

Abstract
Summary
We launch a webserver for RNA structure prediction and design corresponding to tools developed using our RNA-As-Graphs (RAG) approach. RAG uses coarse-grained tree graphs to represent RNA secondary structure, allowing the application of graph theory to analyze and advance RNA structure discovery. Our webserver consists of three modules: (a) RAG Sampler: samples tree graph topologies from an RNA secondary structure to predict corresponding tertiary topologies, (b) RAG Builder: builds three-dimensional atomic models from candidate graphs generated by RAG Sampler, and (c) RAG Designer: designs sequences that fold onto novel RNA motifs (described by tree graph topologies). Results analyses are performed for further assessment/selection. The Results page provides links to download results and indicates possible errors encountered. RAG-Web offers a user-friendly interface to utilize our RAG software suite to predict and design RNA structures and sequences.
Availability and implementation
The webserver is freely available online at: http://www.biomath.nyu.edu/ragtop/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Mathematical and computational methods for RNA structure prediction and design provide an opportunity to gain insight into RNA structure/function relationships by allowing systematic exploration of relevant variables and features. Our lab’s RNA-As-Graphs (RAG) approach represents RNA secondary [2-dimensional (2D)] structures as undirected tree graphs; unpaired loops are represented by vertices and base-paired helices by edges (Gan et al., 2003). This coarse-grained approach reduces 2D structure complexity using graph theory and offers new tools to study RNA structure. Our graph-based structure prediction protocol has shown to be effective (Jain and Schlick, 2017), and experimental testing by chemical mapping has shown the utility of our design approach (Jain et al., 2018).
Here, we present a webserver for RNA structure prediction and design corresponding to our graph-based standalone tools. Three modules are available: RAG Sampler uses our RAG Topology Prediction (RAGTOP) tool to generate candidate tertiary [3-dimensional (3D)] graph topologies from RNA 2D structures (Bayrak et al., 2017; Kim et al., 2014); RAG Builder uses our Fragment Assembly for RAG (F-RAG) (Jain and Schlick, 2017) tool to build 3D atomic models from candidate graphs (generated by RAG Sampler) by assembling atomic fragments of subgraphs, using our RAG-3D database (Zahran et al., 2015); RAG Designer uses F-RAG (Jain et al., 2018) to produce sequences that fold onto target tree graph topologies. These graph-based tools offer more flexibility and modularity than sequence-based approaches. The RAG computational pipeline designs sequences to fold onto a target topology, rather than a specific 2D structure. Our webserver offers a user-friendly interface for utilizing the RAG software suite to predict and design RNA structures and sequences.
2 Webserver features
Figure 1 shows the input/output and the different components for the three modules. For each module, the user provides the input files, runs our programs with either default or specified parameters and obtains notifications via email (optional). After the run is finished, the Results page is updated with results and analyses. Results are available for download, and any error or failure encountered is recorded. Below we briefly describe each module. See Supplementary Material for details and usage guidelines.
Fig. 1.
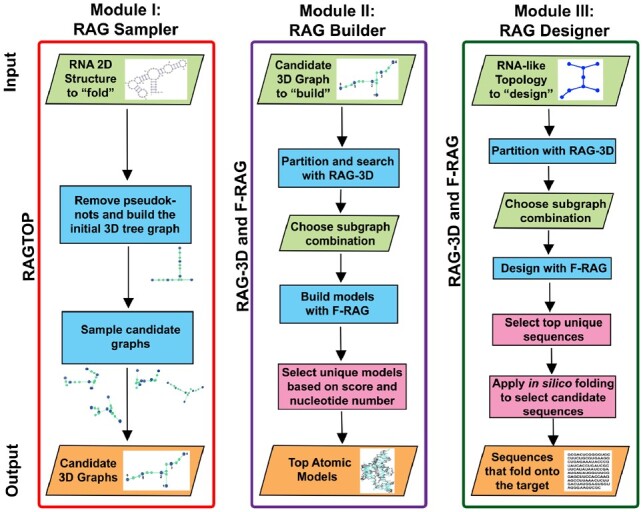
Flowchart for the three webserver modules. RAG Sampler takes RNA sequence and 2D structure as input, and generates corresponding candidate tree graph topologies. RAG Builder takes RNA sequence, 2D structure and target tree graph topology (generated by RAG Sampler) as input and builds corresponding atomic models. RAG Designer takes a target tree graph topology as input, and designs sequences that fold onto target topologies. The input and output information for the three modules is listed in the first and the last rows, respectively. Steps that involve running the three standalone tools (RAGTOP, RAG-3D, and F-RAG) and subsequent result analysis is indicated in rectangular boxes. Parallelograms are used for steps where user input is required during processing
2.1 Module I: RAG Sampler
RAG Sampler takes RNA sequence and 2D structure as input, and uses RAGTOP (Bayrak et al., 2017; Kim et al., 2014) to generate corresponding candidate tree graph topologies. It starts by predicting the helical arrangements for RNA junctions in the given 2D structure by our random forest machine learning approach (Laing et al., 2012, 2013), and converting the 2D tree graph to a scaled 3D tree graph [as described in Kim et al. (2014)]. The bend and twist angles around internal loops are then sampled with Monte Carlo/Simulated Annealing to generate candidate 3D graph topologies, and scored using a knowledge-based potential function generated from known RNA structures. The accepted 3D tree graphs are analyzed, and the user obtains a link to automatically feed the best (lowest) scoring tree graph as the target to our RAG Builder module (next).
