary error number being significantly higher than WT mice in the first and second day of training (Fig. 6D).
Last, mice were subjected to a fear conditioning paradigm to assess how loss of Ftsj1 would affect the formation and retrieval of fear memory. During the conditioning day, WT and Ftsj1 KO mice showed the same degree of freezing responses to the mild foot shock and sound presentation (Fig. 6E). On the next day, the retrieval of fear memory was tested by putting mice in the chamber where they received foot shock (context) and was placed in a new chamber with sound presentation (cue) that they had heard on the conditioning day. Compared to WT mice, Ftsj1 KO mice showed significantly less freezing behavior in response to the context-dependent stimuli and the cue-dependent stimuli (Fig. 6, F and G).
DISCUSSION
Defective tRNA modifications feature in a number of brain disorders (3, 33–35). To date, at least 17 genes encoding cytosolic tRNA modification enzymes, including FTSJ1, have been identified as causal genes for intellectual disabilities, microcephaly, and neurodegeneration (3). However, given their ubiquitous presence throughout the body, understanding why loss of these modifications preferentially affects brain function remains a challenge. In the present study, we have investigated the impact of Ftsj1-deficiency in the mouse brain and uncovered a link to the pathogenesis of FTSJ1-related XLID. Among all tRNA species, hypomodified tRNAPhe was selectively and significantly reduced in the Ftsj1 KO mouse brain, but not in other tissues. Furthermore, decoding at Phe codons was selectively and markedly impaired in the Ftsj1 KO brain, leading to a decrease in TE of genes related to synaptic activity and structure. Thus, our results suggest that the selective decrease in hypomodified tRNAPhe in the brain is associated with the nonsyndromic FTSJ1-related XLID.
Hypomodified tRNAPhe can be susceptible to endoribonuclease-mediated cleavage, which may lead to a decrease in its steady-state level. When preparing RNA without RNase inhibitor, we observed an accumulation of 3′-tRNAPhe fragment in the Ftsj1 KO mouse brain. Concordant with our result, loss of Drosophila Ftsj1 homologs also resulted in an accumulation of 3′-tRNAPhe fragment, although the steady state of full-length tRNAPhe did not change in Drosophila (18). RNase 1 and angiogenin/RNase 5 are the members of ribonuclease A (RNase A) family responsible for the cleavage of tRNA, and their actions can be blocked by 2′-O-methylation of ribose and methylation at nucleotide base. For example, tRNAMet contains 2′-O-methylation at position C34, which is mediated by nucleolar fibrillarin and small nucleolar RNAs (36). Hypomodified tRNAMet at C34 is highly sensitive to angiogenin-mediated cleavage, which efficiently induced generation of 3′-tRNAMet fragment (36). In addition, NOP2/Sun RNA methyltransferase 2 (NSUN2) is a cytosine C5-methyltransferase for both tRNA and mRNA, and a loss-of-function mutation in NSUN2 has been identified in patients with ID (37–39). Notably, loss of NSUN2 resulted in RNase 5/angiogenin–mediated cleavage of hypomodified tRNA, which is possibly involved in the pathogenesis (40). Thus, an RNase A ribonuclease family member is likely involved in the cleavage of hypomodified tRNAPhe in Ftsj1 KO mice. Identification of the responsible RNase in future studies will be important for the development of a therapeutic target.
The cause of brain-specific decrease in tRNAPhe is unclear but might result from tissue-specific differences in the balance of RNase and RNase inhibitor. In mouse and human brain, the major RNase A family member is RNase 1, which has moderate levels of mRNA expression compared to other tissues (41). On the other hand, RNase/angiogenin inhibitor 1, the endogenous inhibitor of both RNase 1 and angiogenin (42, 43), is expressed at lower levels in the brain compared to other tissues such as the liver and pancreas (41). The high RNase-to-RNase inhibitor ratio suggests that the brain contains a relatively high RNase activity, which subsequently induces degradation of hypomodified tRNAPhe. This degradation may lead to the manifestation of ID without other physical abnormalities. In addition, the potentially high brain RNase activity might explain why deficiencies of other cytosolic tRNA modification enzymes such as NSUN2 and elongator acetyltransferase complex subunit 2 (ELP2) also lead to the development of brain malfunction as the primary symptom (40, 44).
Among the 11 Ftsj1 substrate tRNAs, we found that tRNAPhe showed the highest sensitivity to endoribonuclease-mediated cleavage in the Ftsj1 KO mouse brain, despite the fact that 2′-O-methylation was eliminated from positions 32 and 34 of these tRNAs. The reason for the selective cleavage of tRNAPhe, but not other Ftsj1 substrate tRNAs, is currently unknown. One possible explanation may be that the hypermodified G37 of tRNAPhe is associated with loss of the OHyW modification, which is exclusively found at G37 of tRNAPhe and is the bulkiest modification found at position 37 of all Ftsj1 substrate tRNAs. It is conceivable that OHyW modification may prevent access of RNase to tRNAPhe through spatial interference. Consistent with this hypothesis, our tRNA-seq showed that a majority of tRNAPhe fragments found in Ftsj1 KO mouse brain were cleaved at the hypomodified G37. Further studies using OHyW-deficient cells are needed to determine the extent to which loss of OHyW modification induces tRNAPhe cleavage. tRNAPhe, along with tRNATrp and tRNALeu(UAA), is the only three tRNAs that are 2′-O-methylated at both positions 32 and 34 by Ftsj1. Accordingly, tRNAPhe is the only Ftsj1 substrate tRNA that is hypomodified at all three positions (32, 34, and 37) upon Ftsj1 deficiency. Thus, a collective loss of both 2′-O-methylation and OHyW modification may cause the selective cleavage of tRNAPhe in Ftsj1 KO mouse.
Our data from ribosome profiling of the brain at different developmental stages provide clear evidence that ribosome transition is selectively impaired at Phe codons in the Ftsj1 KO mouse brain. Defective decoding at Phe codons was selectively observed in the Ftsj1 KO mouse brain, in which tRNAPhe levels were significantly decreased. These results suggest that the decoding efficiency at Phe codons depends on the quantity of tRNAPhe. Note that decoding at the Phe UUU codon was impaired in Ftsj1 KO mouse brain at both adult and neonatal stages, while decoding at the Phe UUC codon was only impaired at the adult stage. Because tRNAPhe forms wobble binding (G:U) with the third position of the UUU codon and the affinity of tRNAPhe:UUU base pairing is relatively low compared to tRNAPhe:UUC base pairing, it is conceivable that decoding at the Phe UUU codon is more susceptible to the steady-state tRNAPhe level than decoding at the Phe UUC codon. Note that defective modifications of tRNAPhe might directly impair codon-anticodon binding, further contributing to the decoding defect. Previous studies have shown that yW modification at G37 of tRNAPhe is also involved in codon-anticodon recognition (45). Thus, the slow ribosomal transition at Phe codons might be attributed to both quantitative and qualitative defects of hypomodified tRNAPhe in the Ftsj1 KO mouse brain.
Here, we also present a thorough transcriptome and the translational profile from the brain. A previous study has shown that Trm7-deficient yeast exhibited a marked increase in the transcription of genes related to amino acid metabolism induced by Gcn4, the transcription factor related to nutrient stress response (46). In contrast, the Ftsj1 KO mouse brain showed only a limited change in the overall transcriptome, including amino acid metabolism while exhibiting a marked rearrangement of TE. Notably, the TE of a subset of genes related to maintenance of synaptic activity and structure was markedly reduced in the Ftsj1 KO mouse brain. Consequently, Ftsj1 KO mouse hippocampus and cortex exhibited immature spine morphology and decreased synaptic plasticity, ultimately leading to impairment of learning and memory and anxiety-like behavior. These results thus provided molecular, histological, and physiological links to ID related to Ftsj1 deficiency. Other brain regions, such as the amygdala and cerebellum, are also involved in the learning and memory (47, 48) and may also be affected by Ftsj1 deficiency. A detailed and broad inspection of brain region–specific neuron morphology and function will be conducted in future studies.
Note that in addition to the brain dysfunction, Ftsj1 KO mice exhibited a moderate reduction in body weight, which was not apparent in patients having FTSJ1 deficiency (8, 12). Similar to our findings in mice, reduced body weight was observed in Drosophila with deficiency of Ftsj1 homologs (18) and a line of Ftsj1 gene–trapped mice (19). These findings suggest that Ftsj1 might have a broad impact on peripheral tissues. The molecular mechanism underlying the low body weight in mouse and Drosophila is currently unknown. Similar to mouse peripheral tissues, the steady-state level of hypomodified tRNAPhe remains unchanged in Drosophila (18). It is conceivable that the hypomodification of tRNAPhe and other Ftsj1 substrate tRNAs may impair general translational efficiency by affecting base pairing independent of the steady-state tRNAPhe level. In this case, the reduction of general protein translation levels could result in decreased cell growth and cell mass, ultimately leading to reduced body weight. Given the ubiquitous expression of Ftsj1, future studies will be needed to elucidate the general role of Ftsj1 in non-neuronal tissues in eukaryotes.
MATERIALS AND METHODS
Animals
A constitutive Ftsj1 KO mouse line was generated by conventional homologous recombination to replace exons 2 to 5 of the Ftsj1 gene with the neomycin resistance gene (see fig. S1A). Ftsj1 heterozygous female mice were mated to WT littermate male mice to obtain male Ftsj1 KO mice and their littermate controls. Mice were housed at 25°C with 12-hour light and 12-hour dark cycles. To obtain P0 mice, a pair of female and male mice were housed in the same cage for 1 hour (19:00 to 20:00). Subsequently, female mice were placed in a new cage and monitored carefully during pregnancy. When female mice gave birth in the night (20:00 to 7:00), P0 mice were taken the next morning for genotyping and experiments as P0 samples. The Animal Ethics Committee of Kumamoto University reviewed and approved all animal procedures (approval ID: A2019−107 R2).
Human cells
Immortal lymphoblast cells were established from blood samples of a Japanese family as described (12). These blood samples were obtained with informed consent approved by Institute Review Board, National Center of Neurology and Psychiatry (XXXX-115). Control cell lines #1 and #2 were derived from peripheral blood cells of the patient’s father and mother, respectively. The father carried the normal FTSJ1 gene, while the mother was a heterozygous carrier of the c.571+1G>A mutation of the FTSJ1 gene [see (12) for detailed information]. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, and RNA was extracted for quantitative polymerase chain reaction (PCR) and mass spectrometry analysis. Human embryonic kidney (HEK) 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and were used for exogenous expression of FTSJ1-Flag, WDR6-V5, and THADA armadillo repeat containing (THADA)–V5.
Genotyping
To genotype the Ftsj1 KO mice, genomic DNA was extracted from small pieces of tails clipped from 4-week-old mice or P0 mice using phenol/chloroform precipitation. Ten nanograms of DNA was subjected to PCR for genotyping using primers to detect the neomycin resistance gene.
Quantitative PCR
Total RNAs (100 ng) isolated from immortalized lymphoblast cell lines were used for reverse transcription using Prime Script RT Master Mix (TaKaRa) according to the manufacturer’s instructions. Reverse transcription was performed at 37°C for 15 min, followed by enzyme inactivation at 85°C for 5 s. cDNA was mixed using a TB GREEN Premix Ex-Taq2 kit (TaKaRa), and quantitative PCR was performed using Rotor Gene Q (QIAGEN). Quantitative PCR assays were performed using three biological replicates. Target gene expression levels were normalized against that of the 18S ribosomal RNA (rRNA) gene, and relative expression was calculated using the 2–ΔΔCt method.