2.2 Module II: RAG Builder
RAG Builder takes RNA sequence, 2D structure and target tree graph topology (generated by RAG Sampler) as input and uses RAG-3D (Zahran et al., 2015) and F-RAG (Jain and Schlick, 2017) to build corresponding atomic models. The target graph is partitioned into subgraphs, and the best matching atomic fragments are obtained by RAG-3D’s partitioning and search utilities (from our RAG-3D database of subgraphs and corresponding atomic fragments). A combination of two subgraphs that form the full graph is selected from among the various combinations as target for fragment assembly. Atomic fragments corresponding to target subgraphs are assembled by F-RAG using common nucleotides/vertices to generate full models. The length and sequence of the atomic models are adjusted according to the target 2D structure, and the models are scored based on RAGTOP’s knowledge-based potential. The resulting models are screened based on their score and nucleotide number to produce a ranked list of best models. F-RAG has previously generated satisfactory models for the majority of the tested RNA structures as compared to two similar structure prediction programs (Jain and Schlick, 2017).
2.3 Module III: RAG Designer
RAG Designer takes a target tree graph topology as input, and uses RAG-3D (Zahran et al., 2015) and F-RAG (Jain et al., 2018) to design sequences that fold onto the target topology. Similar to RAG Builder, the target topology is partitioned into subgraphs, a subgraph combination is selected and all atomic fragments corresponding to each subgraph ID (from our RAG-3D database) are assembled by F-RAG. The sequences of the fragments are held fixed for the design of novel RNAs. The resulting unique sequences are ranked based on their scores, and top sequences are screened using two 2D structure prediction programs, RNAfold (Lorenz et al., 2011) and NUPACK (Dirks et al., 2007). The sequences that fold onto the target graph topology (the 2D structures can differ) with both in silico programs are generated as successful sequences. We have previously used F-RAG to successfully design sequences to fold onto six RNA-like topologies (Jain et al., 2018). We have also developed an automated approach for mutation to improve the design yield (Jain et al., submitted for publication).
2.4 Limitations
At present, we handle RNAs with no more than 200 nucleotides and tree graph topologies with no more than 13 vertices. Two subgraphs are used for fragment assembly. Energy minimization for RAG Builder and RAG Designer are not presently available. See Supplementary Section S3 and original prediction and design papers.
3 Conclusion
We report a webserver for RNA structure prediction and design using our standalone tools for graph sampling, search and partitioning and fragment assembly (RAGTOP, RAG-3D and F-RAG). The components are integrated, and each module provides the user with analysis tools. We hope that RAG-Web will aid users in RNA structure prediction and design by complementing coarse-grained or atomic modeling studies by RAG-based approaches.
Funding
This work was supported by the National Institute of General Medical Sciences, National Institutes of Health (NIH) [R35GM122562 to T.S.]. Research described in this article was supported (in part) by Philip Morris USA Inc. and Philip Morris International to T.S.
Conflict of Interest: None declared.
Supplementary Material
Contributor Information
Grace Meng, Department of Chemistry, New York University, New York, NY 10003, USA.
Marva Tariq, Department of Chemistry, Smith College, Northampton, MA 01063, USA.
Swati Jain, Department of Chemistry, New York University, New York, NY 10003, USA.
Shereef Elmetwaly, Department of Chemistry, New York University, New York, NY 10003, USA.
Tamar Schlick, Department of Chemistry, New York University, New York, NY 10003, USA; Courant Institute of Mathematical Sciences, New York University, New York, NY 10012, USA; NYU-ECNU Center for Computational Chemistry at New York University Shanghai, Shanghai 3663, China.
References
- Bayrak C. et al. (2017) Using sequence signatures and kink-turn motifs in knowledge-based statistical potentials for RNA structure prediction. Nucleic Acid Res., 45, 5414–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks R. et al. (2007) Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev., 49, 65–88. [Google Scholar]
- Gan H. et al. (2003) Exploring the repertoire of RNA secondary motifs using graph theory; implications for RNA design. Nucleic Acid Res., 31, 2926–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Schlick T. (2017) F-RAG: generating atomic models from RNA graphs using fragment assembly. J. Mol. Biol., 429, 3587–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain et al. , submitted. Inverse folding with RNA-As-Graphs produces a large pool of candidate sequences with target topologies, submitted. [DOI] [PMC free article] [PubMed]
- Jain S. et al. (2018) A pipeline for computational design of novel RNA-like topologies. Nucleic Acid Res., 46, 7040–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N. et al. (2014) Graph-based sampling for approximating global helical topologies of RNA. Proc. Natl. Acad. Sci. USA, 111, 4079–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing C. et al. (2012) Predicting coaxial helical stacking in RNA junctions. Nucleic Acid Res., 40, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing C. et al. (2013) Predicting helical topologies in RNA junctions as tree graphs. PLoS One, 8, e71947.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R. et al. (2011) ViennaRNA package 2.0. Algorith. Mol. Biol., 6, 26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahran M. et al. (2015) RAG-3D: a search tool for RNA 3D substructures. Nucleic Acid Res., 43, 9474–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.