The sequences of DNA oligo primers for examination of expression levels of genes related to general amino acids control pathway are as follows: human ATF4, 5′-ATGACCGAAATGAGCTTCCTG (forward) and 5′-ATGACCGAAATGAGCTTCCTG (reverse); human SCL7A5, 5′-CCGTGAACTGCTACAGCGT (forward) and 5′-CTTCCCGATCTGGACGAAGC (reverse); human SCL1A4, 5′-TGTTTGCTCTGGTGTTAGGAGT (forward) and 5′-CGCCTCGTTGAGGGAATTGAA (reverse); mouse Atf4, 5′-CCTGAACAGCGAAGTGTTGG (forward) and 5′-TGGAGAACCCATGAGGTTTCAA (reverse); mouse Scl7A5, 5′-ATATCACGCTGCTCAACGGTG (forward) and 5′-GCCGCCTGACTTGGAGATG (reverse); mouse Scl1a4, 5′-GTGGCATCGCTGTTGCTTAC (forward) and 5′-TTGCAGACGTAGTGAATGCGG (reverse).
Mass spectrometry analysis of tRNA modification
Total RNA was isolated from the liver of WT and Ftsj1 KO mice at 8 weeks or human lymphoblast cells using TRIzol (Invitrogen) according to the manufacturer’s instructions. Individual mouse tRNA and human tRNA were purified from total RNA using a 5′-amino–modified DNA oligo probe by the reciprocal circulating chromatography (RCC) method (49). For analysis of tRNA fragments, purified tRNA was digested with RNase T1 or RNase A and subjected to mass spectrometry analysis as described (50). For analysis of nucleosides, purified tRNA was digested with 2.5 U of nuclease P1 (Wako) and 0.2 U alkaline phosphatase (TaKaRa, 2250A) in 5 mM ammonium acetate (pH 5.3) and 20 mM Hepes-KOH (pH 7.0) for 3 hours at 37°C. Samples were subject to mass spectrometry analysis as described (50). The sequences of DNA oligo probes for isolation of tRNAs are as follows: tRNAPhe, 5′-GAACCAGGGACCTTTAGATCTTCAGTCTAACGCTC; tRNATrp, 5′-TCTGATCTGGAGTCAGACGCGCTACCGTTGCGCCA; tRNASec, 5′-TTGAAGCCTGCACCCCAGACCACTGAGGATCA; tRNALeu(UAA), 5′-GATCTTAAGTCCAACGCCTTAACCACTCGGCCATCCTGGT; tRNALeu(CAA), 5′-TGGTGTCAGAAGTGGGATTCGAACCCACGCCTCCATCCGG; *tRNALeu(AAG), 5′-TGGTGGCAGCGGTGGGATTCGAACCCACGCCCCCGAAGAG; tRNALeu(CAG), 5′-AGACTGCGACCTGAACGCAGCGCCTTAGACCGCTCGGCCA; tRNAVal(AAC), 5′-ACCTTTCGCGTGTTAGGCGAACGTGATAACCACTACACTA; tRNAGln(CUG), 5′-GTCGCTGGATTCAGAGTCCAGAGTGCTAACCATTACACCAT; tRNAArg(ACG), 5′-AATCTTCTGATCCGTAGTCAGACGCGTTATCCATTGCGCCAC. Asterisk (*) indicates that probe for tRNALeu(AAG) was used to isolate tRNALeu(UAG).
Plasmid construction
The cDNAs of human FTSJ1, WDR6, and THADA were amplified by reverse transcription and PCR from HeLa cells’ total RNA. The primer sequences are as follows: FTSJ1, CACCATGGGACGGACGTCAA (forward) and AGGTGAACAACTCATTTCATTG (reverse); WDR6, CACCATGGGCAGCGCGG (forward) and GTCATACCAGTTGTAAACCTCAAG (reverse); THADA, CACCATGGGTGTAAAGAAGAAGAAAGAAATG (forward) and ACATGCCGCTTCTGTTCTT (reverse). Each amplicon was inserted into a pENTR/D-TOPO (Thermo Fisher Scientific) by directional TOPO cloning and transferred to a modified pDEST12.2 (Invitrogen) with a C-terminal FLAG-tag or a pcDNA-DEST40 (Thermo Fisher Scientific) by Gateway cloning according to the manufacturer’s instruction (Thermo Fisher Scientific).
Protein-protein interaction analysis
The expression vectors of FTSJ1-FLAG and WDR6-V5 (1 μg each per well in a six-well plate) were cotransfected into HEK293T cells (2.5 × 105 cells per well) using polyethyleneimine (51). After 48 hours, the cells were collected and lysed with 0.5 ml of lysis buffer [150 mM KCl, 10 mM tris-HCl (pH 8.0), 2.5 mM MgCl2, 1 mM dithiothreitol (DTT), 0.5% Triton X-100, and 1× complete EDTA-free protease inhibitor cocktail (Roche)] by 20 passages through a 25-gauge syringe needle. The lysate was centrifuged twice at 20,000g for 20 min at 4°C and passed through an Ultrafree 0.45-μm filter (Merck Millipore) to remove cell debris. Immunoprecipitation was conducted by incubating the supernatant with 20 μl of anti-Flag M2 affinity gel (Sigma-Aldrich) for 3 hours at 4°C and washing the gel by lysis buffer three times. The gel was boiled at 95°C for 5 min for SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblotted using anti-Flag-tag polyclonal antibody conjugated with horseradish peroxidase (HRP) (Anti–DDDDK-tag pAb–HRP–DirecT, Medical & Biological Laboratories), or anti-V5-tag polyclonal antibody conjugated with HRP (Anti-V5-tag pAb-HRP-DirecT, Medical & Biological Laboratories) diluted with Can Get Signal Immunoreaction Enhancer Solution 2 (Toyobo) at 1:4000 (v/v). Chemiluminescence was detected using Pierce Western Blotting Substrate Plus (Thermo Fisher Scientific) and an ImageQuant LAS4000 mini system (GE Healthcare).
In vitro 2′-O-methylation assay
For transient expression of FTSJ1-FLAG and WDR6-FLAG, six dishes (10 cm in diameter) of HEK293T (1.5 × 106 cells per dish) were transfected with 10 μg of the FTSJ1-FLAG and WDR6-FLAG vector, respectively, using polyethyleneimine (51). After culturing for 48 hours, the cells were harvested, suspended with 1 ml of lysis buffer [150 mM KCl, 10 mM tris-HCl (pH 8.0), 2.5 mM MgCl2, 1 mM DTT, 0.5% Triton X-100, and 1× complete EDTA-free protease inhibitor cocktail (Roche)], and lysed on ice by 20 passages through a 25-gauge syringe needle. The lysates were centrifuged twice at 20,000g for 20 min at 4°C to remove cell debris.
For immunoprecipitation, FLAG-tagged proteins were captured on 0.1 ml of anti-Flag M2 affinity gel (Sigma-Aldrich), washed three times with wash buffer [300 mM KCl, 10 mM tris-HCl (pH 8.0), 2.5 mM MgCl2, 1 mM DTT, and 0.5% Triton X-100] and extracted with 0.5 ml of elution buffer [150 mM KCl, 10 mM tris-HCl (pH 8.0), 20% glycerol, 1 mM DTT, and FLAG peptide (0.2 mg/ml) (Sigma-Aldrich)]. Amicon Ultra 0.5-ml 3K device (Merck Millipore) was used to concentrate the eluates. The concentration of the collected proteins was determined by band densitometry from an SDS-PAGE gel compared with bovine serum albumin (Sigma-Aldrich) as a standard.
In vitro methylation was conducted in a reaction mixture (50 μl) consisting of 50 mM Hepes-KOH (pH 7.6), 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 1 mM S-adenosyl-l-methionine, 20 μM hypomodified tRNATrp isolated from the patient lymphoblast cells, and 0.1 mM each of the recombinant FTSJ1 and WDR6 for 2 hours at 37°C. The reactant tRNA was collected by PCI treatment [phenol:chloroform:isoamyl alcohol, 25:24:1 mixed (pH 5.2) (nacalai tesque)], chloroform extraction, and ethanol precipitation and subjected to mass spectrometry analysis as described above.
Histology and electron microscopy
Ftsj1 KO and WT mice were euthanized and perfused with 4% paraformaldehyde for histological examination. H&E staining and Nissl staining was performed according to standard protocols. Golgi staining of hippocampal and cortical neurons was performed using the FD Rapid GolgiStain Kit (FD NeuroTechnologies Inc.). Images were acquired using an IX83 Inverted Microscope (Olympus). For immunofluorescent staining, brain slices were stained with anti-Map2 and glial fibrillary acidic protein antibodies, followed by incubation with secondary antibodies conjugated with Alexa Fluor 488. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Images were acquired using a confocal microscope (Olympus FV3000). For transmission electron microscopy, mice were fixed with 2% paraformaldehyde and 2% glutaraldehyde, and hippocampal regions were dissected for downstream processing. Random sections were obtained, and images at a ×5000 magnification were used to calculate the length and the width of the PSD with ImageJ software.
Northern blot
The brain, liver, kidney, and testis of Ftsj1 KO mice or WT littermates were collected, snap-frozen in liquid nitrogen, and stored at −80°C. Right half-brains were used for tRNA and aminoacyl-tRNA analysis, and left half-brains were used for ribosome profiling. Each frozen right half-brain was transferred into 5 ml of TRI Reagent (Molecular Research Center) and homogenized using TissueRuptor (QIAGEN), followed by centrifugation at 12,000g for 10 min at 4°C to remove debris. The brain lysate in TRI Reagent was split into two, one for conventional tRNA Northern blot and the other for aminoacyl-tRNA analysis by acidic Northern blot. For conventional tRNA Northern blot, RNA was extracted according to the manufacturer’s protocol. The liver, kidney, and testis were also homogenized in TRI Reagent, and RNA extraction was performed in the same way as brain. For brain lysate tRNA cleavage analysis, brain lysate was prepared as described in the ribosome profiling method section, and the lysate was incubated on ice for 10 min, followed by RNA extraction using TRI Reagent. For conventional Northern blot, total RNA was resolved using denaturing 7 M urea/tris-borate EDTA/PAGE, and Northern blot was performed using DNA oligo probes that were Digoxigenin (DIG)–labeled using DIG oligonucleotide 3′-end labeling kit second generation (Roche). Aminoacyl-tRNA analysis was performed essentially as described (52). Briefly, after the total RNA pellet was collected from tissue lysate in TRI Reagent by isopropanol precipitation, the RNA pellet was rinsed with 75% ethanol/10 mM Sodium acetate (pH 5.0)/1 mM EDTA and dissolved in 10 mM NaOAc (pH 5.0)/1 mM EDTA. Aminoacylation level was monitored using acidic PAGE/Northern blot, using the same DIG-labeled probes as conventional Northern blot. The sequences of oligo DNA probes are as follows: mouse/human tRNAPhe, 5′-CGAAACCCGGGATCGAACCAGGGACCTTTA;
mouse/human tRNALeu(CAA), 5′-TGTCAGAAGTGGGATTCGAACCCACGCCTC; mouse/human tRNALeu(UAA), 5′-TACCAGAAGTGGGGTTCGAACCCACGCGGA; mouse/human 5.8S rRNA, 5′-GCAAGTGCGTTCGAAGTGTCGATGATCAAT.
Ribosome profiling
To analyze ribosome-contained mRNA fragments, steps such as RNase I treatment, ribosome recovery, footprint fragment purification, 3′ adaptor ligation, rRNA depletion, reverse transcription, circularization, and PCR amplification were performed essentially as described (21), with some modifications as described below. Brain or liver lysis was performed by transferring each frozen left half-brain or liver into polysome buffer (20) and homogenizing using TissueRuptor (QIAGEN). For 3′ adaptor ligation, air-adenylated linker A (BIOO, 5rApp/CTGTAGGCACCATCAAT/3ddC/) was used. cDNA libraries of ribosome-contained mRNA fragments were initially sequenced using MiSeq to perform A site codon analysis using Galaxy (53) and RiboGalaxy (54): First, fastq files were filtered by quality, the 3′ adaptor sequence (CTGTAGGCACCATCAAT) was removed, and rRNA reads were removed by Bowtie2. Ribosome-contained mRNA fragment sequences were acquired by aligning reads to mouse RefSeq mRNA sequences and were analyzed using triplet periodicity and metagene analysis programs in RiboGalaxy, to determine the appropriate footprint length and codon frames. For large-scale ribosome-contained mRNA fragment analysis, cDNA libraries were subjected to Illumina HiSeq2500 sequencing. Using Galaxy, fastq files were filtered by quality, the adaptor sequence (CTGTAGGCACCATCAAT) was removed, and mRNA reads were retrieved by aligning to mouse RefSeq mRNAs. mRNA reads were aligned, counted, and analyzed using HISAT2, HTSeq-count, and DESeq2. Gene ontology analysis was performed using Gene Set Enrichment Analysis (GSEA) website software (55).
Transcriptome analysis
To collect brain or liver total RNA, immediately following ribosome profiling tissue lysis, a fraction of tissue lysate was mixed with TRI Reagent, and total RNA was extracted. Polyadenylate [poly(A)] + RNA was collected from total RNA using the oligotex-dT30 Super mRNA Purification Kit (TaKaRa). Poly(A) + RNA was fragmented and collected by incubation in alkaline buffer (88 mM NaHCO3, 12 mM Na2CO3, and 1 mM EDTA) at 95°C for 45 min, followed by ethanol precipitation. Poly(A) + RNA fragments were resolved by denaturing urea PAGE, and fragments of 20 to 40 nt were gel-excised and extracted. 3′ adaptor ligation, library preparation, and sequencing were performed in the same way as ribosome profiling. Using Galaxy (53), fastq files were filtered by quality, 3′ adaptor sequence (CTGTAGGCACCATCAAT) was removed, and mRNA reads were retrieved by aligning to mouse RefSeq mRNAs. mRNA reads were aligned, counted, and analyzed using HISAT2, HTSeq-count, and DESeq2.
tRNA and tRNA fragment (tRF) sequencing
Total tRNA, ribosome-contained tRNA, and tRF sequencing analyses were performed essentially as described (20). For total tRNA and tRF preparation, total RNA was first prepared from brain lysate (also used for ribosome profiling and transcriptome analysis) using TRI Reagent. Total RNA was resolved by denaturing urea PAGE, and tRNA (60 to 100 nt) and tRF (20 to 40 nt) were collected by gel excision. To collect ribosome-contained tRNA, during gel excision of mRNA fragments (20 to 40 nt) in ribosome profiling, tRNA fragments (60 to 100 nt) were also collected. Total tRNA, ribosome-contained tRNA, and tRF were demethylated using Escherichia coli–derived Alpha-ketoglutarate-dependent dioxygenase (AlkB) to enable reverse transcription (56). Briefly, tRNA or tRF were incubated in 45 mM tris-HCl (pH 8), 0.9 mM α-ketoglutaric acid, 1.8 mM ascorbic acid, 67 μM (NH4)2Fe(SO4)2, and 2.5 μM AlkB, at 37°C for 2 hours. Demethylation was confirmed by performing yeast tRNA demethylation and RNA nucleoside mass spectrometry analysis, as a positive control. Subsequently, library preparation was performed in the same way as mRNA library preparation. After Illumina MiSeq sequencing, tRNA and tRF analysis was performed using Galaxy. Briefly, fastq files were converted to fasta files, 12 nt of adaptor sequence (GGCACCATCAAT) was clipped (leaving 5 nt of 3′ adaptor sequence CTGTA at the tRNA 3′ end to allow mapping of short reads), and tRNA reads were Bowtie2-aligned to mouse tRNA sequences in the Genomic tRNA database (57). Reads were counted using alignment position information in the SAM file and count function in Galaxy.
Electrophysiology
WT and Ftsj1 KO mice at 8 weeks old were used for electrophysiological experiments. Briefly, mice were quickly euthanized by cervical dislocation, and mouse hippocampal slices were acutely prepared at 400 μm in thickness using a vibratome (Leica). Slices were incubated in artificial cerebrospinal fluid (CSF; 123 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, and 26 mM NaHCO3) for 60 min with consistent aeration. A glass micropipette that was filled with artificial CSF was placed on the CA1 region to record the pEPSPs, and a stimulating electrode was placed on the Schaffer collateral fibers. The paired-pulse ratio of the pEPSP response was calculated as the ratio of the second pEPSP to the first pEPSP stimulated at a 50-ms interpulse interval. LTP was induced by TBS consist of five bursts of five pulses with 10-ms intervals, delivered at 200-ms intervals. LTD was induced by LFS that consists of 900-pulse stimulation at 1-Hz frequency.
Behavioral tests
WT and Ftsj1 KO mice were used for a battery of behavioral tests by an experimenter who was blinded to the genotype. Mice at 7 weeks old were handled for 1 week. Mice were then subjected to open-field test, elevated plus maze, Barnes maze, and fear conditioning test with 1-week interval between each test. For open-field test, a 60 cm–by–60 cm–by–60 cm chamber with gray color was used. Mice were placed in the center of the chamber, and the exploring behavior was recorded for 10 min using a video camera above the chamber. Video software (Actimetrics) was used to define a six-by-six grid, and the center two-by-two grid was defined as the center area. Total distance moved in the entire area of chamber, distance moved in the center area of chamber, and time spent in the center area of chamber were analyzed.
Elevated plus maze test was performed as described previously (58). The elevated plus maze used in this study consisted of four arms (two open arms and two arms closed by 10-cm-high walls), 30 cm in length and 6 cm in width. The maze was elevated 50 cm off of the floor using metal legs. Mice were placed at the center of the maze with the face toward open arm opposite to the experimenter. The movement of mice was recorded for 5 min. Total distance moved in both open and closed arms, distance moved in the open arm, and percentage of time spend in the open arm were analyzed using Actimetrics software.
The Barnes maze analysis was performed as described with modifications (59). The Barnes maze consisted of a circular platform (91 cm in diameter) with 20 equally spaced holes (5 cm in diameter) and was elevated 100 cm above the floor. A black box was located under a hole of the platform so the mouse could escape from the open platform surface. Visual cues (triangle, rectangle, circle, and cross) were placed surrounding the maze. On the trial day, mice were place in a dark start chamber in the middle of the platform. After 10 s, the start chamber was lifted, and the movement of each mouse was recorded for 3 min. The trial ended when the mouse entered the goal in 3 min. When the mouse could not find the black box in 3 min, the trial was then forced to end by gently guiding the mouse to the black box. Mice received four trials per day with an intertrial interval of 20 min. Number of pokes to any hole before entering the black box was considered as an error, and the number of errors was used to assess spatial learning ability (Actimetrics).
Fear conditioning test was performed as described previously with modifications (60). For the conditioning of contextual and cued fear memory, mice were placed in the test box and received three identical trials. A trial consists of 120-s habituation period, followed by an auditory cue at 80 dB for 30 s in combination with a mild foot shock (0.5 mA) during the last 2 s of auditory presentation. Next day, mice were placed in the same test box and were monitored for 6 min without auditory presentation. Mice were then removed from the test chamber and placed in their home cage for 60 min before cue testing. Cue testing was performed in a new test box with the floor being changed to plastic board and the ceiling of the test box being changed to triangle. Mice were allowed to freely explore the box for 3 min, followed by auditory presentation for 3 min at 80 dB without foot shock. Freezing behavior was examined using software (Actimetrics) following the instruction.
Quantification and statistical analysis
Data were analyzed using GraphPad Prism 8 software. Unpaired Student’s t tests or Mann-Whitney tests were used to assess differences between two groups, and two-way analysis of variance (ANOVA) tests followed by Tukey’s multiple comparisons were used to examine differences among multiple groups. A two-tailed P value of 0.05 was considered significant unless specified. Data are presented as means ± SEM.
Acknowledgments
We thank N. Maeda for technical assistance. Funding: This work was supported by JSPS KAKENHI grants 18H02599 (to F.-Y.W.), 18K19521 (to F.-Y.W.), 18H02865 (to K.T.), 17905074 (to K.T.), and 18959602 (to K.T.); JST ERATO JPMJER2002 (to F.-Y.W. and Tsutomu S.) and 20H03187 (to T.C.); SAKIGAKE JPMJPR1532 (to F.-Y.W.); the Takeda Science Foundation (to K.T. and F.-Y.W.); the Uehara Memorial Foundation (to F.-Y.W.); the Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP, Japan (to Y.G.); and the RIKEN Aging Project (to M.T.). Research in the laboratory of A.C.N. is supported by the NIH (AG047270, AG062306, AG066508, and DA018343) and the State of Connecticut Department of Mental Health and Addiction Services. Author contributions: F.-Y.W., K.T., Tsutomu S., and A.C.N. contributed to the overall conceptualization, design, and management of the study and analyzed the data. Y.N. performed the morphological and behavioral examination. T.C., C.-W.C., Y.I., and M.Tanaka performed the tRNA-seq, RNA-seq, and ribosome profiling and analyzed the data. S.H., M.Takakura, K.M., and Takeo S. performed the mass spectrometry, in vitro methylation assay, and binding assay and analyzed the data. H.N. and K.N. performed electrophysiological experiments and analyzed the data. B.C.C. and R.R.K. analyzed the ribosome profiling data. F.K. and M.Y. performed the RNA-seq and analyzed the data. Y.G. established and provided patient-derived cell line. F.-Y.W., A.C.N., T.C., and T.K. wrote the paper. Competing interests: C.-W.C. and M.Tanaka. have a pending patent application (JP2017-236458) issued by RIKEN. The authors declare that they have no other competing interests. Data and materials availability: RNA-seq data from this work are available under accession number GSE153758. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All related preliminary experimental data are available from the corresponding author upon request. The genomic DNA and/or Epstein-Barr virus–transformed lymphoblast cells of the patient and his parents can be provided by NCNP Biobank in Japan pending scientific review and a completed material transfer agreement. Requests for the research use should be submitted to biobank@ncnp.go.jp. The Ftsj1 KO mice can be provided by Kumamoto University pending scientific review and a completed material transfer agreement. Requests for the mice should be submitted to K.T. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabf3072/DC1
REFERENCES AND NOTES
- 1.El Yacoubi B., Bailly M., de Crecy-Lagard V., Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Boccaletto P., Machnicka M. A., Purta E., Piatkowski P., Baginski B., Wirecki T. K., de Crécy-Lagard V., Ross R., Limbach P. A., Kotter A., Helm M., Bujnicki J. M., MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Crécy-Lagard V., Boccaletto P., Mangleburg C. G., Sharma P., Lowe T. M., Leidel S. A., Bujnicki J. M., Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 47, 2143–2159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musante L., Ropers H. H., Genetics of recessive cognitive disorders. Trends Genet. 30, 32–39 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Vissers L. E. L. M., Gilissen C., Veltman J. A., Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Neri G., Schwartz C. E., Lubs H. A., Stevenson R. E., X-linked intellectual disability update 2017. Am. J. Med. Genet. A 176, 1375–1388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson R. E., Schwartz C. E., X-linked intellectual disability: Unique vulnerability of the male genome. Dev. Disabil. Res. Rev. 15, 361–368 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Lubs H. A., Stevenson R. E., Schwartz C. E., Fragile X and X-linked intellectual disability: Four decades of discovery. Am. J. Hum. Genet. 90, 579–590 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freude K., Hoffmann K., Jensen L.-R., Delatycki M. B., des Portes V., Moser B., Hamel B., van Bokhoven H., Moraine C., Fryns J.-P., Chelly J., Gécz J., Lenzner S., Kalscheuer V. M., Ropers H.-H., Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine–binding protein cause nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 75, 305–309 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong P., Li J., Dai L., Zhang K., Zheng Z., Gao X., Zhang F., Genetic variations in FTSJ1 influence cognitive ability in young males in the Chinese Han population. J. Neurogenet. 22, 277–287 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Ramser J., Winnepenninckx B., Lenski C., Errijgers V., Platzer M., Schwartz C. E., Meindl A., Kooy R. F., A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9). J. Med. Genet. 41, 679–683 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takano K., Nakagawa E., Inoue K., Kamada F., Kure S., Goto Y.-i.; Japanese Mental Retardation Consortium , A loss-of-function mutation in the FTSJ1 gene causes nonsyndromic X-linked mental retardation in a Japanese family. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 479–484 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Pintard L., Lecointe F., Bujnicki J. M., Bonnerot C., Grosjean H., Lapeyre B., Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J. 21, 1811–1820 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guy M. P., Phizicky E. M., Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes. RNA 21, 61–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guy M. P., Podyma B. M., Preston M. A., Shaheen H. H., Krivos K. L., Limbach P. A., Hopper A. K., Phizicky E. M., Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 18, 1921–1933 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guy M. P., Shaw M., Weiner C. L., Hobson L., Stark Z., Rose K., Kalscheuer V. M., Gecz J., Phizicky E. M., Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum. Mutat. 36, 1176–1187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J., Wang Y.-N., Xu B.-S., Liu Y.-P., Zhou M., Long T., Li H., Dong H., Nie Y., Chen P. R., Wang E.-D., Liu R.-J., Intellectual disability-associated gene ftsj1 is responsible for 2′-O-methylation of specific tRNAs. EMBO Rep. 21, e50095 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angelova M. T., Dimitrova D. G., Silva B. D., Marchand V., Jacquier C., Achour C., Brazane M., Goyenvalle C., Bourguignon-Igel V., Shehzada S., Khouider S., Lence T., Guerineau V., Roignant J.-Y., Antoniewski C., Teysset L., Bregeon D., Motorin Y., Schaefer M. R., Carré C., tRNA 2′-O-methylation by a duo of TRM7/FTSJ1 proteins modulates small RNA silencing in Drosophila. Nucleic Acids Res. 48, 2050–2072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen L. R., Garrett L., Hölter S. M., Rathkolb B., Rácz I., Adler T., Prehn C., Hans W., Rozman J., Becker L., Aguilar-Pimentel J. A., Puk O., Moreth K., Dopatka M., Walther D. J., von Bohlen und Halbach V., Rath M., Delatycki M., Bert B., Fink H., Blümlein K., Ralser M., van Dijck A., Kooy F., Stark Z., Müller S., Scherthan H., Gecz J., Wurst W., Wolf E., Zimmer A., Klingenspor M., Graw J., Klopstock T., Busch D., Adamski J., Fuchs H., Gailus-Durner V., de Angelis M. H., von Bohlen und Halbach O., Ropers H. H., Kuss A. W., A mouse model for intellectual disability caused by mutations in the X-linked 2′-O-methyltransferase Ftsj1 gene. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2083–2093 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Chen C. W., Tanaka M., Genome-wide translation profiling by ribosome-bound tRNA capture. Cell Rep. 23, 608–621 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Ingolia N. T., Brar G. A., Rouskin S., McGeachy A. M., Weissman J. S., The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han L., Guy M. P., Kon Y., Phizicky E. M., Lack of 2′-O-methylation in the tRNA anticodon loop of two phylogenetically distant yeast species activates the general amino acid control pathway. PLOS Genet. 14, e1007288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chenn A., Walsh C. A., Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Rosso S. B., Sussman D., Wynshaw-Boris A., Salinas P. C., Wnt signaling through dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Wang Y., Zeng C., Li J., Zhou Z., Ju X., Xia S., Li Y., Liu A., Teng H., Zhang K., Shi L., Bi C., Xie W., He X., Jia Z., Jiang Y., Cai T., Wu J., Xia K., Sun Z. S., PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell Rep. 24, 2029–2041 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Dong F., Jiang J., McSweeney C., Zou D., Liu L., Mao Y., Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum. Mol. Genet. 25, 2738–2751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuechler A., Willemsen M. H., Albrecht B., Bacino C. A., Bartholomew D. W., van Bokhoven H., van den Boogaard M. J. H., Bramswig N., Büttner C., Cremer K., Czeschik J. C., Engels H., van Gassen K., Graf E., van Haelst M., He W., Hogue J. S., Kempers M., Koolen D., Monroe G., de Munnik S., Pastore M., Reis A., Reuter M. S., Tegay D. H., Veltman J., Visser G., van Hasselt P., Smeets E. E. J., Vissers L., Wieland T., Wissink W., Yntema H., Zink A. M., Strom T. M., Lüdecke H.-J., Kleefstra T., Wieczorek D., De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 134, 97–109 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Montag-Sallaz M., Montag D., Severe cognitive and motor coordination deficits in tenascin-R-deficient mice. Genes Brain Behav. 2, 20–31 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Nithianantharajah J., Komiyama N. H., McKechanie A., Johnstone M., Blackwood D. H., St Clair D., Emes R. D., van de Lagemaat L. N., Saksida L. M., Bussey T. J., Grant S. G. N., Synaptic scaffold evolution generated components of vertebrate cognitive complexity. Nat. Neurosci. 16, 16–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rochefort N. L., Konnerth A., Dendritic spines: From structure to in vivo function. EMBO Rep. 13, 699–708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beutler L. R., Eldred K. C., Quintana A., Keene C. D., Rose S. E., Postupna N., Montine T. J., Palmiter R. D., Severely impaired learning and altered neuronal morphology in mice lacking NMDA receptors in medium spiny neurons. PLOS ONE 6, e28168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ikegami M., Uemura T., Kishioka A., Sakimura K., Mishina M., Striatal dopamine D1 receptor is essential for contextual fear conditioning. Sci. Rep. 4, 3976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki T., Yashiro Y., Kikuchi I., Ishigami Y., Saito H., Matsuzawa I., Okada S., Mito M., Iwasaki S., Ma D., Zhao X., Asano K., Lin H., Kirino Y., Sakaguchi Y., Suzuki T., Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 11, 4269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos J., Fu D., The emerging impact of tRNA modifications in the brain and nervous system. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 412–428 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Arrondel C., Missoury S., Snoek R., Patat J., Menara G., Collinet B., Liger D., Durand D., Gribouval O., Boyer O., Buscara L., Martin G., Machuca E., Nevo F., Lescop E., Braun D. A., Boschat A.-C., Sanquer S., Guerrera I. C., Revy P., Parisot M., Masson C., Boddaert N., Charbit M., Decramer S., Novo R., Macher M.-A., Ranchin B., Bacchetta J., Laurent A., Collardeau-Frachon S., van Eerde A. M., Hildebrandt F., Magen D., Antignac C., van Tilbeurgh H., Mollet G., Defects in t6A tRNA modification due to GON7 and YRDC mutations lead to Galloway-Mowat syndrome. Nat. Commun. 10, 3967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vitali P., Kiss T., Cooperative 2′-O-methylation of the wobble cytidine of human elongator tRNAMet(CAT) by a nucleolar and a Cajal body-specific box C/D RNP. Genes Dev. 33, 741–746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abbasi-Moheb L., Merte S., Gonsior M., Nouri-Vahid L., Kahrizi K., Cirak S., Wieczorek D., Motazacker M. M., Esmaeeli-Nieh S., Cremer K., Weißmann R., Tzschach A., Garshasbi M., Abedini S. S., Najmabadi H., Ropers H. H., Sigrist S. J., Kuss A. W., Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 90, 847–855 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuorto F., Liebers R., Musch T., Schaefer M., Hofmann S., Kellner S., Frye M., Helm M., Stoecklin G., Lyko F., RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 19, 900–905 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Yang X., Yang Y., Sun B.-F., Chen Y.-S., Xu J.-W., Lai W.-Y., Li A., Wang X., Bhattarai D. P., Xiao W., Sun H.-Y., Zhu Q., Ma H.-L., Adhikari S., Sun M., Hao Y.-J., Zhang B., Huang C.-M., Huang N., Jiang G.-B., Zhao Y.-L., Wang H.-L., Sun Y.-P., Yang Y.-G., 5-Methylcytosine promotes mRNA export — NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 27, 606–625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanco S., Dietmann S., Flores J. V., Hussain S., Kutter C., Humphreys P., Lukk M., Lombard P., Treps L., Popis M., Kellner S., Hölter S. M., Garrett L., Wurst W., Becker L., Klopstock T., Fuchs H., Gailus-Durner V., Hrabĕ de Angelis M., Káradóttir R. T., Helm M., Ule J., Gleeson J. G., Odom D. T., Frye M., Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 33, 2020–2039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono H., Ogasawara O., Okubo K., Bono H., RefEx, a reference gene expression dataset as a web tool for the functional analysis of genes. Sci. Data 4, 170105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas S. P., Hoang T. T., Ressler V. T., Raines R. T., Human angiogenin is a potent cytotoxin in the absence of ribonuclease inhibitor. RNA 24, 1018–1027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas S. P., Kim E., Kim J. S., Raines R. T., Knockout of the ribonuclease inhibitor gene leaves human cells vulnerable to secretory ribonucleases. Biochemistry 55, 6359–6362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen J. S., Srivastava S., Farwell K. D., Lu H. M., Zeng W., Lu H., Chao E. C., Fatemi A., ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am. J. Med. Genet. A 167, 1391–1395 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Konevega A. L., Soboleva N. G., Makhno V. I., Semenkov Y. P., Wintermeyer W., Rodnina M. V., Katunin V. I., Purine bases at position 37 of tRNA stabilize codon–anticodon interaction in the ribosomal A site by stacking and Mg2+-dependent interactions. RNA 10, 90–101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou H. J., Donnard E., Gustafsson H. T., Garber M., Rando O. J., Transcriptome-wide analysis of roles for tRNA modifications in translational regulation. Mol. Cell 68, 978–992.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Janak P. H., Tye K. M., From circuits to behaviour in the amygdala. Nature 517, 284–292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sweatt J. D., Neural plasticity and behavior—Sixty years of conceptual advances. J. Neurochem. 139 ( suppl. 2), 179–199 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Miyauchi K., Ohara T., Suzuki T., Automated parallel isolation of multiple species of non-coding RNAs by the reciprocal circulating chromatography method. Nucleic Acids Res. 35, e24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takakura M., Ishiguro K., Akichika S., Miyauchi K., Suzuki T., Biogenesis and functions of aminocarboxypropyluridine in tRNA. Nat. Commun. 10, 5542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reed S. E., Staley E. M., Mayginnes J. P., Pintel D. J., Tullis G. E., Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J. Virol. Methods 138, 85–98 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Janssen B. D., Diner E. J., Hayes C. S., Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Methods Mol. Biol. 905, 291–309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., Blankenberg D., The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michel A. M., Mullan J. P., Velayudhan V., O’Connor P. B., Donohue C. A., Baranov P. V., RiboGalaxy: A browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 13, 316–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng G., Qin Y., Clark W. C., Dai Q., Yi C., He C., Lambowitz A. M., Pan T., Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan P. P., Lowe T. M., GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walf A. A., Frye C. A., The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2, 322–328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pitts M. W., Barnes maze procedure for spatial learning and memory in mice. Bio Protoc. 8, e2744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shoji H., Takao K., Hattori S., Miyakawa T., Contextual and cued fear conditioning test using a video analyzing system in mice. J. Vis. Exp. 85, 50871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabf3072/DC1